

ABSTRACT
Insulin-like growth factor type 2 (IGF2) receptor (IGF2R) recognizes mannose 6-phosphate-containing molecules and IGF2 and plays an important role in many pathophysiological processes, including gut mucosal adaptation. However, the mechanisms that control cellular IGF2R abundance are poorly known. MicroRNAs (miRNAs) and RNA-binding proteins (RBPs) critically regulate gene expression programs in mammalian cells by modulating the stability and translation of target mRNAs. Here we report that miRNA 195 (miR-195) and RBP CUG-binding protein 1 (CUGBP1) jointly regulate IGF2R expression at the posttranscriptional level in intestinal epithelial cells. Both miR-195 and CUGBP1 interacted with the 3′ untranslated region (3′-UTR) of Igf2r mRNA, and the association of CUGBP1 with Igf2r mRNA enhanced miR-195 binding to Igf2r mRNA. Ectopically expressed CUGBP1 and miR-195 repressed IGF2R translation cooperatively without altering the stability of Igf2r mRNA. Importantly, the miR-195- and CUGBP1-repressed levels of cellular IGF2R led to a disruption in the structure of the trans-Golgi network. These findings indicate that IGF2R expression is controlled posttranscriptionally by two factors that associate with Igf2r mRNA and suggest that miR-195 and CUGBP1 dampen IGF signaling by inhibiting IGF2R translation.
KEYWORDS: insulin superfamily, posttranscriptional regulation, trans-Golgi network, intestinal epithelial cells, RNA-binding proteins, noncoding RNAs
INTRODUCTION
Mucosal regeneration/adaptation is an essential process in gut homeostasis and tightly regulated by numerous factors, including insulin-like growth factor type 1 (IGF1) and IGF2 (1, 2). Two structurally distinct types of receptors, the IGF1 receptor (IGF1R) and IGF2R, specifically interact with IGF1 and IGF2 as ligands and regulate diverse biological functions such as cell proliferation, differentiation, and apoptosis (3, 4). Like the insulin receptor, IGF1R is a heterotetrameric transmembrane receptor tyrosine kinase, and its activation results in autophosphorylation of tyrosine residues in the intracellular β-subunits, thus initiating the phosphatidylinositol 3-kinase (PI3) kinase/AKT and/or mitogen-activated protein (MAP) kinase signaling cascade (5). In contrast, IGF2R is a single-transmembrane-domain protein and binds IGF2 with greater affinity than IGF1, although it does not accept insulin as a ligand (4, 6). IGF2R also recognizes, via distinct sites, mannose-6-phosphate (M6P)-containing molecules and can therefore associate with other growth factors and cytokines (4). The majority of IGF2R is localized in the trans-Golgi network (TGN) and endosomal compartments and, to a lesser extent, on the cell surface (6). A subpopulation of the IGF2R on the plasma membrane regulates IGF2 internalization and various M6P-containing ligands for their subsequent clearance or activation (4, 7), whereas surface IGF2R mediates intracellular signaling in response to IGF2 binding (6). The mammalian intestinal epithelium expresses high levels of IGF2R, and its cellular content is dramatically altered in pathological states such as that which follows small-bowel resection (SBR) (1). However, the exact mechanism underlying the control of IGF2R expression, especially at the posttranscriptional level, remains largely unknown.
Although the gene regulatory programs that control the production levels of numerous components of the IGF system are strongly regulated at the transcriptional level, the essential contribution of posttranscriptional events is increasingly recognized (8, 9). In particular, alterations in mRNA stability and translation critically influence the levels of IGFs and IGF receptors in response to environmental signals such as changing blood glucose levels (8, 10). The stability and translation of mRNAs are governed by trans factors, including the RNA-binding proteins (RBPs) and microRNAs (miRNAs) that directly bind to cis elements on target transcripts, frequently present at 3′ untranslated regions (3′-UTRs) (11, 12). The interactions of mRNAs with RBPs and/or miRNAs can alter the production levels of target mRNAs by recruiting the mRNA to specialized cytoplasmic domains such as processing bodies, stress granules, and the exosome, where mRNAs are subjected to translational repression or degradation, or to ribosomes, where they engage in active translation (10). An increasing body of evidence indicates that RBPs interact functionally with miRNAs to jointly regulate shared target mRNAs (13, 14). For example, the RBP HuR recruited the miRNA let-7, along with the RNA-induced silencing complex (RISC), to repress the translation of MYC mRNA (15), whereas RBP Dnd-1 (dead end 1) inhibited access of miRNAs to target mRNAs (16). In other examples, HuR competed with miRNA 195 (miR-195) to modulate Stim1 mRNA stability antagonistically (17), while RBP CUG-binding protein 1 (CUGBP1) and miR-222 repressed Cdk4 mRNA translation synergistically (18).
The evolutionarily conserved miRNA miR-195 is highly abundant in normal gastrointestinal mucosa, but its levels are markedly lower in cancer tissues (19, 20). Our previous genome-wide miRNA profile study showed that miR-195 levels are increased in growth-arrested intestinal epithelial cells (IECs) (14, 18) and under conditions of intestinal mucosal atrophy induced by food starvation or polyamine depletion (21). Our in vitro studies further revealed that increasing the levels of cellular miR-195 repressed IEC migration and proliferation after wounding (17). It has been shown that miR-195 exerts its regulatory functions in different tissues by targeting multiple genes (14). miR-195 inhibits cell proliferation by reducing the levels of cyclin-dependent kinase 4 (CDK4), CCND1 (cyclin D1), CDK6, and WEE1 (19, 20, 22); promotes apoptosis by lowering SIRT1 abundance (23); and affects cell migration and cancer invasion by modulating expression of STIM1 (17) and ActRIIA (24). However, its potential role as a regulator of IGF receptor expression is unknown. In addition, CUGBP1 has also recently emerged as a master regulator of gut epithelial homeostasis by modulating IEC proliferation, apoptosis, and cell-to-cell interaction (18, 25, 26), and low levels of CUGBP1 in mice are associated with crypt hyperplasia in the small intestine (27). Given the presence of sites for predicted binding of miR-195 and CUGBP1 in the Igf2r mRNA, we tested the possibility that miR-195 and CUGBP1 jointly regulate IGF2R expression. Our results show that both miR-195 and CUGBP1 directly interacted with the 3′-UTR of Igf2r mRNA and that they repressed IGF2R translation cooperatively.
RESULTS
miR-195 inhibits IGF2R translation.
To identify new targets of miR-195 in the intestinal epithelium, we examined the effect of overexpressing miR-195 on the expression of IGF receptors, IGF-binding protein (IGFBP), glucagon-like peptide receptors (GLPRs), and epidermal growth factor receptor (EGFR) in cultured IEC-6 IECs. As shown (Fig. 1A, panel a), the levels of cellular miR-195 increased dramatically in cells transfected with a miR-195 precursor (pre-miR-195) compared to cells transfected with scrambled control RNA. Transfection with pre-miR-195 did not alter the abundance of housekeeping noncoding RNA U6 (Fig. 1A, panel b) or miR-222 (data not shown). As determined by Western blotting, ectopically expressed miR-195 specifically inhibited the expression of IGF2R (Fig. 1B, top) but failed to decrease the expression levels of IGF1R, IGFBP5, GLP2R, GLP1R, or EGFR. The levels of IGF2R protein in cells transfected with pre-miR-195 decreased by ∼70% (n = 3; P < 0.05) compared to cells transfected with control scramble small interfering RNA (siRNA).
FIG 1.

Ectopic overexpression of miR-195 inhibits IGF2R translation. (A) Levels of miR-195 (a) and U6 RNA (b) 48 h after transfection of cells with pre-miR-195 as measured by Q-PCR analysis. Values represent means ± standard errors of the means (SEM) of results from three separate experiments. *, P < 0.05 (compared with cells transfected with control scramble oligomer). (B) Representative immunoblots of IGF2R, IGF1R, IGF-binding protein 5 (IGFBP5), glucagon-like peptide 2 receptor (GLP2R), GLP1R, and EGFR as examined by Western blotting of the cells described in the panel A legend. Equal loading was monitored by assessing GAPDH levels. (C) Levels of Igf2r and Igf1r mRNAs in the cells described in the panel A legend. (D) Newly synthesized IGF2R protein in cells overexpressing miR-195. After cells were exposed to l-azidohomoalaine (AHA), cell lysates were incubated with the reaction buffer containing biotin/alkyne reagent; the biotin-alkyne-azide-modified protein complex was pulled down by the use of paramagnetic streptavidin-conjugated dynabeads. (E) Distributions of Igf2r (a) and Gapdh (b) mRNAs in each gradient fraction prepared from the polysomal profile in cells after miR-195 overexpression. Nuclei were pelleted, and the resulting supernatants were fractionated through a 10% to 50% linear sucrose gradient. RNA was isolated from different fractions (fractions 1 to 3, free or/and preinitiation RNA; fractions 4 and 5, monosomes; fractions 6 to 12, polysomes), and the levels of Igf2r and Gapdh mRNAs were measured and plotted as a percentage of each of the total levels of Igf2r and Gapdh mRNA in each sample.
The reduction in IGF2R levels likely occurred at the level of translation, since miR-195 did not decrease the levels of Igf2r mRNA (Fig. 1C) or its stability (see Fig. S1 in the supplemental material) but reduced the rate of nascent IGF2R protein synthesis (Fig. 1D). The levels of newly synthesized IGF2R protein in cells overexpressing miR-195 decreased by >75% (n = 3; P < 0.05) compared to control cells. To further define the role of miR-195 in the regulation of IGF2R translation, we examined the relative levels of distribution of Igf2r mRNA in individual fractions from polyribosome gradients after miR-195 overexpression. Although increasing the levels of miR-195 by transfecting cells with pre-miR-195 did not affect global polysomal profiles (data not shown), the abundance of Igf2r mRNA associated with actively translating components of the gradient (fractions 11 and 12) decreased significantly in pre-miR-195-transfected cells, with a moderate leftward shift of Igf2r mRNA toward low-translating fractions (fractions 9 and 10) (Fig. 1E, panel a). In contrast, Gapdh mRNA, which is not a target of miR-195 and encodes a housekeeping protein, showed similar distributions in the two groups (Fig. 1E, panel b). To test the effect of miR-195 on IGF2R expression in another model, we employed Caco-2 cells, derived from the human colon carcinoma. Similarly to what was observed in IEC-6 cells, ectopic overexpression of miR-195 resulting from transfection of pre-miR-195 also decreased IGF2R expression without affecting Igf2r mRNA levels in Caco-2 cells (Fig. S2).
On the other hand, neutralizing the activity of endogenous miR-195 by transfecting cells with an antagomir targeting miR-195 (anti-miR-195) (Fig. 2A) increased IGF2R translation markedly, as shown by the increased levels of IGF2R protein (Fig. 2B), without changes in Igf2r mRNA levels (Fig. 2C). The levels of IGF2R protein in anti-miR-195-transfected cells were ∼2.2-fold the levels seen in cells transfected with a control oligomer (n = 3; P < 0.05). As expected, miR-195 silencing did not alter the cellular levels of IGF1R, GLP2R, GLP1R, or EGFR proteins. These results indicate that miR-195 specifically represses IGF2R expression at the translation level.
FIG 2.
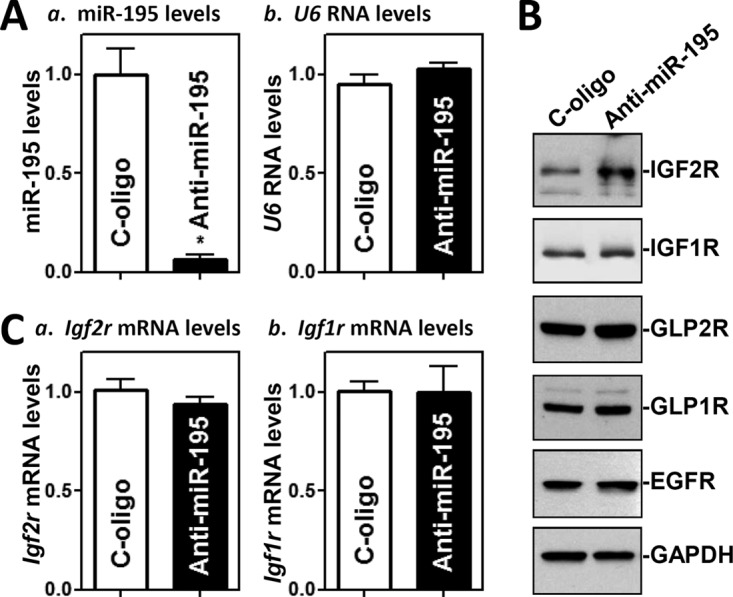
miR-195 silencing enhances IGF2R translation. (A) Levels of miR-195 (a) and U6 RNA (b) 48 h after transfection of cells with specific oligomers targeting miR-195 (anti-miR-195) or control oligomers (C-oligo) as examined by Q-PCR analysis. Values represent means ± SEM of results from three separate experiments. *, P < 0.05 (compared with cells transfected with C-oligo). (B) Representative immunoblots of IGF2R, IGF1R, GLP2R, GLP1R, and EGFR as measured by Western immunoblotting analysis in the cells described in the panel A legend. (C) Levels of Igf2r and Igf1r mRNAs in the cells described in the panel A legend.
miR-195 interacts with the Igf2r mRNA via its 3′-UTR.
To investigate the mechanism underlying the repression of IGF2R translation by miR-195, we examined the association of miR-195 with the Igf2r mRNA by RNA pulldown assays using biotin-labeled miR-195, as the Igf2r mRNA contains one computationally predicted miR-195 binding site in its 3′-UTR (Fig. 3A). Cells were transfected with biotinylated miR-195, and the binding of miR-195 to the Igf2r mRNAs was examined 24 h after the transfection. As shown (Fig. 3B), miR-195 levels increased significantly, although the levels of U6 RNA did not (data not shown). The Igf2r mRNA was enriched in the materials pulled down by biotin-miR-195 but not from cells transfected with control scramble RNA (Fig. 3C, panel a). The association of miR-195 with the Igf2r mRNA was specific, since increasing the levels of biotin-miR-195 did not induce its interaction with the Igfbp5 mRNA. The abundance of Stim1 mRNA was also examined and served as a positive control, since Stim1 mRNA is a known target of miR-195 as reported previously (17). In addition, transfection with biotin-labeled miR-195 did not alter the steady-state levels of Igf2r, Igfbp5, and Stim1 mRNAs (Fig. 3C, panel b). The association of Igf2r mRNA with miR-195 appeared to be specific, since control biotin pulldown experiments revealed that Igf2r mRNA did not interact with a negative-control microRNA, biotin-miR-222 (data not shown).
FIG 3.

miR-195 associates with Igf2r mRNA. (A) Schematic representation of the Igf2r mRNA depicting the target site for miR-195 in its 3′ untranslated region (3′ UTR). Alignment of the Igf2r mRNA sequence with miR-195 is shown. Top strand, Igf2r mRNA; bottom strand, miR-195. (B) Levels of biotinylated miR-195 24 h after transfection. Values represent means ± SEM of results from three separate experiments. *, P < 0.05 (compared with cells transfected with control scramble oligomer). (C) Binding of biotinylated miR-195 to mRNAs encoding IGF2R, IGFBP5, and STIM1: (a) levels of mRNAs in the materials pulled down by biotin-miR-195; (b) levels of total input mRNAs. (D) Levels of reporter activities as measured by analysis of the Igf2r 5′-UTR, various fragments of CR, or 3′-UTR luciferase (Luc) reporters after ectopic overexpression of miR-195. (Left) Schematic of plasmids of different chimeric firefly Luc-Igf2r reporters. At 24 h after transfection with pre-miR-195, cells were transfected with different Igf2r luciferase reporter plasmids. The results were expressed as the mean ± SEM from three separate experiments. *, P < 0.05 (compared with cells transfected with control scrambled oligomer). (E) Effect of deletion of miR-195-binding site (schematic) on luciferase reporter activity after ectopic miR-195 overexpression. WT, wild type; Mut, mutant.
To determine what segment of Igf2r mRNA mediated the repression of IGF2R translation by miR-195, fragments of the Igf2r 5′-UTR, coding region (CR), and 3′-UTR were subcloned into the pmirGLO dual-luciferase miRNA target expression vector to generate reporter constructs pmirGLO-Igf2r-5′UTR, pmirGLO-Igf2r-CR, and pmirGLO-Igf2r-3′UTR (Fig. 3D, schematic). To distinguish translational output from changes in mRNA turnover, the luciferase activities were normalized to luciferase mRNA levels to assess the translational efficiency. miR-195 overexpression induced by transfecting cells with pre-miR-195 selectively decreased the levels of pmirGLO-Igf2r-3′UTR luciferase reporter activity (Fig. 3A, right) but failed to inhibit the activities of pmirGLO-Igf2r-5′-UTR or -CR reporters. Furthermore, when the predicted miR-195-binding site within the Igf2r 3′-UTR was mutated by internal deletion (Fig. 3E, schematic), the miR-195-induced repression was completely prevented (Fig. 3E, right). Taken together, these results indicate that miR-195 represses IGF2R translation by directly interacting with the Igf2r mRNA via its 3′-UTR rather than 5′-UTR and CR.
CUGBP1 associates with the Igf2r mRNA and inhibits its translation.
Since there are several potential hits for CUGBP1 in the Igf2r mRNA, we also elucidated the role of CUGBP1 in the regulation of IGF2R expression and its potential relation to the miR-195-mediated IGF2R repression. Association of the Igf2r mRNA with CUGBP1 was examined by ribonucleoprotein (RNP) immunoprecipitation (IP) assays using anti-CUGBP1 antibody under conditions that preserved RNP integrity (25). As expected, CUGBP1 associated with Igf2r mRNA but not with Igfbp5 mRNA in IEC-6 cells (Fig. 4A). Igf2r PCR products were highly enriched in CUGBP1 IP samples compared with IgG1 IP samples, as were Cdk4 PCR products, included as a positive control, since the Cdk4 mRNA is a known target of CUGBP1 (18). Amplification of Gapdh PCR products, originating from the abundant “contaminating” transcript Gapdh mRNA, which is not a target of CUGBP1, served to monitor the evenness of sample input, as reported previously (28).
FIG 4.
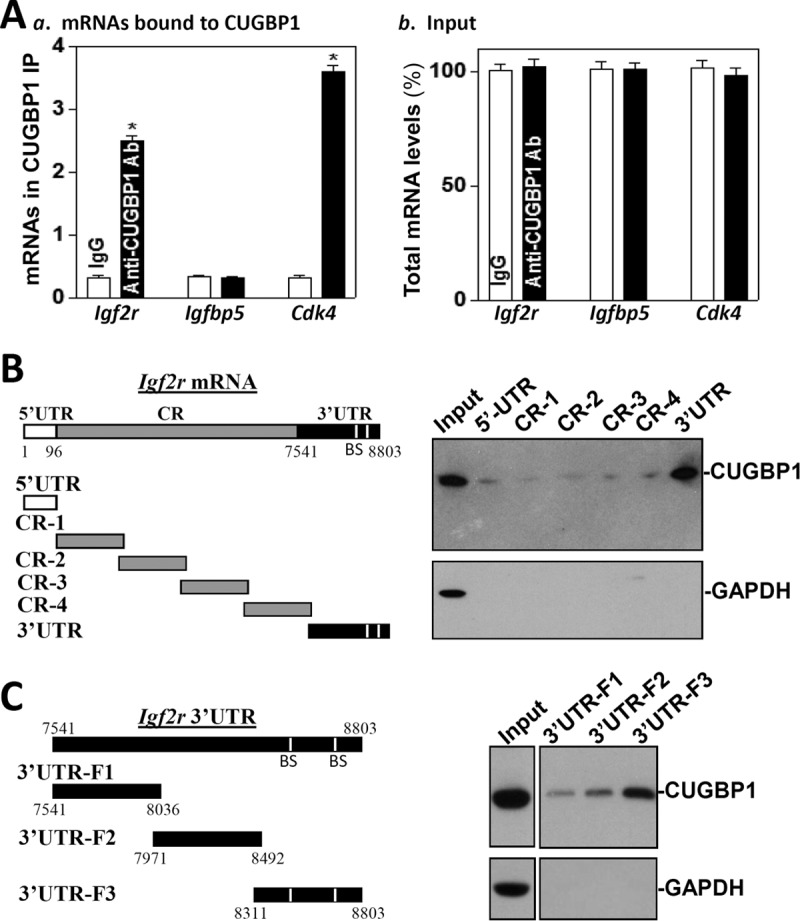
CUGBP1 binds to the Igf2r mRNA via its 3′-UTR. (A) Association of endogenous CUGBP1 with endogenous Igf2r mRNA. After IP of RNA-protein complexes from cell lysates using either anti-CUGBP1 antibody (Ab) or control IgG, RNA was isolated and used in RT reactions. (a) Levels of mRNAs of Igf2r, Igfbp5, or Cdk4 in CUGBP1 or IgG IP materials. (b) Levels of total input mRNAs. Values represent means ± SEM of results from triplicate samples. *, P < 0.05 (compared with IgG IP). (B and C) Representative CUGBP1 results of immunoblotting using the pulldown materials by biotinylated transcripts of Igf2r mRNA (B) and 5′-UTR (CR) and different fragments of CR and 3′-UTR. The left panels show schematic representations of various Igf2r biotinylated transcripts used in this study. Cytoplasmic lysates were incubated with 6 μg of biotinylated Igf2r 5′-UTR, CR, and 3′-UTR for 30 min at 25°C, and the resulting RNP complexes were pulled down by the use of streptavidin-coated beads. The presence of CUGBP1 in the pulldown material was assayed by Western blotting. GAPDH in the pulldown material was also examined and served as a negative control.
Associations of CUGBP1 with Igf2r mRNA were further tested by using biotinylated transcripts spanning the Igf2r 5′-UTR, CR, or 3′-UTR (Fig. 4B, schematic). Following incubation with cytoplasmic lysates, binding of the biotinylated Igf2r transcripts to CUGBP1 was examined by biotin pulldown followed by Western blotting as described previously (29, 30). The Igf2r 3′-UTR transcripts readily associated with CUGBP1 (Fig. 4B, right), but the Igf2r 5′-UTR or CR did not. In addition, none of the Igf2r partial transcripts (5′-UTR, CR, or 3′-UTR) was found to associate with the protein GAPDH (glyceraldehyde-3-phosphate dehydrogenase), included here as a negative control. Further mapping of the association of CUGBP1 with the Igf2r 3′-UTR was determined by testing the interaction of partial biotinylated transcripts spanning the Igf2r 3′-UTR (Fig. 4C, schematic) with CUGBP1 using pulldown assays. CUGBP1 was found to interact specifically with the fragment 3′UTR-F3, which contained several hits of CUGBP1 signature motifs (Fig. 4C, right). On the other hand, there was only marginal activity of binding of CUGBP1 to the fragment 3′UTR-F2, which contained only two GU-rich elements, and CUGBP1 did not bind to the fragment 3′UTR-F1, which contained no predicted binding site. We also examined the association of the Igf2r mRNA with other RBPs and found that neither HuR nor AUF1 interacted with the Igf2r mRNAs (data not shown). These results indicate that CUGBP1 specifically interacts with the Igf2r mRNA via its 3′-UTR.
To determine the functional consequences of CUGBP1 interactions with Igf2r mRNA, we examined the effect of overexpressing CUGBP1 on IGF2R expression. As shown (Fig. 5A), ectopically expressed CUGBP1 also specifically decreased the levels of IGF2R protein by ∼65% (n = 3; P < 0.05) but did not alter the expression levels of GLP1R, GLP2R, and EGFR. This inhibition of IGF2R by CUGBP1 also occurred at the level of translation, since ectopic CUGBP1 overexpression did not lower Igf2r mRNA levels (Fig. 5B) but repressed the rate of nascent IGF2R protein synthesis (Fig. 5C). The levels of newly synthesized IGF2R protein in CUGBP1-transfected cells decreased by ∼70% (n = 3; P < 0.05) compared with vector control-transfected cells. Examining the relative levels of distribution of the Igf2r mRNA in individual fractions from polyribosome gradients after CUGBP1 overexpression, our results showed that the abundance of Igf2R mRNA associated with actively translating components of the gradient (fractions 9 to 12) decreased dramatically, while the distribution of Igf2r mRNA shifted markedly toward low-translating parts of the gradient (fractions 5 to 7) (Fig. 5D, top). In contrast, Gapdh mRNA, which encodes a housekeeping protein and is not a target of CUGBP1, distributed similarly in the two groups (Fig. 5D, bottom). Consistently, ectopic CUGBP1 overexpression decreased the levels of the luciferase reporter transcripts (FL) Luc-Igf2r-3′UTR and Luc-Igf2R-3′UTR-F3, both of which contain CUGBP1 signature motifs (Fig. 5E). In contrast, increasing the levels of CUGBP1 failed to inhibit the production of luciferase from reporter transcripts Luc-Igf2r-3′UTR-F1 and Luc-Igf2r-3′UTR-F2, in which fragment 3′-UTR-F3 was deleted. In addition, the presence of ectopically expressed CUGBP1 did not alter the activity of reporter transcripts Luc-Igf2r-5′UTR and Luc-Igf2r-CR, which bear no CUGBP1 binding sequences (Fig. S3).
FIG 5.

CUGBP1 inhibits IGF2R expression at the translation level. (A) Representative immunoblots of IGF2R, GLP1R, GLP2R, and EGFR in cells overexpressing CUGBP1. After cells were transfected with either CUGBP1 expression vector or control vector for 48 h, whole-cell lysates were harvested for Western blotting. (B) Levels of the Igf2r mRNA in the cells described in the panel A legend. Values represent means ± SEM of data from triplicate experiments. (C) Newly synthesized IGF2R protein in cells overexpressing CUGBP1 as examined by l-azidohomoalaine (AHA) incorporation assays. (D) Distributions of the Igf2r (top) and Gapdh (bottom) mRNAs in each gradient fraction prepared from polysomal profile in cells after CUGBP1 overexpression. (E) Changes in activities of luciferase (Luc) reporters containing Igf2r full-length 3′-UTR (FL-3′UTR) or its different fragments in the cells described in the panel A legend. Values represent means ± SEM of results from triplicate samples. *, P < 0.05 (compared with control vector). (F and G) Effect of CUGBP1 silencing on IGF2R expression. Cells were transfected with either siRNA targeting the Cugbp1 mRNA (siCUGBP1) or control siRNA (C-siRNA) for 48 h (F), and then the levels of IGF2R protein and mRNA were measured (G). Values represent means ± SEM of data from triplicate experiments.
Moreover, CUGBP1 silencing by transfection with siRNA targeting the Cugbp1 mRNA (siCUGBP1) led to increased IGF2R expression. These specific siCUGBP1 nucleotides showed high specificity with the Cugbp1 mRNA and low toxicity, as described previously (25, 26). The levels of CUGBP1 protein decreased by >95% at 48 h after transfection of siCUGBP1, whereas the levels of IGF2R protein increased by ∼2-fold (n = 3; P < 0.05) compared with those in cells transfected with control siRNA (C-siRNA) (Fig. 5F). Decreasing the levels of endogenous CUGBP1 by the use of siCUGBP1 induced IGF2R expression by enhancing its translation, because CUGBP1 silencing did not alter the levels of total Igf2r mRNA (Fig. 5G). These results indicate that CUGBP1 inhibits IGF2R translation by directly interacting with the Igf2r 3′-UTR.
miR-195 and CUGBP1 repress IGF2R translation cooperatively.
As both miR-195 and CUGBP1 downregulate IGF2R translation by interacting with the Igf2r mRNA, it is plausible that miR-195 and CUGBP1 jointly regulate IGF2R expression. To test the possibility, we first examined the effect of altering cellular CUGBP1 levels on miR-195 binding to the Igf2r mRNA, as examined by RNA pulldown assays using biotin-labeled miR-195. Interestingly, transfecting cells with an expression vector to elevate CUGBP1 abundance increased the amount of Igf2r mRNA bound to miR-195 (Fig. 6A, panel a) without affecting total Igf2r mRNA levels (Fig. 5B) or miR-195 abundance (data not shown). The levels of Igf2r mRNA associated with CUGBP1 also increased in cells overexpressing ectopic CUGBP1, as measured by RNP/IP assays using anti-CUGBP1 antibody (Fig. 6B). In contrast, CUGBP1 silencing by transfection with siCUGBP1 decreased the association of Igf2r mRNA with both miR-195 (Fig. 6A, panel b) and CUGBP1. Second, we determined if increasing miR-195 levels altered the association of CUGBP1 with Igf2r mRNA. As shown, expressing miR-195 ectopically by transfection with pre-miR-195 enhanced the association of CUGBP1 with the Igf2r mRNA, and the levels of Igf2r mRNA bound to CUGBP1 in cells overexpressing both CUGBP1 and pre-miR-195 were significantly higher than those observed in cells overexpressing CUGBP1 alone (Fig. 6B). Third, we determined if increasing the levels of both miR-195 and CUGBP1 inhibited IGF2R expression synergistically. Our results showed that the levels of IGF2R protein decreased by >98% (n = 3; P < 0.05) in cells cotransfected with pre-miR-195 and CUGBP1 expression vector and were lower (decreased by ∼70%; n = 3; P < 0.05) than those observed in cells transfected with pre-miR-195 alone (Fig. 6C). Consistently, there were no differences in the levels of Igf2r mRNA in cells cotransfected with pre-miR-195 and CUGBP1 compared with those in control cells (data not shown). Together, our findings suggest a model whereby miR-195 and CUGBP1 inhibit IGF2R translation cooperatively by enhancing their binding to Igf2r mRNA. In this model, both miR-195 and CUGBP1 directly interacted with Igf2r mRNA, whereas the association of CUGBP1 with Igf2r mRNA enhanced miR-195 binding to Igf2r transcript. This cooperative interaction between miR-195 and CUGBP1 with Igf2r mRNA synergistically repressed IGF2R translation, in turn affecting the homeostasis of the gut epithelium.
FIG 6.
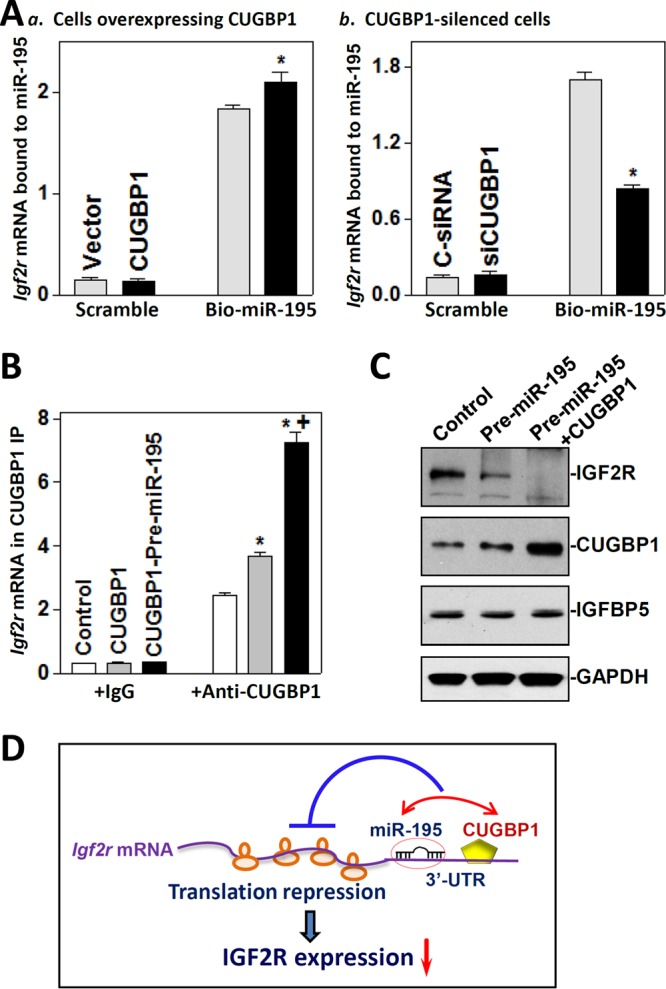
miR-195 and CUGBP1 inhibit IGF2R expression cooperatively. (A) Levels of Igf2r mRNA associated with miR-195 after increasing or decreasing cellular CUGBP1 abundance: (a) cells overexpressing CUGBP1; (b) CUGBP1-silenced cells. At 24 h after transfection with CUGBP1 expression vector or siCUGBP1, cells were transfected with biotinylated miR-195. The levels of Igf2r mRNA in biotin pulldown materials were measured 24 h after transfection with biotinylated miR-195. *, P < 0.05 (compared with cells transfected with control vector or C-siRNA). (B) Levels of the Igf2r mRNA associated with CUGBP1 in cells overexpressing CUGBP1 alone or both miR-195 and CUGBP1. Cells were transfected with CUGBP1 expression vector alone or both CUGBP1 and pre-miR-195, and the levels of Igf2r mRNA in CUGBP1 IP materials were examined 48 h after the transfection. Values represent means ± SEM of data from three separate experiments. * and +, P < 0.05 (compared with control and cells transfected with CUGBP1 alone, respectively). (C) Representative immunoblots of IGF2R, CUGBP1, and IGFBP5 in cells overexpressing both miR-195 and CUGBP1. At 48 h after the cotransfection with the CUGBP1 expression vector and pre-miR-195, the levels of different proteins were examined by Western immunoblotting analysis. (D) Proposed model to explain the cooperative repression of IGF2R translation by miR-195 and CUGBP1. Both miR-195 and CUGBP1 directly interacted with Igf2r mRNA, and the association of CUGBP1 with Igf2r mRNA enhanced miR-195 binding to Igf2r mRNA. This cooperative interaction between miR-195 and CUGBP1 synergistically inhibited IGF2R translation.
miR-195 and CUGBP1 lower IGF2R levels and disrupt the integrity of TGN.
To understand the cellular function of the miR-195/CUGBP1-regulated IGF2R production in the gut epithelium, we examined if decreasing the IGF2R levels mediated by the activity of miR-195 and CUGBP1 affected the integrity of TGN in cultured IECs. As shown (Fig. 7A, panel a), IGF2R was primarily localized to the TGN in control cells, as indicated by IGF2R colocalization with syntaxin 6, a well-established marker for TGN in diverse eukaryotes (21, 31), but not on the cell surface. The Golgi structure in control cells, as revealed by staining for syntaxin 6 and IGF2R, exhibited restricted juxtanuclear localization. Although ectopically expressed miR-195 and CUGBP1 did not alter total cellular levels of syntaxin 6 protein (see Fig. S4), they disrupted the stability and integrity of TGN, causing the disappearance of the typical Golgi morphology in cells with overexpressed levels of pre-miR-195 (Fig. 7A, panel b) or CUGBP1 (Fig. 7A, panel c) or both molecules (Fig. 7A, panel d). The subcellular distribution of syntaxin 6 in IGF2R-deficient cells showed a perinuclear staining pattern without a clear Golgi structure. This disruptive effect of miR-195/CUGBP1-mediated repression of IGF2R on the integrity of TGN was specific, because overexpression of miR-195 and CUGBP1 did not affect the subcellular distribution of cytoskeleton α-tubulin (Fig. 7B). A network of long stress fibers of α-tubulin that were located just beneath the plasma membrane and traversed the cytoplasm was observed in both control cells and cells overexpressing miR-195 and CUGBP1. These results indicate that IGF2R in IECs is essential for maintaining the stability of TGN, in turn regulating distinct cellular processes and functions.
FIG 7.
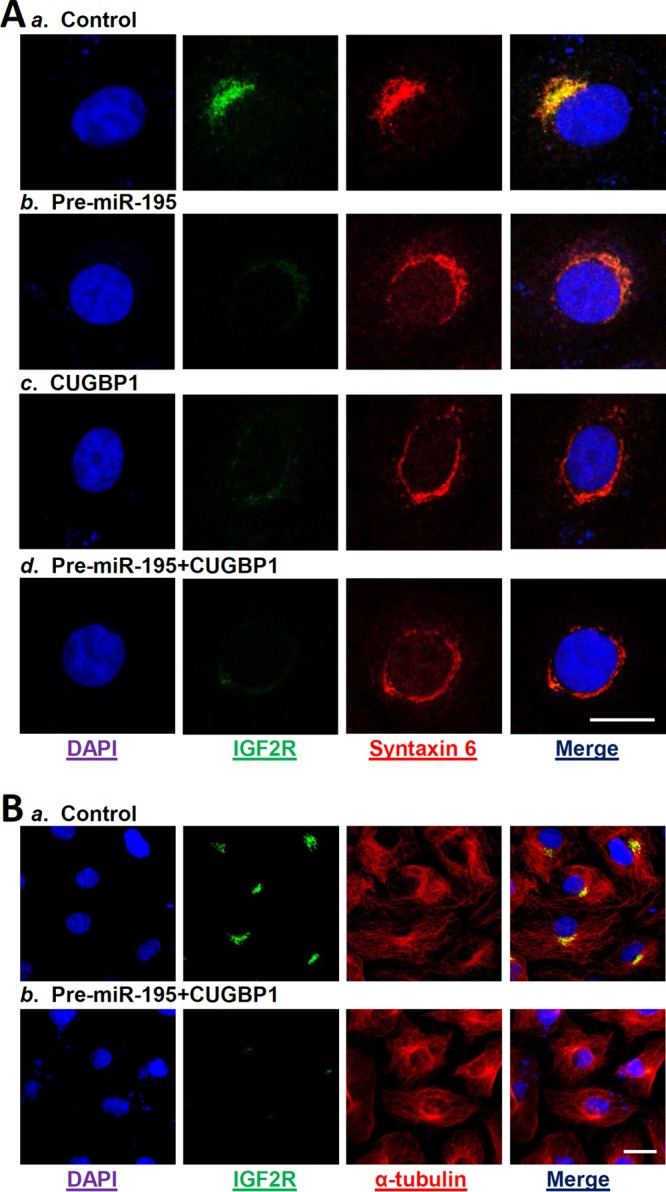
Reduction in IGF2R levels mediated by miR-195 and CUGBP1 alters homeostasis of the Golgi apparatus. (A) Fluorescence analysis of IGF2R colocalization with Golgi structural protein syntaxin 6 after different treatments: (a) control; (b) cells transfected with pre-miR-195 alone; (c) cells transfected with CUGBP1 expression vector alone; (d) cells cotransfected with pre-miR-195 and CUGBP1. At 48 h after transfection, cells were fixed, permeabilized, and incubated with the antibody against IGF2R or syntaxin 6 and then with anti-IgG conjugated with fluorescein isothiocyanate (FITC). Original magnification, ×1,000. Bar, 50 μm. (Left panel) Nuclei were stained by DAPI. (Second panel from left) Green, antibody detecting IGF2R. (Third panel from left) Red, antibody detecting syntaxin 6. (Right panel) Yellow, merge of the two signals. Three separate experiments were performed and showed similar results. (B) Fluorescence analysis of IGF2R and α-tubulin in cells overexpressing both miR-195 and CUGBP1. (a) Control; (b) cells cotransfected with pre-miR-195 and CUGBP1. Original magnification, ×500. Bar, 50 μm.
DISCUSSION
Our recent studies show that miR-195 and CUGBP1 are critical regulators of gut epithelium homeostasis and that elevation of cellular miR-195 and CUGBP1 levels and their binding affinity inhibit IEC migration and proliferation, thus compromising epithelial integrity (14, 17, 25), but the effectors of these actions are not fully known. IGF2R, a 250-kDa multifunctional glycoprotein, recognizes two different classes of ligands, IGF2/IGF1 and M6P-containing molecules, and is implicated in diverse cellular functions (4, 6, 32). In this study, we identified Igf2r mRNA as a novel target of both miR-195 and CUGBP1 in IECs and found that miR-195 and CUGBP1 inhibited IGF2R translation without affecting Igf2r mRNA stability or whole-cell abundance. The binding of Igf2r mRNA to miR-195 was enhanced by elevating CUGBP1 expression and reduced by silencing CUGBP1. Importantly, jointly overexpressing miR-195 and CUGBP1 cooperatively repressed IGF2R translation. These findings provide insight into the control of IGF2R expression at the posttranscriptional level and advance our understanding of the molecular mechanism underlying the homeostasis of the gut mucosal epithelium. Our results suggest that miR-195 and CUGBP1 regulate the growth and adaptation of the gut mucosa at least partially by reducing the levels of IGF2R and thereby modulating IGF signaling. Our results also point to miR-195/CUGBP1-mediated IGF2R reduction as a possible molecular pathway that could be targeted to promote mucosal growth under pathological conditions.
The results presented here indicate that miR-195 and CUGBP1 interacted with the Igf2r mRNA via its 3′-UTR rather than the 5′-UTR and CR. These observations are consistent with other results indicating that miR-195 specifically binds to the 3′-UTRs of Cdk4, Cdk6, WEE1, MO25, ActRIIA, Stim1, and Sirt1 mRNAs to elicit its regulatory actions (17–24). The use of reporters bearing partial transcripts spanning the Igf2r 5′-UTR, CR, or 3′-UTR with or without the predicted miR-195 binding site revealed that the Igf2r 3′-UTR contained the functional sequence through which miR-195 inhibited IGF2R translation. As shown, the repression of IGF2R by miR-195 overexpression was prevented when the binding sequence was mutated from Igf2r 3′-UTR. In some instances, miRNAs are also found to associate with the CRs of target mRNAs for their functions. In this regard, miR-519 represses HuR translation by interacting with the CR but not the 3′-UTR of HuR mRNA (33), while miR-222 inhibits CDK4 translation through association with both the CR and the 3′-UTR of the Cdk4 mRNA (18). In the current paradigm, miR-195 and CUGBP1 interacted with the Igf2r 3′-UTR and jointly repressed IGF2R translation. Although the Igf2r 3′-UTR does not contain typical canonical GU-rich elements (GREs) such as UGUUUGUUUGU, there are many GU repeats and CUG repeats in the Igf2r 3′-UTR which were also recognized as GREs and interacted with CUGBP1 (34–36). We did not further characterize the specific Igf2r 3′-UTR nucleotides with which miR-195 and/or CUGBP1 interact, since those studies would require biochemical, crystallographic, and molecular methods that are more specialized than those used in the present investigation.
Our results also indicate that CUGBP1 and miR-195 jointly regulate IGF2R translation, since CUGBP1 enhanced the binding of miR-195 to the Igf2r mRNA. The cooperative inhibition of IGF2R expression by CUGBP1 and miR-195 was not surprising, as several studies showed that CUGBP1 functionally interacts with miRNAs and/or other RBPs to remodel ribonucleoprotein complexes and influence the posttranscriptional fate of mRNAs positively or negatively (14, 37). For example, CUGBP1 interacts with miR-222 to inhibit translation of the Cdk4 mRNA synergistically (18), but it competes with HuR to regulate translation of the tight junction occludin and transcription factor MYC antagonistically (25, 26). The exact mechanism by which CUGBP1 enhances the association of miR-195 with Igf2r mRNA is unclear at present, but it has been reported that miRNA binding sites are commonly present near RBP binding sites (38, 39), suggesting that in some cases RBP and miRNA actions could be enhanced or could compete via their physical interactions with given mRNAs. However, RBPs and coregulatory miRNAs can also bind at locations that are up to several hundreds or thousands of bases apart in some targets (16, 40, 41). In the present study, we found that both miR-195 and CUGBP1 had high affinity for the Igf2r 3′-UTR. However, the miR-195 binding site was located at the fragment 3′UTR-F1, whereas CUGBP1 predominantly interacted with the fragment 3′UTR-F3. It remains unknown how CUGBP1 and miR-195 interact with the Igf2r 3′-UTR through distinct nonoverlapping binding sites. RNA structure and folding analyses will be needed to determine systematically the process by which binding of CUGBP1 in one area of the Igf2r 3′-UTR enhances interactions with miR-195 in a remote site.
The data obtained in the present study strongly suggest that the miR-195/CUGBP1-elicited repression of IGF2R expression plays a role in the regulation of distinct cellular processes and likely functions by altering the composition of the TGN. Decreasing the levels of cellular IGF2R by overexpressing miR-195 and CUGBP1 disrupted the Golgi structure in IECs, suggesting that IGF2R is essential for TGN formation and stability, whereas reduction in the levels of cellular IGF2R by increasing miR-195 and CUGBP1 disrupts TGN integrity and function. Consistent with our present findings, IGF2R has been shown to facilitate the delivery of nascent lysosomal enzymes from the TGN to endosomes (6, 42), and ectopic overexpression of the IGF2R increases β-amyloid production and affects cell viability in fibroblasts (43). Several studies have also shown that IGF2R is expressed in many tissues, including the gut epithelium, and its expression is known to be altered in response to stressful environments such as after SBR and during the inhibition of intestinal mucosal growth (1, 2). Interestingly, the levels of mucosal miR-195 and CUGBP1 decrease in the small intestine after SBR, along with stimulation of mucosal regeneration, but their levels increase dramatically after food starvation or polyamine depletion, which is associated with an inhibition of intestinal mucosal growth (18, 25, 44). Moreover, IGF2R also regulates cellular processes and functions by (i) activating IGF2 signaling (42, 45); (ii) promoting the cellular uptake and degradation of peptides such as proliferin, leukemia inhibitory factor, and transforming growth factor beta (TGF-β) (4, 6); and (iii) regulating extracellular signal-regulated kinase 1/extracellular signal-regulated kinase 2 (ERK1/2) activity (32). In sum, our results indicate that miR-195 and CUGBP1 repress IGF2R translation cooperatively in IECs, thus compromising the integrity of the gut epithelium in stressful environments involving disruption of Golgi function and impaired IGF signaling. Given that the TGN and IGF systems critically influence gut mucosal regeneration and homeostasis, our findings showing that the combined activity of miR-195 and CUGBP1 represses IGF2R to inhibit mucosal growth under pathological conditions suggest that this system might be exploited in therapies for patients with mucosal atrophy.
MATERIALS AND METHODS
Chemicals and cell culture.
Tissue culture medium and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA), and biochemicals were from Sigma (St. Louis, MO). The antibodies recognizing IGF2R, IGF1R, CUGBP1, IGFBP5, GLP1R, GLP2R, and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), EGFR was from BD Biosciences, and the secondary antibody conjugated to horseradish peroxidase was from Sigma. The pre-miR miRNA precursor and anti-miR miRNA inhibitor of miR-195 were purchased from Ambion (Austin, TX). Biotin-labeled miRNA-195 was custom-made by Dharmacon (Lafayette, CO). The IEC-6 cell line (normal rat intestinal crypt cells) was purchased from the American Type Culture Collection (ATCC) at passage 13 and was maintained under standard culture conditions as described previously (44, 46).
Plasmid construction.
CUGBP1 expression vector was purchased from Origene (Rockville, MD) and was described previously (18, 26). The chimeric firefly luciferase reporter construct containing the entire Igf2r cDNA was constructed as described previously (30, 47). The full-length Igf2r 5′-UTR and 3′-UTR and different CR fragments were amplified and subcloned into pmirGLO Dual-Luciferase miRNA target expression vector (Promega, Madison, WI) to generate pmirGLO-Luc-Igf2R-5′UTR, pmirGLO-Luc-Igf2r-CR, and pmirGLO-Igf2r-3′UTR. DNA sequencing and enzyme digestion were used to confirm the sequence and orientation of the fragment in the luciferase reporter. Transient transfections were performed using Lipofectamine reagent as recommended by the manufacturer (Invitrogen) (48, 49). Luciferase activity was examined using the Dual-Luciferase assay system, and the levels of firefly luciferase activity were normalized to Renilla luciferase activity and were further compared with the levels of luciferase mRNA activity in every experiment. The primer sequences used for generation of these constructs are presented in Table S1 in the supplemental material.
Reverse transcription (RT) and real-time quantitative PCR (Q-PCR) analysis.
Total RNA was isolated by using an RNeasy minikit (Qiagen, Valencia, CA), and RT and PCR amplification reactions were performed as described previously (17). The levels of Gapdh PCR product were examined to monitor the evenness in the RNA inputs in RT-PCR samples. Real-time quantitative PCR (Q-PCR) analysis was conducted using 7500 Fast real-time PCR systems with specific primers, probes, and software (Applied Biosystems, Foster City, CA). For miRNA studies, the levels of miRNA-195 were also quantified by Q-PCR by using TaqMan MicroRNA assay; small nuclear RNA (snRNA) U6 was used as an endogenous control.
Western blot analysis.
Whole-cell lysates were prepared using 2% SDS, sonicated, and centrifuged (12,000 rpm) at 4°C for 15 min. The supernatants were boiled for 5 min and subjected to size fractionation by SDS-PAGE (7.5% acrylamide). After transfer of proteins onto nitrocellulose filters, the blots were incubated with primary antibodies recognizing IGF2R or CUGBP1; following incubations with secondary antibodies, immunocomplexes were developed by using chemiluminescence.
Analysis of newly translated protein.
De novo synthesis of nascent proteins was detected by the use of a Click-iT protein analysis detection kit (Life Technologies, Grand Island, NY) following the manufacturer's instructions with minor modifications as described previously (50). Briefly, cells were incubated in methionine-free medium and then exposed to l-azidohomoalaine (AHA). After the cell lysates were mixed with the reaction buffer containing biotin-alkyne reagent and CuSO4 for 20 min, the biotin-alkyne–azide-modified protein complex was pulled down using paramagnetic streptavidin-conjugated Dynabeads. The pulldown material was resolved by 10% SDS-PAGE and subjected to Western immunoblotting analysis using antibodies that recognized IGF2R or GAPDH proteins.
Polysome analysis was performed as described previously (51). Briefly, cells were incubated at ∼70% confluence for 15 min in 0.1 mg/ml cycloheximide, lifted by scraping in 1 ml of polysome extraction buffer, and lysed on ice for 10 min. Nuclei were pelleted, and the resulting supernatant was processed through a 10% to 50% linear sucrose gradient to fractionate cytoplasmic components according to their molecular weights. The eluted fractions were prepared with a fraction collector (Brandel, Gaithersburg, MD), and their quality was monitored at 254 nm using a UV-6 detector (ISCO, Louisville, KY). After RNA in each fraction was extracted, the levels of each individual mRNA were quantified by Q-PCR in each of the fractions.
Biotin-labeled miR-195 pulldown assays.
Cells were transfected with biotinylated miR-195, and whole-cell lysates were collected 24 h later, mixed with streptavidin-Dynal beads (Invitrogen, Carlsbad, CA), and incubated at 4°C with rotation overnight (52). After the beads were washed thoroughly, the bead-bound RNA was isolated and subjected to RT followed by Q-PCR analysis. Input RNA was extracted and served as a control.
Biotin pulldown assays and RNP IP analysis.
Synthesis of biotin-labeled transcripts and measurement of RBPs bound to biotinylated RNA were performed as previously described (25). The template of cDNA was from IEC-6 cells for PCR amplification of the 5′-UTR, CR, and 3′-UTR of Igf2r mRNA. The 5′ primers contained the T7 RNA polymerase promoter sequence (T7)CCAAGCTTCTAATACGACTCACTATAGGGAGA. The sequences of the oligonucleotides used for preparation of full-length Igf2r 5′-UTR, various fractions of CR, and the 3′-UTR are presented in Table S1. PCR-amplified products were used as the templates to transcribe biotinylated RNAs by using T7 RNA polymerase in the presence of biotin-CTP as described previously (30). Biotin-labeled transcripts (6 μg) were incubated with 120 μg of cytoplasmic lysates for 30 min at room temperature, and RNA/protein complexes were isolated with paramagnetic streptavidin-conjugated Dynabeads (Dynal, Oslo, Norway) and analyzed by Western blotting.
To determine the association of endogenous CUGBP1 with endogenous Igf2r mRNA, immunoprecipitation of RNP complexes was examined as described previously (53, 54). Twenty million cells were collected per sample, and whole-cell lysates were used for IP for 4 h at room temperature in the presence of excess (30 μg) IP antibody (IgG, anti-CUGBP1). RNA in IP materials was used in RT followed by PCR and Q-PCR analysis to detect the presence of Igf2r and Gapdh mRNAs.
Immunofluorescence staining.
The immunofluorescence staining procedure was carried out according to the method described in our previous publications (55, 56). After the cells were fixed in 3.7% formaldehyde–phosphate-buffered saline and rehydrated, they were incubated with the primary antibody against IGF2R or syntaxin 6 in the block buffer (1:300 dilution) at 4°C overnight and then incubated with secondary antibody conjugated with Alexa Fluor 594 (Molecular Probes, Eugene, OR) for 2 h at room temperature. Finally, the slides were washed, mounted, and viewed through a Zeiss confocal microscope (model LSM410). Images were processed using Photoshop software (Adobe, San Jose, CA).
Statistics.
Values represent means ± standard errors (SE) of results from three to six samples. Immunofluorescence staining was repeated three times. Where indicated, P values of <0.05 were considered significant, and an unpaired, two-tailed Student t test was used. In assessing multiple groups, one-way analysis of variance (ANOVA) was utilized with Tukey's post hoc test (57). The statistical software used was SPSS17.1.
Supplementary Material
ACKNOWLEDGMENTS
Yuan Zhang and Yun Zhang performed most experiments, summarized data, and were involved in the preparation of figures. Lan Xiao and Ting-Xi Yu performed experiments dealing with biotin pulldown assays and analyzed the results and participated in the immunofluorescence analysis. Jun-Zhe Li was involved in construction of plasmids and RNP IP analysis. Jaladanki N. Rao contributed to studies analyzing the rate of synthesis of newly synthesized proteins and analyzed results. Douglas J. Turner contributed to data analysis. Myriam Gorospe contributed to experimental design and data analysis. Jian-Ying Wang designed experiments, analyzed data, prepared figures, and drafted the manuscript.
This work was supported by Merit Review Awards (to Jian-Ying Wang, Jaladanki N. Rao, and Douglas J. Turner) from the U.S. Department of Veterans Affairs, grants from the National Institutes of Health (DK57819, DK61972, and DK68491 to Jian-Ying Wang), and funding from the Intramural Research Program in the National Institute on Aging, NIH (to Myriam Gorospe). Jian-Ying Wang is a Senior Research Career Scientist, Biomedical Laboratory Research & Development Service, U.S. Department of Veterans Affairs.
We declare that we have no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00225-17.
REFERENCES
- 1.Kuemmerle JF. 2012. Insulin-like growth factors in the gastrointestinal tract and liver. Endocrinol Metab Clin North Am 41:409–423. doi: 10.1016/j.ecl.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw D, Gohil K, Basson MD. 2012. Intestinal mucosal atrophy and adaptation. World J Gastroenterol 18:6357–6375. doi: 10.3748/wjg.v18.i44.6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kar S, Seto D, Doré S, Hanisch U, Quirion R. 1997. Insulin-like growth factors-I and -II differentially regulate endogenous acetylcholine release from the rat hippocampal formation. Proc Natl Acad Sci U S A 94:14054–14059. doi: 10.1073/pnas.94.25.14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghosh P, Dahms NM, Kornfeld S. 2003. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 5.Riedemann J, Macaulay VM. 2006. GF1R signaling and its inhibition. Endocr Relat Cancer 13:S33–S43. doi: 10.1677/erc.1.01280. [DOI] [PubMed] [Google Scholar]
- 6.Harris LK, Westwood M. 2012. Biology and significance of signaling pathways activated by IGF-II. Growth Factors 30:1–12. doi: 10.3109/08977194.2011.640325. [DOI] [PubMed] [Google Scholar]
- 7.Pfeffer SR. 2009. Multiple routes of protein transport from endosomes to the trans-Golgi network. FEBS Lett 583:3811–3816. doi: 10.1016/j.febslet.2009.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu PC, Kulp SK, Chen CS. 2013. Insulin-like growth factor-I receptor is suppressed through transcriptional repression and mRNA destabilization by a novel energy restriction-mimetic agent. Carcinogenesis 34:2694–2705. doi: 10.1093/carcin/bgt251. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Panda AC, Grammatikakis I, Yoon JH, Abdelmohsen K. 2013. Posttranscriptional regulation of insulin family ligands and receptors. Int J Mol Sci 14:19202–19229. doi: 10.3390/ijms140919202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee EK, Gorospe M. 2010. Minireview: posttranscriptional regulation of the insulin and insulin-like growth factor systems. Endocrinology 151:1403–1408. doi: 10.1210/en.2009-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 12.Cao S, Xiao L, Rao JN, Zou T, Liu L, Zhang D, Turner DJ, Gorospe M, Wang JY. 2014. Inhibition of Smurf2 translation by miR-322/503 modulates TGF-β/Smad2 signaling and intestinal epithelial homeostasis. Mol Biol Cell 25:1234–1243. doi: 10.1091/mbc.E13-09-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srikantan S, Tominaga K, Gorospe M. 2012. Functional interplay between RNA-binding protein HuR and microRNAs. Curr Protein Pept Sci 13:372–379. doi: 10.2174/138920312801619394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JY, Xiao L, Wang JY. 2017. Posttranscriptional regulation of intestinal epithelial integrity by noncoding RNAs. Wiley Interdiscip Rev RNA 8:1399. doi: 10.1002/wrna.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23:1743–1748. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JAF, Slanchev K, Le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Rom UA, Lund AH, Perrakis A, Raz E, Agami R. 2007. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131:1273–1286. doi: 10.1016/j.cell.2007.11.034. [DOI] [PubMed] [Google Scholar]
- 17.Zhuang R, Rao JN, Zou T, Liu L, Xiao L, Cao S, Hansraj NZ, Gorospe M, Wang JY. 2013. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res 41:7905–7919. doi: 10.1093/nar/gkt565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao L, Cui YH, Rao JN, Zou T, Liu L, Smith A, Turner DJ, Gorospe M, Wang JY. 2011. Regulation of cyclin-dependent kinase 4 translation through CUG-binding protein 1 and microRNA-222 by polyamines. Mol Biol Cell 22:3055–3069. doi: 10.1091/mbc.E11-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP, Zhuang SM. 2009. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology 50:113–121. doi: 10.1002/hep.22919. [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharya A, Schmitz U, Wolkenhauer O, Schönherr M, Raatz Y, Kunz M. 2013. Regulation of cell cycle checkpoint kinase WEE1 by miR-195 in malignant melanoma. Oncogene 32:3175–3183. doi: 10.1038/onc.2012.324. [DOI] [PubMed] [Google Scholar]
- 21.Xiao L, Rao JN, Zou T, Liu L, Cao S, Martindale JL, Su W, Chung HK, Gorospe M, Wang JY. 2013. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell 24:3038–3046. doi: 10.1091/mbc.E13-05-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Y, Wu J, Chen H, Mao Y, Liu Y, Mao Q, Yang K, Zheng X, Xie L. 2012. Cyclin-dependent kinase 4 is a novel target in micoRNA-195-mediated cell cycle arrest in bladder cancer cells. FEBS Lett 586:442–447. doi: 10.1016/j.febslet.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Yang Y, Wang Y, Li J, Schiller PW, Peng T. 2011. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res 92:75–84. doi: 10.1093/cvr/cvr145. [DOI] [PubMed] [Google Scholar]
- 24.Bai Y, Yang W, Yang HX, Liao Q, Ye G, Fu G, Ji L, Xu P, Wang H, Li YX, Peng C, Wang YL. 2012. Downregulated miR-195 detected in preeclamptic placenta affects trophoblast cell invasion via modulating ActRIIA expression. PLoS One 7:e38875. doi: 10.1371/journal.pone.0038875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Ouyang M, Rao JN, Zou T, Xiao L, Chung HK, Wu J, Donahue JM, Gorospe M, Wang JY. 2015. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol Biol Cell 26:1797–1810. doi: 10.1091/mbc.E14-11-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu TX, Rao JN, Zou T, Liu L, Xiao L, Ouyang M, Cao S, Gorospe M, Wang JY. 2013. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol Biol Cell 24:85–99. doi: 10.1091/mbc.E12-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung HK, Rao JN, Zou T, Liu L, Xiao L, Gu H, Turner DJ, Yang P, Wang JY. 2014. Jnk2 deletion disrupts intestinal mucosal homeostasis and maturation by differentially modulating RNA-binding proteins HuR and CUGBP1. Am J Physiol Cell Physiol 306:C1167–C1175. doi: 10.1152/ajpcell.00093.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Rao JN, Zou T, Xiao L, Wang PY, Turner DJ, Gorospe M, Wang JY. 2009. Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol Biol Cell 20:4885–4898. doi: 10.1091/mbc.E09-07-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang PY, Rao JN, Zou T, Liu L, Xiao L, Yu TX, Turner DJ, Gorospe M, Wang JY. 2010. Post-transcriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis. Biochem J 426:293–306. doi: 10.1042/BJ20091459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelmohsen K, Srikantan S, Tominaga K, Kang MJ, Yaniv Y, Martindale JL, Yang X, Park SS, Becker KG, Subramanian M, Maudsley S, Lal A, Gorospe . 2012. Growth inhibition by miR-519 via multiple p21-inducing pathways. Mol Cell Biol 32:2530–2548. doi: 10.1128/MCB.00510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu TX, Wang PY, Rao JN, Zou T, Liu L, Xiao L, Gorospe M, Wang JY. 2011. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res 39:8472–8487. doi: 10.1093/nar/gkr567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou T, Rao JN, Liu L, Xiao L, Yu TX, Jiang P, Gorospe M, Wang JY. 2010. Polyamines regulate the stability of JunD mRNA by modulating the competitive binding of its 3′ untranslated region to HuR and AUF1. Mol Cell Biol 30:5021–5032. doi: 10.1128/MCB.00807-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Christodoulou-Vafeiadou E, Rao JN, Zou T, Xiao L, Chung HK, Yang H, Gorospe M, Kontoyiannis D, Wang JY. 2014. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol Biol Cell 25:3308–3318. doi: 10.1091/mbc.E14-03-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao L, Rao JN, Cao S, Liu L, Chung HK, Zhang Y, Zhang J, Liu Y, Gorospe M, Wang JY. 2016. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol Biol Cell 27:617–626. doi: 10.1091/mbc.E15-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui YH, Xiao L, Rao JN, Zou T, Liu L, Chen Y, Turner DJ, Gorospe M, Wang JY. 2012. miR-503 represses CUG-binding protein 1 translation by recruiting CUGBP1 mRNA to processing bodies. Mol Biol Cell 23:151–162. doi: 10.1091/mbc.E11-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zou T, Mazan-Mamczarz K, Rao JN, Liu L, Marasa BS, Zhang AH, Xiao L, Pullmann R, Gorospe M, Wang JY. 2006. Polyamine depletion increases cytoplasmic levels of RNA-binding protein HuR leading to stabilization of nucleophosmin and p53 mRNAs. J Biol Chem 281:19387–19394. doi: 10.1074/jbc.M602344200. [DOI] [PubMed] [Google Scholar]
- 37.Liu L, Zhuang R, Xiao L, Chung HK, Luo J, Turner DJ, Rao JN, Gorospe M, Wang JY. 2017. HuR enhances early restitution of the intestinal epithelium by increasing Cdc42 translation. Mol Cell Biol 37:e00574-16. doi: 10.1128/MCB.00574-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang JY, McCormack SA, Johnson LR. 1996. Role of nonmuscle myosin II in polyamine-dependent intestinal epithelial cell migration. Am J Physiol 270:G355–G362. [DOI] [PubMed] [Google Scholar]
- 39.Zou T, Jaladanki SK, Liu L, Xiao L, Chung HK, Wang JY, Xu Y, Gorospe M, Wang JY. 2016. H19 long noncoding RNA regulates intestinal epithelial barrier function via microRNA 675 by interacting with RNA-binding protein HuR. Mol Cell Biol 36:1332–1341. doi: 10.1128/MCB.01030-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harter JL. 1960. Critical values for Duncan's new multiple range test. Biometrics 16:671–685. doi: 10.2307/2527770. [DOI] [Google Scholar]
- 41.Abdelmohsen K, Pullmann R Jr, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, Furneaux H, Gorospe M. 2007. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell 25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Zou T, Rao JN, Liu L, Xiao L, Wang PY, Cui YH, Gorospe M, Wang JY. 2009. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res 37:7623–7637. doi: 10.1093/nar/gkp755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bock JB, Klumperman J, Davanger S, Scheller RH. 1997. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell 8:1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El-Shewy HM, Lee MH, Obeid LM, Jaffa AA, Luttrell LM. 2007. The insulin-like growth factor type 1 and insulin-like growth factor type 2/mannose-6-phosphate receptors independently regulate ERK1/2 activity in HEK293 cells. J Biol Chem 282:26150–26157. doi: 10.1074/jbc.M703276200. [DOI] [PubMed] [Google Scholar]
- 45.Reverter M, Rentero C, Garcia-Melero A, Hoque M, Vilà de Muga S, Alvarez-Guaita A, Conway JR, Wood P, Cairns R, Lykopoulou L, Grinberg D, Vilageliu L, Bosch M, Heeren J, Blasi J, Timpson P, Pol A, Tebar F, Murray RZ, Grewal T, Enrich C. 2014. Cholesterol regulates Syntaxin 6 trafficking at trans-Golgi network endosomal boundaries. Cell Rep 7:883–897. doi: 10.1016/j.celrep.2014.03.043. [DOI] [PubMed] [Google Scholar]
- 46.Zhang HM, Rao JN, Guo X, Liu L, Zou T, Turner DJ, Wang JY. 2004. Akt kinase activation blocks apoptosis in intestinal epithelial cells by inhibiting caspase-3 after polyamine depletion. J Biol Chem 279:22539–22547. doi: 10.1074/jbc.M314337200. [DOI] [PubMed] [Google Scholar]
- 47.Tsuda K, Kuwasako K, Takahashi M, Someya T, Inoue M, Terada T, Kobayashi N, Shirouzu M, Kigawa T, Tanaka A, Sugano S, Güntert P, Muto Y, Yokoyama S. 2009. Structural basis for the sequence-specific RNA-recognition mechanism of human CUG-BP1 RRM3. Nucleic Acids Res 37:5151–5166. doi: 10.1093/nar/gkp546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rattenbacher B, Beisang D, Wiesner DL, Jeschke JC, von Hohenberg M, St Louis-Vlasova IA, Bohjanen PR. 2010. Analysis of CUGBP1 targets identifies GU-repeat sequences that mediate rapid mRNA decay. Mol Cell Biol 30:3970–3980. doi: 10.1128/MCB.00624-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. 2007. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG pre-mutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao L, Wang JY. 2014. RNA-binding proteins and microRNAs in gastrointestinal epithelial homeostasis and diseases. Curr Opin Pharmacol 19:46–53. doi: 10.1016/j.coph.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N. 2011. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell 43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Tuschl T, Ohler U, Keene JD. 2011. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell 43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 54.Srikantan S, Abdelmohsen K, Lee EK, Tominaga K, Subaran SS, Kuwano Y, Kulshrestha R, Panchakshari R, Kim HH, Yang X, Martindale JL, Marasa BS, Kim MM, Wersto RP, Indig FE, Chowdhury D, Gorospe M. 2011. Translational control of TOP2A influences doxorubicin efficacy. Mol Cell Biol 31:3790–3801. doi: 10.1128/MCB.05639-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Shewy HM, Luttrell LM. 2009. Insulin-like growth factor-2/mannose-6 phosphate receptors. Vitam Horm 80:667–697. doi: 10.1016/S0083-6729(08)00624-9. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Buggia-Prévot V, Zavorka ME, Bleackley RC, MacDonald RG, Thinakaran G, Kar S. 2015. Overexpression of the insulin-like growth factor II receptor increases β-amyloid production and affects cell viability. Mol Cell Biol 35:2368–2384. doi: 10.1128/MCB.01338-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dikkes P, Hawkes C, Kar S, Lopez MF. 2007. Effect of kainic acid treatment on insulin-like growth factor-2 receptors in the IGF2-deficient adult mouse brain. Brain Res 1131:77–87. doi: 10.1016/j.brainres.2006.11.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.