

Abstract
In April 2016, the Food and Drug Administration approved the first biosimilar monoclonal antibody (mAb) – Inflectra/Remsima (Celltrion) based off the original product Remicade (infliximab, Janssen). Biosimilars promise significant cost savings for patients, but the unavoidable differences between innovator and copycat biologics raise questions regarding product interchangeability. In this study, Remicade and Remsima were examined by native mass spectrometry, ion mobility and quantitative peptide mapping. The levels of oxidation, deamidation and mutation of individual amino acids were remarkably similar. We found different levels of C-terminal truncation, soluble protein aggregates and glycation that all likely have a limited clinical impact. Importantly, we identified over 25 glycoforms for each product and observed glycoform population differences, with afucosylated glycans accounting for 19.7% of Remicade and 13,2% of Remsima glycoforms, which translated into a 2-fold reduction in FcγRIIIa binding for Remsima. While this difference was acknowledged in Remsima regulatory filings, our glycoform analysis and receptor binding results appear to be somewhat different from the published values, likely due to methodological differences between laboratories and improved glycoform identification by our laboratory using a peptide map-based method. Our mass spectrometry based analysis provides rapid and robust analytical information vital for biosimilar development. We have demonstrated the utility of our multiple attribute monitoring workflow using the model mAbs Remicade and Remsima, and have provided a template for analysis of future mAb biosimilars.
Keywords: biosimilars, monoclonal antibodies, ion-mobility, glycosylation, mass spectrometry
Graphical abstract
Introduction
In 2014, six of the ten top-selling drugs were monoclonal antibodies (mAbs) with over $50 billion in sales.1 The patents protecting many top-selling mAbs are expiring within the next three years.2 Many pharmaceutical companies have generic or biosimilar versions of these mAbs in development. Biosimilar approvals are expected to provide a 15% to 30% savings for patients that need these life changing treatments.3 Availability of affordable biosimilars will likely change the standard of care by making mAbs accessible to a broader group of patients, including those in earlier disease stages. In order to achieve biosimilarity, products must demonstrate rigorous analytical and clinical comparability in terms of safety and efficacy. All mAbs are produced by recombinant manufacturing processes that inherently yield heterogeneous products; therefore, biosimilars cannot be identical to their template innovator product. Both innovator mAbs and biosimilar are heterogeneous populations of variants characterized by differences in glycosylation, oxidation, deamidation, glycation and aggregation state. Their heterogeneity could potentially affect target protein binding through the Fab domain, receptor binding through the FC domain, and protein aggregation induced immunogenicity.4,5 Hence, analytical characterization of such heterogeneity in the biosimilar product, as well as comparisons to the heterogeneity of the innovator product, is critical to assure a similar level of safety and efficacy of the two products. The concept “totality of evidence” used to assess biosimilarity was emphasized by the FDA for the review process.6,7 The FDA integrates various types of information from state-of-the-art multiple attribute monitoring methods (MAM) which allow for evaluating combinations of analytical attributes from a single method as well as using multiple complementary methods to assess biosimilarity. First, structural assessments are combined with functional studies such as bioactivity and receptor binding assays to evaluate whether the biosimilar is highly similar to the reference product. The critical quality attributes (CQAs) of mAb e.g., oxidation, glycosylation profiles, glycation, and the amount of aggregation along with the results of protein bioactivity assays are quantified in order to assess the biosimilarity of two products. Based on this analytical assessment the extent of required toxicological and clinical evaluations are determined.7 Thus, unequivocally showing complete analytical comparability of biosimilar to the reference product could result in lessening the scope of clinical studies.7,8. Hence, biosimilar developers are incentivized to perform complete analytical comparisons of CQA for their own and innovator products using state-of-the art methodologies.
Infliximab, or Remicade (RC), was developed by Janssen and approved in 1998. Remicade was one of the first therapies to target TNF-α, and its annual sales exceeded $9 billion worldwide in 2014.9 However, patent exclusivity for infliximab has since expired and the first infliximab biosimilar, Remsima (RS), was approved in Europe in 2013 and more recently in the USA in 2016. Remsima, also known as Inflectra or CTP-13, was developed and manufactured by Celltrion and licensed to Hospira/Pfizer.10 RS is the first ever biosimilar mAb approved both in Europe and the US. Currently, several other infliximab biosimilars are either undergoing clinical testing or have already been submitted for FDA/EMA approval, including Flixabi (Samsung), Infimab (Epirus/Ranbaxy) and STI-002 (MabTech).10
In order to demonstrate the biosimilarity between RS and RC, Celltrion generated an extensive data package that included a battery of analytical methods, bioassays, and two clinical studies showing equivalency in efficacy, pharmacokinetics, adverse events, and immunogenicity in ankylosing spondylitis (AS) and rheumatoid arthritis (RA).11,12,14 Given its approval status, the package effectively proved the two antibodies were similar, yet not identical. Among differences, the levels of charged variant species attributed to C-terminal truncation and dimer levels were reported and ultimately deemed non-consequential.10,11,13 Like other IgG1 therapeutics, infliximab is a glycoprotein containing an N-glycosylation site at Asn300 in the Fc region of the protein. Both RC and RS contain a heterogeneous mixture of N-glycans that, when analyzed by orthogonal methods, were assessed overall to be similar, as evidenced in a recent publication by the manufacturer.15 However, released regulatory documentation acknowledged apparent differences in RS glycosylation, specifically, in the levels of fucosylated glycans, and how this difference affected FcγIIIa receptor (FcγRIIIa) binding and antibody-dependent cell-mediated cytotoxicity (ADCC).10,11,13 Mediating ADCC is not thought to be a key component of infliximab's efficacy in the treatment rheumatologic diseases (e.g. RA or AS), but it is presumed to be partially responsible for its activity in inflammatory bowel disease (IBD) through the modulation of local immune cell populations.11,14 Due to glycosylation and ADCC differences, the Canadian regulatory agency did not approve the use of RS for treatment of IBD, and requested additional clinical data in the IBD patients.14 Following RS's approval, various rheumatology and gastroenterology societies, as well as patient advocate groups, issued statements against the biosimilar products.17-19 However, more recent post-approval data suggests RS is just as safe and efficacious as RC, and the biosimilar is rapidly gaining acceptance in Europe.20
As more biosimilar mAbs gain regulatory approval, it is important that a clear framework for a rapid characterization of innovator and biosimilar products exists that could identify clinically relevant differences. Our analysis aims to bring a level of transparency and simplicity for biosimilar characterization. We applied a comprehensive mass spectrometry (MS) based strategy using bottom-up, middle down, and intact strategies. These data were then integrated with ion mobility-mass spectrometry (IM-MS) and collision induced unfolding (CIU) analyses, as well as data from select biophysical techniques and receptor binding assays to comprehensively evaluate biosimilarity. Additionally, we sought to fill in the gaps found in the current literature regarding quantifiable comparisons between RC and RS, specifically quantification of the various glycoform levels and FcγRIIIa binding. Our ultimate goal was to develop a template, which can be applied towards future analytical comparisons of biosimilar mAbs.
Experimental Section
Materials
8 lots of the chimeric antibody infliximab were purchased: 4 lots of RC and 4 lots of RS. Both products are supplied as lyophilized powder of identical composition: infliximab, sucrose, sodium phosphate and polysorbate 80.21,22 All samples were reconstituted using pure water for injection (Thermo-HyClone WFI) to a concentration of 1 mg/mL unless specified otherwise.
Enzymatic Digestion
Tryptic digest
Antibody tryptic digests were prepared according to the procedure for the low pH protein digestion kit (Promega, CAS # CS1895A1). Antibody samples were denatured in 8 M urea, reduced and alkylated with iodoacetamide. The samples were diluted seven-fold and mixed with Trypsin Gold and Lys-C (Promega) at 20:1:1 (w/w/w) ratio. Samples were digested overnight at 37°C and acidified with TFA prior to analysis. Glu-C digest. Antibody samples were denatured, reduced and alkylated as above. The samples were diluted four-fold with 130 mM NH4HCO (pH 7.8)/0.027% ProteaseMAX™ surfactant buffer (Promega). Samples were mixed with Glu-C (Promega) at 5:1 (w/w) ratio, digested overnight at 37°C and acidified with TFA prior to analysis. IdeS digest. Samples were digested with IdeS enzyme (Genovis) to generate Fab and Fc fragments according to manufacturer's protocol. Briefly, antibody ∼1 mg/mL, in 0.55 mM sodium phosphate buffer pH 7.2, was mixed with enzyme at a ratio of 1 μg protein: 1 unit enzyme. The mixture was incubated at 37 °C for 3-4 hours to ensure maximal cleavage and quenched on ice prior to analysis. Glu-C digest. Antibody samples were denatured, reduced and alkylated as above. Then the reactions were diluted four fold with 130 mM ammonium bicarbonate, pH 7.8/0.027% ProteaseMAX™ surfactant buffer and digested with Glu-C (Promega) at 5:1 (w/w) ratio overnight at 37°C. The digests were acidified by addition of TFA to the final concentration of 1% and particulate material was removed with centrifugation at 16,000×g.
Deglycosylation and reduction
For LC-MS analysis, 40 μg of antibody was incubated with 4 μL of PNGase F (Promega) for 3 hours at 37°C. Reduction was performed by adjusting the volume to 100 μL with 10 mM DTT in 25 mM NH4HCO. The sample was acidified with TFA prior to analysis. Deglycosylated mAb samples for native IM-MS analysis were prepared without reduction by incubating 10 μL of ∼1 mg/mL intact or IdeS digested mAb sample with 2 μL of PNGase F (glycerol-free, New England Biolabs) in a total reaction volume of 22 μL under native buffer condition overnight at 37°C.
Mass Spectrometry
Intact mAb analysis
5 μg of each sample was analyzed by LC/MS using a C4 column (Waters Xbridge BEH300 3.5μm) interfaced to a ThermoFisher Q Exactive HF mass spectrometer. Data were acquired in 600-2000 m/z range using the Orbitrap for detection.
Peptide Mapping
500 ng of each digested sample was analyzed by nano UPLC-MS/MS with a Proxeon EASY-nLC 1000 HPLC system interfaced to a ThermoFisher Q Exactive HF mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75 μm × 50 cm analytical column (Thermo Fisher P/N ES-803) at 300 nL/min by using a 2 hour reverse phase gradient; both columns were packed with PepMap LC C18, 2 μm resin. The mass spectrometer was operated in data-dependent mode, with MS and MS/MS performed in the Orbitrap at 70,000 and 17,500 FWHM resolutions, respectively. The fifteen most abundant ions were selected for MS/MS.
Ion Mobility-Mass Spectrometry
Reconstituted antibody samples were dialyzed with 100 mM ammonium acetate using Micro Bio-Spin 30 columns (Bio-Rad, Hercules, CA). Sample aliquots (∼7 μL) were analyzed by IM-MS on a quadrupole-ion mobility-time-of-flight mass spectrometer (Q-IM-ToF MS) instrument (Synapt G2 HDMS, Waters, Milford, MA).23,24 Samples were analyzed in triplicate for each lot. Antibody ions were generated using a nESI source in the positive mode. Capillary voltages of 1.4 kV-1.6 kV were applied and the sampling cone was operated at 60 V. The trap traveling-wave ion guide was pressurized to 3.4 × 10−2 mbar of argon gas. The traveling-wave ion mobility separator was operated at a pressure of ∼2.5 mbar and employed a series of DC voltage waves (40 V wave height traveling at 600 m/s) to generate ion mobility separation. The ToF MS was operated over the m/z range of 1000-10000 at a pressure of 1.7 × 10−6 mbar.
Collision Induced Unfolding
CIU experiments were performed on a Waters Synapt G2 HDMS as previously described.25 The 23+ charge state of the intact mAb was first selected by tandem MS using a quadrupole mass filter. Collision energy was then added to successive packets of stored, selected ions prior to IM separation. The collision voltage, defined as the accelerating potential between the end of the quadrupole mass filter and the beginning of the pre-IM ion trap, was ramped from 5 V to 200 V in 5 V increments in order to construct the fingerprint data shown in this report. IM data were recorded for MS-isolated ions at each collision voltage and the IM data for only those ions corresponding the intact m/z originally isolated were compiled to create the plots and quantitative comparisons shown.
Data Analysis
Peptide Digest Data Analysis
Data analysis for LC-MS/MS analysis of digested specimens was performed with Byonic search software (Protein Metrics Inc., San Carlos, CA, USA).26,27 The MS/MS measures the precursor peptide mass in the first stage (MS1) and the fragments these precursors in order to measure the fragment ion masses in the second stage (MS2). The search software identifies peptide ions by matching the mass of the precursor mass (MS1) and the expected fragment ion masses (MS2) to calculated masses based on an in silico digest of the infliximab sequence. The Byonic search included variable modifications such as mono- and di-oxidation on methionine and tryptophan, deamidation and ammonia loss from asparagine, glycation at lysine residues, site mutations of various amino acids, and a wide range of N-linked glycans.
Quantification of modifications relative to unmodified and other modified peptides was accomplished using the Byologic software (Protein Metrics), which uses a label-free quantification approach with extracted ion chromatogram areas (XIC areas). This software automated the XIC extraction and data organization automatically from the Byonic results and/or in silico generated lists of potentially observed molecular ions. The Byonic search parameters matched infliximab peptides generated by trypsin cleavage and QTOF analysis with fragment tolerances of 20.0 ppm and precursor tolerances of 10.0 ppm. Additional search parameters, e.g. +15.994 at M, W for oxidation, +0.984 at N for deamidation, +162.052 at K for glycation and rarer modifications such as mutations like +58.005 at G for Gly to Asp, were set to identify and assign modifications. The glycan assignments were performed by matching a list of 50 biantennary glycans.
IM-MS and CIU Data Analysis
Mass spectra were calibrated externally using a solution of cesium iodide (100 mg/mL) and processed with Masslynx V4.1 software (Waters, Milford, MA). Exact molecular masses of intact mAb and IdeS digested mAb samples were calculated by assigning the charge states based on the set that gives lowest standard deviation for a given average mass assignment.28
Drift time data was extracted at each collision voltage in Drift Scope (Waters, Milford, MA). Extracted CIU data were analyzed using CIUSuite29 CIU ‘fingerprint’ data were plotted as a 2D contour plot using CIUSuite_gen module. The color scale in these plots indicates the signal intensities recorded which were normalized to a maximum value of 1 at each collision voltage and smoothed using a Savitzky-Golay filter. Statistical analysis of CIU datasets was conducted using the CIUSuite_stats function, in which average CIU fingerprints and standard deviation plots were generated for multiple lots of RC and RS samples.
FcγRIIIa Binding via Biolayer Interferometry (BLI)
The binding of different lots of RC and RS with FcγRIIIa were tested by BLI using a BLITZ instrument (Fortebio, Menlo Park, CA). The procedure used here was adopted from the method reported previously.30,31 Protein G biosensor tips were utilized and the binding measurement was performed at 25 °C. Samples were diluted to 0.8 μM mAb with kinetic buffer (PBS buffer containing 1 mg/mL casein as a blocking agent). The protein G biosensor tips were loaded (120 s) with the RC or RS samples. A baseline (240 s) was established followed by the association (180 s) and dissociation (360 s) of FcγRIIIa measured by dipping the biosensor into solutions of FcγRIIIa (FcγRIIIa-V158 variant) and PBS kinetic buffer, respectively. The biosensor tips were regenerated as described previously31 after each assay cycle. To determine the dissociation constant (KD) for the different lots of RC and RS, a range of FcγRIIIa concentrations from 0.4 μM-3.2 μM were evaluated. Data generated were collected in triplicates for each lot and globally fitted to a 1:1 binding model using BLITZ Pro software to calculate ka, kd and KD.
Size Exclusion Chromatography (SEC)
SEC was performed using a Waters Alliance HPLC system equipped with UV detector set at 220 nm. TSK Gel 3000 SWxl column (Tosoh 7.8 mm × 30 cm 5 μm) was used to perform the separation. The mobile phase, PBS pH=7.4, was delivered at 1 mL/min. Samples were filtered a 0.45 μm filter (Millipore) and 25 μL injection volume was used.
Statistical Analysis
Unless specified otherwise statistics were performed using Prism 6 (GraphPad). Unpaired t-tests were performed without assuming a consistent standard deviation. Statistical significance was determined using the Sidak-Bonferroni method, with alpha=5.0%.
Results and Discussion
Four lots of each product were procured and analyzed within the expiry period. The number of lots analyzed in this study was lower than typically used for biosimilar regulatory filing. For example, the data for 26 lots RS and 36 RC was presented by Celltrion in the FDA briefing document, although some assays were performed on as few as 3 lots for each product.12,13 The methodologies used in this study, quality attributes measured, key findings and some advantages of using MAM quantitative mass spectrometry are summarized in Table 1.
Table 1.
Analytical methodologies used to compare Remicade and Remsima.
| Analytical Method | Sample Processing | Measured Attributes | Method Advantages |
|---|---|---|---|
| Native MS, IM-MS | Buffer exchange, Deglycosylation, IdeS digest | Intact mass, fragments, aggregation, glycation, higher order structure, gas-phase stability | Limited sample preparation, rapid analysis, low detection limit and high information content |
| Denaturing MS | Deglycosylation and reduction | Intact masses of subunits, C-terminal Lys truncation | Information on accurate subunit mass and heterogeneity |
| Peptide Mapping | Trypsin digest,Glu-C digest | Sequence, mutations, oxidations, deamidations, glycosylation Glycations | Single analysis allows for identification and quantification of multiple attributes |
| Size Exclusion Chromatography | None | Level of Aggregation | N.A. |
| Biolayer Interferometry | Buffer exchange | Fc receptor binding KD | N.A. |
N. A. - Not applicable.
Previous published results demonstrated high structural similarities via differential scanning calorimetry, NMR and Fc homology using X-ray crystallography.15 To enhance higher order structural characterization, intact mAbs were first analyzed by IM-MS under native conditions, using only minimal preparation of the as-produced therapeutic samples. Relatively weak electric fields are used to separate gas-phase protein ions accordingly to their orientationally-averaged collision cross sections (CCSs) and charge. RC and RS had similar IM drift times with discrete positions in drift time versus m/z space for antibody fragments, monomers and dimers (Figure 1-A, B).
Figure 1. Ion-mobility mass spectrometry of Remicade and Remsima.
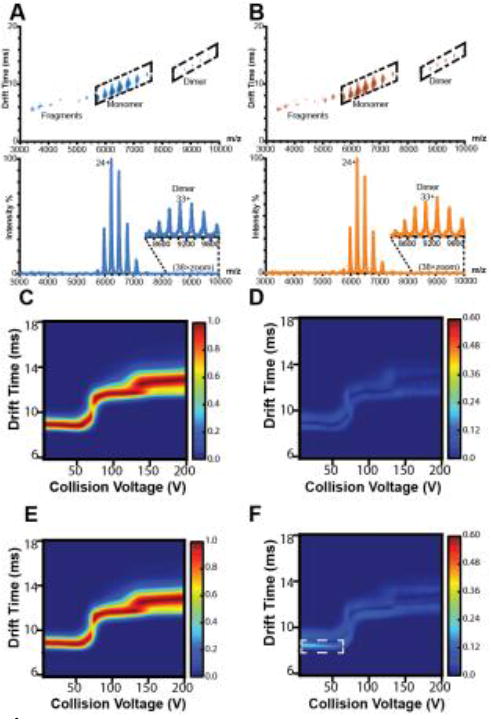
Representative IM-MS spectra of A, RC and B, RS with annotated fragment, monomer, and dimer species. Average collision induced unfolding (CIU) fingerprint of C, RC and E, RS and standard deviations of D, RC and F, RS CIU fingerprints (n = 4 lots).
We further examined the two mAbs structurally using CIU.25,32,33 Collisional heating in the gas-phase is used to generate partially unfolded mAb states, which are subsequently separated by IM and then analyzed by MS. “Fingerprint” plots are created to track the amount of protein unfolding as a function of collisional energy applied, and the unfolding patterns that develop have proven to be highly sensitive to small variations in protein structure that cannot be easily resolved by IM alone.32,33 These fingerprints can be used to rapidly distinguish between antibody isoforms, sensitive to differences in both glycosylation and disulfide bonding.25 An initial CIU analysis of RC and RS revealed remarkably similar structures and, for the first time, quantitatively compared their gas phase unfolding. The CIU fingerprints of mAb 23+ exhibit the strongest degree of similarity at large collision voltages, and the overall variance between RC and RS CIU fingerprints are comparable to levels reported for replicates of mAb reference standard.25 Both mAbs unfold in the gas-phase to produce two unfolded states from an initial compact state over the acceleration voltage range probed, with nearly identical acceleration voltages required to initiate each CIU transition (Figure 1C-F). Conversely, we also observed a marked difference in the variance associated with the CIU fingerprint for RS at collision voltages below 50V which was not observed in RC, highlighted in the white box Figure 1-F. Previous evidence has linked this region of mAb CIU plots to the Fc region and glycoform variation, and we interpret the highlighted variation in Figure 1-F as being related to similar features within RS.25
The intact masses of RC and RS, measured in their fully glycosylated states, after deglycosylation/reduction, and after IdeS digest were all quantitatively compared. The intact masses recorded for glycosylated and deglycosylated mAbs were indistinguishable within our experimental error, 149382 ± 222 and 146066 ± 38 for RC, and 149695± 372 and 146076 ± 51 for RS, respectively (Table S-1). Small levels of misassembled heavy and light chains, resulting in mAb impurities, were found in both products: LC2 or HC, 2.34 ± 0.57% (RC) and 0.09 ± 0.13% (RS) (p<0.001); HC2, 0.83 ± 0.10% (RC) and 1.36 ± 0.25% (RS) (p<0.2); HC-LC, 0.09 ± 0.13% (RC) and 0.88 ± 0.32% (p<0.1) (RS) The presence of misassembled species was also acknowledged in the regulatory filings for RS, with the intact H2L2 98.2% (RC) and 95.1% (RS) as measured by capillary electrophoresis (Table 2).11,13
Table 2.
Key analytical differences between Remicade and Remsima.
| Key attribute | Key new finding (Method) | Reported value (Method)11,12,13,15 |
|---|---|---|
| FcγRIIIa binding | RS: KD=162±18 nM RC: KD=351±48 nM (BLI) |
RS: 101* ± 2.3% RC: 126* ± 7.7% (SPR) |
| Level of afucosylated N-Glycans (%) | RS: 6.46 (Man5), 0.40 (G0), 0.43 (G0GlcNAc) RC: 7.82 (Man5), 3.89 (G0), 2.07 (G0GlcNAc) Sum of 14 detected afucosylated species: RS: 13.2; RC: 19.7 (LC-MS/MS after trypsin digest) |
RS: 4.10 (Man5), 0.72 (G0) RC: 4.31 (Man5), 1.74 (G0) (HPAEC-PAD) |
| Glycation (%) | RS: 3.42; RC: 0.54 (LC-MS/MS, after Glu-C digest) RS: 5.2; RC: 1.7 (Fab+5S, Native MS after IdeS digest) |
RS: 4.0 (HC), 2.4 (LC); RC: 0.8 (HC), 0.8 (LC) (LC-EM-MS after reduction deglycosylation) |
| High molecular weight forms (%) | RS: 1.26; RC: 0.78 (IM-MS) RS: 0.37; RC: 0.06 (SEC) |
RS: 0.5; RC: 0.2 (SEC-MALS) RS: 0.8; RC: 0.2 (SEC) |
| Fragments (%) | RS: 1.97 (HC/LC2); 0.88 (HC-LC); 1.36 (HC2); RC: 2.34 (HC/LC2); 0.09 (HC-LC); 0.83 (HC2); (Native MS) |
RS: 95.1 (Intact H2L2) RC: 98.2 (Intact H2L2) (CE-SDS-NR) |
| Loss of C-terminal Lysine (%) | RS: 72.0 (HC(-)Lys) RC: 62.0 (HC(-)Lys) (LC-MS after reduction and deglycosylation) |
RS: 16.6; RC:26.5 (both Lys are present in H2L2) (IEC) |
Reported as percent relative to RS reference standard. Abbreviations: BLI- Biolayer Interferometry, HPAEC-PAD: High Performance Anion Exchange Chromatography with Pulsed Amperometric Detection, LC-EM-MS: Liquid Chromatography coupled with Electrospray Mass Spectrometry, SEC-MALS: Size Exclusion with Multi-Angle static Light Scattering, CE-SDS-NR: Non-reduced Capillary Electrophoresis based Sodium Dodecyl Sulfate, IEC: Ion Exchange Chromatography, SPR: Surface Plasmon Resonance.
Upon deglycosylation, better MS resolution was achieved for both products (representative curves are shown in Figure 2B-C) and allowed for the identification of further modifications under native conditions. The denaturing MS analysis after deglycosylation and reduction showed that higher levels of C-terminal Lys truncated isoforms were present in RS (72.0±3.5%) relative to RC (62.0±10.8%). Celltrion reported this difference by identifying mAbs species that have two, one and zero C-terminal Lys present by ion-exchange chromatography, with the levels full-length mAbs at 26.5% (RC) and 16.6% (RC).13 The manufacturer also confirmed that the presence of C-terminal Lys has no bearing on infliximab's PK or activity, as it is rapidly cleaved in vivo for both products.13
Figure 2. Intact mass analysis of IdeS digests of Remicade and Remsima.
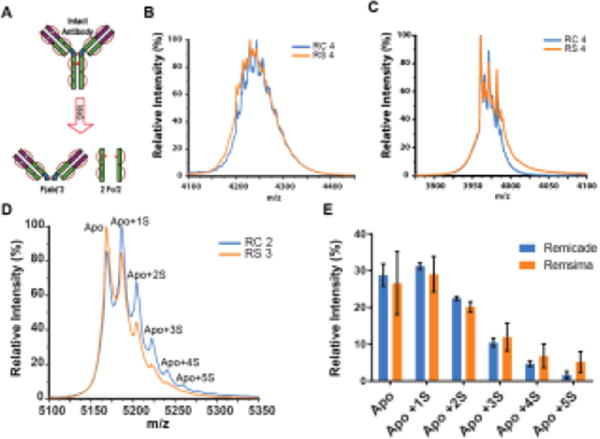
A, Schematic for IdeS enzyme cleavage. Representative intact mass spectra of the 12+ charge state of Fc regions for RC lot#4 and RS lot#4 B, before and C, after deglycosylation. D, representative mass spectra of the 19+ charge state of Fab region of RC lot # 2 and RS lot # 3 resolved peaks of the Apo or unmodified and several sugar-modified variants: Apo+1S, to Apo+5S, corresponding number of glycations). E, Glycation on Fab region of RC and RS (n = 3 lots per product, mean ± SEM).
In order to further evaluate chemical modifications of these mAbs, and map them to either the Fab or Fc portion of the proteins, the two products were subjected to IdeS digestion, which generates F(ab)' 2 and 2 Fc fragments (Figure 2A), and these fragments were analyzed intact by MS. The Fc portion was analyzed before and after deglycosylation (Table S-1, Figure 2B,C). Glycosylated Fc fragments displayed poor MS spectral resolution, especially for the RS samples (Figure 2-B). Alternatively, for F(ab)'2 mass spectra, several peaks were resolved and showed consistent mass shifts of 364 Da. This mass shift corresponds to the addition of two sugar molecules in a process known as glycation, or the non-covalent linkage of sugar to lysine residue.34 Apart from unmodified Apo state we observed Apo+1S to Apo+5S states corresponding to addition of 2 to 10 sugars. The levels of highly glycated Fab (+ 5S) were higher for RS (5.2 ± 2.9%) than RC (1.7 ± 0.9%). Sucrose makes up a significant portion of the drug product and glycation has been observed for other mAb products.21,22 The presence of glycation was confirmed by LC-MS/MS quantitative peptide map analysis using Glu-C for digestion, with an overall glycation levels of 3.42% for RC and 0.54% for RS (Figure S-1). Non-enzymatic glycation was analyzed by the manufacturer using a different method involving deglycosylation/reduction treatment followed by mass analysis and was determined to be 0.8% (HC)/0.8% (LC) and 4.0% (HC)/2.4%(LC) for RC and RS, respectively.13
Next, bottom-up LC-MS analysis was performed and confirmed that the products contained identical sequences with >98% coverage. Trace levels of mutated variants were similar for both products, with a Gly to Asp mutation on the light chains at position 200 (LC200) being the most abundant at ∼0.2% (Figure 3A). Similar levels of individual amino acid oxidation and deamidation were found in both products (Figure 3B-C, Table S-2). Conserved Fc methionine residues (HC-M255 and M431), when oxidized, have been shown to bind inefficiently to neonatal Fc receptors (FcRn), which may result in a shorter mAb plasma circulation half-life. Oxidation has also shown to initiate formation of protein aggregates that could induce immunogenic responses.35,36 The highest extents of oxidation were detected for HC-M255 at 20.8±0.6% (RC) and 21.5±0.6% (RS), as well as HC-M34 at 26.4±2.1% (RC) and 28.7±2.0% (RS). Other methionine residues were oxidized at ∼ 5%, and minor mono- (∼1%) and dioxidized (∼0.5%) tryptophan products were also detected. Observed deamidation levels were similar for both proteins at <3% for any specific residue. Celltrion has also acknowledged the presence of these chemical modifications and the similar levels of these chemical species between the two products suggest these should not clinically impact the products.11,15
Figure 3. LC/MS/MS analysis of trypsin digests of Remicade and Remsima.
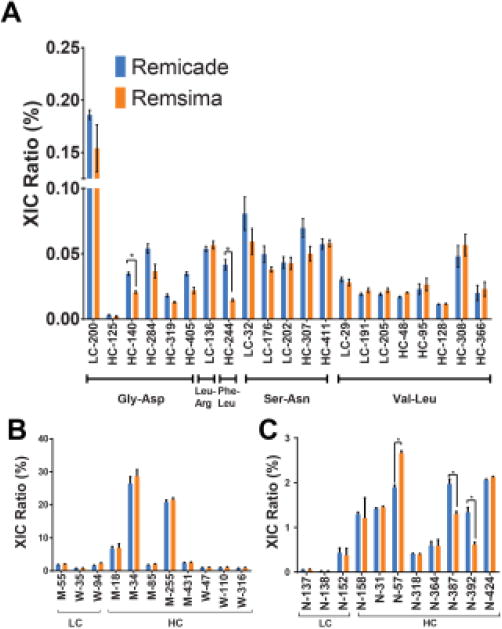
A, sequence variants B, oxidation and C, deamidation (n = 4 lots, mean ± SEM). (*) denotes significance at p<0.05.
Despite these remarkable similarities we also, for the first time, quantified significant differences in the N-glycan distributions for RS and RC. CIU variations detected in RS samples at acceleration voltages less than 50V (Figure 1-F) likely relate to the broader range of glycoforms present in this product when compared to RC. Label free LC-MS analysis of trypsin-digested products identified the presence of over 25 glycoforms for each product (Table S-3, Figure 4). The most abundant were G0F (48.9% RC, 40.7% RS) and G1F (21.6% RC, 37.0% RS), while the least abundant was NGNA (0.03% RC, 0.02% RS). The levels of mannose-terminated glycans, known to reduce the circulation time of mAbs, but possibly also increase FcγRIIIa binding, were also found to be different between the two mAbs (14.9 ± 1.5% for RC and 9.0 ± 3.1% for RS). A total of 14 afucosylated glycoforms were identified in both products with the total afucosylation levels higher for RC (19.7±1.6%) compared to RS (13.2±3.8%). The three most prevalent afucosylated glycoforms were Man5 (7.82% RC, 6.46% RS), Go-Man(3) (3.89% RC, 0.40% RS) and G0-GlcNAc (2.07% RC and 0.43% RS).
Figure 4. Remicade and Remsima glycosylation heterogeneity and FcγR-IIIa binding.
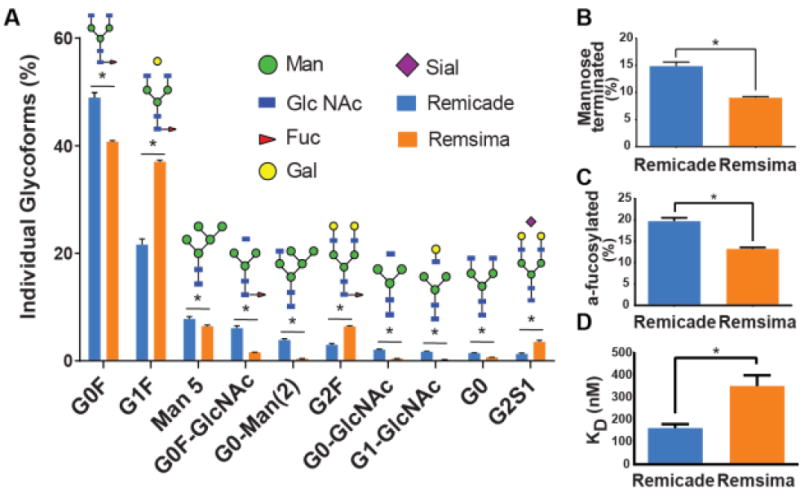
A, Quantification of N-glycans following LC/MS/MS analysis of RC B, total mannose terminated forms C, total afucosylated forms. D, Average KD of RC and RS receptor binding to FcγIIIa as measured by biolayer interferometry (n = 4 lots, mean ± SEM; * denotes significance at p<0.05).
At the time of the original approval of RC, very little was known regarding the effect of antibody glycosylation, and this attribute was not viewed as critical for the action of the therapeutic in neutralizing TNF-α. Since then, however, data have suggested the importance of the conserved Fc glycan profile in determining the efficacy as well as the physicochemical attributes for an antibody.37 Glycosylation is a posttranslational modification occurring in many proteins and the distribution of glycans is a function of cell line used to produce the protein, batch size and cell culture conditions.38 Thus, cell line development and cell culture process optimization for biosimilars, in order to match the glycan distributions of the reference product, present large challenges for the modern biosmilar developer. In the case of RC and RS, murine cell lines are used, but with subtle genetic variants,11 clearly yielding slightly differing glycan populations. General strategies to homogenize polysaccharide profiles include modifying the expression system, fermentation processes, and even enzymatically modifying the purified antibody.39-41 Differences in afucosylation levels of Remsima and Remicade were mentioned by the regulators,12,14,16 but the actual levels were only reported for Man5 (4.32% (RC) and 4.10% (RS) and G0 (1.72% (RC) and 0.72% (RS) (Table 2).13
To assess how this measured difference in afucosylation levels between RC and RS influences mAb biological activity, we measured RC and RS binding to FcγRIIIa using biolayer interferometry (Figure 4D, Figure S-2). Indeed, RC bound to FcγRIIIa with a twofold lower KD compared to RS (162±18 nM versus 351±48 nM). The differences in FcγRIIIa binding between RC and RS have been measured using surface plasmon resonance (SPR) by the biosimilar manufacturer, and were reported in the FDA briefing document.12 The binding of RC and RS to FcγRIIIa were reported to be 126 + 7.7% and 101 + 2.3% relative to reference standard (for a separate lot of RS than what is studied here).12 It is not clear in the FDA briefing document which binding parameters were compared, how many lots of RC and RC and what degree of afucosylation was observed for these lots. In addition, the methodologies used to measure receptor binding by the manufacturer (SPR) and by us (BLI) were different as well as the source of recombinant FcγRIIIa. Nevertheless, afucosylation differences between the two products, quantified by LC-MS in this study, appear to reduce RS binding to FcγRIIIa by approximately 21%, as measured by the biosimilar manufacturer, 12 and 54% as determined by us.
This difference in FcγRIIIa binding for the two products is critical for determination of biosimilarity of RC and RS in IBD, as it affects ADCC,19 and ADCC is speculated to be a mechanism for down regulation of intestinal inflammation. When the Fab domain of infliximab is bound to TNF-α expressed on the membranes of inflammatory macrophages or T-cells, the Fc domain attracts potent natural killer (NK) immune cells. Binding of the Fc domain to FcγRIIIa on NK cells leads to the effective crosslinking of inflammatory cells to NK cells. The lytic synapse is triggered, resulting in the effective “killing” of inflammatory cells by NK cells. Thus, the reduced FcγRIIIa binding by RS could lead to the reduction of ADCC.42 Indeed, a 20% reduction in NK cell-induced ADCC activity of RS over RC is in good agreement with the 21% reduction in FcγRIIIa binding, both values were reported in the FDA briefing document.12 However, 90% of all RS lots tested appeared to be within statistical variability of RC's ADCC values.
Based on this data and the fact that ADCC differences were not apparent in more stringent assays, both the FDA and European regulators approved RS to treat IBD indications. In contrast, Canadian regulators requested additional clinical data in IBD. Our data underscores how chemical analysis of the biosimilar product could be correlated with its bioactivity to define clinical efficacy testing requirements.
To further examine how analytical characterization could be used to foresee potential clinical safety issues, we have used native IM-MS data together with gel permeation chromatography to examine RS and RC aggregation. The presence of protein aggregates has long been attributed to increased immunogenicity with the formation of antidrug antibodies and neutralizing antibodies (NAb).43 IM-MS analysis confirmed the presence of mAb dimers, 0.78 ± 0.22% for RC and 1.26 ± 0.99% for RS (Figure S-3, Table S-4). Size-exclusion chromatography (SEC) corroborated our IM-MS data, detecting small differences in soluble dimer levels of RC (0.06 ± 0.02%) and RS (0.37 ± 0.17%) (Figure S-3, Table S-4). The higher levels of high molecular weight forms were reported in regulatory filing of 0.2% (RC) and 0.8% (RS) as measured by SEC. Another factor that may affect immunogenicity is the presence of sub-visible particulates, and biologic production processes are in place to limit such particulates in the 10-25 μm size range. Regulatory filings suggest both RS and RC have a varied range of particulates sized 1-100 μm, as measured by light obscuration and micro-flow imaging, but the levels across the products were comparable.12 However, infliximab is highly immunogenic due to its chimeric nature and detectable levels of NAb were found in over 50% of RA patients in both the RC and RS treatment arms.11,12,44 Patient response toward the murine portion of infliximab likely dominates its immunogenicity over any small differences in aggregation state.
RC and RS appear to be very similar structurally, yet several important differences between two products were found by our laboratory and are summarized in Table 2, where they are compared with values taken from RS regulatory filings. The important differences were found in FcγR-IIIa binding, glycoform distribution, levels of afucosylation, non-enzymatic glycation, high molecular weight impurities, misassembled mAb species and C-terminal truncation levels. In all cases, our findings were in directional agreement with the reported results, however, specific levels of individual impurities and product variances were different and our results provided higher granularity. Using quantitative MAM peptide map label-free LC-MS/MS methodology, we have obtained a large level of detail regarding populations of glycoforms, specific amino acid oxidations and deamidations, and detected the presence of minor mutations at levels <0.2%. In addition, we identified a higher level of glycation, dimers and miss-assembled mAbs in RS relative to RC. Some of the discrepancies in impurity levels measured in our lab and reported in the regulatory documents are potentially due to the different methodologies used to analyze impurities, different lots of products used for analysis and lab-to-lab variability in experimental procedures and data analyses.
Conclusions
RS is just the first of multiple mAb biosimilar products that are on-track for approval by the FDA. While individual analytical assays are capable of distinguishing product variants and impurities, lab-to-lab variability in analytical methods and equipment makes data comparison cumbersome. This study represents an example of a rapid and thorough examination of two products by an independent laboratory, allowing the identification of clinically relevant differences such as afucosylated glycans levels (RC: 19.7% vs RS: 13.2%) and FcγRIIIa binding (RC: 162 nM vs RS 351 nM). The analytical tools described here can provide extensive data, ranging from primary modifications (levels as low as <0.2%) to intact structural features, and could be a compelling template for emerging MAM workflows.45,46 The ability to rapidly characterize and quantify complex mAb glycoforms will be especially critical for examining biosimilarity of oncology products that are reliant on an ADCC mechanism of action driven by afucosylation levels. Rapid quantification of oxidation, deamidation, glycation and aggregation enhances the general understanding of both the product and process variability inherent in therapeutic development pipelines, and pinpoints “hot spots” leading to protein instability. Such MAM approaches will undoubtedly provide invaluable insights for mAb development and biosimilar comparison, as well as characterization of post-approval innovator product changes, resulting from process modifications, scale-up, and plant transfers.
Supplementary Material
Acknowledgments
Authors would like to acknowledge editorial help of Dr. McCammon and Dr. Azvolinsky.
Funding Sources: The work here is supported by National Institute for Pharmaceutical Technology and Education (NIPTE) U01 FD004275 (AS, SPS, TT), NIGMS R01 GM090080 (TT), NSF CAREER 1253384 (BR), NIGMS R01GM100634 (CB, EC) American Foundation for Pharmaceutical Education Pre-Doctoral Fellowship (KP), NIH Biotechnology Training Grant 5-T32-GM008359 (SZO).
Abbreviations
- ADCC
antibody dependent cell cytotoxicity
- AGE
advanced glycation end products
- AS
ankylosing spondylitis
- BLI
biolayer interferometry
- CCS
collision cross sections
- CIU
collision induced unfolding
- CQA
critical quality attribute
- FcRn
neonatal Fc receptor
- FcγRIIIa
Fc γ receptor III a
- FDA
Food and Drug Administration
- HC
heavy chain
- IBD
inflammatory bowel disorder
- IM-MS
ion mobility mass spectrometry
- LC
light chain
- mAb
monoclonal antibody
- MAM
multiple attribute monitoring
- MS
mass spectrometry
- Nab
neutralizing antibody
- NGNA
N-glycolylneuraminic acid
- NK cells
natural killer cells
- RA
rheumatoid arthritis
- RC
Remicade
- RS
Remsima
- TNF-α
tumor necrosis factor α
- XIC
extracted ion chromatogram
Footnotes
Supporting Information. Additional LC-MS, IM-MS, Fc receptor binding and SEC analyses for RC and RS. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes: The authors declare no competing financial interest.
References
- 1.Walsh G. Nat Biotechnol. 2014;32:992–1000. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 2.Rickwood S, Di Base S. IMS Health. 2013 [Google Scholar]
- 3.Mulcahy A, Predmore Z, Mattke S. Perspective Rand Corporation. 2014 [Google Scholar]
- 4.Schellekens H. NDT Plus. 2008;2:i27–i36. doi: 10.1093/ndtplus/sfn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsiftsoglou AS. Nat Rev Drug Discov. 2007;6 [Google Scholar]
- 6.Kozlowski S, Woodcock J, Midthun K, Behrman Sherman R. N Engl J Med. 2011;365:385–388. doi: 10.1056/NEJMp1107285. [DOI] [PubMed] [Google Scholar]
- 7.US FDA. Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Refrence Product. Guidance for Industry. 2015 [Google Scholar]
- 8.Konara CS, Barnard RT, Hine D, Siegel E, Ferro V. Trends Biotechnol. 2016;34(1):70–83. doi: 10.1016/j.tibtech.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Nisen M. The best selling prescription drugs in the world last year. [accessed October, 2015]; http://qz.com/349929/best-selling-drugs-inthe-world/
- 10.GaBI. Biosimilars of Infliximab. [accessed July, 2016]; http://www.gabionline.net/Biosimilars/General/Biosimilars-of-infliximab.
- 11.European Medicines Agency. Assessment report: Inflectra. 2013 [Google Scholar]
- 12.FDA Arthritis Advisory Committee. FDA Briefing Document Arthritis Advisory Committee Meeting BLA 125544 CTP13 a proposed biosimilar to Remicade (infliximab) Celltrion. 2016 [Google Scholar]
- 13.CT-P13 (infliximab biosimilar) [accessed July, 2016];Briefing Document for the Arthritis Advisory Committee. https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/arthritisadvisorycomm ittee/ucm484860.pdf.
- 14.Health Canada Remsima: Summary Basis of Decision. 2014
- 15.Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim D, Lee SY, Chang SJ. mAbs. 2014;6:1163–1177. doi: 10.4161/mabs.32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ternant D, Berkane Z, Picon L, Gouilleux-Gruart V, Colombel J, Allez M, Louis E, Paintaud G. Clin Pharmacokinet. 2015;54:551–562. doi: 10.1007/s40262-014-0225-3. [DOI] [PubMed] [Google Scholar]
- 17.Danese S, Gomollon F. Journal of Crohn's and Colitis. 2013;7:586–589. doi: 10.1016/j.crohns.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 18.de Ridder L, Waterman M, Turner D, Bronsky J, Hauer AC, Dias JA, Strisciuglio C, Ruemmele FM, Levine A, Lionetti P. J Pediatr Gastroenterol Nutr. 2015;61 doi: 10.1097/MPG.0000000000000903. [DOI] [PubMed] [Google Scholar]
- 19.Dorner T, Kay J. Nat Rev Rheumatol. 2015;11:713–724. doi: 10.1038/nrrheum.2015.110. [DOI] [PubMed] [Google Scholar]
- 20.Danese S, Fiorino G, Michetti P. Has IBD specialists' awareness of biosimilar monoclonal antibodies changed?. Proceedings of 11th congress of ECCO, Amsterdam Netherlands; Mar 16-19 2016. [Google Scholar]
- 21.Janssen Biotech. Remicade ® [package insert] 2015 [Google Scholar]
- 22.Celltrion Inc. Remsima™ [package insert] 2014 [Google Scholar]
- 23.Zhong Y, Hyung S, Ruotolo BT. Analyst. 2011;136:3534–3541. doi: 10.1039/c0an00987c. [DOI] [PubMed] [Google Scholar]
- 24.Giles K, Williams JP, Campuzano I. Rapid Communications in Mass Spectrometry. 2011;25:1559–1566. doi: 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- 25.Tian Y, Han L, Buckner AC, Ruotolo BT. Anal Chem. 2015;87:11509–11515. doi: 10.1021/acs.analchem.5b03291. [DOI] [PubMed] [Google Scholar]
- 26.Bern M, Kil YJ, Becker C. Current Protocols in Bioinformatics. 2012:13.20.1–13.20.14. doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bern M, Cai Y, Goldberg D. Anal Chem. 2007;79:1393–1400. doi: 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- 28.McKay AR, Ruotolo BT, Ilag LL, Robinson CV. J Am Chem Soc. 2006;128:11433–11442. doi: 10.1021/ja061468q. [DOI] [PubMed] [Google Scholar]
- 29.Eschweiler JD, Rabuck-Gibbons JN, Tian Y, Ruotolo BT. Anal Chem. 2015;87:11516–11522. doi: 10.1021/acs.analchem.5b03292. [DOI] [PubMed] [Google Scholar]
- 30.Alsenaidy MA, Okbazghi SZ, Kim JH, Joshi SB, Middaugh CR, Tolbert TJ, Volkin DB. J Pharm Sci. 2014;103:1613–1627. doi: 10.1002/jps.23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okbazghi SZ, More AS, White DR, Duan S, Shah IS, Joshi SB, Middaugh CR, Volkin DB, Tolbert TJ. J Pharm Sci. 2016;105(2):575–87. doi: 10.1016/j.xphs.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong Y, Han L, Ruotolo BT. Angew Chem Int Ed. 2014;53:9209–9212. doi: 10.1002/anie.201403784. [DOI] [PubMed] [Google Scholar]
- 33.Rabuck JN, Hyung S, Ko KS, Fox CC, Soellner MB, Ruotolo BT. Anal Chem. 2013;85:6995–7002. doi: 10.1021/ac4012655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller AK, Hambly DM, Kerwin BA, Treuheit MJ, Gadgil HS. J Pharm Sci. 2011;100:2543–2550. doi: 10.1002/jps.22504. [DOI] [PubMed] [Google Scholar]
- 35.Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, Li L, Brems DN, Remmele RL., Jr Biochemistry. 2008;47:5088–5100. doi: 10.1021/bi702238b. [DOI] [PubMed] [Google Scholar]
- 36.Zhang A, Hu P, MacGregor P, Xue Y, Fan H, Suchecki P, Olszewski L, Liu A. Anal Chem. 2014;86:3468–3475. doi: 10.1021/ac404130a. [DOI] [PubMed] [Google Scholar]
- 37.Hmiel L, Brorson K, Boyne M., II Anal Bioanal Chem. 2015;407:79–94. doi: 10.1007/s00216-014-8108-x. [DOI] [PubMed] [Google Scholar]
- 38.Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Nat Biotech. 2011;29:310–312. doi: 10.1038/nbt.1839. [DOI] [PubMed] [Google Scholar]
- 39.Huang W, Giddens J, Fan S, Toonstra C, Wang L. J Am Chem Soc. 2012;134:12308–12318. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Q, Shankara S, Roy A, Qiu H, Estes S, McVie-Wylie A, Culm-Merdek K, Park A, Pan C, Edmunds T. Biotechnol Bioeng. 2008;99:652–665. doi: 10.1002/bit.21598. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi B, Cook WJ, Cukan M, Houston-Cummings N, Davidson R, Gong B, Hamilton SR, Hoopes JP, Jiang Y, Kim N, Mansfield R, Nett JH, Rios S, Strawbridge R, Wildt S, Gerngross TU. Nat Biotech. 2006;24:210–215. doi: 10.1038/nbt1178. [DOI] [PubMed] [Google Scholar]
- 42.Peake STC, Bernardo D, Mann ER, Al-Hassi H, Knight SC, Hart AL. Inflamm Bowel Dis. 2013;19 doi: 10.1097/MIB.0b013e318281333b. [DOI] [PubMed] [Google Scholar]
- 43.Schellekens H, Jiskoot W. In: Pharmaceutical Biotechnology: Fundamentals and Applications. Crommelin ADJ, Sindelar DR, Meibohm B, editors. Springer New York; New York, NY: 2013. pp. 133–141. [Google Scholar]
- 44.Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, Kovalenko V, Prodanovic N, Abello-Banfi M, Gutierrez-Ureña S, Morales-Olazabal L, Tee M, Jimenez R, Zamani O, Lee SJ, Kim HU, Park W, Müller-Ladner U. Ann Rheum Dis. 2013;72:1613–1620. doi: 10.1136/annrheumdis-2012-203090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Nat Rev Drug Discov. 2012;11:527–540. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pais DAM, Carrondo MJ, Alves PM, Teixeira AP. Curr Opin Biotechnol. 2014;30:161–167. doi: 10.1016/j.copbio.2014.06.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.