

ABSTRACT
For fatty acid biosynthesis, Corynebacterium glutamicum uses two type I fatty acid synthases (FAS-I), FasA and FasB, in addition to acetyl-coenzyme A (CoA) carboxylase (ACC) consisting of AccBC, AccD1, and AccE. The in vivo roles of the enzymes in supplying precursors for biotin and α-lipoic acid remain unclear. Here, we report genetic evidence demonstrating that the biosynthesis of these cofactors is linked to fatty acid biosynthesis through the FAS-I pathway. For this study, we used wild-type C. glutamicum and its derived biotin vitamer producer BFI-5, which was engineered to express Escherichia coli bioBF and Bacillus subtilis bioI. Disruption of either fasA or fasB in strain BFI-5 led to decreased production of biotin vitamers, whereas its amplification contributed to increased production, with a larger impact of fasA in both cases. Double disruptions of fasA and fasB resulted in no biotin vitamer production. The acc genes showed a positive effect on production when amplified simultaneously. Augmented fatty acid biosynthesis was also reflected in pimelic acid production when carbon flow was blocked at the BioF reaction. These results indicate that carbon flow down the FAS-I pathway is destined for channeling into the biotin biosynthesis pathway, and that FasA in particular has a significant impact on precursor supply. In contrast, fasB disruption resulted in auxotrophy for lipoic acid or its precursor octanoic acid in both wild-type and BFI-5 strains. The phenotypes were fully complemented by plasmid-mediated expression of fasB but not fasA. These results reveal that FasB plays a specific physiological role in lipoic acid biosynthesis in C. glutamicum.
IMPORTANCE For the de novo biosynthesis of fatty acids, C. glutamicum exceptionally uses a eukaryotic multifunctional type I fatty acid synthase (FAS-I) system comprising FasA and FasB, in contrast to most bacteria, such as E. coli and B. subtilis, which use an individual nonaggregating type II fatty acid synthase (FAS-II) system. In this study, we reported genetic evidence demonstrating that the FAS-I system is the source of the biotin precursor in vivo in the engineered biotin-prototrophic C. glutamicum strain. This study also uncovered the important physiological role of FasB in lipoic acid biosynthesis. Here, we present an FAS-I enzyme that functions in supplying the lipoic acid precursor, although its biosynthesis has been believed to exclusively depend on FAS-II in organisms. The findings obtained here provide new insights into the metabolic engineering of this industrially important microorganism to produce these compounds effectively.
KEYWORDS: Corynebacterium glutamicum, biotin, fatty acid biosynthesis, lipoic acid, metabolic engineering
INTRODUCTION
Biotin and α-lipoic acid are important sulfur-containing compounds with fatty acid chain-like moieties: biotin is a C, N, S-heterocyclic ring with a C5 (pentanoic) fatty acid chain, and lipoic acid is a C8 (octanoic) fatty acid chain with thiol groups at the C-6 and C-8 carbons. Both compounds are widespread in all three domains of life (1–4) and have crucial functions in cellular metabolism as cofactors. Biotin, also known as vitamin H, is covalently attached to a conserved lysine residue of a biotin-dependent protein by biotin protein ligase, mediating carboxylation and decarboxylation reactions (5–7). Such biotin-dependent carboxylases are known to exist in all domains of life (8). For example, the naturally biotin-auxotrophic Corynebacterium glutamicum has two biotin-dependent enzymes, the anaplerotic enzyme pyruvate carboxylase and the fatty acid biosynthesis enzyme acetyl-coenzyme A (CoA) carboxylase (9, 10), the biotinylation of which was shown to be specified by the biotin protein ligase gene birA (NCgl0679) (11). On the other hand, lipoic acid is essential for the function of several key enzymes involved in oxidative and single-carbon metabolism, including pyruvate dehydrogenase, 2-oxoglutarate dehydrogenase, and the glycine cleavage system (2, 3). Lipoic acid-prototrophic C. glutamicum has two lipoic acid-dependent enzyme complexes, pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase, in its central metabolism (12, 13). In addition to their biological significance, they are important commercially because of their various applications in the pharmaceutical, cosmetic, food, and livestock industries (2, 8, 14, 15). Due to the absence of an efficient method for producing either compound through fermentation, both biotin and lipoic acid are currently synthesized via costly multistep chemical processes (8, 16, 17). However, there is an increasing interest in the development of environmentally friendly fermentation methods that use renewable feedstocks for production.
We have long been working on the amino acid-producing microorganism C. glutamicum. Our current objective is to expand the potential of this bacterium for the production of fatty acids and their derivatives, especially the petroleum-derived high-value chemicals biotin and lipoic acid (18, 19). The first task toward this goal is to establish a route leading to each target metabolite from sugar, based on limited genetic and genomic information (20–24). According to this policy, we recently engineered naturally biotin-auxotrophic C. glutamicum into a biotin prototroph (18). The successful genetic modifications that enabled C. glutamicum to de novo synthesize biotin involve the heterologous expression of the E. coli bioBF genes and the B. subtilis bioI gene (Fig. 1). Likewise, German and Danish groups have developed biotin-prototrophic C. glutamicum by a similar approach with the use of bioI (25, 26). Since the bioI gene product BioI has been shown to be able to generate a C7 pimelate moiety in vitro by catalyzing the oxidative C—C bond cleavage of acyl carrier protein (ACP)-bound long-chain fatty acids, such as oleic acid (C18:1) and palmitic acid (C16:0) (27, 28), the biotin precursor pimeloyl-CoA (or pimeloyl-ACP) in the engineered strain could be supplied from the fatty acid-synthesizing intermediate acyl-CoA (or acyl-ACP) that is destined for incorporation into the membrane lipid (Fig. 1), although this remains speculative. Thus, our next step is to obtain evidence showing that the source of the biotin precursor in vivo is the fatty acid biosynthesis pathway.
FIG 1.

Proposed de novo biosynthetic pathways and the relevant genes of biotin and lipoic acid in C. glutamicum. Fatty acid biosynthesis in this organism begins with the reaction of acetyl-CoA carboxylase consisting of three subunits, AccBC, AccD1, and AccE, and then proceeds to the FAS-I pathway consisting of FasA and FasB. The biotin biosynthesis pathway of C. glutamicum is incomplete due to the lack of bioF and the gene for the de novo synthesis of pimeloyl-CoA (or pimeloyl-ACP). In the previous study, we demonstrated that E. coli bioBF and B. subtilis bioI could bridge the gaps (18). The origin of pimeloyl-CoA (or pimeloyl-ACP) in vivo could be the fatty acid biosynthesis pathway, but this remains speculative. In contrast, lipoic acid is assumed to be synthesized from octanoyl-CoA (or octanoyl-ACP) in a manner similar to that of E. coli (3). The octanoyl moiety is first transferred to the apoprotein (E2) by LipB and is then converted to lipoic acid by LipA to form lipoyl-E2. Also in this case, the origin of octanoyl-CoA (or octanoyl-ACP) remains an enigma. TCA, tricarboxylic acid.
The situation is the same for lipoic acid biosynthesis in C. glutamicum. This organism is assumed to be capable of the de novo synthesis of lipoic acid because of its prototrophy for the cofactor. Studies on E. coli have established that the ACP derivative of octanoic acid (C8) is the precursor of lipoic acid in the de novo biosynthetic pathway (2, 3, 29). Biosynthesis includes the transfer of an octanoyl moiety to the lipoyl domain of a lipoate-dependent apoenzyme (E2) by the LipB reaction, followed by introduction of two sulfur atoms at the C-6 and C-8 positions of the octanoyl moiety by the LipA reaction, resulting in protein-bound lipoic acid (Fig. 1). In addition to the de novo pathway, E. coli has a salvage pathway to utilize exogenous free lipoic acid and octanoic acid through transferring them to the lipoyl domain of the E2 subunit by the lipoate-protein ligase (LplA) reaction (3, 29). As the putative lipA (NCgl2128) and lipB (NCgl2127) genes are present on the C. glutamicum genome to form a cluster with aceF (NCgl2126) encoding the E2 subunit (13), C. glutamicum is thought to de novo synthesize lipoic acid through an octanoic acid derivative in a manner similar to that of E. coli. The putative lplA gene (NCgl1029) also exists on the genome, suggesting that C. glutamicum can utilize exogenous free lipoic acid and octanoic acid, like E. coli. However, in the de novo synthesis of lipoic acid, the origin of the octanoyl moiety remains an enigma (Fig. 1), because the de novo synthesis of medium-chain (8- to 10-carbon) fatty acids has not been observed in C. glutamicum. In this regard, it should be noted that C. glutamicum originally lacks the β-oxidation pathway involving fatty acid degradation (19, 30), which makes it unlikely that the medium-chain fatty acids are generated in the middle of the degradation cycles of long-chain fatty acids.
For the de novo biosynthesis of fatty acids from acetyl-CoA, the Corynebacteriaceae, including C. glutamicum, Corynebacterium ammoniagenes, and Mycobacterium tuberculosis, exceptionally use type I fatty acid synthase (FAS-I) (31–34), a eukaryotic-type multienzyme that performs successive cycles of fatty acid synthesis, into which all activities required for fatty acid elongation are integrated (35). The products of the FAS-I pathway are believed to be CoA-bound long-chain fatty acids, such as oleic acid and palmitic acid (34, 35), both of which represent the majority of fatty acids in the membrane lipid of C. glutamicum (36). In contrast, fatty acid synthesis in most bacteria, such as E. coli and B. subtilis, is catalyzed by individual nonaggregating enzymes (FAS-II), and the products of the FAS-II pathway are ACP derivatives (37). In E. coli, the FAS-II pathway is thought to be the source of the C8 octanoyl moiety required for lipoic acid synthesis (38). More specifically, octanoyl-ACP is believed to be a preferred product of β-ketoacyl-ACP synthase III in vivo (39).
Eukaryotic organisms, including yeast, fungi, plants, and animals, possess the FAS-I pathway in the cytoplasm (40). Recent studies have revealed, however, that they also have the bacterial type FAS-II pathway in mitochondria (41), thus raising the puzzling question of why the FAS-II pathway has been maintained in mitochondria (in addition to the cytosolic FAS-I pathway). On this point, a significant amount of data supports the idea that the mitochondrial FAS-II pathway is involved in the de novo synthesis of the octanoyl moiety required for lipoic acid formation in eukaryotic cells (41–43). This hypothesis seems reasonable, because all of the known lipoic acid-dependent enzymes in eukaryotes are located in mitochondria (44, 45). The known FAS systems in different organisms are summarized in Table 1.
TABLE 1.
FAS systems in organisms
| Organism | Cytosol | Mitochondria | Plastid | Possible source of lipoic acid precursor | Reference(s) |
|---|---|---|---|---|---|
| Animals (mammals) | FAS-I | FAS-II | —a | Mitochondrial FAS-II | 63 |
| Plants (higher plants) | — | FAS-II | FAS-II | Mitochondrial FAS-II | 43, 64, 65 |
| Plastidial FAS-II | |||||
| Fungi (Neurospora crassa) | FAS-I | FAS-II | — | Mitochondrial FAS-II | 66, 67 |
| Yeast (Saccharomyces cerevisiae) | FAS-I | FAS-II | — | Mitochondrial FAS-II | 41 |
| Escherichia coli | FAS-II | — | — | FAS-II | 29 |
| Mycobacterium tuberculosis | FAS-I | — | — | Unknown | 33, 35, 68 |
| FAS-IIb | |||||
| Corynebacterium glutamicum | FAS-I | — | — | Unknown | 31, 33 |
—, not found.
The mycobacterial FAS-II is thought to be incapable of de novo fatty acid synthesis from acetyl-CoA, but it functions in elongating the FAS-I product long-chain fatty acids (12- to 16-carbon) to the very-long-chair mycolic acids (35).
Unlike eukaryotic organisms, C. glutamicum possesses the FAS-I pathway but not the FAS-II pathway (33). How then does this bacterium generate the octanoyl moiety required for lipoic acid synthesis? Our hypothesis is that the FAS-I pathway carried by C. glutamicum is exceptionally responsible for the formation of the octanoyl moiety. On this point, it is worth noting that the FAS-I pathway of C. glutamicum consists of two type I fatty acid synthases, FasA and FasB, encoded by fasA and fasB, respectively (31, 32), just as in the case of the closely related species C. ammoniagenes (46). The expression of fasA is known to be much higher than that of fasB: the fasA transcript accounts for approximately 70% of the sum of both fasA and fasB transcripts in C. glutamicum cells grown on glucose (31, 46). The major FasA enzyme is essential for growth, as it synthesizes membrane lipids consisting mainly of oleic acid and palmitic acid, and its deficiency is known to cause oleic acid auxotrophy (31, 46, 47). The minor FasB enzyme is thought to primarily synthesize palmitic acid, but not oleic acid, and is dispensable for growth (31, 46, 47). In this study, however, we obtained genetic evidence indicating that FasB specifically functions in supplying the octanoyl precursor of lipoic acid, and FasA and FasB thus have different physiological roles in cell growth.
Here, we describe the in vivo roles of the fatty acid biosynthesis enzymes in supplying the precursors for the biosynthesis of biotin and lipoic acid using wild-type C. glutamicum and its derived biotin vitamer producer. Our report shows the direct relationship between the biosynthesis of these cofactors and fatty acid biosynthesis through the FAS-I pathway.
RESULTS
Generation of a C. glutamicum strain with E. coli bioBF and B. subtilis bioI on its genome.
C. glutamicum is a natural biotin auxotroph due to the lack of the bioF gene and the gene(s) for the de novo synthesis of pimeloyl-CoA (or pimeloyl-ACP) (Fig. 1). We have recently constructed the C. glutamicum strain BF-3, which expresses the cotranscribed E. coli bioBF genes on its genome (18). By using this strain as a host, we demonstrated that further expression of the B. subtilis bioI gene by using a plasmid system resulted in a biotin prototroph, BFI-4, that is capable of de novo synthesis of biotin (18). In this study, to facilitate the following strain engineering, we inserted the bioI gene into the noncoding regions of the genome of strain BF-3 to generate strain BFI-5 (Fig. 2). This strain showed both biotin prototrophy and the ability to produce approximately 5 μg per liter of biotin vitamers when cultivated in minimal medium (MM) (1% glucose). If biotin biosynthesis in strain BFI-5 is linked to fatty acid biosynthesis, disruption of the fatty acid biosynthesis gene(s) should affect the biotin-synthesizing ability of the strain.
FIG 2.

Schematic diagram of the creation of strain C. glutamicum BFI-5 carrying E. coli bioBF and B. subtilis bioI on its genome. We previously constructed C. glutamicum BF-3, in which the E. coli genomic region comprising the bioBF gene cluster and its promoter/operator sequence (P/O) was inserted into the wild-type genome (18). Likewise, the B. subtilis bioI gene was inserted in the vicinity of the bioBF genes so as to be constitutively expressed under the promoter (PgapA) of the endogenous gapA gene.
Effect of fas disruption on biotin vitamer production.
Oleic acid and palmitic acid comprise the bulk of the fatty acids found in C. glutamicum membrane lipids, and two functional FAS-I proteins, FasA and FasB (Fig. 1), are considered to play significant roles in controlling the chain length and amounts of these fatty acids (31). To examine the effect of a deficiency of either or both of them on biotin vitamer production by strain BFI-5, we constructed fasA- and fasB-disrupted strains, designated strain BFI ΔfasA and strain BFI ΔfasB, respectively, and their double disruptant, designated strain BFI ΔfasAB, from strain BFI-5. The performances of these three mutant strains were compared with that of the parental strain BFI-5 using MM (1% glucose) supplemented with 10 μg of lipoic acid per liter in 300-ml baffled Erlenmeyer flasks. Since fasA disruption caused a requirement of oleic acid for growth, we evaluated the oleic acid auxotrophic strains BFI ΔfasA and BFI ΔfasAB under the conditions supplemented with the oleic acid surfactant Tween 80. Under these conditions, the oleic acid-auxotrophic strains BFI ΔfasA and BFI ΔfasAB exhibited retarded growth, probably because of the inefficient utilization of Tween 80 as the source of oleoyl-CoA (or oleoyl-ACP), but ultimately led to almost the same growth levels as the control strain (Fig. S1 and 3A). After glucose was consumed, the culture supernatant was subjected to a biotin vitamer bioassay. First of all, we confirmed that supplementation of Tween 80, namely, exogenous oleic acid, had little influence on biotin vitamer production in both the control strain BFI-5 and strain BFI ΔfasB (Fig. 3A). Under such Tween 80-supplemented conditions, strains BFI ΔfasA and BFI ΔfasB showed approximately 80% and 42% decreased yields of biotin vitamers, respectively, compared to the control strain. Strain BFI ΔfasAB produced no detectable biotin vitamer (Fig. 3A). These data suggest that both FasA and FasB play significant roles in supplying carbon into the biotin biosynthesis pathway, and therefore, fas disruption would cause a shortage of the biotin precursor pimeloyl-CoA (or pimeloyl-ACP). In fact, exogenous pimelic acid was shown to improve biotin vitamer production by the fas-disrupted strains to levels comparable to that of the control strain BFI-5: when cultivated under the conditions of supplementation with pimelic acid (10 mg · liter−1), strains BFI ΔfasA, BFI ΔfasB, and BFI ΔfasAB, as well as the control strain BFI-5, produced approximately 30 μg per liter of biotin vitamers.
FIG 3.

Biotin vitamer production by strain BFI-5 with disrupted fatty acid biosynthesis genes (A) and amplified fatty acid biosynthesis genes (B). Cultivations were carried out in 30 ml of biotin-free MM (1% glucose) supplemented with 10 μg of lipoic acid per liter in 300-ml baffled Erlenmeyer flasks. For cultures of strains BFI ΔfasA and BFI ΔfasAB, 1 g of Tween 80 per liter was added to satisfy the oleic auxotrophy. The control strain BFI-5 and strain BFI ΔfasB were cultivated under the conditions both with (+) and without (−) Tween 80 (1 g · liter−1). Plasmid carriers were cultivated in the presence of 10 mg of kanamycin per liter. Under these conditions, the plasmid maintenance rate at the end of cultivation was more than 97.0% in all cultures. Titers of biotin vitamers are shown as the means and standard deviations of the results from three independent cultures. Growth values (■) are means of the results from three independent cultures, which showed <5% differences among them. Data for comparison between groups of the control vector carriers and the pCfasB carriers (*) were analyzed by Student's t test using JMP statistical software version 8.0.1 (SAS Institute, Cary, NC), and the differences were considered statistically significant at P values of <0.03. OD600, OD at 600 nm.
Effect of amplified fatty acid biosynthesis genes on biotin vitamer production.
Based on the above-mentioned results, it could be reasonably expected that the increased carbon flow down the fatty acid biosynthesis pathway results in increased production of biotin vitamers in strain BFI-5. To examine this possibility, we constructed pCaccBC and pCaccD1, high-copy-number plasmids containing the accBC and accD1 genes, respectively, under the strong promoter of the C. glutamicum gapA gene (Fig. S2). In addition, we constructed pCaccBCDE, which carries all subunit genes for the ACC complex under the gapA promoter (Fig. S2). On the other hand, the intact fasA and fasB genes, both of which are approximately 9 kb long, were individually cloned into a high-copy-number vector to generate pCfasA and pCfasB, respectively (Fig. S2). Each plasmid was introduced into strain BFI-5, and the resulting plasmid carriers were compared with the control vector carrier for biotin vitamer production when cultivated in MM (1% glucose). As shown in Fig. 3B, plasmids pCaccBC and pCaccD1, which overexpress one subunit of the ACC complex, had only marginal effects on biotin vitamer production, but plasmid pCaccBCDE, which overexpresses all subunits simultaneously, enhanced the titer by 2.7-fold. Furthermore, plasmids pCfasA and pCfasB brought about increased production by 3.5-fold and 1.6-fold, respectively. These data show that carbon through the biotin biosynthesis pathway originates from the fatty acid biosynthesis pathway in strain BFI-5, and that ACC and FasA have major impacts on the precursor supply for biotin biosynthesis.
Effect of amplified fatty acid biosynthesis genes on pimelic acid production.
Since pimeloyl-CoA (or pimeloyl-ACP) is the precursor of biotin vitamers, the carbon influx into the biotin biosynthesis pathway might be more directly reflected on pimelic acid accumulation if the BioF reaction is blocked. To confirm this hypothesis, we constructed C. glutamicum WTI-1, which expresses only the B. subtilis bioI gene on the wild-type genome. Since the engineered strain originally lacks the bioF gene, carbon flow through the BioI reaction would be arrested at the BioF reaction, thereby causing the accumulation of pimeloyl-CoA (or pimeloyl-ACP) and subsequent excretion of free pimelic acid into the medium. In fact, liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis revealed that when cultivated in fermentation medium LFG1 containing 5% glucose in 300-ml baffled Erlenmeyer flasks, strain WTI-1 and its vector carrier accumulated the expected amounts of pimelic acid (approximately 20 μg per liter) in the culture supernatants, whereas wild-type ATCC 13032 did not. Following this, we introduced plasmids pCaccBCDE, pCfasA, and pCfasB into strain WTI-1, and the resulting plasmid carriers were compared with the control vector carrier for pimelic acid production under the same culture conditions. As expected, all the three plasmids, pCaccBCDE, pCfasA, and pCfasB, brought about increased production by 2.3-fold, 3.0-fold, and 1.8-fold, respectively (Table 2). These results reinforce our conclusion that the fatty acid biosynthesis pathway is the source of the biotin precursor in vivo in our C. glutamicum strains.
TABLE 2.
Pimelic acid production by strain WTI-1 with amplified fatty acid biosynthesis genesa
| Strain (plasmid) | Growth (OD660) | Pimelic acid (μg · liter−1) |
|---|---|---|
| ATCC 13032 | 47.5 ± 1.5 | —b |
| WTI-1 (vector) | 47.1 ± 1.7 | 20.3 ± 0.7 |
| WTI-1 (pCaccBCDE) | 46.3 ± 2.1 | 46.7 ± 2.3 |
| WTI-1 (pCfasA) | 46.5 ± 2.0 | 61.3 ± 2.9 |
| WTI-1 (pCfasB) | 46.1 ± 2.2 | 35.9 ± 1.2 |
Production was carried out in fermentation medium LFG1 containing 5% glucose in 300-ml baffled Erlenmeyer flasks. After glucose was consumed, the culture supernatants were subject to LC-MS/MS analysis to determine the amounts of pimelic acid. The detection limit of pimeric acid is approximately 1.0 μg · liter−1 under our analytical conditions. Values are means and standard deviations of the results from three independent experiments.
—, not detected.
Effect of fas disruption on lipoic acid biosynthesis.
Although fasA deficiency causes oleic acid auxotrophy in C. glutamicum, it has been reported that fasB deficiency does not bring about any detectable auxotrophic phenotype (31). However, since C. glutamicum does not require exogenous lipoic acid for aerobic growth on glucose, the octanoyl moiety required for lipoic acid synthesis needs to be supplied by a specific endogenous biosynthetic route. This prompted us to examine the phenotypes of deficiency in fasA or fasB under the wild-type background. For this purpose, fasA- and fasB-disrupted strains were derived from C. glutamicum wild-type (WT) ATCC 13032 to generate strains WT ΔfasA and WT ΔfasB, respectively. These disruptants, as well as the wild-type strain, were examined for their growth on agar plates under the conditions in the presence and absence of oleate, lipoic acid, and octanoic acid. Figure 4A shows the results when appropriate dilutions of the cultures (approximately 103 cells) were spread onto the plates. Strain WT ΔfasA showed an expected phenotype of oleic acid auxotrophy. In contrast, strain WT ΔfasB exhibited lipoic acid-dependent growth. The requirement of lipoic acid in the fasB mutant could be replaced by octanoic acid. For normal growth on agar plates, the fasB mutant required a very small amount of lipoic acid (0.01 μg · liter−1) or a disproportionately large amount of octanoic acid (1 mg · liter−1) (data not shown). The large requirement of octanoic acid compared to lipoic acid could be due to the limited incorporation into the lipoic acid biosynthesis pathway, because exogenous octanoic acid needs to be taken up into cells and then activated to octanoyl-CoA (or octanoyl-ACP) by an enzyme with octanoyl-CoA (or octanoyl-ACP) synthetase activity or transferred to the apoprotein (E2) by a putative LplA enzyme. It should be noted that the lipoic acid-auxotrophic phenotype of strain WT ΔfasB was not observed on agar plates when a higher concentration of cells (approximately 105 cells and more) was spread on the plates (Fig. S3).
FIG 4.
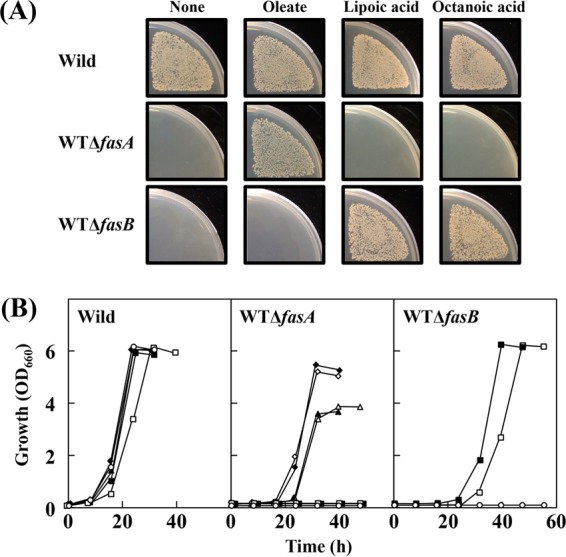
Growth responses of wild-type strain ATCC 13032 and its fasA- and fasB-disrupted strains, WT ΔfasA and WT ΔfasB, respectively, to oleate, lipoic acid, and octanoic acid. (A) After appropriate dilutions of the cultures, an aliquot (approximately 103 cells) was spread onto biotin (100 μg · liter−1)-supplemented MM agar plates with and without 100 mg of sodium oleate, 10 μg of lipoic acid, or 1 mg of octanoic acid per liter and cultured at 30°C for 2 days. The pictures show one representative result from three independent experiments. (B) Cultivations were carried out at 30°C in biotin (100 μg · liter−1)-supplemented MM liquid culture with no additions (○), 50 mg of sodium oleate (▲), 50 mg of sodium oleate plus 10 μg of lipoic acid (△), 100 mg of sodium oleate (◆), 100 mg of sodium oleate plus 10 μg of lipoic acid (♢), 10 μg of lipoic acid (■), or 1 mg of octanoic acid (□) per liter. The inoculum size from the seed culture to the main culture corresponds to 0.01%, as indicated in Materials and Methods. Values are means of the results from three independent cultures, which showed <5% differences among them.
The growth properties were further examined in liquid cultures. Unfortunately, under normal conditions with 1% inoculum from the seed culture to the main culture, we failed to observe lipoic acid auxotrophy for the fasB mutant, although the fasA mutant exhibited oleic acid auxotrophy. However, subsequent investigations revealed that by decreasing the inoculum size to 0.01% or below, real phenotypes of the fasB mutant became evident. For the experiment with results shown in Fig. 4B, which was conducted under conditions with 0.01% inoculum, the phenotypes of the fasB mutant observed on agar plates, namely, the auxotrophy for lipoic acid or octanoic acid, were clearly reproduced in liquid cultures. We also reconfirmed that, in the case of the fasA mutant, lipoic acid and octanoic acid have no stimulating effect on growth in the presence of oleic acid.
Complementation of lipoic acid auxotrophy with cloned fasA and fasB.
To confirm that the lipoic acid auxotrophy of the fasB mutant is actually due to the loss of FasB function, we examined the effects of plasmid-mediated expression of fasA and fasB on the phenotypes in strain WT ΔfasB. Introduction of pCfasB into strain WT ΔfasB resulted in complete recovery of growth even in the absence of lipoic acid and octanoic acid, whereas pCfasA failed to restore the growth (Fig. 5). A series of these results were reproduced under the background of BFI-5 (data not shown). Thus, we concluded that the FasB pathway is the source of the octanoyl moiety required for lipoic acid synthesis under our conditions and that the FasA pathway is unable to produce sufficient amounts of the octanoyl precursor to fulfill the need for cellular lipoic acid synthesis.
FIG 5.
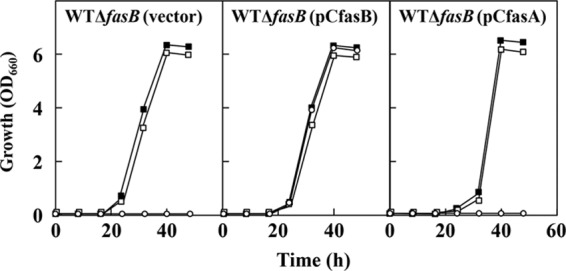
Growth of strains WT ΔfasB carrying the vector plasmids pCS299P, pCfasB, and pCfasA. Cultivations were carried out at 30°C in biotin (100 μg · liter−1)-supplemented MM liquid culture with no additions (○), 10 μg of lipoic acid (■), or 1 mg of octanoic acid (□) per liter. The inoculum size from the seed culture to the main culture corresponds to 0.01%. The plasmid maintenance rate at the end of cultivation was more than 97.0% in all cultures. Values are means of the results from three independent cultures, which showed <5% differences among them.
DISCUSSION
From the 1980s to the early 2000s, a number of biotin-producing strains were developed from various bacterial species, including E. coli, Serratia marcescens, B. subtilis, and Bacillus sphaericus (8). Since the origin of the biotin precursor pimeloyl-CoA (or pimeloyl-ACP) had long been an enigma, previous attempts at strain improvement had focused on the biotin biosynthesis pathway beginning at pimeloyl-CoA (or pimeloyl-ACP) (8). It should be also noted that some of those studies used feeding of relatively costly pimelic acid during fermentation to obtain high production yields (48–50). Only recently, pimeloyl-CoA (or pimeloyl-ACP) synthesis seemed likely to involve fatty acid biosynthesis (2, 7). To the best of our knowledge, however, there have been no reports of increased biotin production studies in which the fatty acid biosynthesis pathway was rationally modified. In this study, we used the engineered biotin vitamer producer C. glutamicum BFI-5 to show that carbon flow down the fatty acid biosynthesis pathway is crucial for the biosynthesis of biotin vitamers. Furthermore, we demonstrated that augmented fatty acid biosynthesis led to increased production of biotin vitamers, thus concluding that the biotin precursor pimeloyl-CoA (or pimeloyl-ACP) originates from the fatty acid biosynthesis pathway in strain BFI-5. In relation to this, Manandhar and Cronan very recently reported that in B. subtilis, the precursor for biotin biosynthesis is free pimelic acid originating from fatty acid biosynthesis, because bioW, encoding pimeloyl-CoA synthetase, was essential for biotin biosynthesis, whereas bioI was dispensable (51). Although the mechanism of formation of free pimelic acid without the BioI function remains to be determined, their hypothesis is that pimeloyl-ACP is directly generated by the FAS-II pathway of B. subtilis and then subjected to thioesterase-catalyzed cleavage to generate free pimelic acid, followed by activation to pimeloyl-CoA by BioW. This hypothesis seems reasonable, because pimeloyl-CoA, but not pimeloyl-ACP, is thought to be the substrate of B. subtilis BioF (51). Nevertheless, this seems not to be the case with our C. glutamicum strain, because C. glutamicum BFI-5 exclusively depends on BioI for de novo biotin biosynthesis. Considering that the products of C. glutamicum FAS-I have been assumed to be CoA derivatives (34), acyl-CoAs rather than acyl-ACPs are likely to be the substrates for BioI in the C. glutamicum strain, whereas the ACP derivatives are thought to be the physiological substrates in B. subtilis (28, 52). Taking these into consideration, it seems reasonable to assume that long-chain fatty acyl-CoAs are subject to BioI-catalyzed oxidative cleavage to directly generate the biotin precursor pimeloyl-CoA in our C. glutamicum strain (Fig. 1).
Both fasA and fasB contributed to biotin vitamer production when amplified in the engineered strain BFI-5. This could be explained as a result of the increased availability of acyl-CoAs (or acyl-ACPs) for the BioI reaction. A larger impact of fasA on production than fasB is reasonable, considering that the expression of fasA is much higher than that of fasB (31). On the other hand, the double disruption of fasA and fasB resulted in a loss of the capability of biotin vitamer synthesis. This means that the FAS-I pathway comprising FasA and FasB is the sole source of the biotin precursor in strain BFI-5. In this regard, it should be noted that the production experiments were carried out under conditions of supplementation with Tween 80. This is not only because fasA disruption caused oleic acid auxotrophy, but also because exogenous oleic acid never affected biotin vitamer production in strain BFI-5 (Fig. 3A). The latter reason raises the question of why exogenous oleic acid failed to contribute to biotin vitamer production. At present, it remains unclear but seems reasonable to speculate as follows, based on the predicted regulatory mechanism of fatty acid biosynthesis in this organism (19, 53). Under the wild-type background with respect to de novo fatty acid biosynthesis, exogenous oleic acid would be taken up into cells and then activated to oleoyl-CoA (or oleoyl-ACP), which would negatively regulate de novo fatty acid biosynthesis so as to maintain the intracellular pool of oleoyl-CoA (or oleoyl-ACP) at steady state. In contrast, against the background of deficiency in the de novo fatty acid biosynthesis, the process of incorporation of exogenous oleic acid into oleoyl-CoA (or oleoyl-ACP), namely, uptake of exogenous oleic acid or its activation to oleoyl-CoA (or oleoyl-ACP), seems to be rate limiting for synthesis of the membrane lipid, judging from the significantly retarded growth of strains BFI ΔfasA and BFI ΔfasAB compared with the parental strain BFI-5 under the conditions of supplementation with Tween 80 (Fig. S1). If so, it is likely that oleoyl-CoA (or oleoyl-ACP) generated through the salvage route would be preferentially incorporated into the membrane lipid instead of being the substrate for the BioI reaction.
Blocking the fatty acid biosynthesis pathway through double disruption of fasA and fasB should make strain BFI-5 incapable of de novo biotin synthesis. Nevertheless, the engineered strain did not show biotin auxotrophy on glucose. This is certainly because biotin is unnecessary for growth on glucose as long as the essential fatty acid oleic acid (or Tween 80) is added to the medium. In contrast, in the case of lipoic acid, a genetic approach is feasible to verify the link between the fatty acid biosynthesis pathway and the source of the lipoic acid precursor, because the cofactor is indispensable for C. glutamicum to grow aerobically on glucose as the sole carbon source. In this study, we showed that disruption of fasB caused auxotrophy for lipoic acid or octanoic acid in the wild-type strain ATCC 13032, and that the phenotypes were fully complemented by plasmid-mediated expression of fasB. These results have proven that the octanoyl moiety of lipoic acid is supplied by FasB in this organism. In this respect, there are two possible mechanisms for the supply of the octanoyl moiety by FasB. The first is that the octanoyl moiety is a direct product of FasB, and the second is that it is derived from the FasB end product palmitic acid or its derivative by an unidentified enzyme with the oxidative C—C bond cleavage activity toward long-chain fatty acids. However, since the long-chain fatty acids made by FasB are also generated by FasA, the second possibility seems unlikely. In fact, the requirement of lipoic acid in the fasB mutant could not be replaced by long-chain oleic acid and palmitic acid, the main products of FasA and FasB, respectively, on MM agar plates (data not shown). Taken together, we conclude that the octanoyl precursor of lipoic acid is a direct product of FasB in this organism, at least under the conditions employed. The octanoyl precursor is likely to be the CoA derivative rather than the ACP derivative (Fig. 1), considering that the products of FasB have been assumed to be CoA derivatives (34). However, this remains speculative because the C. glutamicum LipB catalysis has not been tested with the CoA derivative. The fact that the deficiency of FasA and FasB caused auxotrophy for oleic acid (31, 46) and lipoic acid, respectively, on glucose indicates that the two enzymes have basically different physiological roles.
It is noteworthy that the C. glutamicum FasB, a multifunctional FAS-I enzyme, can function to supply the octanoyl precursor of lipoic acid, since its biosynthesis has been believed to exclusively depend on FAS-II in organisms (29), as mentioned in the introduction. However, this raises the question of what allows FasB to generate the dedicated product octanoyl-CoA. In this respect, the FAS-I of the closely related species C. ammoniagenes has been reported to carry out transacylation of long-chain fatty acids from the enzyme to CoA using its integral palmitoyl transferase activity and to produce long-chain acyl-CoAs, including palmitoyl-, oleoyl-, and stearoyl-CoA (34, 35). Based on this information, one possibility for the synthesis of octanoyl-CoA by the C. glutamicum FasB is that the transacylase in the FasB multienzyme complex may possess the activity of the transfer of the octanoyl moiety to CoA, even at a marginal level. In general, the chain length of its products is considered to be an inherent property of every fatty acid synthase, although the determinants remain elusive (35). Analyses of the structure-activity correlation between FasA and FasB may help to answer the question.
One of the goals of metabolic engineering for product formation is to direct as much carbon as possible from sugar into a desired product. For this goal, the supply of precursors for the relevant terminal biosynthesis pathways is of key importance for successful metabolic engineering. In this study, we demonstrated that the FAS-I pathway is the source of the precursors for both biotin and lipoic acid in C. glutamicum. Furthermore, this study uncovered the important physiological roles of two FAS-I enzymes, FasA and FasB, in the biosynthesis of each cofactor. The findings obtained here provide new insights into the metabolic engineering of this industrially important microorganism to produce these compounds effectively.
MATERIALS AND METHODS
Bacterial strains.
The biotin-auxotrophic wild-type C. glutamicum strain ATCC 13032 was used in this study. A C. glutamicum Δppc mutant was used as an indicator strain for biotin vitamer bioassays, especially under the conditions supplemented with oleic acid. This indicator strain was derived from ATCC 13032 through disruption of the ppc gene encoding phosphoenolpyruvate carboxylase, one of two anaplerotic enzymes carried by this organism. The wild-type strain ATCC 13032, an auxotroph for biotin vitamers, never shows biotin vitamer auxotrophy in the presence of oleic acid. In contrast, the indicator Δppc mutant strain remains auxotrophic for biotin vitamers irrespective of the presence or absence of oleic acid, because ppc deficiency makes cells dependent on the alternative anaplerotic biotin enzyme pyruvate carboxylase, which requires biotin for its activity (54). Since the Δppc mutant strain, like the wild-type strain ATCC 13032, is capable of synthesizing biotin from any of the biotin vitamers, the strain can be used for bioassays for the total biotin vitamers. E. coli K-12 W3110 (55) and B. subtilis RM125 (56) were used as donors of genomic DNA to amplify the biotin biosynthesis genes. E. coli DH5α (57) was used as a host for DNA manipulation.
Plasmids.
Plasmid pCS299P (58), a C. glutamicum-E. coli shuttle vector, was used to clone the PCR products. Plasmid pESB30 (58), which is nonreplicative in C. glutamicum, is a vector for gene replacement in C. glutamicum. Plasmids pCaccBC and pCaccD1 were constructed so that accBC (Cgl0700, NCgl0670) and accD1 (Cgl0708, NCgl0678) were constitutively expressed under the promoter of the endogenous gapA gene (Fig. S2). For the construction of pCaccBC, the coding region of accBC was amplified using primers accBCPgapAFusF and accBCdown100RKpnI, with wild-type ATCC 13032 genomic DNA as the template. On the other hand, the genomic region comprising the gapA promoter was amplified using PgapAKpBgF and accBCPgapAFusR. These two fragments were fused by PCR, digested with KpnI, and then ligated to KpnI-digested pCS299P to yield pCaccBC. Similarly, for the construction of pCaccD1, the coding region of accD1 was amplified using primers accD1PgapAFusF and accD1down150RKpnI, and the genomic region comprising the gapA promoter was amplified using primers PgapAKpBgF and accD1PgapAFusR. These fragments were fused by PCR, digested with KpnI, and then ligated to Kpn-digested pCS299P to yield pCaccD1. Plasmid pCaccBCDE for the constitutive coexpression of modified accBC where a start codon was changed to ATG, accD1, and accE (Cgl0706, NCgl0676) under the gapA promoter, was constructed as follows (Fig. S2). The genomic regions comprising accBC and accE were separately amplified using the primer pairs of InFu-accBCf and InFu-accBCr and of InFu-accEf and InFu-accEr, respectively. On the other hand, pCaccD1 was linearized by inverse PCR using primers InFu-accD1f and InFu-accD1r. The amplified genomic regions and linearized pCaccD1 were fused using the In-Fusion HD cloning kit (Clontech Laboratories, Inc., Mountain View, CA) to yield pCaccBCDE. On this plasmid, the accD1, accBC, and accE genes were tandemly arranged in this order from the gapA promoter. The regions between the accD1 and accBC open reading frames (ORFs) and between the accBC and accE ORFs were identical to the nucleotide sequences from −1 to −30 bp upstream of the accBC gene and from −1 to −23 bp upstream of the accE gene, respectively.
Plasmids pCfasA and pCfasB, which contain the intact fasA gene (Cgl0836 and NCgl0802) and the intact fasB gene (Cgl2495 and NCgl2409), respectively, were constructed as follows (Fig. S2). For pCfasA, the genomic region comprising fasA and its native promoter (from −1 to −332 bp upstream of fasA) was amplified using primers fasAexpFupEcoRV and fasAexpRdownNheI. On the other hand, pCS299P was linearized by inverse PCR using primers InVer-pCS299PfasAf and InVer-pCS299PfasAr. The amplified genomic region and linear pCS299P were fused using the In-Fusion HD cloning kit to yield pCfasA. For plasmid pCfasB, the genomic region comprising fasB and its native promoter (from −1 to −617 bp upstream of fasB) was amplified using primers fasBexpFupBlnI and fasBexpRdownNheI. On the other hand, pCS299P was linearized by inverse PCR using primers InVer-pCS299PfasBf and InVer-pCS299PfasBr. The amplified genomic region and linear pCS299P were fused to yield pCfasB.
The sequences of the primers used in this study are listed in Table 3. All primers were designed based on the genomic sequences of C. glutamicum (accession no. BA000036), B. subtilis (accession no. AL009126), and E. coli (accession no. AP009048), which are publicly available at http://www.genome.jp/kegg/genes.html.
TABLE 3.
Sequences of primers used in this study
| Primer | Sequence (5′ to 3′)a | Purpose |
|---|---|---|
| accBCPgapAFusF | CCTACAATCTTTAGAGGAGACACAACGTGTCAGTCGAGACTAGGAAGATCACCAAG | Expression of accBC |
| accBCdown100RKpnI | CTTGGTACCGAAATCTTGTTGTCGAATG | Expression of accBC |
| PgapAKpBgF | GCGGGTACCAGATCTGAAGATTCCTGATACAAATTCTGTTG | Expression of accBC and accD1 |
| accBCPgapAFusR | CTTGGTGATCTTCCTAGTCTCGACTGACACGTTGTGTCTCCTCTAAAGATTGTAGG | Expression of accBC |
| accD1PgapAFusF | CCTACAATCTTTAGAGGAGACACAACATGACCATTTCCTCACCTTTGATTGACGTC | Expression of accD1 |
| accD1down150RKpnI | TCGGGTACCGGTTATATTAGCCCAGCG | Expression of accD1 |
| accD1PgapAFusR | GACGTCAATCAAAGGTGAGGAAATGGTCATGTTGTGTCTCCTCTAAAGATTGTAGG | Expression of accD1 |
| InFu-accBCf | TGAGTCATCAATTTAAATCAGGAGTTATTAATGTCAGTCGAGACTAGGAAG | Expression of accBC, accD1, and accE |
| InFu-accBCr | TTACTTGATCTCGAGGAGAACAACGC | Expression of accBC, accD1, and accE |
| InFu-accEf | CTCGAGATCAAGTAAAAACTGTTTTTTAAAGGAGAACCATGTCTGAAG | Expression of accBC, accD1, and accE |
| InFu-accEr | TATGGATTCGCCGATCTAGAAGAAATTCACATTCTGAAACGCGC | Expression of accBC, accD1, and accE |
| InFu-accD1f | ATCGGCGAATCCATAAAGGTTCAAAAG | Expression of accBC, accD1, and accE |
| InFu-accD1r | TAAATTGATGACTCATTACAGTGGCATGTTGCCGTGCTTG | Expression of accBC, accD1, and accE |
| fasAexpFupEcoRV | TGGGATATCCTGTGGTGGCTTTTAAGAAG | Expression of fasA |
| fasAexpRdownNheI | TGCGCTAGCAAACTTGAGAAGTTTCATGAG | Expression of fasA |
| InVer-pCS299PfasAf | AAGTTTGCTAGCGCAGGCATGCAAGCTTGGCGTAATCATGG | Expression of fasA |
| InVer-pCS299PfasAr | CCACAGGATATCCCATGCAGGTCGACTCTAGAGGATCC | Expression of fasA |
| fasBexpFupBlnI | AGTCCTAGGCCGGGAGCTGTAGAAAATTGC | Expression of fasB |
| fasBexpRDownNheI | GTTGCTAGCACTAAGTTACCCTCGGTGTGAAG | Expression of fasB |
| InVer-pCS299PfasBf | CTTAGTGCTAGCAACGGCATGCAAGCTTGGCGTAATCATG | Expression of fasB |
| InVer-pCS299PfasBr | TCCCGGCCTAGGACTTGCAGGTCGACTCTAGAGGATCC | Expression of fasB |
| CglfasIA5′BglIIF | ACGAGATCTACGCATTCGTAAGTGG | Deletion of fasA |
| CglfasIAFusR | CAACGGATGCACGTGCCAGGAGGACGGTACCGGTTGCACGTGCCTTGGAAAC | Deletion of fasA |
| CglfasIAFusF | GTTTCCAAGGCACGTGCAACCGGTACCGTCCTCCTGGCACGTGCATCCGTTG | Deletion of fasA |
| CglfasIA3′BglIIR | CAGAGATCTTAGCTATCTAACGTTTAGC | Deletion of fasA |
| fasBupF | CAGTATTCCTGTGCATGTGAATACGC | Deletion of fasB |
| fasBFusR | AGGAGGACTGCAGCTTCAACTTCGTTCCTGCTCAATTCGGTCACGT | Deletion of fasB |
| fasBFusF | ACGTGACCGAATTGAGCAGGAACGAAGTTGAAGCTGCAGTCCTCCT | Deletion of fasB |
| fasBdnR | TCTTGATCAAGGTGCCGGTGGGAA | Deletion of fasB |
| ncrFBam2 | ACTGGATCCACACATAAGTGCTCT | Integration of bioI under gapA promoter into C. glutamicum genome |
| PgapAFusR | CTAAATTTCTTCCAACAAATCTTCCGTCTTGTTTCAGGCCACCACTTAGAAGGC | Integration of bioI under gapA promoter into C. glutamicum genome |
| PgapAFusF | GCCTTCTAAGTGGTGGCCTGAAACAAGACGGAAGATTTGTTGGAAGAAATTTAG | Integration of bioI under gapA promoter into C. glutamicum genome |
| bioIFusR | GCAGTTGACGATGCAATTGTCACGTTGTGTCTCCTCTAAAGATTGTAGG | Integration of bioI under gapA promoter into C. glutamicum genome |
| bioIFusF | CCTACAATCTTTAGAGGAGACACAACGTGACAATTGCATCGTCAACTGC | Integration of bioI under gapA promoter into C. glutamicum genome |
| bioIFusR2 | GACAATTGAATTACGCCCTAGTAGTAGATGTTCACTCCCCTTTTTTATAG | Integration of bioI under gapA promoter into C. glutamicum genome |
| ncrFusF | CTATAAAAAAGGGGAGTGAACATCTACTACTAGGGCGTAATTCAATTGTC | Integration of bioI under gapA promoter into C. glutamicum genome |
| ncrRBam2 | TACGGATCCCAGCATCATGCTTGT | Integration of bioI under gapA promoter into C. glutamicum genome |
KpnI sites are underlined, BglII sites are italicized, the BclI site is underlined and bold, and BamHI sites are in bold.
Media.
BY and BYG (BY medium containing 1% glucose) complete media and minimal medium (MM), not supplemented with biotin, were used as basal media for the growth of C. glutamicum strains (59). The fermentation medium LFG1, containing 5% glucose, was used for pimelic acid production (60). Solid plates were made by the addition of Bacto agar (Difco) to a concentration of 1.5%. For the preparation of MM containing sodium oleic acid, sodium oleic acid was separately autoclaved and then mixed with a magnesium sulfate solution and a solution containing other components to prevent insolubilization of the fatty acid. When required, the agar used for MM plates was washed five times with distilled water to remove unnecessary nutrients in the agar. For cultivation of plasmid carriers, kanamycin was added at a final concentration of 10 mg per liter. For growth of E. coli and B. subtilis, Luria-Bertani broth or agar (57) was used.
Recombinant DNA techniques.
Standard protocols (57) were used for the extraction of B. subtilis and E. coli chromosomal DNA for the construction, purification, and analysis of plasmid DNA and for the transformation of E. coli. The extraction of C. glutamicum chromosomal DNA and transformation of C. glutamicum by electroporation were carried out as described previously (59). PCR was performed using a DNA thermal cycler (GeneAmp PCR system 9700; Applied Biosystems, Foster City, CA, USA) using Phusion high-fidelity DNA polymerase (New England BioLabs, Ipswich, MA, USA). Sequencing to confirm the nucleotide sequences of relevant DNA regions was performed using an ABI Prism 377 DNA sequencer from Applied Biosystems, with an ABI Prism BigDye Terminator cycle sequencing kit (Applied Biosystems). The subsequent electrophoresis analysis was carried out using Pageset SQC-5ALN 377 (Toyobo, Osaka, Japan).
Strain construction.
For the chromosomal deletion of fasA and fasB, plasmids pCΔfasA and pCΔfasB, which contained the corresponding genes with internal deletions, respectively, were used to replace the wild-type chromosomal genes with the deleted genes (Fig. S2). For the construction of pCΔfasA, the 5′ and 3′ regions of fasA were amplified using the primer pairs of CglfasIA5′BglIIF and CglfasIAFusR and of CglfasIAFusF and CglfasIA3′ BglIIR, respectively. These two fragments were fused by PCR, digested with BglII, and then ligated to BamHI-digested pESB30 to yield pCΔfasA. Plasmid pCΔfasA carried the in-frame-deleted fasA gene, which was shortened from 8,910 to 2,181 bp and thus was devoid of a motif sequence for the 3-ketoacyl-ACP synthase active site (Prosite motif PS00606). Similarly, for the construction of pCΔfasB, the 5′ and 3′ regions of the fasB gene were amplified using the primer pairs of fasBupF and fasBFusR and of fasBFusF and fasBdnR, respectively. These fragments were fused by PCR, digested with BclI, and then ligated to BamHI-digested pESB30 to yield pCΔfasB. Plasmid pCΔfasB carried the in-frame-deleted fasB gene, which was shortened from 8,991 to 93 bp. The defined chromosomal deletion of the individual gene in both wild-type and BFI-5 strains was accomplished using each plasmid via two recombination events, as described previously (61).
For the chromosomal insertion of the B. subtilis bioI gene so as to be constitutively expressed under the promoter of the endogenous gapA gene, plasmid pBbioIPgapA was used to insert the B. subtilis gene with the gapA promoter between the nucleotide positions 1827653 and 1827654 of the genomes of C. glutamicum BF-3 and wild-type ATCC 13032 to generate strains BFI-5 (Fig. 2) and WTI-1, respectively. For the construction of pBbioIPgapA, the region from genomic positions 1827654 to 1828204 of the ATCC 13032 genome was amplified using primers ncrFBam2 and PgapAFusR (fragment a). Similarly, the region comprising the gapA promoter was amplified using PgapAFusF and bioIFusR (fragment b). On the other hand, the region comprising the B. subtilis bioI gene was amplified using primers bioIFusF and bioIfusR2 (fragment c). Moreover, the region from 1826948 to 1827653 of the ATCC 13032 genome was amplified using primers ncrFusF and ncrRBam2 (fragment d). Fragments a, b, c, and d were fused by PCR in a stepwise manner, digested with BamHI, and ligated to BamHI-digested pESB30 to yield pBbioIPgapA.
Biotin vitamer production.
A 3-ml sample of the seed culture grown in BYG medium to mid-exponential phase at 30°C was harvested, washed with saline to remove biotin vitamer, and inoculated into a 300-ml baffled Erlenmeyer flask containing 30 ml of biotin-free MM supplemented with 10 μg of lipoic acid per liter, followed by cultivation at 30°C using a rotary shaker at 200 rpm. For the cultures of the oleic acid-auxotrophic strains BFI ΔfasA and BFI ΔfasAB, Tween 80 was added into both BYG medium and MM at a final concentration of 1 g per liter. After glucose was consumed, the culture supernatant was prepared by removing cells through centrifugation and subsequent filtration with a Millex-MA filtrate unit (0.45-μm pore size; Millipore Corporation, Billerica, MA). The resulting solution was subject to a biotin vitamer assay.
Biotin vitamer assays.
Biotin vitamers include not only biotin itself but the intermediates in the biotin biosynthesis pathway, that is, the BioF product 7-keto-8-aminopelargonic acid (KAPA), the BioA product 7,8-diaminopelargonic acid (DAPA), and the BioD product dethiobiotin. The total biotin vitamers in the filtered supernatants were measured, basically as described previously (18), using the Δppc mutant strain as an indicator. It is noted that the growth responses of the indicator strain toward KAPA, DAPA, and dethiobiotin were nearly the same as that to biotin, at least within a range from 1 to 100 μg per liter. The bioassay plates consisted of two layers per plate: 15 ml of biotin-free MM bottom agar (1.5%) and 3 ml of biotin-free MM top agar (0.8%). The MM top agar was supplemented with 0.1 ml of indicator-cell solution that was prepared as described previously (59). The bioassay plates were loaded with sterilized paper disks supplemented with 100 μl of the filtered supernatants. After overnight culture at 30°C, the halos that formed around the disks due to the growth of the indicator strain were measured.
Pimelic acid production.
A 3-ml sample of the seed culture grown in BYG medium to the mid-exponential phase at 30°C was inoculated into a 300-ml baffled Erlenmeyer flask containing 30 ml of LFG1 medium, followed by cultivation at 30°C using a rotary shaker at 200 rpm. After glucose was consumed, the culture supernatant was prepared by removing cells through centrifugation and subsequent filtration with a Millex-MA filtrate unit. The resulting solution was subject to LC-MS/MS analysis.
Quantitative determination of pimelic acid.
The concentration of pimelic acid in culture supernatant was determined using LC-MS/MS system of a Quattro micro API (MS) system with a Waters Acquity ultraperformance liquid chromatograph (UPLC) (Waters Co.). Separation was performed at 40°C using a Chemcobond 5-ODS-W reversed-phase column (4.6 by 250 mm; ChemcoPlus Scientific Co., Ltd., Japan), with isocratic elution. Elution was performed at a flow rate of 0.5 ml · min−1 using 0.1% formic acid containing 25% acetonitrile, and the injection volume was 50 μl. The mass spectrometer was operated in negative-mode electrospray ionization (ESI−) with multiple-reaction monitoring (MRM). The mass transition ion was selected as m/z 159.2→97.0 for pimelic acid. The other optimized MS/MS parameters were as follows: 3,000 V of capillary voltage, 25 V of cone voltage, 600 liters · h−1 of desolvation gas (N2) flow, 50 liters · h−1 of cone gas (N2) flow, 120°C of source temperature, 350°C of desolvation temperature, 9.0 ml · h−1 of argon gas flow (Ar), and 15 V collision voltage. The analytical conditions were determined in preliminary experiments. A linear standard curve was obtained using pimelic acid at a concentration range from 10 to 1,000 μg · liter−1.
Liquid cultures to examine the phenotypes of fas disruption.
A 1-ml sample of the seed culture grown in BYG medium to the mid-exponential phase was harvested, resuspended in 1 ml of saline, and then diluted 10 times with saline. The main culture was started by inoculating 0.005 ml of the 10-times-diluted seed culture into 5 ml of MM supplemented with biotin at a final concentration of 100 μg per liter. In this experiment, the final inoculum size from the seed culture to the main culture corresponds to 0.01%. When required, sodium oleate, lipoic acid, or octanoic acid was added at the indicated concentrations (Fig. 3 to 5). All liquid cultures were incubated at 30°C in L-type test tubes on a Monod shaker at 48 strokes per min. To minimize the influence of the carryover of lipoic acid or other unnecessary nutrients, β-cyclodextrin (Nacalai Tesque, Kyoto, Japan) with clathrate action toward lipoic acid (62) was added to the MM to 1.5%.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shin-ichi Hashimoto, Kazuhiko Tabata, Tetsuya Abe, and Satoshi Mitsuhashi for their encouraging support of our research, and Kozo Nakamura, Masahiro Koyama, and Shohei Yamaguchi for their technical guidance on LC-MS/MS analysis.
This work was supported in part by a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (grant 15K07356).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01322-17.
REFERENCES
- 1.Perham RN. 2000. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- 2.Marquet A, Bui BT, Florentin D. 2001. Biosynthesis of biotin and lipoic acid. Vitam Horm 61:51–101. doi: 10.1016/S0083-6729(01)61002-1. [DOI] [PubMed] [Google Scholar]
- 3.Cronan JE, Zhao X, Jiang Y. 2005. Function, attachment and synthesis of lipoic acid in Escherichia coli. Adv Microb Physiol 50:103–146. doi: 10.1016/S0065-2911(05)50003-1. [DOI] [PubMed] [Google Scholar]
- 4.Lin S, Hanson RE, Cronan JE. 2010. Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat Chem Biol 6:682–688. doi: 10.1038/nchembio.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowles JR. 1989. The mechanism of biotin-dependent enzymes. Annu Rev Biochem 58:195–221. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]
- 6.Jitrapakdee S, Wallace JC. 2003. The biotin enzyme family: conserved structural motifs and domain rearrangements. Curr Protein Pept Sci 4:217–229. doi: 10.2174/1389203033487199. [DOI] [PubMed] [Google Scholar]
- 7.Lin S, Cronan JE. 2011. Closing in on complete pathways of biotin biosynthesis. Mol Biosyst 7:1811–1821. doi: 10.1039/c1mb05022b. [DOI] [PubMed] [Google Scholar]
- 8.Streit WR, Entcheva P. 2003. Biotin in microbes, the genes involved in its biosynthesis, its biochemical role and perspectives for biotechnological production. Appl Microbiol Biotechnol 61:21–31. doi: 10.1007/s00253-002-1186-2. [DOI] [PubMed] [Google Scholar]
- 9.Jäger W, Peters-Wendisch PG, Kalinowski J, Pühler A. 1996. A Corynebacterium glutamicum gene encoding a two-domain protein similar to biotin carboxylases and biotin-carboxyl-carrier proteins. Arch Microbiol 166:76–82. doi: 10.1007/s002030050359. [DOI] [PubMed] [Google Scholar]
- 10.Peters-Wendisch PG, Kreutzer C, Kalinowski J, Pátek M, Sahm H, Eikmanns BJ. 1998. Pyruvate carboxylase from Corynebacterium glutamicum: characterization, expression and inactivation of the pyc gene. Microbiology 144:915–927. doi: 10.1099/00221287-144-4-915. [DOI] [PubMed] [Google Scholar]
- 11.Peters-Wendisch P, Stansen KC, Götker S, Wendisch VF. 2012. Biotin protein ligase from Corynebacterium glutamicum: role for growth and l-lysine production. Appl Microbiol Biotechnol 93:2493–2502. doi: 10.1007/s00253-011-3771-8. [DOI] [PubMed] [Google Scholar]
- 12.Schwinde JW, Hertz PF, Sahm H, Eikmanns BJ, Guyonvarch A. 2001. Lipoamide dehydrogenase from Corynebacterium glutamicum: molecular and physiological analysis of the lpd gene and characterization of the enzyme. Microbiology 147:2223–2231. doi: 10.1099/00221287-147-8-2223. [DOI] [PubMed] [Google Scholar]
- 13.Hoffelder M, Raasch K, van Ooyen J, Eggeling L. 2010. The E2 domain of OdhA of Corynebacterium glutamicum has succinyltransferase activity dependent on lipoyl residues of the acetyltransferase AceF. J Bacteriol 192:5203–5211. doi: 10.1128/JB.00597-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moura FA, de Andrade KQ, dos Santos JC, Goulart MO. 2015. Lipoic acid: its antioxidant and anti-inflammatory role and clinical applications. Curr Top Med Chem 15:458–483. doi: 10.2174/1568026615666150114161358. [DOI] [PubMed] [Google Scholar]
- 15.Rochette L, Ghibu S, Muresan A, Vergely C. 2015. Alpha-lipoic acid: molecular mechanisms and therapeutic potential in diabetes. Can J Physiol Pharmacol 93:1021–1027. doi: 10.1139/cjpp-2014-0353. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg MW, Sternbach LH. November 1949. Synthesis of biotin. US patent 2,489,232.
- 17.Hornberger CS Jr, Heitmiller RF, Gunsalus IC, Schnakenberg GHF, Reed LJ. 1953. Synthesis of dl-alpha-lipoic acid. J Am Chem Soc 75:1273–1277. doi: 10.1021/ja01102a003. [DOI] [Google Scholar]
- 18.Ikeda M, Miyamoto A, Mutoh S, Kitano Y, Tajima M, Shirakura D, Takasaki M, Mitsuhashi S, Takeno S. 2013. Development of biotin-prototrophic and -hyperauxotrophic Corynebacterium glutamicum strains. Appl Environ Microbiol 79:4586–4594. doi: 10.1128/AEM.00828-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeno S, Takasaki M, Urabayashi A, Mimura A, Muramatsu T, Mitsuhashi S, Ikeda M. 2013. Development of fatty acid-producing Corynebacterium glutamicum strains. Appl Environ Microbiol 79:6776–6783. doi: 10.1128/AEM.02003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatakeyama K, Kohama K, Vertès AA, Kobayashi M, Kurusu Y, Yukawa H. 1993. Analysis of the biotin biosynthesis pathway in coryneform bacteria: cloning and sequencing of the bioB gene from Brevibacterium flavum. DNA Seq 4:87–93. doi: 10.3109/10425179309020147. [DOI] [PubMed] [Google Scholar]
- 21.Hatakeyama K, Hohama K, Vertès AA, Kobayashi M, Kurusu Y, Yukawa H. 1993. Genomic organization of the biotin biosynthetic genes of coryneform bacteria: cloning and sequencing of the bioA-bioD genes from Brevibacterium flavum. DNA Seq 4:177–184. doi: 10.3109/10425179309015630. [DOI] [PubMed] [Google Scholar]
- 22.Hatakeyama K, Kobayashi M, Yukawa H. 1997. Analysis of biotin biosynthesis pathway in coryneform bacteria: Brevibacterium flavum. Methods Enzymol 279:339–348. doi: 10.1016/S0076-6879(97)79038-4. [DOI] [PubMed] [Google Scholar]
- 23.Ikeda M, Nakagawa S. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109. doi: 10.1007/s00253-003-1328-1. [DOI] [PubMed] [Google Scholar]
- 24.Fanous A, Weiss W, Görg A, Jacob F, Parlar H. 2008. A proteome analysis of the cadmium and mercury response in Corynebacterium glutamicum. Proteomics 8:4976–4986. doi: 10.1002/pmic.200800165. [DOI] [PubMed] [Google Scholar]
- 25.Peters-Wendisch P, Götker S, Heider SA, Komati Reddy G, Nguyen AQ, Stansen KC, Wendisch VF. 2014. Engineering biotin prototrophic Corynebacterium glutamicum strains for amino acid, diamine and carotenoid production. J Biotechnol 192:346–354. doi: 10.1016/j.jbiotec.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Moslehi-Jenabian S, Solem C, Jensen PR. 2015. Increased expression of pyruvate carboxylase and biotin protein ligase increases lysine production in a biotin prototrophic Corynebacterium glutamicum strain. Eng Life Sci 15:73–82. doi: 10.1002/elsc.201400185. [DOI] [Google Scholar]
- 27.Bower S, Perkins JB, Yocum RR, Howitt CL, Rahaim P, Pero J. 1996. Cloning, sequencing, and characterization of the Bacillus subtilis biotin biosynthetic operon. J Bacteriol 178:4122–4130. doi: 10.1128/jb.178.14.4122-4130.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stok JE, De Voss JJ. 2000. Expression, purification, and characterization of BioI: a carbon-carbon bond cleaving cytochrome P450 involved in biotin biosynthesis in Bacillus subtilis. Arch Biochem Biophys 384:351–360. doi: 10.1006/abbi.2000.2067. [DOI] [PubMed] [Google Scholar]
- 29.Cronan JE. 2016. Assembly of lipoic acid on its cognate enzymes: an extraordinary and essential biosynthetic pathway. Microbiol Mol Biol Rev 80:429–450. doi: 10.1128/MMBR.00073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barzantny H, Brune I, Tauch A. 2012. Molecular basis of human body odour formation: insights deduced from corynebacterial genome sequences. Int J Cosmet Sci 34:2–11. doi: 10.1111/j.1468-2494.2011.00669.x. [DOI] [PubMed] [Google Scholar]
- 31.Radmacher E, Alderwick LJ, Besra GS, Brown AK K, Gibson KJC, Sahm H, Eggeling L. 2005. Two functional FAS-I type fatty acid synthases in Corynebacterium glutamicum. Microbiology 151:2421–2427. doi: 10.1099/mic.0.28012-0. [DOI] [PubMed] [Google Scholar]
- 32.Eggeling L, Besra GS, Alderwick L. 2008. Structure and synthesis of the cell wall, p 267–294. In Burkovski A. (ed) Corynebacteria: genomics and molecular biology. Caister Academic Press, Norwich, United Kingdom. [Google Scholar]
- 33.Gago G, Diacovich L, Arabolaza A, Tsai SC, Gramajo H. 2011. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol Rev 35:475–497. doi: 10.1111/j.1574-6976.2010.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaguchi A, Okuda S. 1977. Fatty acid synthetase from Brevibacterium ammoniagenes: formation of monounsaturated fatty acids by a multienzyme complex. Proc Natl Acad Sci U S A 74:3180–3183. doi: 10.1073/pnas.74.8.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schweizer E, Hofmann J. 2004. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev 68:501–517. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoischen C, Krämer R. 1990. Membrane alteration is necessary but not sufficient for effective glutamate secretion in Corynebacterium glutamicum. J Bacteriol 172:3409–3416. doi: 10.1128/jb.172.6.3409-3416.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cronan JE Jr, Rock CO. 1996. Biosynthesis of membrane lipids. p 612–636. In Neidhardt FC. (ed), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC. [Google Scholar]
- 38.Rock CO. 2009. Opening a new path to lipoic acid. J Bacteriol 191:6782–6784. doi: 10.1128/JB.01151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magnuson K, Jackowski S, Rock CO, Cronan JE Jr. 1993. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev 57:522–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grininger M. 2014. Perspectives on the evolution, assembly and conformational dynamics of fatty acid synthase type I (FAS I) systems. Curr Opin Struct Biol 25:49–56. doi: 10.1016/j.sbi.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Hiltunen JK, Schonauer MS, Autio KJ, Mittelmeier TM, Kastaniotis AJ, Dieckmann CL. 2009. Mitochondrial fatty acid synthesis type II: more than just fatty acids. J Biol Chem 284:9011–9015. doi: 10.1074/jbc.R800068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brody S, Oh C, Hoja U, Schweizer E. 1997. Mitochondrial acyl carrier protein is involved in lipoic acid synthesis in Saccharomyces cerevisiae. FEBS Lett 408:217–220. doi: 10.1016/S0014-5793(97)00428-6. [DOI] [PubMed] [Google Scholar]
- 43.Wada H, Shintani D, Ohlrogge J. 1997. Why do mitochondria synthesize fatty acids? Evidence for involvement in lipoic acid production. Proc Natl Acad Sci U S A 94:1591–1596. doi: 10.1073/pnas.94.4.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujiwara K, Okamura-Ikeda K, Motokawa Y. 1990. cDNA sequence, in vitro synthesis, and intramitochondrial lipoylation of H-protein of the glycine cleavage system. J Biol Chem 265:17463–17467. [PubMed] [Google Scholar]
- 45.Macherel D, Lebrun M, Gagnon J, Neuburger M, Douce R. 1990. cDNA cloning, primary structure and gene expression for H-protein, a component of the glycine-cleavage system (glycine decarboxylase) of pea (Pisum sativum) leaf mitochondria. Biochem J 268:783–789. doi: 10.1042/bj2680783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stuible HP, Wagner C, Andreou I, Huter G, Haselmann J, Schweizer E. 1996. Identification and functional differentiation of two type I fatty acid synthases in Brevibacterium ammoniagenes. J Bacteriol 178:4787–4793. doi: 10.1128/jb.178.16.4787-4793.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stuible HP, Meurer G, Schweizer E. 1997. Heterologous expression and biochemical characterization of two functionally different type I fatty acid synthases from Brevibacterium ammoniagenes. Eur J Biochem 247:268–273. doi: 10.1111/j.1432-1033.1997.00268.x. [DOI] [PubMed] [Google Scholar]
- 48.Yamada H, Tani Y, Izumi Y. January 1986. Process for producing biotin vitamers. US patent 4,563,426.
- 49.Bower SG, Perkins JB, Yocum RR, Pero JG. October 2001. Biotin biosynthesis in Bacillus subtilis. US patent 6,303,377.
- 50.Van Arsdell SW, Perkins JB, Yocum RR, Luan L, Howitt CL, Chatterjee NP, Pero JG. 2005. Removing a bottleneck in the Bacillus subtilis biotin pathway: BioA utilizes lysine rather than S-adenosylmethionine as the amino donor in the KAPA-to-DAPA reaction. Biotechnol Bioeng 91:75–83. doi: 10.1002/bit.20488. [DOI] [PubMed] [Google Scholar]
- 51.Manandhar M, Cronan JE. 2017. Pimelic acid, the first precursor of the Bacillus subtilis biotin synthesis pathway, exists as the free acid and is assembled by fatty acid synthesis. Mol Microbiol 104:595–607. doi: 10.1111/mmi.13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cryle MJ, Schlichting I. 2008. Structural insights from a P450 carrier protein complex reveal how specificity is achieved in the P450BioI ACP complex. Proc Natl Acad Sci U S A 105:15696–15701. doi: 10.1073/pnas.0805983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nickel J, Irzik K, van Ooyen J, Eggeling L. 2010. The TetR-type transcriptional regulator FasR of Corynebacterium glutamicum controls genes of lipid synthesis during growth on acetate. Mol Microbiol 78:253–265. [DOI] [PubMed] [Google Scholar]
- 54.Sato H, Orishimo K, Shirai T, Hirasawa T, Nagahisa K, Shimizu H, Wachi M. 2008. Distinct roles of two anaplerotic pathways in glutamate production induced by biotin limitation in Corynebacterium glutamicum. J Biosci Bioeng 106:51–58. doi: 10.1263/jbb.106.51. [DOI] [PubMed] [Google Scholar]
- 55.Bachmann BJ. 1972. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol Rev 36:525–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uozumi T, Hoshino T, Miwa K, Horinouchi S, Beppu T, Arima K. 1977. Restriction and modification in Bacillus species: genetic transformation of bacteria with DNA from different species, part I. Mol Gen Genet 152:65–69. doi: 10.1007/BF00264941. [DOI] [PubMed] [Google Scholar]
- 57.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 58.Mitsuhashi S, Ohnishi J, Hayashi M, Ikeda M. 2004. A gene homologous to β-type carbonic anhydrase is essential for the growth of Corynebacterium glutamicum under atmospheric conditions. Appl Microbiol Biotechnol 63:592–601. doi: 10.1007/s00253-003-1402-8. [DOI] [PubMed] [Google Scholar]
- 59.Takeno S, Ohnishi J, Komatsu T, Masaki T, Sen K, Ikeda M. 2007. Anaerobic growth and potential for amino acid production by nitrate respiration in Corynebacterium glutamicum. Appl Microbiol Biotechnol 75:1173–1182. doi: 10.1007/s00253-007-0926-8. [DOI] [PubMed] [Google Scholar]
- 60.Takeno S, Murata R, Kobayashi R, Mitsuhashi S, Ikeda M. 2010. Engineering of Corynebacterium glutamicum with an NADPH-generating glycolytic pathway for l-lysine production. Appl Environ Microbiol 76:7154–7160. doi: 10.1128/AEM.01464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M. 2002. A novel methodology employing Corynebacterium glutamicum genome information to generate a new l-lysine-producing mutant. Appl Microbiol Biotechnol 58:217–223. doi: 10.1007/s00253-001-0883-6. [DOI] [PubMed] [Google Scholar]
- 62.Racz CP, Santa S, Tomoaia-Cotisel M, Borodi G, Kacso I, Pirnau A, Bratu I. 2013. Inclusion of α-lipoic acid in β-cyclodextrin. Physical-chemical and structural characterization. J Incl Phenom Macrocycl Chem 76:193–199. [Google Scholar]
- 63.Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. 2014. Lipoic acid biosynthesis defects. J Inherit Metab Dis 37:553–563. doi: 10.1007/s10545-014-9705-8. [DOI] [PubMed] [Google Scholar]
- 64.Gueguen V, Macherel D, Jaquinod M, Douce R, Bourguignon J. 2000. Fatty acid and lipoic acid biosynthesis in higher plant mitochondria. J Biol Chem 275:5016–5025. doi: 10.1074/jbc.275.7.5016. [DOI] [PubMed] [Google Scholar]
- 65.Yasuno R, Wada H. 2002. The biosynthetic pathway for lipoic acid is present in plastids and mitochondria in Arabidopsis thaliana. FEBS Lett 517:110–114. doi: 10.1016/S0014-5793(02)02589-9. [DOI] [PubMed] [Google Scholar]
- 66.Mikolajczyk S, Brody S. 1990. De novo fatty acid synthesis mediated by acyl-carrier protein in Neurospora crassa mitochondria. Eur J Biochem 187:431–437. doi: 10.1111/j.1432-1033.1990.tb15322.x. [DOI] [PubMed] [Google Scholar]
- 67.Jordan SW, Cronan JE Jr. 1997. A new metabolic link. The acyl carrier protein of lipid synthesis donates lipoic acid to the pyruvate dehydrogenase complex in Escherichia coli and mitochondria. J Biol Chem 272:17903–17906. [DOI] [PubMed] [Google Scholar]
- 68.Takayama K, Wang C, Besra GS. 2005. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev 18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.