

Abstract
Recognition of RNA by RNA processing enzymes and RNA binding proteins often involves cooperation between multiple subunits. However, the interdependent contributions of RNA and protein subunits to molecular recognition by ribonucleoproteins are relatively unexplored. RNase P is an endonuclease that removes 5′ leaders from precursor tRNAs and functions in bacteria as a dimer formed by a catalytic RNA subunit (P RNA) and a protein subunit (C5 in E. coli). The P RNA subunit contacts the tRNA body and proximal 5′ leader sequences [N(−1) and N(−2)] while C5 binds distal 5′ leader sequences [N(−3) to N(−6)]. To determine whether the contacts formed by P RNA and C5 contribute independently to specificity or exhibit cooperativity or anti-cooperativity, we compared the relative kcat/Km values for all possible combinations of the six proximal 5′ leader nucleotides (n = 4096) for processing by the E. coli P RNA subunit alone and by the RNase P holoenzyme. We observed that while the P RNA subunit shows specificity for 5′ leader nucleotides N(−2) and N(−1), the presence of the C5 protein reduces the contribution of P RNA to specificity, but changes specificity at N(−2) and N(−3). The results reveal that the contribution of C5 protein to RNase P processing is controlled by the identity of N(−2) in the pre-tRNA 5′ leader. The data also clearly show that pairing of the 5′ leader with the 3′ ACCA of tRNA acts as an anti-determinant for RNase P cleavage. Comparative analysis of genomically encoded E. coli tRNAs reveals that both anti-determinants are subject to negative selection in vivo.
Keywords: RNase P, high-throughput sequencing, kinetics, pre-tRNA processing, enzyme specificity, molecular recognition
INTRODUCTION
Due to the varied and essential roles that RNA plays in gene expression, it is important to understand the specificity of key enzymes that bind and regulate RNA. Numerous studies have focused on identifying binding sites of RNA binding proteins and RNA processing enzymes within the transcriptomes of cells, and defining optimal sequence and structure motifs for association (Licatalosi et al. 2008; Zhao et al. 2010; Ascano et al. 2012; Cook et al. 2015; Jankowsky and Harris 2015). Despite these advances, a quantitative understanding of how multiple enzyme subunits act in concert to achieve molecular recognition of multiple RNA substrates has not been achieved. Differences in the sequence and structure of RNA can affect the processing of alternative substrates by perturbation of local RNA geometry, altering the chemical/electrostatic environment, or by altering the rate-limiting step in the reaction. Dissection of these contributions to affinity is now made possible by quantitative analysis of a complete sampling of large numbers or all possible substrate sequence combinations encompassing the enzyme binding site (Ascano et al. 2013; Buenrostro et al. 2014; Lambert et al. 2014; Jankowsky and Harris 2015; Ozer et al. 2015; Lou et al. 2017).
Ribonuclease P (RNase P) is a ubiquitous and essential tRNA processing endonuclease and a useful model system to investigate how variation in RNA sequence and structure affect shared molecular recognition (Kazantsev and Pace 2006; Klemm et al. 2016). RNase P removes the 5′ leader sequence from all precursor tRNA (pre-tRNAs) in the cell and in Bacteria is composed of a large (approximately 400 nucleotide) catalytic RNA subunit (P RNA) and a smaller (approximately 90 amino acid) protein subunit (termed C5 protein in E. coli). While the P RNA subunit alone can process pre-tRNAs in vitro at high salt concentrations, the protein subunit is necessary for in vivo function and in vitro activity under physiological conditions (Guerrier-Takada et al. 1983, 1984; Harris et al. 1994, 1997; Christian et al. 1998, 2002; Frank and Pace 1998; Christian and Harris 1999; Kurz and Fierke 2000; Zahler et al. 2003; Kazantsev and Pace 2006; Sun et al. 2006, 2010; Klemm et al. 2016). A conserved adenosine in the J5/15 region of P RNA recognizes N(−1) in the 5′ leader, and unidentified nucleotides in the J18/2 region recognize N(−2) relative to the cleavage site at N(1) (Hardt et al. 1993; Brännvall et al. 2002, 2004; Zahler et al. 2005). The C5 protein subunit contacts nucleotides N(−8 to −3) in the distal 5′ leader that contribute to substrate affinity and binding of metal ions important for catalysis (Altman and Guerrier-Takada 1986; Pace et al. 1987; Harris et al. 1994; Christian et al. 2002; Sun et al. 2006; Koutmou et al. 2010; Reiter et al. 2010). Thus, recognition of the pre-tRNA 5′ leader is shared between the RNA and protein subunits of RNase P.
Recently, high-throughput enzymology methods were developed that allow the multiple-turnover and single-turnover kinetics of RNase P processing to be measured for thousands of pre-tRNA substrates simultaneously (Guenther et al. 2013; Niland et al. 2016b). Quantitative analysis of the resulting rate constants revealed that variation in the 5′ leader altered substrate association, not the cleavage step (Niland et al. 2016b). The ability to measure kinetics for all possible RNA substrate variants in an enzyme binding site reveals the effects of mutation at each randomized position in the background of all possible surrounding sequence combinations that can be quantified no other way. Here, we investigate the potential interdependence between 5′ leader interactions with the P RNA and C5 protein subunit (either cooperative or anti-cooperative) by measuring the relative kcat/Km for all possible nucleotide variations of the proximal 5′ leader sequence for processing by the P RNA alone, and comparing these values with the values previously determined for the same substrate pool using the RNase P holoenzyme.
Analyses of the resulting high-density biochemical data sets reveal both familiar and surprising new determinants of RNase P specificity that are confirmed by analysis of individual pre-tRNA sequence variants. As expected, the C5 protein shifts the dependence on the 5′ leader sequence to positions that are more distal to the cleavage site due to direct RNA–protein interactions. The data also clearly show that pairing of the proximal 5′ leader to the tRNA 3′RCCA acts as a strong anti-determinant. Remarkably, we observe that the identity of the nucleotide at N(−2) controls the contribution of C5 protein interactions to kcat/Km for RNase P processing, illustrating energetic coupling between enzyme subunits. Thus, large context-dependent effects contribute to the observed specificity of RNase P for 5′ leader sequences such that variation in nucleotides that contact P RNA modulate the contribution of C5 protein to substrate discrimination. Analysis of naturally occurring pre-tRNAs illustrates how these features shape the observed distribution of nucleotides proximal to the RNase P cleavage sites in the E. coli transcriptome.
RESULTS AND DISCUSSION
Due to the shared molecular recognition of the 5′ leader nucleotides in pre-tRNA by P RNA and C5 protein, we hypothesized that cooperative or anti-cooperative interdependence may exist between their contributions to RNase P specificity. To test this hypothesis, we used HTS-Kin to compare the affinity distributions obtained with the RNase P holoenzyme and with P RNA alone.
To comprehensively investigate the specificity of RNA processing enzymes and RNA binding proteins we developed high-throughput sequencing kinetics (HTS-Kin), which allows the relative kcat/Km of thousands of RNA sequence variants in an RNA processing reaction to be measured simultaneously (Guenther et al. 2013; Niland et al. 2016a). The HTS-Kin technique is outlined in Supplemental Figure S1 and involves the creation of a randomized pre-tRNA substrate pool that is processed by RNase P in vitro. By monitoring the change in concentration of each substrate variant as a function of time (Supplemental Fig. S2), the relative kcat/Km for each species (i) calibrated to a reference sequence [krel = (kcat/Km)i/(kcat/Km)reference] can be calculated using internal competition kinetics (Anderson 2015). By investigating all possible substrate variants in the region of interest, it is possible to comprehensively determine the context-dependent effects of pre-tRNA sequence variation in the 5′ leader.
Comparison of the affinity distributions for P RNA and RNase P processing of pre-tRNAMetN(−6 to −1)
To interrogate the specificity of both subunits of RNase P for the 5′ leader of pre-tRNA, we created a pool of pre-tRNAMet substrate variants that was randomized in the 5′ leader at nucleotides N(−6 to −1) encompassing both protein and RNA contacts (Fig. 1A). The pre-tRNAMetN(−6 to −1) substrate pool was reacted with the P RNA alone or the RNase P holoenzyme (P RNA and C5 protein), and the relative kcat/Km values for all 4096 substrates are shown in Figure 1B. The results of the reaction with RNase P holoenzyme were reported previously (Niland et al. 2016b); however, the analyses reported here are novel.
FIGURE 1.

Sequence determinants for holoenzyme and ribozyme reactions are unique and reveal changes in specificity. (A) Secondary structure of pre-tRNAMetN(−6 to −1) with regions of randomization analyzed in this study indicated by N's. As shown, these regions encompass nucleotides of the 5′ leader that are involved in P RNA and C5 protein contact. (B) The affinity distribution of relative rate constants from holoenzyme (black, published in Niland et al. 2016b) and ribozyme (red) with pre-tRNAMetN(−6 to −1) show that while some substrates are processed with rate constants significantly above the genomically encoded reference in holoenzyme reactions, the same substrate population is significantly slower in the ribozyme reaction with no protein subunit. (C) Comparison of the relative rate constants measured for each substrate variant in HTS-Kin reactions with RNase P holoenzyme (x-axis) and P RNA ribozyme (y-axis). (D) Sequence preference in the 5′ leader of pre-tRNAMetN(−6 to −1) examined by creating sequence probability logos of the fastest reacting substrates in holoenzyme or ribozyme reactions. The position in the 5′ leader is indicated on the bottom of the logo; nucleotide preference at each position is indicated by the identity and size of the letter of the nucleobase.
A large range of relative rate constants spanning about 100-fold is observed in the HTS-Kin reactions of the pre-tRNA pool with the RNase P holoenzyme. In contrast, a reaction with P RNA alone showed a much narrower range of krel values (approximately fivefold range), indicating an overall smaller effect of 5′ leader sequence variation on kcat/Km. The smaller variation in rate constants seen for the P RNA multiple turnover reaction reflects a smaller number of sequence determinants in the binding site. A comparison of the krel values for RNase P and for P RNA alone is shown in Figure 1C. The observed overall linear correlation between the two affinity distributions is consistent with at least a subset of shared sequence specificity determinants.
Optimal sequence logos calculated for the fastest reacting 1% of substrate variants in each data set (Crooks et al. 2004) identify a strong preference for an adenosine at the N(−2) position and uridine at the N(−3) position in the 5′ leader of pre-tRNAMet in RNase P holoenzyme reactions, as reported previously (Fig. 1D). In reactions catalyzed by P RNA alone, sequence preference for an adenosine at the N(−2) position in the 5′ leader is observed, but the preference for uridine at N(−3) is significantly reduced. For the P RNA alone reaction there is weak preference for A at positions N(−4) to N(−6) equivalent to the diminished contribution at N(−1). The apparently larger contribution of distal leader sequences in the P RNA alone reaction does not necessarily indicate a greater contribution to the observed rate constant. The range of effects of sequence variation on krel is significantly smaller for the P RNA alone reaction. In addition, the scale of the logo plots is not equivalent since they express probabilities of nucleobase occurrence in the equivalent number of fast reacting sequence variants. Nonetheless, in both reactions, variation of distal 5′ leader nucleotides has a minimal contribution to the optimal RNAs compared to the nucleotides more proximal to the cleavage site. Thus, for the fastest reacting sequence variants, the C5 protein promotes additional specificity for nucleotides in the 5′ leader but has little effect on optimal N(−2) interactions with P RNA.
Quantitative RNA specificity modeling reveals coupling between adjacent proximal 5′ leader nucleotides
To more fully analyze the quantitative data sets generated by HTS-Kin, we used unbiased modeling approaches to describe the data (Guenther et al. 2013; Lin et al. 2014; Niland et al. 2016b). The krel values for P RNA and RNase P processing were fit to a position weight matrix (PWM) model that also includes terms’ coupling coefficients (IC values) that describe the positive or negative effects between two positions on their contribution to specificity. The IC values thus contain information on effects due to energetic coupling, effects of local environment and secondary structure. A comparison of the observed rate constants to those predicted by a linear fit to the PWM model shows that it can explain over 50% of the effects of sequence variation for both P RNA and the RNase P holoenzyme reactions (Fig. 2A,B).
FIGURE 2.

Plotting the IC values from the PWM+IC model identifies altered energetic coupling in holoenzyme and ribozyme reactions. (A,B) Comparison of the relative rate constants of pre-tRNAMetN(−6 to −1) observed in the ribozyme and holoenzyme HTS-Kin reaction compared to that predicted by the position weight matrix model including coupling coefficients (PWM+IC). Less than 10% of data omitted for clarity. RNase P modeling results published in Niland et al. (2016b). The greater apparent distribution of values for the P RNA reaction is likely due to its reduced specificity for N(−3) to N(−6). (C) Heatmap showing the difference in calculated IC values in the P RNA and RNase P reactions between nucleotides in the randomized region of the 5′ leader, N(−6) to N(−1). The identity and position of the nucleotide is indicated on each axis and the absolute value of the difference of the predicted IC values is indicated by the color at the vertex (black for large difference, white for small difference). (D) Comparison of the IC values calculated from the model between RNase P and P RNA HTS-Kin reactions.
The IC value differences predicted by the models for the P RNA alone versus RNase P holoenzyme reactions are presented in a heatmap in Figure 2C. Notably, there are strong IC value differences in nucleotides proximal to the cleavage site at N(−1), N(−2), N(−3), and N(−4). It is possible that the contribution to specificity of nucleotides distal to the cleavage site could reflect local changes in structure at N(−1) and N(−2). Such effects could also result from differences in sensitivity of the P RNA and RNase P reactions to structure formation of these proximal nucleotides [N(−4) to N(−1)] with the 3′ACCA of the tRNA. Indeed, a pattern is clearly visible from the plot in Figure 2C showing effects due to differences in preferences against neighboring guanosines [i.e., G(−3) and G(−4)] in the RNase P reactions, which would have the potential to pair with the C residues in the 3′ACCA sequence. These data are consistent with the fact that while C5 protein sequence specificity is directed primarily at distal 5′ leader sequences N(−6 to −3), it has a significant effect on the contribution to sequence specificity of nucleotides proximal to the cleavage site. Crosslinking, chemical protection analyses, and X-ray crystallography of the Thermotoga maritima RNase P all support interactions between the protein subunit and nucleotides N(−3) up to approximately N(−8). Importantly, the decrease in magnitude of IC values between these nucleotides and N(−2) to N(−1) is consistent with the suppression of N(−1) specificity revealed by the analyses of optimal sequence logos in Figure 1D.
A direct comparison of the calculated IC values for HTS-Kin reactions with the RNase P ribozyme and holoenzyme is shown in Figure 2D and reveals that the range of IC values is larger in the ribozyme reaction compared to that in the holoenzyme reaction, but there is only a weak correlation between them. This observation supports the interpretation that the presence of the C5 protein subunit results in enzyme specificity for all positions in the 5′ leader that were randomized in the HTS-Kin experiments.
The specificity for N(−6) to N(−3) depends on the identity of proximal 5′ leader nucleotides
To dissect the interdependence of 5′ leader nucleotide interactions with the P RNA and C5 protein subunits, we examined whether the identity of nucleotides at N(−2) and N(−1) influences the observed sequence specificity for positions N(−6) to N(−3) (see Fig. 3A). To extract this information we binned the HTS-Kin data from the holoenzyme reaction into subsets according to the identity of nucleotides at N(−2)N(−1). Next, we used individual dot plots to compare the krel values for different N(−6 to −3) variants for leaders containing different N(−2)N(−1) combinations with the genomically encoded A(−2)U(−1) used as a reference (Fig. 3B). Any arrangement of points other than a line with a positive slope indicates different C5 sequence specificity depending on the identity of the nucleotides at N(−1)N(−2). A linear relationship between the two subsets of HTS-Kin data with a positive slope of unity indicates that the effect of mutation at N(−6 to −3) on RNase P specificity is independent of the identity of N(−2)N(−1). If the identity of the nucleotides at N(−2)N(−1) changes the energetic contribution of distal 5′ leader nucleotides that contact C5, then a change in slope will be observed. Upon inspection it is clear that the effect of variation at N(−6 to −3) is linearly correlated for all combinations of nucleotides at N(−2)N(−1), except for the combinations G(−2)G(−1), C(−2)G(−1), or C(−2)C(−1). This result demonstrates essentially identical C5 protein sequence specificity independent of the geometry of N(−2)N(−1) interactions with the P RNA active site.
FIGURE 3.

Variation of nucleotides in the 5′ leader of pre-tRNA contacting the P RNA subunit of RNase P alters the energetic contribution of nucleotides contacting the C5 protein. (A) The 16 data subsets of HTS-Kin separated by the identity of nucleotides at N(−2) and N(−1) in the RNA binding site. (B) Each subset is compared to the genomically encoded A(−2)U(−1). Each row symbolizes a change in nucleotide identity at the N(−1) position while each column follows a change at N(−2). Each of the 16 boxes represents a scatter plot of the krel for a substrate with a given sequence at N(−6) to N(−3) in the background of the mutant or wild-type nucleotides at N(−2) and N(−1). (C) Each scatter plot was fit to a line with the slope indicated in the graph. Any arrangement of points other than a line with positive slope indicates different C5 sequence specificity depending on the identity of the nucleotides at N(−1)N(−2).
Remarkably, the identity of N(−2)N(−1) has a profound effect on the energetic contribution of distal 5′ leader sequences to RNase P processing. For several combinations of N(−2)N(−1), variation of nucleotides at N(−6 to −3) has little effect on the relative rate constant for RNase P cleavage, which is reflected in slopes for dot plots of krel that approach zero. For comparison, the slopes for each dinucleotide relative to the genomically encoded A(−2)U(−1) reference are summarized in Figure 3C. Changes in the effects of sequence variation in the C5 binding site on RNase P cleavage rate correlate primarily with the identity of N(−2). The most pronounced effects are observed for substrates with a C(−2), where substrates encompassing all possible combinations of nucleotides at N(−3) to N(−6) are processed with similar rate constants. Additionally, pre-tRNAs containing G(−2)U(−1), G(−2)G(−1), and to a lesser extent U(−2)G(−1), are relatively insensitive to variation at positions N(−6 to −3) compared to the reference substrate [A(−2)U(−1)].
Thus, comparing the effect of all possible combinations of sequences at N(−2)N(−1) to the reference sequence A(−2)U(−1) reveals that the identity of proximal nucleotides does not influence the apparent C5 sequence specificity. Rather, the degree to which C5 specificity contributes to kcat/Km is controlled by the identity of the nucleotide at N(−2) and to a lesser extent N(−1). Using the comparative perspective shown in Figure 3, we identified the most prominent interdependence effects indicated by the HTS-Kin data, and tested them by analyzing selected pre-tRNAMet 5′ leader sequence variants using traditional single substrate kinetic assays.
Unfavorable effects on kcat/Km due to pairing between proximal 5′ leader nucleotides and the tRNA 3′ terminal ACCA
It is well documented that the 3′RCCA of pre-tRNA base pairs with nucleotides in the P15 region of the P RNA subunit (G291, G292, and U293 in E. coli P RNA) (Hardt et al. 1993, 1995; Kirsebom and Svärd 1994; Svärd et al. 1996; Brännvall et al. 1998, 2003; Heide et al. 1999; Wegscheid and Hartmann 2006, 2007). Extension of the acceptor stem by engineering paring interactions between the 3′RCCA and the 5′ leader is unfavorable for catalysis by P RNA and can result in mis-cleavage in some substrates (Brännvall et al. 1998, 2003). Additionally, mutational analysis of interactions between the P RNA and 3′RCCA showed defects in the catalytic rate constant and possibly a conformational change step of the reaction (Hardt et al. 1995; Heide et al. 1999; Wegscheid and Hartmann 2006, 2007). However, most of these studies involved experiments using the P RNA subunit alone, or used minimal mini-helix model substrates or a pre-tRNAHis, which naturally contains an additional base pair in its acceptor stem that is not found in other E. coli pre-tRNA.
To confirm the extent to which pairing between the pre-tRNA 5′ and 3′ ends inhibits processing by the RNase P holoenzyme, we measured the processing rate constant of pre-tRNAMet substrates containing increasing numbers of base pairs with the terminal 3′ACCA. We compared these effects in the background of three different N(−6 to −3) sequences (AUAA, UAAA, and GUAA). When each of these sequences is combined with A(−2)A(−1) (i.e., no pairing to the 3′ACCA), the relative kcat/Km values are greater than the native reference sequence (Fig. 4A). Additionally, their relative kcat/Km values are sensitive to N(−6 to −3) sequence variation, consistent with the HTS-Kin results.
FIGURE 4.
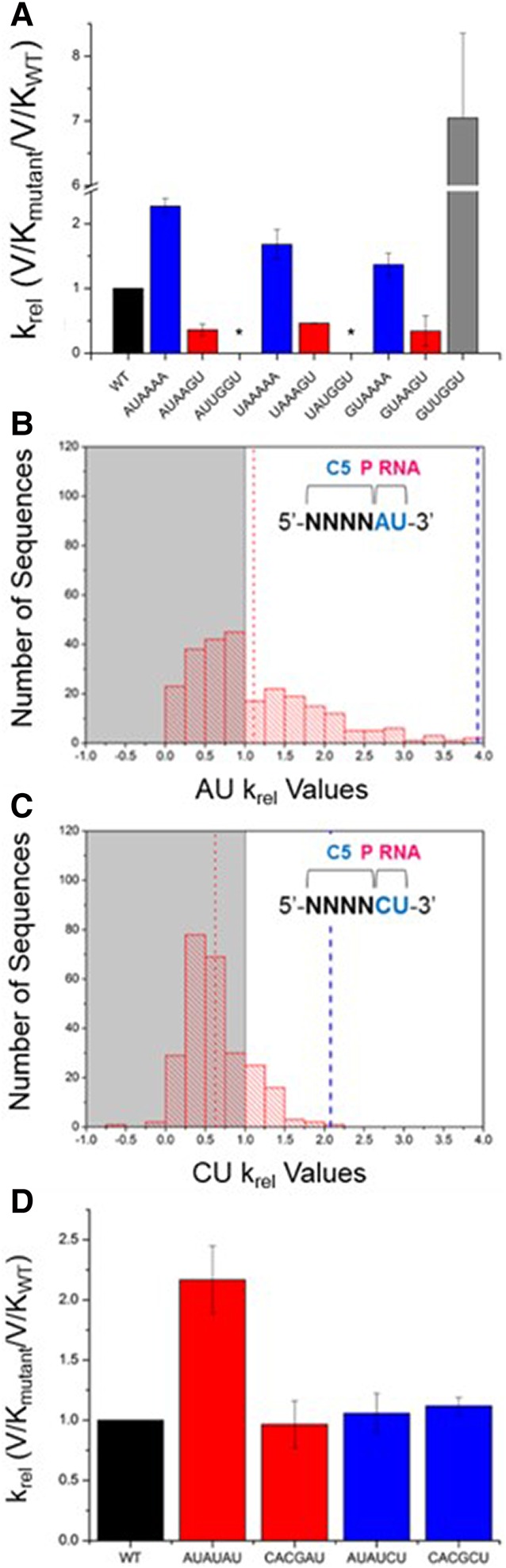
Coupling between nucleotides in the RNA and protein binding sites in the 5′ leader of pre-tRNA is a key component of RNase P specificity. (A) Results of individual substrate assays to test the effect of zero, two, or four base pairs between the 5′ leader and 3′ACCA on the krel. The 5′ leader of each substrate from N(−6) to N(−1) is indicated on the x-axis. Experiments were performed in triplicate and error bars indicate the standard deviation from all experiments. (*) indicates no cleavage was observed. (B) One population of substrates with U(−1) and A(−2) includes 256 sequences; the affinity distribution for these substrates is broad and shows that ptRNA variation in the C5 binding site alters RNase P processing rates. (C) For those substrates with U(−1) and C(−2), the affinity distribution is much more narrow, indicating little effect of protein binding site sequence variation on krel. (D) Single substrate multiple turnover assays measured the absolute kcat/Km for substrates with the indicated 5′ leaders on the x-axis. These reactions contained pre-tRNAMetWT as an internal reference. Experiments were performed in triplicate and error bars indicate the standard deviation from all experiments.
These same pre-tRNAs were engineered to contain a G(−2)U(−1) in order to form complementary base pairs with the A73 and C74 of the 3′ACCA. RNase P processing reactions using individual substrates reveal significantly slower (five- to 10-fold) relative rate constants in accordance with HTS-Kin (Fig. 4A). Interestingly, the same relative rate constant was observed within error for all three pre-tRNAs independent of the identity of N(−6 to −3). This observation contrasts to the sensitivity to N(−6 to −3) sequence variation in the context of 5′ leader sequences with A(−2)A(−1), and further demonstrates the modulation of the energetic contribution of C5 binding by the identity of proximal 5′ leader nucleotides. Substrates designed with perfectly complementary base pairs with the 3′ACCA of the tRNAAsp body contained a 5′-UGGU-3′ at N(−4 to −1) and were inactive in multiple turnover reactions, except when a G(−6)U(−5) was present, which showed a large increase in kcat/Km. Pilot studies of all other combinations at N(−6)N(−5) with 5′-UGGU-3′ at N(−4 to −1) were conducted and only those containing G(−6)U(−5) were appreciably cleaved by RNase P.
A subset of substrates with a G(−2)U(−1) show apparent rate constants measured by HTS-Kin that are slower than the wild-type substrate, and all of these substrates were found to contain a G(−3) (Fig. 3B). The HTS-Kin data indicates that a 5′ leader sequence containing 5′-YGGU-3′ that pairs with the 3′ ACCA does not follow the global trend observed for sequence variants that contain a G(−2)U(−1) compared to the A(−2)U(−1) reference in Figure 3C. For these few substrates, the high-throughput analysis predicts faster rate constants than observed using traditional single substrate assays. This inaccuracy is likely due to nonlinear amplification of these variants during library preparation (Niland et al. 2016b). For these sequence variants, formation of a complete stem involving the 3′ACCA may compete for primer binding in RT-PCR steps required for Illumina sequencing. Nonetheless, the results from validation experiments using individual substrates further reveals how pairing of 5′ leader sequences acts as an anti-determinant for RNase P cleavage. Additionally, the rate constants of substrates containing G(−2)U(−1) are insensitive to sequence variation of distal 5′ leader nucleotides.
N(−2) identity controls the contribution of distal leader sequences that contact C5 protein to specificity
The substrate subpopulation containing the genomically encoded A(−2)U(−1) has a broad affinity distribution arising due to variation in the C5 protein binding site at N(−6 to −3) (Fig. 4B). For substrates with U(−2)U(−1), the apparent sequence specificity in the C5 protein binding site at N(−6 to −3) is essentially unchanged (see Supplemental Fig. S3). In contrast, substrates with C(−2)U(−1) have a narrow distribution of processing rate constants, and thus sequence variation at positions N(−6 to −3) has a small effect on kcat/Km (Fig. 4C). Plotting the observed krel for each possible N(−6 to −3) sequence variant in the background of C(−2)U(−1) versus A(−2)U(−1) (Fig. 3C) reveals a slope of 0.4, demonstrating that substrates with a C(−2) are essentially insensitive to variation in the C5 protein binding site.
These observations are further validated by the results from single substrate assays of individual 5′ leader sequence variants. Mutation of N(−6 to −3) in the C5 protein binding site from AUAU to CACG reduces kcat/Km for RNase P processing by approximately twofold when N(−2) in the RNA binding site is an adenosine. However, there are no measurable differences in rate constant for the same change in the N(−6 to −3) sequence when N(−2) is cytosine (Fig. 4D). These results provide further evidence that the identity of proximal 5′ leader nucleotides that contact P RNA can influence the degree to which nucleotides distal from the cleavage site contribute to specificity.
Conclusion
Previous biochemical and structural studies of RNase P substrate recognition provide a general model for specificity in which the C5 protein contacts 5′ leader nucleotides (Harris et al. 1994; Christian et al. 2002; Reiter et al. 2010; Sun et al. 2010), while the P RNA subunit contacts both the tRNA body and 5′ leader nucleotides proximal to the cleavage site (Hardt et al. 1993; Harris et al. 1994, 1997; Brännvall et al. 2003; Wegscheid and Hartmann 2006). Studies from our laboratory and others identified a direct base-pairing between the U(−1) of pre-tRNAAsp and the A248 residue in J5/15 of the P RNA subunit (Brännvall et al. 2002; Zahler et al. 2003, 2005). Additionally, the nucleotide sequence in the 5′ leader of pre-tRNA was shown to be important for substrate binding by RNase P, particularly for the P RNA subunit (Sun et al. 2006, 2010). Fierke and colleagues identified a specific interaction between tyrosine at position 34 in the C5 protein of Bacillus subtilis and the N(−4) position in the 5′ leader of pre-tRNAAsp that regulates binding affinity (Koutmou et al. 2010). Alanine scanning mutagenesis of the RNase P protein from T. maritima further suggested the importance of N(−4) on holoenzyme catalytic efficiency and provided key biochemical validation of the role of the 5′ leader region as modeled in the T. maritima RNase P holoenzyme-tRNA structure (Reiter et al. 2012). Previous extensive in vitro and in vivo biochemical studies by the Hartmann and Kirsebom laboratories established the essential role of contacts between the P15 region of the P RNA and 3′RCCA of the tRNA body in catalysis and cleavage site recognition. Specifically, pairing between the N(−1) of the 5′ leader and 3′RCCA was shown to be a negative determinant for RNase P processing in vivo (Pettersson and Kirsebom 2008). Recent HTS-Kin analysis on pre-tRNAMetN(−6 to −1) processed by the RNase P holoenzyme (Niland et al. 2016b) showed that the identity of N(−2) and N(−3) primarily controls alternative substrate selection at the level of association, not the cleavage step. As a consequence, the specificity for N(−1), which contacts the active site and contributes to catalysis, is suppressed.
The use of HTS-Kin provides a unique window into the energetic contribution of each residue as it quantifies the effect of mutation of a given nucleotide in the 5′ leader in the background of all possible surrounding sequences. By comparing the results for P RNA and RNase P processing of the pre-tRNAMetN(−6 to −1) random pool, we identified expected and unexpected aspects of RNase P specificity. First, the effect of pairing between proximal 5′ leader nucleotides and the tRNA body was examined and found to be inhibitory in most cases with minor but important exceptions. The significance of this pairing is illustrated by examining all pre-tRNAs from E. coli, which show that while several contain a U(−1) or even both G(−2)U(−1), none contain 5′-UGGU-3′ N(−4 to −1), which argues that this pairing was evolutionarily selected against the large variation in 5′ leader sequences (Fig. 5B). Interestingly, the only endogenous substrates that contain a G(−3)G(−2)U(−1) in E. coli are pre-tRNAAsp, which contain a sequence of 5′-GCCA-3′ at their terminal 3′ end. These substrates would thus be predicted to form a wobble G–U pair between U(−1) and G(73), forming a slightly different but nonetheless complete pairing between the 5′ leader and 3′ end of the tRNA body.
FIGURE 5.

Significance of energetic coupling and base-pairing in vivo. (A) Bar graph of potential pairing between the 5′ leader and 3′RCCA of endogenous pre-tRNA from E. coli K12. Each pre-tRNA was examined for its containing a 5′-UGGU-3′ from N(−4 to −1) and the number calculated based upon sequential containment of this sequence (i.e., 5′-UGGA-3′ has no base pair and 5′-AAAU-3′ has one base pair). (B) Frequency plot of the sequence of 5′ leaders of pre-tRNA from E. coli K12. Each position in the 5′ leader is indicated on the x-axis; the size of the nucleotide letter indicates its portion in the endogenous pre-tRNA population. (C) A diagram for modulation of interdependence of proximal and distal leader sequence specificities. A possible interpretation of the results herein is that there is a change in the rate-limiting step of substrates with either an A(−2) or C(−2), possibly at the conformational change step.
Second, we observe no change in global specificity for nucleotides at N(−6 to −3) in protein–RNA contacts upon variation in sequence at RNA–RNA contacts with N(−1)N(−2). Instead, we observed attenuation effects in which at one extreme the identity of 5′ leader nucleotides contacting P RNA completely eliminates the energetic contribution of nucleotides in the C5 protein-binding site. Particularly, we attribute this regulation to the nucleotide identity at N(−2). Interestingly, few endogenous pre-tRNA substrates from E. coli have a C(−2) (Fig. 5B), which eliminates contribution of the protein to sequence specificity, supporting the idea that the C5 protein is important for RNase P function in vivo. This observed dependence of RNase P protein contribution to 5′ leader sequence specificity on RNA–RNA interactions at the cleavage site could be a result of either altered substrate association or a conformational change of the enzyme upon substrate binding (Fig. 5C).
The idea of coupling between different regions of enzyme or substrate is not unprecedented. It was previously shown for alkaline phosphatase that single mutations of active site residues could not account for the combined rate defect observed when mutated in combination (Sunden et al. 2015). Another example of coupling in RNA processing was shown in tRNA binding by the ribosome, where mutation of the anti-codon stem in the tRNA body resulted in weakened binding of the tRNA to its cognate codon (Olejniczak et al. 2005). While several other examples are found within the literature, the role of energetic coupling in specificity in the case of enzymes with multiple subunits is unexplored.
Overall, this study provides the first foray into a comprehensive and quantitative determination of energetic coupling between individual subunits of RNA processing enzymes. Using RNase P as a simple model for these studies revealed general principles of this energetic coupling that can be applied to understanding more complex enzymes with multiple subunits such as the spliceosome, hnRNPs, ribosome, etc. Given the well-established role of RNA binding proteins in human health, obtaining a complete picture of their substrate specificity is paramount to understanding their potential as drug targets and therapeutics. To fully understand how to target these enzymes using novel small molecules or drugs and to understand their mechanism of action, a quantitative understanding of their substrate recognition is essential.
MATERIALS AND METHODS
RNA and protein preparation
C5 protein was expressed and purified as previously described (Guo et al. 2006). Both P RNA and pre-tRNAMet were prepared by in vitro transcription as described previously (Yandek et al. 2013). Briefly, the genes for P RNA or pre-tRNAMet were cloned into pUC19 vector and linearized to use as a template for T7 RNA polymerase (New England Biolabs). To create the mutant pre-tRNAMetN(−6 to −1)21A substrate pool, DNA primers incorporating mutations at the desired positions were used for PCR amplification of the cloned DNA template, and this PCR product was used for in vitro transcription as previously described (Guenther et al. 2013; Niland et al. 2016a). RNA was purified by polyacrylamide gel electrophoresis with UV shadowing followed by standard phenol–chloroform extraction and ethanol precipitation with a final resuspension in 10 mM Tris–HCl pH 8.0, 1 mM EDTA. A portion of the pre-tRNA population was 5′ end labeled with [γ-32P] using polynucleotide kinase and purified as described above.
RNase P reactions
Multiple turnover substrate reactions were performed in 50 mM Tris–HCl pH 8, 100 mM NaCl, 0.005% Triton X-100, and 17.5 mM MgCl2. For individual substrate reactions, the RNase P holoenzyme was assembled using 2 nM of RNA, heating to 95°C for 3 min followed by 37°C for 10 min before addition of 17.5 mM MgCl2 and 2 nM C5 protein. Substrate pools were prepared separately using 60 nM unlabeled pre-tRNA spiked with a negligible amount of 32P-pre-tRNA with 17.5 mM MgCl2. The reaction was started by mixing equal volumes of enzyme and substrate to give 30 nM substrate and 1 nM enzyme. Aliquots were taken at desired timepoints in order to achieve at least 90% conversion of the substrate and at intervals depending on the rate constant for the reaction and quenched in formamide loading dye with 100 mM EDTA. Polyacyrlamide gel electrophoresis was used to separate substrate and product, and the labeled portion of the substrate population allowed for quantification by phosphorimager and ImageQuant software. For reactions demonstrating coupling, 30 nM of pre-tRNAMetWT with a shortened 5′ leader was included in each reaction as an internal reference for substrate processing and used to derive krel by dividing the processing rate of the mutant substrate by that of the wild-type reference.
High-throughput sequencing kinetics
Reactions were performed exactly as described above except they were scaled up by 10-fold in volume to provide sufficient RNA for Illumina sequencing. Holoenzyme reactions contained 1 µM pre-tRNAMetN(−6 to −1) and 5 nM RNase P holoenzyme. Ribozyme reactions contained 1 µM pre-tRNAMetN(−6 to −1) and 10 nM P RNA. Quantification of the relative processing rate constant was performed as previously described (Guenther et al. 2013; Lin et al. 2014; Niland et al. 2016b) using the final equation:
in which krel is the relative second order rate constant, f is the overall fraction of substrate reacted determined by phosphorimager analysis, and X is the mole fraction obtained from Illumina reads. R indicates a ratio of reads between the mutant and wild-type substrate from Illumina sequencing where the subscript i,0 denotes this ratio before the reaction begins, and the subscript i denotes this ratio at the fraction of substrate reacted f.
RNA sequence specificity modeling
The position weight matrix model with IC values included considered nucleotide identity and position in the randomized region as well as position and identity of other nucleotides in the binding site using the following equation:
where ai, ci, gi, and ui, are integer values (0 or 1) signifying nucleotide identity, and Ai, Ci, Gi, and Ui represent the linear coefficients for that nucleotide at position i. The term βj is the linear coefficient for interaction between two positions and nucleotide identities. Ij is 1 for all substrates with that specific pair of nucleotides and 0 otherwise. Each interaction term that had an absolute t-value >3.5 (P < 0.005) was used in a final model of stepwise regression to obtain predicted krel values.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Paz, M. Pedraza, and K. Simmons for assistance with kinetic experiments and N. Molyneaux for assistance processing high-throughput sequencing data. This work was supported by National Institutes of Health (National Institute of General Medical Sciences) grants RO1 GM056740 (M.E.H.) and T32 GM008056 (C.N.N.).
Author contributions: C.N.N. conducted the experiments; D.R.A. created the mathematical models; C.N.N., E.J., and M.E.H. designed the study; C.N.N. and M.E.H. wrote the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.056408.116.
REFERENCES
- Altman S, Guerrier-Takada C. 1986. M1 RNA, the RNA subunit of Escherichia coli ribonuclease P, can undergo a pH-sensitive conformational change. Biochemistry 25: 1205–1208. [DOI] [PubMed] [Google Scholar]
- Anderson VE. 2015. Multiple alternative substrate kinetics. Biochim Biophys Acta 1854: 1729–1736. [DOI] [PubMed] [Google Scholar]
- Ascano M, Hafner M, Cekan P, Gerstberger S, Tuschl T. 2012. Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdiscip Rev RNA 3: 159–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Gerstberger S, Tuschl T. 2013. Multi-disciplinary methods to define RNA-protein interactions and regulatory networks. Curr Opin Genet Dev 23: 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brännvall M, Mattsson JG, Svärd SG, Kirsebom LA. 1998. RNase P RNA structure and cleavage reflect the primary structure of tRNA genes. J Mol Biol 283: 771–783. [DOI] [PubMed] [Google Scholar]
- Brännvall M, Fredrik Pettersson BM, Kirsebom LA. 2002. The residue immediately upstream of the RNase P cleavage site is a positive determinant. Biochimie 84: 693–703. [DOI] [PubMed] [Google Scholar]
- Brännvall M, Pettersson BM, Kirsebom LA. 2003. Importance of the +73/294 interaction in Escherichia coli RNase P RNA substrate complexes for cleavage and metal ion coordination. J Mol Biol 325: 697–709. [DOI] [PubMed] [Google Scholar]
- Brännvall M, Kikovska E, Kirsebom LA. 2004. Cross talk between the +73/294 interaction and the cleavage site in RNase P RNA mediated cleavage. Nucleic Acids Res 32: 5418–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, Greenleaf WJ. 2014. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat Biotechnol 32: 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian EL, Harris ME. 1999. The track of the pre-tRNA 5′ leader in the ribonuclease P ribozyme-substrate complex. Biochemistry 38: 12629–12638. [DOI] [PubMed] [Google Scholar]
- Christian EL, McPheeters DS, Harris ME. 1998. Identification of individual nucleotides in the bacterial ribonuclease P ribozyme adjacent to the pre-tRNA cleavage site by short-range photo-cross-linking. Biochemistry 37: 17618–17628. [DOI] [PubMed] [Google Scholar]
- Christian EL, Zahler NH, Kaye NM, Harris ME. 2002. Analysis of substrate recognition by the ribonucleoprotein endonuclease RNase P. Methods 28: 307–322. [DOI] [PubMed] [Google Scholar]
- Cook KB, Hughes TR, Morris QD. 2015. High-throughput characterization of protein–RNA interactions. Brief Funct Genomics 14: 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Pace NR. 1998. Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu Rev Biochem 67: 153–180. [DOI] [PubMed] [Google Scholar]
- Guenther UP, Yandek LE, Niland CN, Campbell FE, Anderson D, Anderson VE, Harris ME, Jankowsky E. 2013. Hidden specificity in an apparently nonspecific RNA-binding protein. Nature 502: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada C, McClain WH, Altman S. 1984. Cleavage of tRNA precursors by the RNA subunit of E. coli ribonuclease P (M1 RNA) is influenced by 3′-proximal CCA in the substrates. Cell 38: 219–224. [DOI] [PubMed] [Google Scholar]
- Guo X, Campbell FE, Sun L, Christian EL, Anderson VE, Harris ME. 2006. RNA-dependent folding and stabilization of C5 protein during assembly of the E. coli RNase P holoenzyme. J Mol Biol 360: 190–203. [DOI] [PubMed] [Google Scholar]
- Hardt WD, Schlegl J, Erdmann VA, Hartmann RK. 1993. Role of the D arm and the anticodon arm in tRNA recognition by eubacterial and eukaryotic RNase P enzymes. Biochemistry 32: 13046–13053. [DOI] [PubMed] [Google Scholar]
- Hardt WD, Schlegl J, Erdmann VA, Hartmann RK. 1995. Kinetics and thermodynamics of the RNase P RNA cleavage reaction: analysis of tRNA 3′-end variants. J Mol Biol 247: 161–172. [DOI] [PubMed] [Google Scholar]
- Harris ME, Nolan JM, Malhotra A, Brown JW, Harvey SC, Pace NR. 1994. Use of photoaffinity crosslinking and molecular modeling to analyze the global architecture of ribonuclease P RNA. EMBO J 13: 3953–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ME, Kazantsev AV, Chen JL, Pace NR. 1997. Analysis of the tertiary structure of the ribonuclease P ribozyme-substrate complex by site-specific photoaffinity crosslinking. RNA 3: 561–576. [PMC free article] [PubMed] [Google Scholar]
- Heide C, Pfeiffer T, Nolan JM, Hartmann RK. 1999. Guanosine 2-NH2 groups of Escherichia coli RNase P RNA involved in intramolecular tertiary contacts and direct interactions with tRNA. RNA 5: 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E, Harris ME. 2015. Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol 16: 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AV, Pace NR. 2006. Bacterial RNase P: a new view of an ancient enzyme. Nat Rev Microbiol 4: 729–740. [DOI] [PubMed] [Google Scholar]
- Kirsebom LA, Svärd SG. 1994. Base pairing between Escherichia coli RNase P RNA and its substrate. EMBO J 13: 4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm BP, Wu N, Chen Y, Liu X, Kaitany KJ, Howard MJ, Fierke CA. 2016. The diversity of ribonuclease P: protein and RNA catalysts with analogous biological functions. Biomolecules 6: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutmou KS, Zahler NH, Kurz JC, Campbell FE, Harris ME, Fierke CA. 2010. Protein-precursor tRNA contact leads to sequence-specific recognition of 5′ leaders by bacterial ribonuclease P. J Mol Biol 396: 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz JC, Fierke CA. 2000. Ribonuclease P: a ribonucleoprotein enzyme. Curr Opin Chem Biol 4: 553–558. [DOI] [PubMed] [Google Scholar]
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. 2014. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54: 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. 2008. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Yandek LE, Gjermeni I, Harris ME. 2014. Determination of relative rate constants for in vitro RNA processing reactions by internal competition. Anal Biochem 467: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou TF, Weidmann CA, Killingsworth J, Tanaka Hall TM, Goldstrohm AC, Campbell ZT. 2017. Integrated analysis of RNA-binding protein complexes using in vitro selection and high throughput sequencing and sequence specificity landscapes. Methods 118: 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niland CN, Jankowsky E, Harris ME. 2016a. Optimization of high-throughput sequencing kinetics for determining enzymatic rate constants of thousands of RNA substrates. Anal Biochem 510: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niland CN, Zhao J, Lin HC, Anderson DR, Jankowsky E, Harris ME. 2016b. Determination of the specificity landscape for ribonuclease P processing of precursor tRNA 5′ leader sequences. ACS Chem Biol 11: 2285–2292. [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Dale T, Fahlman RP, Uhlenbeck OC. 2005. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat Struct Mol Biol 12: 788–793. [DOI] [PubMed] [Google Scholar]
- Ozer A, Tome JM, Friedman RC, Gheba D, Schroth GP, Lis JT. 2015. Quantitative assessment of RNA-protein interactions with high-throughput sequencing-RNA affinity profiling. Nat Protoc 10: 1212–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace NR, Reich C, James BD, Olsen GJ, Pace B, Waugh DS. 1987. Structure and catalytic function in ribonuclease P. Cold Spring Harb Symp Quant Biol 52: 239–248. [DOI] [PubMed] [Google Scholar]
- Pettersson BM, Kirsebom LA. 2008. The presence of a C−1/G+73 pair in a tRNA precursor influences processing and expression in vivo. J Mol Biol 381: 1089–1097. [DOI] [PubMed] [Google Scholar]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragón A. 2010. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468: 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter NJ, Osterman AK, Mondragón A. 2012. The bacterial ribonuclease P holoenzyme requires specific conserved residues for efficient catalysis and substrate positioning. Nucleic Acids Res 40: 10384–10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Campbell FE, Zahler NH, Harris ME. 2006. Evidence that substrate-specific effects of C5 protein lead to uniformity in binding and catalysis by RNase P. EMBO J 25: 3998–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Campbell FE, Yandek LE, Harris ME. 2010. Binding of C5 protein to P RNA enhances the rate constant for catalysis for P RNA processing of pre-tRNAs lacking a consensus G(+ 1)/C(+ 72) pair. J Mol Biol 395: 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunden F, Peck A, Salzman J, Ressl S, Herschlag D. 2015. Extensive site-directed mutagenesis reveals interconnected functional units in the alkaline phosphatase active site. eLife 4: e06181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svärd SG, Kagardt U, Kirsebom LA. 1996. Phylogenetic comparative mutational analysis of the base-pairing between RNase P RNA and its substrate. RNA 2: 463–472. [PMC free article] [PubMed] [Google Scholar]
- Wegscheid B, Hartmann RK. 2006. The precursor tRNA 3′-CCA interaction with Escherichia coli RNase P RNA is essential for catalysis by RNase P in vivo. RNA 12: 2135–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegschied B, Hartmann RK. 2007. In vivo and in vitro investigation of bacterial type B RNase P interaction with tRNA 3′-CCA. Nucleic Acids Res 35: 2060–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandek LE, Lin HC, Harris ME. 2013. Alternative substrate kinetics of Escherichia coli ribonuclease P: determination of relative rate constants by internal competition. J Biol Chem 288: 8342–8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler NH, Christian EL, Harris ME. 2003. Recognition of the 5′ leader of pre-tRNA substrates by the active site of ribonuclease P. RNA 9: 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler NH, Sun L, Christian EL, Harris ME. 2005. The pre-tRNA nucleotide base and 2′-hydroxyl at N(−1) contribute to fidelity in tRNA processing by RNase P. J Mol Biol 345: 969–985. [DOI] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. 2010. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40: 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.