

Abstract
HIV-1 Vpu modulates cellular transmembrane proteins to optimize viral replication and provide immune-evasion, triggering ubiquitin-mediated degradation of some targets but also modulating endosomal trafficking to deplete them from the plasma membrane. Interactions between Vpu and the heterotetrameric clathrin adaptors AP-1 and AP-2 have been described, yet the molecular basis and functional roles of such interactions are incompletely defined. To investigate the trafficking signals encoded by Vpu, we fused the cytoplasmic domain (CD) of Vpu to the extracellular and transmembrane domains of the CD8 α-chain. CD8-VpuCD was rapidly endocytosed in a clathrin- and AP-2-dependent manner. Multiple determinants within the Vpu CD contributed to endocytic activity, including phosphoserines of the β-TrCP binding site and a leucine-based ExxxLV motif. Using recombinant proteins, we confirmed ExxxLV-dependent binding of the Vpu CD to the α/σ2 subunit hemicomplex of AP-2 and showed that this is enhanced by serine-phosphorylation. Remarkably, the Vpu CD also bound directly to the medium (μ) subunits of AP-2 and AP-1; this interaction was dependent on serine-phosphorylation of Vpu and on basic residues in the μ subunits. We propose that the flexibility with which Vpu binds AP complexes broadens the range of cellular targets that it can misdirect to the virus’s advantage.
Keywords: Endocytosis, clathrin, adaptor protein, human immunodeficiency virus type-1, HIV-1, Vpu, bone marrow stromal antigen 2, BST-2
Graphical Abstract
INTRODUCTION
Counteraction of host immune defenses is critical to persistent viral infection. The human immunodeficiency virus type 1 (HIV-1) encodes four “accessory” proteins: Vpu, Nef, Vif and Vpr, which provide evasion of host adaptive and innate immune responses. To overcome a diverse range of host defenses, including the intrinsic cellular immunity provided by so-called “restriction factors”, these viral accessory proteins co-opt host degradative and protein-trafficking processes by functioning as molecular adaptors that form ternary complexes with the targeted host factors and components of the host trafficking machinery.
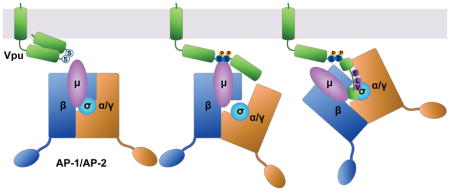
The accessory proteins Vpu and Nef alter the content of the plasma membrane through modulation of cellular transmembrane proteins. Targets of Vpu and Nef include restriction factors such as BST-2, which physically tethers newly budded virions to the plasma membrane 1,2, as well as immuno-receptors such as CD4, which functions as the virus’s primary receptor for entry into cells but which also inhibits viral infectivity 3,4 and sensitizes the viral envelope glycoprotein to host antibodies if unperturbed by Vpu and Nef during virion-production 5,6. Vpu and Nef divide the task of modulating the plasma membrane in a manner that is distinct yet shares certain features, both in terms of the host proteins targeted and in terms of mechanism of action (reviewed by 7). For example, Vpu and Nef each target CD4. In contrast, in a given virus, one or the other protein targets BST-2: Vpu targets BST-2 in most group M HIV-1 strains (the main cause of the pandemic), but in group O (outlier) strains, as well as in most strains of SIV, this activity is subsumed by Nef 8–10. Vpu and Nef are structurally dissimilar, yet both act at least partly via clathrin and clathrin-adaptors. Nef is a 27 kDa N-terminally myristoylated peripheral membrane protein; it consists of three domains: a flexible N-terminal region, a folded core, and a C-terminal loop. The C-terminal loop contains an ExxxLφ motif that binds the heterotetrameric clathrin adaptor protein (AP) complexes; this motif directs the rapid endocytosis of CD4 via AP-2 11, and it is also required for the modulation of BST-2 by Nef proteins that have this activity 12. Vpu is a 17 kDa single-pass type I transmembrane protein; it consists of a short N-terminus, a helical transmembrane domain (TMD), and a C-terminal cytoplasmic domain (CD). The TMD not only participates in the oligomerization of Vpu, but its “alanine-face” mediates the interaction of Vpu with the TMDs of many cellular targets, as exemplified by BST-2 13. The CD, when not complexed with other proteins, consists of two α-helices connected by a flexible linker region. The linker region contains two serines that when phosphorylated constitute a “phosphodegron” that recruits β-TrCP, a substrate adaptor for a cullin1-based E3 ubiquitin ligase complex 14. By this means, Vpu recruits the host ubiquitination machinery to stimulate the degradation of CD4 via an ER-associated degradation (ERAD)-like pathway leading to the proteasome 14–17. Vpu also stimulates the ubiquitination of BST-2 and its degradation within lysosomes 18,19.
The activity of Vpu, at least with respect to BST-2, is also clathrin-dependent 20–22. Vpu-mediated down-regulation of BST-2 from the cell surface is partly dependent on AP-2 20, and surface-downregulation as well as the ability of Vpu to counteract BST-2-mediated restriction of virion-release is reversed by expression of a dominant version of the clathrin assembly factor AP180 21,22. Moreover, group M Vpu proteins of subtype B strains contain an ExxxLV sequence within the membrane distal α-helix; this sequence resembles a variant ExxxLφ motif that is recognized by AP complexes 22. We have shown that this Vpu sequence binds in an extended conformation to the canonical recognition site on the σ (small) and γ (large-specific) subunits of AP-1 and that this interaction is stabilized when the BST-2 CD, which contains a variant Yxxφ AP-binding sequence, is bound to the tyrosine-binding pocket of the AP-1 μ subunit 23. Binding of the Vpu CD to an AP-2 hemi-complex formed by α and σ2 subunits, as well as to the μ2 subunit, was also observed in vitro 23. Recently, interactions between Vpu and both AP-1 and AP-2 have been detected in human cells by biotinylation-proximity and chemical cross-linking/immunoprecipitation assays 24. Like some of the interactions detected using recombinant proteins, these in vivo interactions of Vpu with AP complexes also required the presence of BST-2. Remarkably, these in vivo interactions required not only Vpu’s ExxxLV sequence but also phosphorylation of the key serines noted above, leading to a model in which phosphorylation induces conformational changes that allow binding of the ExxxLV sequence to AP-2 24.
While the effects of Vpu expression on the trafficking of its cellular targets have been extensively studied, little is known about the trafficking itinerary of Vpu itself. Whether previously identified motifs located in the cytoplasmic domain of Vpu function as autonomous sorting signals or instead require target-binding to promote cooperative association with clathrin adaptors or other trafficking machineries is currently unclear. For example, microscopic data suggest that the subcellular distribution of Vpu lacking its ExxxLV sequence is influenced by the presence of BST-2, whereas wild type Vpu is not22. Vpu causes a net surface downregulation of its targets, yet a clear role for stimulation of endocytosis is lacking; rather the preponderance of current data suggest that the primary role of Vpu is to sequester targeted proteins within the endosomal system, either within the biosynthetic pathway or through inhibition of recycling to the plasma membrane following constitutive endocytosis.
In the present investigation, we fused the cytoplasmic domain of Vpu to the extracellular/lumenal and transmembrane domains of the CD8 α-chain and used this chimera to study the trafficking properties of the Vpu CD independently of cellular targets that interact with Vpu via its TMD. We determined that the Vpu CD provides endocytic activity equal to or greater than that of Nef, but that unlike Nef, which relies wholly on its ExxxLL sequence, endocytic activity is distributed among multiple determinants in the Vpu CD. The rapid endocytic rate conferred by the Vpu CD requires clathrin and the clathrin-adaptor AP-2. Finally, we determined that the serines 52 and 56 in the Vpu CD likely contribute to these effects through two mechanisms. Serine phosphorylation enhances binding of the ExxxLV motif to the AP-2 hemicomplex of α/σ2 subunits, consistent with recent structural and functional studies 23,24. In addition, the phosphoserines mediate direct binding to the μ subunit of AP complexes, recognizing basic patches on the μ subunits of both AP-2 and AP-1. We have previously shown that these basic patches (on the μ subunit of AP-1) are recognized by Nef when complexed with the cytoplasmic domain of the class I MHC α chain 25. Thus, our data reveal a striking convergence in how these two very different viral proteins co-opt clathrin adaptors. Our data confirm a duality in which the key serines in Vpu act both as a phosphodegron via the cullin1-based E3 ligase complex and in regulating the association of Vpu with clathrin AP complexes. Moreover, the data reveal a bimodal mechanism for the phosphoserine-dependent interaction of Vpu with AP complexes: the phosphoserines not only facilitate binding of the ExxxLV motif to AP hemicomplexes, they are part of a phosphoserine-acidic cluster that mediates binding to basic patches on the μ subunits.
RESULTS
The cytoplasmic domain of Vpu contains endocytic determinant(s)
To determine whether the cytoplasmic domain (CD) of Vpu contains endocytic determinants that function autonomously, and in particular independently of such determinants that might be present in the CDs of Vpu’s cellular targets, we created a chimeric protein comprising residues 28–81 of HIV-1 NL4.3 Vpu fused to the extracellular/lumenal and transmembrane domains (residues 1–212) of the human CD8 α-chain (as shown in Figure 1A). This and similar strategies have been used previously to evaluate autonomous sorting signals in the CDs of cellular proteins and to evaluate the trafficking activity of HIV-1 Nef 26–29. HeLa-P4.R5 cells, which express CD4, were transiently transfected to express the CD8-VpuCD chimera, a truncated CD8 lacking most of its cytoplasmic domain as a control, CD8-Nef, or CD8-Nef-LL/AA, which due to alanine substitutions of the conserved dileucine motif at Nef-positions 164 and 165 is unable to interact with the clathrin adaptor complex AP-2. Cell surface expression of the chimeric proteins was measured by flow cytometry after staining for CD8 (Figure 1B and 1C). The mean fluorescence intensity of CD8-VpuCD or CD8-Nef at the cell surface was reduced in cells relative to truncated CD8- or CD8-Nef-LL/AA. The high surface expression of CD8-Nef-LL/AA is consistent with previous observations and the critical role of Nef’s dileucine motif in binding to clathrin adaptors, in particular AP-2 28. The expression of the chimeras at steady-state was assessed by western blot (Figure 1D). The addition of the VpuCD or Nef appeared to stabilize expression, while the expression of truncated CD8 was relatively low. CD8-Nef and CD8-Nef-LL/AA were expressed at equal levels. These data demonstrated that the surface expression of the chimeric proteins is independent of total cellular protein concentration and likely depends upon the trafficking properties of their CDs.
Figure 1. The cytoplasmic domain of NL4.3 Vpu contains endocytic determinant(s).

(A) Fusions of human CD8α chain transmembrane and lumenal domains (CD8) with HIV-1 NL4.3 Vpu, Nef or Nef-LL/AA. (B) Surface expression of CD8 constructs in HeLa P4.R5 cells. Cells were co-transfected to express CD8−, CD8-Vpu, CD8-Nef or CD8-Nef-LL/AA, together with a GFP expression construct as a transfection marker. Cell surface CD8 was stained with fluorescent antibody and the cells analyzed by two-color flow cytometry. The graph indicates the mean fluorescence intensity of surface CD8 staining in the GFP-positive cell population, error bars represent the standard deviation of three independent experiments. (C) Representative two-color plots of CD8 expression in GFP-positive cells are shown. (D) Expression of the CD8 chimeras was measured in cell lysates subjected to SDS-PAGE and Western blotting. Control samples contain GFP-transfected cells only. (E) The endocytic rates of the CD8-chimeras were measured in HeLa P4.R5 cells. Cells expressing CD8, CD8-VpuCD, CD8-Nef or CD8-Nef-LL/AA and a GFP expression construct were labeled for surface CD8 using a primary anti-CD8 antibody while on ice. The cells were warmed to 37°C for the indicated time points, and remaining surface CD8 antibody was detected by fluorophore-conjugated secondary antibody staining and two-color flow cytometric analysis. The endocytic rate of endogenous TfnR was also measured using the same method. Data are normalized to the 0-timepoint control (cells not warmed before secondary staining), and presented as the percentage of CD8 remaining on the cell surface over time. Error bars indicate standard deviation of three independent experiments. (F) Two-color plots of the time course of CD8-Vpu surface staining in GFP-positive cells are shown. (G) The rate of surface replenishment of the CD8-chimeras measured in HeLa P4.R5 cells. Surface CD8 was blocked using unconjugated anti-CD8 antibody while the cells were on ice. The cells were warmed to 37°C for the indicated time points and CD8 newly deposited at the cell surface was labeled with fluorophore-conjugated anti-CD8 antibody. Data are the mean fluorescence intensity of CD8 in GFP-positive cells; error bars indicate the standard deviation of two independent experiments. Representative two-color plots of CD8-Vpu surface replenishment over time are shown in (H).
The low surface expression of CD8-VpuCD indicated either that the protein was retained in the biosynthetic pathway or that it was specifically endocytosed from the cell surface, or both. To test the hypothesis that the VpuCD stimulates endocytosis, we compared the internalization rates of CD8−, CD8-VpuCD, CD8-Nef and CD8-Nef-LL/AA in transiently-transfected HeLa-P4.R5 cells. The endocytic rates of the chimeric proteins were also compared to that of transferrin receptor (TfnR), an endogenous marker for clathrin-mediated endocytosis and recycling. For endocytosis assays, surface CD8 or TfnR were labeled at 4°C with primary antibodies before warming to 37°C for the indicated time-points, and antibody remaining at the cell surface was detected by staining with an immunofluorescent secondary antibody. The mean fluorescence intensity of CD8 or TfnR was normalized to the zero time-point and is presented as the percentage remaining on the cell surface over time (Figure 1E and 1F). CD8-VpuCD and CD8-Nef were rapidly endocytosed from the cell surface, with a 61% loss in CD8-VpuCD and a 38% loss in CD8-Nef signal observed after the first 2 minutes of incubation at 37°C. TfnR was also rapidly endocytosed, as anticipated. In contrast, little or no endocytic activity was observed for truncated CD8- and CD8-Nef-LL/AA; these proteins essentially remained at the cell surface. Fusion of CD8 with the CD of clade C Vpu (MJ4 or 96BW clones) resulted in rapid endocytosis, at rates similar to that induced by the CD the Vpu clade B clone NL4.3 (Figure S1).
Although the TMD of Vpu is the main mediator of the protein’s interaction with cellular targets, the CD of Vpu contributes to the interaction with CD4 30. To exclude a contribution of CD4 to the endocytic rate of CD8-Vpu, we repeated these experiments in HeLa-Z24 cells 31, a precursor line to HeLa-P4.R5, which do not express CD4 (Figure S2). The rates of endocytosis of CD8-Vpu were equivalent in the two cell types, indicating that CD4 was not a major determinant of endocytic rate.
The kinetics of surface-deposition of the CD8 chimeras were also studied in transiently-transfected HeLa-P4.R5 cells by flow cytometry. Surface CD8 was labeled at 4°C using unconjugated primary anti-CD8 antibody to block all available sites on the cell surface; the cells were warmed to 37°C for the indicated times; and CD8 newly deposited at the cell surface was detected using fluorophore-conjugated primary anti-CD8 antibody (Figure 1G and H). CD8-VpuCD and CD8-Nef were rapidly deposited at the cell surface, whereas CD8- and CD8-Nef-LL/AA were deposited at a much slower rate.
These data demonstrate that the Vpu cytoplasmic domain, like Nef, contains potent endocytic determinants, evident by its ability to confer trafficking activity to the otherwise inert truncated CD8 α chain. Moreover, the rapid rate of internalization of CD8-VpuCD and its similarity to that of TfnR suggest that the VpuCD, like Nef, drives internalization through a clathrin-mediated endocytic pathway. Finally, the rapid rates of surface-deposition of CD8-VpuCD and CD8-Nef, in contrast to the slow rate of surface-deposition of truncated CD8 and CD8-Nef-LL/AA, suggest that the rapid endocytosis driven by Vpu and Nef is potentially associated with a rapid rate of recycling.
CD8-VpuCD accumulates in recycling- and late-endosomes
The subcellular localization of the CD8-chimeras was investigated in HeLa-P4.R5 cells using immunofluorescence microscopy. Microscopic analysis revealed that CD8-VpuCD and CD8-Nef accumulated in enlarged vesicles, while CD8- and CD8-Nef-LL/AA were restricted to the plasma membrane (Figure 2A). Given the rapid rate of endocytosis and surface-deposition of CD8-VpuCD, we hypothesized that the protein would co-localize with markers of the clathrin-mediated endo-lysosomal pathway as well as with markers of recycling endosomes. To test this, HeLa-P4.R5 cells were either transfected to express CD8-VpuCD alone, or co-transfected to express CD8-VpuCD and DS-Red Clathrin. The cells expressing CD8-VpuCD alone were stained using antibodies to detect either transferrin receptor (TfnR) a marker for clathrin-mediated endocytosis and for recycling endosomes, or CD63, a marker for late endosomes, in addition to CD8. The images shown in Figure 2B indicate that the enlarged vesicles containing CD8-VpuCD were positive for Clathrin, TfnR and CD63. The majority of the vesicles appeared to be positive for CD63, indicating a predominant accumulation of CD8-VpuCD in late endosomes. CD8-Nef, like CD8-Vpu, also colocalized substantially with markers of the clathrin-mediated endo-lysosomal pathway (Figure S3), while CD8- and CD8-Nef-LL/AA show little colocalization and were found predominantly at the plasma membrane.
Figure 2. CD8-Vpu-CD colocalizes with markers of clathrin-mediated endosomal trafficking pathways.

(A) Subcellular localization of CD8 chimeras in HeLa P4.R5 cells. Transfected cells expressing CD8−, CD8-VpuCD, CD8-Nef or CD8-Nef-LL/AA were fixed, permeabilized, and stained using fluorophore-conjugated anti-CD8 antibody. (B) Colocalization of CD8-Vpu with clathrin and endosomal markers. HeLa P4.R5 cells were either co-transfected with plasmids encoding CD8-VpuCD and DSRed-Clathrin, or transfected to express CD8-VpuCD alone. The cells were fixed, permeabilized, blocked and stained for CD8 (green) and the endosomal markers transferrin receptor (TfnR, red) or CD63 (late endosomes, red). The cells were examined by widefield, deconvolution microscopy; images are z-stack projections of the total cell volumes. Scale bars = 10 μm.
Endocytic activity of VpuCD requires clathrin and AP-2
We next examined whether perturbation of the clathrin-mediated endocytic pathway interfered with the trafficking of CD8-VpuCD. HeLa-P4.R5 cells were transfected with siRNAs targeting clathrin heavy chain (siClathrin) or a negative control siRNA (siControl) 48 hours before transfection with plasmids expressing the CD8 chimeras and a plasmid expressing GFP. Endocytosis assays were performed the following day, as described above for the experiments of Figure 1. The mean fluorescence intensity (MFI) of CD8 staining at steady-state (the 0-timepoint control) is shown in Figure 3A. Transfection with siClathrin significantly increased the surface levels of CD8-VpuCD and CD8-Nef at steady state. To analyze internalization kinetics, the data were normalized to the MFI at steady-state and presented as the percentage of CD8 remaining at the cell surface over time in either siControl- or siClathrin-transfected cells (Figure 3B and 3C). The internalization rates of CD8-VpuCD and CD8-Nef were substantially and significantly diminished by clathrin-depletion, whereas CD8- and CD8-Nef-LL/AA remained unaffected. siRNA-mediated depletion of clathrin heavy chain (CHC) was confirmed by western blotting (Figure 3D); clathrin was detectable in cells transfected with siControl but not siClathrin.
Figure 3. Endocytosis of CD8-Vpu-CD is sensitive to siRNA-mediated depletion of clathrin.

(A) The rates of internalization of the truncated CD8−, CD8-VpuCD, CD8-Nef, or CD8-Nef-LL/AA were measured in clathrin-depleted HeLa P4.R5 cells by flow cytometry. The cells were transfected with siRNA oligonucleotides directed against Clathrin (siClathrin) or a non-targeting control (siControl). After 48 hours, the cells were transfected to express the indicated CD8-chimeras and a GFP expression construct. 24 hours later, the internalization rates of the CD8- chimeras were measured by antibody labeling and flow cytometric analysis. Steady-state surface levels of CD8-chimeras are shown (0-timepoint control). (B and C) Data were normalized to the 0-timepoint control and presented as the percentage surface CD8 remaining over time, in either siControl-treated (B) or clathrin-depleted (C) cells. Error bars indicate the standard deviation of three independent experiments. P-values were generated by paired two-tailed t-test, * P<0.05, ** P<0.001, *** P <0.001. (D) Protein from cell lysates was separated by SDS-PAGE and clathrin depletion confirmed by western blotting. (E) The intracellular localization of CD8-VpuCD was determined by widefield microscopy of HeLa P4.R5 cells transfected with negative control or clathrin siRNA. To identify clathrin depleted cells with impaired endocytosis, cells were serum-starved prior to uptake of fluorescent transferrin (red) for 30 minutes; the cells were then fixed and counterstained for CD8 (green). Scale = 10 μm.
To confirm the role of clathrin in trafficking mediated by the Vpu CD, the subcellular localization of CD8-VpuCD with respect to the itinerary of transferrin was examined in HeLa-P4.R5 cells treated with either siControl or siClathrin. Cells were transfected to express CD8-VpuCD 48 hours after treatment with the siRNAs. The following day, the cells were serum-starved for 30 minutes prior to the addition of Alexa-Fluor-555 conjugated transferrin, and incubated for a further 30 minutes before fixation and staining for CD8. Since transferrin binds to TfnR and enters the cell via clathrin-mediated endocytosis, its uptake was used as a control to identify cells in which clathrin was depleted; transferrin should remain at the cell surface in such cells. CD8-VpuCD colocalized with transferrin-positive vesicles in siControl-treated cells (Figure 3E), consistent with the data of Figure 2. In contrast, in the clathrin-depleted cells identified functionally by diminished transferrin uptake, CD8-VpuCD accumulated at the plasma membrane, similar to the staining pattern of truncated CD8. Together, these flow cytometric and microscopic data indicated that clathrin is required for the function of the endocytic determinants in CD of Vpu.
We next investigated whether the clathrin adaptor AP-2 was required for internalization of the CD8-VpuCD chimera. We targeted the AP-2 α subunit for siRNA-mediated knockdown; depletion of any one of the AP complex subunits decreases stability of the remaining subunits and results in inactive, partial complex formation 32–34. HeLa-P4.R5 cells were transfected with siControl or siRNA targeted to AP-2 α (siAP-2α). 48 hours later, the cells were transfected to express the CD8- chimeras and GFP, and steady-state surface-expression and endocytic rate were measured the next day, as described above. The MFI of surface CD8 at steady-state is shown in Figure 4A. Internalization rates normalized to steady-state (the 0-timepoint controls) for each chimera are shown in Figure 4B and 4C. Depletion of AP-2 resulted in a modest, but statistically significant, increase in the surface expression of CD8-VpuCD at steady-state. However, the rate of internalization of CD8-VpuCD was reduced to near that of truncated CD8- and CD8-Nef-LL/AA. In contrast, AP-2 depletion substantially and significantly increased the surface expression of CD8-Nef at steady-state, and the rate of internalization was indistinguishable from that of CD8- or CD8-Nef-LL/AA. AP-2 knockdown was confirmed by western blotting of cell lysates with probing for α-adaptin (Figure 4D). These data indicate that AP-2 is the critical clathrin adaptor co-opted by both the Vpu CD and Nef to mediate endocytosis. The data also indicate that although the surface-expression of CD8-Nef at steady state is mainly determined by AP-2-mediated endocytosis, this is not true of CD8-VpuCD; AP-2 depletion increased the surface levels of CD8-VpuCD less effectively than clathrin-depletion (compare Figure 3 and 4). These findings suggest that a clathrin adaptor in addition to AP-2 contributes to trafficking mediated by the Vpu CD. That adaptor seems unlikely to be AP-1, because suppression of AP-1-expression by siRNA targeting γ-adaptin (the large, specific subunit of AP-1) had no effect on either the surface-expression of CD8-VpuCD at steady state or its rate of endocytosis (Figure S4).
Figure 4. Endocytosis of CD8-Vpu-CD is sensitive to siRNA-mediated depletion of AP-2.

(A) The rates of internalization of the truncated CD8−, CD8-VpuCD, CD8-Nef, or CD8-Nef-LL/AA were measured in AP-2-depleted HeLa P4.R5 cells by flow cytometry. The cells were transfected with siRNA oligonucleotides directed against AP-2 α subunit (siAP-2α) or a non-targeting control (siControl). After 48 hours, the cells were transfected to express the indicated CD8-chimeras and a GFP expression construct. 24 hours later, the internalization rates of the CD8- chimeras were measured by antibody labeling and flow cytometric analysis. Steady-state surface levels of CD8-chimeras are shown (0-timepoint control). Data were normalized to the 0-timepoint control and presented as the percentage surface CD8 remaining over time, in either siControl (B) or siAP-2 α (C) treated P4.R5 cells. Error bars indicate the standard deviation of three independent experiments. P-values were generated by paired two-tailed t-test, * P<0.05, ** P<0.001, *** P <0.001. (D) Protein from cell lysates was separated by SDS-PAGE and AP-2 α depletion confirmed by western blotting. (E) The intracellular localization of CD8-VpuCD was determined by microscopy of HeLa P4.R5 cells transfected with negative control or AP-2α siRNA. Cells were transfected with CD8-VpuCD, and 24 hours later fixed and stained for immunofluorescence. AP-2α depletion in these cells was determined by primary antibody labeling of AP-2α and fluorescent secondary detection (red); CD8 staining is shown in green. (F) The intracellular localization of WT Vpu was determined by microscopy of HeLa P4.R5 cells depleted of clathrin or AP-2α. Cells were transfected with Vpu-FLAG 48hr post-treatment with siRNAs, fixed and immunofluorescently-stained for FLAG. Scale = 10μm.
The subcellular localization of CD8-VpuCD was examined in HeLa-P4.R5 cells transfected with siControl or siAP-2α. The cells were transfected to express CD8-VpuCD 48 hours after treatment with the siRNAs, and then fixed and stained the following day. CD8-VpuCD was detected using antibody to CD8, and cells depleted of AP-2 were identified using antibody to α-adaptin (Figure 4E). The distribution of CD8-VpuCD in the control siRNA-treated cells was predominantly vesicular, as previously noted, with little colocalization with α-adaptin. In AP-2 depleted cells, however, CD8-VpuCD was detectably displaced to the plasma membrane, a result consistent with the role of AP-2 in the endocytosis of the protein but somewhat unexpected given the modest increase in cell-surface expression measured by flow cytometry as discussed above.
Given the discordant effects of AP-2-depletion on the steady-state surface-level of CD8-Vpu (modest) and on the rate of endocytosis of CD8-Vpu (substantial), we examined the role of AP-2 in the subcellular localization of full-length Vpu (Figure 4F). Like CD8-Vpu, native Vpu (modified only by a C-terminal FLAG tag), was only modestly affected by depletion of AP-2, whereas depletion of clathrin caused more substantial re-localization. Clathrin depletion was confirmed by inhibition of Tfn uptake and AP-2 depletion was confirmed by loss of AP-2 staining (data not shown). These data suggested that like CD8-Vpu, full-length Vpu (when expressed in cells that express two of its targets - CD4 and BST-2), is only partly dependent on AP-2 for its endosomal localization.
Combinatorial mutagenesis abrogates endocytosis of the CD8-VpuCD chimera
Several sequences in the Vpu CD have been identified as potential sorting motifs that contribute to the surface-downregulation of cellular proteins affected by Vpu. These motifs include a potential tyrosine-based AP-binding motif Y29xxφ (φ refers to a bulky hydrophobic residue and x to any amino acid) in the membrane-proximal region, which overlaps in some Vpu proteins with a potential leucine-based motif (E/D)xxxLφ, as well as a second leucine-based AP-binding motif in the C-terminal α-helix, ExxxL63V, which we have shown interacts with AP-1 σ and γ subunits in an extended conformation 23. Recently, serines at position 52 and 56, which when phosphorylated bind β-TrCP and recruit a cullin 1-based ubiquitin ligase complex, have been shown to contribute to interactions between Vpu and AP-1 and AP-2; these AP-interactions require not only serine-phosphorylation but reportedly also the interaction of Vpu with one of its cellular targets, BST-2 24. To identify which of these sequences contribute to endocytosis, we introduced mutations to inactivate them, either singly or in combination, in the context of the CD8-VpuCD chimera. Endocytic rates were measured as described above and are presented as the percentage remaining at the cell surface over time (Figure 5A and 5B). Surface levels of CD8 at steady-state are shown in Figure 5C.
Figure 5. Combinatorial mutagenesis abrogates endocytosis of CD8-VpuCD.

(A) Single and combinatorial mutations of potential endocytic motifs Y29A, S52,56N, and ELV/AAA were introduced into the CD8-VpuCD chimera. HeLa P4.R5 cells were transfected with the indicated constructs and endocytic rates measured by flow cytometry assay 24hr later. Internalization rates of the single motif CD8-VpuCD mutants (A) and combination mutants (B) were compared to truncated CD8- control. The data are presented as the percentage of CD8 remaining at the cell surface, following normalization to the 0-timepoint control. Error bars represent the standard deviation of three independent experiments. The mean fluorescence intensity of the surface CD8-VpuCD mutants at steady-state (0-timepoint control) is shown (C). Expression of the CD8-VpuCD constructs was determined by SDS-PAGE and Western blotting (D). The cellular distribution of the CD8-VpuCD chimeras containing multiple mutations was determined by fluorescent staining of CD8 and microscopy (E). Images presented are z-stack projections of total cell volume. Scale = 10μm.
Mutations of single potential motifs modestly affected the surface levels of CD8-VpuCD at steady-state; each sequence contributed a modicum of overall internalization activity. These results regarding the VpuCD are in contrast to Nef, in which the leucine-based motif contributed the entirety of internalization activity. Mutations of single potential motifs also affected only modestly the endocytic rate of CD8-VpuCD. The combination of the Y29A with either the S52,56N or ELV/AAA substitutions significantly inhibited endocytic rate, and the triple mutant Y29A-ELV/AAA-S52,56N was devoid of endocytic activity. Total cellular expression of the chimeras was measured by western blot; all the constructs were similarly expressed, aside from truncated CD8-, as previously observed (Figure 5D). The triple combination mutant was also characterized by immunofluorescence microscopy (Figure 5E). While CD8-VpuCD accumulated in enlarged intracellular vesicles as shown above, the triple mutant Y29A-ELV/AAA-S52,56N was displaced to the plasma membrane, similar to truncated CD8. Together, these data suggested that the ability of the Vpu CD to direct endocytosis requires multiple determinants whose functional roles are additive.
Roles of clathrin and AP-2 in the Vpu-mediated downregulation of surface BST-2
To assess the functional roles of clathrin and clathrin adaptors as co-factors of the complete Vpu protein, we measured the surface levels of BST-2 in HeLa-P4.R5 cells, which express endogenous BST-2, when the expression of clathrin, AP-2, or AP-1 were suppressed (Figure 6A–C). The cells were transfected with siRNAs as described above, then transfected to express Vpu. The next day, the expression of BST-2 at the cell surface was measured by flow cytometry. The data of Figure 6A show the fold-downregulation of BST-2 caused by Vpu under the conditions of depletion of clathrin, AP-2α, or AP-1γ. Primary data from a representative experiment are shown in Figure 6C. Depletion of clathrin inhibited the downregulation of BST-2, which was anticipated, given the ability of a dominant-negative version of the clathrin-assembly cofactor AP180 to inhibit this activity 21,22,24. Depletion of AP-2 inhibited the downregulation of BST-2 to a lesser extent then depletion of clathrin, although these effects were statistically significant (P<0.001). The role of AP-2 in the downregulation of BST-2 has been previously reported 20. Surface BST-2 downregulation appeared insensitive to depletion of AP-1. We also reassessed the functional roles of the Vpu sequences Y29RKL, S52,S56, and EL63V, both alone and in combination (Figure 6D and E). These data largely supported those previously reported, but they also indicated new relationships. The S52,56N and ELV/AAA substitutions each substantially impaired the downregulation of BST-2; moreover, their effects appeared additive (Figure 6D). The Y29A substitution had no effect on the downregulation of BST-2, consistent with previous results, but nonetheless surprising given its contribution to the endocytic rate of the CD8-VpuCD chimera 21. The additive effect of the S52,56N and ELV/AAA substitutions on BST-2 downregulation was lost in the presence of the Y29A substitution. These data suggest that a phenotype for the Y29A substitution might be revealed in certain genetic contexts, but the role played by this residue in Vpu-activity is currently obscure.
Figure 6. Clathrin and AP-2 are required for optimal Vpu-mediated downregulation of cell-surface BST-2.

(A) Vpu-mediated downregulation of surface BST-2 was measured in cells depleted of Clathrin, AP-2α, or AP-1γ subunits. HeLa-P4.R5 cells were transfected with the indicated siRNA oligonucleotides and after 48 hours co-transfected with either empty control plasmid or Vpu-FLAG plasmid, and a GFP expression construct. Surface BST-2 was labeled using fluorophore-conjugated anti-BST-2 antibody and analyzed by two-color flow cytometry. Data presented are the fold effect of Vpu on BST-2 in the presence of the indicated siRNAs. Error bars indicate the standard deviation of four independent experiments. P-values were generated by one-way ANOVA with Bonferroni post hoc test, *** indicates P <0.001. (B) Depletion of Clathrin, AP-2α, and AP-1γ was measured in cell lysates by SDS-PAGE and Western blotting. (C) Representative histograms of fluorescence intensity of BST-2 staining in the GFP-positive cell populations. Shaded histograms represent cells not transfected to express Vpu; unshaded represent cells transfected to express Vpu. The siControl profiles are superimposed on the profiles from the knockdown conditions to facilitate comparisons. (D) Surface BST-2 downregulation was measured in cells transfected to express Vpu-FLAG bearing single or combination mutations S52,56N, ELV/AAA, and Y29A. HeLa P4.R5 cells were co-transfected with either empty plasmid or plasmid expressing the indicated Vpu-FLAG mutants, and a GFP expression construct. The following day, the cells were stained for surface BST-2 and analyzed by two-color flow cytometry. Data presented are the mean fluorescence intensity of BST-2 in the GFP-positive cell population. Error bars indicate the standard deviation of three independent experiments. (E) Cell lysates were subject to SDS-PAGE and western blotting.
Phosphorylation regulates direct interactions of the Vpu CD with subunits of AP-1 and AP-2
We considered that one way in which the Vpu serines and the membrane proximal tyrosine might contribute to clathrin- and AP-2-dependent trafficking would be through the interaction of the Vpu CD with the medium (μ) subunit of the complex. We previously reported a weak interaction between the Vpu CD and the μ subunits of AP-1 (μ1) and AP-2 (μ2), but not AP-3 (μ3), detected using recombinant proteins and size-exclusion chromatography 23. Here, we tested the hypothesis that the Vpu CD bound to μ subunits in a tyrosine- and serine-phosphorylation-dependent manner using partly purified, recombinant glutathione-S-transferase (GST)-Vpu and maltose-binding-protein (MBP)-μ fusion proteins produced in E.coli in pull-down assays (Figure 7). As shown previously, the Vpu-CD bound μ1 and μ2, but not μ3 (Figure 7A). The interactions with μ1 and μ2 required phosphorylation of the Vpu-CD, which we induced by co-expressing both subunits of casein-kinase II (CK-II) along with the GST-Vpu in the E.coli cells and which we confirmed using an antibody specific for detection of phosphorylated Vpu 35 (Figure 7B). The interaction of phosphorylated Vpu-CD with μ2 was surprisingly independent of tyrosine-29 and isoleucine-32; the interaction with μ1 was partly dependent on tyrosine-29 but independent of isoleucine-32 (Figure S5). These results weighed against the hypothesis that Y29 functions as part of an Yxxφ AP-binding motif The interaction of phosphorylated Vpu-CD with μ1 and μ2 was dependent on serines 52 and 56 (Figure 7C), and μ2 binding was independent of the ExxxLV motif, as anticipated (Figure 7D). The Vpu CD also bound to an AP-2 hemi-complex formed of α/σ2 subunits, and this association was enhanced by phosphorylation of serines 52 and 56 (Figure 7D), as proposed previously 24.
Figure 7. Vpu binds AP-1 and AP-2 subunits in a serine-phosphorylation-dependent manner.

(A) Phosphorylated Vpu binds μ1 and μ2 but not μ3 in vitro. Phosphorylated GST-tagged VpuCD recombinant protein was purified from E.Coli co-transformed to express Casein Kinase II (CK-II). The GST-VpuCD was mixed with MBP- μ1, -μ2 or -μ3 before GST-pulldown. Bound proteins were analyzed by SDS-PAGE and Coomassie staining. (B) Phosphorylation of GST-VpuCD by co-expression with CK-II was confirmed by western blot using an antibody which specifically detects Vpu phosphorylated at serines 52 and 56. (C) Phosphorylation of Serines 52 and 56 is required for μ1 and μ2 binding. GST-pulldown of μ1 and μ2 was evaluated using Vpu, Vpu-S52/56N or Vpu and Vpu-S52/56N produced in the presence of Casein Kinase II (CK-II). (D) Phosphorylation of serines 52 and 56 increases Vpu’s binding affinity for both the AP-2 α-σ2 hemicomplex and μ2. GST-pulldown of AP-2 μ2 and α-σ2 was evaluated using either Vpu, or phosphorylated Vpu or the indicated Vpu mutants produced in the presence of Casein Kinase II (CK-II). (E) Analogous basic residues present in μ1 and μ2, but not μ3, as shown in a structure based-sequence alignment. (F) These basic residues occupy similar regions in μ1 and μ2; positively-charged residues are shown in blue, negative in red. (G) Basic residues in μ1 and μ2 are required for binding phosphorylated Vpu. GST-pulldown was performed between phosphorylated Vpu and μ1 or μ2 containing substitutions within the two indicated sets of basic residues; R225, R393/AA and K274, K298, K302, R303/EEED in μ1, and equivalent residues K227, R402/AA and K308, K312/EE in μ2.
The interaction of phosphorylated serines 52 and 56 with μ1 and μ2 supported the hypothesis that these residues, which are surrounded by several aspartates and glutamates, are part of a phosphorylation-regulated acidic cluster AP-binding motif, such as that found in the TGN-resident cellular protease furin 36. The mode of binding of such motifs to AP complexes is controversial; some evidence supports PACS proteins as intermediaries between the acidic cluster and the AP complexes, whereas other evidence supports direct binding of the acidic cluster to the μ subunits, which have a positive (basic) electrostatic potential 37–39. We previously identified two basic patches on μ1 that interact with acidic residues either within HIV-1 Nef or within the CD of one of its cellular targets, the MHC-I α-chain 25. Basic residues at these sites are present in μ2 as well as μ1, but they are not present in μ3 (Figure 7E and 7F). Alanine substitution of these basic residues abrogated the interaction between phospho-Vpu and μ1 and μ2 (Figure 7G). These data support the notion that the S52,S56 region of Vpu plays two roles: in addition to acting as a “phosphodegron” that recruits β-TrCP and a cullin 1-based ubiquitin-ligase complex, this region also supports the interactions of Vpu with clathrin adaptor complex subunits. The latter activity is bimodal: one mode is apparently indirect and through promotion of ExxxLV-mediated binding to the α/σ2 hemicomplex of AP-2; the other mode is direct and reflects activity as an acidic cluster sorting motif that binds to the μ subunits of AP-1 and AP-2. The data also indicate that specific basic patches on the μ subunits are the targets of the Vpu acidic-cluster motif, revealing a remarkable convergence in how Vpu and Nef co-opt clathrin adaptors.
DISCUSSION
We investigated the trafficking itinerary and the clathrin-related interactions of HIV-1 Vpu, a viral accessory transmembrane protein that excludes several cellular immuno-receptors including the restriction factor BST-2 from the plasma membrane. We found that the cytoplasmic domain (CD) of Vpu contains multiple endocytic determinants that contribute to rapid internalization from the plasma membrane and that this process is dependent on clathrin and the clathrin adaptor AP-2. We found that Vpu-mediated surface downregulation of BST-2 requires both the Vpu DpS52GxxpS56 and ExxxL63V motifs and is sensitive to depletion of clathrin and AP-2. We confirmed that Vpu CD binds to the α/σ2 hemicomplex of AP-2 in vitro and that this depends on the leucine-based ExxxL63V motif. We further showed that the ExxxL63V - α/σ2 hemicomplex interaction is enhanced by phosphorylation of serines at the DS52GxxS56 site. Remarkably, we demonstrated that the Vpu CD directly binds the medium (μ) subunits of AP-2 and AP-1 in vitro and that this novel interaction depends on serine phosphorylation of Vpu and on basic residues in the μ subunits. Our data support a model in which the DpS52GxxpS56 motif has a dual role in the downregulation of host cell factors, functioning in the recruitment of both β-TrCP-containing ubiquitin ligase complexes and heterotetrameric clathrin adaptors. Our data also indicate that, like the HIV accessory protein Nef, Vpu has evolved alternative, distinct modes of interaction with AP complexes. We speculate that this flexibility could increase the breadth of cellular targets available to Vpu.
The experimental designs herein, both cell biological and biochemical, are focused on the trafficking and protein-interaction properties of the Vpu CD. By fusing the Vpu CD to the ectodomain and TMD of the CD8α-chain, we aimed to study the autonomous contribution of motifs in the Vpu CD to membrane trafficking activity. This strategy is not without caveats. The missing Vpu TMD renders CD8-VpuCD unlikely to interact with most cellular targets of Vpu, and we experimentally determined that the presence of CD4 did not influence CD8-VpuCD-trafficking, but we cannot exclude that unknown cellular targets of Vpu might influence the behavior of this chimera. Moreover, the oligomerization state of CD8-VpuCD might be different than native Vpu: CD8 is a dimer, and once cross-linked with antibody seems likely to behave as a tetramer, whereas Vpu may exist as a tetramer or pentamer40,41. On the other hand, by fusing the CD of Vpu to GST for pulldown assays using recombinant proteins, we unambiguously studied the autonomous contributions of these motifs to the interaction with subunits and hemicomplexes of AP-1 and AP-2.
The data suggest that the Vpu CD contains determinants that functionally and physically interact with cellular endocytic machineries independently of cooperative interactions with host cell targets. Nonetheless, these motifs also participate in the ultimate ternary interaction between the cellular target, Vpu, and clathrin adaptors. Such cooperative interactions have previously been demonstrated; for example, in current models of the BST-2/Vpu/AP-1 complex, BST-2 and Vpu not only interact with each other, but each protein’s CD contributes to AP-binding 22,23.
What, then, is the relevance of our observations on the Vpu CD when studied in isolation to Vpu function in the context of the complete protein? Notably, the effects of depletion of clathrin and AP-2 on the steady-state surface expression of CD8-Vpu are similar to their effects on the surface expression of BST-2 in Vpu-expressing cells (depletion of clathrin causing a greater up-regulation than depletion of AP-2). These results indicate that the trafficking properties of CD8-Vpu at least partly recapitulate aspects of target-modulation by Vpu, but with a potentially important exception: whereas CD8-Vpu is endocytosed in an AP-2-dependent manner, Vpu is not known to substantially increase the rate of endocytosis of its targets 20,42–44. These considerations lead to the hypothesis that rather than stimulating target-endocytosis, endocytic motifs in the Vpu CD are required to send Vpu along an itinerary appropriate to finding and interacting with its targets within the endosomal system; then, once in complex with those targets, the same motifs participate in their intracellular retention or degradation.
The trafficking properties of the VpuCD were similar but not identical to those of HIV-1 Nef. The surface expression of both CD8-VpuCD and CD8-Nef were comparatively low at steady-state, which was attributable to sequestration in endosomal compartments including late endosomes. The Vpu CD, like Nef, stimulated rapid rates of endocytosis and surface deposition, which occur on a time-scale of minutes. On the other hand, knockdown of AP-2 markedly upregulated the expression of CD8-Nef on the cell surface at steady state, but it had much less an effect on the steady-state surface-expression of CD8-VpuCD.
The association of Vpu with the host membrane trafficking machinery is key to its function. BST-2 antagonism is clathrin-dependent, but exactly how has been unclear 20–22. We reported a role for AP-2 in the Vpu-mediated surface-downregulation of BST-2 20, and a dominant negative version of the clathrin assembly factor AP180 inhibits the ability of Vpu to downregulate cell surface BST-2 and to counteract BST-2-mediated restriction of virion-release 21,22. However, others have reported that Vpu-mediated counteraction of virion-restriction by BST-2 was insensitive to knockdown of AP-1, -2 or -3, and AP-1 γ knockout in mouse fibroblasts had no apparent effect on the ability of Vpu to downregulate BST-2 22. Here, we observed that Vpu-mediated downregulation of surface BST-2 was sensitive to depletion of clathrin and to a much lesser extent AP-2. Thus, our data support the role of clathrin in Vpu function, but they leave unanswered the role of AP-1, and, as elaborated below, they suggest that AP-2 is insufficient as a Vpu-cofactor that explains the protein’s functional dependence on clathrin.
Since the knockdown conditions for both clathrin and AP-2 were sufficient to abrogate the endocytosis of CD8-VpuCD and CD8-Nef, the partial effect of each knockdown in the BST-2 downregulation assay indicates that while clathrin- and AP-2-mediated endocytosis contributes to Vpu function, it is not likely sufficient. Moreover, as noted above, the contribution of clathrin to the reduction in the steady state surface level of CD8-VpuCD relative to CD8 is greater than the contribution of AP-2, and the same disparity is evident in the functional assays of Vpu-mediated downregulation of surface BST-2. These observations lead to the hypothesis that while Vpu activity depends in large part on clathrin, an unidentified clathrin adaptor contributes together with AP-2. Since our data weigh against AP-1 fulfilling this role, candidates include untested members of the heterotetrameric AP complex family or monomeric adaptors such as Hrs or GGA family proteins 45–48. Alternatively, we have not excluded that Vpu binds directly to clathrin, an hypothesis supported by experiments in which a “clathrin-box” appended to the C-terminus of Vpu rescues mutations within the CD that impair counteraction of BST-2-mediated restriction of virion release 24.
Multiple determinants within the CD of Vpu are required to drive clathrin- and AP-2 dependent endocytosis, and the activity of these determinants is additive. This contrasts with endocytosis driven by Nef, which we confirmed relies solely on an intact leucine-based AP-binding motif 28,49. In the Vpu CD, alanine substitution of the membrane-proximal Y29 residue of the potential tyrosine-based AP-binding motif (Yxxφ) had the most prominent effect on endocytic rate of the single mutations tested. The endocytic activity of the Vpu CD was ablated when this mutation was combined with mutation of the leucine-based AP-binding motif, ExxxL63V, or with mutation of serines of the DpS52GxxpS56 motif, or both.
Of these three key endocytic motifs in Vpu, the DSGxxS sequence is conserved throughout all clades of group M HIV-1, the main cause of the pandemic. The Yxxφ motif is not found in clade D viruses, where the consensus sequence has a C instead of a Y, and the ExxxL63V sequence is a variant in clade C viruses, where the consensus sequence has an M instead of an L (see Figure S1C regarding clade C viruses). Despite the variant ExxxLV motif, we observed that the CD of two Vpu proteins from clade C viruses encoded endocytic activity equal to that of the NL4.3 clade B clone used throughout our study.
While tyrosine 29 of Vpu contributed to endocytic activity, neither this residue nor I32, the Y+3 φ residue of the putative Yxxφ motif, appreciably contributed to μ2-binding in vitro. Y29 appeared dispensable for Vpu-function with respect to the downregulation of BST-2, despite the apparent contribution of this residue to endocytic activity, raising the possibility that its activity is restricted to the context of the CD8-Vpu chimera. Nonetheless, this tyrosine motif is relatively well conserved in Vpu across isolates of HIV-1 group M (with the exception of clade D viruses noted above) so it seems likely to be functionally important. While we did not observe a role for Y29 in the surface downregulation of BST-2 by Vpu from the HIV-1 B subtype clone NL4.3, the equivalent TM-proximal YRKL motif in the cytoplasmic domain of clade C Vpu is reportedly important for efficient virion release, a phenotype that is partly a consequence of Vpu’s ability to downregulate cell-surface BST-2 50. An alternative possibility is that the Y29RKI motif contributes to other Vpu activities, such as the downregulation of the immuno-receptors NTB-A, CD1d, or CCR7 43,44,51.
The contribution of the ExxxL63V motif to the endocytic activity of the Vpu CD is consistent with observations that this motif interacts with a canonical recognition site on the γ/σ1 hemicomplex of AP-1 and with the α/σ2 hemicomplex of AP-2 (data herein and 23). The in vitro interaction with the γ/σ1 hemicomplex was previously observed only in the presence of the BST-2 CD, which stabilized the ternary complex of Vpu, BST-2, and AP-1 via the binding of a YxYxxφ sequence in the BST-2 CD to the tyrosine-binding pocket of the AP-1 μ subunit 23. Similarly, the ExxxL63V -dependent interactions of Vpu with AP-1 and AP-2 observed in human cells required the interaction of Vpu with BST-2 24. Since the CD8-VpuCD chimera lacks the transmembrane domain of Vpu and consequently cannot interact with BST-2 13,52, our data indicate that the ExxxL63V motif contributes to the trafficking activity of Vpu independent of co-operative interaction with BST-2. We further demonstrate that in vitro binding between the Vpu CD and the α-σ2 hemicomplex of AP-2 as mediated by the ExxxL63V motif can occur in the absence of BST-2.
How do the phosphoserines in the Vpu CD mediate interactions with the AP complexes? In support of a recent hypothesis that phosphorylation of the serines might lead to conformational changes that render the ExxxLV motif more active, we observed that the ExxxLV-mediated interaction with the α/σ2 hemicomplex of AP-2 was enhanced by serine-phosphorylation 24. But strikingly, our data indicate an alternative and novel mode of binding. We found that the recombinant Vpu CD binds both μ1 and μ2, but not μ3, and that this interaction requires serine-phosphorylation within the DpS52GxxpS56 sequence.
With regard to μ-binding, we hypothesise that these serines in Vpu, which are surrounded by several aspartates and glutamates, are part of a phosphorylation-regulated acidic cluster AP-binding motif, such as that found in the TGN-resident cellular protease furin and mannose-6-phosphate receptors 36,53. Furin, in particular, contains a very similar motif (SDSEED), which regulates localization to the TGN in a phosphorylation-dependent manner 54. Acidic clusters containing CK-II phosphorylation sites are commonly found in cellular proteins which cycle between the TGN and endosomes 55, and we propose that Vpu is a viral protein with similar properties. Direct and indirect associations of these acidic cluster motifs with AP complexes have been proposed, the former through the μ subunits via their positive surface electrostatic potential, and the latter through an intermediate protein, the phosphofurin acidic cluster sorting protein-I (PACS-I) 37. Our in vitro binding studies support the model of direct interaction between the Vpu acidic cluster and μ subunits. In further support and elaboration of this model, we found that two basic patches on μ1 were required for phospho-Vpu-binding. These basic patches are the same ones that we identified in the Nef/MHC-I CD/μ1 complex: one recognizes the “acidic cluster” within Nef (E62–E65), and the other recognizes acidic residues from both Nef and the MHC-I CD 25. The analogous basic patches on μ2 were also essential for interaction with the Vpu CD, and these seem likely to explain the role of the phosphoserine motif in endocytosis. Neither of these basic patches is apparent on the surface of μ3, consistent with the lack of binding of the Vpu CD to μ3. The role of these basic patches on μ1 and μ2 reveals a remarkable convergence in how Nef and Vpu co-opt the clathrin trafficking machinery. Whether these sites are also used by cellular proteins such as furin remains an open question.
The contribution of individual motifs in the Vpu CD to AP-binding, and the different underlying mechanisms of binding that they support, might differentially affect Vpu function with respect to distinct cellular targets. This scenario is exemplified by Nef, in which distinct sequences facilitate distinct modes of interaction with AP complexes that are ultimately stabilized by cooperative interactions with different protein targets. For example, the C-terminal leucine-based motif in Nef binds the AP-2 α/σ2 hemicomplex and is stabilized by formation of the ternary CD4-Nef-AP-2 complex, promoting rapid clathrin-mediated internalization of CD4 from the plasma membrane 11,56. Yet, this leucine-based motif is dispensable for the downregulation of MHC-I. Instead, electrostatic interactions between Nef and the AP-1 μ subunit, together with stabilization of a partial tyrosine-based sorting motif in the MHC-I CD, supports formation of a ternary complex that is diverted into an endo-lysosomal degradative pathway 25,57.
In the case of Vpu, we now have evidence for two distinct modes of interaction with AP complexes: one ExxxLV-based (yet also supported by the phosphoserines) and involving the σ/large specific subunit hemicomplexes, and the other directly mediated by the phosphoserine acidic cluster and basic patches on the μ subunits. The short distance between the phosphoserine and ExxxLV motifs precludes that these modes of interaction are supported simultaneously in the case of a single molecule of Vpu bound to a single AP complex. Consequently, we suspect that these different modes of interaction between Vpu and AP complexes, like the different modes of interaction exemplified by Nef, are involved in the modulation of different targets, perhaps in a manner that reflects the AP binding sequences found in the targets themselves. For example, in the case of the ternary complex formed by the cytoplasmic domains of BST-2 and Vpu with AP-1, BST-2 interacts with the μ subunit via its YxYxxφ motif, while Vpu interacts with the γ/σ1 hemicomplex via its ExxxLV motif 23. Our line of thinking suggests that a converse example might exist: a ternary complex in which the CD of the cellular target binds via its leucine-motif to the AP hemicomplex, while Vpu binds via its phosphoserine motif to the μ subunit. To our knowledge, however, few Vpu targets contain leucine-based AP binding motifs, with CD4 being the primary example. CD4 seems unlikely to fit this model, however, because although the modulation of CD4 by Vpu is completely dependent on the phosphoserines, this activity has been attributed to recruitment of a cullin1-based E3 ligase via β-TrCP rather than to the recruitment of clathrin adaptors 14,16.
In summary, we propose that the serines of the Vpu DpS52GxxpS56 sequence contribute to the modulation of host cellular proteins through two overarching mechanisms: they constitute a phosphodegron that recruits a cullin1-based E3 ubiquitin ligase complex via β-TrCP as shown previously, and they regulate the recruitment of AP complexes. The latter activity is achieved through two distinct binding specificities or modes: indirectly through enhancement of ExxxLV-mediated binding to the σ/large specific subunit hemicomplexes, and directly by contributing to a phosphoserine acidic cluster sorting motif that binds the complexes via basic domains on their μ subunits. Exploring the unusual if not unique duality of this motif in the recruitment of ubiquitin ligase and clathrin adaptor complexes, and testing the model proposed herein regarding the bimodal nature of the Vpu-AP interaction, seems likely to provide insights into Vpu’s activity against not only BST-2 but also other cellular targets.
MATERIALS AND METHODS
Cells, plasmids and reagents
HeLa P4.R5 cells were obtained from the NIH AIDS Research and Reference Reagent program from Nathaniel Landau, and maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10 % fetal bovine serum (FBS), penicillin/streptomycin, and 1 μg/ml puromycin. HeLa Z24 cells were provided by Dr. Chris Aiken, and were maintained in DMEM supplemented with 10% FBS, and penicillin/streptomycin. The C-terminally FLAG-tagged human codon-optimized clade B (NL4.3) Vpu (VpHu) has been previously described 23. The pCG-GFP reporter plasmid 58 was provided by Dr. Jacek Skowronski, Case Western Reserve University, Cleveland, OH. Clathrin-dsRed was a gift of Dr. Tomas Kirchhausen, Harvard Medical School, Boston, MA, USA. pCINeo-CD8-Nef fusion chimera has been previously described 57. This construct contains the first 212 amino acids of human CD8 alpha chain, encompassing the lumenal and transmembrane domains, fused to full-length HIV-1 Nef (clade B clone NL4.3). The control plasmid containing the lumenal and transmembrane domains of CD8 only (pCINeo-CD8), was generated by insertion of a stop codon by QuikChange (Agilent Technologies) site-directed mutagenesis PCR, immediately following the EcoRI site. The cytoplasmic domain (CD, amino acids E28 to L81) of VpHu was inserted into the pCINeo-CD8 vector by In-fusion (Clontech) cloning technique, using the EcoRI and SalI sites for linearization of the parental vector. The same strategy was used to prepare CD8-Vpu fusions with clade C Vpu MJ4 59 and 96BW16B01 60. Single and combination mutations of VpHu in the context of pCINeo-CD8-VpHu-CD or pcDNA3.1-VpHu-FLAG plasmids were generated by QuikChange site-directed mutagenesis PCR. All primers were synthesized by Integrated DNA Technologies and constructs were confirmed by sequencing using Genewiz services.
siRNA targeting AP-2α subunit was obtained from Dharmacon, the siRNA target sequence was AAGAGCAUGUGCACGCUGGCCA 32. Silencer select pre-designed siRNAs targeting Clathrin Heavy Chain (target sequence GGUUGCUCUUGUUACGGAU), AP-1γ1 subunit (target sequence GGUACGAAUUUUGCGGUUA), and non-targeting negative control siRNA were obtained from ThermoFisher Scientific.
Transfections
Plasmid
HeLa P4.R5 cells seeded in 6-well plates were transfected with a total of 1 μg plasmid, 100 ng of which was pCG-GFP transfection control plasmid (for flow cytometry experiments). 200 ng total plasmid was transfected in 24-well plates for immunofluorescence (IF) studies. Plasmids were diluted in Opti-MEM reduced serum medium (Life Technologies) and complexed with Lipofectamine 2000 reagent (ThermoFisher Scientific) following standard procedures. Media was changed 4 hours post-transfection and assays performed the following day.
siRNA
Cells were reverse-transfected in either 6-well plates (6 × 105 cells per well) or 24-well plates (4 × 104 cells per well) using RNAimax transfection reagent (ThermoFisher Scientific), following standard protocols. The siRNAs were diluted in Opti-MEM, and added to wells containing cells in antibiotic-free media at a 10 nM final concentration. Assays were performed 72 hours post-transfection with siRNAs.
Immunofluorescence microscopy
HeLa P4.R5 cells were seeded on 12mm coverslips in 24-well plates 24-hours prior to transfection with plasmids. For siRNA experiments, cells were plated during reverse-transfection with 10 nM siRNAs, and transfected with plasmid 48 hours later. Cells were washed in cold PBS and fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) on ice for 5 minutes, then 15 minutes at room temperature (RT). Prior to fixation, cells to be treated with transferrin were serum-starved for 30 minutes at 37°C before addition of 50μg/ml transferrin-Alexa-555 (Molecular Probes) in serum-free medium. Cells were washed twice with PBS and PFA was quenched with 50 mM ammonium chloride for 5 minutes. Cells were permeabilized with 0.2% Triton X-100 for 5 minutes and blocked with 2 % bovine serum albumin (BSA) for 30 minutes prior to incubation with primary antibodies. CD8 fusions were detected using mouse anti-CD8α conjugated to AlexaFluor-488 (BD Biosciences). For CD8-Vpu colocalization studies, cells were first stained with either mouse anti-γ adaptin (clone 100/3, Sigma-Aldrich), mouse anti-α adaptin (clone AP6, ThermoFisher Scientific), or mouse anti-Transferrin Receptor (TfnR/CD71, Sigma-Aldrich) primary antibodies for 2 hours at RT. The cells were washed and stained with donkey anti-mouse rhodamine-X (RhX) (Jackson ImmunoResearch) for 1 hour at RT. The cells were blocked with 1% BSA in PBS supplemented with 5% normal mouse serum for 30 minutes at RT prior to staining with mouse anti-CD8α AF-488. For CD63 colocalization with CD8-Vpu, CD8-Vpu was detected by indirect staining using mouse anti-CD8 (BD Biosciences) and donkey anti-mouse RhX, followed by blocking with mouse serum, and staining with FITC-conjugated mouse anti-CD63 (BD Pharmingen). Following immunostaining, the cells were washed extensively in PBS before mounting in Mowiol (Polyvinyl alcohol) mounting medium. Images were captured at 100× magnification (1344 ×1024 pixels) using an Olympus IX81 widefield microscope fitted with a Hamamatsu CCD camera. For each field, a Z-series of images was collected, deconvolved using a nearest-neighbor algorithm (Slidebook software, Imaging Innovations, Inc) and presented as Z-stack projections. Image brightness was adjusted using Adobe Photoshop CS4.
Flow Cytometry
To assess levels of surface expression of the CD8-chimeras at steady-state, transfected HeLa P4.R5 cells were washed and resuspended using Acutase dissociation media (Innovative Cell Technologies) before staining with APC-conjugated mouse-anti-CD8 antibody (Clone RPA-T8, BD Pharmingen) or APC-conjugated mouse IgG1 isotype control (BioLegend), for 30 minutes on ice. The cells were washed and fixed in 2% PFA for 15 minutes, prior to analysis using a BD Accuri C6 flow cytometer and CFlow Sampler analysis software. Data presented are mean fluorescence intensity of FL4 (far-red) signal in the GFP-positive (FL1) cell population. The two-color dot-plots presented were prepared using FlowJo software (TreeStar). For surface BST-2 downregulation studies, transfected HeLa P4.R5 cell monolayers were resuspended with Acutase dissociation media and stained with AlexaFluor-647-conjugated anti-human CD317 (BST-2, tetherin; clone RS38E (Biolegend)), or AlexaFluor-647-mouse IgG1, κ isotype control (Biolegend), according to the manufacturer’s instructions. Cells were washed and fixed in 2% PFA before analysis.
Endocytosis assay
HeLa P4.R5 cells were seeded in 6-well plates, 2 wells per condition. The cells were subject to transfection with siRNAs and plasmids as indicated in the Figure legends. The cells were resuspended using Acutase and aliquots stored for Western blotting. The remaining cells were first stained using mouse anti-CD8 antibody (5 μg/ml, BD Pharmingen), mouse anti-TfnR (ABD Serotec) (20 μg/ml) or mouse IgG1, κ isotype control (Biolegend) diluted in 1% FBS in PBS on ice for 30 minutes. The cells were washed with ice-cold FBS-PBS and an aliquot stored on ice as a 0-timepoint control (for measurement of steady-state surface protein levels). The remaining cells were pelleted before transfer to pre-warmed DMEM at 37°C. At 2, 5, and 10 minutes post-warming, aliquots were rapidly cooled in ice-cold FBS-PBS + 0.1% sodium azide to halt endocytosis. The cells were pelleted and resuspended in goat anti-Mouse-APC secondary antibody (2μg/ml) for 30 minutes on ice, to detect CD8 remaining at the cell surface. The cells were washed and fixed in 2% PFA before analysis.
Surface-deposition assay
HeLa-P4.R5 cells were seeded in 6-well plates, 2 wells per condition. The cells were transfected with the indicated CD8-chimeras. 16 hours later, the cells were resuspended using Acutase and aliquots stored for Western blotting. The remaining cells were first stained using mouse anti-CD8 antibody (5 μg/ml) or mouse IgG1, κ isotype control diluted in 1% FBS in PBS on ice for 30 minutes. The cells were washed with ice-cold FBS-PBS and an aliquot stored on ice as a 0-timepoint control (for measurement of background surface staining). The remaining cells were pelleted, cold FBS-PBS removed, and cells warmed to 37°C in DMEM. At 2, 5, and 10 minutes post-warming, aliquots were rapidly cooled in ice-cold FBS-PBS + 0.1% sodium azide to halt endocytosis and recycling. The cells were pelleted and resuspended in APC-conjugated mouse anti-CD8 antibody (according to manufacturer’s instructions), or APC-conjugated isotype control for 30 minutes on ice, in order to detect non-labeled CD8 deposited at the cell surface. The cells were washed and fixed in 2% PFA before analysis.
Western blot
Cell monolayers were washed with PBS and lysed in ice-cold extraction buffer (0.5% Triton X-100, 150 mM NaCl, 25 mM KCl, 25 mM Tris, pH 7.4, 1 mM EDTA) supplemented with a protease inhibitor mixture (Roche Applied Science). Extracts were clarified by centrifugation (12,000 × g for 10 min at 4°C). The sample protein concentration was determined by Bradford assay (BD Biosciences) using standard protocols, and 10 μg denatured by boiling for 5 minutes in SDS sample buffer. Proteins in the extracts were resolved by SDS-PAGE using 12% acrylamide gels, transferred to PVDF membranes, and probed by immunoblotting using mouse anti-Actin (Sigma-Aldrich), mouse anti-AP2α (BD Biosciences), mouse anti-AP1γ (Sigma-Aldrich), rabbit anti-BST-2 (NIH AIDS Reagent Program), rabbit anti-CD8 (Santa Cruz Biotechnology), mouse anti-Clathrin HC (Cell Signaling Technology), mouse anti-FLAG (Sigma-Aldrich), and horseradish peroxidase-conjugated goat anti-Mouse IgG (BioRad) or HRP-donkey anti-Rabbit IgG (BioRad) and Western Clarity detection reagent (BioRad). Apparent molecular mass was estimated using commercial protein standards (PageRulePlus, Thermo Scientific). Chemiluminescence was detected using a BioRad Chemi Doc imager and analyzed using BioRad Image Lab v5.1 software.
Cloning, expression and purification of recombinant proteins
The sequence encoding the HIV-1 Vpu cytoplasmic domain (CD28–81), was PCR amplified and ligated using the BamHI and XhoI sites into the pGEX-4t1 expression vector, producing a fusion with glutathione S-transferase (GST) at the N-terminus of the protein. The plasmid encoding GST- Vpu-CD was transformed into BL21 (DE3) E. coli competent cells to express the fusion protein, and phosphorylated Vpu was expressed by co-transformation with GST-Vpu-CD and a pCDF duet vector encoding both α and β-subunits of Casein Kinase II (CK-II) 23. A single colony of transformed BL21 cells was inoculated into 50 ml of LB-media containing appropriate antibiotics and incubated overnight at 37°C while shaking. 20 ml of the overnight cultures were then inoculated into 2L of LB medium in the presence of antibiotics and grown at 37°C until the OD600 reached 0.600. Protein expression was induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and the cultures grown at 16°C overnight. After overnight induction, cells were harvested by centrifugation at 5000 g for 10 minutes at 4°C. The cell pellets were resuspended in GST-binding buffer (20 mM Tris pH 7.5, 150 mM NaCl containing protease inhibitors). During co-expression of GST-Vpu-CD with CK-II, the cell pellets were resuspended in GST-binding buffer containing phosphatase inhibitor to protect Vpu from dephosphorylation. The cultures were lysed using an EmulsiFlex homogenizer, and the crude lysates were centrifuged at 20,000 g for 1 hour at 4°C. The soluble fractions were first purified using GSTrap- Glutathione Sepharose 4B resin and then using Superdex 200 size-exclusion columns. The purity of the proteins was confirmed by SDS-PAGE. The phosphoryation of Vpu was confirmed using phosphoserine Vpu-specific antibodies 35. Vpu-CD mutants Y29A, I32A and S52,56N were similarly cloned, expressed and purified in both non-phosphorylated and phosphorylated forms. Mutations were introduced using the QuikChange PCR mutagenesis kit and confirmed by DNA sequencing. Expression and purification of μ1 (158–423), μ2 (159–435), and AP-2 hemicomplex α(1–398)/σ2(1–142) proteins was performed as previously described 25. Substitutions were introduced in two regions of basic residues: R225, R393/AA and K274, K298, K302, R303/EEED in μ1, and equivalent residues K227, R402/AA and K308, K312/EE in μ2, using QuikChange PCR mutagenesis. The pMAT9s-μ1 and pMAT9s-μ2 plasmids were co-expressed with the pGro7 vector (Takara Bio), which encodes the groES and groEL chaperone proteins, in E. coli BL21(DE3) cells in Terrific Broth. 2mg/ml L-(+)-Arabinose was added to induce chaperone expression. Expression of MBP-μ1 and MBP-μ2 was induced by 0.1 mM IPTG at A600 of 0.6 and continued at 16 °C overnight. The proteins were purified to homogeneity by using Ni-NTA gravity flow, HiTrap SP cation exchange column and Sephadex 200 gel filtration column.
In vitro binding assays using GST-pulldown
The purified recombinant proteins were mixed in equimolar ratio at 35μM protein concentration in a final volume of 100 μl. These protein mixtures were then incubated with 100 μl Glutathione Sepharose 4B resin overnight at 4°C. The next morning, the resin was washed with GST-binding buffer (20 mM Tris pH 7.5, 150 mM NaCl) to remove unbound proteins. After extensive washing, the bound proteins were eluted using 100 μl of GST elution buffer (50 mM Tris pH 8.0, containing 10 mM reduced glutathione). The proteins were analyzed by SDS-PAGE and Coomassie staining.
Supplementary Material
Figure S1. Similar endocytic activity is observed in both clade B and clade C Vpu. (A) Steady-state surface expression of CD8 constructs in HeLa P4.R5 cells. Cells were co-transfected to express CD8−, CD8−Vpu clade B (NL4.3), CD8-Vpu clade C (MJ4) or CD8-Vpu (96BW), in combination with a GFP expression construct as a transfection marker. Cell surface CD8 was stained and analyzed by two-color flow cytometry. The graph indicates the mean fluorescence intensity of surface CD8 staining in the GFP-positive cell population; error bars represent the standard deviation of two independent experiments. (B) The endocytic rates of the CD8-Vpu chimeras were measured in HeLa P4.R5 cells. Endocytosis assays were performed as detailed in Figure 1. Data are presented as the percentage of surface CD8 remaining over time. Error bars indicate the standard deviation of two independent experiments. (C) Sequence alignment showing location of residues conserved across clade B and C Vpu.
Figure S2. Endocytosis of CD8-Vpu-CD is independent of CD4. (A) Steady-state surface expression of CD8-Vpu-CD was measured in HeLa P4.R5 cells and HeLa Z24 cells, which do not express CD4. Cells were co-transfected to express CD8-Vpu and GFP as a transfection marker. Cell surface CD8 was stained and analyzed by two-color flow cytometry. The graph indicates the mean fluorescence intensity of surface CD8 staining in the GFP-positive cell population; error bars represent the standard deviation of three independent experiments. (B) The endocytic rate of CD8-Vpu was measured in HeLa P4.R5 and Z24 cells. Endocytosis assays were performed as detailed in Figure 1. Data are presented as the percentage of surface CD8 remaining over time. Error bars indicate the standard deviation of three independent experiments. (C) Expression of CD8-Vpu and CD4 was measured in the two cell lines by western blot, control cells were transfected to express GFP only.
Figure S3. Colocalization of CD8-fusion controls with markers of clathrin-mediated endosomal trafficking pathways. HeLa P4.R5 cells were either co-transfected with plasmids encoding CD8−, CD8-Nef, CD8-Nef-LL/AA and DSRed-Clathrin, or transfected to express the CD8-fusion proteins alone. The cells were fixed, permeabilized, blocked and stained for CD8 (green) and the endosomal marker transferrin receptor (TfnR, red). The cells were examined by widefield, deconvolution microscopy; images are z-stack projections of the total cell volumes. Scale bars = 10 μm.
Figure S4. Endocytosis of CD8-Vpu-CD is insensitive to siRNA-mediated depletion of AP-1. (A) The rates of internalization of the truncated CD8−, CD8-VpuCD, CD8-Nef, or CD8-Nef-LL/AA were measured in AP-1-depleted HeLa P4.R5 cells by flow cytometry. The cells were transfected with siRNA oligonucleotides directed against AP-1γ subunit (AP-1γ) or a non-targeting control (siControl). After 48 hours, the cells were transfected to express the indicated CD8-chimeras and a GFP expression construct. 24 hours later, the internalization rates of the CD8- chimeras were measured by antibody labeling and flow cytometric analysis. Steady-state surface levels of CD8-chimeras are shown (0-timepoint control). Data were normalized to the 0-timepoint control and presented as percentage surface CD8 remaining over time, in either siControl (B) or siAP-1γ (C) treated cells. Error bars indicate the standard deviation of three independent experiments. (C) Protein from cell lysates was separated by SDS-PAGE and AP-1γ depletion confirmed by western blotting. (D) The intracellular localization of CD8-VpuCD was determined by widefield microscopy of HeLa P4.R5 cells transfected with negative control or clathrin siRNA. Cells were transfected with CD8-VpuCD, and 24 hours later fixed and stained for immunofluorescence. AP-1γ depletion in these cells was determined by primary antibody labeling of AP-1γ and fluorescent secondary detection (red); CD8 staining is shown in green. Scale = 10μm.
Figure S5. The membrane-proximal Yxxφ motif is dispensable for μ binding. Substitutions Y29A and I32A were introduced into GST-tagged VpuCD. Phosphorylation of serines 52 and 56 was controlled through production of recombinant GST-VpuCD in the absence or presence of Casein Kinase II (CK-II). GST-pulldown was performed between μ1 (A), μ2 (B) and phosphorylated or non-phosphorylated GST-Vpu mutants. Bound proteins were analyzed by SDS-PAGE and Coomassie staining.
SYNOPSIS STATEMENT.
The endosomal coat proteins clathrin and the heterotetrameric clathrin adaptors are cofactors for the immunomodulatory activities of the HIV-1 accessory protein Vpu. Here we show that Vpu interacts with clathrin adaptors in a multifaceted manner: the Vpu cytoplasmic domain binds directly to basic patches on the μ subunit, as well as to a hemi-complex formed by the large, specific (α or γ) and σ subunits. Both interactions are regulated by phosphorylation of serines in the Vpu cytoplasmic domain.
Acknowledgments
We thank the following for reagents: Dr. Akihide Ryo (Yokohama City University School of Medicine, Japan) for the kind gift of the anti-phospho-Vpu (S52,56) antibody, Dr, Jacek Skowronski (Case Western Reserve University, USA) for the pCG-GFP reporter plasmid, Dr. Tomas Kirchhausen (Harvard Medical School, USA) for the Clathrin-dsRed plasmid, and the NIH AIDS Reagent Program for reagents, including rabbit anti-BST-2 antibody.
We also thank members of the Guatelli Laboratory, UC San Diego, and Xiong Laboratory, Yale University, for advice and technical assistance. This work was supported by CFAR grant P30AI036214 to RS, and NIH grants R01AI102778 awarded to JG and YX, and R37AI081668 to JG.
References
- 1.Van Damme N, Goff D, Katsura C, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell host & microbe. 2008 Apr 17;3(4):245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008 Jan 24;451(7177):425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 3.Lama J, Mangasarian A, Trono D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr Biol. 1999 Jun 17;9(12):622–631. doi: 10.1016/s0960-9822(99)80284-x. [DOI] [PubMed] [Google Scholar]
- 4.Ross TM, Oran AE, Cullen BR. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Current biology : CB. 1999 Jun 17;9(12):613–621. doi: 10.1016/s0960-9822(99)80283-8. [DOI] [PubMed] [Google Scholar]
- 5.Pham TN, Lukhele S, Hajjar F, Routy JP, Cohen EA. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology. 2014;11:15. doi: 10.1186/1742-4690-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veillette M, Desormeaux A, Medjahed H, et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. Journal of virology. 2014 Mar;88(5):2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokarev A, Guatelli J. Misdirection of membrane trafficking by HIV-1 Vpu and Nef: Keys to viral virulence and persistence. Cell Logist. 2011 May;1(3):90–102. doi: 10.4161/cl.1.3.16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, Wilson SJ, Landford WC, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell host & microbe. 2009 Jul 23;6(1):54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sauter D, Schindler M, Specht A, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell host & microbe. 2009 Nov 19;6(5):409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia B, Serra-Moreno R, Neidermyer W, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS pathogens. 2009 May;5(5):e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. 2007 Apr;81(8):3877–3890. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang F, Landford WN, Ng M, McNatt MW, Bieniasz PD, Hatziioannou T. SIV Nef proteins recruit the AP-2 complex to antagonize Tetherin and facilitate virion release. PLoS pathogens. 2011 May;7(5):e1002039. doi: 10.1371/journal.ppat.1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skasko M, Wang Y, Tian Y, et al. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. The Journal of biological chemistry. 2012 Jan 2;287(1):58–67. doi: 10.1074/jbc.M111.296772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margottin F, Bour SP, Durand H, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Molecular cell. 1998 Mar;1(4):565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 15.Willey RL, Maldarelli F, Martin MA, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. Journal of virology. 1992 Dec;66(12):7193–7200. doi: 10.1128/jvi.66.12.7193-7200.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Binette J, Dube M, Mercier J, Halawani D, Latterich M, Cohen EA. Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology. 2007;4:75. doi: 10.1186/1742-4690-4-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magadan JG, Perez-Victoria FJ, Sougrat R, Ye Y, Strebel K, Bonifacino JS. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS pathogens. 2010 Apr;6(4):e1000869. doi: 10.1371/journal.ppat.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwabu Y, Fujita H, Kinomoto M, et al. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. The Journal of biological chemistry. 2009 Dec 11;284(50):35060–35072. doi: 10.1074/jbc.M109.058305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Fruh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. Journal of virology. 2009 Aug;83(16):7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell RS, Katsura C, Skasko MA, et al. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS pathogens. 2009 May;5(5):e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau D, Kwan W, Guatelli J. Role of the endocytic pathway in the counteraction of BST-2 by human lentiviral pathogens. Journal of virology. 2011 Oct;85(19):9834–9846. doi: 10.1128/JVI.02633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kueck T, Neil SJ. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS pathogens. 2012;8(3):e1002609. doi: 10.1371/journal.ppat.1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia X, Weber E, Tokarev A, et al. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. eLife. 2014;3:e02362. doi: 10.7554/eLife.02362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kueck T, Foster TL, Weinelt J, Sumner JC, Pickering S, Neil SJ. Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS pathogens. 2015 Aug;11(8):e1005141. doi: 10.1371/journal.ppat.1005141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia X, Singh R, Homann S, Yang H, Guatelli J, Xiong Y. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nature structural & molecular biology. 2012 Jul;19(7):701–706. doi: 10.1038/nsmb.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marks MS, Woodruff L, Ohno H, Bonifacino JS. Protein targeting by tyrosine- and di-leucine-based signals: evidence for distinct saturable components. J Cell Biol. 1996 Oct;135(2):341–354. doi: 10.1083/jcb.135.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangasarian A, Foti M, Aiken C, Chin D, Carpentier JL, Trono D. The HIV-1 Nef protein acts as a connector with sorting pathways in the Golgi and at the plasma membrane. Immunity. 1997 Jan;6(1):67–77. doi: 10.1016/s1074-7613(00)80243-5. [DOI] [PubMed] [Google Scholar]
- 28.Bresnahan PA, Yonemoto W, Ferrell S, Williams-Herman D, Geleziunas R, Greene WC. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Current biology : CB. 1998 Nov 5;8(22):1235–1238. doi: 10.1016/s0960-9822(07)00517-9. [DOI] [PubMed] [Google Scholar]
- 29.Baur AS, Sawai ET, Dazin P, Fantl WJ, Cheng-Mayer C, Peterlin BM. HIV-1 Nef leads to inhibition or activation of T cells depending on its intracellular localization. Immunity. 1994 Aug;1(5):373–384. doi: 10.1016/1074-7613(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 30.Tiganos E, Yao XJ, Friborg J, Daniel N, Cohen EA. Putative alpha-helical structures in the human immunodeficiency virus type 1 Vpu protein and CD4 are involved in binding and degradation of the CD4 molecule. J Virol. 1997 Jun;71(6):4452–4460. doi: 10.1128/jvi.71.6.4452-4460.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clavel F, Charneau P. Fusion from without directed by human immunodeficiency virus particles. Journal of virology. 1994 Feb;68(2):1179–1185. doi: 10.1128/jvi.68.2.1179-1185.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. The Journal of cell biology. 2003 Sep 1;162(5):909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer C, Zizioli D, Lausmann S, et al. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. The EMBO journal. 2000 May 15;19(10):2193–2203. doi: 10.1093/emboj/19.10.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. The Journal of cell biology. 2004 Mar 29;164(7):1065–1076. doi: 10.1083/jcb.200311064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyakawa K, Sawasaki T, Matsunaga S, et al. Interferon-induced SCYL2 limits release of HIV-1 by triggering PP2A-mediated dephosphorylation of the viral protein Vpu. Science signaling. 2012 Oct 9;5(245):ra73. doi: 10.1126/scisignal.2003212. [DOI] [PubMed] [Google Scholar]
- 36.Voorhees P, Deignan E, van Donselaar E, et al. An acidic sequence within the cytoplasmic domain of furin functions as a determinant of trans-Golgi network localization and internalization from the cell surface. EMBO J. 1995 Oct 16;14(20):4961–4975. doi: 10.1002/j.1460-2075.1995.tb00179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crump CM, Xiang Y, Thomas L, et al. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J. 2001 May 1;20(9):2191–2201. doi: 10.1093/emboj/20.9.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teuchert M, Berghofer S, Klenk HD, Garten W. Recycling of furin from the plasma membrane. Functional importance of the cytoplasmic tail sorting signals and interaction with the AP-2 adaptor medium chain subunit. The Journal of biological chemistry. 1999 Dec 17;274(51):36781–36789. doi: 10.1074/jbc.274.51.36781. [DOI] [PubMed] [Google Scholar]
- 39.Teuchert M, Schafer W, Berghofer S, Hoflack B, Klenk HD, Garten W. Sorting of furin at the trans-Golgi network. Interaction of the cytoplasmic tail sorting signals with AP-1 Golgi-specific assembly proteins. The Journal of biological chemistry. 1999 Mar 19;274(12):8199–8207. doi: 10.1074/jbc.274.12.8199. [DOI] [PubMed] [Google Scholar]
- 40.Hussain A, Das SR, Tanwar C, Jameel S. Oligomerization of the human immunodeficiency virus type 1 (HIV-1) Vpu protein--a genetic, biochemical and biophysical analysis. Virol J. 2007;4:81. doi: 10.1186/1743-422X-4-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu JX, Sharpe S, Ghirlando R, Yau WM, Tycko R. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci. 2010 Oct;19(10):1877–1896. doi: 10.1002/pro.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dube M, Paquay C, Roy BB, Bego MG, Mercier J, Cohen EA. HIV-1 Vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic. 2011 Dec;12(12):1714–1729. doi: 10.1111/j.1600-0854.2011.01277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah AH, Sowrirajan B, Davis ZB, et al. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell host & microbe. 2010 Nov 18;8(5):397–409. doi: 10.1016/j.chom.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moll M, Andersson SK, Smed-Sorensen A, Sandberg JK. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood. 2010 Sep 16;116(11):1876–1884. doi: 10.1182/blood-2009-09-243667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janvier K, Pelchen-Matthews A, Renaud JB, Caillet M, Marsh M, Berlioz-Torrent C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011;7(2):e1001265. doi: 10.1371/journal.ppat.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raiborg C, Bache KG, Mehlum A, Stang E, Stenmark H. Hrs recruits clathrin to early endosomes. The EMBO journal. 2001 Sep 3;20(17):5008–5021. doi: 10.1093/emboj/20.17.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirst J, Lui WW, Bright NA, Totty N, Seaman MN, Robinson MS. A family of proteins with gamma-adaptin and VHS domains that facilitate trafficking between the trans-Golgi network and the vacuole/lysosome. The Journal of cell biology. 2000 Apr 3;149(1):67–80. doi: 10.1083/jcb.149.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonifacino JS. The GGA proteins: adaptors on the move. Nature reviews. Molecular cell biology. 2004 Jan;5(1):23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- 49.Greenberg M, DeTulleo L, Rapoport I, Skowronski J, Kirchhausen T. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Current biology : CB. 1998 Nov 5;8(22):1239–1242. doi: 10.1016/s0960-9822(07)00518-0. [DOI] [PubMed] [Google Scholar]
- 50.Ruiz A, Hill MS, Schmitt K, Guatelli J, Stephens EB. Requirements of the membrane proximal tyrosine and dileucine-based sorting signals for efficient transport of the subtype C Vpu protein to the plasma membrane and in virus release. Virology. 2008 Aug 15;378(1):58–68. doi: 10.1016/j.virol.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramirez PW, DePaula-Silva AB, Szaniawski M, Barker E, Bosque A, Planelles V. HIV-1 Vpu utilizes both cullin-RING ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology. 2015 Jul 28;12:65. doi: 10.1186/s12977-015-0192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vigan R, Neil SJ. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. Journal of virology. 2010 Dec;84(24):12958–12970. doi: 10.1128/JVI.01699-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mauxion F, Le Borgne R, Munier-Lehmann H, Hoflack B. A casein kinase II phosphorylation site in the cytoplasmic domain of the cation-dependent mannose 6-phosphate receptor determines the high affinity interaction of the AP-1 Golgi assembly proteins with membranes. The Journal of biological chemistry. 1996 Jan 26;271(4):2171–2178. doi: 10.1074/jbc.271.4.2171. [DOI] [PubMed] [Google Scholar]
- 54.Schapiro FB, Soe TT, Mallet WG, Maxfield FR. Role of cytoplasmic domain serines in intracellular trafficking of furin. Mol Biol Cell. 2004 Jun;15(6):2884–2894. doi: 10.1091/mbc.E03-09-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 56.Chaudhuri R, Mattera R, Lindwasser OW, Robinson MS, Bonifacino JS. A basic patch on alpha-adaptin is required for binding of human immunodeficiency virus type 1 Nef and cooperative assembly of a CD4-Nef-AP-2 complex. Journal of virology. 2009 Mar;83(6):2518–2530. doi: 10.1128/JVI.02227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh RK, Lau D, Noviello CM, Ghosh P, Guatelli JC. An MHC-I cytoplasmic domain/HIV-1 Nef fusion protein binds directly to the mu subunit of the AP-1 endosomal coat complex. PloS one. 2009;4(12):e8364. doi: 10.1371/journal.pone.0008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greenberg ME, Bronson S, Lock M, Neumann M, Pavlakis GN, Skowronski J. Co-localization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 down-regulation. The EMBO journal. 1997 Dec 1;16(23):6964–6976. doi: 10.1093/emboj/16.23.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ndung’u T, Renjifo B, Essex M. Construction and analysis of an infectious human Immunodeficiency virus type 1 subtype C molecular clone. Journal of virology. 2001 Jun;75(11):4964–4972. doi: 10.1128/JVI.75.11.4964-4972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Novitsky VA, Montano MA, McLane MF, et al. Molecular cloning and phylogenetic analysis of human immunodeficiency virus type 1 subtype C: a set of 23 full-length clones from Botswana. Journal of virology. 1999 May;73(5):4427–4432. doi: 10.1128/jvi.73.5.4427-4432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Similar endocytic activity is observed in both clade B and clade C Vpu. (A) Steady-state surface expression of CD8 constructs in HeLa P4.R5 cells. Cells were co-transfected to express CD8−, CD8−Vpu clade B (NL4.3), CD8-Vpu clade C (MJ4) or CD8-Vpu (96BW), in combination with a GFP expression construct as a transfection marker. Cell surface CD8 was stained and analyzed by two-color flow cytometry. The graph indicates the mean fluorescence intensity of surface CD8 staining in the GFP-positive cell population; error bars represent the standard deviation of two independent experiments. (B) The endocytic rates of the CD8-Vpu chimeras were measured in HeLa P4.R5 cells. Endocytosis assays were performed as detailed in Figure 1. Data are presented as the percentage of surface CD8 remaining over time. Error bars indicate the standard deviation of two independent experiments. (C) Sequence alignment showing location of residues conserved across clade B and C Vpu.
Figure S2. Endocytosis of CD8-Vpu-CD is independent of CD4. (A) Steady-state surface expression of CD8-Vpu-CD was measured in HeLa P4.R5 cells and HeLa Z24 cells, which do not express CD4. Cells were co-transfected to express CD8-Vpu and GFP as a transfection marker. Cell surface CD8 was stained and analyzed by two-color flow cytometry. The graph indicates the mean fluorescence intensity of surface CD8 staining in the GFP-positive cell population; error bars represent the standard deviation of three independent experiments. (B) The endocytic rate of CD8-Vpu was measured in HeLa P4.R5 and Z24 cells. Endocytosis assays were performed as detailed in Figure 1. Data are presented as the percentage of surface CD8 remaining over time. Error bars indicate the standard deviation of three independent experiments. (C) Expression of CD8-Vpu and CD4 was measured in the two cell lines by western blot, control cells were transfected to express GFP only.
Figure S3. Colocalization of CD8-fusion controls with markers of clathrin-mediated endosomal trafficking pathways. HeLa P4.R5 cells were either co-transfected with plasmids encoding CD8−, CD8-Nef, CD8-Nef-LL/AA and DSRed-Clathrin, or transfected to express the CD8-fusion proteins alone. The cells were fixed, permeabilized, blocked and stained for CD8 (green) and the endosomal marker transferrin receptor (TfnR, red). The cells were examined by widefield, deconvolution microscopy; images are z-stack projections of the total cell volumes. Scale bars = 10 μm.
Figure S4. Endocytosis of CD8-Vpu-CD is insensitive to siRNA-mediated depletion of AP-1. (A) The rates of internalization of the truncated CD8−, CD8-VpuCD, CD8-Nef, or CD8-Nef-LL/AA were measured in AP-1-depleted HeLa P4.R5 cells by flow cytometry. The cells were transfected with siRNA oligonucleotides directed against AP-1γ subunit (AP-1γ) or a non-targeting control (siControl). After 48 hours, the cells were transfected to express the indicated CD8-chimeras and a GFP expression construct. 24 hours later, the internalization rates of the CD8- chimeras were measured by antibody labeling and flow cytometric analysis. Steady-state surface levels of CD8-chimeras are shown (0-timepoint control). Data were normalized to the 0-timepoint control and presented as percentage surface CD8 remaining over time, in either siControl (B) or siAP-1γ (C) treated cells. Error bars indicate the standard deviation of three independent experiments. (C) Protein from cell lysates was separated by SDS-PAGE and AP-1γ depletion confirmed by western blotting. (D) The intracellular localization of CD8-VpuCD was determined by widefield microscopy of HeLa P4.R5 cells transfected with negative control or clathrin siRNA. Cells were transfected with CD8-VpuCD, and 24 hours later fixed and stained for immunofluorescence. AP-1γ depletion in these cells was determined by primary antibody labeling of AP-1γ and fluorescent secondary detection (red); CD8 staining is shown in green. Scale = 10μm.
Figure S5. The membrane-proximal Yxxφ motif is dispensable for μ binding. Substitutions Y29A and I32A were introduced into GST-tagged VpuCD. Phosphorylation of serines 52 and 56 was controlled through production of recombinant GST-VpuCD in the absence or presence of Casein Kinase II (CK-II). GST-pulldown was performed between μ1 (A), μ2 (B) and phosphorylated or non-phosphorylated GST-Vpu mutants. Bound proteins were analyzed by SDS-PAGE and Coomassie staining.