

Abstract
Methionine residues fulfill a broad range of roles in protein function related to conformational plasticity, ligand binding, and sensing/mediating the effects of oxidative stress. A high degree of internal mobility, intrinsic detection sensitivity of the methyl group, and low copy number have made methionine labeling a popular approach for NMR investigation of selectively labeled protein macromolecules. However, selective labeling approaches are subject to more limited information content. In order to optimize the information available from such studies, we have performed DFT calculations on model systems to evaluate the conformational dependence of 3JCSCC, 3JCSCH, and the isotropic shielding, σiso. Results have been compared with experimental data reported in the literature, as well as data obtained on [methyl-13C]methionine and on model compounds. These studies indicate that relative to oxygen, the presence of the sulfur atom in the coupling pathway results in a significantly smaller coupling constant, 3JCSCC/3JCOCC ~ .7. It is further demonstrated that the 3JCSCH coupling constant depends primarily on the subtended CSCH dihedral angle, and secondarily on the CSCC dihedral angle. Comparison of theoretical shielding calculations with the experimental shift range of the methyl group for methionine residues in proteins supports the conclusion that the intra-residue conformationally-dependent shift perturbation is the dominant determinant of δ13Cε. Analysis of calmodulin data based on these calculations indicates that several residues adopt non-standard rotamers characterized by very large ~ 100° χ3 values. The utility of the δ13Cε as a basis for estimating the gauche/trans ratio for χ3 is evaluated, and physical and technical factors that limit the accuracy of both the NMR and crystallographic analyses are discussed.
Keywords: methionine, [methyl-13C]methionine, calmodulin, NMR, Karplus relation, 3JCSCC, scalar coupling constants
Graphical abstract
Introduction
The methionine residues of proteins fulfill a number of important structural and functional roles. Methionine-rich interaction surfaces in calmodulin, SecA and other proteins are able to accommodate a broad range of structurally diverse, hydrophobic targets (O’Neil and DeGrado 1990; Hunt et al. 2002). The inherent flexibility of the methionine sidechain is an important structural element when conformational plasticity is required (Gellman 1991; Pantoja-Uceda et al. 2004; Strohmeier et al. 2006). Methionine also functions as a metal ligand in heme proteins such as cytochrome c (Santos and Turner 1993) and bacterioferritin (George et al. 1993), as well as in non-heme metalloproteins such as plastocyanin (Bertini et al. 2001). Senn and coworkers have demonstrated that in oxidized cytochrome c, the orientation of the lone pair orbital of the methionine sulfur atom ligated to the heme iron exerts a dominant influence on its electronic structure (Senn and Wuthrich 1983a; b; Senn et al. 1984). The methionine sulfur can also be involved in hydrogen bonding interactions (Gregoret et al. 1991). Recent modeling studies have suggested that in the enzyme dihydrofolate reductase, the proximity of the Met20 sulfur atom to the folate N5 can elevate the pKa, facilitating subsequent hydride transfer chemistry (Khavrutskii et al. 2007). There is also evidence that methionine oxidation can serve as a sensor and a mediator of oxidative stress(Vogt 1995; Yin et al. 2000; Gustavsson et al. 2001; Anbanandam et al. 2005; Schoneich 2005).
The varied roles of methionine residues in DNA polymerases have been subject to intense investigations in recent years (Reha-Krantz and Nonay 1994; Tipples et al. 1996; Shah et al. 2001; Bose-Basu et al. 2004; Niimi et al. 2004; Kirby et al. 2005; Li et al. 2005; Pavlov et al. 2006; Nick McElhinny et al. 2007; Pursell et al. 2007a; Zheng et al. 2009; Zheng et al. 2010). Methionine is a conserved residue in the “primer grip” motif of HIV reverse transcriptase (RT) that positions the primer terminus of the substrate (Ding et al. 1998), and is also a component of the so called “YMDD” motif of the active site in this enzyme(Wakefield et al. 1992). Furthermore, methionine mutations in HIV RT play a critical role in the development of drug-resistance phenotypes (Menendez-Arias 2008). For some polymerases, the introduction of L -> M and M -> L mutations have been shown to confer characteristic infidelity profiles, making it possible to identify the polymerase involved in particular DNA transactions (Nick McElhinny et al. 2007). This approach has proven useful for clarifying the physiological roles of the corresponding polymerases(Pursell et al. 2007b).
As a result of these many roles as well as its favorable relaxation characteristics, [methyl-13C]methionine has proven popular as an NMR label (Jones et al., 1975, 1976; Blakley et al. 1978; Deber et al., 1978; Jaeck & Benz, 1979; Stollery et al., 1980; Wooten et al. 1981; Hardy & Dill, 1982; Rosevear 1988; Seigneuret et al. 1991; Lin et al., 1994; Krudy et al. 1994; Duewel et al. 1995; Howarth et al. 1995; Siivari et al. 1995; Beatty et al. 1996; Kleerekoper et al. 1998; Cox et al. 1999; He et al. 1999; Kleerekoper and Putkey 1999; Yuan et al. 1999; Skrynnikov et al., 2001; Duewel et al. 2001; Bose-Basu et al. 2004; Yuan et al. 2004; Kirby et al. 2005; DellaVecchia et al. 2007; Gelis et al., 2007; Kloiber et al., 2007; Zheng et al. 2009; Zheng et al. 2010; Religa et al., 2010). Nevertheless, there have been few theoretical analyses of the conformational dependence of its NMR parameters. The sulfur-containing sidechain represents a special case to which the extensive results for aliphatic chains are not directly applicable. Furthermore, the limited amount of information available from studies of specifically labeled macromolecules makes it essential to extract the maximum amount of information from these studies. The present studies have been undertaken in order to more fully evaluate the conformational dependence of the methionine NMR parameters, thus allowing a more complete analysis of data derived from methionine labeling studies.
Materials and Methods
Ethyl methyl sulfide ((1-methylsulfanyl)ethane), methyl t-butyl sulfide, and methyl t-butyl ether were obtained from Sigma-Aldrich, and [methyl-13C]-L-methionine was obtained from CIL, Inc. U-[13C,15N]ubiquitin was obtained from VLI Research, Inc. (Malvern, PA).
NMR studies
1D INADEQUATE (Bax et al. 1980) spectra were acquired using Varian’s inadqt experiment. All INADEQUATE experiments were performed using a 13C sweep width of 100.0 ppm, with an acquisition time of 2 s, and a recovery delay of 1.5 s. 1H WALTZ16 decoupling was employed during the acquisition time. The 1/(4Jcc) delay was set to 0.0625 s, corresponding to a carbon-carbon coupling contstant (Jcc) of 4 Hz. The methionine spectrum was acquired on a sample of 50 mM [methyl-13C]-L-lmethionine in D2O, pH 7.1. The t-butyl methyl ether and t-butyl methyl sulfide spectra were acquired with neat samples containing 10% acetone-d6 for 2H lock. The 3JCβCε value for Met1 of ubiquitin was also determined using the INADEQUATE experiment. The sample used in this study was 1 mM U-[13C,15N]ubiquitin in 90% H2O/10% D2O, pH in 10 mM phosphate pH 6.5 buffer containing 1 mM sodium azide, 0.2 mM EDTA, and 10 μM DSS as a chemical shift standard.
Theoretical Calculations
All quantum mechanics (QM) calculations used Gaussian03 (Frisch et al. 2004) and employed the tight self-consistent field option. All reported structures are optimized at, and NMR parameters determined at, the B3LYP/6-311+G(2d,p) level of theory unless otherwise indicated. Variation of the S-CH2 (S-CH3) bond length over the χ3 torsion range is: 1.842 ± 0.008 Å (1.824 ± 0.001). Similar levels of theory have been used in predictions of J-couplings in saccharide models (Tafazzoli and Ghiasi 2007; Zhao et al. 2008) and amino acid sidechains (Chou et al. 2003). 13C shielding calculations used the gauge-independent atomic orbital (GIAO) method (Wolinski et al., 1990; Cheeseman et al., 1996). For sets of calculations scanning a dihedral angle, the torsion in question was held fixed at a given value and all other degrees of freedom were allowed to optimize. Optimal geometry for the gauche conformation of ethyl methyl suflide (EMS) was determined using MP2/6-311+G(2d,p) as discussed previously(Butterfoss and Hermans 2003).
Theoretical 3JCSCC and 3JCSCH coupling constants for EMS determined as a function of dihedral angle using the QM models were subsequently fit to Karplus relations of the form:
| [1] |
using the non-linear least squares fitting routine of Mathematica (Wolfram Research, Champaign, IL). Numerical fits of the theoretical 2JCSC coupling and the theoeretical shielding data obtained for EMS were made to a Fourier cosine series of the form:
| [2] |
The chemical shielding tensor is a symmetric, second-rank tensor and can thus be described by three principal components: σ11, σ22, and σ33 and their orientations. The isotropic chemical shielding, σiso, is given by σiso = Trσ = (σ11 + σ22 + σ33) and by convention is reported in parts per million from the bare 13C nucleus.
Molecular Modeling
The structure of the calmodulin-M13 peptide complex was modeled using the Amber force field. The initial structure used in the molecular modeling work was based on the best NMR structure given in pdb entry: 2BBM. The initial positions of the 4 calcium ions were the same as in the pdb structure and the system including peptide plus the calcium ions was solvated in a bath of water (15,974 water molecules) with water molecules extending a minimum of 15 Å away from any protein atom in a given direction. Prior to equilibration, the system was subjected to energy minimizations at various levels followed by a low temperature constant pressure molecular dynamics period to ensure a reasonable starting density. Step-wise heating at constant volume to bring the temperature to 300° K followed by a 2 ns constant volume molecular dynamics simulation completed the equilibration. Final unconstrained trajectories for 30 ns were calculated at 300° K under constant pressure of 1 atm (1 fs time step) using the particle mesh Ewald molecular dynamics (PMEMD; Amber.10) to accommodate long range interactions. The interaction parameters were taken from the FF03 force field of the Amber suite (Case et al. 2008).
Nomenclature
Following Lovell et al. (2000) we have used “p”, “m” and “t” to refer to methionine χi values near +60 −60° and 180°, respectively. For example, the pmt conformation indicates χ1 ~ +60, χ2 ~ −60, and χ3 ~ 180°. In studies referring to the complex between calmodulin and a 26-residue peptide, “M13”, derived from myosin light chain kinase, we have italicized the name of the peptide in order to avoid confusion with a methionine residue. The subscript M13 is used to indicate residues located on the peptide; for example, residue alanine-13 on the peptide is referred to as A13M13.
Results
Coupling constant analysis
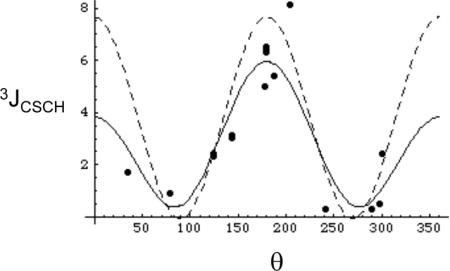
Measurements of three bond coupling constants provide the most consistent and reliable basis for rotamer analysis (Chou et al. 2003; Contrera and Peralta 2000). We are unaware of previous analyses of the dihedral angle dependence of 3JCSCC for methionine or related compounds, however, two previous studies have investigated the conformational dependence of 3JCSCH (Tvaroska et al. 1992; Tafazzoli and Ghiasi 2007). In the present study, we utilized density functional theory (DFT) employing several different basis sets in order to determine the dihedral angle dependence for the model compound ethyl methyl sulfide (EMS), which we believe provides a reasonable model for the 3JCSCC and 3JCSCH coupling interactions involving the methionine methyl group. The results obtained using the B3LYP/6-311+G(2d,p) level of theory were readily interpreted based on a Karplus-type dependence on the subtended dihedral angle, yielding the following parameters (Figure 1):
| [3] |
Some comparisons with results obtained using other basis sets are included as Supplementary Material. For completeness, the 2JCSC results obtained from the same calculation are shown in Figure 1b. The smooth curve corresponds to a fit of the data with a Fourier cosine series:
| [4] |
Based on this calculation, the 2JCSC value is expected to vary between −0.7 and −0.9 Hz over the energetically favorable range.
Figure 1.
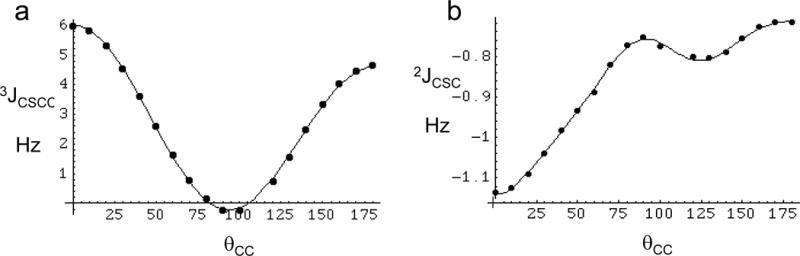
Conformational dependence of scalar coupling constants in ethylmethyl sulfide. a) The filled circles (③) correspond to the calculated 3JCSCC values, and the smooth curve to the fitted values, Eq. 3; b) the filled circles (③) correspond to the calculated 2JCSC values, and the smooth curve to the fitted values, Eq. 4.
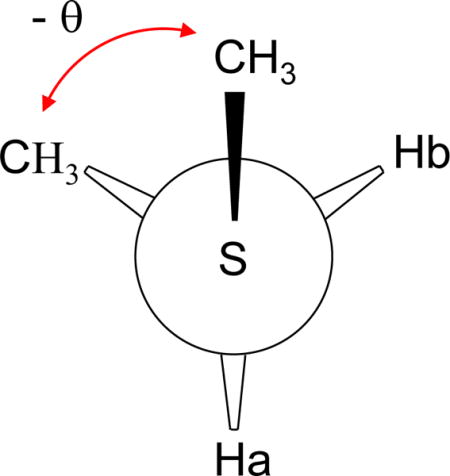
In contrast with the 3JCSCC calculations, analysis of the dihedral angle dependence of the 3JCSCH coupling interaction exhibited more complex behavior. Analysis of the calculated coupling constants between the methyl carbon and either of the methylene protons indicates that there is significant asymmetry about the CSCH dihedral angle, i.e., 3JCSCH(+θ) ≠ 3JCSCH(-θ), with the discrepancy reaching ~ 1 Hz for some values of θ. This behavior is most clearly illustrated by a plot of either 3JCSCHa or 3JCSCHb vs. the absolute value of θ (Supplementary Material). A more comprehensive analysis reveals that the 3JCSCH curve for each proton is shifted in the opposite sense relative to the best fits obtained using standard Karplus relations (Eq. 1), (Figure 2a, b). Based on the simplicity of the model compound, the asymmetry characterizing the 3JCSCH coupling interaction can be described mathematically by utilizing terms that depend on both the subtended dihedral angle between the coupled nuclei, θ, as well as on the angle describing the relative orientation of the two methyl carbon atoms, θ′. The geometric constraint of the molecule requires that θ′ = θ ±120°, with the sign dependent on which proton is being observed. The coupling constant data for both Ha and Hb were thus fit to a relation of the form:
| [5] |
where the dihedral angle describing the relationship of the S-methyl carbon with Ha (Hb) is equal to θCC +120° (θCC−120°). Figure 2a,b illustrates the best fits obtained for both the 3JCSCHa and 3JCSCHb calculations using a Karplus equation (Eq. 1), and Figure 2c,d shows the fits obtained using Eq. 5, which yields the parameters indicated below:
| [6] |
where the negative (positive) sign in the last term of Eq. 6 corresponds to 3JCSCHa (3JCSCHb). Inclusion of a Cosθ term did not improve the fits of the offset curves and so this term was not included. To summarize, the 3JCSCH value will be dependent on both the CSCH and CSCC dihedral angles, θ and θ′ = θ ± 120°. As another way of viewing this result, the use of a single coupling constant, Jg, to describe the coupling constant of the 1H-13C pair in a gauche orientation must be replaced by the use of two coupling constants, Jg,gCC and Jg,tCC, which also depend on the orientation of the two carbon nuclei. As a result of the shifted coupling constant curves illustrated in Figure 2, the values will generally differ substantially, with Jg,gCC > Jg,tCC, i.e., the gauche 3JCSCH coupling is reduced if the CSCC bond angle is trans. In some situations, it may be useful to include an expression for the average coupling constant that neglects this additional complexity:
which after simplification using trigonometric identities becomes:
| [7] |
Figure 2.
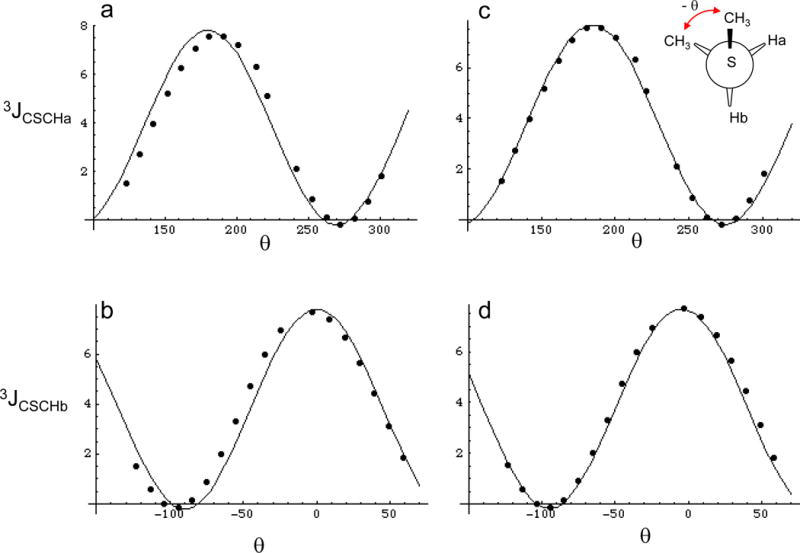
Conformational dependence of 3JCSCH. Panels a and b show the calculated coupling constants at discrete values represnted by the closed circles, and the Karplus relations that best fit the data for 3JCSCHa and 3JCSCHb. Panels c and d were fit using equation 6, where the - value corresponds to Ha and the + value to Hb. 360° was added to negative values of θ in order to make the data cover a continuous range, however the resulting Karplus relations are not altered by this procedure.
The above relations may be compared with the relation obtained by Tvaroska et al. (1992) based on studies of a set of conformationally-constrained thioglycosides:
| [8] |
and to the result given by Tafazzoli and Ghiasi (2007):
| [9] |
There are substantial differences between the values predicted by Equations 7 and 8 or 9 near θ = 0, but no data is available near this value, while the trans coupling constant obtained using Eq. 7 is ~ 1.5 Hz larger than the value obtained using Eq. 8 or 9. In general, the greater magnitude of the 3JCSCH values obtaining using Eq. 6 or 7 is consistent with the fact that the model compounds investigated by Tvaroska et al. are more highly substituted than the methyl ethyl sulfide used for the present calculations. The lower values obtained on model compounds containing additional substituents in the coupling pathway has been discussed by Zhao et al. (2008), and a similar effect is observed in the results for the t-butyl compounds discussed below. A comparison of the coupling data of Tvaroska et al. (1992) with Eq. 7 is included in the Supplementary Material.
Conformational dependence of 13C shielding
Recent evaluations of the relationships between sidechain 13C shift behavior and residue conformation indicated that gauche conformations of methionine χ3 are associated with an upfield shift of −1.8 ppm (London et al. 2008). Such an upfield shift would presumably have the same origin as the extensively studied γ-gauche effect in hydrocarbons (Grant and Cheney 1967; Tonelli 1984; Tonelli et al. 1984). As noted in our recent study, the interpretation of such crystallographic/magnetic resonance correlations is limited by the higher temperatures used in most NMR studies so that the NMR observation corresponds to a thermal average rather than to the unique conformations identified in the crystal. Solid state NMR studies of methionine crystals also have demonstrated the existence of a significant upfield shift for the gauche conformers (Diaz et al. 1986). In the study of Diaz et al. (1986), the 13C shifts for both enantiomerically pure methionine as well as for D,L-methionine mixtures were correlated with conformational data from crystal structures. The results, summarized in Table 1, include 13C shift data for crystallographically inequivalent A and B methionine molecules in L-methionine crystals, as well as for the α and β crystalline forms of D,L methionine; upfield 13C shifts for the gauche conformers of magnitudes −2.0 ppm and −2.9 ppm are obtained from the two comparisons.
Table 1.
Solid State NMR and crystallographic data for Methioninea
| Crystal Structure | δCγ | δCε | χ3 |
|---|---|---|---|
| L-methionine, molecule A | 32.0 | 18.3 | 179.7° |
| L-methionine, molecule B | 32.0 | 16.3 | 73.6° |
| α-D,L-methionine | 31.7 | 15.8 | 80.5 |
| β-D,L-methionine | 32.3 | 18.7 | 190.4 |
Assembled from data in Table 1 and Figures 3 and 5 of Diaz et al. (1986). In contrast with Cε, the shift of Cγ varies by only 0.6 ppm.
The ethyl methyl sulfide model used for the calculation of scalar coupling constants is also expected to provide a useful model for calculating the effect of conformation on shielding of Cε. In contrast with the nuclei of amino acid sidechains located closer to the peptide backbone, e.g. valine Cγ (Pearson et al. 1997), the position of methionine Cε should result in shielding parameters that are much less dependent on backbone torsional angles.
Calculated isotropic shielding values for the SCH3 carbon as a function of dihedral angle were fit to a Fourier cosine series (shielding anisotropy values are included as Supplementary Material):
| [10] |
A plot of σiso-σref is shown in Figure 3, along with the methionine Cε shift values reported in the solid state NMR studies of Diaz et al. (1986). Including σref allows comparison with experimental data using a particular chemical shift reference. In the figure, we set the reference shift σref = 178.8 ppm, which yielded optimal agreement with the solid state NMR values. The consistency between the experimental data and the theoretical curve supports the conclusion that the conformational dependence of the 13Cε shift in the methionine crystals can be adequately described by the approximations inherent in the theoretical analysis of the EMS model compound.
Figure 3.
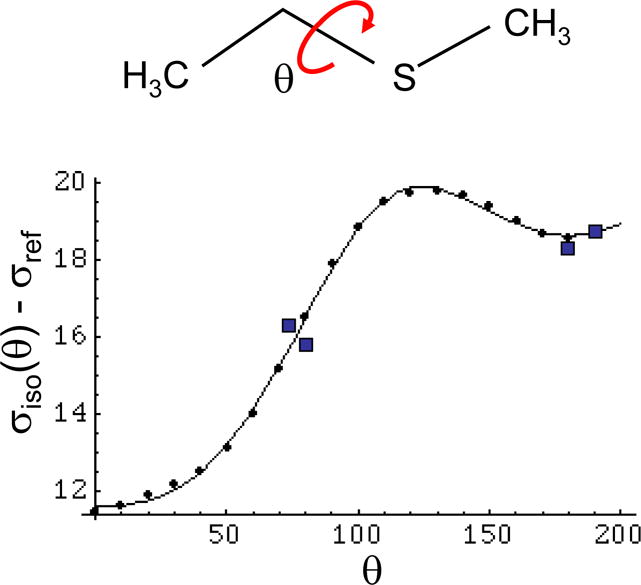
Isotropic shielding of the S-CH3 carbon. Theoretical determination of the isotropic shielding for the S-CH3 group of methyl ethyl sulfide as a function of dihedral angle. The discrete points correspond to the calculated values, and the smooth curve is the Fourier series approximation given in Equation 10. The value of σref = 178.8 ppm was chosen to give the best agreement with the solid state NMR values obtained by Diaz et al. (1986) for methionine (Table 1), which are indicated by squares.
A slightly different value of σref was used in order to fit the data for free methionine in solution. This value was selected in order to optimize the agreement with the 13Cε shift for free methionine measured relative to DSS, in combination with the theoretical MP2 calculations for EMS, which indicate a gauche/trans energy difference of 0.39 kcal/mol and(Butterfoss and Hermans 2003), leading to the conclusion that free methionine has a 21% probability of adopting a trans conformation with χ3 = 180°, and a 79% of adopting a gauche conformation with χ3 = 67°. This approach leads to a value for σref = 179.9, 1.1 ppm greater than the value using the solid state data. In combination with Eq. 10, this value for σref indicates that the gauche conformation of methionine defined by χ3 = 67°, is associated with a shift of ~ 15.85 ppm compared with a trans shift of 19.49 ppm (χ = 180°), giving Δgt = 3.64 ppm. As expected, this predicted shift difference is larger than the statistical value determined from a comparison of NMR shift data with the most probable conformation identified in a set of protein crystal structures, due to the fact that in proteins, methionine residues generally adopt a conformational mixture rather than the single conformation usually observed in protein crystals.
Relationship of Shielding with Coupling constant
Multiple approaches are available for interpreting shift and coupling data in terms of methionine conformation. The issue of whether methionine residues in proteins exhibit rotameric χ3 behavior has been controversial, however the analysis by Word et al. (1999) suggests that this is the case, particularly if consideration is limited to residues with lower B-factors. Assuming that the methionine χ3 can be described as a mixture of gauche and trans conformers, the observed shift and coupling constant can be expressed as weighted averages according to:
| [11] |
In combination, the above relations also imply a linear dependence of δobs on 3Jβε:
| [12] |
The validity of these relations is dependent on two important assumptions that are discussed more fully below: 1) there are no other significant contributions to the 13Cε shift, and 2) the conformational behavior of χ3 is adequately represented as a simple gauche/trans equilibrium in which the two gauche conformations exhibit similar shift and coupling constant values. Calculations for the ethyl methyl sulfide model indicate conformational minima at χ3 = ±67°, 180°. However, the barrier heights are significantly lower than those for the corresponding aliphatic chain so that, for example, the trans conformation is actually an energy-weighted average of values that may extend over a range of ± 15° or greater (Butterfoss et al. 2005), and nominal gauche conformations may deviate significantly from the 67° minimum predicted for the model compound using the ab initio MP2 calculation presented previously (Butterfoss and Hermans 2003). This conclusion is consistent with evaluations of protein crystal structures by Word et al. (1999), who find that, even selecting for structures with the lowest B-factors, methionine χ3 values deviate from the ideal staggered conformation by as much as 36°. Equations 11 and 12 remain valid, however, if the equilibrium involves any two normalized states or, as in the above example, three states with two of them being equivalent. However, it will not be possible to apply the above relations to a group of methionine residues if the nominal gauche and trans states differ significantly from one residue to the next.
Utilizing the calculations given above and assuming gauche and trans minima correspond to 67° and 180°, the parameter values for Equations 11 and 12 are:
| [13] |
| [14] |
Unfortunately, the exact values of the slopes and intercepts used in these equations is highly sensitive to value of σref and the angles selected for the gauche and trans conformers.
Further insight into the relationship between shift and coupling constants is obtained from a parametric plot of the calculated shielding (Equation 10) vs. 3JCSCC values (Equation 3) as a function of θ (Figure 4). The dihedral angles are indicated for some of the data points calculated for EMS; each point corresponds to a 10° increment in χ3, with the exception of missing values for χ3 = 110°. In this figure, we have indicated the generously allowed trans and gauche conformers with red ovals. From the figure, it becomes clear that a substantial deviation of the χ3 value from 180° in the trans rotamer will not result in a large change of the predicted shift, but can significantly reduce the 3JCSCC value. In contrast, significant deviations from the ideal gauche geometry can dramatically alter both the shift and coupling constant. For example, for θg = 100°, the 13Cε shift of the gauche conformer becomes larger than that of the trans conformer. Analysis of experimental data for calmodulin presented below indicates that in some instances, such large deviations are required to explain the observed parameters.
Figure 4.
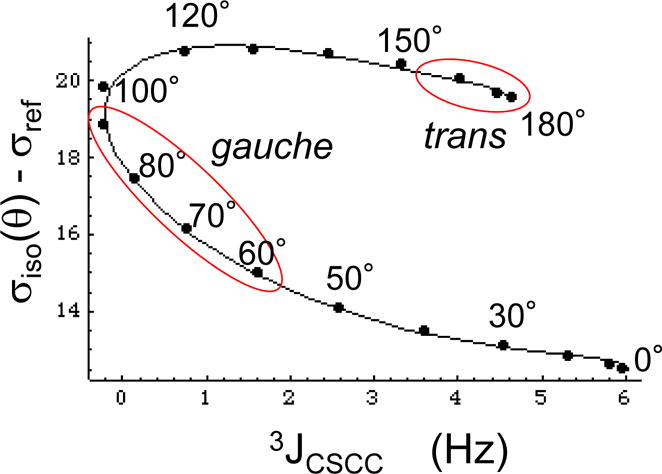
Parametric plot of the theoretical relationship between shielding and vicinal coupling as a function of the dihedral angle θ. The figure shows a parametric plot of 3JCSCC vs. σiso – σref determined for the model compound methyl ethyl sulfide. The points correspond to the values calculated for EMS in 10° increments, while the smooth curves are obtained from Equations 3 and 10. Generously defined trans and gauche parameter ranges are indicated by red ovals. For this calculation, we set σref = 179.9 ppm, as discussed in the text.
Studies of model compounds
Experimental 3JCC data were obtained for methionine, and for the model compounds t-butyl methyl sulfide and t-butyl methyl ether using the INADEQUATE experiment (Bax et al. 1980). This set of compounds is ideally suited to this type of analysis due to the absence of 1JCC interactions for the S-CH3 groups which may be incompletely suppressed using this approach. We considered t-butyl methyl sulfide a useful model since it has only one conformation and known dihedral angles (Figure 5a). The experimental value of 3JCSCC = 1.85 Hz for t-butyl methyl sulfide was compared with values calculated for θ = ± 60°, 180° using the methyl ethyl sulfide model and four different basis sets. The calculated value using B3LYP/6-311+G(2d,p) exceeds the experimental value by ~ 40 %:
| [15] |
It has long been established, however, that additional substituents can influence, and often reduce, vicinal coupling constants (Zhao et al. 2008; Barfield et al. 1980), indicating that EMS might be an inadequate theoretical model for t-buthyl methyl sulfide. We therefore performed an analogous set of calculations directly on the t-butyl methyl sulfide, B3LYP/6-311+G(2d,p), which yielded the values: 3JCSCC(60°) = 1.22 Hz; 3JCSCC(180°) = 2.83 Hz. The calculated weighted mean obtained using these values is within 5 % of the experimental value of 1.85 Hz:
| [16] |
In this example, the addition of two methyl groups that are internal to the coupling pathway reduces both the Jg and Jt values by 21 % and 38 %, respectively. The effect of additional internal substituents is qualitatively similar to that noted by Zhao et al. (2008) in studies of carbohydrate coupling constants.
Figure 5.
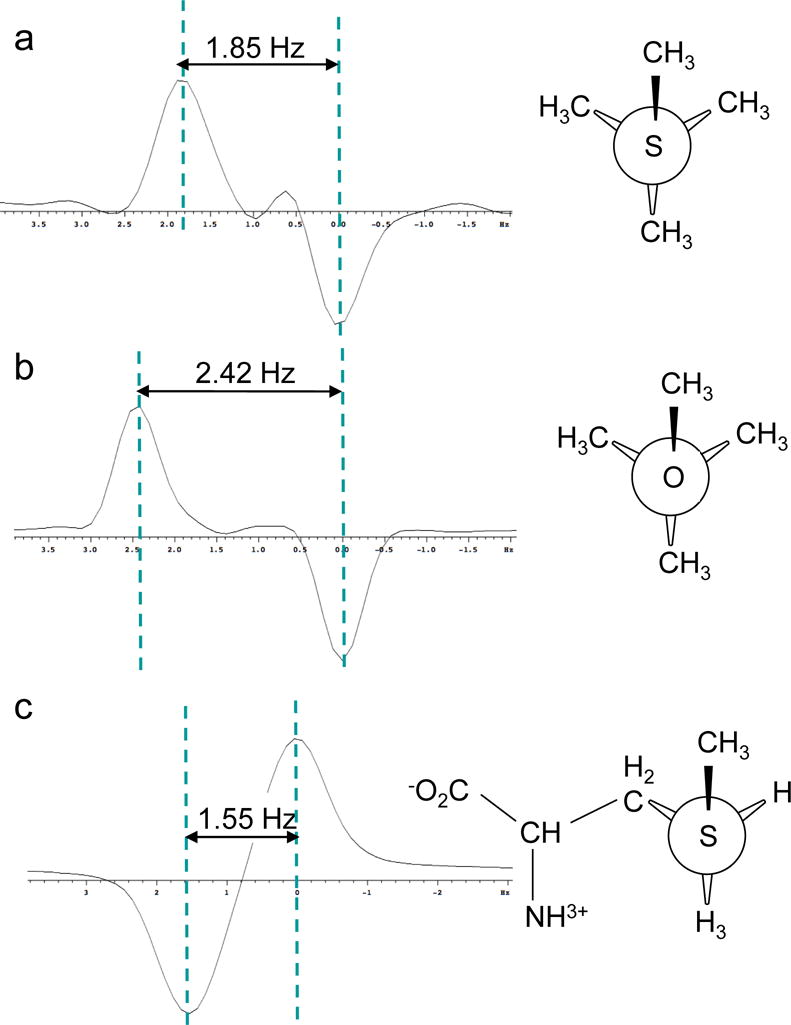
3JCSCC measurements on S-CH3 groups obtained using the INADEQUATE pulse sequence. A) methyl t-butyl sulfide; b) methyl t-butyl ether; c) [methyl-13C]methionine. The first two samples used 10% dissolved acetone-d6 for the lock, and the last was measured in D2O.
In order to further evaluate the effect of the heteroatom, we performed analogous calculations and measurements on t-butyl methyl ether. A weighted average 3JCOCC coupling constant of 2.42 Hz was measured using the INADEQUATE pulse sequence as in the above example. The theoretical coupling constants for the gauche and trans interactions calculated using the same B3LYP model are: 3JCOCC(60°) = 1.75 Hz, and 3JCOCC(180°) = 3.69 Hz, resulting in a predicted weighted average in excellent agreement with the measured value:
| [17] |
On the basis of these model compound studies, we can therefore conclude that the DFT calculations used provide excellent fits for the vicinal 3JCSCC and 3JCOCC coupling interactions involving S or O heteroatoms, and that the couplings are significantly reduced by the presence of methyl groups internal to the coupling pathway. Based on the results for the model compounds, replacement of oxygen with sulfur substantially reduces both the trans and gauche vicinal carbon-carbon coupling constants, with a ratio 3JCSCC/3JCOCC ~ 0.7.
We next consider the conformation of free methionine. An INADEQUATE experiment on [methyl-13C]methionine gave long range coupling constants of 3JCβCε = 1.55 Hz, 2JCγCε = 0.9 Hz. Assuming a simple gauche trans model, the fractional trans probability is given by:
| [18] |
where 3Jβεexp is the experimental value of 1.55 Hz and the 3JCSCC(θ) values are obtained using Eq. 3. The MP2 calculations on EMS (Butterfoss and Hermans 2003) indicate a gauche energy minimum at θg = 67° and a pt value of 0.2. Using equation 3, we obtain pt = 20.4 % corresponding to θg = 69°. In fact, it is reasonable to expect that free methionine would have a somewhat larger value for χ3, since in a few conformations steric interactions with the amino acid backbone result in a significantly larger χ3 value (Butterfoss et al. 2005; Lovell et al. 2003). A more detailed discussion of the conformation of free methionine is included as Supplementary Material. The measured 2JCγCε value is consistent with the calculation of Figure 1b and with a predominantly gauche conformation.
As discussed below in connection with the data on calmodulin, we also sought a model system in which the methionine χ3 angle was held in a trans orientation. We ultimately selected the M1 residue of ubiquitin, for which both crystallographic and NMR structural data are available (Vijaykumar et al. 1987; Ramage et al. 1994; Cornilescu et al. 1998). In the two crystallographic studies, The χ3 for M1 has values of 177.5° (pdb: 1UBQ) and −177.9° (pdb: 1UBI). All of the 10 NMR structures reported for the 1D3Z ensemble also correspond to a trans conformation for χ3 of Met1, however, the values are significantly lower, averaging −145.3°. The 3JCβCε value for M1 determined using the INADEQUATE experiment was 3.2 Hz, well below the theoretical value of 4.6 Hz predicted by Equation 3 with a dihedral angle of 180°. This result can be explained either on the basis of a different dihedral angle for the nominally trans structure, or as an indication of a conformational mixture, and is discussed further in a later section.
Analysis of Calmodulin Data
To the best of our knowledge, calmodulin is the only protein for which a significant amount of methionine coupling data for Cε has been determined(Bax et al. 1994). The data, corresponding to the complex formed between [U-13C] calmodulin and a 26-residue peptide fragment of skeletal muscle myosin light chain kinase, M13, include 3JCεCβ, 3JCεHγ, and 2JCεCγ values for nine methionine residues. For four of these residues (M72, M109, M124, and M144), 3JCβCε is 0.5–0.6 Hz, and a value of 0.8 Hz was obtained for two others (Met71 and Met76). These very low values cannot be fit within the context of the gauche/trans equilibrium model for χ3 unless we increase the value of θg and require that the fractional probability of the trans conformer is near 0. For example, using Equation 3, 3JCSCC = 0.5 Hz can be obtained for a single conformation with θ = 73°. At the other extreme, the largest 3Jβε values of 2.0 and 2.2 Hz for residues M51 and M145 are still much lower than the calculated value of 4.6 Hz for a pure trans conformation with χ3 = 180°. Overall, we can conclude from the experimental data that the conformational ensemble describing the calmodulin methionine residues, which make up a significant fraction of the ligand interaction surface of the protein, is heavily biased toward χ3 gauche conformations, and probably also contains several residues with significant populations having θg χ3 values > 70°.
The shifts of the methionine methyl resonances are well dispersed. Although the methyl group of M145, which exhibits the largest coupling constant, exhibits a relatively large shift that is consistent with a relatively greater fraction of a trans χ3 conformer (Eq. 12), several of the residues with very low 3JCβCε values also exhibit large shifts. For some of these residues, these values probably result from gauche conformers with angles in the 80 – 100° range. As illustrated in Figure 4, gauche conformations with larger dihedral angles correspond to small coupling constants and larger downfield shifts (reduced shielding). The presence of such conformational states for methionine is supported by the analysis of crystallographic structures by Word et al. (1999), who noted that the methionine methyl groups deviate by as much as 36° away from staggered, while the largest deviation found for alanine methyl groups was only 16°.
Consistent with the location of most of the calmodulin methionine residues on the peptide interaction surface, large shift perturbations have been observed to result from formation of the M13 peptide complex (Elshorst et al. 1999). Residues M71, M124, M145 undergo large upfield 1Hε shifts, M109 and M144 exhibit smaller upfield 1Hε shift perturbations, and residue M72 undergoes a dramatic −2.8 ppm upfield 13C shift change. However, application of the program SHIFTX to the NMR-determined structure of the calmodulin-M13 complex (pdb code: 2BBM; Ikura et al. 1992) fails to predict most of these 1H shifts (Figure 6). In order to better understand the behavior of these methionine residues, we performed a set of AMBER simulations on the structure of the calmodulin-M13 complex.
Figure 6.
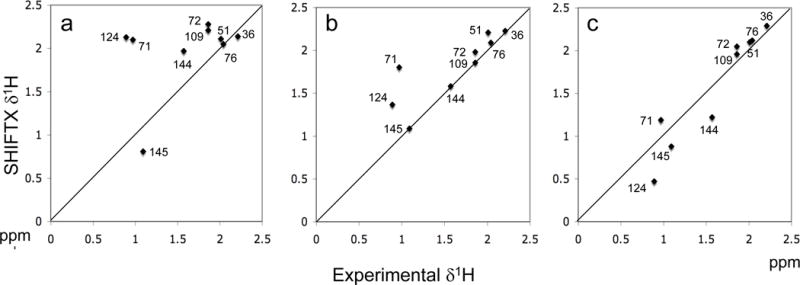
Calculated vs. experimental δ1H shift values for the methionine methyl resonances of calmodulin complexed with the M13 peptide. Panel a used the 2BBM structure for the SHIFTX calculations; panel b represents an average of 15 SHIFTX calculations for Amber-generated structures covering the period from 14 to 30 ns; panel c corresponds to the best of these calculations obtained for the 22 ns simulation. The correlation coefficients were determined to be r=0.41, r=0.83, and r=0.95, respectively.
Amber modeling of the calmodulin-M13 complex
Since as noted above, a simple gauche/trans model restricted to approximately staggered conformations for χ3 was found to be inadequate to allow interpretation of much of the calmodulin methionine data, we performed more extensive molecular modeling calculations using the program AMBER. The time dependent values for χ1, χ2, and χ3 of each methionine residue determined during the simulation period between 14 and 30 ns are included as Supplementary Material. In general, each methionine residue exhibits unique behavior. Some residues, e.g. the linker residue M76, rapidly sample multiple conformations, others change conformation much more infrequently, and still others exhibit very strong preferences for one or two conformational states. For example, the M145 conformation is predominantly mmt.
As noted above, the methyl resonance of calmodulin residue M72 exhibits a −2.8 ppm shift upon formation of the M13 complex. Since this shift change involves a comparison of the uncomplexed and complexed structures, we looked for changes in the conformational distribution of M72. In ligand-free Ca-calmodulin, represented by crystal structures 3CLN (Babu et al. 1988) and 1CLL(Chattopadhyaya et al. 1992), M72 exhibits a trans χ3 conformation, with values of −166° and 174°, respectively. In the NMR structure of the M13 complex, pdb code 2BBM, χ3 has an intermediate value of −136.5°, suggesting an average of gauche and trans conformations. However, in the Amber-minimized structures, the M72 methyl group moves much closer to the M13 peptide backbone and to the methyl group of A13M13. Although NOE restraints were observed between M72 Hε and the Ha and Hb protons of A13M13, the distances of the corresponding carbon nuclei are ~ 6 Å in the 2BBM structure and in most of the structures of the 2BBN ensemble. The close approach of the M72 methyl group to A13M13 in the Amber simulations is sufficient so that in many of the structures, M72 χ3 cannot be rotated into a trans conformation without additional compensatory conformational adjustments. Thus, the available structural data and Amber simulations indicate that peptide complexation is accompanied by a transition away from a conformation that contains a significant fraction of trans χ3 rotamer to a conformation in which interactions with the peptide ligands limit or preclude the adoption of a trans rotamer. These changes are consistent with the large upfield 13C shift of M72 observed by Elshorst et al. (1999) upon formation of the M13 peptide complex.
It was also of interest to identify the presence of mmp conformations for methionine, since the analysis of crystal structures by Word et al. (1999) indicates that the χ3 values for the mmp conformation are frequently very large. Residues M36, M72 and, to a lesser extent, M76 spend limited periods of the simulation in the mmp conformation during which the χ3 value is elevated to ~ 100° (Supplementary Material). A significant occupancy of the mmp state provides a basis for understanding the combination of a downfield shifted 13C resonance with a small 3JCβCε value, and is consistent with the data for M36 and M76, but less so for M72, which exhibits a δ13C = 15.0 ppm. These instances of the mmp conformation were the only simulation periods during which the amplitude of χ3 exceeded ~ 80° for a sustained period.
Throughout the entire period of the Amber simulations, residue M109 showed a strong preference for only two conformations: mmt and mtt, so that χ3 was almost always trans. This result is consistent with the 2BBM structure, and with the downfield δ13C = 18.2 ppm. However, it is inconsistent with the extremely small 3JCβCε = 0.5 Hz. Although the basis for this inconsistency is not known, the small, 0.2 ppm shift difference between M109 Cβ and Cγ suggests that one possible factor could be the high order perturbation discussed in the following section.
Application of the SHIFTX program to the structure of the Amber-minimized complex produced a significant improvement in the methionine δ1Hε values, with all of the complexation-induced upfield methionine HE shifts at least qualitatively predicted on the basis of this structure. The correlation coefficient increased from r = 0.41 for the SHIFTX calculations using the 2BBM structure to r = 0.83 for an average of 15 SHIFTX calculations that used the simulations obtained between 14 and 29 ns (Figure 6). A plot of the calculated vs. experimental shift values is also given for the best simulation, corresponding to the AMBER simulation at 22 ns (Figure 6c). Analysis of the structures resulting from the Amber minimization indicates that these 1Hε shift perturbations result primarily from closer contacts between the methionine sidechains and aromatic residues of the M13 peptide. More specifically, the methyl group of M71 moves closer to F17M13, M124 moves closer to W4M13, and M145 moves closer to F8M13. The two methyl resonances that experience smaller upfield shifts also move closer to aromatic peptide residues: M109-W4M13. And M144-W4M13. With the exception of M71-F17M13, intermolecular NOEs were observed between each of the corresponding methionine Hε protons and protons on the nearby aromatic residues (Ikura et al. 1992). Interestingly, the Amber minimization moved the methyl groups closer to the nearby aromatic groups, resulting in more accurate shift predictions as determined by the SHIFTX calculations, and in greater agreement with the intermolecular methionine NOE data for the calmodulin-M13 complex, although the NOE data were not included in the Amber minimization. This exercise also supports the value of using SHIFTX sidechain shift predictions as constraints in the determination of solution structures using NMR data.
High Order effects in the analysis of methionine coupling constants
One important limitation of the analysis of the reported coupling data results from the high order effects expected to characterize many of the observed coupling interactions for the calmodulin methionine resonances. For more than half of the methionine residues in the calmodulin-M13 complex the shift difference between Cβ and Cγ was measured as 0.1 or 0.2 ppm. This small shift difference leads to significant high order perturbations for 3Jβε and 2Jγε. Modeling Cβ, Cγ, Cε as an ABX spin system (Bernstein et al. 1957), the apparent coupling constants can be approximated as:
| [19] |
These relations predict that as νβ− νγ → 0, the multiplet structure of the Cε methyl group will change from a doublet of doublets to a 1:2:1 triplet. As an illustration of this perturbation, we have calculated the apparent coupling constants defined in the above equation for values.
as a function of νβ− νγ (Figure 7). For Δβγ = νβ− νγ values less than ~ 0.3 ppm, the apparent 3Jβεapp value is significantly reduced, while the apparent 2Jγεapp value is increased. Analogous effects will also be present for some of the 1H coupling interactions involving Hγ1 and Hγ2. Since the very small 3JCC values are observed for the same residues for which νβ− νγ is 0.1 – 0.2 ppm, it seems likely that these very low values may be reduced as a consequence of high order effects, and thus underreport the true coupling constant values. Surprisingly, the measured 2JCγCε values in calmodulin are all below the values determined for free methionine and predicted from the DFT calculations (Figure 1b) although, as indicated by the simulation of Figure 7, high order effects would be expected to increase the apparent 2JCγCε values. Although the reason for this discrepancy is not clear, these very small coupling constants are difficult to measure accurately.
Figure 7.
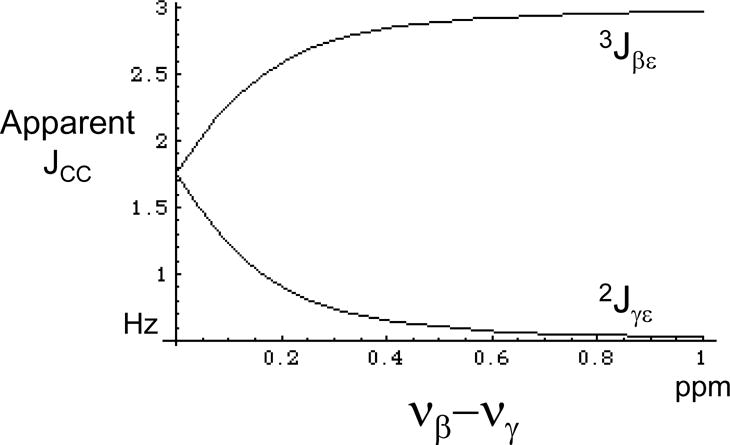
Effect of high order interaction on the apparent 3JCC and 2JCC coupling constants. The apparent coupling constants between Cε and Cβ, and between Cε and Cγ are plotted as a function of the chemical shift difference between Cβ and Cγ, Δβγ = νβ − νγ (expressed in ppm for Ho = 14.1 T). Values used for the simulation are: 1JCβCγ = 33 Hz, 3JCβCε = 3.0 Hz, and 2JCγCε = 0.5 Hz. At zero shift difference, the virtual coupling effect is complete and the Cε resonance appears as a triplet with apparent coupling constant = ½(3JCβCε + 2JCγCε).
Other contributions to the methionine 13Cε chemical shifts in proteins
According to the calculation of Figure 3, conformationally-dependent “γ-effect” shift contributions span a total shift range of up to 8.4 ppm, although ~ 2 ppm of this range corresponds to values below 50° which are likely to be rarely encountered; there are no entries for 0 ± 40° in the methionine χ3 data set with B-factors < 30 (Word et al. 1999). Our previous analysis of assigned methionine residues yielded a similar range of ~ 6 – 7 ppm, after discarding two extreme outliers (London et al. 2008). This comparison suggests that the intra-residue conformationally-dependent shift contributions included in the calculations on the EMS model compound are the dominant determinant of the methionine 13Cε shift dispersion in proteins, and also supports the conclusion that the shift is primarily dependent on the χ3 conformation.
One important factor that reduces the contribution of other environmental influences on the methionine methyl shift is the very low order parameter that generally characterizes this group. Analysis of the methyl relaxation behavior for eight proteins by Mittermaier et al. (2003) reported a mean value of Saxis2 = 0.22 for the methionine methyl group, less than half the mean value of 0.47 for the leucine and isoleucine Cδ methyls. Since intermolecular shift contributions will depend on the relative orientations of each perturbing influence and the methionine sidechain, it is reasonable to conclude that in general, the relative significance of intermolecular shift contributions as a source of 13Cε shift dispersion will be lower for methionine than for other methyl resonances.
Averaging of the electric field effects was observed for several small molecules. We compared the 13C titration shifts determined for two sulfur-containing analogs of aliphatic compounds previously studied by Batchelor et al. (1975) in their analysis of electric field shift contributions: methylthioacetate, an analog of butyrate, and 2-thiomethyl ethylamine, an analog of n-butylamine (Supplementary Material). For the aliphatic compounds, increasing the pH (deprotonation) produces an upfield shift of the methyl carbon resonance. The corresponding titration shifts for the sulfur-containing analogs are significantly lower, despite the larger polarizability of the S-CH3 bond. This behavior results from the greater averaging of the polarizing effects of the charge, which in turn results from the conformational heterogeneity of the sulfur-containing analogs, corresponding to much larger gauche/trans ratios about the CH2-S bond. Paradoxically, the above analysis suggests that shift analyses for the methionine residues that are most conformationally pure will also be subject to the largest errors if environmental shift perturbations are neglected.
Methionine conformational analysis in other proteins
Although careful analysis of crystallographic data has demonstrated that a small but significant percentage of the χ3 torsional angles for the C-S bond in well ordered methionine residues deviate by a full 60° from a perfectly staggered geometry (Butterfoss et al. 2005), the overall behavior of χ3 is statistically consistent with a significant preference for rotameric states (Word et al. 1999; Lovell et al. 2003). From a technical standpoint, NMR data are limited by the paucity of 3JCβCε data, problems of resolving coupling interactions in large proteins, and by the potential of high order perturbations resulting from the small νβ− νγ shift differences that sometimes occur. Crystallographic data also are prone to error due to the large electron density of the sulfur atom, which tends to obscure the position of the terminal methyl group. Additionally, the low rotamer interconversion barriers also increase the χ3 deviation from canonical rotameric states. Although in some cases eclipsed methionine χ3 rotamers can result from the local protein environment (Butterfoss et al. 2005), the large number of reported χ3 values near 120°–140° suggests that there is a general over-reliance on the use of a single rotamer model in situations that correspond to a mixture of at least two methionine χ3 rotamers.
Despite these limitations, we have found that analysis of the methionine Cε NMR data, and particularly interpretation of the 13C shift, can provide useful insight if sufficient caution is used in the interpretation. If a gauche/trans model is assumed, then using the calculated shift values derived above, Equation 10 can be written as:
The accuracy of this approach can be somewhat improved by introducing a correction for the ring current shift contributions of aromatic residues. The simplest empirical approach is to assume that the measured δ1H for the methyl protons is dominated by the ring current contribution (Perkins and Dwek 1980), and that on a ppm scale, δ13C ~ δ1H. The latter approximation improves for methyl groups that are further from the ring. Taking the unshifted δ1H value for the methionine methyl protons as 2.10 ppm (Wishart et al. 1995), we have:
| [20] |
where in addition to the 1H shift correction, we have used the theoretical trans shift value of 19.5 ppm obtained above for EMS, and reduced δg to 15 ppm allowing for a smaller gauche angle. This approximation can also be viewed as allowing for small shift contributions arising from other mechanisms, and also indicates the approximate nature of the trans probability predicted using Equation 20. The above formula must be viewed as a probability of a trans conformation. It fails primarily for conformations characterized by unusually large gauche angles, which, e.g., characterize the MMP conformation, and in instances where significant shift contributions arise from other mechanisms. Shift values falling outside the allowed range need to be evaluated on a case by case basis.
As noted in the section on model compounds, we initially considered ubiquitin M1 as a useful model for a methionine residue with a trans χ3 conformation. However, the 3JCβCε value of 3.2 Hz determined for ubiquitin M1 is significantly below the value of 4.59 Hz expected for a methionine residue with a trans χ3 orientation. Application of Eq. 18 using θg = 70° and θt = 180° results in pt =0.64:
Application of Equation 20 to ubiquitin M1, with shift parameters δ(1H,13C) =(1.63,17.8), gives pt = 0.73, and exact agreement can be obtained obtained if the nominally trans conformation is assumed to deviate from 180°, as suggested by the NMR structure. Methionine M1 has a significant fractional solvent exposure, ~ 0.23, consistent with the possibility that at the measurement temperature it may adopt conformations that differ significantly from the PTT conformation observed in the crystals.
The above relation provides a useful basis for estimating the gauche/trans conformational behavior of other methionine-labeled proteins under investigation in our laboratory: UvrB, HIV reverse transcriptase, and DNA polymerase β (Bose-Basu et al. 2004; DellaVecchia et al. 2007; Zheng et al. 2009). In the spectrum of [methyl-13C] methionine-labeled UvrB derived from B. caldotenax, M551 is the most downfield-shifted 13C resonance, with δ(1H,13C) =(1.9,18.7) ppm (DellaVecchia et al. 2007) (Figure 8). Equation 20 predicts a predominantly trans conformation for M551. Although, the χ3 values for M551 of UvrB in both the A and B molecules in the unit cell for structures 1T5L and 2FDC (uncomplexed and DNA-complexed structures) indicate an extreme gauche conformation characterized by very low θ values, analysis of the closely related UvrB structure from B. subtilis (sequence identity = 82%), M551 gives χ3 = 177.9°.
Figure 8.
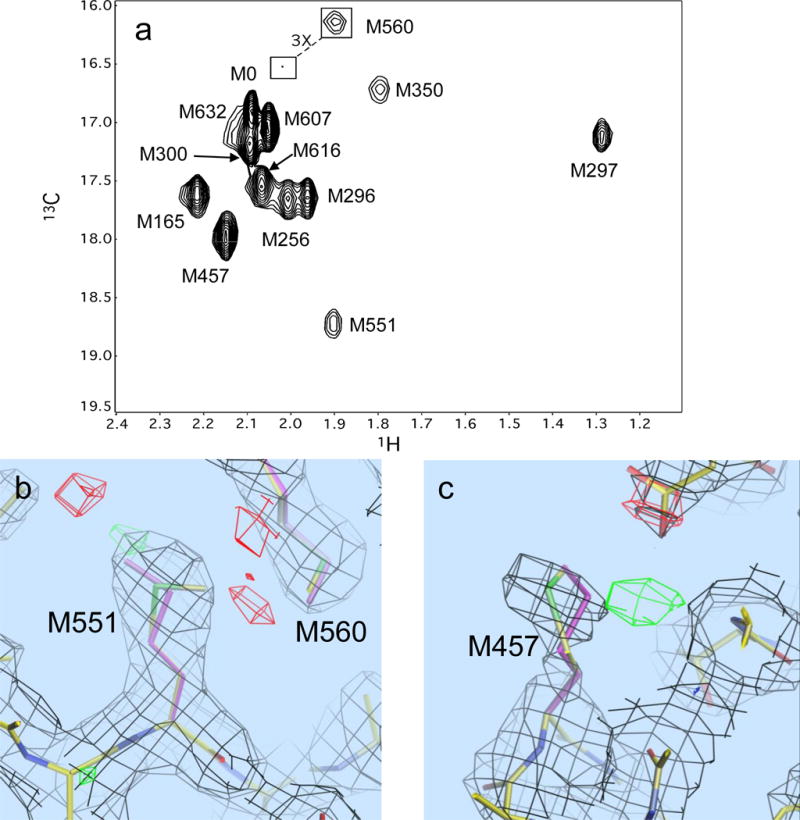
Behavior of the methionine resonances of [methyl-13C]methionine in UvrB from B. caldotenax. Panel a shows the 1H-13C HSQC spectrum with assigned methionine resonances; panel b shows the electron density near M551 of chain B, the originally selected methionine rotamer (green and yellow), and the revised conformation (purple); panel c shows the electron density for M457, the original conformation (green and yellow), and the optimal single rotamer (purple). The inadequacy of the original conformation is indicated by the region in green. For M457, the best fitting single rotamer (purple) provides a poorer fit to the density than a mixture of rotamers (not shown). The red and green polygons correspond to regions of excessive and insufficient electron density predicted by the original conformational selection.
This variation led to a re-evaluation of the B. caldotenax UvrB data. Crystallography-based methionine conformations were re-evaluated by systematically refining the structure starting from each of the 13 common methionine rotamer conformations compiled by Lovell et al. (2000). An Fobs-Fcalc density difference map was scored by summing the absolute value squared, Σ │Fobs – Fcalc│2 over grid points within 2.5 Å of each methionine sidechain atom. Regions of deficient density are contoured as green polygons, and regions of overestimated density are shown as red polygons; significant red/green regions of space surrounding the residue are indicative of suboptimal fits. Following this procedure clearly indicates that the preferred χ3 torsion for M551 of UvrB is trans, as illustrated for chain B in pdb file 1T5L in Figure 8b. A similar analysis also indicates that M457, with the second largest δ13C value, 18.0 ppm, can be fit more accurately using a mixture of gauche and trans rotamers (Figure 8c). Interestingly, older commonly used used rotamer databases lacked a trans rotamer for methionine χ3 (Ponder and Richards 1987; Jones et al. 1991).
For lower resolution crystal structures the electron density is often insufficient to allow a reliable χ3 value to be extracted. The resolution of most of the structures of HIV reverse transcriptase (RT) is generally low; only a single pdb file has a resolution below 2.0 Å. The intense M16 resonance on the p66 subunit exhibits a 13C shift value of 18.3 ppm which, according to equation 20, is consistent with a predominantly trans conformation (Figure 9) (Zheng et al. 2009). Better resolved crystal structures indicate χ3 values near 140°, suggesting a gauche/trans equilibrium. Significantly, structure 1DLO, with resolution 2.70 Å, has the lowest average B-factor for the four heavy atoms defining the M16 χ3 torsion of any of the structures evaluated, Bav = 30.0, and corresponds to χ3 = 173.3°. Examination of the environment of this residue in multiple RT structures indicates that there are significant steric clashes with residues K13 and R83 in either of the possible gauche χ3 conformations (Figure 9), consistent with the predominance of a trans χ3 value.
Figure 9.
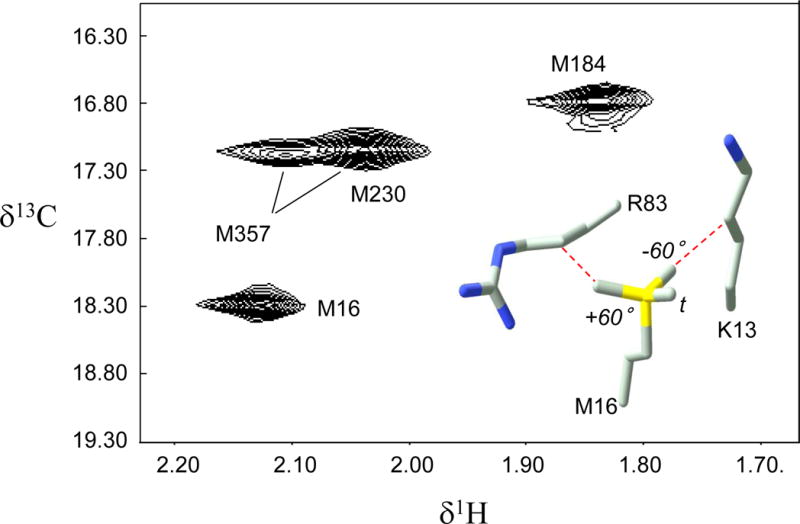
1H-13C spectrum of [methyl-13C]methionine66 HIV-1 reverse transcriptase (RT), where the subscript indicates labeling in the p66 subunit of the RT heterodimer. Assignments are from Zheng et al.29. The insert shows M16 and two nearby residues, K13 and R83, that constrain the conformation of M16. The three possible staggered conformations for M16 χ3 are also shown.
As a third example, we consider the methionine shifts observed for DNA polymerase β, an enzyme involved in base excision repair. It has been demonstrated by crystallographic and NMR studies that formation of an abortive ternary complex results in conformational activation, which is associated with multiple shift perturbations of the methionine resonances (Bose-Basu et al. 2004). The 13C shift of the M158 methyl resonance is increased by ~ 0.5 ppm in this complex, while several crystal structures suggest only small conformational changes of this residue. However, as indicated by Figures 3 and 4, for gauche conformations with χ3 near 70°, small changes in χ3 are associated with relatively large changes in shielding, so that the magnitude of the 13C shift/χ3 ratio > 0.1 ppm/°. Thus the observed 13C shift perturbations can be extremely sensitive to conformation, as illustrated by this example. Two residues with larger 13C shifts, M282 and M191, are predicted to exhibit higher fractional trans probabilities. M282 χ3 adopts a trans conformation in nearly all crystal structures, however, in most crystal structures M191 adopts a gauche χ3 conformation. Interestingly, a recent high resolution structure of Pol ß with a resolution of 1.65 Å has two alternate conformations for this residue, corresponding to χ3 = −77.5° and −154.1° (pdb code 2FMP, Batra et al. 2006). We note here that the shift assignments for M158 and M191 have been interchanged based on recent NOE measurements (unpublished results).
Lattice contacts can perturb the conformations of residues located on the protein surface, however there are numerous examples in which lattice contacts can alter molecular conformation to a more significant degree. The N-terminal lobe of human transferrin provides a relevant example in which crystal packing alters the conformation of an exposed loop, which in turn influences the conformation of the buried methionine residue, M109. This effect is observed in a comparison of structures 1A8E and 1A8F, corresponding to two different space groups (MacGillivray et al. 1998). The change in position of residue L135 which immediately proceeds a loop involved in a lattice contact alters the interaction with M109 in structure 1A8F, changing M109 χ3 from trans to gauche (Supplementary Material). The structure of the full transferrin molecule is characterized by a trans M109 χ3 conformation, as observed in 1A8E, consistent with the conclusion that lattice packing results in the perturbed gauche conformation in structure 1A8F. Based on the reported shifts values of He et al. (1999), application of Equation 20 gives pt = 0.6 for M109, more consistent with the 1A8E structure. This example provides another illustration of the limits of crystallographic studies to the analysis of solution conformation.
As a final example, M247 of the protein synaptotagmin exhibits one of the largest δ13Cε values of 20.86 ppm included in our previous analysis (London et al. 2008), although the crystal structure 1RSY(Sutton et al. 1995) indicates a gauche χ3 conformation. A more detailed examination of the structure indicates that this residue adopts an MMP conformation and exhibits the large θg value that is correlated with this conformational state. This conformational state is estimated to correspond to ~ 3 % of all methionine residues (Butterfoss and Hermans 2003), and hence does not significantly influence the statistical analysis. However, this example illustrates the limits of applicability of the gauche/trans model and the value of additional structural information for the interpretation of the NMR data when this is available.
Conclusions
Methionine labeling provides a useful approach for studies of the conformational behavior of large, complex molecules. As a result of the distance and effective isolation of Cε from the protein backbone, theoretical shielding and Karplus curves determined for ethyl methyl sulfide are useful for interpreting the conformational dependence of the 13C shift and coupling constants for methionine residues in proteins. The validity of the DFT analysis of 13C shielding and scalar coupling constants was also confirmed for several model compounds. The atypical behavior of the methionine χ3 conformation limits the general applicability of a simple gauche/trans model, however useful results can be obtained if sufficient caution is used and/or additional data can be introduced to further constrain the conformational possibilities. Application of this approach to the calmodulin-M13 complex indicates that all of the methionine residues at the interaction surface show a significant preference for gauche conformations. Significantly improved fits of the methionine methyl δ1Hε values were obtained after Amber minimization of the structure, despite the omission of intermolecular NOE restraints, and the results also support the conclusion that the large upfield 13C shift for M72 results from a change of χ3 from a predominantly trans to a predominantly gauche conformation. Application of these analyses to other proteins has in some cases led to a reassessment of crystallographically-determined methionine χ3 values.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences (NIEHS). The contributions of E.F.D. and J.M.K. were funded in whole with Federal funds from NIEHS, under Delivery Order HHSN273200700046U to SRA International, Inc. This research was supported by Research Project Z01- ES050111 (R.E.L.) in the Intramural Research Program of the National Institutes of Health.
Footnotes
SUPPLEMENTARY INFORMATION
Supporting information incudes: 1) Evaluation of 3JCSCC using alternate basis sets; 2) calculated 3JCSCH coupling constants as a function of the absolute value of the subtended dihedral angle; 3) calculated isotropic and anisotropic shielding parameters for EMS; 4) plots illustrating comparisons with Tvaroska Data and Tafazzoli calculations; 5) analysis of free methionine conformation using both 3JCεCβ and 3JCεHγ coupling constants; 6) pH titration results for compounds containing C-CH3 groups; 7) solvent effect on the shift of the methyl resonance of methylthiopropanol; 8) effect of lattice contacts on the conformation of a buried methionine residue – transferrin residue M109; 9) graphical representation of methionine χi values during the Amber simulation of the calmodulin-M13 complex.
References
- Anbanandam A, Bieber Urbauer RJ, Bartlett RK, Smallwood HS, Squier TC, Urbauer JL. Mediating molecular recognition by methionine oxidation: conformational switching by oxidation of methionine in the carboxyl-terminal domain of calmodulin. Biochemistry. 2005;44:9486–9496. doi: 10.1021/bi0504963. [DOI] [PubMed] [Google Scholar]
- Babu YS, Bugg CE, Cook WJ. Structure of calmodulin refined at 2.2 A resolution. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- Barfield M, Marshall JL, Canada ED. Nuclear Spin-Spin Coupling Via Nonbonded Interactions .2. Gamma-Substituent Effects for Vicinal Coupling-Constants Involving C-13. Journal of the American Chemical Society. 1980;102:7–12. [Google Scholar]
- Batchelor JG, Feeney J, Roberts GCK. C-13 Nmr Protonation Shifts of Amines, Carboxylic-Acids and Amino-Acids. Journal of Magnetic Resonance. 1975;20:19–38. [Google Scholar]
- Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax A, Delaglio F, Grzesiek S, Vuister GW. Resonance assignment of methionine methyl groups and chi 3 angular information from long-range proton-carbon and carbon-carbon J correlation in a calmodulin-peptide complex. J Biomol NMR. 1994;4:787–797. doi: 10.1007/BF00398409. [DOI] [PubMed] [Google Scholar]
- Bax A, Freeman R, Kempsell SP. Natural Abundance C-13-C-13 Coupling Observed Via Double-Quantum Coherence. Journal of the American Chemical Society. 1980;102:4849–4851. [Google Scholar]
- Beatty EJ, Cox MC, Frenkiel TA, Tam BM, Mason AB, MacGillivray RT, Sadler PJ, Woodworth RC. Interlobe communication in 13C-methionine-labeled human transferrin. Biochemistry. 1996;35:7635–7642. doi: 10.1021/bi960684g. [DOI] [PubMed] [Google Scholar]
- Bernstein HJ, Pople JA, Schneider WG. The Analysis of Nuclear Magnetic Resonance Spectra .1. Systems of 2 and 3 Nuclei. Canadian Journal of Chemistry. 1957;35:65–81. [Google Scholar]
- Bertini I, Ciurli S, Dikiy A, Fernandez CO, Luchinat C, Safarov N, Shumilin S, Vila AJ. The first solution structure of a paramagnetic copper(II) protein: the case of oxidized plastocyanin from the cyanobacterium Synechocystis PCC6803. J Am Chem Soc. 2001;123:2405–2413. doi: 10.1021/ja0033685. [DOI] [PubMed] [Google Scholar]
- Blakley RL, Cocco L, London RE, Walker TE, Matwiyoff NA. Nuclear magnetic resonance studies on bacterial dihydrofolate reductase containing [methyl-13C]methionine. Biochemistry. 1978;17:2284–2293. doi: 10.1021/bi00605a005. [DOI] [PubMed] [Google Scholar]
- Bose-Basu B, DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE. Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-13C]methionine-labeled DNA polymerase beta. Biochemistry. 2004;43:8911–8922. doi: 10.1021/bi049641n. [DOI] [PubMed] [Google Scholar]
- Butterfoss GL, Hermans J. Boltzmann-type distribution of side-chain conformation in proteins. Protein Sci. 2003;12:2719–2731. doi: 10.1110/ps.03273303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfoss GL, Richardson JS, Hermans J. Protein imperfections: separating intrinsic from extrinsic variation of torsion angles. Acta Crystallographica Section D-Biological Crystallography. 2005;61:88–98. doi: 10.1107/S0907444904027325. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, I, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, et al. AMBER 10. University of California; San Francisco: 2008. [Google Scholar]
- Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. Calmodulin Structure Refined at 1.7 Angstrom Resolution. Journal of Molecular Biology. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- Cheeseman JR, Trucks GW, Keith TA, Frisch MJ. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J Chem Phys. 1996;104:5497–5509. [Google Scholar]
- Chou JJ, Case DA, Bax A. Insights into the mobility of methyl-bearing side chains in proteins from (3)J(CC) and (3)J(CN) couplings. Journal of the American Chemical Society. 2003;125:8959–8966. doi: 10.1021/ja029972s. [DOI] [PubMed] [Google Scholar]
- Contrera RH, Peralta JE. Angular dependence of spin–spin coupling constants. Progress in Nuclear Magnetic Resonance Spectroscopy. 2000;37:321–425. [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. Journal of the American Chemical Society. 1998;120:6836–6837. [Google Scholar]
- Cox MC, Barnham KJ, Frenkiel TA, Hoeschele JD, Mason AB, He QY, Woodworth RC, Sadler PJ. Identification of platination sites on human serum transferrin using (13)C and (15)N NMR spectroscopy. J Biol Inorg Chem. 1999;4:621–631. doi: 10.1007/s007750050386. [DOI] [PubMed] [Google Scholar]
- Deber CM, Moscarello MA, Wood DD. Conformational Studies on 13C-Enriched Human and Bovine Myelin Basic Protein, in Solution and Incorporated into Liposomes. Biiochemistry. 1978;17:898–903. doi: 10.1021/bi00598a024. [DOI] [PubMed] [Google Scholar]
- DellaVecchia MJ, Merritt WK, Peng Y, Kirby TW, DeRose EF, Mueller GA, Van Houten B, London RE. NMR analysis of [methyl-13C]methionine UvrB from Bacillus caldotenax reveals UvrB-domain 4 heterodimer formation in solution. J Mol Biol. 2007;373:282–295. doi: 10.1016/j.jmb.2007.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LE, Morin F, Mayne CL, Grant DM, Chang CJ. Conformational-Analysis of Dl-Methionine, L-Methionine and D-Methionine by Solid-State C-13 Nmr-Spectroscopy. Magnetic Resonance in Chemistry. 1986;24:167–170. [Google Scholar]
- Ding J, Das K, Hsiou Y, Sarafianos SG, Clark AD, Jr, Jacobo-Molina A, Tantillo C, Hughes SH, Arnold E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J Mol Biol. 1998;284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- Duewel HS, Daub E, Honek JF. Investigations of the interactions of saccharides with the lysozyme from bacteriophage lambda. Biochim Biophys Acta. 1995;1247:149–158. doi: 10.1016/0167-4838(94)00207-w. [DOI] [PubMed] [Google Scholar]
- Duewel HS, Daub E, Robinson V, Honek JF. Elucidation of solvent exposure, side-chain reactivity, and steric demands of the trifluoromethionine residue in a recombinant protein. Biochemistry. 2001;40:13167–13176. doi: 10.1021/bi011381b. [DOI] [PubMed] [Google Scholar]
- Elshorst B, Hennig M, Forsterling H, Diener A, Maurer M, Schulte P, Schwalbe H, Griesinger C, Krebs J, Schmid H, et al. NMR solution structure of a complex of calmodulin with a binding peptide of the Ca2+ pump. Biochemistry. 1999;38:12320–12332. doi: 10.1021/bi9908235. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J, Jr, Vreven T, Kudin KN, Burant JC, et al. Gaussian 03, Revision D.02. Wallingford, CT: Gaussian, Inc.; 2004. [Google Scholar]
- Gelis I, Bonvin AMJJ, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG. Structural Basis for Signal-Sequence Recognition by the Translocase Motor SecA as Determined by NMR. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellman SH. On the role of methionine residues in the sequence-independent recognition of nonpolar protein surfaces. Biochemistry. 1991;30:6633–6636. doi: 10.1021/bi00241a001. [DOI] [PubMed] [Google Scholar]
- George GN, Richards T, Bare RE, Gea YJ, Prince RC, Stiefel EI, Watt GD. Direct Observation of Bis-Sulfur Ligation to the Heme of Bacterioferritin. Journal of the American Chemical Society. 1993;115:7716–7718. [Google Scholar]
- Grant DM, Cheney BV. Carbon-13 Magnetic Resonance .7. Steric Perturbation of Carbon-13 Chemical Shift. Journal of the American Chemical Society. 1967;89:5315–&. [Google Scholar]
- Gregoret LM, Rader SD, Fletterick RJ, Cohen FE. Hydrogen bonds involving sulfur atoms in proteins. Proteins. 1991;9:99–107. doi: 10.1002/prot.340090204. [DOI] [PubMed] [Google Scholar]
- Gustavsson N, Kokke BP, Anzelius B, Boelens WC, Sundby C. Substitution of conserved methionines by leucines in chloroplast small heat shock protein results in loss of redox-response but retained chaperone-like activity. Protein Sci. 2001;10:1785–1793. doi: 10.1110/ps.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RE, Dill K. Magnetic resonance study of glycophorin A-containing 13C-enriched methionines. FEBS Lett. 1982;143:327–331. doi: 10.1016/0014-5793(82)80127-0. [DOI] [PubMed] [Google Scholar]
- He QY, Mason AB, Tam BM, MacGillivray RT, Woodworth RC. [13C]Methionine NMR and metal-binding studies of recombinant human transferrin N-lobe and five methionine mutants: conformational changes and increased sensitivity to chloride. Biochem J. 1999;344(Pt 3):881–887. [PMC free article] [PubMed] [Google Scholar]
- Howarth JW, Krudy GA, Lin X, Putkey JA, Rosevear PR. An NMR and spin label study of the effects of binding calcium and troponin I inhibitory peptide to cardiac troponin C. Protein Sci. 1995;4:671–680. doi: 10.1002/pro.5560040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- Jaeck G, Benz FW. Synthesis of ribonuclease labeled with 13C on Methionine-29. Biochem Biophys Res Commun. 1979;86:885–892. doi: 10.1016/0006-291x(79)91795-9. [DOI] [PubMed] [Google Scholar]
- Jones WC, Jr, Rothgeb TM, Gurd FRN. Specific Enrichment with 13C of the Methionine Methyl Groups of Sperm Whale Myoglobin’. J Amer Chem Soc. 1975;97:3875–3877. doi: 10.1021/ja00846a085. [DOI] [PubMed] [Google Scholar]
- Jones WC, Jr, Rothgeb TM, Gurd FRN. Nuclear Magnetic Resonance Studies of Sperm Whale Myoglobin Specifically Enriched with 13C in the Methionine Methyl Groups. J Biol Chem. 1976;251:7452–7460. [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Khavrutskii IV, Price DJ, Lee J, Brooks CL., 3rd Conformational change of the methionine 20 loop of Escherichia coli dihydrofolate reductase modulates pKa of the bound dihydrofolate. Protein Sci. 2007;16:1087–1100. doi: 10.1110/ps.062724307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby TW, DeRose EF, Beard WA, Wilson SH, London RE. A thymine isostere in the templating position disrupts assembly of the closed DNA polymerase beta ternary complex. Biochemistry. 2005;44:15230–15237. doi: 10.1021/bi0511742. [DOI] [PubMed] [Google Scholar]
- Kleerekoper Q, Liu W, Choi D, Putkey JA. Identification of binding sites for bepridil and trifluoperazine on cardiac troponin C. J Biol Chem. 1998;273:8153–8160. doi: 10.1074/jbc.273.14.8153. [DOI] [PubMed] [Google Scholar]
- Kleerekoper Q, Putkey JA. Drug binding to cardiac troponin C. J Biol Chem. 1999;274:23932–23939. doi: 10.1074/jbc.274.34.23932. [DOI] [PubMed] [Google Scholar]
- Kloiber K, Fischer M, Ledolter K, Nagl M, Schmid W, Konrat R. Generation and relaxation of high rank coherences in AX3 systems in a selectively methionine labelled SH2 domain. J Biomolec NMR. 2007;38:125–131. doi: 10.1007/s10858-007-9153-y. [DOI] [PubMed] [Google Scholar]
- Krudy GA, Kleerekoper Q, Guo X, Howarth JW, Solaro RJ, Rosevear PR. NMR studies delineating spatial relationships within the cardiac troponin I-troponin C complex. J Biol Chem. 1994;269:23731–23735. [PubMed] [Google Scholar]
- Li L, Murphy KM, Kanevets U, Reha-Krantz LJ. Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase delta in Saccharomyces cerevisiae. Genetics. 2005;170:569–580. doi: 10.1534/genetics.104.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Krudy GA, Howarth J, Brito RMM, Rosevear PR, Putkey JA. Assignment and Calcium Dependence of Methionyl εC and εH Resonances in Cardiac Troponin C. Biochemistry. 1994;33:14434–14442. doi: 10.1021/bi00252a009. [DOI] [PubMed] [Google Scholar]
- London RE, Wingad BD, Mueller GA. Dependence of amino acid side chain 13C shifts on dihedral angle: application to conformational analysis. J Am Chem Soc. 2008;130:11097–11105. doi: 10.1021/ja802729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- MacGillivray RT, Moore SA, Chen J, Anderson BF, Baker H, Luo Y, Bewley M, Smith CA, Murphy ME, Wang Y, et al. Two high-resolution crystal structures of the recombinant N-lobe of human transferrin reveal a structural change implicated in iron release. Biochemistry. 1998;37:7919–7928. doi: 10.1021/bi980355j. [DOI] [PubMed] [Google Scholar]
- Menendez-Arias L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008;134:124–146. doi: 10.1016/j.virusres.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Mittermaier A, Davidson AR, Kay LE. Correlation between 2H NMR side-chain order parameters and sequence conservation in globular proteins. J Am Chem Soc. 2003;125:9004–9005. doi: 10.1021/ja034856q. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny SA, Stith CM, Burgers PM, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 2007;282:2324–2332. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A, Limsirichaikul S, Yoshida S, Iwai S, Masutani C, Hanaoka F, Kool ET, Nishiyama Y, Suzuki M. Palm mutants in DNA polymerases alpha and eta alter DNA replication fidelity and translesion activity. Mol Cell Biol. 2004;24:2734–2746. doi: 10.1128/MCB.24.7.2734-2746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil KT, DeGrado WF. How calmodulin binds its targets: sequence independent recognition of amphiphilic alpha-helices. Trends Biochem Sci. 1990;15:59–64. doi: 10.1016/0968-0004(90)90177-d. [DOI] [PubMed] [Google Scholar]
- Pantoja-Uceda D, Shewry PR, Bruix M, Tatham AS, Santoro J, Rico M. Solution structure of a methionine-rich 2S albumin from sunflower seeds: relationship to its allergenic and emulsifying properties. Biochemistry. 2004;43:6976–6986. doi: 10.1021/bi0496900. [DOI] [PubMed] [Google Scholar]
- Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol. 2006;16:202–207. doi: 10.1016/j.cub.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Pearson JG, Le HB, Sanders LK, Godbout N, Havlin RH, Oldfield E. Predicting chemical shifts in proteins: Structure refinement of valine residues by using ab initio and empirical geometry optimizations. Journal of the American Chemical Society. 1997;119:11941–11950. [Google Scholar]
- Perkins SJ, Dwek RA. Comparisons of ring-current shifts calculated from the crystal structure of egg white lysozyme of hen with the proton nuclear magnetic resonance spectrum of lysozyme in solution. Biochemistry. 1980;19:245–258. doi: 10.1021/bi00543a001. [DOI] [PubMed] [Google Scholar]
- Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Regulation of B family DNA polymerase fidelity by a conserved active site residue: characterization of M644W, M644L and M644F mutants of yeast DNA polymerase epsilon. Nucleic Acids Res. 2007a;35:3076–3086. doi: 10.1093/nar/gkm132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007b;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage R, Green J, Muir TW, Ogunjobi OM, Love S, Shaw K. Synthetic, structural and biological studies of the ubiquitin system: the total chemical synthesis of ubiquitin. Biochem J. 1994;299(Pt 1):151–158. doi: 10.1042/bj2990151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reha-Krantz LJ, Nonay RL. Motif A of bacteriophage T4 DNA polymerase: role in primer extension and DNA replication fidelity. Isolation of new antimutator and mutator DNA polymerases. J Biol Chem. 1994;269:5635–5643. [PubMed] [Google Scholar]
- Religa TL, Sprangers R, Kay LE. Dynamic Regulation of Archaeal Proteasome Gate Opening As Studied by TROSY NMR. Science. 2010;328:98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- Rosevear PR. Purification and NMR studies of [methyl-13C]methionine-labeled truncated methionyl-tRNA synthetase. Biochemistry. 1988;27:7931–7939. doi: 10.1021/bi00420a052. [DOI] [PubMed] [Google Scholar]
- SANTOS H, TURNER DL. Determination of the methionine haem ligand conformation in cytochrome cH from Methylophilus methylotrophus by two-dimensional 1H NMR. Magnetic resonance in chemistry. 1993;31:S90–S95. [Google Scholar]
- Schoneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer’s disease. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2005;1703:111–119. doi: 10.1016/j.bbapap.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Seigneuret M, Neumann JM, Levy D, Rigaud JL. High-resolution 13C NMR study of the topography and dynamics of methionine residues in detergent-solubilized bacteriorhodopsin. Biochemistry. 1991;30:3885–3892. doi: 10.1021/bi00230a012. [DOI] [PubMed] [Google Scholar]
- Senn H, Billeter M, Wuthrich K. The spatial structure of the axially bound methionine in solution conformations of horse ferrocytochrome c and Pseudomonas aeruginosa ferrocytochrome c551 by 1H NMR. Eur Biophys J. 1984;11:3–15. doi: 10.1007/BF00253853. [DOI] [PubMed] [Google Scholar]
- Senn H, Wuthrich K. A new spatial structure for the axial methionine observed in cytochrome c5 from Pseudomonas mendocina. Correlations with the electronic structure of heme c. Biochim Biophys Acta. 1983a;747:16–25. doi: 10.1016/0167-4838(83)90115-2. [DOI] [PubMed] [Google Scholar]
- Senn H, Wuthrich K. Conformation of the Axially Bound Ligands of the Heme Iron and Electronic-Structure of Heme-C in the Cytochromes-C-551 from Pseudomonas-Mendocina and Pseudomonas-Stutzeri and in Cytochrome-C2 from Rhodospirillum-Rubrum. Biochimica Et Biophysica Acta. 1983b;746:48–60. [Google Scholar]
- Shah AM, Conn DA, Li SX, Capaldi A, Jager J, Sweasy JB. A DNA polymerase beta mutator mutant with reduced nucleotide discrimination and increased protein stability. Biochemistry. 2001;40:11372–11381. doi: 10.1021/bi010755y. [DOI] [PubMed] [Google Scholar]
- Siivari K, Zhang M, Palmer AG, 3rd, Vogel HJ. NMR studies of the methionine methyl groups in calmodulin. FEBS Lett. 1995;366:104–108. doi: 10.1016/0014-5793(95)00504-3. [DOI] [PubMed] [Google Scholar]
- Skrynnikov NR, Mulder FAA, Hon B, Dahlquist FW, Kay LE. Probing slow time scale dynamics at methyl-containing side chains in proteins by Relaxation Dispersion NMR Measurements: Application to Methionine Residues in a Cavity Mutant of T4 Lysozyme. J Amer Chem Soc. 2001;123:4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- Stollery JG, Boggs JM, Moscarello MA, Deber CM. Direct Observation by Carbon-13 Nuclear Magnetic Resonance of Membrane-Bound Human Myelin Basic Protein. Biochemistry. 1980;19:2391–2396. doi: 10.1021/bi00552a017. [DOI] [PubMed] [Google Scholar]
- Strohmeier M, Raschle T, Mazurkiewicz J, Rippe K, Sinning I, Fitzpatrick TB, Tews I. Structure of a bacterial pyridoxal 5′-phosphate synthase complex. Proc Natl Acad Sci U S A. 2006;103:19284–19289. doi: 10.1073/pnas.0604950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- Tafazzoli M, Ghiasi M. New Karplus equations for 2JHH, 3JHH, 2JCH, 3JCH, 3JCOCH, 3JCSCH, and 3JCCCH in some aldohexopyranoside derivatives as determined using NMR spectroscopy and density functional theory calculations. Carbohydr Res. 2007;342:2086–2096. doi: 10.1016/j.carres.2007.05.032. [DOI] [PubMed] [Google Scholar]
- Tipples GA, Ma MM, Fischer KP, Bain VG, Kneteman NM, Tyrrell DLJ. Mutation in HBV RNA-dependent DNA polymerase confers resistance to lamivudine in vivo. Hepatology. 1996;24:714–717. doi: 10.1002/hep.510240340. [DOI] [PubMed] [Google Scholar]
- Tonelli AE. C-13-Nmr Chemical-Shifts and the Conformations of Rigid Polypeptides. Biopolymers. 1984;23:819–829. [Google Scholar]
- Tonelli AE, Schilling FC, Bovey FA. Conformational Origin of the Nonequivalent C-13 Nmr Chemical-Shifts Observed for the Isopropyl Methyl Carbons in Branched Alkanes. Journal of the American Chemical Society. 1984;106:1157–1158. [Google Scholar]
- Tvaroska I, Mazeau K, Blancmuesser M, Lavaitte S, Driguez H, Taravel FR. Karplus-Type Equation for Vicinal Carbon Proton Coupling-Constants for the C-S-C-H Pathway in 1-Thioglycosides. Carbohydrate Research. 1992;229:225–231. [Google Scholar]
- Vijaykumar S, Bugg CE, Cook WJ. Structure of Ubiquitin Refined at 1.8 a Resolution. Journal of Molecular Biology. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- Wakefield JK, Jablonski SA, Morrow CD. In vitro enzymatic activity of human immunodeficiency virus type 1 reverse transcriptase mutants in the highly conserved YMDD amino acid motif correlates with the infectious potential of the proviral genome. J Virol. 1992;66:6806–6812. doi: 10.1128/jvi.66.11.6806-6812.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- Wolinski K, Hinton JF, Pulay P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc. 1990;112:8251–8260. [Google Scholar]
- Wooten JB, Cohen JS, Vig I, Schejter A. pH-induced conformational transitions of ferricytochrome c: a carbon-13 and deuterium nuclear magnetic resonance study. Biochemistry. 1981;20:5394–5402. doi: 10.1021/bi00522a007. [DOI] [PubMed] [Google Scholar]
- Word JM, Lovell SC, LaBean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- Yin D, Kuczera K, Squier TC. The sensitivity of carboxyl-terminal methionines in calmodulin isoforms to oxidation by H(2)O(2) modulates the ability to activate the plasma membrane Ca-ATPase. Chem Res Toxicol. 2000;13:103–110. doi: 10.1021/tx990142a. [DOI] [PubMed] [Google Scholar]
- Yuan T, Gomes AV, Barnes JA, Hunter HN, Vogel HJ. Spectroscopic characterization of the calmodulin-binding and autoinhibitory domains of calcium/calmodulin-dependent protein kinase I. Arch Biochem Biophys. 2004;421:192–206. doi: 10.1016/j.abb.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Yuan T, Ouyang H, Vogel HJ. Surface exposure of the methionine side chains of calmodulin in solution. A nitroxide spin label and two-dimensional NMR study. J Biol Chem. 1999;274:8411–8420. doi: 10.1074/jbc.274.13.8411. [DOI] [PubMed] [Google Scholar]
- Zhao H, Carmichael I, Serianni AS. Oligosaccharide trans-glycoside 3JCOCC Karplus curves are not equivalent: effect of internal electronegative substituents. J Org Chem. 2008;73:3255–3257. doi: 10.1021/jo702288h. [DOI] [PubMed] [Google Scholar]
- Zheng X, Mueller GA, Cuneo MJ, Derose EF, London RE. Homodimerization of the p51 subunit of HIV-1 reverse transcriptase. Biochemistry. 2010;49:2821–2833. doi: 10.1021/bi902116z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Mueller GA, DeRose EF, London RE. Solution characterization of [methyl-(13)C]methionine HIV-1 reverse transcriptase by NMR spectroscopy. Antiviral Res. 2009;84:205–214. doi: 10.1016/j.antiviral.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.