

ABSTRACT
Poxviruses replicate within the cytoplasm and encode proteins for DNA and mRNA synthesis. To investigate poxvirus replication and transcription from a new perspective, we incorporated 5-ethynyl-2′-deoxyuridine (EdU) into nascent DNA in cells infected with vaccinia virus (VACV). The EdU-labeled DNA was conjugated to fluor- or biotin-azide and visualized by confocal, superresolution, and transmission electron microscopy. Nuclear labeling decreased dramatically after infection, accompanied by intense labeling of cytoplasmic foci. The nascent DNA colocalized with the VACV single-stranded DNA binding protein I3 in multiple puncta throughout the interior of factories, which were surrounded by endoplasmic reticulum. Complexes containing EdU-biotin-labeled DNA cross-linked to proteins were captured on streptavidin beads. After elution and proteolysis, the peptides were analyzed by mass spectrometry to identify proteins associated with nascent DNA. The known viral replication proteins, a telomere binding protein, and a protein kinase were associated with nascent DNA, as were the DNA-dependent RNA polymerase and intermediate- and late-stage transcription initiation and elongation factors, plus the capping and methylating enzymes. These results suggested that the replicating pool of DNA is transcribed and that few if any additional viral proteins directly engaged in replication and transcription remain to be discovered. Among the host proteins identified by mass spectrometry, topoisomerases IIα and IIβ and PCNA were noteworthy. The association of the topoisomerases with nascent DNA was dependent on expression of the viral DNA ligase, in accord with previous proteomic studies. Further investigations are needed to determine possible roles for PCNA and other host proteins detected.
IMPORTANCE Poxviruses, unlike many well-characterized animal DNA viruses, replicate entirely within the cytoplasm of animal cells, raising questions regarding the relative roles of viral and host proteins. We adapted newly developed procedures for click chemistry and iPOND (Isolation of proteins on nascent DNA) to investigate vaccinia virus (VACV), the prototype poxvirus. Nuclear DNA synthesis ceased almost immediately following VACV infection, followed swiftly by the synthesis of viral DNA within discrete cytoplasmic foci. All viral proteins known from genetic and proteomic studies to be required for poxvirus DNA replication were identified in the complexes containing nascent DNA. The additional detection of the viral DNA-dependent RNA polymerase and intermediate and late transcription factors provided evidence for a temporal coupling of replication and transcription. Further studies are needed to assess the potential roles of host proteins, including topoisomerases IIα and IIβ and PCNA, which were found associated with nascent DNA.
KEYWORDS: DNA replication, DNA binding proteins, iPOND, poxvirus, transcription factors, vaccinia virus
INTRODUCTION
Poxviruses have linear double-stranded DNA genomes of approximately 200,000 bp that are covalently linked at each end by an incompletely base-paired hairpin that is 104 nucleotides long in the case of vaccinia virus (VACV), the prototype member of the family (1). DNA replication occurs exclusively in the cytoplasm, forming discrete juxtanuclear sites known as factories (2, 3). The mechanism of poxvirus genome replication is uncertain, and two models have been proposed. One version, based on the rolling-hairpin model of parvovirus replication (4), posits only leading-strand DNA synthesis starting at a nick near one or both covalently closed ends of the genome and continuing by strand displacement to the other end, generating concatemers that are subsequently resolved into unit genomes (5, 6). Although mechanistically simple, a weakness of this model is the failure to identify the putative nickase. The other version, like replication in prokaryotes and eukaryotes, involves leading- and lagging-strand synthesis at replication forks and is consistent with viral encoding of a primase (7), requirement for a DNA ligase (8), evidence for Okazaki fragments (9, 10), suggestive electron microscopic images (11), and identification of a putative bidirectional origin (12). Missing from the identified replication proteins, however, is a flap endonuclease for removing RNA from Okazaki fragments.
Because poxvirus replication occurs in the cytoplasm and even in enucleated cells (3, 13), it has been generally thought that most or even all of the replication proteins are virus encoded. Currently, 10 VACV proteins are known to be involved in DNA replication, another 4 in precursor metabolism, and 2 more in packaging DNA in virus particles (14). The proteins required for DNA replication include the DNA polymerase holoenzyme, consisting of a DNA polymerase with a C-terminal 3′-5′ proofreading exonuclease domain (15–18); a processivity factor (19, 20); and a uracil DNA glycosylase that has a function in DNA synthesis, in addition to repair (21–23). A multifunctional scaffold protein (24) and a primase/helicase (7, 25) interact with DNA polymerase and form a multicomponent DNA replication-repair complex. Additional proteins involved in VACV genome replication are a single-stranded DNA binding protein (26–30), DNA ligase (8, 31, 32), serine-threonine protein kinase (33), a nuclease homologous to Fen-1 (34), and a Holiday junction resolvase (35, 36). The above-mentioned proteins, except for the resolvase, are synthesized at the early stage of infection prior to the initiation of DNA synthesis. The resolvase is not required for DNA synthesis, as it functions to convert concatemeric replicative intermediates into unit length genomes with hairpin ends.
Although the identified viral proteins seem to fulfill many of the basic requirements for VACV genome replication, uncertainty remains as to whether additional viral or host proteins might enhance replication or supply some missing functions. Indeed, there is some evidence for roles of cellular topoisomerases IIα and IIβ (37), DNA ligase I (8) and DNA damage response proteins, the single-stranded DNA binding protein RPA, and the DNA clamp protein PCNA (38). In addition, BAF1 acts as a negative regulator of VACV genome replication (39) and DNA-PK as a sensor of cytoplasmic DNA (40). Although some of the host proteins have been shown to localize in viral-factory areas of the cytoplasm, none has yet been demonstrated to associate directly with the VACV genome in vivo.
There are also open questions regarding the proteins required for transcription of the VACV genome. The virus-encoded proteins include a multisubunit DNA-dependent RNA polymerase (41, 42), factors specific for the early intermediate and late stages of transcription (43–45), type 1 topoisomerase (46–48), an elongation factor (49), capping and methylating enzymes (50, 51), poly(A) polymerase (52, 53), and transcription termination factors (54–56). Although these viral proteins seem sufficient for transcription, there is evidence, mainly from in vitro experiments, for roles of host proteins, including cellular heterogeneous nuclear ribonucleoproteins (57), G3BP and caprin I (58), TATA binding protein (59), and YY1 (60).
To address gaps in knowledge of the VACV DNA replication and transcription machinery, we adapted recently developed click chemistry methods for visualizing newly synthesized chromosomal DNA (61, 62) and for isolating and identifying protein components of nuclear eukaryotic (63–65) and herpesvirus (66, 67) replisomes. The method is based on the in vivo incorporation of ethynyl deoxynucleoside analogs, such as 5-ethynyl-2′-deoxyuridine (EdU), into replicating DNA. The principal advantage of this approach is the availability of the ethynyl group for the click chemistry reaction, with a fluorescent-azide conjugate for visualization by microscopy or biotin-azide for affinity purification of DNA. The DNA-associated proteins can be covalently but reversibly cross-linked to the DNA in vivo with formaldehyde and then copurified with biotin-labeled DNA under stringent conditions to minimize nonspecific protein interactions, a procedure known as iPOND (Isolation of proteins on nascent DNA). Here, we identify viral and cellular proteins that associate with nascent DNA following infection of human cells with wild-type VACV and a DNA ligase deletion mutant.
RESULTS
Localization of newly synthesized viral DNA by confocal and superresolution microscopy.
By labeling infected cells with [3H]thymidine, Cairns (2) provided autoradiographic evidence for VACV DNA replication in discrete areas of the cytoplasm. Subsequent in situ hybridization studies confirmed the presence of viral DNA in these factory areas (3). However, much remains to be learned about the formation and development of the factories. The incorporation of the clickable nucleoside EdU offers an advantageous way of localizing nascent DNA by fluorescence microscopy. In a recent study, both nuclei and cytoplasmic factories were labeled when cells were incubated with EdU for 24 h and then infected with VACV in the continuous presence of the nucleoside (68). We followed up those experiments first by visualizing EdU incorporated during 1-h intervals before and after infection of ∼80% confluent human A549 cells. Incorporation of EdU was determined by click chemistry with the fluorescent probe Alexa Fluor 488 azide; the cells were also stained for total DNA with DAPI (4′,6-diamidino-2-phenylindole) and with a monoclonal antibody (MAb) to the VACV-encoded I3 single-stranded DNA binding protein (26, 29, 69). In Fig. 1A, DAPI, EdU, and I3 staining are shown in the upper left, upper right, and lower left quadrants, respectively, with the merged image in the lower right quadrant. Fluorescence due to EdU was undetectable in controls when the click chemistry reaction was prevented by omission of its essential catalyzer, CuSO4 (not shown). In the absence of infection, bright EdU fluorescence was detected in the nuclei of about one-third of the unsynchronized cells, and the pattern of granular perinuclear and intranuclear labeling (Fig. 1A) was typical of mid-S phase (70, 71). Nuclear EdU fluorescence intensity decreased markedly after infection, although it was still detectable at a low level in some cells. Occasional cytoplasmic EdU foci were detected at 1 to 2 h, were more numerous at 2 to 3 h, and were abundant at 3 to 4 h and 4 to 5 h (Fig. 1A). The pulse-labeled cytoplasmic DNA colocalized with the I3 protein, which was detectable at 2 h and increased substantially at subsequent times (Fig. 1A). Incorporation of EdU into DNA depends on diffusion through the cell membrane and conversion to the triphosphate. To determine whether shorter pulse-labeling times were feasible, infected cells were incubated with EdU for 10, 20, or 30 min. Fluorescence increased greatly between the 10- and 20-min time intervals (Fig. 1B).
FIG 1.

Visualization of newly labeled nuclear and cytoplasmic DNA by confocal microscopy. (A) Approximately 80% confluent A549 cells were infected with VACV at a multiplicity of infection of 10 PFU per cell. Uninfected (un) cells and infected cells at the indicated times were incubated for 1 h with 10 μM EdU and then fixed, permeabilized, and reacted with Alexa Fluor 488 azide. The I3 single-strand binding protein was visualized by staining with a MAb, followed by an anti-mouse secondary antibody conjugated to Alexa Fluor 594. DAPI was used to stain total DNA. The cells were viewed with a confocal microscope, and maximum projections are shown. Each subpanel is divided into quadrants: upper left, DAPI (blue); upper right, EdU (green); lower left, I3 (red); and lower right, merge. (B) Cells were infected for 3.5 h and incubated for 10, 20, or 30 min with EdU as for panel A.
VACV factories are surrounded by endoplasmic reticulum (ER), and it has been suggested that the ER is specifically recruited to enable viral DNA synthesis in association with membranes (72, 73). We took advantage of the rapid incorporation of EdU to localize nascent DNA in relation to the ER. At 3.5 h after infection, the cells were incubated with EdU for 10 min, which, as discussed below, labels about 30,000 bp, which is equivalent to less than 20% of a full-length genome in this time interval. The cells were then fixed, permeabilized, and incubated with the fluorescent probe Alexa Fluor 488 azide. The cells were also stained with antibody to the ER-resident protein calnexin and with DAPI. In Fig. 2, DAPI, EdU, and calnexin staining are shown in the upper left, upper right, and lower left quadrants, respectively, with the merged image in the lower right quadrant. EdU staining appeared throughout the factory, whereas calnexin was peripheral and cytoplasmic. The intense calnexin staining at the periphery of the factories suggested to us that the membranes may be displaced from these sites by crowding due to macromolecular synthesis. The images did not suggest replication in close proximity to membranes, although small amounts of ER or membranes with little or no calnexin may not have been discerned within the factories.
FIG 2.

Localization of nascent viral DNA relative to the ER. A549 cells were infected with VACV at a multiplicity of infection of 10 PFU/cell. At 3.5 h after infection, the cells were incubated for 10 min with EdU and then fixed, permeabilized, and reacted with Alexa Fluor 488 azide. The cells were then incubated with rabbit polyclonal antibody to calnexin (Santa Cruz), followed by secondary antibody conjugated to Alexa Fluor 594 and DAPI. The image was made with Huygens deconvolution software and analyzed with Imaris 8.4.1.
Superresolution microscopy was employed for further analysis of the EdU- and I3-labeled bodies. The procedure was like that used for confocal microscopy, except that DAPI staining was omitted and images were collected with a stimulated emission depletion (STED) microscope. When the EdU was added for 10 min at 3.5 h after infection, the nascent DNA appeared as puncta that did not entirely colocalize with I3 (Fig. 3A), likely because some protein was associated with genomes that had been synthesized before the pulse. The smallest I3-labeled puncta may represent the association of the protein with uncoated DNA released from the core. The image in Fig. 3B shows an infected cell incubated for 1 h with EdU in which both nucleus and cytoplasm were labeled. The nuclear pattern was like that of uninfected cells but much lower in intensity, and I3 localized only with the intensely labeled cytoplasmic foci. Such low-level nuclear staining was detected in about 25% of infected cells at this time.
FIG 3.

Superresolution images of nascent DNA. A549 cells infected with VACV for 3.5 h were incubated for 10 (A) or 60 (B) min with EdU, fixed, permeabilized, and reacted with Alexa Fluor 488 azide. In addition, the cells were stained with a MAb to I3, followed by a secondary antibody conjugated to Alexa Fluor 594. The images were made using Huygens deconvolution software and analyzed with Imaris 8.4.1. Scale bars are shown. A higher-magnification image is shown in the inset.
Localization of newly synthesized DNA by electron microscopy.
To our knowledge, EdU labeling and click chemistry have not previously been employed to visualize DNA by electron microscopy. We modified the procedure used for light microscopy by substituting biotin-azide, followed by streptavidin–6-nm gold for the fluor-azide. Transmission electron microscopy of thin sections revealed that the streptavidin-gold was distributed throughout the viral factory, which was recognized at this early time by the juxtanuclear location, paucity of cellular organelles, relatively homogeneous electron density, occasional crescent structures (not shown), and membranes at the periphery (Fig. 4A). There was virtually no background of streptavidin-gold when biotin-azide was omitted (Fig. 4B). A similar labeling pattern was obtained with an alternative procedure in which the EdU-labeled cells were fixed, permeabilized with 0.05% saponin, and incubated with biotin-azide, followed by cryosectioning and then streptavidin–10-nm gold (Fig. 4C). Again, the nascent DNA was distributed throughout the factory. Absence of background streptavidin-gold is shown in Fig. 4D. In a parallel experiment, protein disulfide isomerase was localized with specific antibody and protein A-gold mainly in the membranes surrounding the virus factory (data not shown).
FIG 4.

Transmission electron microscopy of EdU-labeled cells. (A) A549 cells were infected with VACV and at 3.5 h were incubated with EdU for 30 min. The cells were fixed and permeabilized with 0.1% Triton X-100 and reacted with biotin-azide, followed by streptavidin–6-nm gold. (B) Same as panel A, except that biotin-azide was omitted as a control. (C) A549 cells were infected and labeled with EdU as for panel A. The cells were then permeabilized with 0.05% saponin and reacted with biotin-azide. The cells were cryosectioned and incubated with streptavidin–10-nm gold. (D) Same as panel C, except that biotin-azide was omitted as a control. Nu, nucleus; Go, Golgi apparatus; Mi, mitochondria.
Kinetics of VACV DNA synthesis in the absence and presence of EdU.
A detailed analysis of the kinetics of VACV genome synthesis was carried out by Joklik and Becker (74). In their study, the incorporation of [14C]deoxyribosylthymine (dT) into cytoplasmic DNA was detected at 1.5 h, was maximal between 2 and 2.5 h, and decreased appreciably at 4 h in a suspension of HeLa cell cultures. Although the rate of DNA synthesis was not calculated directly, they estimated that it was up to 3 times that of nuclear DNA in uninfected HeLa cells. From the data of Painter and Schaefer and of Collins (75, 76), the rate of replication of a single replicon of a suspension of HeLa cells corresponds to 60 bp/s. Based on this and the data of Joklik and Becker (74), VACV replication was estimated to be about 180 bp/s. Using confluent A549 cell monolayers, we detected by droplet digital PCR (ddPCR) VACV DNA synthesis that was maximal between 4 and 5 h (Fig. 5). As described by Dembowski et al. (67), we calculated a rate of 86 bp/s from 3.5 to 4 h and 130 bp/s from 4 to 5 h, assuming a single replicon per genome. The estimated rate of an individual polymerase would be lower if there were multiple origins. From these calculations, it would take 23 to 35 min to replicate a full-length genome. We also used ddPCR to determine the number of actin genes in the DNA sample. If we assume that A549 cells are at least diploid for the actin gene, then approximately 500 VACV genomes per cell accumulated by 6 h (Fig. 5). To determine the effect of EdU on ongoing viral DNA synthesis, the nucleoside was added at a concentration of 10 μM at 3.5 h after infection. Based on ddPCR data, we calculated a diminished rate of 52 bp/s from 3.5 h to 4 h and 95 bp/s from 4 to 5 h.
FIG 5.

Kinetics of VACV DNA synthesis in the absence (−) and presence (+) of EdU. A549 cells were infected with VACV for 6 h in duplicate. At 3.5 h, EdU (10 μM) was added to one portion of the cells. At the time points shown, the cells were harvested, and the amounts of VACV DNA and host actin were determined by ddPCR.
Adaptation of iPOND for isolation of VACV DNA with cross-linked proteins.
The basic iPOND protocol consists of labeling cells with EdU, cross-linking DNA and proteins by mild formaldehyde treatment, permeabilizing the cells, incubating with biotin-azide, shearing the DNA into small fragments, and isolating the nascent DNA-protein complexes on streptavidin beads. Our modified protocol developed for analysis of proteins associated with VACV DNA is shown in Fig. 6 and described below. Labeling with EdU was performed for 20 min at 3.5 h after infection, since confocal microscopy had shown that EdU was predominantly incorporated into DNA located in cytoplasmic viral factories at that time. After cross-linking the intact cells with formaldehyde, the cells were lysed with 0.5% Triton X-100 and divided into two equal parts to provide an experimental sample (click) that was incubated with biotin-azide and a control sample (nonclick) that was treated identically except for omission of biotin-azide. Because of the cytoplasmic location of VACV DNA, low-speed centrifugation is routinely used to enrich viral DNA in the supernatant fraction of lysates, although some also pellets with nuclei (74). Under the conditions used for iPOND, the bulk of viral DNA pelleted with nuclei at 800 × g, presumably because of the formaldehyde cross-linking. We therefore discarded the low-speed supernatant and incubated the pellet with 1% SDS at room temperature. The latter was separated by centrifugation at 8,000 × g into an insoluble fraction containing the bulk of the chromatin and a soluble fraction. The DNA complexes were sheared by sonication so that the DNA was approximately 100 to 300 bp, as determined by gel electrophoresis after deproteinization. The biotin-linked DNA with associated proteins was captured on streptavidin-agarose beads in the presence of SDS. Following washes with SDS at room temperature, the proteins were eluted by boiling the beads in 4% SDS buffer to reverse the heat-labile formaldehyde cross-links.
FIG 6.

iPOND protocol. Confluent A549 cells were infected with VACV, incubated with EdU, and reacted with biotin-azide, and DNA-protein complexes were isolated as depicted. TX-100, Triton X-100; RT, room temperature.
The amounts of experimental (click) and control (nonclick) viral DNA bound to streptavidin from the various fractions were determined by ddPCR (Table 1). Only small amounts of viral DNA were in the 800 × g supernatant fraction, up to 75% was in the SDS 8,000 × g supernatant fraction, and the remainder was in the pellet. Importantly, nearly 30-fold more viral DNA was associated with the streptavidin-agarose beads than with the controls when click chemistry was carried out, indicating selective isolation of nascent DNA. We further analyzed the 1% SDS supernatant (soluble) and pellet (insoluble) fractions. The DNA fragment sizes after sonication were similar (Fig. 7A). Western blots showed that more of the single-stranded DNA binding protein I3 was associated with DNA from the SDS-soluble fraction than with the insoluble fraction and that there was none in the no-click controls (Fig. 7B). I3 migrated as a 30-kDa monomer and a 60-kDa dimer, evidently due to incomplete reversal of the formaldehyde cross-linking. The self-association of I3 was previously shown using the yeast two-hybrid system (77) and by in vitro experiments (29). Taking the above-described findings into account, we focused on the SDS-soluble fractions for mass spectrometry analysis.
TABLE 1.
Recovery of nascent VACV DNA
| Cell fractiona | Relative amount of VACV DNAb |
||
|---|---|---|---|
| Click− | Click+ | Click+/click− | |
| 800 × g supt | 0.1 | 1.4 | 14 |
| 8,000 × g supt | 3.6 | 100 | 28 |
| 8,000 × g pellet | 1.2 | 32.2 | 27 |
Supt, supernatant.
Click+, with biotin-azide; click−, without biotin-azide.
FIG 7.

Detection of I3 by Western blotting of proteins associated with affinity-purified EdU-biotin complexes. (A) Gel electrophoresis of deproteinized DNA from the 1% SDS-insoluble and -soluble fractions as shown in Fig. 6. (B) Proteins eluted from streptavidin beads were resolved by SDS-PAGE, electroblotted onto a nitrocellulose membrane, probed with a MAb to the VACV I3 protein, and detected by chemiluminescence. The control (−click) and experimental (+click) samples are indicated. The masses of marker proteins are shown on the right, with the position of monomeric I3 indicated by the arrow. The band of about 62 kDa is presumed to be an I3 dimer.
Analysis of viral proteins associated with nascent DNA by mass spectrometry.
Confluent A549 cells were infected either with wild-type VACV or with an A50 DNA ligase deletion mutant, and nascent DNA was labeled with EdU for 20 min starting at 3.5 h, corresponding to peak viral DNA replication, as well as active early and intermediate viral protein synthesis. At the determined replication rate in the presence of EdU, this would result in synthesis of about 62,400 bp or about 1/3 of the VACV genome. Following the protocol outlined in Fig. 6, proteins eluted from streptavidin-agarose were subjected to electrophoresis on 2-cm-long SDS-polyacrylamide gels. Peptides produced by tryptic digestion were analyzed by label-free nano-liquid chromatography-tandem mass spectrometry (nano-LC–MS-MS), in some experiments with the Velos mass spectrometer (MS) and in others with the Fusion MS, which provided a greater depth of peptide identification due to its speed. Altogether, there were 10 independent experiments in which click and nonclick controls were analyzed. We required at least two peptides for positive identification of a protein. The TOP3 method (78), in which the extracting ion current (XIC) peak areas of the three most abundant peptides (or two when only two peptides were detected) were averaged and used for protein quantification, was employed. A value of 5 × 104 was used as the threshold for further analysis. For significance, the protein had to be absent from the nonclick control or the abundance of the protein in the experimental (click) sample had to be at least 8-fold greater than that in the nonclick control sample.
A total of 30 viral proteins were identified using the above-mentioned criteria in at least two independent experiments (Table 2). Peptides were undetected in the control samples for 26 of these proteins, and the abundances of the peptides for the other 4 proteins were at least 8-fold higher in the experimental samples than in the controls (see Table S1 in the supplemental material). Seven (H5, I3, A50, A20, E9, D5, and D4) of the proteins were known to be required for DNA replication; one (G5) for homologous recombination, double-strand break repair, and full-size genome formation; one (B1) for phosphorylating the BAF1 DNA binding protein; and one (I1) for telomere binding and morphogenesis. The genes encoding these proteins all have early promoters except for I1, which has an intermediate promoter. Each of these proteins was detected in both analyses with the Fusion MS. Except for D4, they were also detected in one or both analyses with the Velos MS. The concatemer resolution protein (A22), which is not required for DNA synthesis, was undetected by either the Fusion or the Velos MS. No protein already known to be required for VACV DNA synthesis was missing, indicating good sensitivity.
TABLE 2.
VACV proteins associated with nascent DNA
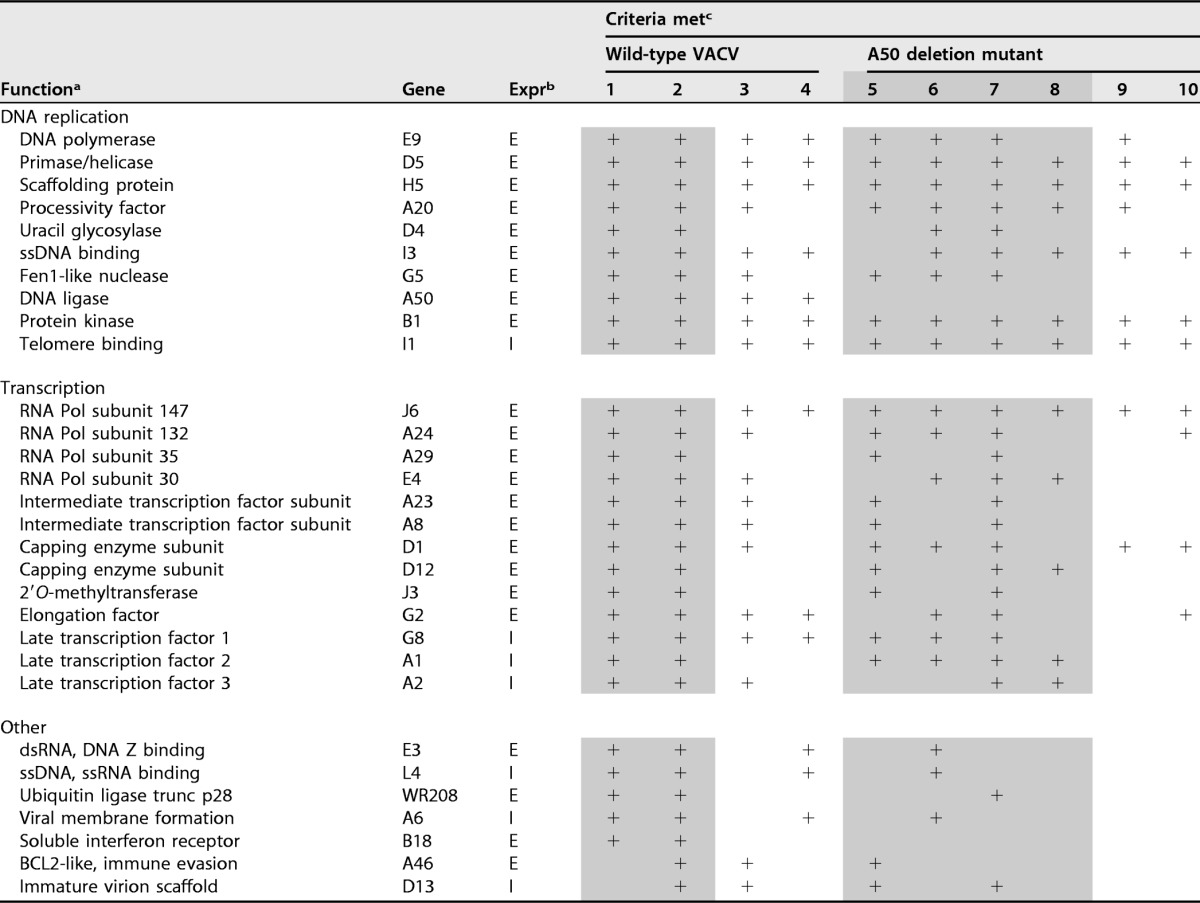
aAbbreviations: trunc, truncated; ssDNA, single-stranded DNA; dsRNA, double-stranded RNA.
bExpr, expression; E, early, or I, intermediate.
cThe numbers refer to independent experiments. Shading indicates experiments were performed with Fusion MS, and lack of shading indicates experiments were performed with Velos MS. The plus signs indicate that the inclusion criteria were met.
Thirteen of the identified proteins are involved in transcription, including the four largest of the eight DNA-dependent RNA polymerase subunits (J6, A24, A29, and E4), the intermediate transcription factor complex (A23 and A8), the late transcription factor complex (G8, A1, and A2), the transcription elongation factor (G2), the two capping enzyme subunits (D1 and D12), and the cap 2′O-methyltransferase (J3). These proteins were detected in at least four experiments but not always with the Velos MS, evidently because of the lower depth of peptide identification (Table 2). The genes encoding these proteins all have early promoters, except for the three late transcription factors, which have intermediate promoters. The A18 protein, a putative helicase with a role in the release of RNA from transcription complexes (79, 80), was detected, but the peptide abundance was below the threshold that we chose for significance. A transient association with the transcription complex, as well as other technical factors, could explain this result. The absence of the poly(A) polymerase was not unexpected, since it interacts with RNA posttranscriptionally. Similarly, the proteins expressed late in infection and required specifically for early transcription within virus cores, including the two subunits of the early transcription factor, RAP94, NPH-I, NPH-II, and the type I topoisomerase, were not associated with nascent DNA, which may occur at later times in concert with genome packaging.
The relative abundances of the individual replication and transcription proteins on nascent viral DNA are plotted in Fig. 8. The replication proteins, except G5 and D4, were among the 10 most abundant proteins, whereas the transcription proteins, except for the largest RNA polymerase subunit J6 and the late transcription factor G8, were lower in abundance. H5, I1, and I3 were consistently the highest-abundance proteins, consistent with noncatalytic roles. The ratio of replication to transcription proteins would likely be affected by the duration of EdU labeling.
FIG 8.

Relative abundances of viral and host proteins. The average XIC peptide areas for each protein were determined from experiment 1 in Tables 2 and 3. Blue, viral DNA replication proteins; yellow, viral transcription proteins; green, host proteins. The diameters of the circles are proportional to the number of experiments with wild-type virus in which the proteins met the inclusion criteria.
Of the 30 viral proteins that met the cutoff for significance, only 7 were not in the DNA replication or transcription functional category (Table 2). Two such proteins, E3 and L4, have DNA binding properties, possibly accounting for their isolation with DNA. However, the known information regarding the other five proteins does not provide a basis for such an association and should be considered nonspecific until demonstrated otherwise.
We also carried out iPOND using cells infected with a VACV DNA ligase deletion mutant that has been shown to require cellular DNA ligase 1 for replication. Viral DNA replication was slightly reduced in the cells infected with the mutant virus compared to the wild type, and the experiments were repeated several times, initially with the Velos MS and then with the Fusion MS (Table 2) for greater depth of analysis. In one Fusion MS experiment (number 7), we identified all the viral DNA replication and transcription proteins that were found in infection with wild-type VACV, except, of course, A50, and large subsets of those proteins were found in the other experiments (Table 2). Therefore, the absence of the viral ligase did not impact the association of other viral proteins with the nascent DNA.
Analysis of host proteins recovered with nascent DNA by mass spectrometry.
No experiments to identify host proteins associated with nascent VACV DNA have been reported previously. Two candidates, however, were topoisomerase IIα (TOP2A) and topoisomerase IIβ (ΤΟP2B), which are recruited to DNA factories, bind to the VACV DNA ligase, and are responsible for the sensitivity of VACV DNA synthesis to etoposide (37). We considered that the detection of these proteins in association with nascent DNA from cells infected with wild-type VACV but not from cells infected with a VACV DNA ligase deletion mutant would further validate the sensitivity and specificity of the analysis. Seventeen proteins were identified from wild-type VACV infection using the Velos MS. These proteins were undetected in 16 of the 17 controls, and the ratio of experimental to control samples was 8 to 1 (see Table S2 in the supplemental material). Ten of the 17 proteins belonged to the Gene Ontology (GO) categories of chromatin organization, chromatin binding, and chromosome, which translated into significant enrichment for each of these categories (P values of 0.009, 0.000149, and 000299, respectively). In addition, 3 of the 10 proteins (Top2A, Top2B, and PCNA) also belong to the DNA-dependent DNA replication category, which also corresponds to significant enrichment (P = 0.03). A much larger number of host proteins were detected with the Fusion MS (see Table S3 in the supplemental material). The greater depth of analysis revealed mostly contaminants, as neither chromatin organization, DNA replication, nor DNA repair terms were enriched to a statistically significant extent. To reveal proteins that could be involved in DNA replication or repair, we manually removed ribosomal proteins and ribonucleoproteins, heat shock proteins, histones, actins, tubulins, proteins involved in ER trafficking, translation elongation factors, tRNA ligases, and mitochondrial proteins, even though they met the inclusion criteria. Of the remaining proteins, 10 were in the GO category chromosome organization and included those detected with the Velos MS (Table 3). From the combined Velos and Fusion MS list, 5 proteins were in the GO category DNA replication and a partially overlapping group of 5 were in the DNA repair category (Table 3). Top2A and -2B were identified in both the Velos and Fusion MS analyses of proteins from wild-type-virus-infected cells in the abundance range of viral replication proteins (Fig. 8), but not from cells infected with the VACV DNA ligase mutant. In contrast PCNA, which was in lower abundance than TOP2A or -2B (Fig. 8), was detected in both wild-type and mutant virus infections and was absent from controls (see Table S2 in the supplemental material). The other candidate DNA replication/repair proteins, HMGA1, TOP1, PARP1, and RUVBL2, were less consistently found (Table 3).
TABLE 3.
Host proteins associated with nascent DNA
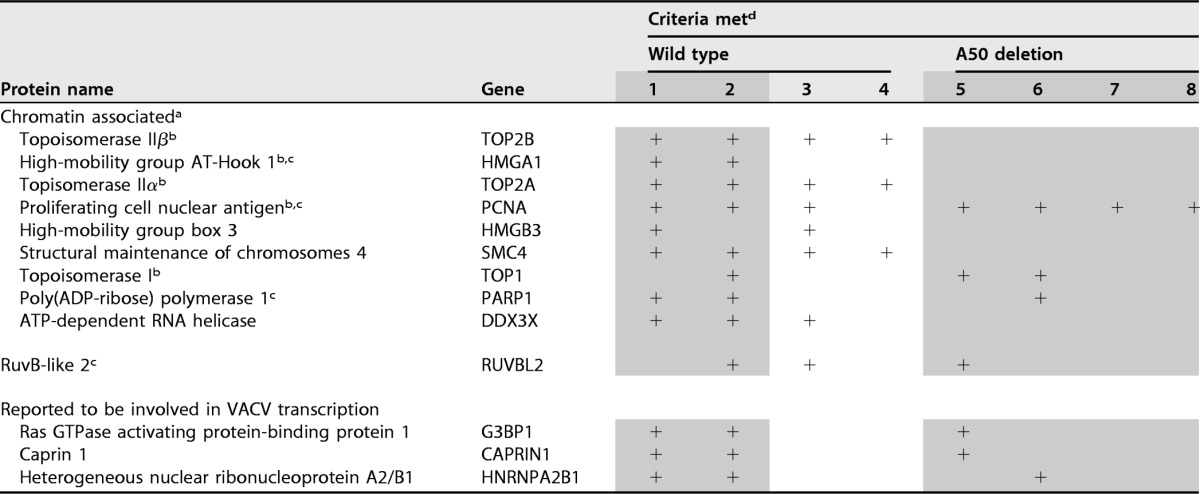
aListed in order of abundance for the first six proteins in experiment 1.
bGO term DNA replication.
cGO term DNA repair.
dThe numbers refer to independent experiments. Shading indicates experiments were performed with Fusion MS, and lack of shading indicates experiments were performed with Velos MS. The plus signs indicate that the inclusion criteria were met.
Several host proteins were suggested to have roles in intermediate and late transcription based on in vitro studies (57–60). Of those, G3BP1 and CAPRIN1 (58) and HNRPA2B1 (57) met the inclusion criteria in three experiments (Table 3).
DISCUSSION
We used EdU labeling and click chemistry to address several aspects of poxvirus DNA replication, including the inhibition of nuclear DNA synthesis, the intracellular localization of nascent VACV DNA, and the identification of viral and cellular proteins associated with nascent DNA. First, we confirmed that the thymidine analog EdU was rapidly incorporated into VACV DNA and had a modest effect on synthesis at the concentration employed. The rate of VACV DNA synthesis, determined by ddPCR, was approximately 86 to 130 bp/s assuming one replisome per genome, which was several times faster than that calculated for herpes simplex virus (67). At that rate, it would take 25 to 35 min for a DNA polymerase to synthesize a complete VACV genome. At 6 h, there were approximately 500 genomes per A549 cell, consistent with exponential replication.
EdU labeling and attachment of a fluor-azide by click chemistry coupled with fluorescence confocal and superresolution microscopy provided an advantageous way to study the intracellular location of nascent VACV DNA compared to other methods, such as [3H]thymidine and 5-bromouridine labeling. Prior to infection, about 30% of the unsynchronized cells displayed intense nuclear labeling, which decreased markedly during the first few hours of infection. The decreased labeling was consistent with previous reports of inhibition of host DNA synthesis by VACV (74, 81). Further studies employing EdU may be useful to determine the parameters necessary for this inhibition. Regardless of the mechanism, the decrease in nuclear labeling with EdU facilitated subsequent iPOND analysis. Labeling times with EdU as short as 10 min, which is much less than the time to complete a genome, were sufficient for detection by fluorescence microscopy. Consistent with previous studies (29, 82), the I3 single-stranded-DNA binding protein colocalized with pulse-labeled DNA. However, the superresolution of STED microscopy revealed that the colocalization was incomplete, likely because I3 was also associated with genomes that were replicated prior to EdU labeling.
Virus factories are surrounded by ER that was suggested to be actively recruited (72) or, as suggested here, passively pushed out from the interior, along with other cellular organelles, as the factory expands due to macromolecular synthesis. To determine the spatial relationship between the nascent viral DNA and ER, infected cells were pulse-labeled for 10 min with EdU, followed by attachment of fluor-azide, and stained with an antibody to calnexin, a resident ER membrane protein. There was intense ER staining around the factory, whereas the nascent DNA was distributed throughout the interior. We also adapted EdU labeling and reaction with biotin-azide for transmission electron microscopy. With labeling times of 30 min, streptavidin-gold spheres, which attach to biotin, were distributed throughout the factory rather than at the periphery, where most of the ER was located. With these techniques, we did not find evidence that VACV DNA synthesis occurs in close association with recognizable ER membranes.
The main purpose of the study was to use EdU labeling and click chemistry for isolating nascent DNA with cross-linked proteins. By labeling cells with EdU at 3.5 h after infection, we took advantage of the dramatic decrease in EdU incorporation into nuclear DNA to preferentially label viral DNA. With some modifications that further enriched viral DNA, the protocol closely followed that previously used (63–65). We chose iPOND over the alternative AniPOND procedure (83) because formaldehyde cross-linking allows stringent washing of the beads with SDS. Cross-linking using formaldehyde is particularly useful for studying protein-DNA interactions because of its small size, linkage of groups that are 2 Å apart, ability to penetrate the cell membrane, and preferential reactivity with accessible lysines that are frequently in close contact with nucleic acids because of their charge (84). Formaldehyde also cross-links proteins to each other if reactive groups are sufficiently close.
iPOND demonstrated the association of the complete set of viral replication proteins with nascent DNA. The seven proteins (H5, I3, A50, A20, E9, D5, and D4) previously shown by genetic and biochemical methods to be necessary for DNA synthesis (14, 18) were all found to be physically associated with nascent DNA. In addition, the Fen1-like nuclease G5, which is involved in homologous recombination, double-strand break repair, and full-size genome formation (34); the I1 DNA telomere-binding protein, which is required for morphogenesis rather than DNA replication (85, 86); and the B1 kinase, which phosphorylates cellular BAF1, thereby preventing the latter from binding to VACV DNA and inhibiting replication (33), were also recovered with the EdU-labeled DNA. Protein-protein interactions, in addition to protein-DNA interactions, may contribute to the association of these proteins with nascent DNA. For example, D4 can bind DNA (87, 88) and, together with A20 (which has not been shown to bind DNA) and E9, forms a holoenzyme complex (23). H5 has affinity for DNA and interacts with both A20 (77) and the B1 protein kinase (77, 89). The three most abundant viral proteins detected by iPOND were H5, I1, and I3. This correlates with their direct DNA binding properties, as well as the amounts of their mRNAs at 4 h after infection (90). Since I1 is a virion protein (85), some may have been released from the core, though most was probably expressed from a strong intermediate promoter (91). Unexpectedly, the least abundant viral DNA replication protein was D4, even though it can bind directly to DNA and form a 1:1:1 complex with E9 and A20 (23). However, there are many factors that contribute to the amount of peptide detected by mass spectrometry.
We also identified the viral proteins known to be involved in intermediate and late transcription on nascent DNA, indicating that the pool of replicating DNA was being transcribed. A similar observation was recently made with herpesvirus (67). In contrast to herpesvirus, VACV encodes its own DNA-dependent RNA polymerase, which consists of eight subunits. The two large subunits of Pol II interact with DNA and RNA, and the homologous VACV subunits RPO147 (J6) and RPO132 (A24) were recovered with nascent DNA. The next two subunits in size were lower in abundance, and the smaller ones were not detected. The finding of the intermediate (A8 and A23) and late (A1, A2, and G8) transcription factor complexes, which are expressed by early and intermediate genes, respectively, was interesting, because they have not been shown to directly interact with DNA or the RNA polymerase. The two-subunit (D1 and D12) capping enzyme, which has been shown to associate with the RNA polymerase (92), and the cap 2′O-methyltransferase (J3) were also recovered. In addition to its catalytic role in methylation, J3 is also a noncatalytic subunit of the poly(A) polymerase (93) and a positive transcription elongation factor (49). The absence of the large catalytic subunit of poly(A) polymerase (E1) from the DNA-associated proteins is consistent with its posttranscriptional activity and association with mRNA rather than DNA (94). H5, which was discussed above with regard to its role in DNA replication, is also involved in transcription (95–98) and interacts with G2, another positive transcription elongation factor associated with nascent DNA (99, 100).
Relatively few viral proteins that did not fall into the known DNA replication or transcription group were found, suggesting that additional ones may not exist. One of the proteins in this “other” category was the E3 double-stranded RNA binding protein, which also has a Z-DNA binding domain (101) that could account for its association with nascent DNA. E3 is a host defense protein that is not required for either DNA replication or transcription. L4, a DNA binding protein thought to be the major component of the nucleocapsid (102), was also detected by mass spectrometry. The few remaining proteins could be contaminants or hitchhikers, as they have no obvious relation to replication or transcription.
An important reason for undertaking iPOND experiments was to search in an unbiased fashion for host proteins that could be involved in poxvirus DNA replication. The two important concerns were sensitivity and nonspecific binding (103). The recovery of TOP2A and TOP2B associated with nascent DNA from cells infected with wild-type virus but not from cells infected with the VACV DNA ligase mutant supports findings from the Evans laboratory (37). Nevertheless, the role that the type II topoisomerases play in VACV DNA replication remains uncertain, as the DNA ligase deletion mutant replicated even though topoisomerase II was not found associated with the VACV replisome under these conditions. We found PCNA in multiple experiments; although present at a low abundance level, it was undetected in all the controls. PCNA is involved in eukaryotic DNA replication by increasing the processivity of the DNA polymerase during elongation of the leading strand. However, the VACV proteins A20 and D4 form a complex with the VACV DNA polymerase to provide a similar function (18). Nevertheless, a possible role for PCNA in replication of VACV was suggested indirectly by a report that two human PCNA-associated proteins are inhibitors of a host range mutant (104). To ascertain whether to pursue this lead in the future, we investigated the effect of the specific PCNA inhibitor T2AA (105). When added either before or after adsorption of VACV, T2AA inhibited VACV DNA replication in a concentration-dependent manner (data not shown). Very recently, Postigo and coworkers (38) reported the inhibition of VACV DNA synthesis by T2AA, as well as by small interfering RNAs (siRNAs) specific for PCNA, emphasizing the importance of further investigations into the role of PCNA in VACV replication.
Several additional host DNA replication/repair proteins (HMGA1, TOP1, PARP1, and RUVBL2) were identified with average peptide abundances above that of controls in several independent experiments. Although one could postulate functions for some of these proteins, such as DNA unwinding for TOP1, there is no evidence as yet that they have essential roles in viral DNA replication. The DEAD box helicase DDX3X, which has been identified as a target of the VACV K7 protein and an activator of IRF3 (106, 107), was detected with nascent DNA, but the specificity of this is uncertain. On the other hand, we were surprised that cellular DNA ligase 1 was not detected by iPOND when cells were infected with the VACV A50 deletion mutant, as this host protein is required for viral DNA replication in the absence of the viral DNA ligase and localizes in cytoplasmic factories (8). DNA ligase 1 is cell cycle dependent, and synchronization of the cells might improve recovery. The DNA damage response proteins (ATR, CHK1, Rhino, INTS7, and TOPBP1) and the DNA replication protein RPA, which have been reported to be involved in VACV DNA replication based on inhibitor, cytoplasmic localization, and protein interaction studies (38), were not detected in our experiments even below cutoff levels. The proposal that cellular RPA rather than I3 is the replicative single-stranded-DNA binding protein associated with the VACV replisome (38) was not affirmed by our finding of large amounts of I3 associated with nascent VACV DNA and failure to detect any of the RPA subunits.
We also specifically looked for the association of several host proteins that were reported to have roles in VACV transcription based on in vitro studies. G3BP1 and CAPRIN1, which form a heterodimer, were isolated from cell extracts in a search for factors necessary for transcription of intermediate genes (58) and were subsequently shown to colocalize with viral mRNA in virus factories (108). Both proteins were recovered in association with nascent DNA (Table 3). Similarly, the RNA binding proteins HNRPA2B1 and RBM3 were found to stimulate late gene transcription (57), and the former was associated with nascent DNA (Table 3). On the negative side, the TATA binding protein (59) and YY1 (60), which were reported to up- and downregulate intermediate and late VACV transcription by promoter binding, respectively, were not found.
In conclusion, we demonstrated the value of EdU and click chemistry to investigate the intracellular localization of nascent VACV DNA and to identify associated proteins. Multiple puncta of pulse-labeled DNA visualized by confocal, superresolution, and transmission microscopy were present throughout the interior of cytoplasmic factories, which were surrounded by ER. This first implementation of iPOND to study VACV provided strong support for the major roles of viral proteins in DNA replication and transcription, suggested that transcription begins on nascent DNA, confirmed the association of cellular topoisomerase II with nascent DNA, and hinted at a possible role of PCNA. In future studies, we plan to use short pulse and chase protocols to characterize the proteins in the replication fork, to determine the kinetics of binding of transcription factors and core proteins to DNA, and to employ additional mutant viruses and drugs.
MATERIALS AND METHODS
Cells and viruses.
Standard procedures for preparation and maintenance of cells and propagation and titration of VACV were used (109). Human A549 cell monolayers were infected (or mock infected) with the WR strain of VACV (ATCC VR-1354) or its derivative, vΔA50gfp (8), which was propagated on BS-C-1 cells. As the titer of vΔA50gfp was usually lower than that of WR, it was concentrated by purification through a sucrose cushion (109).
Confocal and STED microscopy.
A549 cells plated on coverslips were infected with 10 PFU per cell of VACV and incubated for 1 h at room temperature for virus adsorption. After three washes with Eagle's minimum essential medium (EMEM), the incubation was continued at 37°C as indicated for each experiment, and EdU was added to a final concentration of 10 μM. The cells were then washed twice with EMEM and once with phosphate-buffered saline (PBS) with Mg2+ and Ca2+ and fixed with 3.7% formaldehyde in PBS for 15 min at room temperature. The cells were permeabilized with 0.1% Triton X-100 in PBS for 20 min at room temperature and subjected to click reaction with Alexa Fluor 488 azide according to the Click-it Plus EdU imaging kit protocol (Thermo Fisher). In control reactions, CuSO4 was omitted. The cells were incubated with anti-I3 mouse MAb (37) or anti-calnexin rabbit polyclonal antibody (sc-11397; Santa Cruz) in 10% fetal calf serum (FCS), followed by the corresponding secondary antibody conjugated to Alexa Fluor 594 and by DAPI staining, which was omitted for STED experiments. Coverslips were mounted on slides using ProLong Gold (Life Technologies). Images were collected on a Leica DMI6000 confocal microscope (Leica Microsystems) or a Leica TCS SP5 STED microscope. STED images were deconvoluted with Huygens deconvolution software and analyzed with Imaris 8.4.1 (Bitplane AG, Zurich).
Electron microscopy.
A549 cells were incubated with 10 PFU/cell of VACV for 1 h at room temperature for adsorption, washed three times with EMEM, and then incubated at 37°C. EdU (10 μM) was added at 3.5 h after infection for 30 min. For embedding in Epon, the cells were fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100. The cells were washed twice with PBS-3% bovine serum albumin (BSA) and reacted with biotin-azide as described for iPOND, except that it was done on cell monolayers rather than after releasing the cells by scraping. In controls, biotin-azide was omitted. The cells were washed with 3% BSA-PBS 3 times and with PBS twice and incubated with streptavidin–6-nm gold (with gelatin as a blocking agent) (Electron Microscopy Sciences, Hatfield, PA) for 1 h with rotation, washed three times with 3% BSA-PBS and twice with PBS, fixed again with 2% glutaraldehyde-0.1 M sodium cacodylate buffer for 1 h, washed extensively 0.1 M sodium cacodylate buffer, and left at 4°C overnight. The samples were postfixed with reduced osmium tetroxide, washed with sodium cacodylate buffer, dehydrated in a series of ethyl alcohol incubations, and then treated with propylene oxide. Samples were embedded in Embed 812 (Electron Microscopy Sciences), and sections were cut on a Leica EM UC7 ultramicrotome (Leica Microsystems Inc., Buffalo Grove, IL). The sections were stained with 7% uranyl acetate in 50% ethanol and then 0.01% lead citrate. The sections were reviewed and photographed on a Tecnai G2 Spirit transmission electron microscope (Thermo Fisher Scientific, FEI, Hillsboro, OR) fitted with a Gatan (Warrendale, PA) charge-coupled-device (CCD) camera.
For cryosectioning, infected cells were fixed with 4% paraformaldehyde-0.05% glutaraldehyde-0.1 M phosphate buffer for 30 min at room temperature, washed three times with PBS, permeabilized in PBS-0.05% saponin, washed with PBS, and subjected to click reaction with or without biotin-azide as for iPOND. The cell monolayers were washed extensively with PBS and left in PBS overnight at 4°C. Cells were scraped, pelleted, resuspended, and incubated in 10% gelatin at 37°C for 15 min. The cells were repelleted, solidified on ice, cut at 4°C into small cubes, infiltrated with 2.3 M sucrose in 0.1 M phosphate buffer, frozen on pins in liquid nitrogen, and cut into 80-nm cryosections on a Leica EM UC7 ultramicrotome. The cryosections were picked up on Formvar-coated grids, thawed, washed free of sucrose, and stained with streptavidin–10-nm gold.
ddPCR.
DNA for kinetic experiments was prepared from duplicate sets of 24 wells of A549 cells infected with 10 PFU/cell of VACV for 1 h at room temperature, washed three times with EMEM, and incubated for the indicated times at 37°C. At each time point, duplicate samples were collected by cell scraping, pelleted by centrifugation, and frozen at −80°C. DNA was extracted with a QIAamp DNA blood kit (Qiagen) and eluted in 200 μl of H2O. ddPCR was performed using a QX200 droplet digital PCR system (Bio-Rad) according to the manufacturer's protocols. Forward (F) and reverse (R) primers (F, 5′-GTCTCCGATACGAACGCTAAACTCTAG, and R, 5′-CATAAGAAAGTGAGTTACGAAGATATCATCGGTTC) were used for VACV DNA amplification; primers for the human actin beta gene, ACTB, were purchased from Bio-Rad (catalog number 10031258).
Preparation and isolation of DNA-protein complexes.
For each iPOND experiment, confluent A549 cells (∼8 × 107) were infected with 10 PFU/cell of VACV or vΔA50gfp. Virus adsorption was for 1 h at room temperature with rotation, and the cells were then washed 3 times with EMEM and incubated at 37°C for 3.5 h. EDU was added to a final concentration of 10 μM, and the cells were incubated for 20 min more. The cells were then washed 3 times with EMEM and incubated with 1% formaldehyde in PBS for 30 min at room temperature. Unreacted formaldehyde was quenched with 0.125 M glycine for 10 min at room temperature, and the cell monolayers were washed 3 times with PBS. Cells were collected by scraping, washed twice with PBS, and permeabilized with 0.5% Triton X-100 with protease inhibitor cocktail (Roche) in PBS for 15 min at 4°C. The cells were then washed twice with PBS–0.5% Triton X-100, suspended in PBS, and divided into two tubes for experimental and control samples. The click reaction components were added in the following order to the following final concentrations in 1 ml: biotin-azide, 10 mM; CuSO4, 20 mM; sodium l-ascorbate, 100 mM. For the control (−click) sample, DMSO was added instead of biotin-azide. Samples were incubated for 1 h at room temperature with rotation in the presence of protease inhibitors. The cells were washed with PBS to remove unincorporated biotin-azide and resuspended in 50 mM Tris, pH 8, 1% SDS. The cell extract was incubated with rotation for 30 min in the presence of protease inhibitors at room temperature and then centrifuged at 8,000 × g for 10 min. The supernatant was collected, sonicated with a Bioruptor sonicator (Diagenode Inc.) for 30 s on and 30 s off for 45 min, and centrifuged at 20,000 × g for 30 min. The supernatant was diluted 1:1 with PBS, preadsorbed on protein A agarose beads for 30 min, and loaded on streptavidin agarose beads (Novagen) at 4°C overnight with rotation. The beads were washed with 1% SDS-50 mM Tris, pH 8.0, 1 M NaCl, and proteins were eluted in 4% SDS gel loading buffer at 95°C for 25 min. The eluted proteins were separated by SDS-PAGE on 2-cm-long gels, which were cut into 4 slices, and the proteins were digested and analyzed by mass spectrometry.
Western blotting.
Proteins were analyzed on NuPAGE gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). The membranes were incubated with antibodies in 5% nonfat milk in PBS-0.1% Tween 20 for various times at room temperature or 4°C and then with secondary horseradish peroxidase-conjugated antibodies. The blot was developed with SuperSignal West Pico, Dura, or Femto chemiluminescent substrate (Pierce, Rockford, IL), depending on the luminescence intensity.
Mass spectrometry.
Identification of proteins was performed on reduced and alkylated, trypsin-digested samples prepared by standard mass spectrometry protocols. The supernatant and two washes (5% formic acid in 50% acetonitrile) of the gel digests were pooled and concentrated with a Speed Vac (Labconco, Kansas, MO) to dryness directly in 200 μl polypropylene autosampler vials (Sun Sri, Rockwood, TN). The recovered peptides were resuspended in 5 μl of solvent A (0.1% formic acid, 2% acetonitrile, and 97.9% water).
Mass spectrometry analysis was performed with either an LTQ-Velos Orbitrap (Thermo Fisher Scientific, West Palm Beach, FL) or an Orbitrap Fusion Tribrid with in-line chromatography without trap cleanup. For the Velos Orbitrap acquisitions, nano-LC–MS-MS was performed with a ProXeon Easy-nLC II multidimensional liquid chromatograph and a temperature-controlled dynamic nanospray ion source (ThermoFisher Scientific). Peptides were separated at 500 nl/min using Magic aQC reverse-phase medium (3-μm particle size; 75-μm inside diameter [i.d.]; 15-cm length) packed in a pulled-tip nanochromatography column (Precision Capillary Columns, San Clemente, CA). For the Orbitrap Fusion acquisitions, nano-LC–MS-MS was performed with a ProXeon Easy-nLC 1000 multidimensional liquid chromatograph and a temperature-controlled Nanospray Flex ion source (ThermoFisher Scientific). Peptides were separated at 200 nl/min using Reprosil-Pur C18 reverse-phase medium (3-μm particle size; 75-μm i.d.; 30-cm length) packed in a pulled-tip nanochromatography column (Precision Capillary Columns). In both acquisition protocols, the mobile phase consisted of a linear gradient prepared from solvent A and solvent B (0.1% formic acid, 2% water, and 97.9% acetonitrile) at room temperature. Computer-controlled data-dependent automated switching to MS-MS by Xcalibur software was used for data acquisition and provided the peptide sequence information.
Protein identification.
Data processing and data bank searching were performed with Peaks Studio 8 (Bioinformatics Solution Inc., Waterloo, ON, Canada). The data were searched against the human and vaccinia virus (Western Reserve) proteins deposited in the UniProt KB (June 2016) and the common Repository of Adventitious Proteins (http://theGPM.org). The peptides were filtered at a 0.5% false-discovery rate (FDR) calculated using a decoy sequence approach with a 2 peptide per protein minimum. The TOP3 method was used for quantification (78), with the XIC peptide areas determined using Peaks Studio 8.
Supplementary Material
ACKNOWLEDGMENTS
We thank Catherine Cotter for cells and virus propagation and titrations, Jeffery Americo for help with ddPCR, Juraj Kabat for help with confocal microscopy, Margery Smelkinson for acquiring STED images, and Alison McBride for deconvolution of STED images. David Evans generously provided the MAb to I3.
Support for research was provided by the Division of Intramural Research, NIAID, NIH.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01015-17.
REFERENCES
- 1.Baroudy BM, Venkatesan S, Moss B. 1982. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell 28:315–324. doi: 10.1016/0092-8674(82)90349-X. [DOI] [PubMed] [Google Scholar]
- 2.Cairns J. 1960. The initiation of vaccinia infection. Virology 11:603–623. doi: 10.1016/0042-6822(60)90103-3. [DOI] [PubMed] [Google Scholar]
- 3.Minnigan H, Moyer RW. 1985. Intracellular localization of rabbit poxvirus nucleic acid within infected cells as determined by in situ hybridization. J Virol 55:634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tattersall P, Ward DC. 1976. Rolling hairpin model for replication of parvovirus and linear chromosomal DNA. Nature 263:106–109. doi: 10.1038/263106a0. [DOI] [PubMed] [Google Scholar]
- 5.Moyer RW, Graves RL. 1981. The mechanism of cytoplasmic orthopoxvirus DNA replication. Cell 27:391–401. doi: 10.1016/0092-8674(81)90422-0. [DOI] [PubMed] [Google Scholar]
- 6.Baroudy BM, Venkatesan S, Moss B. 1983. Structure and replication of vaccinia virus telomeres. Cold Spring Harbor Symp Quant Biol 47:723–729. doi: 10.1101/SQB.1983.047.01.083. [DOI] [PubMed] [Google Scholar]
- 7.De Silva FS, Lewis W, Berglund P, Koonin EV, Moss B. 2007. Poxvirus DNA primase. Proc Natl Acad Sci U S A 104:18724–18729. doi: 10.1073/pnas.0709276104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paran N, De Silva FS, Senkevich TG, Moss B. 2009. Cellular DNA ligase I is recruited to cytoplasmic vaccinia virus factories and masks the role of the vaccinia ligase in viral DNA replication. Cell Host Microbe 6:563–569. doi: 10.1016/j.chom.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olgiati DD, Pogo BG, Dales S. 1976. Evidence for RNA linked to nascent DNA in HeLa cells. J Cell Biol 68:557–566. doi: 10.1083/jcb.68.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esteban M, Holowczak JA. 1977. Replication of vaccinia DNA in mouse L cells. I. In vivo DNA synthesis. Virology 78:57–75. [DOI] [PubMed] [Google Scholar]
- 11.Esteban M, Flores L, Holowczak JA. 1977. Model for vaccinia virus DNA replication. Virology 83:467–473. doi: 10.1016/0042-6822(77)90197-0. [DOI] [PubMed] [Google Scholar]
- 12.Senkevicha TG, Bruno D, Martens C, Porcella SF, Wolf YI, Moss B. 2015. Mapping vaccinia virus DNA replication origins at nucleotide level by deep sequencing. Proc Natl Acad Sci U S A 112:10908–10913. doi: 10.1073/pnas.1514809112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prescott DM, Kates J, Kirkpatrick JB. 1971. Replication of vaccinia virus DNA in enucleated L-cells. J Mol Biol 59:505–508. doi: 10.1016/0022-2836(71)90313-5. [DOI] [PubMed] [Google Scholar]
- 14.Moss B. 2013. Poxvirus DNA replication. Cold Spring Harb Perspect Biol 5:a010199. doi: 10.1101/cshperspect.a010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Challberg MD, Englund PT. 1979. Purification and properties of the deoxyribonucleic acid polymerase induced by vaccinia virus. J Biol Chem 254:7812–7819. [PubMed] [Google Scholar]
- 16.McDonald WF, Klemperer N, Traktman P. 1997. Characterization of a processive form of the vaccinia virus DNA polymerase. Virolgy 234:168–175. doi: 10.1006/viro.1997.8639. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton MD, Nuara AA, Gammon DB, Buller RM, Evans DH. 2007. Duplex strand joining reactions catalyzed by vaccinia virus DNA polymerase. Nucleic Acids Res 35:143–151. doi: 10.1093/nar/gkl1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czarnecki MW, Traktman P. 2017. The vaccinia virus DNA polymerase and its processivity factor. Virus Res 234:193–206. doi: 10.1016/j.virusres.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klemperer N, McDonald W, Boyle K, Unger B, Traktman P. 2001. The A20R protein is a stoichiometric component of the processive form of vaccinia virus DNA polymerase. J Virol 75:12298–12307. doi: 10.1128/JVI.75.24.12298-12307.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyle KA, Stanitsa ES, Greseth MD, Lindgren JK, Traktman P. 2011. Evaluation of the role of the vaccinia virus uracil DNA glycosylase and A20 proteins as intrinsic components of the DNA polymerase holoenzyme. J Biol Chem 286:24702–24713. doi: 10.1074/jbc.M111.222216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuart DT, Upton C, Higman MA, Niles EG, McFadden G. 1993. A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J Virol 67:2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Silva FS, Moss B. 2003. Vaccinia virus uracil DNA glycosylase has an essential role in DNA synthesis that is independent of its glycosylase activity: catalytic site mutations reduce virulence but not virus replication in cultured cells. J Virol 77:159–166. doi: 10.1128/JVI.77.1.159-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanitsa ES, Arps L, Traktman P. 2006. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J Biol Chem 281:3439–3451. doi: 10.1074/jbc.M511239200. [DOI] [PubMed] [Google Scholar]
- 24.Boyle KA, Greseth MD, Traktman P. 2015. Genetic confirmation that the H5 protein is required for vaccinia virus DNA replication. J Virol 89:6312–6327. doi: 10.1128/JVI.00445-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyle KA, Arps L, Traktman P. 2007. Biochemical and genetic analysis of the vaccinia virus D5 protein: multimerization-dependent ATPase activity is required to support viral DNA replication. J Virol 81:844–859. doi: 10.1128/JVI.02217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rochester SC, Traktman P. 1998. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J Virol 72:2917–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welsch S, Doglio L, Schleich S, Locker JK. 2003. The vaccinia virus I3L gene product is localized to a complex endoplasmic reticulum-associated structure that contains the viral parental DNA. J Virol 77:6014–6028. doi: 10.1128/JVI.77.10.6014-6028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gammon DB, Evans DH. 2009. The 3′-to-5′ exonuclease activity of vaccinia virus DNA polymerase is essential and plays a role in promoting virus genetic recombination. J Virol 83:4236–4250. doi: 10.1128/JVI.02255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greseth MD, Boyle KA, Bluma MS, Unger B, Wiebe MS, Soares-Martins JA, Wickramasekera NT, Wahlberg J, Traktman P. 2012. Molecular genetic and biochemical characterization of the vaccinia virus 13 protein, the replicative single-stranded DNA binding protein. J Virol 86:6197–6209. doi: 10.1128/JVI.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison ML, Desaulniers MA, Noyce RS, Evans DH. 2016. The acidic C-terminus of vaccinia virus I3 single-strand binding protein promotes proper assembly of DNA-protein complexes. Virology 489:212–222. doi: 10.1016/j.virol.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 31.Sekiguchi J, Shuman S. 1997. Domain structure of vaccinia DNA ligase. Nucleic Acids Res 25:727–734. doi: 10.1093/nar/25.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parks RJ, Winchcombe-Forhan C, DeLange AM, Xing X, Evans DH. 1998. DNA ligase gene disruptions can depress viral growth and replication in poxvirus-infected cells. Virus Res 56:135–147. doi: 10.1016/S0168-1702(98)00055-0. [DOI] [PubMed] [Google Scholar]
- 33.Wiebe MS, Traktman P. 2007. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe 1:187–197. doi: 10.1016/j.chom.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senkevich TG, Koonin EV, Moss B. 2009. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc Natl Acad Sci U S A 106:17921–17926. doi: 10.1073/pnas.0909529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia AD, Moss B. 2001. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J Virol 75:6460–6471. doi: 10.1128/JVI.75.14.6460-6471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia AD, Aravind L, Koonin EV, Moss B. 2000. Bacterial-type DNA Holliday junction resolvases in eukaryotic viruses. Proc Natl Acad Sci U S A 97:8926–8931. doi: 10.1073/pnas.150238697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin YC, Li J, Irwin CR, Jenkins H, DeLange L, Evans DH. 2008. Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J Virol 82:5922–5932. doi: 10.1128/JVI.02723-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Postigo A, Ramsden AE, Howell M, Way M. 2017. Cytoplasmic ATR activation promotes vaccinia virus genome replication. Cell Rep 19:1022–1032. doi: 10.1016/j.celrep.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nichols RJ, Wiebe MS, Traktman P. 2006. The vaccinia-related kinases phosphorylate the N′ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol Biol Cell 17:2451–2464. doi: 10.1091/mbc.E05-12-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. 2012. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baroudy BM, Moss B. 1980. Purification and characterization of a DNA-dependent RNA polymerase from vaccinia virions. J Biol Chem 255:4372–4380. [PubMed] [Google Scholar]
- 42.Ahn B-Y, Moss B. 1992. RNA polymerase-associated transcription specificity factor encoded by vaccinia virus. Proc Natl Acad Sci U S A 89:3536–3540. doi: 10.1073/pnas.89.8.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broyles SS, Yuen L, Shuman S, Moss B. 1988. Purification of a factor required for transcription of vaccinia virus early genes. J Biol Chem 263:10754–10760. [PubMed] [Google Scholar]
- 44.Sanz P, Moss B. 1999. Identification of a transcription factor, encoded by two vaccinia virus early genes, that regulates the intermediate stage of viral gene expression. Proc Natl Acad Sci U S A 96:2692–2697. doi: 10.1073/pnas.96.6.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keck JG, Baldick CJ, Moss B. 1990. Role of DNA replication in vaccinia virus gene expression: a naked template is required for transcription of three late transactivator genes. Cell 61:801–809. doi: 10.1016/0092-8674(90)90190-P. [DOI] [PubMed] [Google Scholar]
- 46.Shuman S, Moss B. 1987. Identification of a vaccinia virus gene encoding a type I DNA topoisomerase. Proc Natl Acad Sci U S A 84:7478–7482. doi: 10.1073/pnas.84.21.7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klemperer N, Traktman P. 1993. Biochemical analysis of mutant alleles of the vaccinia virus topoisomerase I carrying targeted substitutions in a highly conserved domain. J Biol Chem 268:15887–15899. [PubMed] [Google Scholar]
- 48.Da Fonseca F, Moss B. 2003. Poxvirus DNA topoisomerase knockout mutant exhibits decreased infectivity associated with reduced early transcription. Proc Natl Acad Sci U S A 100:11291–11296. doi: 10.1073/pnas.1534874100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Latner DR, Thompson JM, Gershon PD, Storrs C, Condit RC. 2002. The positive transcription elongation factor activity of the vaccinia virus J3 protein is independent from its (nucleoside-2 ′-O-) methyltransferase and poly(A) polymerase stimulatory functions. Virology 301:64–80. doi: 10.1006/viro.2002.1538. [DOI] [PubMed] [Google Scholar]
- 50.Martin SA, Paoletti E, Moss B. 1975. Purification of mRNA guanylyltransferase and mRNA (guanine 7-)methyltransferase from vaccinia virus. J Biol Chem 250:9322–9329. [PubMed] [Google Scholar]
- 51.Barbosa E, Moss B. 1978. mRNA (nucleoside-2′-)-methyltransferase from vaccinia virus. Purification and physical properties. J Biol Chem 253:7692–7697. [PubMed] [Google Scholar]
- 52.Moss B, Rosenblum EN, Paoletti E. 1973. Polyadenylate polymerase from vaccinia virions. Nat New Biol 245:59–63. [DOI] [PubMed] [Google Scholar]
- 53.Gershon PD. 1998. mRNA 3′ end formation by vaccinia virus: mechanism of action of a heterodimeric poly(A) polymerase. Semin Virol 8:343–350. doi: 10.1006/smvy.1997.0137. [DOI] [Google Scholar]
- 54.Shuman S, Broyles SS, Moss B. 1987. Purification and characterization of a transcription termination factor from vaccinia virions. J Biol Chem 262:12372–12380. [PubMed] [Google Scholar]
- 55.Christen LM, Sanders M, Wiler C, Niles EG. 1998. Vaccinia virus nucleoside triphosphate phosphohydrolase I is an essential viral early gene transcription termination factor. Virology 245:360–371. doi: 10.1006/viro.1998.9177. [DOI] [PubMed] [Google Scholar]
- 56.Deng L, Shuman S. 1998. Vaccinia NPH-I, a DExH-box ATPase, is the energy coupling factor for mRNA transcription termination. Genes Dev 12:538–546. doi: 10.1101/gad.12.4.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright CF, Oswald BW, Dellis S. 2001. Vaccinia virus late transcription is activated in vitro by cellular heterogeneous nuclear ribonucleoproteins. J Biol Chem 276:40680–40686. doi: 10.1074/jbc.M102399200. [DOI] [PubMed] [Google Scholar]
- 58.Katsafanas GC, Moss B. 2004. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem 279:52210–55217. doi: 10.1074/jbc.M411033200. [DOI] [PubMed] [Google Scholar]
- 59.Knutson BA, Liu X, Oh J, Broyles SS. 2006. Vaccinia virus intermediate and late promoter elements are targeted by the TATA-binding protein. J Virol 80:6784–6793. doi: 10.1128/JVI.02705-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knutson BA, Oh J, Broyles SS. 2009. Downregulation of vaccinia virus intermediate and late promoters by host transcription factor YY1. J Gen Virol 90:1592–1599. doi: 10.1099/vir.0.006924-0. [DOI] [PubMed] [Google Scholar]
- 61.Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zessin PJ, Finan K, Heilemann M. 2012. Super-resolution fluorescence imaging of chromosomal DNA. J Struct Biol 177:344–348. doi: 10.1016/j.jsb.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 63.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kliszczak AE, Rainey MD, Harhen B, Boisvert FM, Santocanale C. 2011. DNA mediated chromatin pull-down for the study of chromatin replication. Sci Rep 1:95. doi: 10.1038/srep00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Munoz J, Fernandez-Capetillo O. 2013. A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep 3:1105–1116. doi: 10.1016/j.celrep.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dembowski JA, DeLuca NA. 2015. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog 11:e1004939. doi: 10.1371/journal.ppat.1004939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dembowski JA, Dremel SE, DeLuca NA. 2017. Replication-coupled recruitment of viral and cellular factors to herpes simplex virus type 1 replication forks for the maintenance and expression of viral genomes. PLoS Pathog 13:e1006166. doi: 10.1371/journal.ppat.1006166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang IH, Suomalainen M, Andriasyan V, Kilcher S, Mercer J, Neef A, Luedtke NW, Greber UF. 2013. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 14:468–480. doi: 10.1016/j.chom.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Tseng M, Palaniyar N, Zhang WD, Evans DH. 1999. DNA binding and aggregation properties of the vaccinia virus I3L gene product. J Biol Chem 274:21637–21644. doi: 10.1074/jbc.274.31.21637. [DOI] [PubMed] [Google Scholar]
- 70.Nakayasu H, Berezney R. 1989. Mapping replicational sites in the eucaryotic cell nucleus. J Cell Biol 108:1–11. doi: 10.1083/jcb.108.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raulf A, Spahn CK, Zessin PJ, Finan K, Bernhardt S, Heckel A, Heilemann M. 2014. Click chemistry facilitates direct labelling and super-resolution imaging of nucleic acids and protein. RSC Adv 4:30462–30466. doi: 10.1039/C4RA01027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schramm B, Locker JK. 2005. Cytoplasmic organization of poxvirus DNA replication. Traffic 6:839–846. doi: 10.1111/j.1600-0854.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- 73.Tolonen N, Doglio L, Schleich S, Locker JK. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell 12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Joklik WK, Becker Y. 1964. The replication and coating of vaccinia DNA. J Mol Biol 10:452–474. doi: 10.1016/S0022-2836(64)80066-8. [DOI] [PubMed] [Google Scholar]
- 75.Painter RB, Schaefer AW. 1969. Rate of synthesis along replicons of different kinds of mammalian cells. J Mol Biol 45:467–479. doi: 10.1016/0022-2836(69)90306-4. [DOI] [PubMed] [Google Scholar]
- 76.Collins JM. 1978. Rates of DNA synthesis during the S-phase of HeLa cells. J Biol Chem 253:8570–8577. [PubMed] [Google Scholar]
- 77.McCraith S, Holtzman T, Moss B, Fields S. 2000. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc Natl Acad Sci U S A 97:4879–4884. doi: 10.1073/pnas.080078197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. 2006. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics 5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 79.Xiang Y, Simpson DA, Spiegel J, Zhou A, Silverman RH, Condit RC. 1998. The vaccinia virus A18R DNA helicase is a postreplicative negative transcription elongation factor. J Virol 72:7012–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lackner CA, Condit RC. 2000. Vaccinia virus gene A18R DNA helicase is a transcript release factor. J Biol Chem 275:1485–1494. doi: 10.1074/jbc.275.2.1485. [DOI] [PubMed] [Google Scholar]
- 81.Jungwirth C, Launer J. 1968. Effects of poxvirus infection on host cell deoxyribonucleic acid synthesis. Virology 2:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Domi A, Beaud G. 2000. The punctate sites of accumulation of vaccinia virus early proteins are precursors of sites of viral DNA synthesis. J Gen Virol 81:1231–1235. doi: 10.1099/0022-1317-81-5-1231. [DOI] [PubMed] [Google Scholar]
- 83.Leung KH, Abou El Hassan M, Bremner R. 2013. A rapid and efficient method to purify proteins at replication forks under native conditions. Biotechniques 55:204–206. doi: 10.2144/000114089. [DOI] [PubMed] [Google Scholar]
- 84.Hoffman EA, Frey BL, Smith LM, Auble DT. 2015. Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem 290:26404–26411. doi: 10.1074/jbc.R115.651679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klemperer N, Ward J, Evans E, Traktman P. 1997. The vaccinia virus I1 protein is essential for the assembly of mature virions. J Virol 71:9285–9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeMasi J, Du S, Lennon D, Traktman P. 2001. Vaccinia virus telomeres: interaction with the viral I1, I6, and K4 proteins. J Virol 75:10090–10105. doi: 10.1128/JVI.75.21.10090-10105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schormann N, Banerjee S, Ricciardi R, Chattopadhyay D. 2015. Binding of undamaged double stranded DNA to vaccinia virus uracil-DNA glycosylase. BMC Struct Biol 15:10. doi: 10.1186/s12900-015-0037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burmeister WP, Tarbouriech N, Fender P, Contesto-Richefeu C, Peyrefitte CN, Iseni F. 2015. Crystal structure of the vaccinia virus uracil-DNA glycosylase in complex with DNA. J Biol Chem 290:17923–17934. doi: 10.1074/jbc.M115.648352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beaud G, Beaud R, Leader DP. 1995. Vaccinia virus gene H5R encodes a protein that is phosphorylated by the multisubstrate vaccinia virus B1R protein kinase. J Virol 69:1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc Natl Acad Sci U S A 107:11513–11518. doi: 10.1073/pnas.1006594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 85:9899–9908. doi: 10.1128/JVI.05446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katsafanas GC, Moss B. 1999. Histidine codons appended to the gene encoding the RPO22 subunit of vaccinia virus RNA polymerase facilitate the isolation and purification of functional enzyme and associated proteins from virus-infected cells. Virology 258:469–479. doi: 10.1006/viro.1999.9744. [DOI] [PubMed] [Google Scholar]
- 93.Schnierle BS, Gershon PD, Moss B. 1992. Cap-specific mRNA (nucleoside-O2′-)-methyltransferase and poly(A) polymerase stimulatory activities of vaccinia virus are mediated by a single protein. Proc Natl Acad Sci U S A 89:2897–2901. doi: 10.1073/pnas.89.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gershon PD, Ahn BY, Garfield M, Moss B. 1991. Poly(A) polymerase and a dissociable polyadenylation stimulatory factor encoded by vaccinia virus. Cell 66:1269–1278. doi: 10.1016/0092-8674(91)90048-4. [DOI] [PubMed] [Google Scholar]
- 95.Kovacs GR, Vasilakis N, Moss B. 2001. Regulation of viral intermediate gene expression by the vaccinia virus B1 protein kinase. J Virol 75:4048–4055. doi: 10.1128/JVI.75.9.4048-4055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cresawn SG, Condit RC. 2007. A targeted approach to identification of vaccinia virus postreplicative transcription elongation factors: genetic evidence for a role of the H5R gene in vaccinia transcription. Virology 363:333–341. doi: 10.1016/j.virol.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.D'Costa SM, Bainbridge TW, Kato SE, Prins C, Kelley K, Condit RC. 2010. Vaccinia H5 is a multifunctional protein involved in viral DNA replication, postreplicative gene transcription, and virion morphogenesis. Virology 401:49–60. doi: 10.1016/j.virol.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jamin A, Ibrahim N, Wicklund A, Weskamp K, Wiebe MS. 2015. Vaccinia virus B1 kinase is required for postreplicative stages of the viral life cycle in a BAF-independent manner in U2OS cells. J Virol 89:10247–10259. doi: 10.1128/JVI.01252-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Condit RC, Xiang Y, Lewis JI. 1996. Mutation of vaccinia virus gene G2R causes suppression of gene A18R ts mutants: implication for control of transcription. Virology 220:10–19. doi: 10.1006/viro.1996.0280. [DOI] [PubMed] [Google Scholar]
- 100.Black EP, Moussatche N, Condit RC. 1998. Characterization of the interactions among vaccinia virus transcription factors G2R, A18R, and H5R. Virology 245:313–322. doi: 10.1006/viro.1998.9166. [DOI] [PubMed] [Google Scholar]
- 101.Kwon JA, Rich A. 2005. Biological function of the vaccinia virus Z-DNA-binding protein E3L: gene transactivation and antiapoptotic activity in HeLa cells. Proc Natl Acad Sci U S A 102:12759–12764. doi: 10.1073/pnas.0506011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jesus DM, Moussatche N, Condit RC. 2014. Vaccinia virus mutations in the L4R gene encoding a virion structural protein produce abnormal mature particles lacking a nucleocapsid. J Virol 88:14017–14029. doi: 10.1128/JVI.02126-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. 2013. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288:31458–31467. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Panda D, Fernandez DJ, Lal M, Buehler E, Moss B. 2017. Triad of human cellular proteins, IRF2, FAM111A, and RFC3, restrict replication of orthopoxvirus SPI-1 host-range mutants. Proc Natl Acad Sci U S A 114:3720–3725. doi: 10.1073/pnas.1700678114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Punchihewa C, Inoue A, Hishiki A, Fujikawa Y, Connelly M, Evison B, Shao Y, Heath R, Kuraoka I, Rodrigues P, Hashimoto H, Kawanishi M, Sato M, Yagi T, Fujii N. 2012. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J Biol Chem 287:14289–14300. doi: 10.1074/jbc.M112.353201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schroder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Benfield CTO, Ren HW, Lucas SJ, Bahsoun B, Smith GL. 2013. Vaccinia virus protein K7 is a virulence factor that alters immune response to infection. J Gen Virol 94:1647–1657. doi: 10.1099/vir.0.052670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cotter CA, Earl PL, Wyatt LS, Moss B. 2017. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Mol Biol 117:16.16.1–16.16.18. doi: 10.1002/cpmb.33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.