

Abstract
Discovered in the beginning of the 20th century, nicotinamide adenine dinucleotide (NAD +) has evolved from a simple oxidoreductase cofactor to being an essential cosubstrate for a wide range of regulatory proteins that include the sirtuin family of NAD +‐dependent protein deacylases, widely recognized regulators of metabolic function and longevity. Altered NAD + metabolism is associated with aging and many pathological conditions, such as metabolic diseases and disorders of the muscular and neuronal systems. Conversely, increased NAD + levels have shown to be beneficial in a broad spectrum of diseases. Here, we review the fundamental aspects of NAD + biochemistry and metabolism and discuss how boosting NAD + content can help ameliorate mitochondrial homeostasis and as such improve healthspan and lifespan.
Keywords: aging, metabolic disorders, neurodegeneration, nicotinamide adenine dinucleotide, poly ADP‐ribose polymerase
Subject Categories: Metabolism, Molecular Biology of Disease
Introduction
The first cofactor ever described, nicotinamide adenine dinucleotide (NAD+), was discovered by the British biochemists Arthur Harden and William John Young in 1906 (Harden & Young, 1906). They observed that adding boiled yeast extracts to non‐boiled yeast extracts significantly accelerated alcoholic fermentation, suggesting that the boiled yeast fraction contained something capable of promoting the fermentation reaction. They named this heat‐stable, but yet unidentified factor, “cozymase”. Almost 25 years later, Hans von Euler‐Chelpin established the chemical composition of the cozymase as an adenine, a reducing sugar group and a phosphate (von Euler & Myrbäck, 1930). Finally, in 1936, Otto Heinrich Warburg discovered the capability of the cozymase to transfer hydride from one molecule to another and identified nicotinamide base as the site of redox reactions (Warburg & Christian, 1936). Together with its reduced counterpart, NADH, NAD+ has since been known for being involved in reactions that required the transfer of electrons from one molecule to another. As such, the redox couple NAD+/NADH has been reported to participate in numerous reactions requiring an electron exchange, such as glycolysis, pyruvate‐to‐lactate and pyruvate‐to‐acetyl‐CoA interconversions, β‐oxidation, citric acid cycle (TCA cycle), and oxidative phosphorylation. Moreover, addition of a phosphate to the adenosine ribose of NAD+ by NAD+ kinases (NADKs) leads to a formation of nicotinamide adenine dinucleotide phosphate (NADP+). NADP+ and its reduced form, NADPH, play a key role in cellular defense against oxidative stress, as well as in the synthesis of fatty acids, cholesterol, and DNA. Detailed description of the physiological roles of the NADP+/NADPH redox couple is reviewed elsewhere (Ying, 2008). Although the role of NAD+ in redox reactions is now rather well understood, NAD+ biology underwent a renaissance when NAD+ was reported to influence the activity of the sirtuins (Imai et al, 2000), a family of NAD+‐dependent deacylases, implicated in the regulation of metabolism and mitochondrial function (Haigis & Sinclair, 2010; Houtkooper et al, 2012). Besides sirtuins, other enzymes, such as the poly ADP‐ribose polymerase (PARP) protein family and the cyclic ADP‐ribose (cADPR) synthases, such as CD38 and CD157, are currently known to require NAD+ as a cosubstrate to perform their function. The dependence of these important metabolic enzymes on NAD+ levels provides an attractive possibility to modulate their activity and thereby achieve health benefits and has led to an increased interest in NAD+ metabolism over the last decade. The therapeutic potential of NAD+ boosting techniques to activate the sirtuins has now been explored in a large spectrum of preclinical disease models that mimic rare genetic disorders, such as the Cockayne syndrome, as well as pandemic‐like contemporary diseases, such as obesity or non‐alcoholic fatty liver disease (NAFLD). The near future will hopefully see these studies translate from the bench to the bedside.
Biosynthesis of NAD+
Intracellular NAD+ can be produced through either de novo synthesis or via salvage pathways from precursor molecules, naturally occurring vitamins: nicotinamide (NAM), nicotinic acid (NA), and nicotinamide riboside (NR) (Bogan & Brenner, 2008; Houtkooper et al, 2010) (Fig 1). The NAD+ de novo synthesis pathway starts from the amino acid tryptophan (Bender, 1983; Houtkooper et al, 2010) and most likely takes place in the cytosol, since all the enzymes catalyzing the different steps of this process are localized there (Houtkooper et al, 2010).
Figure 1. Pathways modulating NAD+ content in mammals.
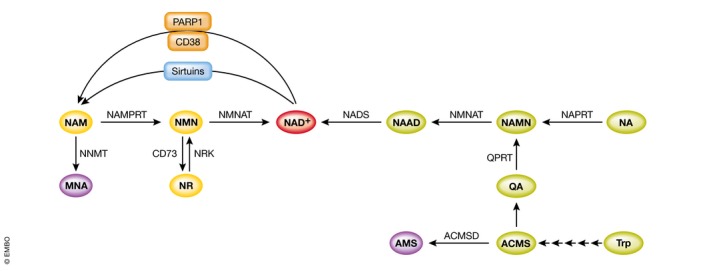
Intermediates of the amidated and deamidated routes are depicted in yellow and green, respectively. NAD +‐consuming enzymes competing with sirtuins for NAD + availability are depicted in orange. Purple color indicates metabolites not recycled in the NAD + synthesis pathway.
NAD+ synthesis from NAM requires only two steps: NAM gets first converted by nicotinamide phosphoribosyltransferase (NAMPRT) into NAM mononucleotide (NMN), which in its turn leads to the production of NAD+ in a reaction catalyzed by nicotinamide mononucleotide adenylyltransferase (NMNAT) (Fig 1). Three different isoforms of NMNAT have been reported, each of them possessing a specific subcellular localization: NMNAT1 is a nuclear enzyme (Emanuelli et al, 2001; Yalowitz et al, 2004), NMNAT2 is located in the cytosol and Golgi apparatus (Yalowitz et al, 2004; Berger et al, 2005), while NMNAT3 was detected in the cytosol and mitochondria (Zhang et al, 2003; Berger et al, 2005; Yang et al, 2007). NR also gets converted into NMN by nicotinamide riboside kinase (NRK) (Bieganowski & Brenner, 2004). Mammals possess two isoforms of NRK: an ubiquitously expressed NRK1 and NRK2, whose expression was mainly detected in heart, skeletal muscle, brown adipose tissue (BAT), and liver (Bogan & Brenner, 2008). Interestingly, it has been recently reported that NRK1 is required for NAD+ synthesis not only from the exogenously administered NR, but also NMN (Ratajczak et al, 2016). Both NAM and NR operate via the “amidated” route to produce NAD+ (Fig 1).
Nicotinic acid, in its turn, initiates the “deamidated” route (Fig 1). Conversion of NA into NA mononucleotide (NAMN) constitutes the first step of this route, which most often is referred as the Preiss–Handler pathway (Preiss & Handler, 1958). The NMNATs recognize both NAMN and NMN as substrates; however, in the case of NAMN the conversion results in NA adenine dinucleotide (NAAD), and therefore, one additional step, catalyzed by NAD synthetase (NADS), is required to produce NAD+ (Fig 1). Interestingly, it has been recently reported that NR leads to the production of NAAD via a yet‐unknown mechanism (Trammell et al, 2016a).
The de novo NAD+ synthesis pathway, which converts tryptophan into NAD+, consists of eight steps. The first reaction of this pathway constitutes of a conversion of tryptophan into N‐formylkynurenine, which in mammals can be catalyzed by two different enzymes: tryptophan‐2,3‐dioxygenase (TDO) and indoleamine 2,3‐dioxygenase (IDO). This conversion is considered to be the first rate‐limiting step for the pathway. TDO is the major contributor to NAD+ biosynthesis in liver, while IDO is ubiquitously expressed in extrahepatic tissues, with the highest activity detected in lung, intestine, and spleen (Yamazaki et al, 1985; Kudo & Boyd, 2000). TDO is induced by tryptophan and glucocorticoids (Comings et al, 1995), while IDO is induced by inflammatory stimuli (Yoshida & Hayaishi, 1978; Yoshida et al, 1979; Takikawa et al, 1986; Heyes et al, 1992; Reinhard, 1998; Sanni et al, 1998; Daubener & MacKenzie, 1999). N‐formylkynurenine gets converted by formamidase (KFase) into kynurenine. Kynurenine in its turn leads to 3‐OH kynurenine in a reaction catalyzed by kynurenine 3‐hydroxylase (K3H). Kynureninase (Kyase) then forms 3‐hydroxyanthranilate, which gets transformed into α‐amino‐β‐carboxymuconate‐ε‐semialdehyde (ACMS) by 3‐hydroxyanthranilate 3,4‐dioxygenase (3HAO). The formation of this unstable ACMS constitutes a branching point of the de novo NAD+ synthesis pathway (Bender, 1983; Houtkooper et al, 2010). ACMS can either undergo cyclization forming quinolinic acid (QA), which is then converted by quinolinate phosphoribosyltransferase (QPRT) into NAMN and from this point fuses with the Preiss–Handler pathway to produce NAD+ (Fig 1). Otherwise, the carbon group of ACMS can be removed, which either leads to the production of picolinic acid or is directed to total oxidation to CO2 and H2O. While the cyclization of ACMS is a spontaneous reaction, the transformation of ACMS into α‐amino‐β‐muconate‐ε‐semialdehyde (AMS) is catalyzed by the enzyme α‐amino‐β‐carboxymuconate‐ε‐semialdehyde decarboxylase (ACMSD) (Fig 1).
Preferential source for NAD+ production
The existence of different pathways leading to NAD+ production raises questions on the relative importance of each pathway and which of them possess the highest potential to boost NAD+ levels. The preferable precursor for NAD+ production within the organism is hence still a matter of debate. There is evidence that NAM possesses a higher NAD+ boosting capability when compared to NA in different organs in mice (Collins & Chaykin, 1971, 1972; Mori et al, 2014; Yang et al, 2014). Additionally, in human plasma, levels of NAM were reported to be fivefold higher than NA levels (Jacobson et al, 1995). However, several other studies claim the opposite: NA is a more effective NAD+ precursor than NAM (Ijichi et al, 1966; Hagino et al, 1968; Lin & Henderson, 1972; Williams et al, 1985; Jackson et al, 1995; Hara et al, 2007). It is important to mention that in Mori et al (2014) the authors quantified the activity of NMNAT and NADS; therefore, the comparison was rather made between the “deamidated” (e.g., from NA) and “amidated” route, which includes both NAM and NR. And even if the authors of this study claim that NAM is the main precursor for NAD+ synthesis, the possibility of a significant contribution of other precursors using the amidated NAD+ biosynthesis route (e.g., NR) cannot be discounted. In support of this, a very recent study showed that NR has a greater capacity over NA and NAM to boost hepatic NAD+ levels (Trammell et al, 2016a). It is also important to mention that both NA and NAM have reported side effects, whereas no adverse effects are currently reported for NR. NA activates the G protein‐coupled receptor, GPR109A and causes flushing, characterized by vasodilation and a burning sensation (Benyo et al, 2006). While NAM raises health concerns for treatment of diabetic patients, as high doses of NAM can be hepatotoxic (Knip et al, 2000).
As for tryptophan, its administration to humans has been used as treatment for pain, sleep disorders, depression, hyperactivity, and bulimia (Richard et al, 2009). No severe adverse effects have been reported for tryptophan administration, even with doses going as high as 20 g/day in schizophrenic patients (Sidransky, 2001). A large number of reviews attribute a marginal role to the de novo NAD+ synthesis pathway. However, a solid support for this claim is lacking. One of the studies frequently cited to sustain this point of view reports that tryptophan alone is not sufficient to maintain the physiological NAD+ concentration of the cell (Nikiforov et al, 2011). However, this conclusion was exclusively based on the observation that supplementation with tryptophan is not sufficient to protect cells from the death induced by NAMPRT inhibitor FK866 and no NAD+ quantification was performed in this study. In addition, some studies show that, at least in the liver, tryptophan constitutes the preferable substrate for NAD+ production. Rat primary hepatocytes, treated with NA, NAM, or tryptophan, were reported to use exclusively tryptophan for their NAD+ biosynthesis, even though they were still able to take up NA and NAM from the culture medium (Bender & Olufunwa, 1988). Administration of tryptophan, NA, or NAM to rats showed that tryptophan resulted in the highest hepatic NAD+ concentrations (Bender et al, 1982). Moreover, it has been shown that in rat liver, NA and NAM have a very limited capacity for NAD+ production, probably due to the saturation of the involved phosphoribosyltransferases, whereas no such limitations were detected for the NAD+ synthesis from tryptophan (Williams et al, 1950; Bender et al, 1982; McCreanor & Bender, 1986).
NAD+ consuming enzymes
Sirtuin proteins require NAD+ as a cosubstrate for their activity. A detailed description of their role in the regulation of metabolism and aging is beyond the scope of this review, but has been extensively covered elsewhere (Haigis & Sinclair, 2010; Satoh et al, 2011; Houtkooper et al, 2012; Chang & Guarente, 2014). Besides sirtuins, two different protein families are well known to use NAD+ as a cofactor for their enzymatic activities. These include the PARPs and the cADPR synthases, CD38 and CD157. PARPs are involved in DNA repair, maintenance of genomic integrity, and cell death, with PARP1 accounting for more than 85% of NAD+ consumption of this protein family (Bai & Canto, 2012). cADPR, which is generated by CD38 and CD157, is a signaling molecule that controls intracellular calcium fluxes. The catalytic efficiency of CD38 is significantly higher than that of CD157 (Quarona et al, 2013). While CD38 expression was initially considered to be limited to the immune system, it was later found to be ubiquitously distributed. CD38 is an important NAD+ consumer, as its loss of function (LOF) in mice led up to a 30‐fold increase in NAD+ levels in different tissues (Barbosa et al, 2007). Combined increases in PARP1 (Braidy et al, 2011; Mouchiroud et al, 2013) and CD38 (Camacho‐Pereira et al, 2016) activities upon aging were reported to cause age‐associated reduction in NAD+ content. For more extensive coverage of these NAD+ consuming enzymes, we refer the readers to a few reviews on PARPs (Bai & Canto, 2012; Canto et al, 2013; Jubin et al, 2016) and cADPR synthases (Malavasi et al, 2008; Quarona et al, 2013).
Regulation of NAD+ content
Regulation of NAD+ content by diet and aging
An increase of NAD+ levels followed by sirtuin activation is observed in situations of energy deficit, such as fasting (Rodgers et al, 2005; Chen et al, 2008; Cantó et al, 2010), calorie restriction (CR) (Qin et al, 2006; Chen et al, 2008; Cantó et al, 2010) or low glucose feeding (Fulco et al, 2008), and exercise (Canto et al, 2009; Cantó et al, 2010; Costford et al, 2010). On the contrary, multiple studies reported that high‐fat (HF)/high‐fat high‐sucrose (HFHS) feeding diminishes NAD+ content in liver (Yoshino et al, 2011; Gariani et al, 2016, 2017; Trammell et al, 2016b), skeletal muscle (Canto et al, 2012), BAT (Canto et al, 2012), and white adipose tissue (WAT) (Yoshino et al, 2011). Orotic acid administration (Fukuwatari et al, 2002) or feeding a methionine‐/choline‐deficient (MCD) diet (Gariani et al, 2017) also leads to the concomitant appearance of liver fat accumulation and a drop in hepatic NAD+ levels. A recent study, however, reported that administration of a HF diet for 11 weeks led to an increase in NAD+ content in mouse liver, accompanied by enhanced Sirt1 activity (Penke et al, 2015). The stimulation of the NAD+ biosynthesis could represent an initial compensatory attempt to maintain energy homeostasis (Penke et al, 2015; Drew et al, 2016), whereas more prolonged exposure to HF diet reduces NAD+ content, resulting in functional damage (Drew et al, 2016). Interestingly, in Saccharomyces cerevisiae NAD+ content is affected by the carbon source used: Yeast grown on ethanol contain practically double the amount of NAD+ compared to yeast grown on glucose (Agrimi et al, 2011).
Finally, a decrease in NAD+ content was also reported to be associated with aging in Caenorhabditis elegans, mice, rats, and humans (Braidy et al, 2011; Yoshino et al, 2011; Massudi et al, 2012; Gomes et al, 2013; Mouchiroud et al, 2013; Camacho‐Pereira et al, 2016; Guan et al, 2017).
Regulation by circadian rhythm
Circadian rhythm is another important regulator of NAD+ content. The key regulators of the mammalian circadian clock machinery are the transcription factors CLOCK and BMAL1, which act together as a heterodimer. Their transcriptional targets, PER and CRY, form a negative feedback loop by repressing CLOCK‐BMAL1 activity. Hepatic NAD+ levels oscillate in a diurnal manner (Nakahata et al, 2009; Ramsey et al, 2009). Mice with LOF mutations in the circadian activator genes, Clock and Bmal1, show reduced NAD+ content, while NAD+ levels were elevated in mice with mutations in the clock repressor genes Cry1 and Cry2 (Ramsey et al, 2009). The circadian clock‐controlled expression of Namprt is thought to be responsible for this fine‐tuning of the NAD+ availability (Nakahata et al, 2009; Ramsey et al, 2009). Importantly, clock‐driven oscillations of NAD+ were claimed to regulate the activity of both SIRT1 (Nakahata et al, 2009; Ramsey et al, 2009) and SIRT3 (Peek et al, 2013). It is possible that modulation of the redox state of NAD+ can reciprocally impact on the circadian clock. In vitro NADH was shown to enhance binding of the CLOCK‐BMAL1 heterodimer to DNA, whereas NAD+ was inhibiting this process (Rutter et al, 2001). On their turn, the NAD+‐dependent enzymes SIRT1, SIRT6, and PARP1 were reported to control the circadian clock machinery via post‐translational modifications of the core clock transcription factors and via regulation of their transcription (Asher et al, 2008, 2010; Chang & Guarente, 2013; Masri et al, 2014).
NAD+ boosting strategies
NAD+ levels can be increased either by promoting its synthesis—by enhancing the enzymes involved in NAD+ biosynthesis or administration of NAD+ precursor molecules—or by limiting its consumption. Supplementation with NA, NAM, NR, NMN, or tryptophan can increase NAD+ content (Canto et al, 2015). Overexpressing or activating enzymes catalyzing the rate‐limiting steps of NAD+ biosynthesis also are efficient to boost NAD+ levels (Araki et al, 2004; Sasaki et al, 2006; Hsu et al, 2009; Wang et al, 2014a; Williams et al, 2017). However, the translational potential of this approach may be lower compared to the other strategies.
Pharmacological or genetic inhibition of non‐sirtuin NAD+ consumers, such as PARP‐1 or CD38 (Fig 1), can help to preserve NAD+ levels for sirtuin activation (Aksoy et al, 2006a; Barbosa et al, 2007; Bai et al, 2011; Pirinen et al, 2014), especially in situations when non‐sirtuin NAD+ consumers are overactivated (Bai et al, 2011; Braidy et al, 2011; Mouchiroud et al, 2013; Fang et al, 2014; Mukhopadhyay et al, 2014; Ryu et al, 2016; Gariani et al, 2017). For instance, DNA damage is known to cause a dramatic decline in NAD+ intracellular levels, which is due to PARP activation (Berger, 1985), and overexpression of CD38 in cells leads to a ~35% decrease in NAD+ levels (Hu et al, 2014). On the contrary, Cd38 −/− and PARP‐1 −/− mice have increased NAD+ content in different organs (Aksoy et al, 2006a,b; Young et al, 2006; Bai et al, 2011).
Increased nicotinamide methyl transferase (NNMT) expression was reported in obesity and type 2 diabetes (Lee et al, 2005; Yaguchi et al, 2005; Salek et al, 2007; Kraus et al, 2014). NNMT is the enzyme catalyzing the transformation of NAM into methylnicotinamide (MNA) (Fig 1) and is highly expressed in liver and adipose tissue (Aksoy et al, 1994; Riederer et al, 2009). Inhibiting NNMT in these tissues should increase NAD+ content, since NAM would not be degraded but exclusively reconverted into NAD+ (Fig 1). In line, the knockdown of NNMT increased NAD+ levels in adipose tissue, but not in the liver (Kraus et al, 2014).
Resveratrol, a natural polyphenol found in red wine, activates AMPK and thereby increases NAD+ levels (Fulco et al, 2008; Canto et al, 2009; Price et al, 2012; Desquiret‐Dumas et al, 2013), which promotes sirtuin activation.
Therapeutic potential of NAD+
Pellagra
Pellagra, a disease that was epidemic in the XVII–XIX centuries in several rural areas of Europe and the United States (Bender, 1983), owes its name to the Italian “pelle” = skin and “agra” = sour or rough, which describes its most noticeable feature. Otherwise, it is also called the disease of “the three Ds”: Diarrhea, dermatitis, and dementia, which, if untreated, can lead to the fourth “D”, i.e. death. In 1915, Joseph Goldberger demonstrated that pellagra was not an infectious disease, as previously thought, but is due to poor nutrition and could be prevented by consumption of fresh meat and milk (Bender, 1983). Administration of both NAM and NA (Elvehjem et al, 1937), and later tryptophan (Krehl et al, 1945) were also reported to prevent pellagra.
Aging
Sirtuins are well‐known longevity regulators, and their decreased function with age might at least be partially explained by a systemic decline in NAD+ levels upon aging (Mouchiroud et al, 2013) [reviewed in (Imai & Guarente, 2014; Menzies et al, 2016)]. Rising NAD+ content, followed by sirtuin activation, has been reported to increase lifespan in yeast (Lin et al, 2004; Belenky et al, 2007; Easlon et al, 2008), worms (Mouchiroud et al, 2013), and mice (Zhang et al, 2016). Administration of NR, NMN, or NAM recovered NAD+ content and protected against aging‐related complications, such as mitochondrial dysfunction (Gomes et al, 2013; Mouchiroud et al, 2013; Mills et al, 2016), decline in physical performance (Mills et al, 2016; Zhang et al, 2016) and muscle regeneration (Zhang et al, 2016), arterial dysfunction (de Picciotto et al, 2016), decline in vision (Lin et al, 2016; Mills et al, 2016), including glaucoma (Williams et al, 2017), and age‐associated insulin resistance (Mills et al, 2016).
The most striking benefits of NAD+ supplementation on aging were observed in several rare diseases linked to abnormal DNA repair that are typified by accelerated aging, such as the Cockayne syndrome group B (CSB), xeroderma pigmentosum group A (XPA), or ataxia‐telangiectasia (A‐T). In a mouse model of CSB, neurons show mitochondrial defects, which have an impact on the cerebellum and inner ear. Administration of PARP inhibitors or the NAD+ precursor, NR, to csb −/− animals attenuated many of the phenotypes of CSB and restored altered mitochondrial function in their neurons (Scheibye‐Knudsen et al, 2014). Another DNA damage repair disorder is XPA, which is also characterized by mitochondrial alterations and reduced NAD+‐SIRT1 signaling due to the overactivation of PARP1 (Fang et al, 2014). Treatment with NAD+ precursors, NR and NMN, or with the PARP inhibitor, Olaparib, rescued the XPA phenotype in cells and worms. Similar observations of increased PARylation, NAD+ depletion, and mitochondrial dysfunction were made in mouse and worm models of another progressive neurodegenerative disease, A‐T (Fang et al, 2016). Restoring the NAD+/SIRT1 pathway, by NR and NMN administration to C. elegans and mice, improved A‐T neuropathology (Fang et al, 2016).
Metabolic disorders
The importance of NAD+ as a metabolic regulator has been demonstrated by its efficacy to attenuate many features of the metabolic syndrome, a cluster of pathologies including insulin resistance, fatty liver, dyslipidemia, and hypertension, with increased risk of developing type 2 diabetes and heart failure. Different approaches aiming to raise NAD+ levels were shown to provide protection against obesity, such as (i) inhibition of NAD+ consumers, PARPs (Bai et al, 2011; Gariani et al, 2017) and CD38 (Barbosa et al, 2007), (ii) administration of NAD+ precursors, such as NR (Canto et al, 2012; Gariani et al, 2016; Trammell et al, 2016b) or NMN (Yoshino et al, 2011), (iii) or inhibition of NNMT (Kraus et al, 2014). NAD+ boosting was also efficient to improve glucose homeostasis in obese, prediabetic, and T2DM animals (Barbosa et al, 2007; Bai et al, 2011; Yoshino et al, 2011; Canto et al, 2012; Kraus et al, 2014; Gariani et al, 2016, 2017; Trammell et al, 2016b). Likewise, re‐establishing NAD+ levels with NR or PARP inhibitors also protected from non‐alcoholic steatohepatitis (NASH) (Gariani et al, 2016, 2017; Mukhopadhyay et al, 2017) as well as alcoholic steatohepatitis (ASH) (Mukhopadhyay et al, 2017).
Muscle function
Increase in muscle NAD+ content, resulting from NR administration or PARP inhibition, improved muscle function and exercise capacity in mice (Canto et al, 2012; Pirinen et al, 2014), including in aged animals (Zhang et al, 2016). Interestingly, muscular dystrophy is characterized by a dramatic drop in NAD+ in the muscle (Ryu et al, 2016). NR administration to the mdx mouse, a model for muscular dystrophy, improved muscle function by enhancing bioenergetics, attenuating inflammation and fibrosis (Ryu et al, 2016), as well as, by favoring regeneration and preventing the exhaustion and senescence of muscle stem cells, typical to the mdx mice (Zhang et al, 2016).
The beneficial effects of improving muscle bioenergetics are also illustrated in models of mitochondrial myopathies. Increasing muscle NAD+ levels by the administration of NR or a PARP inhibitor preserved muscle function in two different models of mitochondrial myopathy (Cerutti et al, 2014; Khan et al, 2014). Similar benefits on mitochondrial myopathy were seen with the AMPK agonist, AICAR (Viscomi et al, 2011), which may at least in part be due to the recovery of NAD+ content upon AMPK activation.
Cardiac function
Exposing the heart to different types of stresses was reported to result in a decline in cardiac NAD+ content (Pillai et al, 2005, 2010; Karamanlidis et al, 2013; Yamamoto et al, 2014). For instance, cardiomyocyte hypertrophy is characterized by a drop in cellular NAD+ levels. Supplementation with NAD+ was hence protective against cardiac hypertrophy in mice, and these anti‐hypertrophic effects were in part attributed to the activation of SIRT3 (Pillai et al, 2010).
Cardiac ischemia is another condition causing a steep decrease in NAD+ levels. NMN administration protected the mice from ischemic injury via the recovery of cardiac NAD+ content and subsequent SIRT1 activation (Yamamoto et al, 2014). Similarly, cardiac‐specific overexpression of NAMPRT in mice increased NAD+ content and reduced the extent of myocardial infarction and apoptosis in response to prolonged ischemia and ischemia/reperfusion (Hsu et al, 2009). Maintaining NAD+ levels in pressure‐overloaded hearts is crucial for myocardial adaptation and protection from heart failure, as demonstrated by NMN administration to mice treated with the NAMPRT inhibitor FK866 (Yano et al, 2015) and to cardiac‐specific mitochondrial complex I‐deficient mice (Lee et al, 2016). In a mouse model of heart failure caused by iron deficit, reconstituting NAD+ content also improved mitochondrial quality, protected cardiac function, and increased lifespan (Xu et al, 2015). Similarly, NR administration improved cardiac function in aged mdx mice, which, like muscular dystrophy patients, display cardiomyopathy (Ryu et al, 2016).
Renal function
Multiple studies demonstrated the loss of SIRT1 and SIRT3 activity as a key feature of kidney dysfunction, including kidney abnormalities linked with aging (Koyama et al, 2011; Zhuo et al, 2011; Morigi et al, 2015; Ugur et al, 2015; Guan et al, 2017). Acute kidney injury (AKI) is characterized by a reduction in NAD+ content and NAMPRT expression (Morigi et al, 2015; Ugur et al, 2015). Promoting NAD+ synthesis via NAM or NMN supplementation was reported to mitigate AKI in ischemia/reperfusion‐ and cisplatin‐induced mouse models of AKI (Tran et al, 2016; Guan et al, 2017). Furthermore, administration of the AMPK agonist, AICAR, which positively impacts on NAD+ levels (Canto et al, 2009), was protective against cisplatin‐induced AKI in SIRT3‐dependent manner (Morigi et al, 2015). Although no NAD+ quantification was performed in this particular study, the involvement of SIRT3, as well as the increase in Namprt expression detected upon AICAR administration, points toward a potential increase in NAD+ levels (Morigi et al, 2015). Kidney mesangial cell hypertrophy is also characterized by a depletion of NAD+ content (Zhuo et al, 2011) and restoring intracellular NAD+ levels via supplementation with exogenous NAD+ prevented its onset by activating SIRT1 and SIRT3 (Zhuo et al, 2011).
Neurodegeneration
NAD+ boosting has also been shown to be neuroprotective. Raising NAD+ levels protects against neuronal death induced by ischemic brain (Klaidman et al, 2003; Sadanaga‐Akiyoshi et al, 2003; Kabra et al, 2004; Feng et al, 2006; Kaundal et al, 2006; Zheng et al, 2012) or spinal cord injuries (Xie et al, 2017). Axonal degeneration is considered as an early pathological mechanism in this type of neurodegeneration. An accumulating amount of data indicates that axonal degeneration is not only limited to ischemic brain and spinal cord injuries, but constitutes a hallmark process, preceding neuronal death, in a much larger spectrum of disease states, including traumatic brain injury, inflammatory disorders, like multiple sclerosis, and degenerative disorders, such as Alzheimer's and Parkinson's diseases (Lingor et al, 2012; Johnson et al, 2013). Degenerating axons show a decrease in NAD+ content (Wang et al, 2005; Gerdts et al, 2015), while replenishing NAD+ by supplementing NAM (Wang et al, 2005), NR and NMN (Sasaki et al, 2006), and high doses of NAD+ (Araki et al, 2004), or overexpressing enzymes involved in NAD+ biosynthesis (Araki et al, 2004; Sasaki et al, 2006) delayed axonal degeneration. In line with this, supplementation with NAM, NMN, or NR was neuroprotective in rodent models of Alzheimer disease (Qin et al, 2006; Gong et al, 2013; Liu et al, 2013; Turunc Bayrakdar et al, 2014; Wang et al, 2016a), and supplementation with NAM or LOF of PARP were protective in Drosophila models of Parkinson's disease (Lehmann et al, 2017).
NAD+ depletion is also involved in the neurodegeneration induced by highly toxic misfolded prion protein (Zhou et al, 2015). Replenishment of intracellular NAD+ stocks, either by providing NAD+ or NAM, rescued the neurotoxic effects of protein aggregates (Zhou et al, 2015). Importantly, restoring NAD+ content is not exclusively protecting neurons, since it has also been reported to prevent the death of astrocytes (Alano et al, 2004).
P7C3, a compound that enhances neurogenesis (Pieper et al, 2010) and that was neuroprotective in mouse models of Parkinson's disease (De Jesus‐Cortes et al, 2012), amyotrophic lateral sclerosis (Tesla et al, 2012) and brain injury (Yin et al, 2014), was subsequently identified as an NAMPRT activator (Wang et al, 2014a). Therefore, the beneficial effects of P7C3 on neuron preservation seem at least in part to be due to a NAMPRT‐mediated increase in NAD+ levels (Wang et al, 2014a).
Nicotinamide riboside supplementation recovered depressed sensory and motor neuron conduction velocities and thermal insensitivity in T2DM mice (Trammell et al, 2016b) and alleviated chemotherapy‐induced peripheral neuropathy in rats (Hamity et al, 2017), indicating that NAD+ also is beneficial in the peripheral neuronal system.
NAD+ boosting was also able to protect mice from loss of vision and hearing (Shindler et al, 2007; Brown et al, 2014). Intravitreal injections of NR in mice attenuated optic neuritis in a dose‐dependent manner (Shindler et al, 2007). Even if no NAD+ quantification was performed in this study, SIRT1 activity was necessary for the neuroprotective effects of NR, since the protection was blunted in the presence of sirtinol, a SIRT1 inhibitor (Shindler et al, 2007). Furthermore, systemic administration of NAM and overexpression of Nmnat1 had spectacular effects on vision in DBA/2J mice, which are prone to glaucoma (Williams et al, 2017). Noise exposure results in degeneration of the neurons innervating the cochlear hair cells. Increase in NAD+ levels induced by NR administration prevented against noise‐induced hearing loss and neurite degeneration (Brown et al, 2014). In line with this, CR was shown to protect against cochlear cell death and aging‐associated hearing loss in a Sirt3‐dependent manner (Someya et al, 2010). It is therefore tempting to speculate that this improvement could also be associated with increased NAD+ levels upon CR, though no direct measurements of NAD+ levels were performed in this study.
Future challenges and perspectives
Although it was thought that the NAD+ biosynthetic pathways were entirely understood, we still continue to discover new actors of NAD+ metabolism. For instance, whereas the conversion of NR into NMN by NRK was established some time ago (Bieganowski & Brenner, 2004), the opposite reaction, that is, the transformation of NMN into NR by CD73, was only recently shown to occur in humans (Garavaglia et al, 2012). Another example is the recent description that NR not only induced NAAD concentrations by 45‐fold, but that it is also a direct biosynthetic precursor of NAAD (Trammell et al, 2016a). The biochemical basis of this conversion cannot be explained with our current state of knowledge, as NAD+ synthetase, which catalyzes the transformation of NAAD into NAD+, works unidirectionally (Bieganowski et al, 2003; Wojcik et al, 2006) and can therefore not catalyze the reverse reaction from NAD+ into NAAD.
Additionally, it is possible that we still ignore some functions that NAD+ might accomplish within the cell. For instance, very recently NAD+ was found to be linked to RNA in bacteria (Chen et al, 2009). By forming a cap at the 5′‐terminus of bacterial RNA molecule, it is not only increasing its stability (Cahova et al, 2015), but also serves as non‐canonical initiation nucleotides for de novo transcription initiation (Bird et al, 2016). Initially thought to be prokaryote‐specific, this RNA modification appears to be also conserved in eukaryotic systems (Jiao et al, 2017; Walters et al, 2017). Similarly to bacteria, in eukaryotic cells NAD+ addition seems to occur during transcription initiation (Bird et al, 2016; Walters et al, 2017). Intriguingly, a subset of eukaryotic non‐coding RNAs have also been reported to possess a NAD+‐cap. Since these RNAs are formed exonucleolytically, NAD+ cap addition in their case would occur post‐transcriptionally (Jiao et al, 2017). Oppositely to prokaryotes, in mammalian cells the NAD+ cap was reported to rather promote mRNA decay (Jiao et al, 2017). The full physiological significance of NAD+‐capping is yet to be discovered. It is, however, tempting to speculate that the proportion of cellular mRNA possessing NAD+ cap might be influenced by intracellular NAD+ content and thus by the energy state of the cell.
Devising better NAD+ quantification methods is a critical challenge in the field. Measurements based on UV–Vis methods are less accurate and sensitive than mass spectrometry methods (Trammell & Brenner, 2013). Moreover, accurate NAD+ quantification in different subcellular compartments is challenging due the complexity of subcellular fractionations and the NAD+ isolation procedures. Over the last few years, a new generation of NAD+ biosensors was developed, allowing NAD+ quantification in intact cells as well as within specific subcellular compartments (Hung et al, 2011; Bilan et al, 2014; Cambronne et al, 2016). Further development and wider application of these biosensors combined with strategies to explore the kinetics of NAD+ biosynthesis and metabolism, using flux studies, will hence be important for future research. Besides, according to a recent study NADP and NADPH were more significantly deregulated in T2DM and obesity than NAD+ and less correctable by NR supplementation (Trammell et al, 2016b). Monitoring of the entire NAD+ metabolome could hence help our further understanding of its role in metabolism, which might extend far beyond NAD+–sirtuin or NAD+–PARP axis.
As reviewed here, manipulations of NAD+ concentrations have demonstrated multiple beneficial effects in a large spectrum of diseases in animal models (Fig 2). Translating these effects into clinical benefits now becomes one of the main challenges. The fact that the long‐term administration of the NAD+ precursor molecules showed no deleterious effects in animals should be considered promising. As such, administration of NMN for 12 months demonstrated no toxicity in mice (Mills et al, 2016). Similarly, administration of NR to mice for a duration of 5–6 months (Gong et al, 2013), 10 months (Zhang et al, 2016), and 12 months (Tummala et al, 2014) showed no obvious adverse effects. Moreover, 2016 was marked by the first report on the effects of NR in humans, showing that the oral administration of NR led to a dose‐dependent increase in NAD+ levels in blood in healthy volunteers (Trammell et al, 2016a). Another NAD+ precursor, NAM, has also been already tested in humans and protected β‐cell function in type 1 diabetes patients (Olmos et al, 2006) and even though the clinical trials for NAM as a treatment for type 1 diabetes failed, no adverse effects of NAM were detected (Gale et al, 2004; Cabrera‐Rode et al, 2006). Furthermore, a slow release form of NA (acipimox) was effective in inducing mitochondrial activity in skeletal muscle of type 2 diabetic patients (van de Weijer et al, 2015).
Figure 2. Therapeutic potential of NAD + boosting in humans based on findings in animal studies.
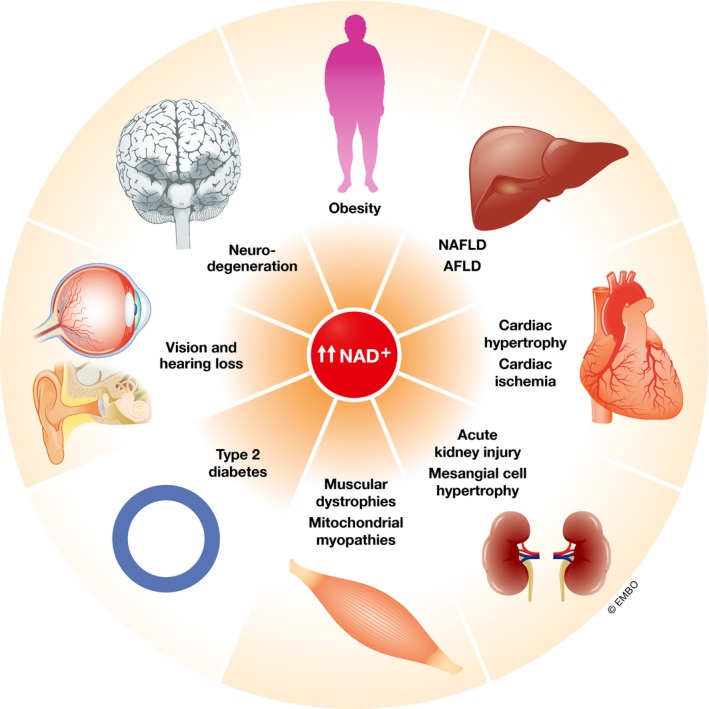
NAFLD, non‐alcoholic fatty liver disease; AFLD, alcoholic fatty liver disease.
In theory, all this bodes well for the use of NAD+ precursors in the clinic. The fact that many of these NAD+ precursors are rightfully considered vitamins (which are generally regarded as safe (GRAS)), and that they are widely available to the public at large, however, also poses some issues. Despite the many health benefits that are inferred from their use in animal disease models, these NAD+ precursors need still to undergo rigorous clinical testing in diseases setting, before one can recommend their widespread use. Therefore, some caution is required, so that the overuse or improper use in uncontrolled settings does not hamper their clinical development as nutra‐ or pharmaceutical agents.
It will also be important to identify which pharmacological strategies aiming to boost NAD+ content would be the most appropriate in patients. Various monoclonal antibodies targeting CD38 have been developed as a treatment for hematological malignancies, some of them being in preclinical, and some even in late clinical, studies (van de Donk et al, 2016). Many flavonoids were reported to inhibit human CD38 at low micromolar concentrations (Kellenberger et al, 2011; Escande et al, 2013), some of them showing promising therapeutic effects in mice (Escande et al, 2013; Boslett et al, 2017). Thiazoloquin(az)olinones have recently been described as potent CD38 inhibitors, able to elevate NAD+ in plasma, liver, and muscle in mice (Haffner et al, 2015). Moreover, several reports have disclosed different small molecules blocking CD38 activity at low micromolar concentrations (Zhou et al, 2012; Moreau et al, 2013; Swarbrick et al, 2014; Wang et al, 2014b; Becherer et al, 2015). Testing and exploring their therapeutic potential in animal models is now just a matter of time.
Several PARP inhibitors are currently either marketed (Olaparib) or undergoing advanced clinical trials for the treatment of cancers in patients with BRCA mutations (Wang et al, 2016b). The drawback of these PARP inhibitors is that none of them is selective for a specific PARP family member (Wang et al, 2016b). For instance, the clinically approved compound, Olaparib, inhibits PARP‐1, PARP‐2, PARP‐3, and PARP‐4 (Wahlberg et al, 2012), increasing chances of adverse effects. As a case in point, the loss of both PARP‐1 and PARP‐2 in mice was reported to cause embryonic lethality (Menissier de Murcia et al, 2003), while a T cell‐specific deficiency in both them leads to highly aggressive lymphomas (Navarro et al, 2017). Moreover, all current PARP inhibitors are genotoxic (Ito et al, 2016), which raises concerns about their use for non‐oncologic indications. Indeed, some side effects that could be tolerated in case of the treatment of life‐threatening diseases such as advanced cancers (Brown et al, 2016) may not be tolerated for non‐oncologic indications. All these evidences indicate that the development of safer and more selective PARP inhibitors is necessary.
The growing literature on the beneficial effects of raising and maintaining NAD+ levels in different disease models and the high evolutionary conservation of the NAD+–sirtuin signaling axis suggests that strategies that increase cellular NAD+ content may have a preventive and/or therapeutic potential in a large number of human diseases (Fig 2). With the first reports on human trials of various NAD+ boosting techniques that start to appear, we are entering in the exciting era of NAD+ therapeutics. While there is no certitude that NAD+ boosting will be able to extend lifespan in humans, such strategies definitely possess the potential to delay age‐associated physiological decline, and therefore, we predict that they will be useful to manage aging‐related diseases and extend healthspan.
Conflict of interest
JA is a founder and SAB member of Mitobridge, a company that develops NAD+ boosting therapeutics.
Acknowledgements
JA is supported by the École Polytechnique Fédérale de Lausanne, the Swiss National Science Foundation (31003A‐140780), AgingX of the Swiss Initiative for Systems Biology (51RTP0‐151019), and the NIH (R01AG043930).
The EMBO Journal (2017) 36: 2670–2683
References
- Agrimi G, Brambilla L, Frascotti G, Pisano I, Porro D, Vai M, Palmieri L (2011) Deletion or overexpression of mitochondrial NAD+ carriers in Saccharomyces cerevisiae alters cellular NAD and ATP contents and affects mitochondrial metabolism and the rate of glycolysis. Appl Environ Microbiol 77: 2239–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy S, Szumlanski CL, Weinshilboum RM (1994) Human liver nicotinamide N‐methyltransferase. cDNA cloning, expression, and biochemical characterization. J Biol Chem 269: 14835–14840 [PubMed] [Google Scholar]
- Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN (2006a) Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem Biophys Res Comm 349: 353–359 [DOI] [PubMed] [Google Scholar]
- Aksoy P, White TA, Thompson M, Chini EN (2006b) Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Comm 345: 1386–1392 [DOI] [PubMed] [Google Scholar]
- Alano CC, Ying W, Swanson RA (2004) Poly(ADP‐ribose) polymerase‐1‐mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 279: 18895–18902 [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305: 1010–1013 [DOI] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U (2008) SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134: 317–328 [DOI] [PubMed] [Google Scholar]
- Asher G, Reinke H, Altmeyer M, Gutierrez‐Arcelus M, Hottiger MO, Schibler U (2010) Poly(ADP‐ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell 142: 943–953 [DOI] [PubMed] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier‐de Murcia J, Auwerx J (2011) PARP‐1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Canto C (2012) The role of PARP‐1 and PARP‐2 enzymes in metabolic regulation and disease. Cell Metab 16: 290–295 [DOI] [PubMed] [Google Scholar]
- Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN (2007) The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet‐induced obesity. FASEB J 21: 3629–3639 [DOI] [PubMed] [Google Scholar]
- Becherer JD, Boros EE, Carpenter TY, Cowan DJ, Deaton DN, Haffner CD, Jeune MR, Kaldor IW, Poole JC, Preugschat F, Rheault TR, Schulte CA, Shearer BG, Shearer TW, Shewchuk LM, Smalley TL Jr, Stewart EL, Stuart JD, Ulrich JC (2015) Discovery of 4‐amino‐8‐quinoline carboxamides as novel, submicromolar inhibitors of NAD‐hydrolyzing enzyme CD38. J Med Chem 58: 7021–7056 [DOI] [PubMed] [Google Scholar]
- Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C (2007) Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ . Cell 129: 473–484 [DOI] [PubMed] [Google Scholar]
- Bender DA, Magboul BI, Wynick D (1982) Probable mechanisms of regulation of the utilization of dietary tryptophan, nicotinamide and nicotinic acid as precursors of nicotinamide nucleotides in the rat. Br J Nutr 48: 119–127 [DOI] [PubMed] [Google Scholar]
- Bender DA (1983) Biochemistry of tryptophan in health and disease. Mol Aspects Med 6: 101–197 [DOI] [PubMed] [Google Scholar]
- Bender DA, Olufunwa R (1988) Utilization of tryptophan, nicotinamide and nicotinic acid as precursors for nicotinamide nucleotide synthesis in isolated rat liver cells. Br J Nutr 59: 279–287 [DOI] [PubMed] [Google Scholar]
- Benyo Z, Gille A, Bennett CL, Clausen BE, Offermanns S (2006) Nicotinic acid‐induced flushing is mediated by activation of epidermal langerhans cells. Mol Pharmacol 70: 1844–1849 [DOI] [PubMed] [Google Scholar]
- Berger NA (1985) Poly(ADP‐ribose) in the cellular response to DNA damage. Radiat Res 101: 4–15 [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M (2005) Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280: 36334–36341 [DOI] [PubMed] [Google Scholar]
- Bieganowski P, Pace HC, Brenner C (2003) Eukaryotic NAD+ synthetase Qns1 contains an essential, obligate intramolecular thiol glutamine amidotransferase domain related to nitrilase. J Biol Chem 278: 33049–33055 [DOI] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C (2004) Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss‐Handler independent route to NAD+ in fungi and humans. Cell 117: 495–502 [DOI] [PubMed] [Google Scholar]
- Bilan DS, Matlashov ME, Gorokhovatsky AY, Schultz C, Enikolopov G, Belousov VV (2014) Genetically encoded fluorescent indicator for imaging NAD(+)/NADH ratio changes in different cellular compartments. Biochim Biophys Acta 1840: 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird JG, Zhang Y, Tian Y, Panova N, Barvik I, Greene L, Liu M, Buckley B, Krasny L, Lee JK, Kaplan CD, Ebright RH, Nickels BE (2016) The mechanism of RNA 5′ capping with NAD+, NADH and desphospho‐CoA. Nature 535: 444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan KL, Brenner C (2008) Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 28: 115–130 [DOI] [PubMed] [Google Scholar]
- Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL (2017) Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD(P)(H). J Pharmacol Exper Ther 361: 99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan‐Ling T, Poljak A, Grant R (2011) Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6: e19194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, Jaffrey SR (2014) Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise‐induced hearing loss. Cell Metab 20: 1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, Kaye SB, Yap TA (2016) PARP inhibitors: the race is on. Br J Cancer 114: 713–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera‐Rode E, Molina G, Arranz C, Vera M, Gonzalez P, Suarez R, Prieto M, Padron S, Leon R, Tillan J, Garcia I, Tiberti C, Rodriguez OM, Gutierrez A, Fernandez T, Govea A, Hernandez J, Chiong D, Dominguez E, Di Mario U et al (2006) Effect of standard nicotinamide in the prevention of type 1 diabetes in first degree relatives of persons with type 1 diabetes. Autoimmunity 39: 333–340 [DOI] [PubMed] [Google Scholar]
- Cahova H, Winz ML, Hofer K, Nubel G, Jaschke A (2015) NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 519: 374–377 [DOI] [PubMed] [Google Scholar]
- Camacho‐Pereira J, Tarrago MG, Chini CC, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN (2016) CD38 dictates age‐related NAD decline and mitochondrial dysfunction through an SIRT3‐dependent mechanism. Cell Metab 23: 1127–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne XA, Stewart ML, Kim D, Jones‐Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH (2016) Biosensor reveals multiple sources for mitochondrial NAD(+). Science 352: 1474–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez‐Marcos PJ, Yamamoto H, Andreux PA, Cettour‐Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J (2012) The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high‐fat diet‐induced obesity. Cell Metab 15: 838–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 11: 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Sauve AA, Bai P (2013) Crosstalk between poly(ADP‐ribose) polymerase and sirtuin enzymes. Mol Aspects Med 34: 1168–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, Auwerx J (2015) NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, Zeviani M (2014) NAD(+)‐dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab 19: 1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Guarente L (2013) SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153: 1448–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Guarente L (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25: 138–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L (2008) Tissue‐specific regulation of SIRT1 by calorie restriction. Genes Dev 22: 1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Kowtoniuk WE, Agarwal I, Shen Y, Liu DR (2009) LC/MS analysis of cellular RNA reveals NAD‐linked RNA. Nat Chem Biol 5: 879–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins PB, Chaykin S (1971) Comparative metabolism of nicotinamide and nicotinic acid in mice. Biochem J 125: 117P [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins PB, Chaykin S (1972) The management of nicotinamide and nicotinic acid in the mouse. J Biol Chem 247: 778–783 [PubMed] [Google Scholar]
- Comings DE, Muhleman D, Dietz G, Sherman M, Forest GL (1995) Sequence of human tryptophan 2,3‐dioxygenase (TDO2): presence of a glucocorticoid response‐like element composed of a GTT repeat and an intronic CCCCT repeat. Genomics 29: 390–396 [DOI] [PubMed] [Google Scholar]
- Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, Church TS, Jubrias SA, Conley KE, Smith SR (2010) Skeletal muscle NAMPT is induced by exercise in humans. Am J Physiol Endocrinol Metab 298: E117–E126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubener W, MacKenzie CR (1999) IFN‐gamma activated indoleamine 2,3‐dioxygenase activity in human cells is an antiparasitic and an antibacterial effector mechanism. Adv Exp Med Biol 467: 517–524 [DOI] [PubMed] [Google Scholar]
- De Jesus‐Cortes H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, Melito LM, Wang G, Williams NS, Ready JM, McKnight SL, Pieper AA (2012) Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc Natl Acad Sci USA 109: 17010–17015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desquiret‐Dumas V, Gueguen N, Leman G, Baron S, Nivet‐Antoine V, Chupin S, Chevrollier A, Vessieres E, Ayer A, Ferre M, Bonneau D, Henrion D, Reynier P, Procaccio V (2013) Resveratrol induces a mitochondrial complex I‐dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J Biol Chem 288: 36662–36675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, Lokhorst HM, Parren PW (2016) Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev 270: 95–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew JE, Farquharson AJ, Horgan GW, Williams LM (2016) Tissue‐specific regulation of sirtuin and nicotinamide adenine dinucleotide biosynthetic pathways identified in C57Bl/6 mice in response to high‐fat feeding. J Nutr Biochem 37: 20–29 [DOI] [PubMed] [Google Scholar]
- Easlon E, Tsang F, Skinner C, Wang C, Lin SJ (2008) The malate‐aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction‐mediated life span extension in yeast. Genes Dev 22: 931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvehjem CA, Madden RJ, Strong FM, Woolley DW (1937) Relation of nicotinic acid and nicotinic acid amide to canine black tongue. J Am Chem Soc 59: 1767–1768 [Google Scholar]
- Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, Magni G (2001) Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J Biol Chem 276: 406–412 [DOI] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O'Neil L, White TA, Sinclair DA, Chini EN (2013) Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 62: 1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Euler H, Myrbäck K (1930) Co‐Zymase. XVII. Hoppe‐Seyler's Zeitschrift für physiologische Chemie 190: 93–100 [Google Scholar]
- Fang EF, Scheibye‐Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA (2014) Defective mitophagy in XPA via PARP‐1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157: 882–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Kassahun H, Croteau DL, Scheibye‐Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, Iser WB, Wollman BN, Morevati M, Li J, Kerr JS, Lu Q, Waltz TB, Tian J, Sinclair DA, Mattson MP et al (2016) NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 24: 566–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Paul IA, LeBlanc MH (2006) Nicotinamide reduces hypoxic ischemic brain injury in the newborn rat. Brain Res Bull 69: 117–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuwatari T, Morikawa Y, Sugimoto E, Shibata K (2002) Effects of fatty liver induced by niacin‐free diet with orotic acid on the metabolism of tryptophan to niacin in rats. Biosci Biotechnol Biochem 66: 1196–1204 [DOI] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V (2008) Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK‐mediated regulation of Nampt. Dev Cell 14: 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale EA, Bingley PJ, Emmett CL, Collier T, European Nicotinamide Diabetes Intervention Trial Group (2004) European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 363: 925–931 [DOI] [PubMed] [Google Scholar]
- Garavaglia S, Bruzzone S, Cassani C, Canella L, Allegrone G, Sturla L, Mannino E, Millo E, De Flora A, Rizzi M (2012) The high‐resolution crystal structure of periplasmic Haemophilus influenzae NAD nucleotidase reveals a novel enzymatic function of human CD73 related to NAD metabolism. Biochem J 441: 131–141 [DOI] [PubMed] [Google Scholar]
- Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, Moullan N, Zhang H, Perino A, Lemos V, Kim B, Park YK, Piersigilli A, Pham TX, Yang Y, Ku CS, Koo SI, Fomitchova A, Canto C, Schoonjans K et al (2016) Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63: 1190–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariani K, Ryu D, Menzies KJ, Yi HS, Stein S, Zhang H, Perino A, Lemos V, Katsyuba E, Jha P, Vijgen S, Rubbia‐Brandt L, Kim YK, Kim JT, Kim KS, Shong M, Schoonjans K, Auwerx J (2017) Inhibiting poly ADP‐ribosylation increases fatty acid oxidation and protects against fatty liver disease. J Hepatol 66: 132–141 [DOI] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J (2015) SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 348: 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell 155: 1624–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Pan Y, Vempati P, Zhao W, Knable L, Ho L, Wang J, Sastre M, Ono K, Sauve AA, Pasinetti GM (2013) Nicotinamide riboside restores cognition through an upregulation of proliferator‐activated receptor‐gamma coactivator 1alpha regulated beta‐secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol Aging 34: 1581–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Wang SR, Huang XZ, Xie QH, Xu YY, Shang D, Hao CM (2017) Nicotinamide mononucleotide, an NAD+ precursor, rescues age‐associated susceptibility to AKI in a sirtuin 1‐dependent manner. J Am Soc Nephrol 28: 2337–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, Jeune MR, Kaldor I, Milliken N, Petrov KG, Preugschat F, Schulte C, Shearer BG, Shearer T, Smalley TL Jr, Stewart EL et al (2015) Discovery, synthesis, and biological evaluation of Thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J Med Chem 58: 3548–3571 [DOI] [PubMed] [Google Scholar]
- Hagino Y, Lan SJ, Ng CY, Henderson LM (1968) Metabolism of pyridinium precursors of pyridine nucleotides in perfused rat liver. J Biol Chem 243: 4980–4986 [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5: 253–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL (2017) Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel‐induced peripheral neuropathy in female rats. Pain 158: 962–972 [DOI] [PubMed] [Google Scholar]
- Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, Tsuchiya M (2007) Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J Biol Chem 282: 24574–24582 [DOI] [PubMed] [Google Scholar]
- Harden A, Young WJ (1906) The alcoholic ferment of yeast‐juice. Proc Biol Sci 77: 405–420 [Google Scholar]
- Heyes MP, Saito K, Jacobowitz D, Markey SP, Takikawa O, Vickers JH (1992) Poliovirus induces indoleamine‐2,3‐dioxygenase and quinolinic acid synthesis in macaque brain. FASEB J 6: 2977–2989 [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J (2010) The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J (2009) Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res 105: 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wang H, Wang Q, Deng H (2014) Overexpression of CD38 decreases cellular NAD levels and alters the expression of proteins involved in energy metabolism and antioxidant defense. J Proteome Res 13: 786–795 [DOI] [PubMed] [Google Scholar]
- Hung YP, Albeck JG, Tantama M, Yellen G (2011) Imaging cytosolic NADH‐NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab 14: 545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi H, Ichiyama A, Hayaishi O (1966) Studies on the biosynthesis of nicotinamide adenine dinucleotide. 3. Comparative in vivo studies on nicotinic acid, nicotinamide, and quinolinic acid as precursors of nicotinamide adenine dinucleotide. J Biol Chem 241: 3701–3707 [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD‐dependent histone deacetylase. Nature 403: 795–800 [DOI] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Murphy CG, Doubrovina E, Jasin M, Moynahan ME (2016) PARP inhibitors in clinical use induce genomic instability in normal human cells. PLoS One 11: e0159341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TM, Rawling JM, Roebuck BD, Kirkland JB (1995) Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP‐ribose) levels but do not affect diethylnitrosamine‐induced altered hepatic foci in Fischer‐344 rats. J Nutr 125: 1455–1461 [DOI] [PubMed] [Google Scholar]
- Jacobson EL, Dame AJ, Pyrek JS, Jacobson MK (1995) Evaluating the role of niacin in human carcinogenesis. Biochimie 77: 394–398 [DOI] [PubMed] [Google Scholar]
- Jiao X, Doamekpor SK, Bird JG, Nickels BE, Tong L, Hart RP, Kiledjian M (2017) 5′ end nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO‐mediated deNADding. Cell 168: 1015–1027.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246: 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubin T, Kadam A, Jariwala M, Bhatt S, Sutariya S, Gani AR, Gautam S, Begum R (2016) The PARP family: insights into functional aspects of poly (ADP‐ribose) polymerase‐1 in cell growth and survival. Cell Prolif 49: 421–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabra DG, Thiyagarajan M, Kaul CL, Sharma SS (2004) Neuroprotective effect of 4‐amino‐1,8‐napthalimide, a poly(ADP ribose) polymerase inhibitor in middle cerebral artery occlusion‐induced focal cerebral ischemia in rat. Brain Res Bull 62: 425–433 [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia‐Menendez L, Kolwicz SC, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaundal RK, Shah KK, Sharma SS (2006) Neuroprotective effects of NU1025, a PARP inhibitor in cerebral ischemia are mediated through reduction in NAD depletion and DNA fragmentation. Life Sci 79: 2293–2302 [DOI] [PubMed] [Google Scholar]
- Kellenberger E, Kuhn I, Schuber F, Muller‐Steffner H (2011) Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett 21: 3939–3942 [DOI] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A (2014) Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med 6: 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaidman L, Morales M, Kem S, Yang J, Chang ML, Adams JD Jr (2003) Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology 69: 150–157 [DOI] [PubMed] [Google Scholar]
- Knip M, Douek IF, Moore WP, Gillmor HA, McLean AE, Bingley PJ, Gale EA, European Nicotinamide Diabetes Intervention Trial Group (2000) Safety of high‐dose nicotinamide: a review. Diabetologia 43: 1337–1345 [DOI] [PubMed] [Google Scholar]
- Koyama T, Kume S, Koya D, Araki S, Isshiki K, Chin‐Kanasaki M, Sugimoto T, Haneda M, Sugaya T, Kashiwagi A, Maegawa H, Uzu T (2011) SIRT3 attenuates palmitate‐induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med 51: 1258–1267 [DOI] [PubMed] [Google Scholar]
- Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB (2014) Nicotinamide N‐methyltransferase knockdown protects against diet‐induced obesity. Nature 508: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krehl WA, Teply LJ, Sarma PS, Elvehjem CA (1945) Growth‐retarding effect of corn in nicotinic acid‐low rations and its counteraction by tryptophane. Science 101: 489–490 [DOI] [PubMed] [Google Scholar]
- Kudo Y, Boyd CA (2000) Human placental indoleamine 2,3‐dioxygenase: cellular localization and characterization of an enzyme preventing fetal rejection. Biochim Biophys Acta 1500: 119–124 [DOI] [PubMed] [Google Scholar]
- Lee YH, Nair S, Rousseau E, Allison DB, Page GP, Tataranni PA, Bogardus C, Permana PA (2005) Microarray profiling of isolated abdominal subcutaneous adipocytes from obese vs non‐obese Pima Indians: increased expression of inflammation‐related genes. Diabetologia 48: 1776–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CF, Chavez JD, Garcia‐Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, Tian R (2016) Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134: 883–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann S, Loh SH, Martins LM (2017) Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson's disease. Biol Open 6: 141–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LF, Henderson LM (1972) Pyridinium precursors of pyridine nucleotides in perfused rat kidney and in the testis. J Biol Chem 247: 8023–8030 [PubMed] [Google Scholar]
- Lin SJ, Ford E, Haigis M, Liszt G, Guarente L (2004) Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev 18: 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, Nakamura R, Zapata N, Kubota M, Tsubota K, Yoshino J, Imai S, Apte RS (2016) NAMPT‐mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep 17: 69–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingor P, Koch JC, Tonges L, Bahr M (2012) Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res 349: 289–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Pitta M, Jiang H, Lee JH, Zhang G, Chen X, Kawamoto EM, Mattson MP (2013) Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34: 1564–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S (2008) Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 88: 841–886 [DOI] [PubMed] [Google Scholar]
- Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, Roqueta‐Rivera M, Deng C, Osborne TF, Mostoslavsky R, Baldi P, Sassone‐Corsi P (2014) Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158: 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ (2012) Age‐associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7: e42357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreanor GM, Bender DA (1986) The metabolism of high intakes of tryptophan, nicotinamide and nicotinic acid in the rat. Br J Nutr 56: 577–586 [DOI] [PubMed] [Google Scholar]
- Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G (2003) Functional interaction between PARP‐1 and PARP‐2 in chromosome stability and embryonic development in mouse. EMBO J 22: 2255–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies KJ, Zhang H, Katsyuba E, Auwerx J (2016) Protein acetylation in metabolism—metabolites and cofactors. Nat Rev Endocrinol 12: 43–60 [DOI] [PubMed] [Google Scholar]
- Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai SI (2016) Long‐term administration of nicotinamide mononucleotide mitigates age‐associated physiological decline in mice. Cell Metab 24: 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau C, Liu Q, Graeff R, Wagner GK, Thomas MP, Swarbrick JM, Shuto S, Lee HC, Hao Q, Potter BV (2013) CD38 structure‐based inhibitor design using the N1‐Cyclic inosine 5′‐diphosphate ribose template. PLoS One 8: e66247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N, Orsomando G (2014) Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One 9: e113939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A (2015) Sirtuin 3‐dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 125: 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J (2013) The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154: 430–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Cao Z, Horváth B, Park O, Wang H, Erdelyi K, Holovac E, Wang Y, Liaudet L, Hamdaoui N, Lafdil F, Haskó G, Szabo C, Boulares AH, Gao B, Pacher P (2014) Poly (ADP‐ribose) polymerase‐1 is a key mediator of liver inflammation and fibrosis. Hepatology 59: 1998–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horvath B, Rajesh M, Varga ZV, Gariani K, Ryu D, Cao Z, Holovac E, Park O, Zhou Z, Xu MJ, Wang W, Godlewski G, Paloczi J, Nemeth BT, Persidsky Y, Liaudet L, Hasko G, Bai P, Boulares AH et al (2017) PARP inhibition protects against alcoholic and non‐alcoholic steatohepatitis. J Hepatol 66: 589–600 [DOI] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone‐Corsi P (2009) Circadian control of the NAD+ salvage pathway by CLOCK‐SIRT1. Science 324: 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro J, Gozalbo‐Lopez B, Mendez AC, Dantzer F, Schreiber V, Martinez C, Arana DM, Farres J, Revilla‐Nuin B, Bueno MF, Ampurdanes C, Galindo‐Campos MA, Knobel PA, Segura‐Bayona S, Martin‐Caballero J, Stracker TH, Aparicio P, Del Val M, Yelamos J (2017) PARP‐1/PARP‐2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci Rep 7: 41962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov A, Dolle C, Niere M, Ziegler M (2011) Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 286: 21767–21778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos PR, Hodgson MI, Maiz A, Manrique M, De Valdes MD, Foncea R, Acosta AM, Emmerich MV, Velasco S, Muniz OP, Oyarzun CA, Claro JC, Bastias MJ, Toro LA (2006) Nicotinamide protected first‐phase insulin response (FPIR) and prevented clinical disease in first‐degree relatives of type‐1 diabetics. Diabetes Res Clin Pract 71: 320–333 [DOI] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J (2013) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342: 1243417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penke M, Larsen PS, Schuster S, Dall M, Jensen BA, Gorski T, Meusel A, Richter S, Vienberg SG, Treebak JT, Kiess W, Garten A (2015) Hepatic NAD salvage pathway is enhanced in mice on a high‐fat diet. Mol Cell Endocrinol 412: 65–72 [DOI] [PubMed] [Google Scholar]
- de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S, Seals DR (2016) Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15: 522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen CH, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J et al (2010) Discovery of a proneurogenic, neuroprotective chemical. Cell 142: 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai JB, Isbatan A, Imai S, Gupta MP (2005) Poly(ADP‐ribose) polymerase‐1‐dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem 280: 43121–43130 [DOI] [PubMed] [Google Scholar]
- Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP (2010) Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3‐LKB1‐AMP‐activated kinase pathway. J Biol Chem 285: 3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies KJ, Williams EG, Mouchiroud L, Moullan N, Hagberg C, Li W, Timmers S, Imhof R, Verbeek J, Pujol A, van Loon B, Viscomi C, Zeviani M, Schrauwen P, Sauve AA et al (2014) Pharmacological inhibition of poly(adp‐ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab 19: 1034–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss J, Handler P (1958) Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J Biol Chem 233: 488–492 [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJ, Duarte FV, Martin‐Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R et al (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15: 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM (2006) Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem 281: 21745–21754 [DOI] [PubMed] [Google Scholar]
- Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, Funaro A, Horenstein AL, Malavasi F (2013) CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom 84: 207–217 [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, Takahashi JS, Si Imai, Bass J (2009) Circadian Clock feedback cycle through NAMPT‐mediated NAD+ biosynthesis. Science 324: 651–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C, Canto C (2016) NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun 7: 13103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard JF Jr (1998) Altered tryptophan metabolism in mice with herpes simplex virus encephalitis: increases in spinal cord quinolinic acid. Neurochem Res 23: 661–665 [DOI] [PubMed] [Google Scholar]
- Richard DM, Dawes MA, Mathias CW, Acheson A, Hill‐Kapturczak N, Dougherty DM (2009) L‐Tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int J Tryptophan Res 2: 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer M, Erwa W, Zimmermann R, Frank S, Zechner R (2009) Adipose tissue as a source of nicotinamide N‐methyltransferase and homocysteine. Atherosclerosis 204: 412–417 [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC‐1alpha and SIRT1. Nature 434: 113–118 [DOI] [PubMed] [Google Scholar]
- Rutter J, Reick M, Wu LC, McKnight SL (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293: 510–514 [DOI] [PubMed] [Google Scholar]
- Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Canto C, Conley KE, Walter L, Lovering RM, Chin ER et al (2016) NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med 8: 361ra139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadanaga‐Akiyoshi F, Yao H, Tanuma S, Nakahara T, Hong JS, Ibayashi S, Uchimura H, Fujishima M (2003) Nicotinamide attenuates focal ischemic brain injury in rats: with special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochem Res 28: 1227–1234 [DOI] [PubMed] [Google Scholar]
- Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, Cheeseman M, Nunez D, Sweatman BC, Haselden JN, Cox RD, Connor SC, Griffin JL (2007) A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol Genomics 29: 99–108 [DOI] [PubMed] [Google Scholar]
- Sanni LA, Thomas SR, Tattam BN, Moore DE, Chaudhri G, Stocker R, Hunt NH (1998) Dramatic changes in oxidative tryptophan metabolism along the kynurenine pathway in experimental cerebral and noncerebral malaria. Am J Pathol 152: 611–619 [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J (2006) Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci 26: 8484–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Stein L, Imai S (2011) The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb Exp Pharmacol 206: 125–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye‐Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan‐Olive MM, Mangerich A, Wilson MA, Mattson MP, Bergersen LH, Cogger VC, Warren A, Le Couteur DG, Moaddel R, Wilson DM III, Croteau DL et al (2014) A high‐fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20: 840–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A (2007) SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci 48: 3602–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky H (2001) Tryptophan: biochemical and health implications. Boca Raton, FL: CRC Press; [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA (2010) Sirt3 mediates reduction of oxidative damage and prevention of age‐related hearing loss under caloric restriction. Cell 143: 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarbrick JM, Graeff R, Zhang H, Thomas MP, Hao Q, Potter BV (2014) Cyclic adenosine 5′‐diphosphate ribose analogs without a “southern” ribose inhibit ADP‐ribosyl cyclase‐hydrolase CD38. J Med Chem 57: 8517–8529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa O, Yoshida R, Kido R, Hayaishi O (1986) Tryptophan degradation in mice initiated by indoleamine 2,3‐dioxygenase. J Biol Chem 261: 3648–3653 [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, Starwalt R, Morlock L, Naidoo J, Williams NS, Ready JM, McKnight SL, Pieper AA (2012) Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 109: 17016–17021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Brenner C (2013) Targeted, LCMS‐based metabolomics for quantitative measurement of NAD(+) metabolites. Comput Struct Biotechnol J 4: e201301012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C (2016a) Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 7: 12948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, Brenner C (2016b) Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep 6: 26933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, Parikh SM (2016) PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala KS, Gomes AL, Yilmaz M, Graña O, Bakiri L, Ruppen I, Ximénez‐Embún P, Sheshappanavar V, Rodriguez‐Justo M, Pisano DG, Wagner EF, Djouder N (2014) Inhibition of de novo NAD(+) synthesis by oncogenic URI Causes liver tumorigenesis through DNA damage. Cancer Cell 26: 826–839 [DOI] [PubMed] [Google Scholar]
- Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A (2014) Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP‐1 activity in Abeta(1‐42)‐induced rat model of Alzheimer's disease. Free Radical Res 48: 146–158 [DOI] [PubMed] [Google Scholar]
- Ugur S, Ulu R, Dogukan A, Gurel A, Yigit IP, Gozel N, Aygen B, Ilhan N (2015) The renoprotective effect of curcumin in cisplatin‐induced nephrotoxicity. Ren Fail 37: 332–336 [DOI] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M (2011) In vivo correction of COX deficiency by activation of the AMPK/PGC‐1alpha axis. Cell Metab 14: 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell A, Ekblad T, Oncu D, Kull B, Robertson GM, Pellicciari R, Schuler H, Weigelt J (2012) Family‐wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol 30: 283–288 [DOI] [PubMed] [Google Scholar]
- Walters RW, Matheny T, Mizoue LS, Rao BS, Muhlrad D, Parker R (2017) Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 114: 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z (2005) A local mechanism mediates NAD‐dependent protection of axon degeneration. J Cell Biol 170: 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL (2014a) P7C3 neuroprotective chemicals function by activating the rate‐limiting enzyme in NAD salvage. Cell 158: 1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhu W, Wang X, Li J, Zhang K, Zhang L, Zhao YJ, Lee HC, Zhang L (2014b) Design, synthesis and SAR studies of NAD analogues as potent inhibitors towards CD38 NADase. Molecules 19: 15754–15767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hu X, Yang Y, Takata T, Sakurai T (2016a) Nicotinamide mononucleotide protects against beta‐amyloid oligomer‐induced cognitive impairment and neuronal death. Brain Res 1643: 1–9 [DOI] [PubMed] [Google Scholar]
- Wang YQ, Wang PY, Wang YT, Yang GF, Zhang A, Miao ZH (2016b) An update on poly(ADP‐ribose)polymerase‐1 (PARP‐1) inhibitors: opportunities and challenges in cancer therapy. J Med Chem 59: 9575–9598 [DOI] [PubMed] [Google Scholar]
- Warburg O, Christian W (1936) Pyridine, the hydrogen transfusing component of fermentative enzymes. Helv Chim Acta 19: 79–88 [Google Scholar]
- van de Weijer T, Phielix E, Bilet L, Williams EG, Ropelle ER, Bierwagen A, Livingstone R, Nowotny P, Sparks LM, Paglialunga S, Szendroedi J, Havekes B, Moullan N, Pirinen E, Hwang JH, Schrauwen‐Hinderling VB, Hesselink MK, Auwerx J, Roden M, Schrauwen P (2015) Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 64: 1193–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JN, Feigelson P, Elvehjem CA (1950) Relation of tryptophan and niacin to pyridine nucleotides of tissue. J Biol Chem 187: 597–604 [PubMed] [Google Scholar]
- Williams GT, Lau KM, Coote JM, Johnstone AP (1985) NAD metabolism and mitogen stimulation of human lymphocytes. Exp Cell Res 160: 419–426 [DOI] [PubMed] [Google Scholar]
- Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, John SW (2017) Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355: 756–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik M, Seidle HF, Bieganowski P, Brenner C (2006) Glutamine‐dependent NAD+ synthetase. How a two‐domain, three‐substrate enzyme avoids waste. J Biol Chem 281: 33395–33402 [DOI] [PubMed] [Google Scholar]
- Xie L, Wang Z, Li C, Yang K, Liang Y (2017) Protective effect of nicotinamide adenine dinucleotide (NAD+) against spinal cord ischemia‐reperfusion injury via reducing oxidative stress‐induced neuronal apoptosis. J Clin Neurosci 36: 114–119 [DOI] [PubMed] [Google Scholar]
- Xu W, Barrientos T, Mao L, Rockman HA, Sauve AA, Andrews NC (2015) Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep 13: 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi H, Togawa K, Moritani M, Itakura M (2005) Identification of candidate genes in the type 2 diabetes modifier locus using expression QTL. Genomics 85: 591–599 [DOI] [PubMed] [Google Scholar]
- Yalowitz JA, Xiao S, Biju MP, Antony AC, Cummings OW, Deeg MA, Jayaram HN (2004) Characterization of human brain nicotinamide 5′‐mononucleotide adenylyltransferase‐2 and expression in human pancreas. Biochem J 377: 317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J (2014) Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 9: e98972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki F, Kuroiwa T, Takikawa O, Kido R (1985) Human indolylamine 2,3‐dioxygenase. Its tissue distribution, and characterization of the placental enzyme. Biochem J 230: 635–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming Dudley W, Souza‐Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve Anthony A, Sinclair DA (2007) Nutrient‐sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130: 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SJ, Choi JM, Kim L, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, Park C‐Y (2014) Nicotinamide improves glucose metabolism and affects the hepatic NAD‐sirtuin pathway in a rodent model of obesity and type 2 diabetes. J Nutr Biochem 25: 66–72 [DOI] [PubMed] [Google Scholar]
- Yano M, Akazawa H, Oka T, Yabumoto C, Kudo‐Sakamoto Y, Kamo T, Shimizu Y, Yagi H, Naito AT, Lee JK, Suzuki J, Sakata Y, Komuro I (2015) Monocyte‐derived extracellular Nampt‐dependent biosynthesis of NAD(+) protects the heart against pressure overload. Sci Rep 5: 15857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin TC, Britt JK, De Jesus‐Cortes H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, Wassink C, McDaniel L, Newell EA, Dutca LM, Naidoo J, Cui H, Bassuk AG, Harper MM, McKnight SL, Ready JM et al (2014) P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep 8: 1731–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10: 179–206 [DOI] [PubMed] [Google Scholar]
- Yoshida R, Hayaishi O (1978) Induction of pulmonary indoleamine 2,3‐dioxygenase by intraperitoneal injection of bacterial lipopolysaccharide. Proc Natl Acad Sci USA 75: 3998–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida R, Urade Y, Tokuda M, Hayaishi O (1979) Induction of indoleamine 2,3‐dioxygenase in mouse lung during virus infection. Proc Natl Acad Sci USA 76: 4084–4086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, S‐i Imai (2011) Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 14: 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GS, Choleris E, Lund FE, Kirkland JB (2006) Decreased cADPR and increased NAD+ in the Cd38‐/‐ mouse. Biochem Biophys Res Commun 346: 188–192 [DOI] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H (2003) Structural characterization of a human cytosolic NMN/NAMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem 278: 13503–13511 [DOI] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J (2016) NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352: 1436–1443 [DOI] [PubMed] [Google Scholar]
- Zheng CB, Han J, Xia WL, Shi ST, Liu JR, Ying WH (2012) NAD(+) administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett 512: 67–71 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Ting KY, Lam CM, Kwong AK, Xia J, Jin H, Liu Z, Zhang L, Cheung Lee H, Zhang L (2012) Design, synthesis and biological evaluation of noncovalent inhibitors of human CD38 NADase. ChemMedChem 7: 223–228 [DOI] [PubMed] [Google Scholar]
- Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, Brantley AF, Lasmezas CI (2015) Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 138: 992–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Fu B, Bai X, Zhang B, Wu L, Cui J, Cui S, Wei R, Chen X, Cai G (2011) NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins‐AMPK‐mTOR pathway. Cellular Physiol Biochem 27: 681–690 [DOI] [PubMed] [Google Scholar]