

Abstract
A new formulation of the Reaction Ensemble Monte Carlo technique (RxMC) combined with the Continuous Fractional Component Monte Carlo method is presented. This method is denoted by serial Rx/CFC. The key ingredient is that fractional molecules of either reactants or reaction products are present and that chemical reactions always involve fractional molecules. Serial Rx/CFC has the following advantages compared to other approaches: (1) One directly obtains chemical potentials of all reactants and reaction products. Obtained chemical potentials can be used directly as an independent check to ensure that chemical equilibrium is achieved. (2) Independent biasing is applied to the fractional molecules of reactants and reaction products. Therefore, the efficiency of the algorithm is significantly increased, compared to the other approaches. (3) Changes in the maximum scaling parameter of intermolecular interactions can be chosen differently for reactants and reaction products. (4) The number of fractional molecules is reduced. As a proof of principle, our method is tested for Lennard-Jones systems at various pressures and for various chemical reactions. Excellent agreement was found both for average densities and equilibrium mixture compositions computed using serial Rx/CFC, RxMC/CFCMC previously introduced by Rosch and Maginn (Journal of Chemical Theory and Computation, 2011, 7, 269–279), and the conventional RxMC approach. The serial Rx/CFC approach is also tested for the reaction of ammonia synthesis at various temperatures and pressures. Excellent agreement was found between results obtained from serial Rx/CFC, experimental results from literature, and thermodynamic modeling using the Peng–Robinson equation of state. The efficiency of reaction trial moves is improved by a factor of 2 to 3 (depending on the system) compared to the RxMC/CFCMC formulation by Rosch and Maginn.
1. Introduction
Substantial efforts have been made by scientists and engineers to study chemical reactions in nonideal environments.1−3 An optimal design and operation of many chemical processes relies on accurate information regarding reaction equilibria.4 Reaction equilibria vary as the operating conditions of a reactor change. As a result, an approach is required which can provide information regarding chemical equilibria for a wide range of operating conditions. In an ideal gas, chemical equilibria are determined by the difference between the standard Gibbs free energies of formation of reactants and reaction products.5 Due to interactions of the reacting molecules with surrounding molecules, the chemical equilibrium may significantly differ from the ideal gas situation as the medium formed by the surrounding molecules may stabilize reactant and reaction product molecules differently.4 It is not always possible to measure reaction equilibria experimentally. The main reasons for this are as follows: (1) Extreme conditions may not be accessible experimentally. (2) Kinetic limitation may prohibit reaching chemical equilibrium on accessible time scales. (3) Large-scale experimental screening of solvents for chemical reactions may not be feasible. Therefore, there is a demand for theoretical methods that can accurately predict reaction equilibria. Molecular simulation is a natural tool for this as interactions between atoms and molecules are explicitly taken into account. One can perform Molecular Dynamics (MD) with a force field that can handle chemical reactions, e.g., DFT-based,6 Car–Parrinello,7,8 or ReaxFf based MD.9,10 The main disadvantage of these approaches is that reactions may not occur within the limited time scale of MD simulations. Therefore, advanced simulation techniques such as metadynamics11−13 or transition path sampling14−21 may be required. These types of simulation techniques are not considered further in this paper. One of the most commonly used approaches in molecular simulation is to simulate the reaction equilibria in the reaction ensemble (RxMC).1,4,22−27 In this approach, the chemical reaction is carried out by a Monte Carlo (MC) trial move. Beside thermalization (translation, rotation, etc.), trial moves are carried out in which reactants are removed and reaction products are inserted in the system, in such a way that an equilibrium distribution of reactants and reaction products is obtained. The mechanism and the transition state of the reaction are not considered as this approach is purely thermodynamic and reaction kinetics are not considered. As a result, the efficiency of this simulation technique is not affected by the height of the activation energy barrier of the reaction as reaction kinetics are not considered. The RxMC method requires the ideal gas partition functions of all reactant and reaction product molecules, a list of all possible chemical reactions in the system, and an appropriate force field accurately describing interactions between molecules.4 The ideal gas partition function can be obtained from Quantum Mechanics5,26,28 or standard thermochemical tables, e.g., the JANAF tables.29 Computing ideal gas partition functions using quantum packages is well established.28 However, due to lack of experimental data, it is not always straightforward to compute partition functions from QM software, especially for large molecules or ions.30,31 For a detailed review of RxMC techniques, the reader is referred to ref (4).
Just like many other ensembles that rely on sufficient molecule insertions/removals (e.g., the Gibbs ensemble and grand-canonical ensemble),32 RxMC struggles when insertions/removals of molecules are difficult, e.g., at low temperatures and high densities. During the past few years, significant progress has been made in Monte Carlo techniques for the insertion and deletion of molecules. There are two types of solutions to overcome low acceptance probabilities for molecule insertions/removals in RxMC: methods such as Configurational-Bias Monte Carlo (CBMC) or related methods26,27,33−39 that try to insert whole molecules in a single Monte Carlo trial move and methods based on the idea of expanded ensembles40−42 such as the Continuous Fractional Component Monte Carlo (CFCMC) method first developed by Shi and Maginn.43,44 The main advantage of the latter approach is that instead of inserting whole molecules in a single trial move, molecules are inserted gradually, so that the surrounding can easily adapt to the inserted/deleted molecules. This is particularly important at high densities.45 Therefore, CFCMC does not rely on the spontaneous occurrence of cavities in the system that are large enough to accommodate a large molecule. The CFCMC technique is frequently used for simulations that suffer from low acceptance probability of molecule insertions/removals.1,45−55 Applications of this approach include computation of the loading and enthalpy of adsorption of guest molecules in porous materials near the saturation loading,38,45 reaction equilibria of complex systems,1 and solubilities of small molecules in ionic liquids.43,44,46,47,56−59 For more details on the challenges of Monte Carlo simulations in open ensembles, the reader is referred to refs (60−62). The combination of CFCMC in RxMC was first proposed by Rosch and Maginn55 (from now on referred to as “parallel Rx/CFC”). Balaji et al. used parallel Rx/CFC to compute the equilibrium concentrations of the different species in CO2/monoethanolamine solutions for different CO2 loadings.1 In this method, fractional molecules of reaction products are gradually changed into whole reaction product molecules, while the fractional molecules of reactants are gradually removed and vice versa. This algorithm is shown schematically in Figure 1a. A key ingredient of parallel Rx/CFC is that the fractional molecules of both all reactants and reaction products are always present in the system. This CFCMC version of RxMC improves the acceptance probability of molecule insertions/removals significantly compared to the conventional RxMC algorithm.55 It does not allow direct calculation of chemical potentials, and it is not possible to directly check if the reaction is at equilibrium. Additional free energy calculations are needed to compute the chemical potentials of reactant and reaction product molecules. The fractional molecules of reactants and reaction products have to adapt to their surroundings simultaneously, which reduces the efficiency of the algorithm.
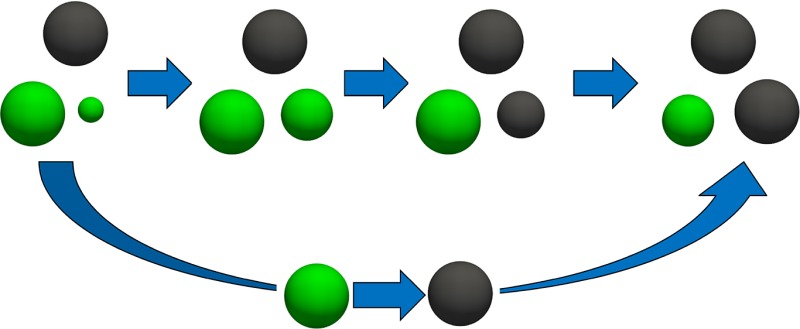
Figure 1.

(a) Schematic representation of parallel Rx/CFC for the combination of RxMC with CFCMC (denoted by parallel Rx/CFC).55 The conventional RxMC is expanded with fractional molecules of each component participating in the reaction. The number of fractional molecules of each component is equal to its stoichiometric coefficient νi. The coupling parameters for intermolecular interactions of fractional molecules of reactants and reaction products are constrained by λR + λP = 1. (b) Schematic representation of serial Rx/CFC for the combination of RxMC with CFCMC (the method described in this paper). In serial Rx/CFC, either fractional molecules of reactants or fractional molecules of reaction products are present in the system. In both figures, we consider the reaction A ⇌ B, in which A = green and B = black. The dashed spheres represent fractional molecules.
Recently, a more efficient approach to combine CFCMC with the Gibbs ensemble was introduced by us.63,64 Only one fractional molecule per component is present, which can be in either of the simulation boxes, and chemical potentials of all components are obtained without any additional calculations. Inspired by this, a new formulation for RxMC combined with CFCMC is introduced (denoted by serial Rx/CFC). The crucial difference with the parallel Rx/CFC is that either fractional molecules of reactants or reaction products are present in the system. The chemical potentials of reactants/reaction products are directly obtained without using Widom’s test particle insertion (or related) method. Therefore, one can directly check for the condition of chemical equilibrium.
This paper is organized as follows. In Section 2, the conventional RxMC ensemble and its combination with CFCMC are reviewed. Our formulation of RxMC with CFCMC (denoted by serial Rx/CFC) is introduced in Section 3. The partition function, types of trial moves, and computation of chemical potentials are also discussed in this section. Simulation details and systems are described in Section 4. In Section 5, the performance of serial Rx/CFC is compared to conventional RxMC and parallel Rx/CFC for Lennard-Jones (LJ) molecules. We considered various model reactions and pressures for which ideal gas free energy changes are specified in advance. Our approach is also tested for the reaction of ammonia synthesis at various temperatures and pressures. Compared to parallel Rx/CFC, serial Rx/CFC is more efficient, faster, and allows for the computation of chemical potentials of all components without any additional computation. Our findings are summarized in Section 6.
2. Conventional RxMC and Parallel Rx/CFC
In RxMC simulations, the number of atoms is conserved and not the number of molecules of individual species.4 Usually, the temperature is constant and either pressure or volume is imposed. The constant pressure version is more interesting for practical applications. In the Supporting Information, first the partition function and acceptance rules are derived for the constant volume case and extended to the constant pressure version by adding a term exp[−β PV] to the partition function.32 In this section, the partition function and acceptance rules are discussed in detail for the constant pressure version. In addition to Monte Carlo trial moves for thermalization and volume changes, attempts are made to remove reactants and insert reaction product molecules and vice versa. These are the so-called reaction trial moves. Here, for simplicity, we only consider systems with a single reaction, as extension to systems with multiple reactions is straightforward.1,32 The partition function for the constant pressure version of conventional RxMC equals4,23
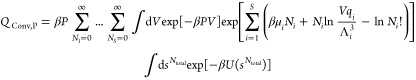 |
1 |
where S is the number of components, β = 1/(kBT), kB is the Boltzmann constant, s are reduced coordinates, V is the volume of the simulation box, P is the pressure, Ntotal is the total number of molecules in the simulation box, and U is the total potential energy. Here, qi, μi, Ni, and Λi are the ideal gas partition function excluding the translational part, the chemical potential, the number of molecules, and the thermal wavelength of component (molecule type) i, respectively. The ensemble of eq 1 is subject to the constraints that the total number of atoms of each type is constant and that chemical reactions converting reactants into reaction products are in equilibrium. This sets limits on the values of μi. Sampling of configurations in this ensemble requires (1) sampling of the degrees of freedom of the interacting molecules (e.g., translation, rotation (for chain molecules), and sampling the internal configuration of molecules), (2) sampling the volume fluctuations, and (3) sampling of chemical reactions subject to the constraint that the total number of atoms of each component is constant, as well as that the reaction is at chemical equilibrium. The latter is obtained by performing reaction trial moves. The reaction trial move is attempted to remove randomly selected reactants and insert reaction product molecules, simultaneously. According to the partition function of conventional RxMC (eq 1), the probabilities of being in the old and new configurations for the reaction trial move in the forward direction are
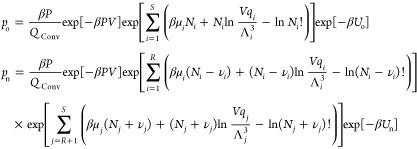 |
2 |
where νi is the stoichiometric coefficient of component i in the reaction. Here, n and o denote the new and old configurations, respectively. We choose the convention that νi is positive if component i participates in the reaction, and νi is zero otherwise. Here, R is the number of reactant components, and P is the number of reaction product components. As only systems with a single reaction where all components are either reactants or reaction products are considered here, one can write R + P = S. Therefore, the reaction product components are ranging from R + 1 to S, with S being the total number of components. The summation ∑i = 1R is a sum over all reactant types, and ∑j = R+1 is the sum over all reaction product types. Therefore, the ratio of the probabilities of being in the new and old configurations equals
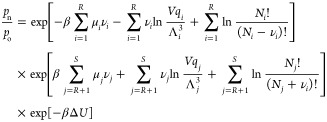 |
3 |
here ΔU = Un – U0 is the total change in the potential energy of the system. Reaction equilibrium implies ∑i = 1Rμiνi = ∑j = R+1μjνj. Consequently, the acceptance rule for the reaction trial move is4,32
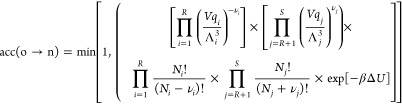 |
4 |
Due to simultaneous insertion of the molecules in a single step, the efficiency of this algorithm can be very low at high densities. This is also the case when Configurational-bias Monte Carlo is used for inserting/deleting molecules.26
In parallel Rx/CFC,55 the conventional RxMC is expanded with fractional molecules of each component participating in the reaction (Figure 1a). The number of fractional molecules of each component is equal to its stoichiometric coefficient. Interactions of the fractional molecules are scaled with a coupling parameter λj. The value λj = 0 corresponds to no interactions with the surrounding molecules (the fractional molecule acts as an ideal gas molecule), and λj = 1 corresponds to full interactions with the surrounding molecules (the fractional molecule has the same interactions as whole molecules of the same component). There are two coupling parameters per reaction, one for all reactants (λR) and one for all reaction products (λP). The coupling parameters for the fractional molecules of reactants and reaction products are constrained by λR + λP = 1. Attempts are made to change the coupling parameters by λn,R = λo,R + Δλ with Δλ ∈ [−Δλmax, + Δλmax]. Due to the constraint λR + λP = 1, the coupling parameter of the fractional molecules of reaction products also changes according to λn,P = λo,P – Δλ. When λn,R > 1 or λn,R < 0, an attempt is made to perform a chemical reaction. The acceptance rule for performing a chemical reaction in this ensemble is the same as eq 4. For more details, we refer the reader to the original publication by Maginn and Rosch.55
3. Serial Rx/CFC
3.1. Partition Function
In serial Rx/CFC, either fractional molecules of the reactants or reaction products are present in the system, in sharp contrast to parallel Rx/CFC where fractional molecules of both reactants and reaction products are always present (Figure 1b). Besides trial moves for thermalization and volume changes, there are three additional trial moves to facilitate the sampling of chemical reactions subject to the constraint that the total number of atoms of each component is constant, as well as chemical equilibrium. As derived in the Supporting Information, the partition function for the constant pressure version of the ensemble equals (not yet taking into account the conservation of atoms)
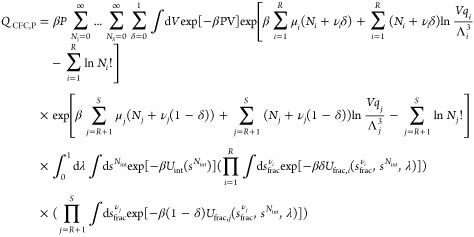 |
5 |
where Nint is the total number of whole molecules (regardless the component type), and Ni is the number of whole molecules of component i. When δ = 1, fractional molecules of reactants are present in the simulation box (νi fractional molecule for component i), and when δ = 0, fractional molecules of reaction products are present. Here, a system with a single reaction is considered. Also, Uint is the total internal energy of whole molecules, and Ufrac,i is the interaction energy of fractional molecules of component i with the other molecules, including other fractional molecules. The interactions of the fractional molecules with the surroundings are such that λ = 0 means no interactions and λ = 1 means full interactions, and the value of λ is restricted to λ ∈ [0,1].
Since fractional molecules are always distinguishable from whole molecules, the term Ni! only counts for whole indistinguishable molecules. The main difference between eq 5 and eq 1 is the integration over λ in eq 5. This is an immediate consequence of expanding the conventional RxMC with fractional molecules. In the Supporting Information, we show that for systems without intermolecular interactions (ideal gas phase) the partition functions of eq 5 and eq 1 are proportional. Therefore, these ensembles result in identical average numbers of molecules for each component, provided that fractional molecules are not counted when computing ensemble averages. The fact that one should not count fractional molecules when computing the average number of molecules is in line with earlier studies in the Gibbs ensemble and in the grand-canonical ensemble.45,64
3.2. Trial Moves
In addition to Monte Carlo trial moves for thermalization and volume changes, there are three trial moves in this ensemble to mimic the chemical reaction. A detailed description of these trial moves and the derivation of the acceptance rules is provided in the Supporting Information.
3.2.1. Changing the Value of λ
This trial move is used to change the value of λ either for reactants or reaction products, depending on the value of δ (Figure 2). The value of λ is changed according to λn = λo + Δλ, in which Δλ is a uniformly distributed random number between −Δλmax and +Δλmax. Note that Δλmax can be different for reactants and reaction products. When the new value of λ is not in the range λ ∈ [0,1], this trial move is automatically rejected. In this trial move, the value of δ, all positions of molecules, and the number of whole molecules and fractional molecules remain the same. By changing the value of λ, only the interactions between the fractional molecules and other molecules are changed. In the Supporting Information, it is shown that the acceptance rule for this trial move is
| 6 |
in which ΔU = Un – U0 is the change in the total internal energy of the system.
Figure 2.

Schematic representation of the trial move attempting to change the coupling parameter λ for serial Rx/CFC. In this trial move, δ and the positions of all molecules remain the same. We consider the reaction A ⇌ B in which A = green and B = black. The dashed spheres represent fractional molecules.
3.2.2. Reaction for Fractional Molecules
In this trial move, fractional molecules of reactants/reaction products are removed, and fractional molecules of reaction products/reactants are inserted at random positions (Figure 3). In this trial move, essentially the value of δ is changed, so if δo = 1 then δn = 0 and vice versa. The number of whole molecules and also the value of λ are constant. This trial move basically mimics a chemical reaction for the fractional molecules. Here, the acceptance rule for the forward reaction (reactants → reaction products) is shown. The direction of the chemical reaction is defined by the value of δ for the old configuration (if we have the fractional molecules of reactants or reaction products). In the Supporting Information, it is derived that the acceptance rule for converting the reactants into reaction products equals
| 7 |
Since the number of whole molecules remains
constant during this move, the terms 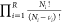 and
and 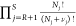 are not present in eq 7. The acceptance rule for
the reverse reaction
(reaction products → reactants) simply follows by swapping
the labels of the reactants and reaction products. The acceptance
probability for this trial move is large when λ is small. The
reason for this is that fractional molecules have very limited interactions
with the surrounding molecules, and therefore, the term ΔU is nearly zero. For the limiting case of λ ↓ 0, the
acceptance rule reduces to
are not present in eq 7. The acceptance rule for
the reverse reaction
(reaction products → reactants) simply follows by swapping
the labels of the reactants and reaction products. The acceptance
probability for this trial move is large when λ is small. The
reason for this is that fractional molecules have very limited interactions
with the surrounding molecules, and therefore, the term ΔU is nearly zero. For the limiting case of λ ↓ 0, the
acceptance rule reduces to
| 8 |
Figure 3.

Schematic representation of the trial move attempting to perform the reaction for fractional molecules for serial Rx/CFC. In this trial move, the number of the whole molecules and also the value of λ are constant. We consider the reaction A ⇌ B, in which A = green and B = black. The dashed spheres represent fractional molecules. The fractional molecule of A is removed, and a fractional molecule of B is inserted at a randomly selected position.
3.2.3. Reaction for Whole Molecules
In this trial move, fractional molecules of reactants/reaction products are transformed into whole molecules of reactants/reaction products, while simultaneously, randomly selected whole molecules of reaction products/reactants are transformed into fractional molecules of reaction products/reactants. In this trial move, all molecule positions and the value of λ stay the same. The value of δ is changed as follows: if δo = 1, then δn = 0 and vice versa. This trial move is illustrated in Figure 4 and can be seen as a reaction for whole molecules. In the forward reaction, whole molecules of reactants are transformed into fractional molecules, and fractional molecules of reaction products are turned into whole molecules. Essentially, the number of whole molecules of reactants is reduced, and the number of whole molecules of reaction products is increased, according to their stoichiometric coefficients. Trial moves are automatically rejected when there are not enough whole molecules to turn into fractional molecules. Here, the acceptance rule for the forward reaction (reactants → reaction products) is shown. The direction of the reaction eventually depends on the value of δ for the old configuration (if we have fractional molecules of reactants or reaction products). As derived in the Supporting Information, the acceptance rule for this trial move is
| 9 |
in which ΔU = Un – U0 is the change in the total internal energy of the system including the interaction of fractional molecules as result of the trial move. The acceptance rule for the reverse reaction (reaction products → reactants) simply follows by swapping the labels. Since the total number of whole and fractional molecules for each component remains constant, ideal gas partition functions are not present in eq 9. This trial move has a very high acceptance probability when the value of λ is close to 1. The reason for this is that fractional molecules have almost the same interactions as whole molecules, and therefore, the term ΔU is nearly zero. For the limiting case of λ ↑ 1, the acceptance rule reduces to
| 10 |
Figure 4.

Schematic representation of the trial move attempting to perform the reaction for whole molecules for serial Rx/CFC. In this trial move, the value of λ and all positions of all molecules remain the same. We consider the reaction A ⇌ B in which A = green and B = black. The dashed spheres represent fractional molecules. The fractional molecule of A is transformed into a whole molecule of A, while at the same time a randomly selected whole molecule of B is transformed into a fractional molecule of B.
3.2.4. Volume Changes
This trial move is only used for the case where the temperature and external pressure are imposed. In this trial move, the volume of the simulation box is changed, while the number and relative coordinates of the whole molecules and fractional molecules stay the same. Here, the random walk is performed in V and not ln(V).32 The acceptance rule for this trial move is32
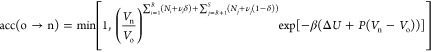 |
11 |
3.2.5. Efficient Selection of Trial Moves
As discussed in the previous section, the reaction trial move for fractional molecules has a very high acceptance probability at low values of λ, and the reaction trial move for the whole molecules has a very high acceptance probability at high values of λ. In Figure 5, typical acceptance probabilities of these trial moves as a function of λ are shown. Therefore, one may attempt reaction trial moves for fractional molecules only at values of λ close to 0, and reaction trial moves for the whole molecules only at values of λ close to 1. In this way, each trial move is used where it is efficient, and the overall efficiency of the algorithm is improved. This is done as follows: one can define a switching point for the value of λ (λsec). The probabilities of selecting a trial move [thermalization, volume change, or changing the value of λ (Section 3.2.1)] are always constant. For selection of the remaining trial moves (Sections 3.2.2 and 3.2.3), one has a choice: selecting these with fixed probability or always selecting the reaction trial move for fractional molecules (Section 3.2.2) when λ < λsec and always selecting the reaction trial move for whole molecules (Section 3.2.3) when λ > λsec. In the latter approach, the reaction trial moves are selected when they have a higher acceptance probability. Since the value of λ remains constant during either of these trial moves, the probabilities for selecting the trial moves also remain constant. Therefore, the condition of detailed balance is not violated.
Figure 5.
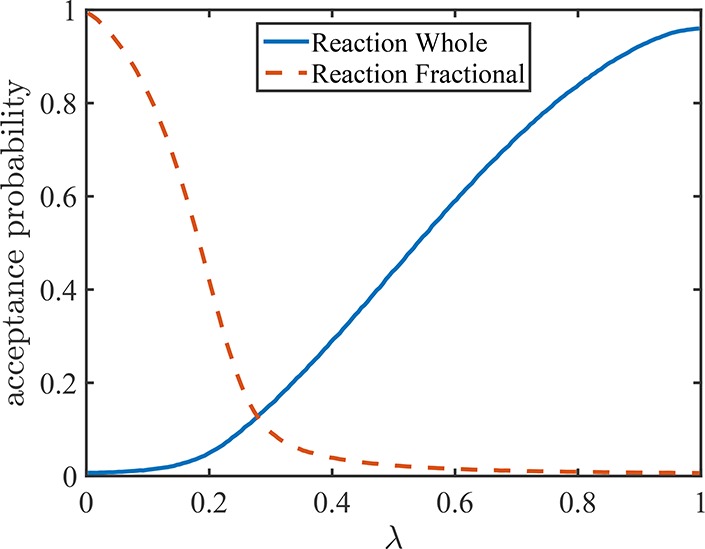
Acceptance probabilities for trial moves attempting to perform reactions for fractional molecules (dashed line, Figure 3) and for trial moves attempting to perform reactions for whole molecules (solid line, Figure 4) for serial Rx/CFC. Simulation conditions are reduced temperature T = 2 and constant reduced pressure P = 3.0 for the reaction A ⇌ B. Similar results are obtained for the other reactions and at other conditions.
3.3. Biasing the Probability Distribution p(λ, δ)
It is important to bias the probability distribution of p(λ, δ) (δ indicates whether fractional molecules of reactants or reaction products are in the simulation box) in such a way that the sampled probability distributions p(λ, δ) is flat and that it is equally likely to have the fractional molecules of reactants (δ = 1) or reaction products (δ = 0). By using an adequate biasing function, one can overcome the problem of being “stuck” in free energy minima and can easily diffuse through the λ space. This is obtained by multiplying the statistical weight of each system state by a factor exp[W(λ, δ) ]. For parallel Rx/CFC,55 since fractional molecules of both reactants and reaction products are always present in the system, one would only need a one-dimensional weight function to obtain flat probability distribution of p(λ). It is important to note that in serial Rx/CFC the weight function W(λ, δ) is a two-dimensional function that depends both on λ and the identity of the fractional molecules (δ). By using this biased sampling, additional terms exp[W(λn, δn) – W(λo, δo)] will be present in the acceptance rules of eqs 6,7, and 9. For example, the acceptance rule for the trial move attempting to mimic a reaction for the fractional molecules (eq 7) will become
| 12 |
To remove this bias, the Boltzmann averages of any observable X should be computed using
| 13 |
The weight function W(λ, δ) can be obtained using the Wang–Landau algorithm65,66 or iteratively.55 To compute ensemble averages corresponding to the conventional RxMC while performing simulation with serial Rx/CFC, one should exclude the contribution of fractional molecules. By doing this, one can analytically show that for an ideal gas case the ensemble averages computed using the serial Rx/CFC and the conventional RxMC are identical (see Supporting Information). Including the contribution of the fractional molecules in the ensemble averages leads to small differences between the ensemble averages compute in the serial Rx/CFC and those computed in the conventional RxMC.64
3.4. Free Energy Calculations
In serial Rx/CFC, chemical potentials can be computed without any additional computational efforts. As shown in the Supporting Information, one can show that
| 14 |
where qi is the ideal gas partition function of component i excluding the translational part. One can obtain the sum of the chemical potentials of reaction products in a similar way. Equation 14 allows for an independent check of reaction equilibria without any additional calculations (e.g., test molecules). The chemical potential of component i for a nonideal gas equals4,5
| 15 |
in which φi and yi are the fugacity coefficient and mole fraction of component i, respectively. Here, P0 is the reference pressure (1 bar), and P is the pressure of the mixture. The first term on the right-hand side of eq 15 is the standard reference chemical potential (μi0(T)). Therefore, we have
| 16 |
Combining this with eq 14 immediately leads to
| 17 |
where λR = λ when we have the fractional molecules of reactants (δ = 1). In this equation, p(λR ↑ 1) is the probability that λR approaches 1, and p(λR ↓ 0) is the probability that λR approaches 0. To compute the chemical potential of individual components, one should couple the interactions of different components in a smart way. The two limiting cases are well defined: at λ = 0, fractional molecules of reactants (or reaction products) do not interact, and at λ = 1, fractional molecules of reactants have full interactions with the surrounding molecules. However, for intermediate values of λ, one has a choice. One can choose different paths to scale the interactions of fractional molecules from no interactions to full interactions. The interactions can be scaled atom by atom, or molecule by molecule, or in any other way. By scaling the interactions of the fractional molecules of only one of the components from no interactions to full interactions when the value of λ is changed from 0 to A, one can write
| 18 |
The first term on the right-hand side accounts for the ideal gas part.The second term on the right-hand side accounts for the excess part of the chemical potential (due to interactions with surrounding molecules). Similar to eq 17, one can write for φi
| 19 |
When νi > 1 and interactions of fractional molecules are scaled sequentially (one by one), fractional molecules that have interactions with surrounding molecules later (at higher values of λ) experience the effect of the fractional molecules which were inserted earlier (at lower values of λ). For sufficiently large system sizes, this will not affect the calculated chemical potentials. However, for small system sizes, there may be minor differences between the chemical potential of molecules that are inserted at lower values of λ and those inserted at at higher values of λ. Although these differences are expected to be small, one should be aware of them.
4. Simulation Details
As a proof of principle, simulations are performed for different systems of LJ molecules. The ammonia synthesis reaction at various pressures (100–1000 bar) and temperatures (573–873 K) is also considered as a practically important application. For different systems of LJ molecules, simulations are performed at constant pressure and temperature using conventional RxMC, parallel Rx/CFC,55 and serial Rx/CFC. Various model reactions of LJ molecules are studied. For these simulations, all properties are defined in reduced units. LJ interactions are truncated and shifted at 2.5σ. The reduced temperature is set to T = 2.0, and simulations are always started with 400 molecules of component A. For simulations of LJ molecules using parallel Rx/CFC and serial Rx/CFC, the maximum molecule displacements, maximum volume change, and maximum change in the value of λ are set to achieve 50% acceptance for these trial moves. For simulations using serial Rx/CFC, the switching point for the value of λ is set to λsec = 0.3 (Section 3.2.4). In each Monte Carlo step, a trial move is selected at random with the following probabilities: 49.5% molecule displacements, 1% volume changes, and 49.5% reaction trial moves.
For the ammonia synthesis reaction, simulations are performed at constant pressure and temperature using serial Rx/CFC. All simulations start with a random configuration of 120 N2, 360 H2 molecules, and no ammonia molecules. Fractional molecules are added to this configuration. All molecules are rigid and interact only through LJ and electrostatic interactions. Force field parameters for N2, H2, and NH3 are taken from the literature.67−70 The Wolf method is used to compute electrostatic interactions.71 Details regarding the force field parameters, scaling of the electrostatic interactions, and the Wolf method are provided in the Supporting Information. The ideal gas partition functions for this system are listed in Table S3 of the Supporting Information. In this table, a detailed comparison is made between ideal gas partition functions from experiments and QM computations using Gaussian09.72 In each Monte Carlo step, a trial move is selected at random with the following probabilities: 33% molecule displacements, 33% molecule rotation, 1% volume changes, and 33% reaction trial moves. For the ammonia synthesis reaction, LJ interactions are switched on for λ ∈ [0,0.9], and electrostatic interactions are switched on for λ ∈ [0.9,1].
For all simulations using parallel Rx/CFC and serial Rx/CFC, the weight function is determined using the Wang–Landau algorithm.65 In serial Rx/CFC, the weight function W(λ, δ) is determined such that the observed two-dimensional probability distribution p(λ, δ) in the proposed ensemble is flat. Here, 200 bins are used to store the probability distribution of λ for reactants or reaction products. All simulations are started with 0.2 million Monte Carlo cycles to equilibrate the system, followed by 1 million production trial moves. The number of Monte Carlo steps per cycle equals the total number of molecules initially in the system, with a minimum of 20. LJ interactions are scaled according to48
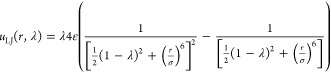 |
20 |
In the conventional method and parallel Rx/CFC, there is only one type of reaction trial move. In contrast, serial Rx/CFC requires three types of trial moves for facilitating molecule transfers: 50% changes in the λ space (Figure 2), 50% reaction for the fractional molecules (Figure 3) when λ < λsec, or 50% reaction for the whole molecules (Figure 4) when λ > λsec. In serial Rx/CFC, the chemical potentials are computed from eq 14. The contribution of fractional molecules are excluded while computing ensemble averages.64
To compare the efficiencies of the three methods, a fair way to define the efficiency of each method is required. In this work, the efficiency for all three methods is defined as the number of accepted trial moves (either forward or backward) resulting in a change in the number of whole molecules due to the reaction divided by the total number of reaction trial moves. For parallel Rx/CFC, this means the number of accepted λ trial moves that resulted in λn > 1 or λn < 0 divided by the total number of λ trial moves. For serial Rx/CFC, this means the number of accepted reaction trial moves for whole molecules (Figure 4) divided by the total number of all reaction trial moves, including changing the value of λ, reaction for fractional molecules, and reaction for whole molecules. It should be noted that reaction trial moves in serial Rx/CFC are computationally cheaper compared to parallel Rx/CFC. This is due to the reduction in the number of fractional molecules, and therefore, the number of interacting molecule pairs is reduced. Simulations performed using serial Rx/CFC require less CPU time compared to similar simulations when parallel Rx/CFC is used. The CPU time depends on the programming of the algorithms, the compiler, and CPUs used to perform the calculations. In this paper, different approaches are only compared in terms of efficiency and not the CPU time. This can be considered as the worst-case scenario for serial Rx/CFC.
5. Results
To ensure that serial Rx/CFC is implemented correctly, the equilibrium composition for different reactions of LJ molecules are computed and compared for the three methods. The LJ parameters and partition function for LJ molecules used in this study are listed in Table 1. The equilibrium composition obtained with different methods at reduced pressures P = 0.3, P = 1.0, P = 3.0, and P = 5.0 are shown in Tables 2–5, respectively. Equilibrium compositions obtained for the three methods are the same for all reactions and conditions (Tables 2–5). This confirms the validity of the expressions used for the partition function and acceptance rules of serial Rx/CFC and indicates that this method is implemented correctly. The efficiencies of the three methods for different reactions are also shown in Tables 2–5. The conventional method has a very high efficiency for all reactions at the lowest pressure (P = 0.3). Since in this case the density of the system is very low and therefore interactions between the molecules are limited, there is almost no energy penalty for the reaction trial moves, and most of the attempts to perform reaction trial moves for whole molecules are accepted. Therefore, the method which attempts to directly replace the reactants with reaction products and vice versa has a high efficiency. For the conventional method, reaction trial moves for the whole molecules are the only reaction trial move, and this trial move is accepted with a high probability for the low pressure case. As a result, this method has high efficiencies for this case. In parallel Rx/CFC,55 many trial moves are spent diffusing through the entire λ-space, and less attempts are made to perform a reaction. Therefore, this method has the lowest efficiency for the low pressure case. Already at P = 1.0, the efficiency of the conventional method is much lower than its efficiency at P = 0.3. At higher pressures (P = 3.0, P = 5.0), the efficiency of the conventional method drops below 0.01 even for the simplest reaction (A ⇌ B). When the density is high, most of the reaction trial moves in the conventional method result in an overlap between the newly inserted molecules and molecules that are already in the system. Therefore, this move has very low acceptance probability. In this case, the efficiency of parallel Rx/CFC varies between 0.06 to 0.1, while the efficiency of serial Rx/CFC varies between 0.1 to 0.2 depending on the reaction. Due to the efficient use of the three trial moves in serial Rx/CFC, this method has a better performance compared to the conventional method and parallel Rx/CFC.
Table 1. Interaction Parameters (Lennard-Jones) and Partition Functions (q/Λ3) for Different Molecule Typesa.
| Molecule type | σ | ϵ | q/Λ3 |
|---|---|---|---|
| A | 1.0 | 1.0 | 0.002 |
| B | 1.0 | 1.0 | 0.002 |
| C | 1.1 | 0.9 | 0.002 |
| D | 1.0 | 1.0 | 0.02 |
| E | 1.1 | 0.9 | 0.02 |
| F | 1.0 | 1.0 | 0.02 |
Note that there are several molecule types with exactly the same interaction parameters. This was done to show the effect of (in)distinguishability of the molecules in the reactions.
Table 2. Average Number of Molecules and Density at Equilibrium for Different Reactions for Different Methodsa.
| Reaction | ⟨NA⟩ | ⟨NProduct 1⟩ | ⟨NProduct 2⟩ | ⟨ρtot⟩ | Efficiency | Method |
|---|---|---|---|---|---|---|
| 200.00(3) | 200.00(3) | 0.162(0) | 0.40 | Conventional | ||
| A ⇌ B | 199.99(7) | 200.01(7) | 0.161(0) | 0.11 | Parallel Rx/CFC | |
| 199.98(6) | 200.02(6) | 0.161(0) | 0.30 | Serial Rx/CFC | ||
| 206.62(2) | 193.38(2) | 0.155(0) | 0.37 | Conventional | ||
| A ⇌ C | 206.63(3) | 193.37(2) | 0.154(0) | 0.098 | Parallel Rx/CFC | |
| 206.62(5) | 193.38(5) | 0.155(0) | 0.30 | Serial Rx/CFC | ||
| 192.59(6) | 414.8(2) | 0.162(0) | 0.26 | Conventional | ||
| A ⇌ 2D | 192.27(6) | 415.5(2) | 0.161(0) | 0.097 | Parallel Rx/CFC | |
| 192.36(4) | 415.27(8) | 0.161(0) | 0.25 | Serial Rx/CFC | ||
| 202.75(5) | 394.5(1) | 0.153(0) | 0.21 | Conventional | ||
| A ⇌ 2E | 202.35(6) | 395.3(2) | 0.152(0) | 0.086 | Parallel Rx/CFC | |
| 202.47(4) | 395.06(8) | 0.152(0) | 0.25 | Serial Rx/CFC | ||
| 91.52(3) | 308.48(3) | 308.48(3) | 0.162(0) | 0.26 | Conventional | |
| A ⇌ D + F | 91.22(9) | 308.78(9) | 308.78(9) | 0.161(0) | 0.097 | Parallel Rx/CFC |
| 91.33(3) | 308.67(3) | 308.67(3) | 0.161(0) | 0.25 | Serial Rx/CFC | |
| 95.57(3) | 304.43(3) | 304.43(3) | 0.156(0) | 0.23 | Conventional | |
| A ⇌ D + E | 95.28(4) | 304.72(4) | 304.72(4) | 0.155(0) | 0.094 | Parallel Rx/CFC |
| 95.39(2) | 304.61(2) | 304.61(2) | 0.155(0) | 0.25 | Serial Rx/CFC |
The efficiency is defined in Section 4. The reduced pressure and temperature are set to P = 0.3 and T = 2.0, respectively. Simulations are started with 400 molecules of type A. The interaction parameters of different molecules are listed in Table 1. The numbers between brackets denote the uncertainty in the last digit.
Table 5. Average Number of Molecules and Density at Equilibrium for Different Reactions for Different Methodsa.
| Reaction | ⟨NA⟩ | ⟨NProduct 1⟩ | ⟨NProduct 2⟩ | ⟨ρtot⟩ | Efficiency | Method |
|---|---|---|---|---|---|---|
| 199.8(3) | 200.2(3) | 0.766(0) | 1 × 10–3 | Conventional | ||
| A ⇌ B | 199(1) | 201(1) | 0.764(0) | 0.096 | Parallel Rx/CFC | |
| 200.1(4) | 199.9(4) | 0.766(0) | 0.20 | Serial Rx/CFC | ||
| 298.5(5) | 101.5(5) | 0.718(0) | 9 × 10–4 | Conventional | ||
| A ⇌ C | 298.5(8) | 101.5(8) | 0.716(0) | 0.079 | Parallel Rx/CFC | |
| 298.6(4) | 101.4(4) | 0.718(0) | 0.20 | Serial Rx/CFC | ||
| 372.5(3) | 54.9(6) | 0.766(0) | 3 × 10–5 | Conventional | ||
| A ⇌ 2D | 372.1(4) | 55.8(7) | 0.764(0) | 0.063 | Parallel Rx/CFC | |
| 372.4(2) | 55.2(4) | 0.765(0) | 0.11 | Serial Rx/CFC | ||
| 390.6(3) | 18.8(5) | 0.757(1) | 6 × 10–6 | Conventional | ||
| A ⇌ 2E | 390.6(2) | 18.9(3) | 0.755(0) | 0.048 | Parallel Rx/CFC | |
| 390.5(2) | 19.0(4) | 0.756(0) | 0.11 | Serial Rx/CFC | ||
| 345.2(5) | 54.8(5) | 54.8(2) | 0.766(0) | 3 × 10–5 | Conventional | |
| A ⇌ D + F | 345.4(6) | 54.6(6) | 54.6(6) | 0.764(0) | 0.067 | Parallel Rx/CFC |
| 345.3(6) | 54.7(6) | 54.7(6) | 0.765(0) | 0.12 | Serial Rx/CFC | |
| 368.1(6) | 31.9(6) | 31.9(6) | 0.752(0) | 1 × 10–5 | Conventional | |
| A ⇌ D + E | 368.2(4) | 31.8(4) | 31.8(4) | 0.749(1) | 0.063 | Parallel Rx/CFC |
| 368.1(5) | 31.9(5) | 31.9(5) | 0.751(0) | 0.11 | Serial Rx/CFC |
The efficiency is defined in Section 4. The reduced pressure and temperature are set to P = 5.0 and T = 2.0, respectively. Simulations are started with 400 molecules of type A. The interaction parameters of different molecules are listed in Table 1. The numbers between brackets denote the uncertainty in the last digit.
Table 3. Average Number of Molecules and Density at Equilibrium for Different Reactions for Different Methodsa.
| Reaction | ⟨NA⟩ | ⟨NProduct 1⟩ | ⟨NProduct 2⟩ | ⟨ρtot⟩ | Efficiency | Method |
|---|---|---|---|---|---|---|
| 200.00(4) | 200.00(4) | 0.433(0) | 0.077 | Conventional | ||
| A ⇌ B | 200.0(2) | 200.0(2) | 0.431(0) | 0.095 | Parallel Rx/CFC | |
| 200.01(8) | 199.99(8) | 0.432(0) | 0.26 | Serial Rx/CFC | ||
| 226.48(4) | 173.52(4) | 0.392(0) | 0.068 | Conventional | ||
| A ⇌ C | 226.4(2) | 173.6(2) | 0.390(0) | 0.079 | Parallel Rx/CFC | |
| 226.45(9) | 173.55(9) | 0.391(0) | 0.26 | Serial Rx/CFC | ||
| 273.05(5) | 253.89(9) | 0.433(0) | 0.017 | Conventional | ||
| A ⇌ 2D | 272.8(2) | 254.5(4) | 0.430(0) | 0.074 | Parallel Rx/CFC | |
| 272.8(2) | 254.3(3) | 0.431(0) | 0.17 | Serial Rx/CFC | ||
| 300.57(6) | 198.9(2) | 0.395(0) | 0.011 | Conventional | ||
| A ⇌ 2E | 300.3(1) | 199.4(2) | 0.393(0) | 0.059 | Parallel Rx/CFC | |
| 300.4(2) | 199.3(3) | 0.394(0) | 0.17 | Serial Rx/CFC | ||
| 177.73(5) | 222.27(5) | 222.27(5) | 0.433(0) | 0.017 | Conventional | |
| A ⇌ D + F | 177.4(3) | 222.6(3) | 222.6(3) | 0.431(0) | 0.075 | Parallel Rx/CFC |
| 177.5(2) | 222.5(2) | 222.5(2) | 0.431(0) | 0.17 | Serial Rx/CFC | |
| 197.92(7) | 202.08(7) | 202.08(7) | 0.401(0) | 0.014 | Conventional | |
| A ⇌ D + E | 197.6(3) | 202.4(3) | 202.4(3) | 0.399(0) | 0.070 | Parallel Rx/CFC |
| 197.6(2) | 202.4(2) | 202.4(2) | 0.399(0) | 0.17 | Serial Rx/CFC |
The efficiency is defined in Section 4. The reduced pressure and temperature are set to P = 1.0 and T = 2.0. Simulations are started with 400 molecules of type A. The interaction parameters of different molecules are listed in Table 1. The numbers between brackets denote the uncertainty in the last digit.
Table 4. Average Number of Molecules and Density at Equilibrium for Different Reactions for Different Methodsa.
| Reaction | ⟨NA⟩ | ⟨NProduct 1⟩ | ⟨NProduct 2⟩ | ⟨ρtot⟩ | Efficiency | Method |
|---|---|---|---|---|---|---|
| 200.1(2) | 199.9(2) | 0.667(0) | 7 × 10–3 | Conventional | ||
| A ⇌ B | 199.9(4) | 200.1(4) | 0.665(0) | 0.096 | Parallel Rx/CFC | |
| 199.9(2) | 200.1(2) | 0.667(0) | 0.20 | Serial Rx/CFC | ||
| 268.7(2) | 131.3(2) | 0.614(0) | 5 × 10–3 | Conventional | ||
| A ⇌ C | 268.8(2) | 131.2(2) | 0.612(0) | 0.076 | Parallel Rx/CFC | |
| 268.7(2) | 131.3(2) | 0.614(0) | 0.20 | Serial Rx/CFC | ||
| 345.2(2) | 109.5(4) | 0.667(0) | 3 × 10–4 | Conventional | ||
| A ⇌ 2D | 345.0(3) | 110.0(5) | 0.665(0) | 0.066 | Parallel Rx/CFC | |
| 344.8(4) | 110.5(8) | 0.666(0) | 0.11 | Serial Rx/CFC | ||
| 373.0(2) | 54.0(3) | 0.646(0) | 1 × 10–4 | Conventional | ||
| A ⇌ 2E | 372.9(2) | 54.1(3) | 0.643(0) | 0.051 | Parallel Rx/CFC | |
| 372.9(2) | 54.3(4) | 0.645(0) | 0.11 | Serial Rx/CFC | ||
| 293.5(3) | 106.5(3) | 106.5(3) | 0.667(0) | 3 × 10–4 | Conventional | |
| A ⇌ D + F | 293.1(6) | 106.9(6) | 106.9(6) | 0.665(0) | 0.068 | Parallel Rx/CFC |
| 293.3(5) | 106.7(5) | 106.7(5) | 0.666(0) | 0.11 | Serial Rx/CFC | |
| 324.2(2) | 75.8(2) | 75.8(2) | 0.641(0) | 2 × 10–4 | Conventional | |
| A ⇌ D + E | 324.2(5) | 75.8(5) | 75.8(5) | 0.638(0) | 0.064 | Parallel Rx/CFC |
| 324.1(4) | 75.9(1) | 75.9(1) | 0.639(0) | 0.11 | Serial Rx/CFC |
The efficiency is defined in Section 4. The reduced pressure and temperature are set to P = 3.0 and T = 2.0, respectively. Simulations are started with 400 molecules of type A. The interaction parameters of different molecules are listed in Table 1. The numbers between brackets denote the uncertainty in the last digit.
In Figure 6, the
(unbiased) probability distributions p(λ, δ)
for two different reactions (A ⇌ B and A ⇌ D + E) and
the weight functions to make p(λ, δ)
flat are shown. For the reaction A ⇌ B, the probability distributions
and the weight functions for the reactants and reaction products are
identical, as A and B have a similar interaction with the surrounding
molecules. For the reaction A ⇌ D + E, one reactant molecule
is replaced with two product molecules. For this reaction, the interactions
of the first product molecule are scaled from no interactions (ideal
gas molecule) to full interactions (whole molecule) when the value
of λ is changed from 0 to 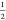 . For the second molecule, the interactions
are scaled from no interactions (ideal gas molecule) to full interactions
(whole molecule) when the value of λ is changed from
. For the second molecule, the interactions
are scaled from no interactions (ideal gas molecule) to full interactions
(whole molecule) when the value of λ is changed from 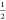 to 1. This can also be clearly seen in
the shape of the probability distribution of λ and the weight
function of the reactant molecules. In this way, according to eq 18, one can obtain the
excess chemical potential of the first reactant molecule using p(λR ↓ 0) and
to 1. This can also be clearly seen in
the shape of the probability distribution of λ and the weight
function of the reactant molecules. In this way, according to eq 18, one can obtain the
excess chemical potential of the first reactant molecule using p(λR ↓ 0) and 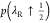 and
the excess chemical potential of the
second reactant molecule using
and
the excess chemical potential of the
second reactant molecule using 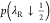 and p(λR ↑ 1). The values obtained for the excess
chemical potential
of the first and second reactant molecules were very close to each
other.
and p(λR ↑ 1). The values obtained for the excess
chemical potential
of the first and second reactant molecules were very close to each
other.
Figure 6.

(a) and (c) Probability distributions p(λ, δ) for reactants (δ = 1) and reactions products (δ = 0) for reaction (a) A ⇌ B and (c) A ⇌ D + E at a reduced temperature T = 2 and constant reduced pressure P = 3.0. (b) and (d) Weight functions (in units of kBT) to flatten the corresponding probability distributions of λ and to ensure that it is equally likely to have fractional molecules of reactants and reaction products for reactions (b) A ⇌ B and (d) A ⇌ D + E.
In Table 6, the sum of the total and excess chemical potentials times the stoichiometric coefficients are shown forthe reactants and reaction products for different pressures and reactions. These values can only be directly computed in serial Rx/CFC according to eq 14. The data provided in Table 6 shows that for the reaction A ⇌ B, where the reactant and reaction products have identical LJ interactions, the values obtained for the chemical potentials of the reactants and reaction products are equal. Since molecules of A and B are identical (Table 1), this is exactly what is expected. This case is included because it is trivial and can serve as an additional check on the implementation and on convergence of the simulation. It is verified that the computed excess chemical potentials are identical to those obtained from Widom’s test particle insertion method in the conventional NPT ensemble at the same conditions (data not shown).32
Table 6. Chemical Potentials of Reactants and Reaction Products for Different Reactions of the Lennard-Jones System at Different Pressures Obtained with Serial Rx/CFCa.
| Reaction | P | ∑reactants νiμiexcess | ∑reactants νiμitot | ∑products νiμiexcess | ∑products νiμitot |
|---|---|---|---|---|---|
| 0.3 | –0.344(9) | 7.036(9) | –0.344(6) | 7.036(6) | |
| A ⇌ B | 1.0 | 0.07(1) | 9.42(1) | 0.066(6) | 9.421(6) |
| 3.0 | 2.73(1) | 12.95(1) | 2.727(6) | 12.953(6) | |
| 5.0 | 5.23(1) | 15.74(1) | 5.23(2) | 15.73(1) | |
| 0.3 | –0.265(8) | 7.101(8) | –0.133(6) | 7.101(6) | |
| A ⇌ C | 1.0 | 0.25(1) | 9.66(1) | 0.79(1) | 9.67(1) |
| 3.0 | 2.887(6) | 13.541(6) | 4.32(1) | 13.53(1) | |
| 5.0 | 5.35(2) | 16.53(2) | 7.51(2) | 16.52(2) | |
| 0.3 | –0.34(1) | 6.13(1) | –0.68(1) | 6.12(1) | |
| A ⇌ 2D | 1.0 | 0.07(1) | 9.49(1) | 0.13(1) | 9.48(1) |
| 3.0 | 2.73(1) | 13.79(1) | 5.43(4) | 13.74(2) | |
| 5.0 | 5.23(2) | 16.84(2) | 10.45(3) | 16.74(2) | |
| 0.3 | –0.240(9) | 6.253(8) | –0.205(9) | 6.245(9) | |
| A ⇌ 2E | 1.0 | 0.247(8) | 9.791(7) | 1.553(9) | 9.772(9) |
| 3.0 | 2.81(1) | 14.08(1) | 8.45(3) | 13.98(1) | |
| 5.0 | 5.25(3) | 17.02(3) | 14.78(8) | 16.67(6) | |
| 0.3 | –0.340(8) | 4.319(8) | –0.68(1) | 4.32(1) | |
| A ⇌ D + F | 1.0 | 0.07(1) | 8.30(2) | 0.13(1) | 8.29(1) |
| 3.0 | 2.724(9) | 13.246(6) | 5.44(2) | 13.213(8) | |
| 5.0 | 5.23(1) | 16.57(1) | 10.45(4) | 16.52(2) | |
| 0.3 | –0.270(7) | 4.418(7) | –0.41(1) | 4.43(1) | |
| A ⇌ D + E | 1.0 | 0.220(9) | 8.578(9) | 0.96(1) | 8.57(1) |
| 3.0 | 2.809(9) | 13.573(9) | 7.04(2) | 13.528(6) | |
| 5.0 | 5.27(2) | 16.80(2) | 12.69(6) | 16.69(3) |
It is instructive to repeat this test case for systems with a full LJ potential. The use of tail correction is dictated by legacy use of some force field like TraPPE, as these were developed using Monte Carlo methodology. It tries to approximate the “full” Lennard-Jones interactions using a finite (small) cutoff to keep computations tractable. However, for many other system, especially for use in MD, the discontinuity such a cutoff would create in the forces is problematic. In the SI, it is also shown that for the case of LJ molecules (truncated interactions and using analytic tail correction) identical values for the excess chemical potentials are obtained from the serial Rx/CFC, Widom’s test particle insertion method in the conventional NPT, and EOS modeling using a full LJ potential.73
Reaction equilibrium implies ∑i = 1Rμiνi = ∑j = R+1μjνj. It can be clearly seen that this condition is satisfied for all reactions at all pressures within the error bars. This indicates that simulations have reached the condition of chemical equilibrium, and one can trust the results obtained from the simulations. Moreover, one can directly compute the excess chemical potential of individual components according to eq 18.
To test the suitability of serial Rx/CFC simulations for practical systems and molecules with partial charges, the ammonia synthesis reaction (N2 + 3H2 ⇌ 2NH3) is considered. Equilibrium compositions obtained from serial Rx/CFC are validated with the RASPA software.61,62 In Figure 7, the mole fractions of ammonia at equilibrium obtained form serial Rx/CFC simulations at different temperatures and pressures are compared with experimental results74 and results using equation of state modeling (Peng–Robinson (PR) equation of state75). Excellent agreement is observed between the equilibrium mixture compositions obtained using the three different approaches. This validates the applicability of serial Rx/CFC for systems including molecules with electrostatic interactions. In Figure 8, fugacity coefficients of ammonia at chemical equilibrium computed using serial Rx/CFC simulations are compared with the results of thermodynamic modeling (using the PR equation of state) at different temperatures and pressures. It is well known that cubic equations of state fail to provide accurate estimates for the fugacity coefficient at very high pressures.76 For pressures below 600 bar, fugacity coefficients computed using serial Rx/CFC simulations are in very good agreement with those calculated from equation of state modeling. No experimental data was found to compare with the values obtained for fugacity coefficients.
Figure 7.
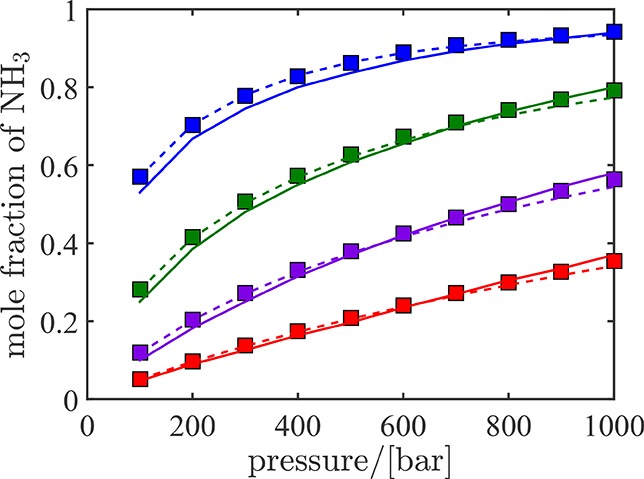
Mole fractions of ammonia at equilibrium obtained from serial Rx/CFC simulations (symbols), experiments (solid lines),74 and equation of state modeling using the Peng–Robinson equation of state (dashed lines) at 573 K (blue), 673 K (green), 773 K (purple), and 873 K (red) as a function of pressure. All simulations start with a random configuration of 120 N2, 360 H2 molecules, and no ammonia molecules.
Figure 8.
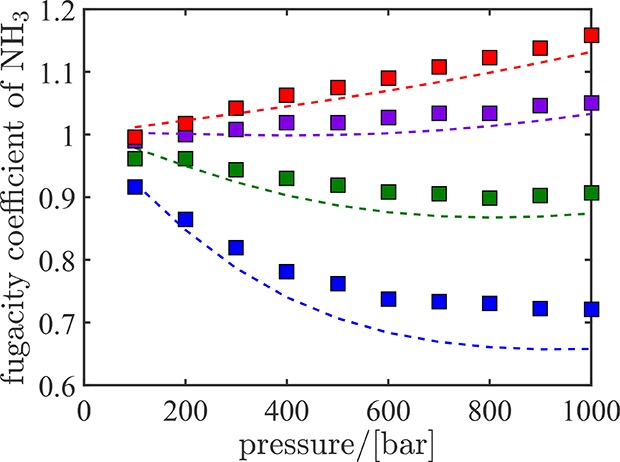
Fugacity coefficients of ammonia at equilibrium obtained from serial Rx/CFC simulations (symbols) and equation of state modeling using the Peng–Robinson equation of state (dashed lines) at 573 K (blue), 673 K (green), 773 K (purple), and 873 K (red) as a function of pressure. All simulations start with a random configuration of 120 N2, 360 H2 molecules, and no ammonia molecules.
6. Conclusion
An improved formulation of the Reaction Ensemble combined with Continuous Fractional Component Monte Carlo is presented (serial Rx/CFC). The main difference between serial Rx/CFC and parallel Rx/CFC55 is that in serial Rx/CFC either the fractional molecules of the reactants or the fractional molecules of the reaction products are present in the system. In serial Rx/CFC, there are three trial moves to facilitate a chemical reaction: (1) changing the value of λ, (2) reaction for fractional molecules, and (3) reaction for whole molecules. As a proof of principle, serial Rx/CFC is compared to the conventional formulation of RxMC and parallel Rx/CFC for systems of LJ molecules at different reduced pressures. Moreover, equilibrium mixture compositions obtained for the ammonia synthesis reaction using serial Rx/CFC are compared with experimental results and mixture compositions computed using equation of state modeling. The equilibrium compositions obtained with serial Rx/CFC are in excellent agreement with those obtained from the conventional RxMC and parallel Rx/CFC. For the ammonia synthesis reaction, excellent agreement between the results of serial Rx/CFC and experimental measured mixture compositions74 was found as well. For systems at high pressures, the acceptance probability of the reaction trial move is improved by a factor of 2 to 3 (depending on the system under study) compared to parallel Rx/CFC. Serial Rx/CFC has the following advantages: (1) One directly obtains chemical potentials of all reactants and reaction products. These chemical potentials can directly be used as an independent check to ensure that chemical equilibrium is achieved. (2) Independent biasing is applied to the fractional molecules of reactants and reaction products; therefore, the efficiency of the algorithm is increased. (3) Changes in the maximum scaling parameter of intermolecular interactions can be chosen differently for reactants and reaction products. Serial Rx/CFC can be easily extended to molecules with intramolecular degrees of freedom. The trial moves of Figure 3 can be performed by inserting fractional molecules at random positions with random orientations. The internal configuration of the molecule can be generated randomly or using the Rosenbluth scheme.32 The trial moves of Figure 4 can be performed by keeping the internal configuration of the molecule the same as in the old configuration. For ergodic sampling, trial moves that attempt to change the internal configuration of flexible molecules should be added to the MC method.32 The serial Rx/CFC method could also be used for reactions involving ions. One can calculate the potential energy of a periodic system with a net charge by placing a dummy charge at the center of charges. Although it is difficult to interpret computed partial molar properties of ions (such as the chemical potential or the partial molar volume)77 by using serial Rx/CFC, one can still benefit from other advantages of the method such as efficient reaction trial moves.
Acknowledgments
This work was sponsored by NWO Exacte Wetenschappen (Physical Sciences) for the use of supercomputer facilities, with financial support from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research, NWO). The authors also gratefully acknowledge the financial support from Shell Global Solutions B.V. and The Netherlands Research Council for Chemical Sciences (NWO/CW) through a VIDI grant (David Dubbeldam) and a VICI grant (Thijs J. H. Vlugt).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.7b00092.
Expressions for the partition function, acceptance rules for additional trial moves, and chemical potentials of components in serial Rx/CFC are derived. Details regarding the calculation of equilibrium mixture compositions by thermodynamic modeling using the Peng–Robinson equation of state. (PDF)
Author Contributions
A.P. and T.J.H.V. conceived the idea of serial Rx/CFC and derived the partition function and acceptance rules. R.H. coded all MC algorithms and performed all simulations. A.R. validated the results for the ammonia synthesis reaction with RASPA, performed all computations with Gaussian09, and contributed to the initial development of serial Rx/CFC. A.R. and M.R. performed the PR-EOS modeling and solved the reaction equilibria equations. All authors contributed to the scientific discussions and writing of the manuscript. The first draft was written by A.P.
The authors declare no competing financial interest.
Supplementary Material
References
- Balaji S. P.; Gangarapu S.; Ramdin M.; Torres-Knoop A.; Zuilhof H.; Goetheer E. L. V.; Dubbeldam D.; Vlugt T. J. H. Simulating the Reactions of CO2 in Aqueous Monoethanolamine Solution by Reaction Ensemble Monte Carlo Using the Continuous Fractional Component Method. J. Chem. Theory Comput. 2015, 11, 2661–2669. 10.1021/acs.jctc.5b00160. [DOI] [PubMed] [Google Scholar]
- Ross M. A high-density fluid-perturbation theory based on an inverse 12th-power hard-sphere reference system. J. Chem. Phys. 1979, 71, 1567–1571. 10.1063/1.438501. [DOI] [Google Scholar]
- Brenner D. W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B: Condens. Matter Mater. Phys. 1990, 42, 9458. 10.1103/PhysRevB.42.9458. [DOI] [PubMed] [Google Scholar]
- Turner H. C.; Brennan J. K.; Lisal M.; Smith W. R.; Johnson K. J.; Gubbins K. E. Simulation of chemical reaction equilibria by the reaction ensemble Monte Carlo method: a review. Mol. Simul. 2008, 34, 119–146. 10.1080/08927020801986564. [DOI] [Google Scholar]
- McQuarrie D. A.; Simon J. D.. Physical Chemistry: A Molecular Approach, 1st ed.; University Science Books: Sausalito, CA, 1997. [Google Scholar]
- Car R.; Parrinello M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471. 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- Laasonen K.; Sprik M.; Parrinello M.; Car R. Abinitio”liquid water. J. Chem. Phys. 1993, 99, 9080–9089. 10.1063/1.465574. [DOI] [Google Scholar]
- Iannuzzi M.; Laio A.; Parrinello M. Efficient exploration of reactive potential energy surfaces using Car-Parrinello molecular dynamics. Phys. Rev. Lett. 2003, 90, 238302. 10.1103/PhysRevLett.90.238302. [DOI] [PubMed] [Google Scholar]
- van Duin A. C.; Dasgupta S.; Lorant F.; Goddard W. A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. 10.1021/jp004368u. [DOI] [Google Scholar]
- Chenoweth K.; van Duin A. C.; Goddard W. A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. 10.1021/jp709896w. [DOI] [PubMed] [Google Scholar]
- Laio A.; Gervasio F. L. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. 10.1088/0034-4885/71/12/126601. [DOI] [Google Scholar]
- Bussi G.; Laio A.; Parrinello M. Equilibrium free energies from nonequilibrium metadynamics. Phys. Rev. Lett. 2006, 96, 090601. 10.1103/PhysRevLett.96.090601. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bonomi M.; Parrinello M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. 10.1002/wcms.31. [DOI] [Google Scholar]
- Dellago C.; Bolhuis P. G.; Csajka F. S.; Chandler D. Transition path sampling and the calculation of rate constants. J. Chem. Phys. 1998, 108, 1964–1977. 10.1063/1.475562. [DOI] [Google Scholar]
- Bolhuis P. G.; Dellago C.; Chandler D. Sampling ensembles of deterministic transition pathways. Faraday Discuss. 1998, 110, 421–436. 10.1039/a801266k. [DOI] [Google Scholar]
- Bolhuis P. G.; Chandler D.; Dellago C.; Geissler P. L. Transition path sampling: Throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318. 10.1146/annurev.physchem.53.082301.113146. [DOI] [PubMed] [Google Scholar]
- Rogal J.; Bolhuis P. G. Multiple state transition path sampling. J. Chem. Phys. 2008, 129, 224107. 10.1063/1.3029696. [DOI] [PubMed] [Google Scholar]
- van Erp T. S.; Bolhuis P. G. Elaborating transition interface sampling methods. J. Comput. Phys. 2005, 205, 157–181. 10.1016/j.jcp.2004.11.003. [DOI] [Google Scholar]
- Moroni D.; van Erp T. S.; Bolhuis P. G. Investigating rare events by transition interface sampling. Phys. A 2004, 340, 395–401. 10.1016/j.physa.2004.04.033. [DOI] [Google Scholar]
- Moroni D.; Bolhuis P. G.; van Erp T. S. Rate constants for diffusive processes by partial path sampling. J. Chem. Phys. 2004, 120, 4055–4065. 10.1063/1.1644537. [DOI] [PubMed] [Google Scholar]
- Moroni D.; van Erp T. S.; Bolhuis P. G. Simultaneous computation of free energies and kinetics of rare events. Phys. Rev. E 2005, 71, 056709. 10.1103/PhysRevE.71.056709. [DOI] [PubMed] [Google Scholar]
- Smith W.; Triska B. The reaction ensemble method for the computer simulation of chemical and phase equilibria. I. Theory and basic examples. J. Chem. Phys. 1994, 100, 3019–3027. 10.1063/1.466443. [DOI] [Google Scholar]
- Johnson J. K.; Panagiotopoulos A. Z.; Gubbins K. E. Reactive canonical Monte Carlo: a new simulation technique for reacting or associating fluids. Mol. Phys. 1994, 81, 717–733. 10.1080/00268979400100481. [DOI] [Google Scholar]
- Glotzer S. C.; Stauffer D.; Jan N. Monte Carlo simulations of phase separation in chemically reactive binary mixtures. Phys. Rev. Lett. 1994, 72, 4109. 10.1103/PhysRevLett.72.4109. [DOI] [PubMed] [Google Scholar]
- Johnson J. K. Reactive canonical monte carlo. Adv. Chem. Phys. 1999, 105, 461–481. 10.1002/9780470141649.ch15. [DOI] [Google Scholar]
- Hansen N.; Jakobtorweihen S.; Keil F. J. Reactive Monte Carlo and grand-canonical Monte Carlo simulations of the propene metathesis reaction system. J. Chem. Phys. 2005, 122, 164705. 10.1063/1.1884108. [DOI] [PubMed] [Google Scholar]
- Jakobtorweihen S.; Hansen N.; Keil F. Combining reactive and configurational-bias Monte Carlo: Confinement influence on the propene metathesis reaction system in various zeolites. J. Chem. Phys. 2006, 125, 224709. 10.1063/1.2404658. [DOI] [PubMed] [Google Scholar]
- Ungerer P.; Collell J.; Yiannourakou M. Molecular modeling of the volumetric and thermodynamic properties of kerogen: Influence of organic type and maturity. Energy Fuels 2015, 29, 91–105. 10.1021/ef502154k. [DOI] [Google Scholar]
- Chase M. W.JANAF Thermochemical Tables; American Chemical Society; National Bureau of Standards: New York, 1986. [Google Scholar]
- Fetisov E. O.; Kuo I.-F. W.; Knight C.; VandeVondele J.; Van Voorhis T.; Siepmann J. I. First-Principles Monte Carlo Simulations of Reaction Equilibria in Compressed Vapors. ACS Cent. Sci. 2016, 2, 409–415. 10.1021/acscentsci.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghahremanpour M. M.; van Maaren P. J.; Ditz J. C.; Lindh R.; van der Spoel D. Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity. J. Chem. Phys. 2016, 145, 114305. 10.1063/1.4962627. [DOI] [Google Scholar]
- Frenkel D.; Smit B.. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, 2002. [Google Scholar]
- Bai P.; Siepmann J. I. Assessment and optimization of configurational-bias Monte Carlo particle swap strategies for simulations of water in the Gibbs ensemble. J. Chem. Theory Comput. 2017, 13, 431–440. 10.1021/acs.jctc.6b00973. [DOI] [PubMed] [Google Scholar]
- Siepmann J. I. A method for the direct calculation of chemical potentials for dense chain systems. Mol. Phys. 1990, 70, 1145–1158. 10.1080/00268979000101591. [DOI] [Google Scholar]
- Siepmann J. I.; Karaborni S.; Smit B. Vapor-liquid equilibria of model alkanes. J. Am. Chem. Soc. 1993, 115, 6454–6455. 10.1021/ja00067a088. [DOI] [Google Scholar]
- Consta S.; Vlugt T. J. H.; Wichers Hoeth J.; Smit B.; Frenkel D. Recoil growth algorithm for chain molecules with continuous interactions. Mol. Phys. 1999, 97, 1243–1254. 10.1080/00268979909482926. [DOI] [Google Scholar]
- Houdayer J. The wormhole move: A new algorithm for polymer simulations. J. Chem. Phys. 2002, 116, 1783–1787. 10.1063/1.1433500. [DOI] [Google Scholar]
- Poursaeidesfahani A.; Torres-Knoop A.; Rigutto M.; Nair N.; Dubbeldam D.; Vlugt T. J. H. Computation of the Heat and Entropy of Adsorption in Proximity of Inflection Points. J. Phys. Chem. C 2016, 120, 1727–1738. 10.1021/acs.jpcc.5b11606. [DOI] [Google Scholar]
- Combe N.; Vlugt T. J. H.; Wolde P. R. T.; Frenkel D. Dynamic pruned-enriched Rosenbluth method. Mol. Phys. 2003, 101, 1675–1682. 10.1080/0026897031000094461. [DOI] [Google Scholar]
- Escobedo F. A.; de Pablo J. J. Monte Carlo simulation of the chemical potential of polymers in an expanded ensemble. J. Chem. Phys. 1995, 103, 2703–2710. 10.1063/1.470504. [DOI] [Google Scholar]
- Lyubartsev A. P.; Martsinovski A. A.; Shevkunov S. V.; Vorontsov-Velyaminov P. N. New approach to Monte Carlo calculation of the free energy: Method of expanded ensembles. J. Chem. Phys. 1992, 96, 1776–1783. 10.1063/1.462133. [DOI] [Google Scholar]
- Rane K. S.; Murali S.; Errington J. R. Monte Carlo Simulation Methods for Computing Liquidâ”Vapor Saturation Properties ofModel Systems. J. Chem. Theory Comput. 2013, 9, 2552–2566. 10.1021/ct400074p. [DOI] [PubMed] [Google Scholar]
- Shi W.; Maginn E. J. Continuous Fractional Component Monte Carlo: An Adaptive Biasing Method for Open System Atomistic Simulations. J. Chem. Theory Comput. 2007, 3, 1451–1463. 10.1021/ct7000039. [DOI] [PubMed] [Google Scholar]
- Shi W.; Maginn E. J. Improvement in molecule exchange efficiency in Gibbs ensemble Monte Carlo: Development and implementation of the continuous fractional component move. J. Comput. Chem. 2008, 29, 2520–2530. 10.1002/jcc.20977. [DOI] [PubMed] [Google Scholar]
- Torres-Knoop A.; Poursaeidesfahani A.; Vlugt T. J. H.; Dubbeldam D. Behavior of the Enthalpy of Adsorption in Nanoporous Materials close to Saturation Conditions. J. Chem. Theory Comput. 2017, 13, 3326–3339. 10.1021/acs.jctc.6b01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdin M.; Balaji S. P.; Torres-Knoop A.; Dubbeldam D.; de Loos T. W.; Vlugt T. J. H. Solubility of Natural Gas Species in Ionic Liquids and Commercial Solvents: Experiments and Monte Carlo Simulations. J. Chem. Eng. Data 2015, 60, 3039–3045. 10.1021/acs.jced.5b00469. [DOI] [Google Scholar]
- Shi W.; Maginn E. J. Atomistic Simulation of the Absorption of Carbon Dioxide and Water in the Ionic Liquid 1-n-Hexyl-3-methylimidazolium Bis(trifluoromethylsulfonyl)imide ([hmim][Tf2N]. J. Phys. Chem. B 2008, 112, 2045–2055. 10.1021/jp077223x. [DOI] [PubMed] [Google Scholar]
- Maginn E. J. Atomistic Simulation of the Thermodynamic and Transport Properties of Ionic Liquids. Acc. Chem. Res. 2007, 40, 1200–1207. 10.1021/ar700163c. [DOI] [PubMed] [Google Scholar]
- Kelkar M. S.; Shi W.; Maginn E. J. Determining the Accuracy of Classical Force Fields for Ionic Liquids: Atomistic Simulation of the Thermodynamic and Transport Properties of 1-Ethyl-3-methylimidazolium Ethylsulfate ([emim][EtSO4]) and Its Mixtures with Water. Ind. Eng. Chem. Res. 2008, 47, 9115–9126. 10.1021/ie800843u. [DOI] [Google Scholar]
- Zhang X.; Huo F.; Liu Z.; Wang W.; Shi W.; Maginn E. J. Absorption of CO2 in the Ionic Liquid 1-n-Hexyl-3-methylimidazolium Tris(pentafluoroethyl)trifluorophosphate ([hmim][FEP]): A Molecular View by Computer Simulations. J. Phys. Chem. B 2009, 113, 7591–7598. 10.1021/jp900403q. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Balaji S. P.; Ramdin M.; Gutierrez-Sevillano J. J.; Bardow A.; Goetheer E. L. V.; Vlugt T. J. H. Validation of the CO2/N2O Analogy Using Molecular Simulation. Ind. Eng. Chem. Res. 2014, 53, 18081–18090. 10.1021/ie503488n. [DOI] [Google Scholar]
- Ramdin M.; Balaji S. P.; Vicent-Luna J. M.; Gutierrez-Sevillano J. J.; Calero S.; de Loos T. W.; Vlugt T. J. H. Solubility of the Precombustion Gases CO2, CH4, CO, H2, N2, and H2S in the Ionic Liquid [bmim][Tf2N] from Monte Carlo Simulations. J. Phys. Chem. C 2014, 118, 23599–23604. 10.1021/jp5080434. [DOI] [Google Scholar]
- Shi W.; Siefert N. S.; Morreale B. D. Molecular Simulations of CO2, H2, H2O, and H2S Gas Absorption into Hydrophobic Poly(dimethylsiloxane) (PDMS) Solvent: Solubility and Surface Tension. J. Phys. Chem. C 2015, 119, 19253–19265. 10.1021/acs.jpcc.5b05806. [DOI] [Google Scholar]
- Torres-Knoop A.; Balaji S. P.; Vlugt T. J. H.; Dubbeldam D. A Comparison of Advanced Monte Carlo Methods for Open Systems: CFCMC vs CBMC. J. Chem. Theory Comput. 2014, 10, 942–952. 10.1021/ct4009766. [DOI] [PubMed] [Google Scholar]
- Rosch T. W.; Maginn E. J. Reaction Ensemble Monte Carlo Simulation of Complex Molecular Systems. J. Chem. Theory Comput. 2011, 7, 269–279. 10.1021/ct100615j. [DOI] [PubMed] [Google Scholar]
- Jamali S. H.; Ramdin M.; Becker T. M.; Torres-Knoop A.; Dubbeldam D.; Buijs W.; Vlugt T. J. H. Solubility of sulfur compounds in commercial physical solvents and an ionic liquid from Monte Carlo simulations. Fluid Phase Equilib. 2017, 433, 50–55. 10.1016/j.fluid.2016.11.015. [DOI] [Google Scholar]
- Ramdin M.; Balaji S. P.; Vicent-Luna J. M.; Torres-Knoop A.; Chen Q.; Dubbeldam D.; Calero S.; de Loos T. W.; Vlugt T. J. H. Computing bubble-points of CO2/CH4 gas mixtures in ionic liquids from Monte Carlo simulations. Fluid Phase Equilib. 2016, 418, 100–107. 10.1016/j.fluid.2015.09.041. [DOI] [Google Scholar]
- Ramdin M.; Chen Q.; Balaji S. P.; Vicent-Luna J. M.; Torres-Knoop A.; Dubbeldam D.; Calero S.; de Loos T. W.; Vlugt T. J. H. Solubilities of CO2, CH4, C2H6, and SO2 in ionic liquids and Selexol from Monte Carlo simulations. J. Comput. Sci. 2016, 15, 74–80. 10.1016/j.jocs.2015.09.002. [DOI] [Google Scholar]
- Maginn E. J. Atomistic simulation of the thermodynamic and transport properties of ionic liquids. Acc. Chem. Res. 2007, 40, 1200–1207. 10.1021/ar700163c. [DOI] [PubMed] [Google Scholar]
- Theodorou D. N. Progress and Outlook in Monte Carlo Simulations. Ind. Eng. Chem. Res. 2010, 49, 3047–3058. 10.1021/ie9019006. [DOI] [Google Scholar]
- Dubbeldam D.; Torres-Knoop A.; Walton K. S. On the inner workings of Monte Carlo codes. Mol. Simul. 2013, 39, 1253–1292. 10.1080/08927022.2013.819102. [DOI] [Google Scholar]
- Dubbeldam D.; Calero S.; Ellis D. E.; Snurr R. Q. RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 2016, 42, 81–101. 10.1080/08927022.2015.1010082. [DOI] [Google Scholar]
- Poursaeidesfahani A.; Torres-Knoop A.; Dubbeldam D.; Vlugt T. J. H. Direct Free Energy Calculation in the Continuous Fractional Component Gibbs Ensemble. J. Chem. Theory Comput. 2016, 12, 1481–1490. 10.1021/acs.jctc.5b01230. [DOI] [PubMed] [Google Scholar]
- Poursaeidesfahani A.; Rahbari A.; Torres-Knoop A.; Dubbeldam D.; Vlugt T. J. H. Computation of Thermodynamic Properties in the Continuous Fractional Component Monte Carlo Gibbs Ensemble. Mol. Simul. 2017, 43, 189–195. 10.1080/08927022.2016.1244607. [DOI] [Google Scholar]
- Wang F.; Landau D. P. Efficient, Multiple-Range Random Walk Algorithm to Calculate the Density of States. Phys. Rev. Lett. 2001, 86, 2050–2053. 10.1103/PhysRevLett.86.2050. [DOI] [PubMed] [Google Scholar]
- Poulain P.; Calvo F.; Antoine R.; Broyer M.; Dugourd P. Performances of Wang-Landau algorithms for continuous systems. Phys. Rev. E 2006, 73, 056704. 10.1103/PhysRevE.73.056704. [DOI] [PubMed] [Google Scholar]
- Turner C. H.; Johnson J. K.; Gubbins K. E. Effect of confinement on chemical reaction equilibria: The reactions 2NO⇆(NO)2 and N2+ 3H2 ⇆ 2NH3 in carbon micropores. J. Chem. Phys. 2001, 114, 1851–1859. 10.1063/1.1328756. [DOI] [Google Scholar]
- Gao J.; Xia X.; George T. F. Importance of bimolecular interactions in developing empirical potential functions for liquid ammonia. J. Phys. Chem. 1993, 97, 9241–9247. 10.1021/j100138a028. [DOI] [Google Scholar]
- Talbot J.; Tildesley D.; Steele W. Molecular-dynamics simulation of fluid N2 adsorbed on a graphite surface. Faraday Discuss. Chem. Soc. 1985, 80, 91–105. 10.1039/dc9858000091. [DOI] [Google Scholar]
- Murthy C.; Singer K.; Klein M.; McDonald I. Pairwise additive effective potentials for nitrogen. Mol. Phys. 1980, 41, 1387–1399. 10.1080/00268978000103611. [DOI] [Google Scholar]
- Wolf D.; Keblinski P.; Phillpot S.; Eggebrecht J. Exact method for the simulation of Coulombic systems by spherically truncated, pairwise r–1 summation. J. Chem. Phys. 1999, 110, 8254–8282. 10.1063/1.478738. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Egidi F. L. F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J. Gaussian 09, Revision C.02. Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Kolafa J.; Nezbeda I. The Lennard-Jones fluid: an accurate analytic and theoretically- based equation of state. Fluid Phase Equilib. 1994, 100, 1–34. 10.1016/0378-3812(94)80001-4. [DOI] [Google Scholar]
- Gillespie L. J.; Beattie J. A. The thermodynamic treatment of chemical equilibria in systems composed of real gases. I. An approximate equation for the mass action function applied to the existing data on the Haber equilibrium. Phys. Rev. 1930, 36, 743. 10.1103/PhysRev.36.743. [DOI] [Google Scholar]
- Peng D.-Y.; Robinson D. B. A new two-constant equation of state. Ind. Eng. Chem. Fundam. 1976, 15, 59–64. 10.1021/i160057a011. [DOI] [Google Scholar]
- Poling B. E.; Prausnitz J. M.; Oćonnell J. P.. The Properties of Gases and Liquids; Mcgraw-Hill, New York, 2001; Vol. 5. [Google Scholar]
- Schnell S. K.; Englebienne P.; Simon J.-M.; Krüger P.; Balaji S. P.; Kjelstrup S.; Bedeaux D.; Bardow A.; Vlugt T. J. H. How to apply the Kirkwood-Buff theory to individual species in salt solutions. Chem. Phys. Lett. 2013, 582, 154–157. 10.1016/j.cplett.2013.07.043. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.