

Abstract
Consistent with a tentative diagnosis of neuronal ceroid lipofuscinosis (NCL), autofluorescent cytoplasmic storage bodies were found in neurons from the brains of 2 related Shiba Inu dogs with a young‐adult onset, progressive neurodegenerative disease. Unexpectedly, no potentially causal NCL‐related variants were identified in a whole‐genome sequence generated with DNA from 1 of the affected dogs. Instead, the whole‐genome sequence contained a homozygous 3 base pair (bp) deletion in a coding region of HEXB. The other affected dog also was homozygous for this 3‐bp deletion. Mutations in the human HEXB ortholog cause Sandhoff disease, a type of GM2 gangliosidosis. Thin‐layer chromatography confirmed that GM2 ganglioside had accumulated in an affected Shiba Inu brain. Enzymatic analysis confirmed that the GM2 gangliosidosis resulted from a deficiency in the HEXB encoded protein and not from a deficiency in products from HEXA or GM2A, which are known alternative causes of GM2 gangliosidosis. We conclude that the homozygous 3‐bp deletion in HEXB is the likely cause of the Shiba Inu neurodegenerative disease and that whole‐genome sequencing can lead to the early identification of potentially disease‐causing DNA variants thereby refocusing subsequent diagnostic analyses toward confirming or refuting candidate variant causality.
Keywords: Autofluorescent storage bodies, Lysosomal storage disease, Neuronal ceroid lipofuscinosis, Sandhoff disease, Whole‐genome sequence
Abbreviations
- NCL
neuronal ceroid lipofuscinosis
- PCR‐RFLP
PCR‐restriction fragment length polymorphism
The clinical records of 2 young‐adult Shiba Inu dogs that exhibited similar signs of progressive, fatal, neurodegeneration came to our attention from independent sources. Pedigree inspection indicated that both dogs shared at least 1 common maternal and paternal ancestor, as illustrated in Fig. 1. Thus, it appeared that the same recessively inherited neurodegenerative disease affected both dogs. We used whole‐genome sequencing to efficiently identify a genetic variant that was most likely responsible for this disease and to reach a highly probable, but unexpected, diagnosis.
Figure 1.
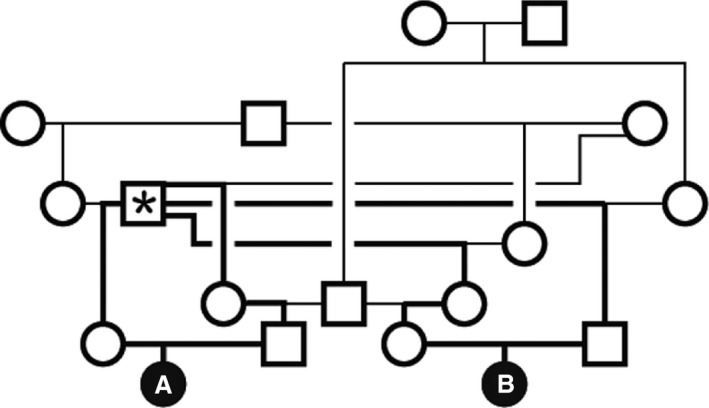
Pedigree shows consanguineous familial relationships for the two 2 affected Shiba Inu, Dog A and Dog B. The dog marked with an asterisk contributed genetic material to both parents of both affected dogs. Bold lines show the shortest possible pathway for the inheritance of the causal allele from an ancestor to the homozygous affected dogs. Nonetheless, we do not have DNA from the ancestors to verify this pathway, and additional consanguineous relationships among earlier ancestors are likely. Thus, Dog A and Dog B may have obtained their homozygous mutant allele by alternative inheritance pathways.
Methods
The DNA from 1 affected Shiba Inu (Dog A) was submitted to a commercial sequencing center1 for PCR‐free library preparation and sequencing with a commercial sequencer2 that produced paired‐end 150‐bp reads. Details about the pipeline used to process and analyze this data set are available in the supplementary material. The PCR primers 5′‐AAATAGTATAATCATGTGCAA‐3′ and 5′‐CCCAAATACATTTACTGCCTA‐3′ were used to produce amplicons containing a candidate variant identified in the whole‐genome sequence. These amplicons were analyzed by Sanger sequencing for validation and by PCR‐restriction fragment length polymorphism (PCR‐RFLP) with MnlI for genotyping archived Shiba Inu DNA. Previously described procedures were used for fluorescence and electron microscopy1 and for thin‐layer chromatography and enzymology.2
Clinical Findings
Dog A, a spayed female Shiba Inu, exhibited progressive neurologic signs starting with generalized tremors and altered tail carriage (tail down) when approximately 12 months old. As the disease progressed, the severity of the tremors increased and the dog exhibited marked cerebellar ataxia, spastic tetraparesis, anxiety, decreased appetite, decreased responsiveness to verbal commands, trance‐like behavior, loss of ability to climb up and down stairs, and impaired vision. Magnetic resonance imaging of the head showed a diffuse loss of gray‐white matter distinction on T2‐weighted imaging with white matter appearing isointense to marginally hypointense relative to gray matter. Because of the progression of neurologic signs, the dog was euthanized at approximately 17 months of age.
Dog B also was a spayed female Shiba Inu. Similar to Dog A, the first clinical signs in Dog B appeared at about 12 months of age and included situational anxiety, inability to climb up or down stairs, a wide‐based stance, severe generalized tremors, and the same alteration in tail carriage shown by Dog A. During a neurologic examination at 15 months of age, Dog B had normal proprioception but showed intention tremors and severe cerebellar ataxia with frequent falls. The menace response was decreased to absent. Brain magnetic resonance imaging, including T2‐ and T1‐weighted sequences before and after administration of a contrast agent, showed decreased gray‐white matter distinction in both the cerebrum and cerebellum similar to Dog A. In the cerebellum, arbor vitae could not be identified on the sagittal T2‐weighted sequence. The ventricular system was symmetrical and normal in size. The neurologic signs progressed to include anxiety, compulsive panting, trance‐like behavior, intolerance to loud noise, and loss of training including house training. Dog B developed severe muscle stiffness and weakness that spread from the hind limbs resulting in ataxia and frequent falls. In addition, Dog B showed severe visual impairment in both bright and dim light. A sleep disturbance with leg movements mimicking running started at about 18 months of age. Near the end of its life, Dog B was unable to walk or eat without assistance. Dog B died while asleep when approximately 20 months old. Commercial laboratories3 , 4 determined that neither affected dog had a GLB1 frameshift mutation, chr23:3,796,317delC (CanFam 3.1), known to cause GM1 gangliosidosis in the Shiba Inu breed (OMIA 000402‐9615).3
Because the clinical signs suggested neuronal ceroid lipofuscinosis (NCL) as a possible cause, blood and other tissue samples were shipped to the University of Missouri for further analysis. Fluorescence microscopy of unstained tissue sections from both affected dogs identified massive accumulations of yellow‐emitting autofluorescent storage bodies in neurons of the cerebellum (Fig 2A), the cerebral cortex (Fig 2B), and the retina (Fig 2C).
Figure 2.
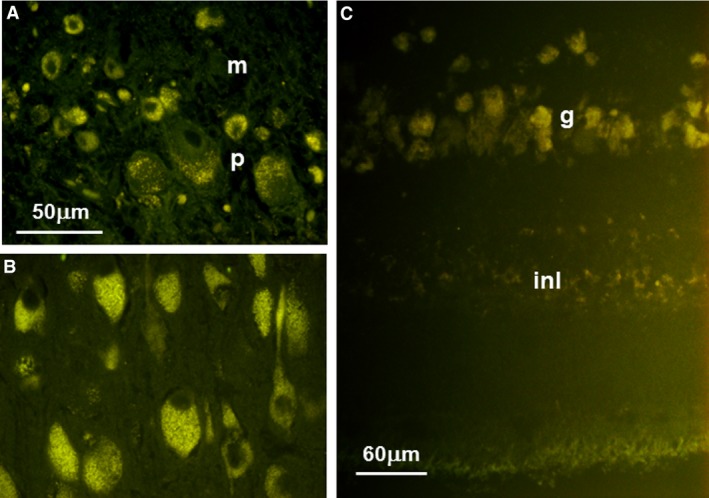
Fluorescence micrographs of unstained sections of cerebellum (A), cerebral cortex (B), and retina (C) from an affected Shiba Inu. In the cerebellum, autofluorescent intracellular inclusions were present in both the Purkinje cell (p) and molecular (m) layers. Neurons throughout the cerebral cortex gray matter had abundant autofluorescent inclusions. In the retina, the most pronounced accumulation of autofluorescent material was seen in the ganglion cell layer (g), but some accumulation was also present in the inner nuclear layer (inl). Bar in (A) indicates the magnification for micrographs (A) and (B).
Molecular‐Genetic Diagnosis
The clinical signs and fluorescent microscopic results were consistent with a diagnosis of NCL. Because whole‐genome sequencing previously had proven to be an effective strategy for identifying genetic variants responsible for NCL in dogs,1, 4, 5, 6, 7, 8, 9 a whole‐genome sequence was generated with DNA from Dog A. The resulting sequence data had 31‐fold average coverage of the CanFam3.1 reference assembly and are available in the National Center for Biotechnology Information Sequence Read Archives (accession SRS1692083). As we have done previously,1, 4, 5, 7, 8 the aligned reads from the whole‐genome sequence were scanned for rare variants in or near the coding exons of canine orthologs of the 13 genes currently associated with human NCL: PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, DNAJC5, CTSF, ATP13A2, GRN, and KCTD7.10 Unexpectedly, we did not find plausible candidate variants. In addition, the alignment was checked for a GLB1 frameshift mutation, chr23:3,796,317delC (CanFam 3.1), known to cause a GM1 gangliosidosis in the Shiba Inu,3 but only the reference allele was found confirming antemortem DNA test results. The search for a causal genetic variant was expanded by filtering the millions of sequence variants found in the Shiba Inu whole‐genome sequence to retain only those that were (1) predicted to affect the primary structure of a gene product, (2) homozygous in the affected Shiba Inu, and (3) absent from the 105 whole‐genome sequences of normal dogs or dogs with other diseases. Only 80 variants in 74 different genes met these criteria (see Table S1). Brief searches for the known biologic functions of these 74 genes in OMIM (https://www.ncbi.nlm.nih.gov/omim) and in PubMed (https://www.ncbi.nlm.nih.gov/pubmed) identified a 3‐bp deletion in HEXB as the sequence variant considered most likely to be responsible for the Shiba Inu disorder because it was the only variant in a gene that has previously been associated with lysosomal storage disease. The chromosomal location of this deletion is chr2:57,243,656_57,243,658delCCT (CanFam 3.1).
Sanger sequencing confirmed that both affected Shiba Inu were homozygous for the 3‐bp HEXB deletion. The absence of this deletion from our 105 control whole‐genome sequences suggested that it is uncommon in dogs in general. To estimate the frequency of the 3‐bp HEXB deletion among the Shiba Inu, all 40 Shiba Inu represented in the University of Missouri DNA repository were genotyped by PCR‐RFLP for this deletion. Thirty‐seven of the tested Shiba Inu were homozygous for the reference allele; the other 3 were heterozygotes. These heterozygotes were born in 2002, 2004, and 2007. There are no known familial relationships among them or between them and the 2 affected Shiba Inu described here. This observation suggests that the 3‐bp HEXB deletion may be rare but widely distributed in the Shiba Inu breed.
Biochemical and Electron Microscopic Confirmation
The HEXB deletion predicts the loss of a leucine residue (p.Leu317del; XP_005617969.1) in the encoded β‐subunit of the β‐hexosaminidase enzyme complex. A leucine residue occurs at a homologous position in β‐subunits from 53 other mammalian species (see Figure S1). A functional β‐subunit provides essential glycosidic activity to the β‐hexosaminidase enzyme complex that removes the terminal N‐acetyl‐galactosamine from GM2 ganglioside, an essential step in the lysosomal degradation of gangliosides. Genetic variants in human HEXB can result in a β‐hexosaminidase deficiency called Sandhoff disease and cause the accumulation of GM2 ganglioside.11
Thin‐layer chromatography was used to determine whether GM2 ganglioside had accumulated in brain gray matter from 1 of the affected Shiba Inu (Dog B). The resulting thin‐layer chromatogram is shown in Fig. 3. An extract from the brain of a human patient with GM2 gangliosidosis known to contain GM2 ganglioside as the major component was fractionated in lane 1. The brain extract in lane 3 was from a human GM1 gangliosidosis patient. GM1 ganglioside was the major component in this extract. Lane 4 contained a normal bovine brain extract and served to show that GM1 and GM2 gangliosides do not accumulate in healthy brains. The extract in lane 2 was from one of the affected Shiba Inu (Dog B). The migration of the major component in this lane matched that in lane 1, indicating that it is also GM2 ganglioside and that the dog had a GM2 gangliosidosis.
Figure 3.

Thin‐layer chromatographic fractionation of brain gangliosides extracted from the gray matter of a human patient who died from Tay–Sachs disease (lane 1), the gray matter of Dog B, the affected Shiba Inu dog (lane 2), the gray matter of a human patient who died with GM1 gangliosidosis (lane 3) and gangliosides extracted from a normal bovine brain obtained from a commercial source5 (lane 4). The most abundant component in lane 1 is GM2 ganglioside (GM2). In lane 2, the most abundant component co‐migrated with the GM2 ganglioside in lane 1 and not with GM1 ganglioside (GM1), the most abundant component in lane 3. Only trace amounts of GM2 ganglioside were detected in Lane 3, and none was detected in extracts from the normal brain in lane 4.
Besides HEXB, GM2 gangliosidosis can be caused by genetic variants in 2 other genes: HEXA that encodes the α‐subunit of the β‐hexosaminidase enzyme complex and GM2A that encodes GM2 activator protein. Although the activator protein does not possess intrinsic enzymatic activity, its presence is required for efficient enzymatic degradation of GM2 ganglioside by β‐hexosaminidase.12 The α‐ and β‐subunits of β‐hexosaminidase are enzymatically active as homo‐ or heterodimers. The 3 genetic causes of GM2 gangliosidosis can be enzymologically distinguished with synthetic substrates that identify the source of the residual hexosaminidase activities in patients’ tissues.11 Patients with GM2 gangliosidosis caused by a GM2A deficiency have residual activity from both the α‐ and β‐subunits. Thus, they are classified as AB variants. Human patients with HEXA deficiency (known as Tay–Sachs disease) are considered B variants because they have only β‐subunit activity. Homodimers of the α‐subunit are unstable and heat labile. Patients with Sandhoff disease (HEXB deficiency) who lack the β‐subunit necessary to form stable heterodimers can only form unstable α‐subunit homodimers and thus have subnormal activities from both the α‐ and β‐subunits.11 These patients are considered to be 0 (zero) variants. Table 1 contains a summary of the genetic causes, the subunit activities, and the associated nomenclature for the 3 causes of GM2 gangliosidosis.
Table 1.
Types of GM2 Gangliosidosis
| Disease | Genea | Encoded Protein | Residual Activity | Variantb |
|---|---|---|---|---|
| Tay–Sachs Disease | HEXA | α‐Subunit of β‐Hexosaminidase | β‐Subunit | B Variant |
| Sandhoff disease | HEXB | β‐Subunit of β‐Hexosaminidase | Minimalc | 0 Variant |
| GM2 Activator Deficiency | GM2A | GM2 Activator | α‐ and β‐Subunits | AB variant |
Harboring causal variants.
based on residual enzymatic activity.
minimal because α‐subunit dimers are unstable.
We used previously described procedures2 and the synthetic substrate, 4‐methylumbelliferyl n‐acetyl‐β‐d‐glucosaminide, to estimate total β‐hexosaminidase activity in cerebral cortical gray and white matter extracts. We also measured β‐galactosidase activity in these extracts as a control. As indicated in Table 2, the total β‐hexosaminidase activities in the extracts from Dog B were markedly decreased compared to 2 control dogs. Furthermore, the β‐hexosaminidase activities in the extracts from Dog B were completely heat labile as would be expected for α‐subunit homodimers. These results are similar to those observed with 0 variant or Sandhoff disease patients and provide further evidence that the HEXB deletion identified by whole‐genome sequencing is responsible for the progressive neurodegeneration in the 2 affected Shiba Inu dogs.
Table 2.
Enzyme Activities
| Enzyme | Enzymatic Activities (nmol/h/mg protein) | |||||
|---|---|---|---|---|---|---|
| Affected Dog B | Control Dog 1 | Control Dog 2 | ||||
| Gray Matter | White Matter | Gray Matter | White Matter | Gray Matter | White Matter | |
| β‐galactosidasea | 269 | 174 | 67.1 | 25.5 | 58.9 | 31.8 |
| β‐hexosaminidase (% Heat labile) | 15.1 (100%) | 11.3 (100%) | 663 (77%) | 363 (72%) | 669 (79%) | 427 (79%) |
Run as control.
Electron microscopic examination of the storage bodies in the brain and retina showed that many neurons contained membrane‐like structures arranged in loosely associated concentric whorls (Fig 4A, C and D). Inclusions with this ultrastructural feature traditionally are called membranous cytoplasmic granules.11, 12, 13, 14 Other less common neuronal inclusions appeared to be vacuoles with only a small amount of dispersed electron‐dense material (Fig 4B). Abnormal inclusions with the membranous cytoplasmic granules ultrastructure are common in human patients with GM2 gangliosidosis.13, 15, 16 In addition, membranous storage granules have been found in neurons from patients with GM1 gangliosidosis or mucopolysaccharidosis.16, 17 However, they have not been reported in NCL‐affected brains, which instead typically contain inclusions with granular, curvilinear, rectilinear, or fingerprint profiles, either singularly, or in combination.18
Figure 4.
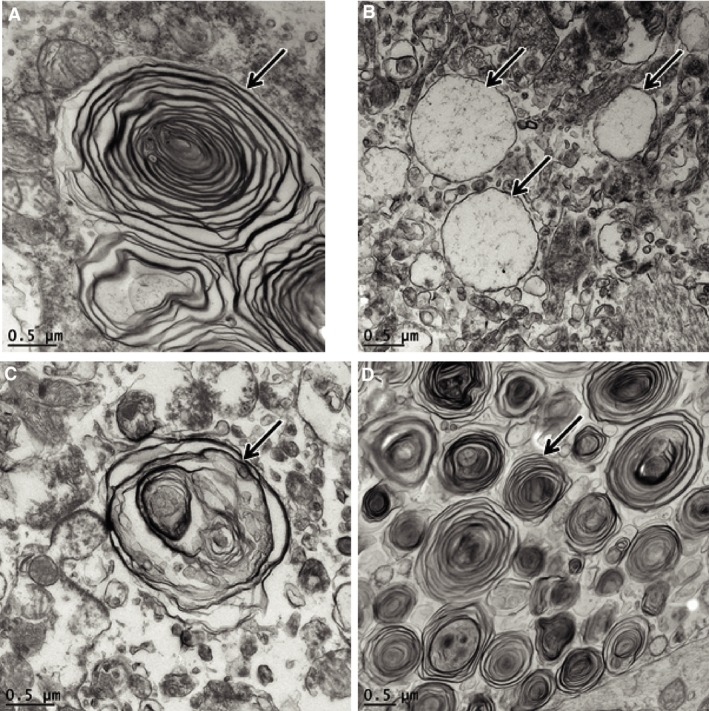
Electron micrographs of storage material in the cerebral cortex (A and B), cerebellar molecular layer cell (C), and retinal ganglion cells (D) of Dog B. Representative storage bodies are indicated by arrows. In cerebral cortical neurons, the storage material consisted of both concentric whorls of membrane‐like structures (A) and membrane‐bound vacuole‐like inclusion bodies (B). In the cerebellum (C), the storage bodies contained more irregularly arranged membrane‐like material. The ultrastructural appearance of the cerebellar storage bodies was similar in both the molecular layer and the Purkinje cells. In the retinal ganglion cells (D), the storage material consisted primarily of concentric whorls of membrane‐like structures.
Autofluorescent Storage Bodies in NCL and Other Diseases
Before we evaluated the whole‐genome sequence, we suspected that the affected Shiba Inu dogs had NCL because of their clinical signs of progressive neurodegeneration and the presence of autofluorescent storage bodies. Progressive neurodegeneration and accumulation of autofluorescent lysosomal storage bodies within neurons are widely recognized disease characteristics required for inclusion among the diverse diseases classified as NCLs.10 Nonetheless, in a few cases, disorders with these characteristics have been classified as other types of lysosomal storage diseases because of the biochemical composition of the associated storage granules or the nature of the associated enzyme deficiency or both. Among these are GM1 gangliosidosis and mucopolysaccharidosis IIIA in humans and α‐mannosidosis in mice.14, 17, 19, 20 In addition, there is an example in dogs. When American Staffordshire Terriers and Pit Bull Terriers with a recessively inherited, adult‐onset, progressive neurodegenerative disease were found to accumulate autofluorescent storage bodies in hippocampal and cerebellar neurons, their disease was classified as an NCL.21 This classification remained unchanged with the identification of the causal genetic variant in ARSG, a gene with a biologic function that had not yet been determined.22 Subsequently, it was shown using Arsg‐knockout mice that the natural substrate for the encoded enzyme was heparan sulfate, a mucopolysaccharide, and that the storage bodies were rich in partially degraded heparan sulfate.23 Thus, the disease caused by ARSG deficiency is now considered to be a mucopolysaccharidosis. Similarly, the Shiba Inu of our case report were thought to have NCL until we determined that brain extracts from 1 of the dogs contained accumulated GM2 ganglioside and had a β‐hexosaminidase deficiency, thus classifying the disorder as a gangliosidosis.
Ours may be the first report that storage bodies associated with GM2 gangliosidosis are autofluorescent. In fact, at least 1 report states that storage bodies from human patients with GM2 gangliosidosis do not auto‐fluoresce.13 Thus, we examined tissue from a previously reported case of GM2 gangliosidosis in a Japanese Chin dog caused by a HEXA mutation (OMIA 001461‐9615).2 As with the results reported here, the storage bodies in the tissue from the dog with the HEXA mutation were autofluorescent (Fig. 5).
Figure 5.
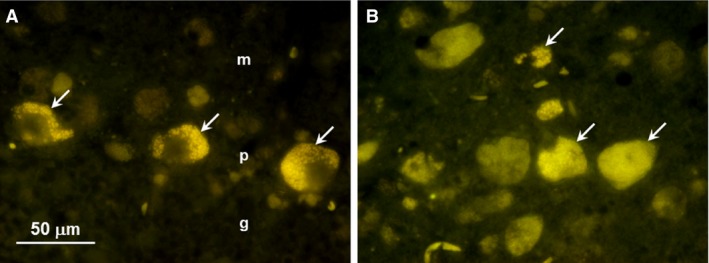
Fluorescence micrographs of unstained cryostat sections of cerebellum (A) and cerebral cortex (B) from a Japanese Chin dog with GM2 gangliosidosis due to a homozygous HEXA nonsynonymous substitution. In the cerebellum, autofluorescent inclusions were present primarily in the Purkinje cells (arrows in A) in the Purkinje cell layer (p), although weak fluorescent cells were also present in the molecular layer (m). No autofluorescence was observed in the granular cell layer (g). Neurons throughout the cerebral cortex gray matter of the parietal lobe had abundant autofluorescent inclusions (arrows in B). Bar in (A) indicates the magnification for micrographs (A) and (B).
Comparisons with Other Gangliosidoses in Dogs
Although a GM1 gangliosidosis has been recognized in the Shiba Inu for >15 years,3, 24, 25 ours is the first report of a GM2 gangliosidosis in this breed. GM2 gangliosidoses have been reported in a mixed breed dog and in members of other dog breeds including German Shorthaired Pointer, Japanese Chin, Golden Retriever, and Toy Poodle.2, 26, 27, 28, 29, 30 The Toy Poodle, Golden Retriever, and the mixed breed dog were classified as 0 variants (OMIA 001462‐9615).28, 29, 30 The 1‐bp HEXB frameshift variant responsible for the 0‐variant gangliosidosis in Toy Poodles31 is different from the 3‐bp in‐frame deletion that we found in the affected Shiba Inu dogs. The molecular‐genetic causes for the 2 other 0‐variant gangliosidoses have not been reported.
Conclusions
Although not yet routine, the sequencing and analysis of whole genomes generated from patient DNA is becoming an increasingly important strategy for diagnosing inherited diseases in humans.32 Technical improvements in massively parallel DNA sequencing have resulted in the development of a sequencing instrument2 capable of generating deep sequence coverages of mammalian genomes for <$2,000.33 Although initially licensed exclusively for the sequencing of human genomes,34, 35 this instrument can now be used to sequence domestic animal genomes. With access to this technology, it became economically feasible for us to make use of whole‐genome sequencing early in our efforts to identify the molecular‐genetic cause of the neurodegeneration in these Shiba Inu. Analysis of the whole‐genome sequence from 1 of the affected dogs identified a potentially causal candidate variant. This finding enabled us to proceed with more traditional diagnostic techniques (e.g., thin‐layer chromatography, enzymology, electron microscopy) that were chosen specifically to confirm or refute the causality of the candidate variant. Thus, in this case study, whole‐genome sequencing was an efficient and cost‐effective procedure that led to the identification of the Shiba Inu disease as a GM2 gangliosidosis caused by a 3‐bp deletion in HEXB. Once impediments such as long turn‐around times, low causal variant discovery rates, and inadequate data storage facilities have been overcome, we expect whole‐genome sequencing to become an increasingly important tool for diagnosing heritable animal diseases.
Supporting information
Figure S1. Alignments of β‐hexosaminidase β‐subunit amino acids sequences from 54 mammalian species predicted from codons surrounding the deleted Leu137 codon in the affected Shiba Inu.
Table S1. Rare variants predicted to change the primary sequence of the gene product.
Acknowledgments
We thank Dr. Andrew Miller, Cornell University, for sending us postmortem tissue and Paw Print Genetics for supplying us with DNA from Dog B. This study was supported in part by AKC Canine Health Foundation grant 02257.
Conflict of Interest Declaration
Authors declare no conflict of interest.
Off‐label Antimicrobial Declaration
Authors declare no off‐label use of antimicrobials.
The work was done at the University of Missouri and at Jefferson Medical College.
Supported in part by AKC Canine Health Foundation grant 02257.
The research has not been reported at a meeting.
Footnotes
McDonnell Genome Institute, Washington University, St Louis, MO 63108
HiSeq X Ten Sequencing System, Illumina, Inc., San Diego, CA 92122
Paw Print Genetics, 850 E. Spokane Falls Blvd. Suite 200, Spokane, WA 99202
VetGen, 3728 Plaza Drive, Suite 1, Ann Arbor, MI 48108
Sigma‐Aldrich, 3300 S 2nd St, St. Louis, MO 63118
References
- 1. Guo J, Johnson GS, Brown HA, et al. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Mol Genet Metab 2014;112:302–309. [DOI] [PubMed] [Google Scholar]
- 2. Sanders DN, Zeng R, Wenger DA, et al. GM2 gangliosidosis associated with a HEXA missense mutation in Japanese Chin dogs: A potential model for Tay Sachs disease. Mol Genet Metab 2013;108:70–75. [DOI] [PubMed] [Google Scholar]
- 3. Yamato O, Endoh D, Kobayashi A, et al. A novel mutation in the gene for canine acid beta‐galactosidase that causes GM1‐gangliosidosis in Shiba dogs. J Inherit Metab Dis 2002;25:525–526. [DOI] [PubMed] [Google Scholar]
- 4. Guo J, O'Brien DP, Mhlanga‐Mutangadura T, et al. A rare homozygous MFSD8 single‐base‐pair deletion and frameshift in the whole genome sequence of a Chinese Crested dog with neuronal ceroid lipofuscinosis. BMC Vet Res 2014;10:960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilliam D, Kolicheski A, Johnson GS, et al. Golden Retriever dogs with neuronal ceroid lipofuscinosis have a two‐base‐pair deletion and frameshift in CLN5. Mol Genet Metab 2015;115:101–109. [DOI] [PubMed] [Google Scholar]
- 6. Faller KM, Bras J, Sharpe SJ, et al. The Chihuahua dog: A new animal model for neuronal ceroid lipofuscinosis CLN7 disease? J Neurosci Res 2016;94:339–347. [DOI] [PubMed] [Google Scholar]
- 7. Kolicheski A, Johnson GS, O'Brien DP, et al. Australian cattle dogs with neuronal ceroid lipofuscinosis are homozygous for a CLN5 nonsense mutation previously identified in border collies. J Vet Intern Med 2016;30:1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolicheski A, Barnes Heller HL, Arnold S, et al. Homozygous PPT1 splice donor mutation in a Cane Corso dog with neuronal ceroid lipofuscinosis. J Vet Intern Med 2017;31:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirz M, Drogemuller M, Schanzer A, et al. Neuronal ceroid lipofuscinosis (NCL) is caused by the entire deletion of CLN8 in the Alpenlaendische Dachsbracke dog. Mol Genet Metab 2017;120:1149–1158. [DOI] [PubMed] [Google Scholar]
- 10. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta 2015;1852:2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lawson CA, Martin DR. Animal models of GM2 gangliosidosis: Utility and limitations. Appl Clin Genet 2016;9:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandhoff K, Harzer K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J Neurosci 2013;33:10195–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brownstein S, Carpenter S, Polomeno RC, et al. Sandhoff's disease (GM2 gangliosidosis type 2). Histopathology and ultrastructure of the eye. Arch Ophthalmol 1980;98:1089–1097. [DOI] [PubMed] [Google Scholar]
- 14. Lowden JA, Callahan JW, Gravel RA, et al. Type 2 GM1 gangliosidosis with long survival and neuronal ceroid lipofuscinosis. Neurology 1981;31:719–724. [DOI] [PubMed] [Google Scholar]
- 15. Fontaine G, Resibois A, Tondeur M, et al. Gangliosidosis with total hexosaminidase deficiency: Clinical, biochemical and ultrastructural studies and comparison with conventional cases of Tay‐Sachs disease. Acta Neuropathol 1973;23:118–132. [DOI] [PubMed] [Google Scholar]
- 16. Suzuki K. Neuropathology of late onset gangliosidoses. A review. Dev Neurosci 1991;13:205–210. [DOI] [PubMed] [Google Scholar]
- 17. Oldfors A, Sourander P. Storage of lipofuscin in neurons in mucopolysaccharidosis. Report on a case of Sanfilippo's syndrome with histochemical and electron‐microscopic findings. Acta Neuropathol 1981;54:287–292. [DOI] [PubMed] [Google Scholar]
- 18. Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta 1852;2015:2262–2266. [DOI] [PubMed] [Google Scholar]
- 19. Wisniewski K, Rudelli R, Laure‐Kamionowska M, et al. Sanfilippo disease, type A with some features of ceroid lipofuscinosis. Neuropediatrics 1985;16:98–105. [DOI] [PubMed] [Google Scholar]
- 20. Damme M, Stroobants S, Walkley SU, et al. Cerebellar alterations and gait defects as therapeutic outcome measures for enzyme replacement therapy in alpha‐mannosidosis. J Neuropathol Exp Neurol 2011;70:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siso S, Navarro C, Hanzlicek D, et al. Adult onset thalamocerebellar degeneration in dogs associated to neuronal storage of ceroid lipopigment. Acta Neuropathol 2004;108:386–392. [DOI] [PubMed] [Google Scholar]
- 22. Abitbol M, Thibaud JL, Olby NJ, et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc Natl Acad Sci U S A 2010;107:14775–14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kowalewski B, Lamanna WC, Lawrence R, et al. Arylsulfatase G inactivation causes loss of heparan sulfate 3‐O‐sulfatase activity and mucopolysaccharidosis in mice. Proc Natl Acad Sci U S A 2012;109:10310–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamato O, Ochiai K, Masuoka Y, et al. GM1 gangliosidosis in shiba dogs. Vet Rec 2000;146:493–496. [DOI] [PubMed] [Google Scholar]
- 25. Yamato O, Masuoka Y, Yonemura M, et al. Clinical and clinico‐pathologic characteristics of Shiba dogs with a deficiency of lysosomal acid beta‐galactosidase: A canine model of human GM1 gangliosidosis. J Vet Med Sci 2003;65:213–217. [DOI] [PubMed] [Google Scholar]
- 26. Karbe E, Schiefer B. Familial amaurotic idiocy in male German shorthair pointers. Pathol Vet 1967;4:223–232. [DOI] [PubMed] [Google Scholar]
- 27. Cummings JF, Wood PA, Walkley SU, et al. GM2 gangliosidosis in a Japanese spaniel. Acta Neuropathol 1985;67:247–253. [DOI] [PubMed] [Google Scholar]
- 28. Yamato O, Matsuki N, Satoh H, et al. Sandhoff disease in a golden retriever dog. J Inherit Metab Dis 2002;25:319–320. [DOI] [PubMed] [Google Scholar]
- 29. Tamura S, Tamura Y, Uchida K, et al. GM2 gangliosidosis variant 0 (Sandhoff‐like disease) in a family of toy poodles. J Vet Intern Med 2010;24:1013–1019. [DOI] [PubMed] [Google Scholar]
- 30. Kohyama M, Yabuki A, Kawasaki Y, et al. GM2 gangliosidosis variant 0 (Sandhoff Disease) in a mixed‐breed dog. J Am Anim Hosp Assoc 2015;51:396–400. [DOI] [PubMed] [Google Scholar]
- 31. Rahman MM, Chang HS, Mizukami K, et al. A frameshift mutation in the canine HEXB gene in toy poodles with GM2 gangliosidosis variant 0 (Sandhoff disease). Vet J 2012;194:412–416. [DOI] [PubMed] [Google Scholar]
- 32. Precone V, Del Monaco V, Esposito MV, et al. Cracking the code of human diseases using next‐generation sequencing: Applications, challenges, and perspectives. Biomed Res Int 2015;2015:161648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park ST, Kim J. Trends in next‐generation sequencing and a new era for whole genome sequencing. Int Neurourol J 2016;20:S76–S83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hall N. It's only human. Genome Biol 2014;15:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watson M. Illuminating the future of DNA sequencing. Genome Biol 2014;15:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Alignments of β‐hexosaminidase β‐subunit amino acids sequences from 54 mammalian species predicted from codons surrounding the deleted Leu137 codon in the affected Shiba Inu.
Table S1. Rare variants predicted to change the primary sequence of the gene product.