

Abstract
Hedgehog (HH) signaling critically regulates embryonic and postnatal development as well as adult tissue homeostasis, and its perturbation can lead to developmental disorders, birth defects, and cancers. Neuropilins (NRPs), which have well-defined roles in Semaphorin and VEGF signaling, positively regulate HH pathway function, although their mechanism of action in HH signaling remains unclear. Here, using luciferase-based reporter assays, we provide evidence that NRP1 regulates HH signaling specifically at the level of GLI transcriptional activator function. Moreover, we show that NRP1 localization to the primary cilium, a key platform for HH signal transduction, does not correlate with HH signal promotion. Rather, a structure–function analysis suggests that the NRP1 cytoplasmic and transmembrane domains are necessary and sufficient to regulate HH pathway activity. Furthermore, we identify a previously uncharacterized, 12-amino acid region within the NRP1 cytoplasmic domain that mediates HH signal promotion. Overall, our results provide mechanistic insight into NRP1 function within and potentially beyond the HH signaling pathway. These insights have implications for the development of novel modulators of HH-driven developmental disorders and diseases.
Keywords: cell signaling, cell surface receptor, cilia, Hedgehog signaling pathway, PKA, signal transduction, Neuropilin
Introduction
Hedgehog (HH)4 signaling is essential for tissue patterning and organ formation during embryonic and postnatal development as well as tissue homeostasis, renewal, and repair in adult animals (1–3). Deregulation of the HH pathway causes a wide range of developmental abnormalities (4, 5) as well as a growing number of pediatric and adult cancers (6, 7). However, despite the widespread importance of HH signaling, our understanding of the mechanisms that regulate HH signal transduction remains incomplete.
HH signaling is tightly regulated by a number of inputs that together control the function of glioma-associated oncogene homolog (GLI) proteins, the transcriptional effectors of the mammalian HH pathway (8). In the absence of HH ligands, the cell surface protein Patched 1 (PTCH1) inhibits the activity of Smoothened (SMO), a putative G protein–coupled receptor that mediates intracellular HH signal transduction (9–12). In this “off” state, GLI2 and GLI3 are phosphorylated by PKA, GSK3β, and CK1 (8). As a consequence of this phosphorylation, GLI2 is largely degraded, whereas GLI3 is processed into a transcriptional repressor (13, 14). HH ligand binding to PTCH1 results in derepression of SMO, which initiates a signal transduction cascade that culminates in GLI processing into transcriptional activators that modulate target gene expression in a context-specific manner (8, 15–17).
Multiple cohorts of cell surface proteins regulate HH pathway activity by binding to HH ligand. DISP1 and SCUBE2 tightly control ligand secretion (18–20), whereas trafficking and turnover are regulated by LRP2 and GPCs (21–23). The cell surface components GAS1, CDON, and BOC function as essential co-receptors at the level of signal reception (24, 25). Additionally, ligand interactions with PTCH1, PTCH2, and HHIP1 result in pathway antagonism (26–28). Together, these and other cell surface proteins regulate HH signaling in a multitude of tissues throughout embryonic and postnatal development.
The Neuropilins (NRPs), a small family comprised of NRP1 and NRP2, have well-established roles in axon guidance and vascular patterning (29–34) and act to positively regulate HH signaling at the cell surface (35, 36). NRPs are expressed in a variety of HH-responsive tissues during critical periods of HH-related developmental patterning (37, 38). Importantly, loss-of-function experiments demonstrated that Nrp1a knockdown in zebrafish disrupts HH-dependent somite development (35), whereas genetic deletion of Nrp1 and Nrp2 in the mouse suppresses HH-driven cerebellar granular neuron progenitor proliferation (36). Additionally, NRPs exacerbate HH-related cancers, suggesting that they impact both HH-dependent development and HH-driven disease (36, 39–42) Notably, NRPs are thought to act downstream of HH ligands (36), distinguishing their mode of action from most other cell surface regulators of the HH pathway. Previous reports suggest that NRP modulation of HH signaling occurs at the level of suppressor of fused (SUFU) (35). More recent data, however, suggest that NRPs act downstream of SUFU, regulating GLI phosphorylation by interacting with phosphodiesterase 4D (PDE4D), which inhibits PKA (36). However, the precise mechanism of NRP function in HH signal transduction remains unclear.
Here we provide data defining a novel mechanism of NRP action in HH signaling. Specifically, we find that NRPs promote HH signaling selectively at the level of GLI activation, independent of PKA phosphorylation. We also demonstrate that NRP1, but not NRP2, traffics to the primary cilium, a highly regulated subcellular compartment required for vertebrate HH signal transduction. Strikingly, NRP1 ciliary localization does not correlate with the promotion of HH pathway activity. Instead, we find that membrane-anchored NRP1 cytoplasmic domain (CD) is both necessary and sufficient to promote HH pathway activation. Further, we map the region in the NRP1 CD that is critical for HH signal promotion to a 12-amino acid motif not previously implicated in NRP function. Overall, these data characterize NRPs as a novel class of cell surface HH pathway regulators that act downstream of ligand binding through cytoplasmic effectors to control HH pathway function.
Results
NRP1 and NRP2 promote HH signaling by modulating GLI activator function
A previous study showed that NRP1 overexpression increases ligand-stimulated HH pathway activity in HH-responsive fibroblasts (35). To confirm and extend these findings, we first tested whether NRP1 and NRP2 promote HH signaling using a luciferase-based reporter assay system in NIH-3T3 fibroblasts (43). Although the addition of HH ligand is sufficient to induce a transcriptional response, we found that NRP1 and NRP2 both significantly increase ligand-activated HH pathway activity, as detected by GLI-dependent luciferase output (Fig. 1A and supplemental Fig. S1), consistent with a previous report (35). Notably, co-expression of Nrp1 and Nrp2 does not significantly change the level of NRP-mediated HH pathway promotion (Fig. 1A). Western blot analysis confirmed that HA-tagged NRP1 and NRP2 are expressed at similar levels in NIH-3T3 cells (Fig. 1B). Although NRP1 significantly promoted HH signaling in ∼90% of assays (n = 8, average -fold change = 2.04), NRP2 significantly promoted HH signaling in only 40% of assays (n = 8, average -fold change = 1.36, supplemental Fig. S1). Because of this variability, we decided to focus on NRP1 for further analysis.
Figure 1.

NRP1 promotes HH signaling at the level of GLI activation. A, HH-dependent luciferase reporter activity measured in NIH-3T3 cells transfected with the indicated constructs and stimulated with NSHH-conditioned medium (+NSHH). Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t test. n.s., not significant. B, top, Western blot analysis of NIH-3T3 lysates collected from cells expressing the indicated HA-tagged proteins. Bottom, quantitation of NRP levels relative to β-tubulin. a-β-Tub, anti-β-tubulin. C, schematic of various modes of HH pathway activation at the level of ligand (HH), small-molecule SMO agonist (SAG), oncogenic SMO mutations (SMOM2), and constitutive GLI activator (GLI2ΔN). D–F, luciferase reporter activity similar to A in NIH-3T3 cells stimulated with SAG (D) or co-transfected with SmoM2 (E) or GLI2ΔN (F). G and H, luciferase reporter activity similar to A in NIH-3T3 cells transfected with the Gli3 repressor.
To determine whether HH ligand is required for NRP1-mediated HH signal promotion, we activated HH signaling by adding exogenous smoothened agonist (SAG), co-transfecting a constitutively active form of Smoothened (SmoM2), or co-transfecting a constitutively active form of GLI (GLI2ΔN; Fig. 1, C–F) (44–46). Strikingly, NRP1 significantly increases the HH-dependent luciferase output, regardless of the means of pathway activation (Fig. 1, D–F). In contrast, NRP1 does not alter GLI3-mediated repression of Hedgehog signaling (Fig. 1, G and H). Together, these data support a model in which NRP1 acts to selectively regulate GLI activator function downstream of HH ligand.
The membrane-anchored NRP1 CD is necessary and sufficient to promote HH signaling
To determine the domain requirements for NRP-mediated promotion of HH signaling, we generated a Nrp1 construct lacking the cytoplasmic domain, Nrp1ΔCD (Fig. 2A). Strikingly, NRP1ΔCD does not promote HH signaling (Fig. 2B). Western blot analyses in NIH-3T3 cells confirmed equal expression of NRP1 and NRP1ΔCD (Fig. 2C). These data are consistent with recent results suggesting that NRP1 utilizes its cytoplasmic domain to promote HH pathway activity (36). However, in contrast to previous work, a version of NRP1 that lacks all functional extracellular domains (NRP1ΔECD) is sufficient to promote HH signaling (Fig. 2, A and B). Western blot analysis confirmed NRP1ΔECD expression (Fig. 2C). Immunofluorescent analysis of NRP1, NRP1ΔCD, and NRP1ΔECD under permeabilizing and non-permeabilizing conditions using dual extracellular and intracellular antibody staining confirmed the cell surface localization of NRP1 and NRP1ΔCD (Fig. 2D and supplemental Fig. S2). To further explore the requirement for the NRP1 CD in HH signaling, we generated a cytosolic version of the CD that is not membrane-tethered (NRP1CD). Notably, transfection of Nrp1CD is not sufficient to promote HH signaling in NIH-3T3 cells (supplemental Fig. S3), suggesting a role for the NRP1 transmembrane (TM) domain in HH signal promotion. Together, these data suggest that the membrane-anchored NRP1 CD is both necessary and sufficient to promote HH signaling.
Figure 2.

NRP1 cytoplasmic and transmembrane domains are necessary and sufficient to promote HH signaling. A, schematic of full-length NRP1, NRP1ΔCD, and NRP1ΔECD. Dotted lines indicate regions that were deleted in each construct. B, HH-dependent luciferase reporter activity measured in NIH-3T3 cells transfected with the indicated constructs and stimulated with NSHH-conditioned medium (+NSHH). Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t tests. n.s., not significant. C, Western blot analysis of HA-tagged protein expression in NIH-3T3 cells. Anti-β-tubulin (α-β-Tub) was used as loading control. Right, quantitation of NRP1 and NRP1ΔCD levels relative to β-tubulin. D, antibody detection of an extracellular NRP1 epitope (α-NRP1, red) and an intracellular HA tag (α-HA, red) in permeabilized (left panels) and non-permeabilized (center and right panels) conditions to assess cell surface localization of NRP1, NRP1ΔCD, and NRP1ΔECD. Nuclear GFP (green) indicates transfected cells, whereas DAPI (blue) marks all nuclei. Shown are diagrams of each construct, with brackets indicating antibody-binding sites. Scale bar = 10 μm.
NRP1 transmembrane dimerization is not required for HH signal promotion
Neuropilin TM dimerization is mediated by a double GXXXG motif in the TM domain that stabilizes signaling complexes for both Semaphorin and VEGF ligands (47). Mutating the three glycine residues within the double GXXXG motif to valines completely disrupts dimerization and blocks NRP1 function in Semaphorin signaling (47). To determine whether NRP1 TM dimerization is required for HH signal promotion, we recreated these three glycine mutations in both NRP1 (Fig. 3A) and NRP1ΔECD (Fig. 3B). Strikingly, these mutations do not impair the ability of either construct to promote HH signaling (Fig. 3), suggesting that NRP1 membrane attachment, but not TM dimerization, is required for its function in HH signaling.
Figure 3.
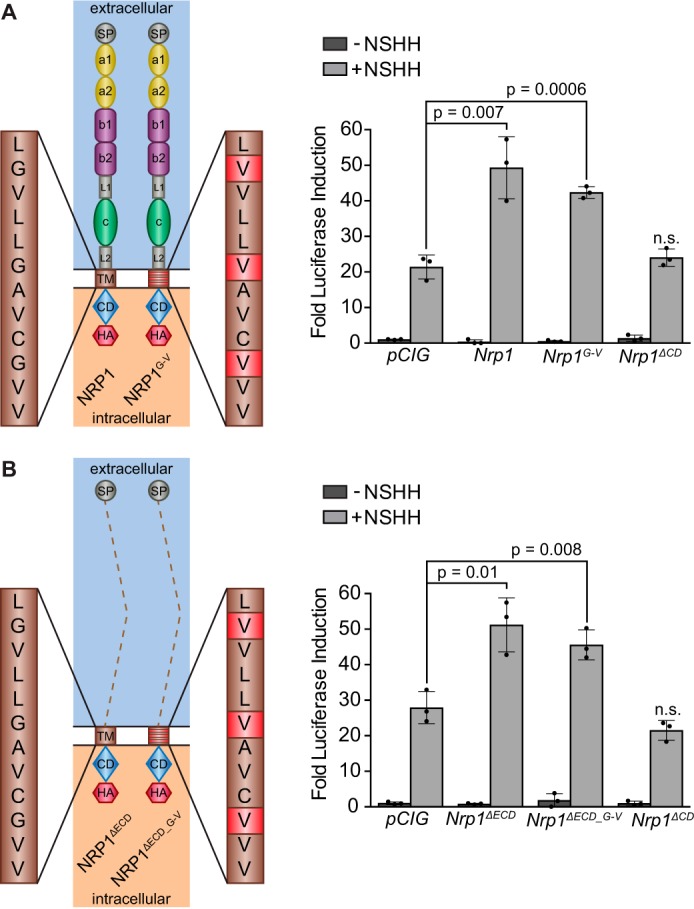
NRP1 TM dimerization is not required to promote HH signaling. A, left, schematic of full-length NRP1 and NRP1G-V, in which three glycine residues are mutated to valines. Right, HH-dependent luciferase reporter activity measured in NIH-3T3 cells transfected with the indicated constructs and stimulated with NSHH-conditioned medium (+NSHH). Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t tests. n.s., not significant. B, left, schematic of NRP1ΔECD and NRP1ΔECD_G-V. Right, HH-dependent luciferase reporter activity measured in NIH-3T3 cells transfected with the indicated constructs and stimulated with NSHH-conditioned medium. Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t tests.
NRP1 promotes HH signaling independently of GLI2 phosphorylation by PKA
Previous work has suggested that NRPs regulate HH signaling by recruiting PDE4D to the cell membrane (36). PDE4D negatively regulates PKA activity by locally reducing levels of cAMP (48). PKA phosphorylates GLI transcription factors at a number of consensus and non-consensus sites to regulate their activity, including six consensus sites within the activation domain of GLI2 that are sufficient to repress GLI2 activity (13, 14, 49). To test whether the NRP1 CD modulates HH activity through PKA-dependent GLI phosphorylation, we generated serine-to-alanine mutations at the six consensus PKA phosphorylation sites critical for GLI2 repression (13, 14, 49) (GLI2P1–6, Fig. 4A). As expected, GLI2P1–6 expression results in a significant increase in HH signaling compared with WT GLI2 (Fig. 4B). Although GLI2 stimulates HH signaling less effectively than its constitutively active counterpart GLI2ΔN, we still observed a reliable increase in activity with Nrp1 co-expression (Fig. 4B). This increase was not observed when we co-expressed Nrp1ΔCD, consistent with previous results (Fig. 4B, cf. Fig. 2B). Surprisingly, Nrp1 still promotes HH signaling when co-expressed with GLI2P1–6, suggesting that NRP1 regulates GLI activity independently of PKA phosphorylation. Importantly, Nrp1ΔCD does not promote signaling when co-expressed with GLI2P1–6, indicating that this PKA-independent promotion of HH signaling still requires the NRP1 CD (Fig. 4B).
Figure 4.
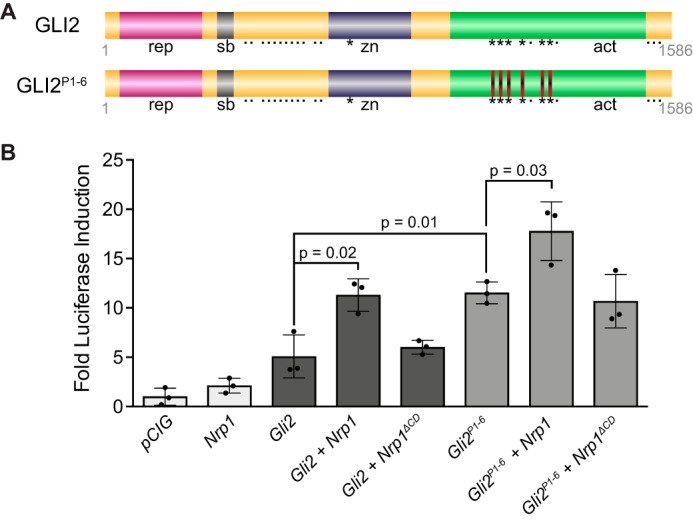
GLI2 phosphorylation by PKA is not required for NRP1-mediated HH promotion. A, diagram of GLI2 indicating the repressor domain (rep), SUFU binding region (sb), zinc finger binding domain (zn), and activation domain (act). Asterisks mark consensus PKA phosphorylation sites, whereas dots mark non-consensus sites. Red bars indicate locations of serine-to-alanine mutations to disrupt PKA phosphorylation at six key repressive sites. B, luciferase reporter activity in NIH-3T3 cells transfected with Nrp and GLI2 constructs as indicated. Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t tests.
Identification of a novel NRP1 cytoplasmic motif that mediates HH signal promotion
To elucidate which region of the NRP1 CD promotes HH pathway activation, we initially targeted a highly conserved, C-terminal SEA motif described previously to bind PDZ-containing proteins, as this is the only region of the NRP1 CD with any previously ascribed function (50). Notably, adding a C-terminal HA tag to NRP1 itself could block PDZ binding at the SEA motif. However, NRP1 was able to promote HH signaling equally well, regardless of whether we placed the tag at its C terminus or N terminus (Fig. 2; data not shown). Furthermore, deleting the NRP1 SEA (NRP1Δ920–922, Fig. 5A) did not impair NRP1-mediated promotion of HH signaling in NIH-3T3 cells (Fig. 5B). To narrow the region of the NRP1 CD that mediates HH signaling, we deleted the N-terminal 20 amino acids of the NRP1 CD (NRP1Δ883–902, Fig. 5A) and assessed function in NIH-3T3 HH signaling assays. Strikingly, NRP1Δ883–902 failed to promote HH signaling (Fig. 5C), suggesting that the residues required for NRP regulation of HH activity are located in the membrane-proximal half of the NRP1 CD. Western blot analyses confirmed expression of NRP1Δ883–902 (Fig. 5F), and immunofluorescent staining under non-permeabilizing conditions confirmed that NRP1Δ883–902 properly localizes to the cell surface (Fig. 5G). We then asked whether restoring part of this region would rescue NRP1 function in HH signaling; however, NRP1Δ890–922 still failed to promote signaling (Fig. 5D). Ultimately, adding back 12 additional residues from amino acid 890–902 (NRP1Δ902–922) rescued NRP1-mediated promotion of HH signaling equivalently to full-length NRP1 (Fig. 5E), confirming the importance of this region to NRP1 function in HH signaling. Although this 12-amino acid region has no previously described function, we noted the presence of two serine residues and a tyrosine residue in this motif. To investigate whether phosphorylation at these sites might regulate NRP function, we mutated these residues to alanine. Remarkably, alanine mutagenesis of these residues does not alter NRP1 promotion of HH signaling (supplemental Fig. S4). Together, these data suggest that a conserved, 12-amino acid region of the NRP1 CD between amino acids 890 and 902 plays an essential role in HH signal promotion through selective regulation of GLI activator function.
Figure 5.

Identification of a 12-amino acid motif in the NRP1 CD required for HH signal promotion. A, diagram of the NRP cytoplasmic domain, with amino acid number indicated (top) and deletions indicated by dotted lines. B–E, luciferase reporter activity in NIH-3T3 cells transfected with NRP constructs as indicated and stimulated with NSHH-conditioned medium (+NSHH). Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t tests. F, top, Western blot analysis of HA-tagged protein levels in NIH-3T3 cell lysates with detection of β-tubulin (β-Tub) as a loading control. Bottom, quantitation of NRP levels relative to β-tubulin. G, antibody detection of an extracellular NRP1 epitope (α-NRP1, red) in non-permeabilized NIH-3T3 cells to assess cell surface localization of NRP1, NRP1ΔECD, NRP1Δ883–902, and NRP1Δ890–922. Nuclear GFP (green) indicates transfected cells, whereas DAPI (blue) marks all nuclei. Note that NRP1ΔECD lacks the NRP1 antibody epitope and served as a negative control. Scale bar = 10 μm.
NRP1, but not NRP2, localizes to primary cilia in HH-responsive fibroblasts
The primary cilium is a highly regulated subcellular compartment into which molecules over 40 kDa cannot freely diffuse (51), and an intact cilium is important for HH signaling to proceed normally (52). Given that NRP1 regulates HH signaling through the modulation of GLI activity, and that GLI proteins localize to cilia and require intact cilia for their processing and function (53), we asked whether NRPs localize to the primary cilium.
To assess primary cilia localization, we expressed Nrp1 and Nrp2 in WT and Dynein-mutant (Dync2h1lln/lln) mouse embryonic fibroblasts (MEFs) (Fig. 6). Dynein motors mediate retrograde transport of ciliary components; thus, cilia-localized proteins accumulate within the primary cilium of Dynein-mutant MEFs, allowing for more robust detection (54). We found that NRP1, but not NRP2, localizes to primary cilia (identified with anti-acetylated tubulin, AcTub) in WT and Dynein-mutant MEFs (Fig. 6). NRP1 was detected in 51% of cilia in WT MEFs and further enriched in dynein-mutant MEFs, with 68% of cilia positive for NRP1 (Fig. 6, A, E, I, and J). NRP2, on the other hand, was only detected in primary cilia in 9% of Dynein-mutant MEFs, with no ciliary localization observed in WT MEFs (Fig. 6, B, F, I, and J). As a positive control, SMOM2 robustly localizes to the primary cilium in both WT and Dynein-mutant MEFs (98% of cilia in each group; Fig. 6, C, G, I, and J), consistent with previous findings (55). In contrast, BOC, a cell surface–localized HH co-receptor (56, 57), was detected broadly throughout the cell surface but was not observed in primary cilia (Fig. 6, D and H–J). Importantly, no HA staining was observed in the cilia of vector-transfected cells (supplemental Fig. S5). To further confirm these data, we stained WT and Dynein-mutant MEFs for endogenous NRP1 and detected NRP1 localization to primary cilia (Fig. 6K). These results suggest that NRP1, but not NRP2, localizes to the primary cilium of HH-responsive fibroblasts.
Figure 6.

NRP1, but not NRP2, localizes to primary cilia in HH-responsive fibroblasts. A–H, antibody detection of HA/YFP (green) and primary cilia (red, AcTub) in WT (top) and Dynein-mutant (Dync2h1lln/lln) MEFs (bottom). Dync2h1lln/lln MEFs exhibit impaired retrograde transport out of primary cilia. Arrows indicate the location of primary cilia. Insets show higher-magnification views of primary cilia in individual (left and center) and merged (right) channels. DAPI indicates nuclei (blue). I and J, quantitation of data represented in images from WT (left) and Dync2h1lln/lln (right) MEFs, reported as mean ± S.D., with scatterplot values indicating averages of three independent experiments and the total number of cells analyzed listed below each bar. Data are reported from at least three independent experiments. K, antibody detection of endogenous NRP1 in WT (left panel) and Dync2h1lln/lln (right panel) MEFs. Scale bar = 10 μm, inset scale bar = 1 μm.
NRP1 cilia localization does not correlate with HH signal promotion
Although both NRP1 and NRP2 promote HH signaling, our data indicate that NRP1 functions more consistently than NRP2 in our cell signaling assays (supplemental Fig. S1). Given that NRP1 preferentially localizes to primary cilia, we assessed the requirement for NRP1 cilia localization in HH signal promotion, taking advantage of two of the deletion constructs described previously, Nrp1ΔECD and Nrp1Δ902–922. Notably, both of these constructs robustly promoted HH signaling (Figs. 2B, 5E, and 7E). We performed immunofluorescent staining to examine the ciliary localization of NRP1, NRP1ΔECD, or NRP1Δ902–922 in NIH-3T3 cells (Fig. 6). Although NRP1 localizes to primary cilia in roughly 40% of transfected cells (Fig. 7, A and D), both NRP1ΔECD and NRP1Δ902–922 displayed significantly reduced localization to primary cilia (Fig. 7, B–D). Taken together, these results suggest that cilia localization does not correlate with NRP1-mediated promotion of HH signal transduction.
Figure 7.
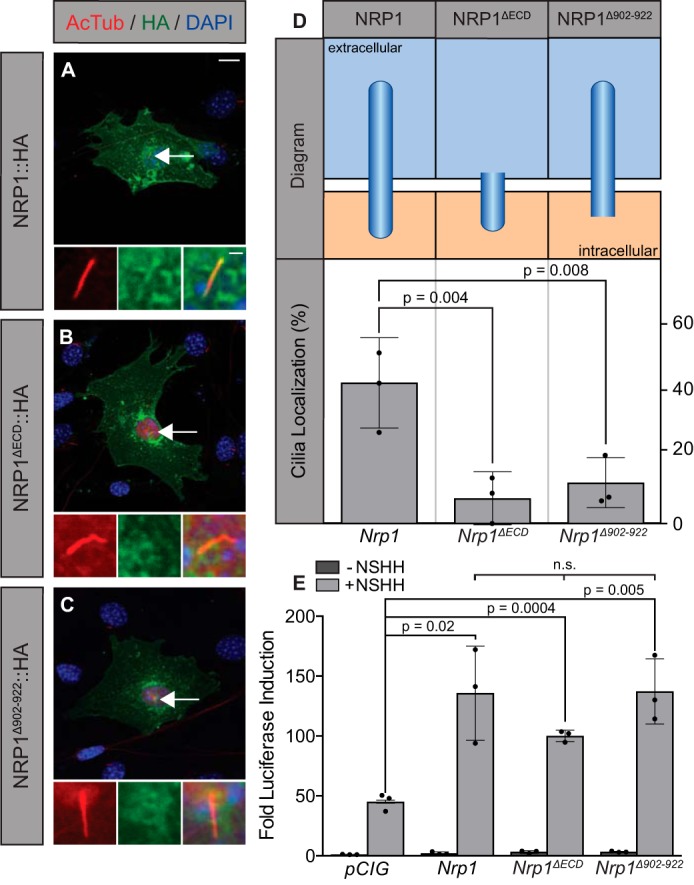
Ciliary localization of NRP1 does not correlate with HH signal promotion. A–C, antibody detection of NRP1 (A), NRP1ΔECD (B), and NRP1Δ902–922 (C) in NIH-3T3 cells (HA, green; AcTub, red; DAPI, blue). D, chart summarizing the structure (top) and cilium localization (bottom) of each construct imaged on the left. n = 165, 166, and 162 cells for Nrp1, NRP1ΔECD, and NRP1Δ902–922, respectively. Scatterplot values indicate averages of three independent experiments, with column height indicating the overall average ± S.D. E, luciferase reporter activity in NIH-3T3 cells transfected with constructs as indicated and stimulated with NSHH-conditioned medium (+NSHH). Data are reported as mean -fold induction ± S.D., with p values calculated using two-tailed Student's t tests. n.s., not significant. Scale bar = 10 μm, inset scale bar = 1 μm.
Discussion
Cell surface regulation of the HH signaling pathway is essential for proper tissue patterning during embryonic and postnatal development as well as adult tissue homeostasis, repair, and regeneration (4, 24, 25, 58–60). Conversely, deregulation of HH cell surface components contributes to HH-driven birth defects and cancers (61–66). NRPs are also implicated in numerous human cancers (40, 67). Notably, Nrp2 knockdown increases survival in a HH-dependent mouse model of medulloblastoma (39). Here we present evidence that NRPs promote HH signaling intracellularly by regulating GLI activator function. Further, we report that NRP1 localizes to the primary cilium; however, this localization does not correlate with NRP1-mediated promotion of HH signaling. Instead, we determine that the NRP1 CD and TM domains are necessary and sufficient to promote HH signal transduction. Finally, we identify a novel region of the NRP1 CD as essential for this process, a region not previously implicated in NRP1 function. Taken together, these findings identify the membrane-tethered NRP1 CD as a key positive regulator of HH signal transduction via selective regulation of GLI activator function.
NRPs as a novel class of ligand-independent HH cell surface regulators
Numerous cell surface proteins promote HH pathway activity through interactions with HH ligands (23–28, 65, 68, 69). Our data suggest that, unlike these proteins, cell surface–localized NRPs act downstream of ligand to regulate HH signaling. Indeed, NRP1 promotes HH pathway activity even when signaling is stimulated by GLI2ΔN, a constitutive transcriptional activator, strongly suggesting that NRPs function at the level of GLI regulation. More specifically, our data suggest that NRPs regulate GLI activator function selectively, failing to impair GLI3 repressor activity.
Although the precise mechanism of NRP-mediated regulation of GLI function remains unclear, our data are consistent with NRPs acting downstream of SUFU at the level of GLI function, since GLI2ΔN is not regulated by SUFU (70). Importantly, we find that Nrp1 still promotes HH pathway activity when co-transfected with GLI2P1–6, a version of GLI2 that cannot be phosphorylated by PKA at six critical repressive sites. Therefore, in contrast to previous work (36), our data suggest that NRP1 promotion of HH signaling is independent of PKA-mediated phosphorylation of GLI2. It is possible that NRP binding to PDE4D could impact PKA-dependent phosphorylation at non-consensus sites (49) or affect GSK3β activity, which is also regulated by cAMP (71); further experiments are required to investigate these possibilities. Also worth considering is that NRP knockdown does not change the amount of GLI in the primary cilium (35), suggesting that NRPs may regulate GLI proteins after they have been processed in the cilium, perhaps by regulating GSK3β, affecting degradation of GLI activators, or impacting endocytosis. Overall, our findings suggest that the NRP1 CD regulates GLI proteins intracellularly, independently of HH ligand binding and independently of PKA-mediated GLI phosphorylation.
Although NRPs promote signaling downstream of HH ligand, it remains unclear whether Semaphorin ligands can contribute to HH signal promotion. Class 3 Semaphorin ligands interact with the extracellular domains of NRP1 and NRP2 (72, 73). Although two previous studies present contrasting results regarding Semaphorin ligand involvement in HH signal regulation (35, 36), our data suggest that Semaphorin ligand binding is not required because the NRP1 extracellular domain is dispensable for HH signal promotion. We cannot exclude the possibility that Semaphorins or other NRP-binding ligands might still modulate HH activity. In addition to Semaphorins and VEGFs, NRPs interact with a wide variety of other proteins, including PIGF-2, heparan sulfate, TFG-β1, HGF, PDGF, FGF, L1-CAM, Plexins, and integrins (67). It is possible that NRP interactions with these or other binding partners also contribute to the promotion of HH signaling.
Identification of a cytoplasmic motif in NRP1 that mediates HH signal transduction
Our data indicate that the membrane-attached NRP1 CD is necessary and sufficient to promote HH signaling. Notably, this contrasts with the HH co-receptors CDON and BOC, whose cytoplasmic domains are dispensable for HH signal promotion (57, 74). NRP1 and NRP2 share several areas of conservation, including a carboxyl-terminal SEA motif that binds the PDZ-containing protein GIPC1 (50, 75–77). Strikingly, our data indicate that this motif is not required for HH signal promotion. Instead, we present evidence that a previously uncharacterized region of the NRP1 cytoplasmic domain between amino acids 890 and 902 is required for HH signal promotion (Fig. 8). Notably, this motif is highly conserved across vertebrate species, including chicken, frog, zebrafish, mouse, rat, and human. This region is also only partially conserved between NRP1 and NRP2, suggesting potential differences in the way the two proteins function in HH signaling. Further analyses will be needed to narrow this region to the exact amino acids necessary for HH regulation and determine the degree of overlap between NRP1 and NRP2 function. Others have reported that genetic deletion of the NRP1 cytoplasmic domain in mice results in defective spatial separation between arteries and veins (78), although no HH-dependent phenotypes have been reported. It is likely that redundancy with NRP2 functionally compensates for NRP1 loss in the promotion of HH signaling, as has been reported in both zebrafish and mice (35, 36).
Figure 8.

Summary and model of NRP function in HH signal transduction. Top left panel, chart summarizing requirements for the NRP1 extracellular domain, dimerization domain, cytoplasmic domain, and cilia localization in HH and Semaphorin signaling. Bottom panel, schematic of NRP expression throughout the cell surface, including in primary cilia. Top right panel, cell surface–localized NRP1 mediates HH signaling through its cytoplasmic domain, specifically requiring amino acids 890–902. This contrasts with the extracellular domain, which is required for Semaphorin signaling, and a conserved SEA motif that is required for VEGF signaling. The NRP1 cytoplasmic domain regulates HH signaling at the level of GLI activity, increasing GLI transcriptional activation through an unknown mechanism.
It remains unclear exactly how this 12-amino acid cytoplasmic region mediates NRP1 function in HH signaling. We have mutated several conserved serine and tyrosine residues located in this region, ruling out the possibility that phosphorylation of these residues affects downstream signaling. One possibility is that this region interacts directly or indirectly with PDE4D, which regulates PKA and could modify GLI proteins through non-consensus phosphorylation sites or through other kinases, as discussed previously. Alternatively, a recent publication identifies a suite of additional intracellular molecules that interact with the NRP cytoplasmic domain, including MYH9, MYH10, DYHC1, FLNA, EF1α1, and ENO1 (79). These molecules may also interact with amino acids 890–902 to mediate GLI regulation, although significant future studies will be required to analyze their potential roles in HH signal transduction. It is also possible that this motif could regulate the conformation or subcellular localization of NRP1 or perhaps play a role in regulating endocytosis of other proteins (see below).
Another aspect to consider in NRP1 function is its ability to homodimerize and heterodimerize with NRP2 (47, 80). Our data suggest that mutation of the dimerization motif in the NRP1 TM domain does not impact its ability to promote HH pathway function, in contrast to an important role for NRP TM dimerization in Semaphorin signaling (47). Similar to the involvement of Semaphorin ligands, the possibility remains that NRP TM dimerization is not required but may somehow modulate HH signaling. It is also possible that NRP interactions with other TM proteins, such as VEGF receptors, Plexins, FGF receptors, or PDGF receptors, may contribute to HH signal promotion (67, 81), although many of these receptors interact with NRP1 through its extracellular domain, which, as our data indicate, is dispensable for NRP1 function in HH signaling.
NRP1 ciliary localization does not correlate with increased HH signal promotion
Our data suggest that, although NRP1 can localize to primary cilia, mutant constructs with reduced cilia localization still promote HH signal transduction. Although actual entry into the highly regulated ciliary compartment does not correlate with NRP-mediated promotion of HH signaling, we cannot exclude the possibility that the low levels of cilia localization we observe may be sufficient to impact HH signal transduction. Alternatively, NRPs may play an important role elsewhere in the cell, perhaps even at the ciliary base. Accordingly, NRP1, NRP2, NRP1ΔECD, and NRP1Δ902–922 were all detected broadly throughout the cell membrane, including at the base of the cilium. Although cytosolic splice variants of NRP1 do exist (82–84), expression of a cytosolic version of the NRP1 cytoplasmic domain fails to promote HH signaling, suggesting that NRP1 must reach the cell surface to impact HH signaling. NRPs are commonly internalized through endocytosis (85, 86); thus, it is possible that endocytic vesicle-associated NRPs affect PKA or GLI function.
Together, these data raise the question of why NRP1 localizes to the primary cilium. One possibility is that NRP1 (compared with NRP2) preferentially binds to another protein that mediates its localization to primary cilia. Alternatively, some parallels have been drawn between cilia and dendritic spines, including the presence of a regulated diffusion barrier (87). It is possible that NRP localization to primary cilia is a byproduct of its role in dendritic spines or other similarly regulated structures. Perhaps the same sequences or mechanisms that allow NRP entry into dendrites and axons also allow their entry into the primary cilium, with or without functional consequence. NRP1 plays well-defined roles in many different signaling pathways (67), any of which might rely on cilium localization of NRP1 for proper function. Interestingly, NRP1 has several roles that differ from NRP2, including axon guidance in response to specific class 3 Semaphorins and angiogenesis (88). Furthermore, NRP1 and NRP2 are expressed in overlapping and distinct cell types during development, some of which rely on primary cilia for proper function. For example, NRP1 is expressed predominantly in arterial endothelial cells, which are thought to rely on mechanosensory cilia for homeostasis (89), whereas NRP2 is expressed predominantly in venous and lymphatic endothelial cells (67). These or potentially other undiscovered functions may result from differential subcellular localization of NRP1 and NRP2 relative to the cilium.
Experimental procedures
Neuropilin and GLI constructs
Nrp and GLI constructs were derived from full-length cDNAs using standard molecular biology techniques. All constructs were cloned into the pCIG vector, which contains a CMV enhancer, chicken β actin promoter, and an internal ribosome entry site with a nuclear enhanced GFP reporter (3XNLS-EGFP) (90). C-terminal or N-terminal HA tags (YPYDVPDYA) were added to the constructs as indicated. Subsequent deletion and mutation variants were generated using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, 200521). To mutagenize the dimerization motif within the NRP1 TM domain, primer sequences were as follows: gtacagcacaactacacatactgcaaccaggagtaccaccagggcactcat (forward) and atgagtgccctggtggtactcctggttgcagtatgtgtagttgtgctgtac (reverse). To mutagenize the serines and tyrosine within the membrane-proximal half of the NRP1 CD, primer sequences were as follows: gttctccagggcagctaggttcgcttccgccatcccattgtgcc (forward), atccacaagttcaaagttagcgttctccagggcagctagg (forward), ggcacaatgggatggcggaagcgaacctagctgccctggagaac (reverse), and cctagctgccctggagaacgctaactttgaacttgtggat (reverse). The GLI2P1–6 construct was created by synthesizing a 1.4-kb portion of human GLI2 containing serine-to-alanine mutations at residues 808, 824, 836, 867, 941, and 970 using Invitrogen GeneArt gene synthesis (Thermo Fisher Scientific) and cloning into full-length human GLI2 using endogenous AgeI and NheI sites.
Cell culture
Cell lines were maintained in DMEM (Life Technologies, 1965) supplemented with 10% bovine calf serum (ATCC, 30-2030) and 1× penicillin—streptomycin–l-glutamine (Life Technologies, 10378016). Cultures were kept at 37 °C with 5% CO2 and 95% humidity.
Cell signaling assays
Luciferase-based reporter assays to assess HH signaling in NIH-3T3 cells were performed as described previously using a ptcΔ136-GL3 reporter construct (43). Briefly, cells were seeded at 2.5 × 104 cells/well into gelatin-coated 24-well plates. The next day, cells were transfected with empty vector (pCIG) or experimental constructs along with a luciferase reporter construct and β-galactosidase transfection control (pSV-β-galactosidase, Promega, E1081). Transfections were performed using Lipofectamine 2000 (Invitrogen, 11668) and Opti-MEM reduced serum medium (Invitrogen, 31985). 48 h after transfection, the culture medium was replaced with low-serum medium (0.5% bovine calf serum, 1% penicillin—streptomycin—l-glutamine) containing either control or N-terminal SHH (NSHH)-conditioned medium. Alternatively, SAG (Sigma-Aldrich, SML1314) was added at a concentration of 300 ng/μl to activate HH signaling. Luciferase reporter activity and β-galactosidase activity were measured 48 h later on a Spectramax M5e plate reader (Molecular Devices) using the luciferase assay system (Promega, E1501) and the Betafluor β-galactosidase assay kit (EMD Millipore, 70979), respectively. Luciferase values were divided by β-galactosidase activity to control for transfection, and data were reported as -fold induction relative to the vector-transfected control. All treatments were performed in triplicate and averaged, with error bars representing the standard deviation between triplicate wells. Student's t tests were used to determine whether each treatment was significantly different from the control, with p values of 0.05 or less considered statistically significant.
Immunofluorescent analysis
Dynein-mutant (Dync2h1lln/lln) and wild-type littermate control MEFs (generously provided by Dr. Kathryn V. Anderson, Memorial Sloan Kettering (54)), were plated at 1.5 × 105 cells/well in a 6-well dish with a coverslip at the bottom of each well. Cells were transfected 24 h after plating using Lipofectamine 2000 (Invitrogen, 11668) and Opti-MEM reduced-serum medium (Invitrogen, 31985). Approximately 6 h after transfection, cells were placed in low-serum medium (0.5% bovine calf serum, 1% penicillin—streptomycin–l-glutamine) for 48 h. Cells were then fixed in 4% paraformaldehyde for 10 min at room temperature, washed with PBS, and permeabilized with 0.2% Triton X-100 in PBS for 5 min before antibodies were added. Primary antibodies included mouse IgG1 anti-HA.11 (1:1000, Biolegend, 901502), goat IgG anti-NRP1 (1:100, R&D Systems, AF566), and mouse IgG2b anti-acetylated tubulin (1:2500, Sigma-Aldrich, T7451). Coverslips were incubated with primary antibodies overnight, followed by a 10-min DAPI stain (1:30,000 at room temperature, Invitrogen, D1306) and 1-h incubation with secondary antibodies, including Alexa Fluor 546 goat anti-mouse IgG1 (γ1), Alexa Fluor 488 donkey anti-goat IgG, Alexa Fluor 488 goat anti-mouse IgG2b, and Alexa Fluor 555 goat anti-mouse IgG2b (1:500, Invitrogen, A21123, A21202, A21141, and A21147, respectively). Coverslips were mounted to glass slides using Shandon Immu-Mount mounting medium (Fisher, 9990412). Immunofluorescent analysis and imaging were performed on a Leica SP5X upright two-photon confocal microscope using LAS AF software (Leica) and a Leica ×63 (type, HC Plan Apochromat CS2; NA1.2) water immersion objective. Cilium counts were performed in a single-blind fashion. Control constructs included Boc and SmoM2.
Western blot analysis
COS-7 or NIH-3T3 cells were transfected using Lipofectamine 2000 (Invitrogen, 11668) and Opti-MEM reduced-serum medium (Invitrogen, 31985). Cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.2), 150 mm NaCl, 0.1% Triton X-100, 1% sodium deoxycholate, and 5 mm EDTA) 48 h after transfection, sonicated using a Fisher Scientific sonic dismembrator, model 500 (four pulses at 20%), and centrifuged at 14,000 × g for 25 min at 4 °C to remove the insoluble fraction. Protein concentrations were determined using a BCA protein assay kit (Fisher, PI23225). After boiling for 10 min, 50 μg of protein from each sample was separated using SDS-PAGE with 7.5–12.5% gels and transferred onto Immun-Blot PVDF membranes (Bio-Rad, 162-0177). Membranes were washed in TBS with 0.5% OmniPur Tween 20 (TBST, EMD Millipore, 9480) and blocked in Western blocking buffer (30 g/liter bovine serum albumin with 0.2% NaN3 in TBST) for 1 h to overnight. Blots were probed with the following antibodies: mouse IgG1 anti-HA.11 (1:1000, Covance, MMS-101P-200), goat IgG anti-Neuropilin1 (1:100, R&D Systems, AF566), and mouse IgG1 anti-β-tubulin (1:10,000, generously provided by Dr. Kristen J. Verhey, University of Michigan). Secondary antibodies were diluted 1:10,000 and included peroxidase-conjugated AffiniPure goat anti-mouse IgG, light chain–specific (Jackson ImmunoResearch, 115-035-174), and peroxidase-conjugated AffiniPure donkey anti-goat IgG, light chain–specific (Jackson ImmunoResearch, 705-035-147). Immobilon Western Chemiluminescent HRP substrate (EMD Millipore, WBKLS0500) was added for 10 min before membranes were exposed to HyBlot CL autoradiography film (Denville, E3018) and developed using a Konica Minolta SRX-101A medical film processor.
Author contributions
The experiments were conceived and designed by J. M. P., B. L. A., and R. J. G. J. M. P., A. N. M., and N. E. F. performed the experiments, and J. M. P. compiled the data. J. M. P. and B. L. A. analyzed the data and wrote and edited the manuscript. R.J.G. provided reagents, technical assistance, and assistance with manuscript preparation and editing.
Supplementary Material
Acknowledgments
We thank Dr. A. L. Kolodkin (Johns Hopkins University) for providing Neuropilin constructs and Dr. K. V. Anderson (Memorial Sloan Kettering Cancer Center) for providing wild-type and Dync2h1lln/lln MEFs. Members of the B. L. A. and R. J. G. laboratories contributed technical assistance, insightful comments, and helpful suggestions. We also thank Drs. K. Sue O'Shea, K. J. Verhey, J. D. Engel, K. F. Barald, S. Barolo, and J. R. Spence for sharing equipment and reagents. Confocal imaging was performed in the Microscopy and Image Analysis Laboratory at the University of Michigan.
This work was supported by the NCI, National Institutes of Health Grant P30CA046592. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S5.
- HH
- Hedgehog
- GLI
- glioma-associated oncogene homolog
- SMO
- Smoothened
- NRP
- Neuropilin
- SUFU
- suppressor of fused
- CD
- cytoplasmic domain
- ECD
- extracellular domain
- SAG
- Smoothened agonist
- TM
- transmembrane domain
- MEF
- mouse embryonic fibroblast
- AcTub
- acetylated tubulin
- NSHH
- N-terminal Sonic Hedgehog.
References
- 1. McMahon A. P., Ingham P. W., and Tabin C. J. (2003) Developmental roles and clinical significance of Hedgehog signaling. Curr. Top. Dev. Biol. 53, 1–114 [DOI] [PubMed] [Google Scholar]
- 2. Briscoe J., and Thérond P. P. (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 [DOI] [PubMed] [Google Scholar]
- 3. Petrova R., and Joyner A. L. (2014) Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 141, 3445–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murdoch J. N., and Copp A. J. (2010) The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. A Clin. Mol. Teratol. 88, 633–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schachter K. A., and Krauss R. S. (2008) Chapter 3: murine models of holoprosencephaly. Curr. Top. Dev. Biol. 84, 139–170 [DOI] [PubMed] [Google Scholar]
- 6. Barakat M. T., Humke E. W., and Scott M. P. (2010) Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 16, 337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Teglund S., and Toftgård R. (2010) Hedgehog beyond medulloblastoma and basal cell carcinoma, Biochim. Biophys. Acta 1805, 181–208 [DOI] [PubMed] [Google Scholar]
- 8. Hui C. C., and Angers S. (2011) Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513–537 [DOI] [PubMed] [Google Scholar]
- 9. Byrne E. F. X., Sircar R., Miller P. S., Hedger G., Luchetti G., Nachtergaele S., Tully M. D., Mydock-McGrane L., Covey D. F., Rambo R. P., Sansom M. S. P., Newstead S., Rohatgi R., and Siebold C. (2016) Structural basis of smoothened regulation by its extracellular domains. Nature 535, 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ogden S. K., Fei D. L., Schilling N. S., Ahmed Y. F., Hwa J., and Robbins D. J. (2008) G protein Gαi functions immediately downstream of smoothened in hedgehog signalling. Nature 456, 967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Riobo N. A., Saucy B., Dilizio C., and Manning D. R. (2006) Activation of heterotrimeric G proteins by smoothened. Proc. Natl. Acad. Sci. U.S.A. 103, 12607–12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luchetti G., Sircar R., Kong J. H., Nachtergaele S., Sagner A., Byrne E. F., Covey D. F., Siebold C., and Rohatgi R. (2016) Cholesterol activates the G-protein coupled receptor smoothened to promote hedgehog signaling. eLife 5, e20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang B., Fallon J. F., and Beachy P. A. (2000) Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 [DOI] [PubMed] [Google Scholar]
- 14. Pan Y., Bai C. B., Joyner A. L., and Wang B. (2006) Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peterson K. A., Nishi Y., Ma W., Vedenko A., Shokri L., Zhang X., McFarlane M., Baizabal J.-M., Junker J. P., van Oudenaarden A., Mikkelsen T., Bernstein B. E., Bailey T. L., Bulyk M. L., Wong W. H., and McMahon A. P. (2012) Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning, Genes Dev. 26, 2802–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vokes S. A., Ji H., McCuine S., Tenzen T., Giles S., Zhong S., Longabaugh W. J., Davidson E. H., Wong W. H., McMahon A. P. (2007) Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development 134, 1977–1989 [DOI] [PubMed] [Google Scholar]
- 17. Vokes S. A., Ji H., Wong W. H., and McMahon A. P. (2008) A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 22, 2651–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. D'Angelo G., Matusek T., Pizette S., and Thérond P. P. (2015) Endocytosis of Hedgehog through dispatched regulates long-range signaling. Dev. Cell 32, 290–303 [DOI] [PubMed] [Google Scholar]
- 19. Creanga A., Glenn T. D., Mann R. K., Saunders A. M., Talbot W. S., and Beachy P. A. (2012) Scube/You activity mediates release of dually lipid-modified Hedgehog signal in soluble form. Genes Dev. 26, 1312–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tukachinsky H., Kuzmickas R. P., Jao C. Y., Liu J., and Salic A. (2012) Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2, 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Willnow T. E., Christ A., and Hammes A. (2012) Endocytic receptor-mediated control of morphogen signaling. Development 139, 4311–4319 [DOI] [PubMed] [Google Scholar]
- 22. Ortmann C., Pickhinke U., Exner S., Ohlig S., Lawrence R., Jboor H., Dreier R., and Grobe K. (2015) Sonic hedgehog processing and release are regulated by glypican heparan sulfate proteoglycans. J. Cell Sci. 128, 4462. [DOI] [PubMed] [Google Scholar]
- 23. Li F., Shi W., Capurro M., and Filmus J. (2011) Glypican-5 stimulates rhabdomyosarcoma cell proliferation by activating Hedgehog signaling. J. Cell Biol. 192, 691–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allen B. L., Song J. Y., Izzi L., Althaus I. W., Kang J. S., Charron F., Krauss R. S., and McMahon A. P. (2011) Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 20, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Izzi L., Lévesque M., Morin S., Laniel D., Wilkes B. C., Mille F., Krauss R. S., McMahon A. P., Allen B. L., and Charron F. (2011) Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 20, 788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holtz A. M., Peterson K. A., Nishi Y., Morin S., Song J. Y., Charron F., McMahon A. P., and Allen B. L. (2013) Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development 140, 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holtz A. M., Griffiths S. C., Davis S. J., Bishop B., Siebold C., and Allen B. L. (2015) Secreted HHIP1 interacts with heparan sulfate and regulates Hedgehog ligand localization and function, J. Cell Biol. 209, 739–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jeong J., and McMahon A. P. (2005) Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development 132, 143–154 [DOI] [PubMed] [Google Scholar]
- 29. Kawasaki T., Kitsukawa T., Bekku Y., Matsuda Y., Sanbo M., Yagi T., and Fujisawa H. (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126, 4895–4902 [DOI] [PubMed] [Google Scholar]
- 30. Gu C., Rodriguez E. R., Reimert D. V., Shu T., Fritzsch B., Richards L. J., Kolodkin A. L., and Ginty D. D. (2003) Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 5, 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gelfand M. V., Hagan N., Tata A., Oh W.-J., Lacoste B., Kang K.-T., Kopycinska J., Bischoff J., Wang J.-H., and Gu C. (2014) Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. eLife 3, e03720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takashima S., Kitakaze M., Asakura M., Asanuma H., Sanada S., Tashiro F., Niwa H., Miyazaki J. J., Hirota S., Kitamura Y., Kitsukawa T., Fujisawa H., Klagsbrun M., and Hori M. (2002) Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 3657–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujisawa H. (2004) Discovery of semaphorin receptors, neuropilin and plexin, and their functions in neural development. J. Neurobiol. 59, 24–33 [DOI] [PubMed] [Google Scholar]
- 34. Giger R. J., Cloutier J. F., Sahay A., Prinjha R. K., Levengood D. V., Moore S. E., Pickering S., Simmons D., Rastan S., Walsh F. S., Kolodkin A. L., Ginty D. D., and Geppert M. (2000) Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron 25, 29–41 [DOI] [PubMed] [Google Scholar]
- 35. Hillman R. T., Feng B. Y., Ni J., Woo W.-M., Milenkovic L., Hayden Gephart M. G., Teruel M. N., Oro A. E., Chen J. K., and Scott M. P. (2011) Neuropilins are positive regulators of Hedgehog signal transduction. Genes Dev. 25, 2333–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ge X., Milenkovic L., Suyama K., Hartl T., Purzner T., Winans A., Meyer T., and Scott M. P. (2015) Phosphodiesterase 4D acts downstream of Neuropilin to control Hedgehog signal transduction and the growth of medulloblastoma. eLife 4, e07068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gomez C., Burt-Pichat B., Mallein-Gerin F., Merle B., Delmas P. D., Skerry T. M., Vico L., Malaval L., and Chenu C. (2005) Expression of Semaphorin-3A and its receptors in endochondral ossification: potential role in skeletal development and innervation. Dev. Dyn. 234, 393–403 [DOI] [PubMed] [Google Scholar]
- 38. Mauti O., Sadhu R., Gemayel J., Gesemann M., and Stoeckli E. T. (2006) Expression patterns of plexins and neuropilins are consistent with cooperative and separate functions during neural development. BMC Dev. Biol. 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hayden Gephart M. G., Su Y. S., Bandara S., Tsai F. C., Hong J., Conley N., Rayburn H., Milenkovic L., Meyer T., and Scott M. P. (2013) Neuropilin-2 contributes to tumorigenicity in a mouse model of Hedgehog pathway medulloblastoma. J. Neurooncol. 115, 161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Snuderl M., Batista A., Kirkpatrick N. D., Ruiz de Almodovar C., Riedemann L., Walsh E. C., Anolik R., Huang Y., Martin J. D., Kamoun W., Knevels E., Schmidt T., Farrar C. T., Vakoc B. J., Mohan N., et al. (2013) Targeting placental growth factor/Neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pan Q., Chanthery Y., Liang W.-C., Stawicki S., Mak J., Rathore N., Tong R. K., Kowalski J., Yee S. F., Pacheco G., Ross S., Cheng Z., Le Couter J., Plowman G., Peale F., et al. (2007) Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 11, 53–67 [DOI] [PubMed] [Google Scholar]
- 42. Goel H. L., Pursell B., Chang C., Shaw L. M., Mao J., Simin K., Kumar P., Vander Kooi C. W., Shultz L. D., Greiner D. L., Norum J. H., Toftgard R., Kuperwasser C., and Mercurio A. M. (2013) Gli1 regulates a novel neuropilin–2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol. Med. 5, 488–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nybakken K., Vokes S. A., Lin T.-Y., McMahon A. P., and Perrimon N. (2005) A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signalling pathway. Nat. Genet. 37, 1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen J. K., Taipale J., Young K. E., Maiti T., and Beachy P. A. (2002) Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. U.S.A. 99, 14071–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xie J., Murone M., Luoh S. M., Ryan A., Gu Q., Zhang C., Bonifas J. M., Lam C. W., Hynes M., Goddard A., Rosenthal A., Epstein E. H. Jr., and de Sauvage F. J. (1998) Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92 [DOI] [PubMed] [Google Scholar]
- 46. Roessler E., Ermilov A. N., Grange D. K., Wang A., Grachtchouk M., Dlugosz A. A., and Muenke M. (2005) A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 14, 2181–2188 [DOI] [PubMed] [Google Scholar]
- 47. Roth L., Nasarre C., Dirrig-Grosch S., Aunis D., Crémel G., Hubert P., and Bagnard D. (2008) Transmembrane domain interactions control biological functions of neuropilin-1. Mol. Biol. Cell 19, 646–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beavo J. A., and Brunton L. L. (2002) Cyclic nucleotide research: still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3, 710–718 [DOI] [PubMed] [Google Scholar]
- 49. Niewiadomski P., Kong J. H., Ahrends R., Ma Y., Humke E. W., Khan S., Teruel M. N., Novitch B. G., and Rohatgi R. (2014) Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 6, 168–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cai H., and Reed R. R. (1999) Cloning and characterization of neuropilin-1-interacting protein: a PSD-95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J. Neurosci. 19, 6519–6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kee H. L., Dishinger J. F., Blasius T. L., Liu C.-J., Margolis B., and Verhey K. J. (2012) A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol. 14, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goetz S. C., and Anderson K. V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wong S. Y., Seol A. D., So P.-L., Ermilov A. N., Bichakjian C. K., Epstein E. H. Jr., Dlugosz A. A., and Reiter J. F. (2009) Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 15, 1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ocbina P. J., Eggenschwiler J. T., Moskowitz I., and Anderson K. V. (2011) Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet. 43, 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., and Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 56. Kang J.-S., Mulieri P. J., Hu Y., Taliana L., and Krauss R. S. (2002) BOC, an Ig superfamily member, associates with CDO to positively regulate myogenic differentiation. EMBO J. 21, 114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tenzen T., Allen B. L., Cole F., Kang J. S., Krauss R. S., and McMahon A. P. (2006) The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev. Cell 10, 647–656 [DOI] [PubMed] [Google Scholar]
- 58. Hsu Y.-C., Li L., and Fuchs E. (2014) Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell 157, 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hooper J. E., and Scott M. P. (2005) Communicating with hedgehogs. Nat. Rev. Mol. Cell Biol. 6, 306–317 [DOI] [PubMed] [Google Scholar]
- 60. Han Y.-G., Spassky N., Romaguera-Ros M., Garcia-Verdugo J.-M., Aguilar A., Schneider-Maunoury S., and Alvarez-Buylla A. (2008) Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 11, 277–284 [DOI] [PubMed] [Google Scholar]
- 61. Mille F., Tamayo-Orrego L., Lévesque M., Remke M., Korshunov A., Cardin J., Bouchard N., Izzi L., Kool M., Northcott P. A., Taylor M. D., Pfister S. M., and Charron F. (2014) The Shh receptor Boc promotes progression of early medulloblastoma to advanced tumors. Dev. Cell 31, 34–47 [DOI] [PubMed] [Google Scholar]
- 62. Lee E. Y., Ji H., Ouyang Z., Zhou B., Ma W., Vokes S. A., McMahon A. P., Wong W. H., and Scott M. P. (2010) Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 9736–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mathew E., Zhang Y., Holtz A. M., Kane K. T., Song J. Y., Allen B. L., and Pasca di Magliano M. (2014) Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 9, 484–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang W., Kang J.-S., Cole F., Yi M.-J., and Krauss R. S. (2006) Cdo functions at multiple points in the Sonic Hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephaly. Dev. Cell 10, 657–665 [DOI] [PubMed] [Google Scholar]
- 65. Milenkovic L., Goodrich L. V., Higgins K. M., and Scott M. P. (1999) Mouse patched1 controls body size determination and limb patterning. Development 126, 4431–4440 [DOI] [PubMed] [Google Scholar]
- 66. Mo R., Freer A. M., Zinyk D. L., Crackower M. A., Michaud J., Heng H. H., Chik K. W., Shi X. M., Tsui L. C., Cheng S. H., Joyner A. L., and Hui C. (1997) Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123 [DOI] [PubMed] [Google Scholar]
- 67. Prud'homme G. J., and Glinka Y. (2012) Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity, Oncotarget 3, 921–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Christ A., Christa A., Kur E., Lioubinski O., Bachmann S., Willnow T. E., and Hammes A. (2012) LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev. Cell 22, 268–278 [DOI] [PubMed] [Google Scholar]
- 69. Kwong L., Bijlsma M. F., and Roelink H. (2014) Shh-mediated degradation of Hhip allows cell autonomous and non-cell autonomous Shh signalling. Nat. Commun. 5, 4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Han Y., Shi Q., and Jiang J. (2015) Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. U.S.A. 112, 6383–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Khaled M., Larribere L., Bille K., Aberdam E., Ortonne J.-P., Ballotti R., and Bertolotto C. (2002) Glycogen synthase kinase 3β is activated by cAMP and plays an active role in the regulation of melanogenesis. J. Biol. Chem. 277, 33690–33697 [DOI] [PubMed] [Google Scholar]
- 72. Neufeld G., and Kessler O. (2008) The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 8, 632–645 [DOI] [PubMed] [Google Scholar]
- 73. Gu C., Limberg B. J., Whitaker G. B., Perman B., Leahy D. J., Rosenbaum J. S., Ginty D. D., and Kolodkin A. L. (2002) Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J. Biol. Chem. 277, 18069–18076 [DOI] [PubMed] [Google Scholar]
- 74. Song J. Y., Holtz A. M., Pinskey J. M., and Allen B. L. (2015) Distinct structural requirements for CDON and BOC in the promotion of Hedgehog signaling. Dev. Biol. 402, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. De Vries L., Lou X., Zhao G., Zheng B., and Farquhar M. G. (1998) GIPC, a PDZ domain containing protein, interacts specifically with the C terminus of RGS-GAIP. Proc. Natl. Acad. Sci. U.S.A. 95, 12340–12345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gao Y., Li M., Chen W., and Simons M. (2000) Synectin, syndecan-4 cytoplasmic domain binding PDZ protein, inhibits cell migration. J. Cell. Physiol. 184, 373–379 [DOI] [PubMed] [Google Scholar]
- 77. Prahst C., Héroult M., Lanahan A. A., Uziel N., Kessler O., Shraga-Heled N., Simons M., Neufeld G., and Augustin H. G. (2008) Neuropilin-1-VEGFR-2 complexing requires the PDZ binding domain of neuropilin-1. J. Biol. Chem. 283, 25110–25114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fantin A., Schwarz Q., Davidson K., Normando E. M., Denti L., and Ruhrberg C. (2011) The cytoplasmic domain of neuropilin 1 is dispensable for angiogenesis, but promotes the spatial separation of retinal arteries and veins. Development 138, 4185–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Seerapu H. R., Borthakur S., Kong N., Agrawal S., Drazba J., Vasanji A., Fantin A., Ruhrberg C., Buck M., and Horowitz A. (2013) The cytoplasmic domain of neuropilin-1 regulates focal adhesion turnover. FEBS Lett. 587, 3392–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sawma P., Roth L., Blanchard C., Bagnard D., Crémel G., Bouveret E., Duneau J.-P., Sturgis J. N., and Hubert P. (2014) Evidence for new homotypic and heterotypic interactions between transmembrane helices of proteins involved in receptor tyrosine kinase and neuropilin signaling. J. Mol. Biol. 426, 4099–4111 [DOI] [PubMed] [Google Scholar]
- 81. Muhl L., Folestad E. B., Gladh H., Wang Y., Moessinger C., Jakobsson L., and Eriksson U. (2017) Neuropilin 1 binds platelet-derived growth factor (PDGF)-D and is a co-receptor in PDGF-D/PDGF receptor β signaling. J. Cell Sci. 130, 1365–1378 [DOI] [PubMed] [Google Scholar]
- 82. Cackowski F. C., Xu L., Hu B., and Cheng S. Y. (2004) Identification of two novel alternatively spliced Neuropilin-1 isoforms. Genomics 84, 82–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rossignol M., Gagnon M. L., and Klagsbrun M. (2000) Genomic organization of human Neuropilin-1 and Neuropilin-2 genes: identification and distribution of splice variants and soluble isoforms. Genomics 70, 211–222 [DOI] [PubMed] [Google Scholar]
- 84. Gagnon M. L., Bielenberg D. R., Gechtman Z., Miao H. Q., Takashima S., Soker S., and Klagsbrun M. (2000) Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: in vivo expression and antitumor activity. Proc. Natl. Acad. Sci. U.S.A. 97, 2573–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pang H.-B., Braun G. B., Friman T., Aza-Blanc P., Ruidiaz M. E., Sugahara K. N., Teesalu T., and Ruoslahti E. (2014) An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 5, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lanahan A., Zhang X., Fantin A., Zhuang Z., Rivera-Molina F., Speichinger K., Prahst C., Zhang J., Wang Y., Davis G., Toomre D., Ruhrberg C., and Simons M. (2013) The Neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 25, 156–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nechipurenko I. V., Doroquez D. B., and Sengupta P. (2013) Primary cilia and dendritic spines: different but similar signaling compartments. Mol. Cell 36, 288–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hatanaka Y., Matsumoto T., Yanagawa Y., Fujisawa H., Murakami F., and Masu M. (2009) Distinct roles of neuropilin 1 signaling for radial and tangential extension of callosal axons. J. Comp. Neurol. 514, 215–225 [DOI] [PubMed] [Google Scholar]
- 89. Nauli S. M., Jin X., and Hierck B. P. (2011) The mechanosensory role of primary cilia in vascular hypertension. Int. J. Vasc. Med. 2011, 376281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Megason S. G., and McMahon A. P. (2002) A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development 129, 2087–2098 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.