

Abstract
Apolipoprotein E (apoE) is a lipid carrier in both periphery and the central nervous system (CNS). Lipid-loaded apoE lipoprotein particles bind to several cell surface receptors to support membrane homeostasis and injury repair in the brain. Considering prevalence and relative risk magnitude, the ε4 allele of the APOE gene is the strongest genetic risk factor for late-onset Alzheimer’s disease (AD). ApoE4 contributes to AD pathogenesis by modulating multiple pathways including but not limited to the metabolism, aggregation, and toxicity of amyloid-β (Aβ) peptide, tauopathy, synaptic plasticity, lipid transport, glucose metabolism, mitochondrial function, vascular integrity, and neuroinflammation. Emerging knowledge on apoE-related pathways in the pathophysiology of AD presents new opportunities for AD therapy. In this Review, we describe the biochemical and biological features of apoE and apoE receptors in the CNS. We also discuss the evidence and mechanisms addressing differential effects of apoE isoforms and the role of apoE receptors in AD pathogenesis, with a particular emphasis on the clinical and preclinical studies related to Aβ pathology. Finally, we summarize the current strategies of AD therapy targeting apoE, and postulate that effective strategies require an apoE isoform-specific approach.
Keywords: Apolipoprotein E, low-density lipoprotein receptor family, Alzheimer’s disease, amyloid-β, tauopathy, synaptic plasticity
Alzheimer’s disease (AD), the most common form of dementia, is a neurodegenerative disorder characterized by the appearance of extracellular senile plaques and intracellular neurofibrillary tangles, accompanying a progressive loss of memory, thinking and language skills, as well as behavioral changes (1). Mounting genetic and biochemical evidence support that the accumulation and aggregation in the brain of amyloid-β (Aβ), either 40 or 42 amino acids in length derived from proteolytic processing of amyloid precursor protein (APP), is a central and defining event in the pathogenesis of AD (2). The toxic soluble Aβ oligomers, intraneuronal Aβ, and extracellular amyloid plaques disrupt normal synaptic functions and trigger downstream toxic pathways which ultimately lead to neurodegeneration (1). The toxicity of Aβ-induced neuronal dysfunction likely depends on the presence of microtubule-associated protein tau, which aggregates and deposits in AD brains as neurofibrillary tangles (3).
Only a small percentage (<5%) of AD cases develop symptoms before the age of 65 commonly referred to as early-onset AD (EOAD). A portion of EOAD known as familiar AD (FAD) is caused by genetic mutations in APP, presenilin 1 (PSEN1) or PSEN2 gene leading to Aβ overproduction (2). The vast majority of AD cases occur late in life (age 65 and older) known as late-onset AD (LOAD) (2). The impairment of Aβ clearance appears to be the leading cause of Aβ accumulation in LOAD (4). Considering prevalence and relative risk magnitude, the strongest genetic risk factor for LOAD is apolipoprotein E (APOE) gene allele, with the ε4 allele increasing AD risk and the ε2 allele being protective compared with the common ε3 allele (5–7). Although studies indicate that apoE4 enhances AD risk likely by both gain of toxic function and loss of neuroprotective function through several distinct pathways when compared to apoE2 and apoE3, the exact mechanisms by which apoE isoforms differentially contribute to AD pathogenesis are still not completely understood (6, 8). In this review, we will discuss the current status of clinical and basic evidences on how apoE and apoE receptors modulate AD pathogenic pathways with a particular focus on Aβ-related pathways, and update the recent advances in targeting apoE and apoE receptors for AD therapy.
Biochemical features of apoE
ApoE is a glycoprotein of 299 amino acids with a molecular mass of ~34 kDa. It transports and delivers cholesterol and other lipids in the plasma and the central nervous system (CNS) via binding to cell surface apoE receptors (Figure 1) (9, 10). The receptor-binding domain of apoE is in the N-terminal domain (residues 136–150), whereas the major lipid-binding region (residues 244–272) resides in the C-terminal domain (6, 8). The three isoforms, apoE2, apoE3 and apoE4, differ at position 112 and 158. ApoE2 has Cys residues at both positions 112 and 158; apoE3 has a Cys residue at 112 and an Arg residue at 158, and apoE4 has Arg residues at both positions (11).
Figure 1. Schematic illustration of the structure of human apoE.
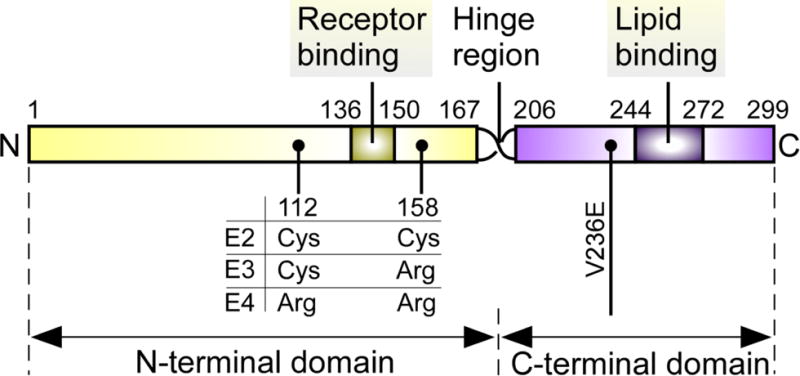
ApoE can be subdivided into two major domains (6–8). The amino-terminal domain includes the receptor-binding region (residues 136–150), which is responsible for the interaction of apoE and its receptors. The carboxyl-terminal domain contains the major lipid-binding region (residues 244–272), which is responsible for lipid binding. The middle region of apoE (residues 167–206) is a hinge region that joins the two domains together. The apoE2, apoE3, and apoE4 isoforms differ from one another at amino acid residues 112 and/or 158 (11). ApoE2 has Cys at both positions, apoE3 has Cys at position 112 and Arg at position 158, and apoE4 has Arg residues at both positions. The V236E variance just outside the lipid-binding region, which reduces the risk of late-onset of Alzheimer’s disease (LOAD) (122), is marked.
ApoE is mainly produced by hepatocytes, macrophages and adipocytes in the peripheral tissues (12). In the CNS, apoE is highly expressed in astrocytes, as well as in microglia, vascular mural cells, and choroid plexus, and in neurons only under stress or injury conditions (12–14). The secreted apoE is lipidated by ATP-binding cassette transporter A1 (ABCA1) and ABCG1, which transport the cellular cholesterol and phospholipids to nascent apoE forming lipoprotein particles (15). ApoE in the plasma is preferentially associated with very low-density lipoprotein (VLDL) particles, whereas it is found in HDL-like particles in the CNS (16, 17). Brain apoE is primarily derived from de novo synthesis because the blood-brain barrier (BBB) limits the transport of apoE into and out of brain (18). Consistent with this notion, liver transplantation changes only the plasma apoE isoform in the recipient to that of the donor but not in the cerebral spinal fluid (CSF) (19). Interestingly, it was recently reported that plasma apoE influences cognitive functions (20). Further investigation is needed to understand whether this effect is apoE isoform-dependent, and to identify the mechanism by which peripheral apoE impacts CNS functions.
The levels of plasma apoE vary with APOE genotypes with APOE ε2/2 being the highest and APOE ε4/4 being the lowest (21, 22). This apoE isoform-dependent difference in apoE levels are also shown in CSF (23) and interstitial fluid (ISF) (24). ApoE4 preferentially forms a molten globule intermediate that makes it less stable than apoE3 (25). The domain interaction of apoE4 through an ionic interaction between arginine-61 and glutamic acid-255 may also increase hepatic clearance and enhance astrocyte degradation of apoE4 (22, 26).
ApoE receptors
The low-density lipoprotein receptor (LDLR) family, including LDLR, LDLR-related protein 1 (LRP1), very-low-density lipoprotein receptor (VLDLR) and apoE receptor 2 (apoER2), are the major apoE receptors which exhibit distinct binding affinity to apoE with different isoforms and lipidation status (6). LRP1 preferentially binds to recombinant apoE or apoE aggregates, whereas LDLR and VLDLR bind to either lipidated apoE particles or lipid-free apoE, respectively (6, 7, 27). In addition to the LDLR family, both lipidated and non-lipidated apoE bind to cell surface heparan sulfate proteoglycans (HSPGs) (28, 29). ApoE2 has a poor binding affinity to the LDLR (1–2% of apoE3 binding) which contributes to an increased risk of a small percent of APOE2 homozygous individuals for type III hyperlipoproteinemia characterized by increased plasma cholesterol and triglycerides (30, 31). The decreased affinity of apoE2 to LRP1 compared with apoE3 or apoE4 seems to be less severe (40% of apoE3 binding) (31). VLDLR (27)and HSPGs (10) recognize all apoE isoforms with similar affinity. Additionally, LRP1 can form a complex with HSPG in mediating apoE binding, suggesting a cooperative function for these apoE receptors at the cell surface (28).
ApoE in Aβ metabolism and pathology
Effects of apoE on brain Aβ deposition
Using 18F-florbetapir-PET or 11C-Pittsburgh compound B (PiB)-PET imaging, clinical studies showed that cerebral Aβ deposition is positively associated with APOE4 genotype in cognitively normal subjects, mild cognitive impairment (MCI) cases, and symptomatic AD patients (32–34). Additionally, amyloid imaging positivity appears to begin earlier in cognitively intact APOE4 carriers (near age 56 years) than APOE4 non-carriers (at age 76 years) (35). These observations suggest that APOE4 likely increases the risk of AD by initiating and accelerating Aβ accumulation, aggregation and deposition in the brain. APOE2 is associated with milder Aβ pathology and slower cognitive decline compared to APOE3 and APOE4, suggesting a neuroprotective effect of APOE2 in AD (36). Interestingly, in the oldest-old population (90 years and older), the presence of APOE2 reduces dementia risk but increases AD neuropathology (37), implying that the mechanism by which apoE2 protects the cognitive function is independent of AD pathology. The levels of oligomeric Aβ, potentially the most neurotoxic Aβ species, are 2.7 times higher in AD brains with APOE ε4/4 than those with APOE ε3/3, suggesting that apoE4 impacts the metabolism of Aβ oligomers (38). In amyloid mouse models, the presence of human apoE4 is associated with accelerated oligomerization and deposition of Aβ in the brain. By crossing human apoE-targeted replacement (TR) mice to murine apoE(−/−)/APP mice, it was shown that apoE4/APP mice have more severe amyloid deposition compared with apoE3/APP mice (39). Viral-mediated expression of apoE4 in APP/PS1 mouse brain increases oligomeric Aβ within the ISF and exacerbates plaque deposition, whereas expression of apoE2 leads to an opposite effect (40, 41). In transgenic mice expressing five familial AD mutations and human apoE isoforms (EFAD), the levels of plaque deposition and oligomeric Aβ are greater in E4FAD mice in which the plaques are more compact compared to those in E2FAD or E3FAD mice (42). A recent study showed that the effects of apoE on the aggregation of Aβ40 depend on the apoE isoforms, with apoE4 possessing the greatest effects (43), although a conflicting finding was reported previously (44). Whether apoE4 promotes seeding and aggregation of Aβ in vivo warrants further investigation.
Effects of apoE on CSF Aβ
CSF Aβ is one of the promising biomarkers with high accuracy to differentiate patients with AD from control subjects or patients with other neurologic conditions (45). In AD patients, the CSF Aβ42 concentration is decreased about 50% to that of control individuals likely due to amyloid deposition in the brain (45). Although the APOE4 allele is associated with enhanced cerebral Aβ deposition, how it influences CSF Aβ is still controversial. It was previously shown that APOE4 is strongly associated with reduced CSF Aβ42 levels in both AD and control populations (46, 47). However, a recent study from 4 centers in different countries revealed that CSF Aβ42 levels are strongly associated with the diagnosis of AD and cerebral Aβ accumulation, but are independent of APOE genotype (48). Other studies focusing on the levels of apoE and Aβ42 in CSF revealed that apoE protein levels are positively associated with those of Aβ42 and phosphorylated tau in CSF, making apoE levels a potential biomarker for AD (49, 50).
Mechanisms of apoE and apoE receptors in modulating Aβ metabolism
The Aβ level in the brain represents the net balance of Aβ production and clearance (2); thus, Aβ accumulation in AD brains may reflect overproduction, inefficient clearance, or both. Mounting evidence demonstrates that apoE and apoE receptors play important roles in these processes which will be discussed in more detail below (Figure 2).
Figure 2. Major Aβ clearance pathways and the roles of apoE and apoE receptors.
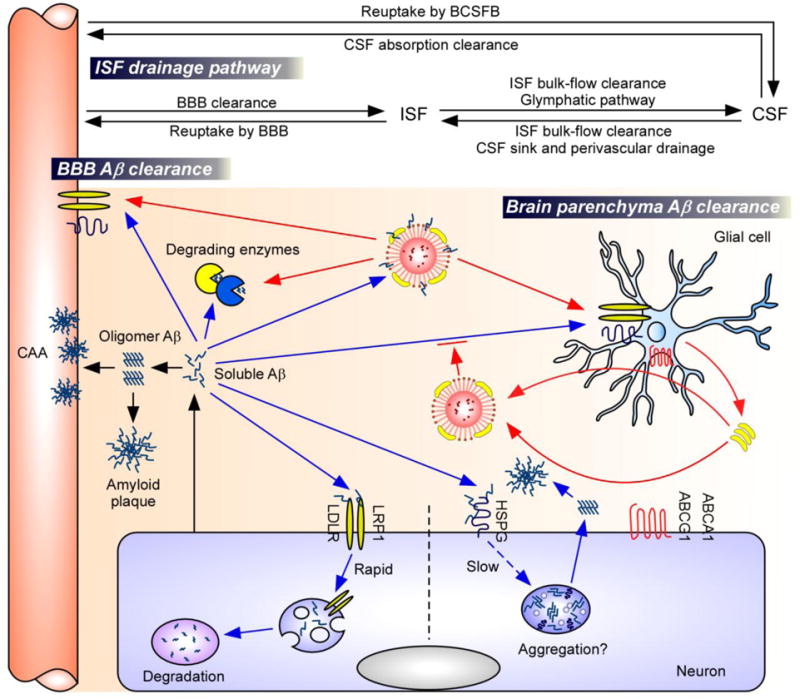
Aβ is predominantly produced by neurons (6–8). The accumulation of soluble Aβ in the brain parenchyma leads to the formation of Aβ oligomers and amyloid plaques, whereas its accumulation in the perivascular region leads to CAA (6–8). Soluble Aβ can be removed from the brain by various clearance pathways closely linked to each other: soluble Aβ in ISF can be cleared in brain parenchyma, transported to the blood through BBB, or is degraded by vascular mural cells. Aβ can also traffic along the ISF bulk flow into CSF sink where it could be absorbed into the circulatory and lymphatic systems (6, 60). Major Aβ clearance pathways include receptor-mediated clearance (e.g., LRP1, LDLR) by cells in the brain parenchyma (neurons and glia), uptake from the perivascular space by vascular mural cells, or proteolytic degradation by endopeptidases (e.g., NEP, IDE) (6–8). Binding of Aβ to neuronal HSPGs on the cell surface hinders proteolytic degradation of Aβ, and promotes Aβ oligomerization and aggregation (75). ApoE is generated mainly by the glial cells and lipidated by ABCA1 and ABCG1 transporters, forming lipoprotein particles (15). Lipidated apoE could either bind to Aβ and facilitates Aβ clearance through cell-surface receptors (e.g., LRP1, LDLR) (apoE2 > apoE3 > apoE4) (77–80). Alternatively, apoE might also suppress Aβ clearance by competing with Aβ for receptor binding in astrocytes (apoE4 > apoE3) (81).
Abbreviations: Aβ, amyloid-β; BBB, blood-brain barrier; BCSFB, blood-CSF barrier; CSF, cerebrospinal fluid; ISF, interstitial fluid; CAA, cerebral amyloid angiopathy; NEP, neprilysin; IDE, insulin-degrading enzyme; LRP1, low-density lipoprotein receptor-related protein 1; LDLR, low-density lipoprotein receptor; HSPG, heparan sulfate proteoglycan; ABCA1, ATP-binding cassette A1; ABCG1, ATP-binding cassette G1.
ApoE and apoE receptors in Aβ production
APP localizes on the surface of the plasma membrane where it is subjected to proteolytic processing by α-secretase. In the amyloidogenic pathway, APP is internalized into endocytic compartments and cleaved by β-secretase and γ-secretase to generate Aβ peptides (Aβ40 and Aβ42) (51). Several apoE receptors, including LRP1, interact with APP and modulate its trafficking and processing to Aβ (15, 52, 53). For instance, due to the fast endocytosis, LRP1 accelerates APP endocytic trafficking and thus increases the production of Aβ (54, 55). LRP6, an apoE receptor that mediates Wnt signaling, is shown to reduce APP endocytic trafficking and Aβ production. Consistently, conditional knockout of neuronal LRP6 increases amyloid deposition and associated neuroinflammation (56). In addition, studies show that apoE regulates APP trafficking and Aβ production in an isoform-specific manner. ApoE4 promotes APP internalization which increases Aβ production to a greater extent than apoE3 in a neuronal cell line and this effect is mediated by LRP1 (57). Furthermore, the increase of Aβ production by apoE4 was also reported to be modulated by an apoE-binding protein TMCC2 (58). Most recently, it was reported that apoE isoforms (apoE4>apoE3>apoE2) differentially affect APP transcription and Aβ production through stimulating a non-canonical mitogen-activated protein (MAP) kinase signaling pathway in human neurons (59). Whether the effect of apoE on increasing APP through these signaling pathways contributes to AD risk and its in vivo relevance in the brains of apoE-TR mice or humans require further studies.
ApoE and apoE receptors in Aβ clearance
Increasing evidence indicates that impaired Aβ clearance is a major pathogenic event for LOAD (4). Soluble Aβ can be removed from the brain by various clearance pathways (Figure 2) including receptor-mediated cellular clearance (neurons, glial cells and vascular mural cells), cerebrovascular clearance (via the perivascular drainage, BBB and glymphatic system), and proteolytic degradation by Aβ degrading enzymes (6, 60).
LRP1 in Aβ clearance
Among the apoE receptors, LRP1 is the most extensively studied for its role in brain Aβ clearance. An in vitro study revealed that overexpression of a functional LRP1 mini-receptor increases Aβ trafficking to lysosomes, whereas knockdown of LRP1 decreases neuronal Aβ uptake (61). Conditional knockout of neuronal Lrp1 gene in the forebrain of APP/PS1 mice leads to impaired ISF Aβ clearance in the cortex without affecting Aβ production (62). However, reduction of LRP1 in the hippocampus of APP/PS1 mice has little effect on the rate of Aβ accumulation and deposition, indicating that different brain regions may eliminate Aβ by distinct pathways (63).
Aβ can also be cleared locally in the cerebrovasculature by an LRP1-dependent pathway. Conditional deletion of Lrp1 in vascular smooth muscle cell in APP/PS1 mice accelerates brain Aβ accumulation and exacerbates Aβ deposition without affecting Aβ production (64). Primary mouse brain capillary endothelial cells derived from mice harboring a LRP1 mutation in endocytosis/sorting domain exhibit a reduced Aβ transport from brain-to-blood and blood-to-brain compared with cells with wild-type LRP1, implying that LRP1 might modulate Aβ across the BBB (65). A recent study showed that deletion of LRP1 in brain endothelial cells leads to a reduced brain efflux of injected 125I-Aβ42 and elevated soluble brain Aβ in 5×FAD mice (66). Whether LRP1 has a direct or accelerative role in transporting Aβ across BBB requires further investigation.
LDLR in Aβ clearance
LDLR is another apoE receptor that is involved in Aβ clearance (67). Deletion of LDLR in astrocytes leads to a decrease in Aβ uptake, whereas increasing LDLR levels significantly enhances the uptake and cellular degradation of Aβ (68). LDLR-deficient mice have accelerated Aβ deposition and Aβ-related neurotoxicity without affecting the APP expression (69, 70). Overexpression of LDLR in mice dramatically reduces Aβ aggregation and enhances Aβ clearance, as well as attenuates plaque-associated neuroinflammation (71). LDLR overexpression also enhances BBB-mediated clearance of exogenously-administered Aβ and promotes brain-to-blood efflux transport of endogenous Aβ (72).
HSPG in Aβ clearance
HSPG, expressed on all cell types including neurons, has been implicated in several features in the pathogenesis of AD (73–75). HSPG binds to Aβ on the cell surface and functions with LRP1 in a cooperative manner to mediate neuronal Aβ uptake (74). Removal of neuronal heparan sulfate (HS) by conditional deletion of the Ext1 gene in APP/PS1 mice decreases Aβ oligomers and amyloid deposition, as well as accelerating Aβ clearance rate in the brain ISF. This result suggests that neuronal HSPG either inhibits or represents an inefficient pathway for Aβ clearance (75). It is postulated that binding of Aβ to neuronal HSPG on the cell surface promotes Aβ oligomerization/aggregation, thus depletion of neuronal HS leads to more efficient clearance of Aβ (Figure 2). However, it is still unclear whether HSPG also regulates Aβ clearance in other brain cell types. Additionally, an in vitro study showed that apoE particles inhibit HSPG-dependent cellular Aβ uptake without isoform difference (76). Further investigations are required to elucidate the role of apoE isoforms in HSPG-related Aβ metabolism in vivo.
ApoE isoform effects on receptor-mediated Aβ clearance
Using in vivo microdialysis, a study showed that clearance of Aβ from brain ISF is apoE isoform-dependent (apoE2>apoE3>apoE4) in the amyloid model mice (47). In addressing potential mechanism, early studies have shown that apoE can bind to Aβ and form a complex, which in turns is internalized by the cells through cell surface LRP1 and LDLR (77). The majority of endocytosed apoE is recycled, while the endocytosed Aβ is typically delivered to lysosomes for degradation (7). Since apoE4-Aβ complex is less stable than apoE3-Aβ, apoE4 is less effective in promoting Aβ clearance than apoE3 (78, 79). Other studies have shown that apoE disrupts Aβ clearance across the BBB in an isoform-dependent manner; Aβ binding to apoE4 redirects the rapid clearance of Aβ from LRP1 to the VLDLR, which internalizes apoE4-Aβ complex at the BBB more slowly than LRP1, whereas apoE2-Aβ and apoE3-Aβ complexes are cleared via both VLDLR and LRP1 in a faster speed than apoE4-Aβ complex (80). In addition, apoE may compete with Aβ for binding to common cell surface receptors, LRP1 and LDLR. By infusing apoE to the brain through reverse microdialysis, it was shown that apoE competes with Aβ for LRP1-dependent clearance pathway in astrocytes, with apoE4 exhibiting the strongest blocking effect relative to other apoE isoforms (81).
ApoE isoform effects on protease-mediated Aβ clearance
Aβ can be removed from the brain by proteolytic degradation via Aβ-degrading proteases, including neprilysin (NEP), insulin-degrading enzyme (IDE), endothelin-converting enzyme (ECE), angiotensin-converting enzyme (ACE), plasminogen activators, and the matrix metalloproteinases 2 (MMP2) and MMP9 (82, 83). NEP and IDE are the two most extensively studied Aβ proteases that are expressed in neurons, glia, and vascular mural cells (84–86). ApoE has been shown to facilitate the proteolytic clearance of soluble Aβ from the brain. Interestingly, hippocampal IDE protein is reduced by approximately 50% in APOE4 AD patients compared to non-APOE4 individuals (87), perhaps by down-regulating IDE expression in neurons (88). Studies revealed that apoE enhances both the endolytic degradation of Aβ within microglia (89) and astrocytes by NEP (90), and Aβ degradation extracellularly by IDE (89), with apoE3 functioning more efficiently than apoE4.
ApoE and apoE receptors in modulating synaptic integrity and plasticity
Synaptic failure, including synaptic dysfunction and synapse loss, is an early pathological feature of AD (91–93). It was reported that apoE receptors may be involved in modulating synaptic plasticity. ApoER2 and LRP1 are reported to interact with N-methyl-D-aspartate (NMDA) receptor subunits (94), modulate the internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, and regulate synaptic plasticity (95). LRP6 and related Wnt signaling are important for the regulation of synaptic integrity and cognition (56). In addition, apoE likely in an isoform-dependent manner modulates synaptic integrity and plasticity (Table 1). ApoE4 suppresses the expression of synaptic proteins and impairs dendritic morphology, synaptic transmission and plasticity in an age-dependent manner; however, the underlying mechanism is not well-established. In vitro studies indicate that the protective effects of apoE3 on synaptic loss caused by toxic Aβ are dependent on LRP1 (96, 97). It was also reported that apoE4 reduces cell surface apoER2, leading to impaired Reelin-induced glutamate receptor trafficking and long-term potentiation (LTP) (98). Depending on brain circuits, the strength of LTP differs in mice with different apoE isoforms (Table 1). However, it is still unclear whether long-term depression (LTD), another important synaptic mechanism associated with learning and memory (99), is affected by apoE isoforms.
Table 1.
The effects of apoE isoforms on synaptic integrity and plasticity
| Synaptic protein | |
|---|---|
| Synaptophysin Sytaxin1 PSD95 |
Specific synaptic proteins are altered in the superior temporal cortex of normal aged human brains in an apoE isoform-specific manner (apoE2>apoE3 >apoE4) (130). |
| Glutamate receptors | Glutamate receptors are decreased in APOE4 carriers of human AD brains (131) and aged female apoE4-TR mice (132). |
| Dendritic morphology | |
| Dendritic spine density | APOE4 dose inversely correlates with dendritic spine density of DG neurons in both AD patients and aged normal controls (133). ApoE4-TR mice showed age-dependent decrease of spine density in DG and cortex (apoE4<apoE3) (133, 134). |
| Dendrite complexity | ApoE4-TR mice show progressive dendritic spine deficits with aging (apoE4<apoE3) (135, 136). |
| EI balance (transmission) | |
| EPSC vs IPSC | Young apoE4-TR mice exhibit deficits in EPSC (apoE4<apoE3) (135, 136), whereas EI balance in aged apoE4-TR mice shows a bias toward inhibition (137). |
| Synaptic plasticity | |
| LTP | Young apoE4-TR mice exhibit higher LTP strength in the Schaffer collateral pathway of hippocampus (apoE4>apoE3) (98, 138, 139) but lower strength in the perforant pathway than apoE3 (apoE4<apoE3) (140). Under Aβ stress, perforant LTP was severely impaired in apoE4 mice compared with apoE2-TR mice (141). |
| LTD | Unknown |
Abbreviations: DG, dental gyrus; EPSC, excitatory postsynaptic potential; IPSC, inhibitory postsynaptic potential; LTP, long-term potentiation; LTD, long-term depression; Aβ, amyloid-β.
ApoE in tauopathy, lipid transport, glucose metabolism, vascular function, and neuroinflammation
ApoE is a therapeutic target for AD
ApoE determines therapeutic responses
Several clinical trials in AD therapies demonstrated APOE genotype-dependent responses. For example, recent trials revealed that the beneficial effects of intranasal insulin treatment on cognition are modulated by APOE genotype status in AD patients (3, 79, 100). Acute treatment of short-acting intranasal insulin leads to cognitive improvement in AD patients who are APOE4 non-carriers, but not in APOE4 carriers (78). Long-acting intranasal insulin improves cognition in APOE4 AD patients but worsens the outcome for APOE4 non-carriers (79). In addition, a Phase 3 trial using passive immunization of a humanized anti-Aβ antibody, bapineuzumab, prevents the increase of Aβ deposition and reduces CSF phosphorylated tau in AD patients who are APOE4 carriers, but not in APOE4 non-carriers, although this antibody did not benefit the clinical outcomes (101). As some therapies are mechanism-based, which potentially impact apoE isoform-specific functions, it will be crucial to consider APOE genotype status to identify clinical trial participants, and determine the therapeutic windows and evaluate treatment efficacy against AD.
Modulation of apoE levels and/or lipidation
The strong association of APOE4 with AD emphasizes apoE as a critical target for therapy. Here, we briefly discuss several approaches that are currently being explored (Figure 3). There is still debate regarding whether increasing or decreasing apoE is the best strategy in regard to AD therapeutics. Overexpressing ABCA1, which increases the lipidation of apoE, reduces Aβ deposition in an amyloid mouse model (102). Treatment of amyloid model mice with liver X receptor (LXR) agonists (TO901317 and GW3965) or retinoid X receptor (RXR) agonists (bexarotene) improves memory and reduces Aβ burden by increasing apoE level and lipidation (103–105), though conflicting effects on amyloid pathology were reported (106, 107). Bexarotene also restores aging-related synaptic loss mediated by LRP1, and reverses the apoE4-associated cognitive deficits in the absence of amyloid (108, 109). However, the therapeutic application of these agents should be carefully examined in human clinical trials due to their adverse effects in the liver (110). Quercetin, a flavonoid widely distributed in nature, reduces AD-related pathology by stabilizing apoE in an amyloid mouse model (111), suggesting that modulating the stability of apoE might represent an alternative strategy. In addition to pharmacological approaches, nanoparticles for apoE delivery (112) and adeno-associated virus (AAV)-mediated gene delivery of apoE into the brains are also promising strategies to regulate apoE levels (113).
Figure 3. ApoE-targeting strategies for treatment of Alzheimer’s disease.
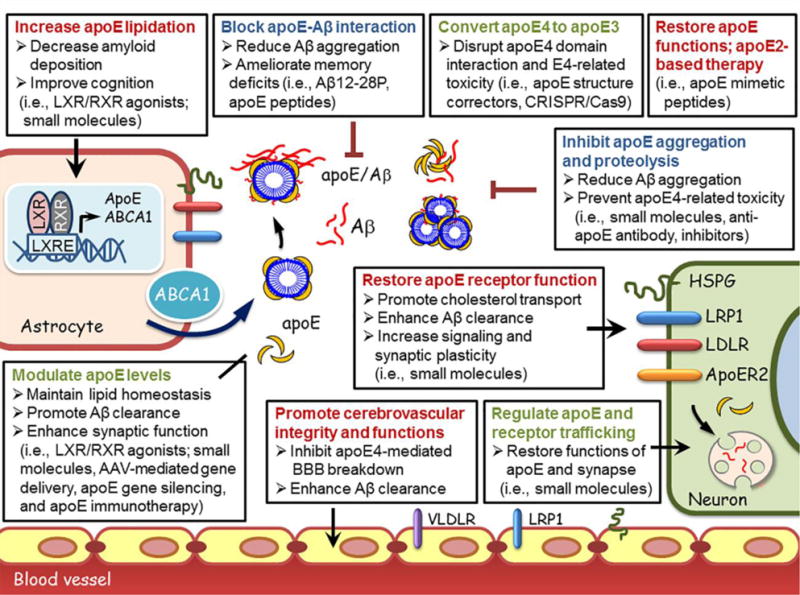
ApoE is primarily synthesized by astrocytes, and is lipidated by the ABCA1 transporter, forming apoE lipoprotein particles. Lipidated apoE binds soluble Aβ and facilitates Aβ uptake through cell surface receptors including LRP1, LDLR, VLDLR, and HSPG (6). ApoE and apoE receptors play critical roles in both Aβ-dependent and Aβ-independent AD pathogenic pathways (8). ApoE-directed approaches that are currently explored are categorized as below: 1) Modulating apoE level, stability and lipidation (ie., LXR/RXR agonists (103–105, 108), apoE stabilizing compounds (111), nanoparticles or AAV-mediated gene delivery of apoE (40, 113), gene silencing approaches and anti-apoE immunotherapy (115, 116); 2) Modulating apoE properties by converting apoE4 to apoE3 (i.e., structure correctors, CRISPR/Cas9 genome-editing) (118–121), suppressing apoE aggregation (122) and proteolysis (118, 119) and blocking apoE-Aβ interaction (126, 127); 3) Regulating the levels, intracellular trafficking and functions of apoE and apoE receptors (6, 72, 75, 98); 4) Restoring normal apoE functions (i.e., apoE-mimetic peptides) (128); 5) Restoring apoE4-mediated defects by apoE2-based treatment (40, 113), and 6) Promoting cerebrovascular functions (i.e., physical exercise, healthy diet and lifestyle), particularly in APOE4 carriers (142).
Abbreviations: Aβ, amyloid-β; apoE, apolipoprotein E; LXR, liver X receptor; RXR, retinoid X receptor; ABCA1, ATP-binding cassette A1; HSPG, heparan sulphate proteoglycan; LDLR, low-density lipoprotein receptor; LRP1, low-density lipoprotein receptor-related protein 1; BBB, blood-brain barrier; ApoER2, apolipoprotein E receptor 2; AAV, adeno-associated virus.
Conversely, decreasing apoE levels by overexpressing LDLR or haploinsufficiency of apoE has also been shown to reduce amyloid pathology (17, 114). ApoE immunotherapy through intraperitoneal administration improves cognitive performance, suppresses Aβ deposition, and enhances Aβ clearance in amyloid mouse models by reducing brain apoE level (115, 116). However, due to the critical functions of apoE in lipid homeostasis and neuronal functions (117), careful assessments and optimization will be required when modulating apoE levels in AD treatment. Importantly, lowering apoE4 expression might be an effective therapy for AD in APOE4 carriers to reduce the toxic functions associated with apoE4. Although further studies are required, anti-apoE4 immunotherapy and gene silencing approaches against apoE4 (e.g., antisense oligonucleotide) may be therapeutically beneficial.
Modulation of apoE properties
Domain-domain interaction has been suggested to confer apoE4 susceptible to proteolytic cleavage and the generation of neurotoxic fragments (118, 119) though the full-length structure of apoE remains to be determined. Small molecules used as apoE structure correctors to block such interactions have been shown to blunt the effects of apoE4 in neurons (120). In addition, targeting apoE proteolysis to reduce apoE neurotoxic fragments may suppress the detrimental effects of apoE4. With recent advances in CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9 technology (121), conversion of apoE4 to apoE3- or apoE2-like structure through genome-editing may also be an attractive approach to increase the normal functions of apoE while reducing the toxicity of apoE4.
A recent study identifies APOE3-V236E variant to be protective against AD with a population carrier frequency of 0.3% (Figure 1) (122). It is the first LOAD-associated apoE variant that locates within the C-terminal region that is critical for apoE oligomerization and lipidation (122, 123). ApoE4 was shown to be easily self-aggregated (124) which might reduce its lipidation capacity and promote amyloid deposition (125). Thus, inhibition of apoE aggregation will facilitate apoE-mediated functions and suppress amyloidosis which holds therapeutic potential for AD treatment. In addition, studies show that blocking apoE-Aβ interaction by a synthetic peptide (Aβ12–28P) decreases amyloid deposition in amyloid mouse models (126). Aβ12–28P treatment also reduces brain Aβ accumulation, neuritic degeneration, and prevents memory deficit in amyloid model mice expressing apoE2 and apoE4 (127), suggesting that targeting the apoE/Aβ interaction might serve as an alternative therapeutic approach for AD.
Other apoE-related therapeutic approaches
LRP1, LDLR, and HSPG are cell surface receptors for both apoE and Aβ in the brain and have been shown to regulate lipid metabolism and Aβ clearance (6, 72, 75). Identifying small molecules to promote the functions of LRP1 and LDLR or to specifically block HSPG-Aβ interaction could serve as therapeutic approaches for AD treatment. Restoring the intracellular trafficking of apoER2 may ameliorate apoE4-mediated synaptic deficits (98). Moreover, treatment of amyloid model mice with apoE-mimetic peptides (apoE133–149) displays anti-inflammatory and neuroprotective activities (128), indicating that restoring the normal apoE functions may be beneficial. In addition, AAV-mediated delivery of apoE2 increases apoE-associated cholesterol and decreases Aβ pathology, emphasizing the therapeutic potential of apoE2-based treatment (40, 113). Finally, improving vascular health (i.e., physical exercise, healthy diet, and lifestyle) could be beneficial in reducing the risk of AD and cognitive decline, particularly in APOE4 carriers.
Although apoE-targeted therapies remain in early phase of development they hold great promises in the fight against AD. It is likely that up-regulation of apoE3 benefits synapses and other apoE-related functions, whereas down-regulation of apoE4 reduces its toxic effects and should reduce amyloid deposition when treated early. Due to the differential roles of apoE isoforms in AD pathogenesis, isoform-specific targeting approach will be an encouraging strategy for treating AD. With recent advances in the technology of induced-pluripotent stem cell (iPSC), patient-derived iPSC with different apoE isoforms can be differentiated to various brain cell types for disease modeling, drug screening, and potentially replacement therapy (129). Testing therapeutic efficacy with better preclinical study design in animal models with apoE isoforms would be an instrumental tool to improve the translational potential of AD therapeutics targeting apoE.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant R01AG046205, R01AG035355, R01AG027924, RF1AG051504, P01NS074969, and P50AG016574 (to G.B.), a grant from the Cure Alzheimer’s Fund (to G.B.), a fellowship from the BrightFocus Foundation (to C.-C.L.), and a pilot grant from Mayo Clinic Alzheimer’s Disease Research Center (to N.Z.). We thank Lindsey Felton for carefully reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Reger MA, Watson GS, Frey WH, 2nd, Baker LD, Cholerton B, Keeling ML, et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 2006;27:451–458. doi: 10.1016/j.neurobiolaging.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 6.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81:740–754. doi: 10.1016/j.neuron.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 10.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 11.Weisgraber KH, Rall SC, Jr, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- 12.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casey CS, Atagi Y, Yamazaki Y, Shinohara M, Tachibana M, Fu Y, et al. Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-mediated Pathway. J Biol Chem. 2015;290:14208–14217. doi: 10.1074/jbc.M114.625251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelmus MM, Otte-Holler I, Davis J, Van Nostrand WE, de Waal RM, Verbeek MM. Apolipoprotein E genotype regulates amyloid-beta cytotoxicity. J Neurosci. 2005;25:3621–3627. doi: 10.1523/JNEUROSCI.4213-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- 16.Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;31:1503–1511. [PubMed] [Google Scholar]
- 17.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lane-Donovan C, Wong WM, Durakoglugil MS, Wasser CR, Jiang S, Xian X, et al. Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice. J Neurosci. 2016;36:10141–10150. doi: 10.1523/JNEUROSCI.1054-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linton MF, Gish R, Hubl ST, Butler E, Esquivel C, Bry WI, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88:270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker-Nigh AT, Mawuenyega KG, Bollinger JG, Ovod V, Kasten T, Franklin EE, et al. Human Central Nervous System (CNS) ApoE Isoforms are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared to Plasma. J Biol Chem. 2016;291:27204–27218. doi: 10.1074/jbc.M116.721779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol. 2015;77:301–311. doi: 10.1002/ana.24326. [DOI] [PubMed] [Google Scholar]
- 22.Mahley RW. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305–1315. doi: 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28:11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ulrich JD, Burchett JM, Restivo JL, Schuler DR, Verghese PB, Mahan TE, et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegener. 2013;8:13. doi: 10.1186/1750-1326-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, et al. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J Biol Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 26.Ramaswamy G, Xu Q, Huang Y, Weisgraber KH. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25:10658–10663. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruiz J, Kouiavskaia D, Migliorini M, Robinson S, Saenko EL, Gorlatova N, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res. 2005;46:1721–1731. doi: 10.1194/jlr.M500114-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Ji ZS, Brecht WJ, Miranda RD, Hussain MM, Innerarity TL, Mahley RW. Role of heparan sulfate proteoglycans in the binding and uptake of apolipoprotein E-enriched remnant lipoproteins by cultured cells. J Biol Chem. 1993;268:10160–10167. [PubMed] [Google Scholar]
- 29.Saito H, Dhanasekaran P, Nguyen D, Baldwin F, Weisgraber KH, Wehrli S, et al. Characterization of the heparin binding sites in human apolipoprotein E. J Biol Chem. 2003;278:14782–14787. doi: 10.1074/jbc.M213207200. [DOI] [PubMed] [Google Scholar]
- 30.Schneider WJ, Kovanen PT, Brown MS, Goldstein JL, Utermann G, Weber W, et al. Familial dysbetalipoproteinemia. Abnormal binding of mutant apoprotein E to low density lipoprotein receptors of human fibroblasts and membranes from liver and adrenal of rats, rabbits, and cows. J Clin Invest. 1981;68:1075–1085. doi: 10.1172/JCI110330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowal RC, Herz J, Weisgraber KH, Mahley RW, Brown MS, Goldstein JL. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J Biol Chem. 1990;265:10771–10779. [PubMed] [Google Scholar]
- 32.Gonneaud J, Arenaza-Urquijo EM, Fouquet M, Perrotin A, Fradin S, de La Sayette V, et al. Relative effect of APOE epsilon4 on neuroimaging biomarker changes across the lifespan. Neurology. 2016;87:1696–1703. doi: 10.1212/WNL.0000000000003234. [DOI] [PubMed] [Google Scholar]
- 33.Kantarci K, Lowe V, Przybelski SA, Weigand SD, Senjem ML, Ivnik RJ, et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology. 2012;78:232–240. doi: 10.1212/WNL.0b013e31824365ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy KR, Landau SM, Choudhury KR, Hostage CA, Shpanskaya KS, Sair HI, et al. Mapping the effects of ApoE4, age and cognitive status on 18F-florbetapir PET measured regional cortical patterns of beta-amyloid density and growth. Neuroimage. 2013;78:474–480. doi: 10.1016/j.neuroimage.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fleisher AS, Chen K, Liu X, Ayutyanont N, Roontiva A, Thiyyagura P, et al. Apolipoprotein E epsilon4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2013;34:1–12. doi: 10.1016/j.neurobiolaging.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 36.Serrano-Pozo A, Qian J, Monsell SE, Betensky RA, Hyman BT. APOEepsilon2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. 2015;77:917–929. doi: 10.1002/ana.24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berlau DJ, Corrada MM, Head E, Kawas CH. APOE epsilon2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology. 2009;72:829–834. doi: 10.1212/01.wnl.0000343853.00346.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. 2012;32:15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tai LM, Youmans KL, Jungbauer L, Yu C, Ladu MJ. Introducing Human APOE into Abeta Transgenic Mouse Models. Int J Alzheimers Dis. 2011;2011:810981. doi: 10.4061/2011/810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5:212ra161. doi: 10.1126/scitranslmed.3007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao W, Dumanis SB, Tamboli IY, Rodriguez GA, Jo Ladu M, Moussa CE, et al. Human APOE genotype affects intraneuronal Abeta1–42 accumulation in a lentiviral gene transfer model. Hum Mol Genet. 2014;23:1365–1375. doi: 10.1093/hmg/ddt525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, et al. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garai K, Verghese PB, Baban B, Holtzman DM, Frieden C. The binding of apolipoprotein E to oligomers and fibrils of amyloid-beta alters the kinetics of amyloid aggregation. Biochemistry. 2014;53:6323–6331. doi: 10.1021/bi5008172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood SJ, Chan W, Wetzel R. Seeding of A beta fibril formation is inhibited by all three isotypes of apolipoprotein E. Biochemistry. 1996;35:12623–12628. doi: 10.1021/bi961074j. [DOI] [PubMed] [Google Scholar]
- 45.Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2:605–613. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 46.Prince JA, Zetterberg H, Andreasen N, Marcusson J, Blennow K. APOE epsilon4 allele is associated with reduced cerebrospinal fluid levels of Abeta42. Neurology. 2004;62:2116–2118. doi: 10.1212/01.wnl.0000128088.08695.05. [DOI] [PubMed] [Google Scholar]
- 47.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lautner R, Palmqvist S, Mattsson N, Andreasson U, Wallin A, Palsson E, et al. Apolipoprotein E genotype and the diagnostic accuracy of cerebrospinal fluid biomarkers for Alzheimer disease. JAMA Psychiatry. 2014;71:1183–1191. doi: 10.1001/jamapsychiatry.2014.1060. [DOI] [PubMed] [Google Scholar]
- 49.Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 2012;21:4558–4571. doi: 10.1093/hmg/dds296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toledo JB, Da X, Weiner MW, Wolk DA, Xie SX, Arnold SE, et al. CSF Apo-E levels associate with cognitive decline and MRI changes. Acta Neuropathol. 2014;127:621–632. doi: 10.1007/s00401-013-1236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 52.Waldron E, Heilig C, Schweitzer A, Nadella N, Jaeger S, Martin AM, et al. LRP1 modulates APP trafficking along early compartments of the secretory pathway. Neurobiol Dis. 2008;31:188–197. doi: 10.1016/j.nbd.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 53.Pietrzik CU, Yoon IS, Jaeger S, Busse T, Weggen S, Koo EH. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci. 2004;24:4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, et al. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A. 2004;101:1075–1080. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 2002;21:5691–5700. doi: 10.1093/emboj/cdf568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron. 2014;84:63–77. doi: 10.1016/j.neuron.2014.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, et al. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci U S A. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hopkins PC, Sainz-Fuertes R, Lovestone S. The impact of a novel apolipoprotein E and amyloid-beta protein precursor-interacting protein on the production of amyloid-beta. J Alzheimers Dis. 2011;26:239–253. doi: 10.3233/JAD-2011-102115. [DOI] [PubMed] [Google Scholar]
- 59.Huang YA, Zhou B, Wernig M, Sudhof TC. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell. 2017;168:427–441 e421. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuentealba RA, Liu Q, Zhang J, Kanekiyo T, Hu X, Lee JM, et al. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS One. 2010;5:e11884. doi: 10.1371/journal.pone.0011884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanekiyo T, Cirrito JR, Liu CC, Shinohara M, Li J, Schuler DR, et al. Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J Neurosci. 2013;33:19276–19283. doi: 10.1523/JNEUROSCI.3487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu G, Green CC, Fromholt SE, Borchelt DR. Reduction of low-density lipoprotein receptor-related protein (LRP1) in hippocampal neurons does not proportionately reduce, or otherwise alter, amyloid deposition in APPswe/S1dE9 transgenic mice. Alzheimers Res Ther. 2012;4:12. doi: 10.1186/alzrt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J Neurosci. 2012;32:16458–16465. doi: 10.1523/JNEUROSCI.3987-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pflanzner T, Janko MC, Andre-Dohmen B, Reuss S, Weggen S, Roebroek AJ, et al. LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol Aging. 2011;32:2323 e2321–2311. doi: 10.1016/j.neurobiolaging.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 66.Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, et al. Endothelial LRP1 transports amyloid-beta(1–42) across the blood-brain barrier. J Clin Invest. 2016;126:123–136. doi: 10.1172/JCI81108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abdulkarim Y, Hameed Z. Is the LDL receptor involved in cortical amyloid protein clearance? Neurochem Res. 2006;31:839–847. doi: 10.1007/s11064-006-9084-0. [DOI] [PubMed] [Google Scholar]
- 68.Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J Biol Chem. 2012;287:13959–13971. doi: 10.1074/jbc.M111.288746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Oliveira J, Moreira EL, dos Santos DB, Piermartiri TC, Dutra RC, Pinton S, et al. Increased susceptibility to amyloid-beta-induced neurotoxicity in mice lacking the low-density lipoprotein receptor. J Alzheimers Dis. 2014;41:43–60. doi: 10.3233/JAD-132228. [DOI] [PubMed] [Google Scholar]
- 70.Katsouri L, Georgopoulos S. Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer’s disease mouse model. PLoS One. 2011;6:e21880. doi: 10.1371/journal.pone.0021880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, et al. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A. 2012;109:15502–15507. doi: 10.1073/pnas.1206446109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reynolds MR, Singh I, Azad TD, Holmes BB, Verghese PB, Dietrich HH, et al. Heparan sulfate proteoglycans mediate Abeta-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol Neurodegener. 2016;11:9. doi: 10.1186/s13024-016-0073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci. 2011;31:1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu CC, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM, et al. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci Transl Med. 2016;8:332ra344. doi: 10.1126/scitranslmed.aad3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu Y, Zhao J, Atagi Y, Nielsen HM, Liu CC, Zheng H, et al. Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol Neurodegener. 2016;11:37. doi: 10.1186/s13024-016-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 78.Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis. 2008;13:323–331. doi: 10.3233/jad-2008-13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, et al. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis. 2015;44:897–906. doi: 10.3233/JAD-141791. [DOI] [PubMed] [Google Scholar]
- 80.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leissring MA. The AbetaCs of Abeta-cleaving proteases. J Biol Chem. 2008;283:29645–29649. doi: 10.1074/jbc.R800022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Turner AJ, Nalivaeva NN. New insights into the roles of metalloproteinases in neurodegeneration and neuroprotection. Int Rev Neurobiol. 2007;82:113–135. doi: 10.1016/S0074-7742(07)82006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 86.Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, et al. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, et al. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 allele. Am J Pathol. 2003;162:313–319. doi: 10.1016/s0002-9440(10)63822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du J, Chang J, Guo S, Zhang Q, Wang Z. ApoE 4 reduces the expression of Abeta degrading enzyme IDE by activating the NMDA receptor in hippocampal neurons. Neurosci Lett. 2009;464:140–145. doi: 10.1016/j.neulet.2009.07.032. [DOI] [PubMed] [Google Scholar]
- 89.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mulder SD, Veerhuis R, Blankenstein MA, Nielsen HM. The effect of amyloid associated proteins on the expression of genes involved in amyloid-beta clearance by adult human astrocytes. Exp Neurol. 2012;233:373–379. doi: 10.1016/j.expneurol.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 91.Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer’s disease: synapses gone cold. Mol Neurodegener. 2011;6:63. doi: 10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu DS, Pan XD, Zhang J, Shen H, Collins NC, Cole AM, et al. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol Neurodegener. 2015;10:7. doi: 10.1186/s13024-015-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hoe HS, Pocivavsek A, Chakraborty G, Fu Z, Vicini S, Ehlers MD, et al. Apolipoprotein E receptor 2 interactions with the N-methyl-D-aspartate receptor. J Biol Chem. 2006;281:3425–3431. doi: 10.1074/jbc.M509380200. [DOI] [PubMed] [Google Scholar]
- 95.Nakajima C, Kulik A, Frotscher M, Herz J, Schafer M, Bock HH, et al. Low density lipoprotein receptor-related protein 1 (LRP1) modulates N-methyl-D-aspartate (NMDA) receptor-dependent intracellular signaling and NMDA-induced regulation of postsynaptic protein complexes. J Biol Chem. 2013;288:21909–21923. doi: 10.1074/jbc.M112.444364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sen A, Alkon DL, Nelson TJ. Apolipoprotein E3 (ApoE3) but not ApoE4 protects against synaptic loss through increased expression of protein kinase C epsilon. J Biol Chem. 2012;287:15947–15958. doi: 10.1074/jbc.M111.312710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hayashi H, Campenot RB, Vance DE, Vance JE. Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J Neurosci. 2007;27:1933–1941. doi: 10.1523/JNEUROSCI.5471-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci U S A. 2010;107:16697–16702. doi: 10.1073/pnas.1008200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, et al. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J Alzheimers Dis. 2013;35:789–797. doi: 10.3233/JAD-122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 104.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 105.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924-f. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]
- 107.Tesseur I, Lo AC, Roberfroid A, Dietvorst S, Van Broeck B, Borgers M, et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340:924-e. doi: 10.1126/science.1233937. [DOI] [PubMed] [Google Scholar]
- 108.Tachibana M, Shinohara M, Yamazaki Y, Liu CC, Rogers J, Bu G, et al. Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Exp Neurol. 2016;277:1–9. doi: 10.1016/j.expneurol.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Boehm-Cagan A, Michaelson DM. Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J Neurosci. 2014;34:7293–7301. doi: 10.1523/JNEUROSCI.5198-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, et al. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem. 2014;289:30538–30555. doi: 10.1074/jbc.M114.600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang X, Hu J, Zhong L, Wang N, Yang L, Liu CC, et al. Quercetin stabilizes apolipoprotein E and reduces brain Abeta levels in amyloid model mice. Neuropharmacology. 2016;108:179–192. doi: 10.1016/j.neuropharm.2016.04.032. [DOI] [PubMed] [Google Scholar]
- 112.Zensi A, Begley D, Pontikis C, Legros C, Mihoreanu L, Wagner S, et al. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J Control Release. 2009;137:78–86. doi: 10.1016/j.jconrel.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 113.Hu J, Liu CC, Chen XF, Zhang YW, Xu H, Bu G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener. 2015;10:6. doi: 10.1186/s13024-015-0001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Stewart FR, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med. 2012;209:2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci. 2014;34:7281–7292. doi: 10.1523/JNEUROSCI.0646-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mak AC, Pullinger CR, Tang LF, Wong JS, Deo RC, Schwarz JM, et al. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol. 2014;71:1228–1236. doi: 10.1001/jamaneurol.2014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement. 2008;4:179–192. doi: 10.1016/j.jalz.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mahley RW, Huang Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J Med Chem. 2012;55:8997–9008. doi: 10.1021/jm3008618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–5266. doi: 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Medway CW, Abdul-Hay S, Mims T, Ma L, Bisceglio G, Zou F, et al. ApoE variant V236E is associated with markedly reduced risk of Alzheimer’s disease. Mol Neurodegener. 2014;9:11. doi: 10.1186/1750-1326-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Frieden C, Garai K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109:8913–8918. doi: 10.1073/pnas.1207022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. 2006;361:932–944. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 125.Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, et al. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, et al. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez S, Sullivan PM, et al. Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. 2014;2:75. doi: 10.1186/s40478-014-0075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vitek MP, Christensen DJ, Wilcock D, Davis J, Van Nostrand WE, Li FQ, et al. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener Dis. 2012;10:122–126. doi: 10.1159/000334914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- 130.Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27:797–803. doi: 10.1016/j.neurobiolaging.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 131.Sweet RA, MacDonald ML, Kirkwood CM, Ding Y, Schempf T, Jones-Laughner J, et al. Apolipoprotein E*4 (APOE*4) Genotype Is Associated with Altered Levels of Glutamate Signaling Proteins and Synaptic Coexpression Networks in the Prefrontal Cortex in Mild to Moderate Alzheimer Disease. Mol Cell Proteomics. 2016;15:2252–2262. doi: 10.1074/mcp.M115.056580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yong SM, Lim ML, Low CM, Wong BS. Reduced neuronal signaling in the ageing apolipoprotein-E4 targeted replacement female mice. Sci Rep. 2014;4:6580. doi: 10.1038/srep06580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience. 2003;122:305–315. doi: 10.1016/j.neuroscience.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 134.Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Klein RC, Mace BE, Moore SD, Sullivan PM. Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience. 2010;171:1265–1272. doi: 10.1016/j.neuroscience.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18:390–398. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 137.Klein RC, Acheson SK, Mace BE, Sullivan PM, Moore SD. Altered neurotransmission in the lateral amygdala in aged human apoE4 targeted replacement mice. Neurobiol Aging. 2014;35:2046–2052. doi: 10.1016/j.neurobiolaging.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kitamura HW, Hamanaka H, Watanabe M, Wada K, Yamazaki C, Fujita SC, et al. Age-dependent enhancement of hippocampal long-term potentiation in knock-in mice expressing human apolipoprotein E4 instead of mouse apolipoprotein E. Neurosci Lett. 2004;369:173–178. doi: 10.1016/j.neulet.2004.07.084. [DOI] [PubMed] [Google Scholar]
- 139.Korwek KM, Trotter JH, Ladu MJ, Sullivan PM, Weeber EJ. ApoE isoform-dependent changes in hippocampal synaptic function. Mol Neurodegener. 2009;4:21. doi: 10.1186/1750-1326-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Ye GL, et al. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 2004;15:2655–2658. doi: 10.1097/00001756-200412030-00020. [DOI] [PubMed] [Google Scholar]
- 141.Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Stine WB, et al. ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-beta1–42. Neurobiol Aging. 2005;18:75–82. doi: 10.1016/j.nbd.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 142.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.