

Abstract
ADP-ribosyltransferases transfer the ADP-ribose moiety of βNAD+ to an acceptor molecule, usually a protein that modulates the function of the acceptor. Pierisin-1 is an ADP-ribosyltransferase from the cabbage butterfly Pieris rapae and is composed of N-terminal catalytic and C-terminal ricin B–like domains. Curiously, it ADP-ribosylates the DNA duplex, resulting in apoptosis of various cancer cells, which has raised interest in pierisin-1 as an anti-cancer agent. However, both the structure and the mechanism of DNA ADP-ribosylation are unclear. Here, we report the crystal structures of the N-terminal catalytic domain of pierisin-1, its complex with βNAD+, and the catalytic domain with the linker connecting it to the ricin B–like domains. We found that the catalytic domain possesses a defined, positively charged region on the molecular surface but that its overall structure is otherwise similar to those of protein-targeting ADP-ribosyltransferases. Electrophoretic mobility shift assays and site-directed mutagenesis indicated that pierisin-1 binds double-stranded but not single-stranded DNA and that Lys122, Lys123, and Lys124, which are found in a loop, and Arg181 and Arg187, located in a basic cleft near the loop, are required for DNA binding. Furthermore, the structure of the catalytic domain with the linker revealed an autoinhibitory mechanism in which the linker occupies and blocks both the βNAD+- and DNA-binding sites, suggesting that proteolytic cleavage to remove the linker is necessary for enzyme catalysis. Our study provides a structural basis for the DNA-acceptor specificity of pierisin-1 and reveals that a self-regulatory mechanism is required for its activity.
Keywords: ADP-ribosylation, apoptosis, crystal structure, DNA-protein interaction, toxin, X-ray crystallography, ADP-ribosyltransferase, DNA ADP-ribosylation, autoinhibition, pierisin
Introduction
ADP-ribosyltransferases that target proteins as acceptor molecules are widely distributed in bacteria and eukaryotes (1–3). Hitherto known target amino acid residues are lysine, arginine, glutamate, aspartate, cysteine, diphthamide, phosphorylated serine, and asparagine (3). ADP-ribosyltransferases are classified into poly- and mono-ADP-ribosyltransferases. Poly-ADP-ribosyltransferases are mainly found in multicellular eukaryotes and are involved in genome stability. Mono-ADP-ribosyltransferases are distributed throughout eukaryotes, prokaryotes, and bacteriophages. The bacterial mono-ADP-ribosyltransferases form a large family of mono-ADP-ribosylating toxins, including cholera toxin, diphtheria toxin, and pertussis toxin, which target specific cellular proteins to impair the function of the protein (1, 2). 3D structures of various mono ADP-ribosyltransferases and their complexes with βNAD+ have been determined by X-ray crystallography and reveal that the core structures, key residues for catalysis, and βNAD+ binding motifs are conserved. Some of these core structures and key residues are also conserved in poly-ADP-ribosyltransferases (3). Mutation analysis based on the 3D structure has also been used to clarify important residues for target recognition. The catalytic mechanisms of exotoxin A and ι toxin have also been proposed based on their crystal structures in complex with target proteins (4–6).
Pierisin-1 is an ADP-ribosyltransferase produced by the last stage larvae and pupae of the cabbage butterfly Pieris rapae and induces apoptosis of various types of cancer cell lines, such as HeLa and TMK-1 cells (7–10). The enzyme is composed of 850 amino acid residues and consists of an N-terminal catalytic domain (amino acid residues 1–233) and a C-terminal ricin B–like HA33 domain (residues 267–850) (Fig. 1A) (11). The N-terminal domain is homologous with ADP-ribosyltransferases of various bacterial toxins and transfers the ADP-ribose moiety of βNAD+ to an acceptor molecule to modulate the molecular function. The C-terminal ricin B–like domain has binding activity toward cell surface glycosphingolipid receptors, such as globotriaosylceramide and globotetraosylceramide (12, 13). Binding of the C-terminal domain to the cell surface receptors triggers the internalization of pierisin-1, where proteolytic cleavage between the N- and C- terminal domains induces apoptosis of the cells (12). Unlike the protein ADP-ribosyltransferases mentioned above, pierisin-1 is a unique mono-ADP-ribosyltransferase that targets DNA as the acceptor molecule, with the N-terminal catalytic domain transferring the ADP-ribose moiety to the N2 amino group of guanine (14). Pierisin-1 has 30% sequence identity with mosquitocidal toxin (MTX)2 of Bacillus sphaericus, which also belongs to the ADP-ribosylating toxin family and has a similar domain structure to pierisin-1 (Fig. 1A) (11, 15, 16). Although the in vivo target of MTX has not been identified, it is likely to be a protein (17), and thus despite their similarity, the acceptor molecules of MTX and pierisin-1 are different. To date, six pierisins (pierisin-1, -1b, and -2–5) and CARP-1 have been found in the butterfly Pieridae family and in a clam, respectively, and have been identified as DNA-targeting ADP-ribosyltransferases (18–24). To understand the acceptor recognition and catalytic mechanisms of pierisin-1, we determined the crystal structures of the N-terminal catalytic domain of the enzyme with and without a linker (residues 234–267) (pierisin(1–267) and pierisin(1–233)) and βNAD+-bound pierisin(1–233) and demonstrate that pierisin-1 has a binding activity toward dsDNA, not ssDNA. We also identified the amino acid residues important for dsDNA binding by mutation analysis using EMSA. We demonstrate that the linker between the N- and C-terminal domains controls catalysis by regulating how βNAD+ and DNA access their binding sites.
Figure 1.

Structure of pierisin-1. A, the domain structure of pierisin-1 and MTX. B, the structures of pierisin(1–233)E165Q (form 1) (left), βNAD+-bound pierisin (1–233)E165Q (form 1) (center), and pierisin(1–267)E165Q (right). The linker of pierisin (1–267)E165Q is colored orange, with the disordered region shown as a dashed line. The βNAD+ molecule is shown as a stick model. α and β are α helix and β strand, respectively. C, close-up view of important motifs for βNAD+ binding and catalysis in pierisin(1–233)E165Q. The (Q/E)XE motif (Gln163 and Glu165), STS motif (Ser114, Thr115, and Thr116), and the conserved Arg residue (Arg70) are shown as stick models colored red, magenta, and blue, respectively. D, conformations of the PN loop in an asymmetric unit of pierisin(1–233)E165Q form 1 and 2 crystals. Each conformation is differently colored. The disordered region of the PN loop is depicted as a dashed line.
Results
Overall structure
To understand the DNA-binding mechanism, the catalytic domain of pierisin-1, pierisin(1–233), and the catalytic domain with a linker between the catalytic and Ricin B–like domains, pierisin(1–267), were produced in Escherichia coli and subjected to X-ray crystallographic analyses. To reduce the cytotoxicity of pierisin-1, Glu165, a catalytic residue, was replaced with glutamine. We determined the crystal structures of pierisin(1–233)E165Q and pierisin(1–233)E165Q in complex with βNAD+ at 1.9 and 2.1 Å resolution, respectively (form 1 crystal) (Table 1 and Fig. 1B (left and center)). We also determined those structures in another space group at 1.8 and 1.9 Å (form 2 crystal) (Table 1). The structures in two forms were essentially the same (supplemental Fig. S1), and the electron density around βNAD+ is clearer for form 1 than for form 2 (supplemental Fig. S2). Thus, we treat the form 1 structure as the pierisin(1–233)E165Q. Pierisin(1–233)E165Q possesses two β-sheets (one consisting of β4, β6, and β7 and the other consisting of β3, β5, β8, β9, and β10) surrounded by six α-helices (Fig. 1B). β3 and β4 are twisted relative to each other by 90°. We define β3, β5, β4, and β7 across the two β-sheets as a core structure.
Table 1.
Data collection and refinement statistics
Values in parentheses are for highest resolution shell. asu, asymmetric unit; RMSD, root mean square deviation.
| Crystal form | Pierisin(1–233)E165Q (form 1) | Pierisin(1–233)E165Q + βNAD+ (form 1) | Pierisin(1–233)E165Q (form 2) | Pierisin(1–233)E165Q + βNAD+ (form 2) | Pierisin(1–267)E165Q |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P3221 | P3221 | P43212 | P43212 | P21 |
| Cell dimensions | |||||
| a, b, c (Å) | 134.7, 134.7, 73.1 | 134.7, 134.7, 73.1 | 93.4, 93.4, 120.6 | 93.4, 93.4, 120.6 | 52.1, 110.5, 133.6 |
| α, β, γ ° | 90, 90, 120 | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 | 90, 91.3, 90 |
| No. of protein molecules/asu | 2 | 2 | 2 | 2 | 4 |
| X-ray source | PF BL17A | PF BL17A | PF BL5A | PF BL17A | PF BL5A |
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| Resolution (Å) | 49.54 to 1.90 (1.94 to 1.90) | 45.67 to 2.10 (2.16 to 2.10) | 50.00 to 1.80 (1.86 to 1.80) | 43.52 to 1.90 (1.94 to 1.90) | 30.00 to 1.80 (1.83 to 1.80) |
| No. of observations | 726,225 | 479,548 | 223,980 | 552,439 | 496,050 |
| No. of unique reflections | 60,377 | 45,134 | 47,586 | 42,294 | 132,652 |
| Redundancy | 12.0 (11.7) | 10.6 (9.0) | 4.7 (2.2) | 13.1 (10.8) | 3.7 (2.2) |
| Rmerge (%)a | 6.3 (35.6) | 13.1 (35.0) | 7.7 (40.9) | 8.7 (71.2) | 7.1 (53.0) |
| Rpim (%)b | 1.9 (11.3) | 4.1 (12.4) | 2.4 (20.2) | 2.5 (27.2) | 4.5 (41.8) |
| CC½c | 0.999 (0.996) | 0.995 (0.958) | 0.998 (0.864) | 0.999 (0.826) | 0.996 (0.664) |
| Completeness | 99.9 | 99.6 | 95.0 | 98.9 | 94.9 |
| 〈I/σ(I)〉 | 26.5 (6.8) | 13.5 (5.4) | 16.7 (1.7) | 20.9 (1.9) | 9.6 (1.2) |
| Refinement | |||||
| Rworkd/Rfreee | 20.3/21.9 | 24.3/25.9 | 23.1/26.0 | 26.3/28.0 | 22.4/24.1 |
| No. of non-H atoms | 4053 | 3910 | 3862 | 4019 | 8473 |
| Protein/solvent atoms | 3655/397 | 3639/271 | 3602/260 | 3641/378 | 7885/588 |
| Average B-factor (Å2) | |||||
| Overall/βNAD+ | 24.4/—f | 37.5/40.9 | 32.1/— | 31.4/52.5 | 31.4/— |
| RMSD from ideality | |||||
| Bond lengths (Å)/bond angles (degrees) | 0.006/1.147 | 0.007/1.031 | 0.006/1.155 | 0.007/1.028 | 0.006/1.122 |
| Ramachandran plot | |||||
| Favored (%)/outlier (%) | 99.8/0.0 | 99.1/0.0 | 99.3/0.0 | 99.1/0.0 | 98.0/0.1 |
a Rmerge = ΣhΣi|Ii(h) − 〈I(h)〉|/ΣhΣiIi(h), where Ii(h) is the ith measurement.
b Rpim = Σh(1/(N(h) − 1))½ Σi|Ii(h) − 〈I(h)〉|/ΣhΣiIi(h).
c CC½ = Σ(x − 〈x〉)(y − 〈y〉)/(Σ(x − 〈x〉)2Σ(y − 〈y〉)2)½.
d Rwork is the crystallographic R-factor (Rcryst) for the working set used for the refinement. Rcryst = Σh‖Fobs(h)| − |Fcalc(h)‖/Σh|Fobs(h)|, where Fobs(h) and Fcalc(h) are the observed and calculated structure factors.
e Rfree is the Rcryst calculated for the test set consisting of 5% of reflections excluded from the refinement.
f —, no data.
Homology searching using the Dali server (25) demonstrates that pierisin(1–233)E165Q is similar to mono-ADP-ribosyltransferases, such as MTX (PDB code 2VSE), cholera toxin (PDB code 2A5D), heat-labile enterotoxin IIB (PDB code 1TII), and pertussis toxin (PDB code 1BCP) with Z scores of 25.7, 11.5, 10.8, and 9.3, respectively, and less similar to poly(ADP-ribose) polymerase 1 (PARP1; PDB code 4HHZ) with a Z score of 4.0. From a large number of biochemical and crystallographic studies of mono- and poly-ADP-ribosyltransferases, the core structure, important motifs, and key residues have been identified (2, 3, 26). Of these, the core structure is conserved among all ADP-ribosyltransferases. Particularly in the R-S-E type of ADP-ribosyltransferases, an Arg residue, a Ser-Thr-Ser (STS) motif, and a Gln- or Glu-X-Glu ((Q/E)XE) motif play crucial roles in βNAD+ binding and catalysis and are conserved (3, 26). As shown in Fig. 1C, an Arg residue (Arg70), STS motif (Ser114-Thr115-Thr116), and (Q/E)XE motif (Gln163 and Glu165) are located on β3, β4, and a loop, termed the ADP-ribosylating turn-turn loop (ARTT loop) (27), which precedes β7, respectively. The crystal structure of the catalytic domain with a linker region (pierisin(1–267)E165Q) was also determined (Table 1 and Fig. 1B (right)) at 1.8 Å resolution. The crystal of pierisin(1–267)E165Q and the form 1 and 2 crystals of pierisin(1–233)E165Q contain four and two molecules in the asymmetric unit, respectively. The structures in each asymmetric unit were essentially the same, with a root mean square deviation of 1.1 Å, with the exception of a portion of the phosphate–nicotinamide loop (PN loop; Lys117–Gly135) (Fig. 1D and supplemental Fig. S1). Moreover, no substantial structural difference was observed between pierisin(1–233)E165Q, βNAD+-bound pierisin(1–233)E165Q, and pierisin (1–267)E165Q, indicating that neither βNAD+ binding nor the presence of the linker affects the structure of the catalytic domain.
Recognition of βNAD+
The binding of βNAD+ induced no significant change around the βNAD+ binding pocket as described above. The interaction between pierisin(1–233)E165Q and βNAD+ is shown in Fig. 2. The nicotinamide moiety of βNAD+ is buried deep in the βNAD+ binding pocket, which is formed by the STS motif (residues 114–116), the main chain of Trp71, and the side chains of Arg70, Trp127, Gln163, and Gln165. In the absence of βNAD+, this pocket was occupied by water molecules and an ethylene glycol molecule used for cryoprotection in X-ray diffraction experiments at 100 K (supplemental Fig. S3A). The carboxyamide group of the nicotinamide moiety is recognized by backbone carbonyl and amide groups of Trp71. The 2′-OH group of the nicotinamide ribose moiety is bound to the side chain of Gln165, which replaces the catalytic Glu165, through a hydrogen bond. The two phosphate groups are bound to the guanidinium group of Arg70 by ionic interactions and also to the OH group of Ser114 in the STS motif by hydrogen bonds. The 2′-OH group of adenine ribose contacts the carboxyl group of Asp72 by van der Waals interactions. The adenine moiety of βNAD+ interacts with the guanidinium group of Arg74 via π-π stacking, and the NH2 group of the adenine moiety (AN6) is bound to the main chain of Val85 by a hydrogen bond.
Figure 2.
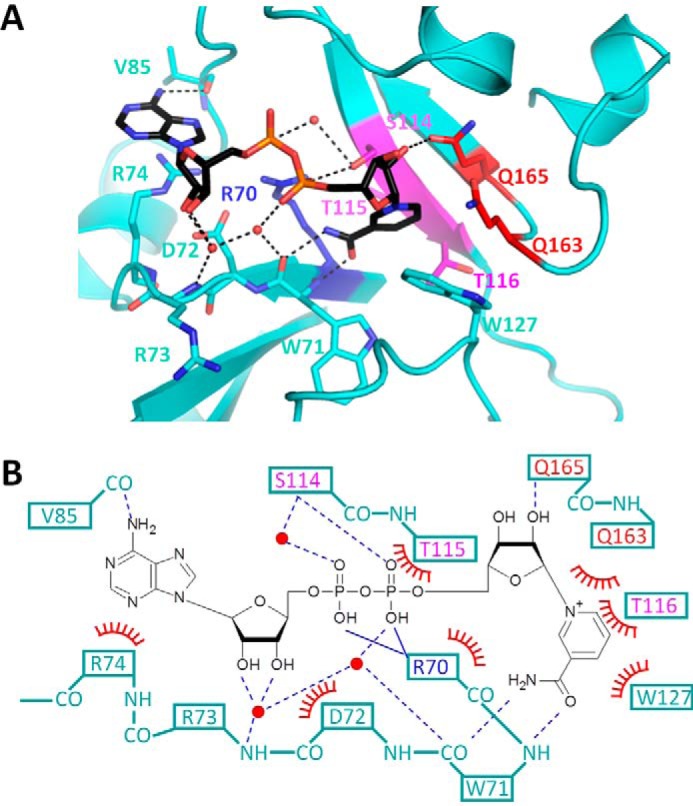
Interaction between catalytic domain and βNAD+ molecule. A, close-up views of the βNAD+ binding pockets in βNAD+-bound pierisin(1–233)E165Q. The catalytic domain is colored cyan. The (Q/E)XE motif, STS motif, and conserved Arg residue are colored in the same manner as in Fig. 1C. Water molecules are represented as red spheres. Hydrogen bonds are represented by dashed black lines. βNAD+ molecules are depicted as stick models. B, schematic drawing of the interaction between βNAD+ and pierisin(1–233)E165Q. Solid and dashed blue lines represent ionic interactions and hydrogen bonds, respectively. Red arcs with spokes represent van der Waals interactions or π-π interactions.
Many interactions observed between pierisin-1 and βNAD+ are conserved in cholera toxin (28) (supplemental Fig. S3, B and C). However, the hydrogen bond network mediated through water molecules is different in pierisin-1 and cholera toxin.
Putative DNA-binding region
We studied the interaction between pierisin-1 and DNA by EMSA. EMSA with pierisin(1–233)E165Q and dsDNA shows a clearly shifted band, but this is not the case with ssDNA (Fig. 3A), indicating that the binding affinity for ssDNA is much lower than that for dsDNA. This result is compatible with the fact that pierisin-1 and -2 prefer dsDNA to ssDNA as a substrate (14, 20, 29). Most surprisingly, pierisin(1–267)E165Q has no binding activity toward dsDNA with or without βNAD+, whereas pierisin(1–233)E165Q does bind (Fig. 3B). This indicates that the linker region (residues 234–267) inhibits dsDNA binding.
Figure 3.

Interaction between pierisin-1 and DNA. A, EMSA of 15-bp dsDNA or 15-nt ssDNA for pierisin(1–233)E165Q. 0.155 μm fluorescence dye (6-FAM)-labeled DNA was mixed to interact with varying concentrations (0, 0.155, 0.31, 0.62, 1.24, and 2.48 μm) of pierisin(1–233)E165Q. The bands of DNA on the gel were detected by fluorescence of 6-FAM. B, EMSA of 14-bp dsDNA for pierisin(1–233)E165Q or pierisin(1–267)E165Q in the presence or absence of βNAD+. 6.67 μm dsDNA was mixed to interact with 20 μm of pierisin(1–233)E165Q in the presence or absence of 4 mm βNAD+. The bands of DNA on the gel were detected by ethidium bromide staining.
To identify the dsDNA-binding region, a series of pierisin(1–233)E165Q mutants were prepared and then subjected to EMSA to test dsDNA-binding activity. Probable candidates for mutation are positively charged residues on the molecular surface. The electrostatic surface potential of pierisin(1–233)E165Q shows a characteristic positively charged surface suitable for dsDNA binding (Fig. 4A). Another candidate is the ARTT loop that has been identified as a substrate binding site for a protein-targeting mono-ADP-ribosyltransferases (30). Likewise, an area around the βNAD+ binding pocket is also a candidate dsDNA-binding site. These candidate amino acids are mapped onto Fig. 4B. Lys117, Arg120, Lys122, Lys123, and Lys124 are positioned in the PN loop, and Arg181 and Arg187 are components of a basic surface identified by electrostatic surface potential calculation. Trp160 is located in the ARTT loop. Arg73, His108, Trp127, Arg130, and Arg134 are positioned around the βNAD+ binding pocket, with Arg73, Arg130, and Arg134 forming a basic knob. SDS-PAGE of these mutant preparations gave single bands, indicating that the preparations are homogeneous (supplemental Fig. S4). The results of the mutagenesis demonstrated that DNA-binding activity was slightly reduced by the mutations R120S, W127A, and R187A but not significantly reduced by R73A, H108A, K117A, R130A, R134A, and W160A (Fig. 4C). A single mutation of R181A considerably reduced the DNA-binding activity, and double mutagenesis of R181A and R187A completely abolished the binding activity. Furthermore, triple mutation of K122N, K123N, and K124N also completely abolished the DNA-binding activity (Fig. 4C). However, among single mutations of K122A, K123A, and K124A, K122A and K124A significantly reduced the DNA binding activity, whereas K123A did not measurably affect it (Fig. 4C, bottom). In addition, these three Lys residues in the loop are conserved among the group of all six pierisin members as described below (supplemental Fig. S5), suggesting a contribution of Lys123 to the DNA binding. Taken together, these results indicated that the two residues Lys122 and Lys124, and probably Lys123 on the PN loop and Arg181 and Arg187 on a basic cleft near the PN loop, are required for dsDNA binding. Thus, pierisin-1 binds dsDNA via electrostatic interactions. This is also supported by a gel filtration chromatography analysis of the pierisin-1 interacting with dsDNA (supplemental Fig. S6). Under high-salt conditions, pierisin(1–233)E165Q and dsDNA elute separately, and an elution peak for a complex between pierisin(1–233)E165Q and dsDNA is not observed. In contrast, in low-salt conditions, pierisin(1–233)E165Q and dsDNA associate and elute as a single complex.
Figure 4.

Identification of dsDNA-binding region. A, electrostatic potentials depicted on the surface structures. Positive and negative potentials are colored blue and red, respectively. The electrostatic potential of MTX without a linker region is also shown. The 15-bp dsDNA shown as green lines can be modeled onto the surface of the pierisin-1 catalytic domain to fit the basic cleft, but not onto MTX. B, mutation sites mapped onto the ribbon diagram (left) and the surface structure of pierisin(1–233)E165Q (center and right). DNA binding of mutants was compared with that of (1–233)E165Q as a control. The amino acid residues essential for maintaining full binding affinity are colored blue, and the residues exhibiting weak or no effect on binding are gray. The ribbon diagram (left) is drawn in the same orientation for A. C, interaction between mutants of pierisin(1–233)E165Q and 201-bp dsDNA. 10 nm 201-bp dsDNA was mixed to interact with varying concentrations (0, 25, 50, 100, 200, and 400 nm) of pierisin(1–233) and subjected to EMSA. DNA on the gels was located by SYBR Gold staining. E165Q was taken as a control. All other mutants indicated above the gels were derivatives of E165Q. The mutant K122–124N represents the triple mutation of Lys122, Lys123, and Lys124 to Asn. R181/187A represents the double mutant R181A and R187A.
Mutational analysis of DNA binding of pierisin(1–233)E165Q and its derivatives (DNA-binding mutants) revealed the amino acid residues responsible for DNA binding. However, what effect they have on the DNA ADP-ribosylating activity was not examined because they lack the DNA ADP-ribosylating activity. A useful plasmid containing a wild-type pierisin-1 (residue Glu165) cDNA to express it in E. coli cells has not been established because of the instability of its toxic gene during a cloning procedure and other unknown factors.
Autoinhibition by the linker region
The linker (residues 234–267) is positioned between the catalytic and ricin B–like domains and masks the basic cleft of the catalytic domain (Fig. 4A, center). The segment 245–256 of the linker region interacts with the PN loop and induces a β strand β4′ to form a β-sheet (β4′ and β11). The PN loop, which has a flexible nature in the structure of pierisin(1–233)E165Q, was stabilized by this interaction (Figs. 1B (right) and 5 (A and B)). The side chain of Asp252 in the linker interacts with the side chains of Arg67 and Gln139 in the catalytic domain through hydrogen bonds. The main-chain carbonyl group of Phe254 and the side chain of Asp256 in the linker interact with the side chain of Arg181 in the catalytic domain through hydrogen bonds. In addition to these interactions, a hydrogen bond network mediated by a number of water molecules was observed (Fig. 5, A and B). Thus, the segment 245–256 masks the putative DNA-binding region and inhibits DNA binding.
Figure 5.

Interaction between catalytic domain and linker region. A, close-up view of the segment 245–256 of pierisin-1. The linker is represented as an orange stick. The catalytic domain, water molecules, and hydrogen bonds are shown as in the same manner as in Fig. 2A. B, schematic drawing of interactions between the catalytic domain and the segment 245–256 of pierisin-1. C and E, close-up view of the segment 239–244 of pierisin-1 (C) and 271–278 of MTX (E) (PDB code 2VSE) around the βNAD+ binding pocket. The catalytic domain and linker region of MTX are colored gray and light orange, respectively. The catalytic domains are drawn in the same orientation for Fig. 2A. The disordered region of the linker is depicted as an orange or light orange dashed line. The (Q/E)XE motif, STS motif, and conserved Arg residue are colored in the same manner as in Figs. 1C and 2A. D and F, schematic drawing of interactions between the βNAD+ binding pocket and segment 239–244 of pierisin-1 (B) and between the βNAD+ binding pocket and segment 271–278 of MTX (C). B, D, and F are depicted in the same manner as in Fig. 2B.
The linker region (residues 234–267) masks not only the putative DNA-binding region but also the βNAD+ binding pocket. The βNAD+ binding pocket was masked by a segment (residues 239–244) of the linker in pierisin(1–267)E165Q (Fig. 5C). The crystal structure of pierisin(1–267)E165Q clearly shows that the segment is stabilized mainly by van der Waals interactions (Fig. 5, C and D). This segment is also observed in MTX in a region (residues 273–278) between the catalytic and ricin B–like domains and masks the βNAD+ binding pocket (Fig. 5, E and F) (15, 31). However, the amino acid sequence of the segment is not conserved between pierisin-1 and MTX. Indeed, the interactions of the segment with pierisin-1 in the binding pocket are significantly different from those in MTX. In fact, Asp275 of MTX makes hydrogen bonds with the guanidinium group of Arg100 and hydroxyl group of Ser162. Asp275, Arg100, and Ser162, which correspond to Leu241, Arg73, and Asn131 in pierisin-1, are in contact with each other by van der Waals interactions. Furthermore, the hydrogen bonds between Gln163 and the main chain of Met242 in pierisin-1 are not observed between Met276 and Glu195 of MTX. In contrast, the interaction between the side chain of Met242 and the βNAD+ binding pocket of pierisin-1 is conserved in MTX. Met242 of pierisin-1 is buried deep by van der Waals interactions with Thr115 of the STS motif, Trp127, and Trp71, disturbing the interaction with the nicotinamide moiety. These residues correspond to Met276, Thr143, Trp158, and Trp98, respectively, in MTX, where they form several similar contacts. Thus, despite the sequence differences in the linker region of pierisin-1 and MTX, the segment 239–244 of pierisin-1 adopts a similar conformation to that of MTX through the above interactions and prevents βNAD+ from binding to the pocket.
The electron density of a segment (residues 231–238), which precedes amino acids 239–244 and which contains three C-terminal residues (Ser231–Arg233) of the catalytic domain, is missing in pierisin(1–267)E165Q, most likely due to the disordered nature of the segment (Fig. 1B, right). Because the flexible unstructured segment protrudes from the protein surface, proteases can target and cleave the site. It is reasonable to propose that the segment is cleaved by proteolysis and is followed by dissociation of the C-terminal ricin B–like domain, together with the linker region from the catalytic domain, resulting in enzyme activity in vitro and in vivo (12). Pierisin-1 is specifically cleaved at Arg233-Ser234 by trypsin in vitro. In vivo proteolytic cleavage might occur in intracellular lysosomes after pierisin is ingested into cells, and then the excised catalytic domain is transported to the nucleus to bind to DNA (Fig. 6). However, little is known about these processes. A similar activation mechanism is also suggested for MTX (15, 16, 31). In fact, the segment 263–269 of MTX is disordered and is subject to protease digestion in the activation of its catalytic activity (32, 33).
Figure 6.
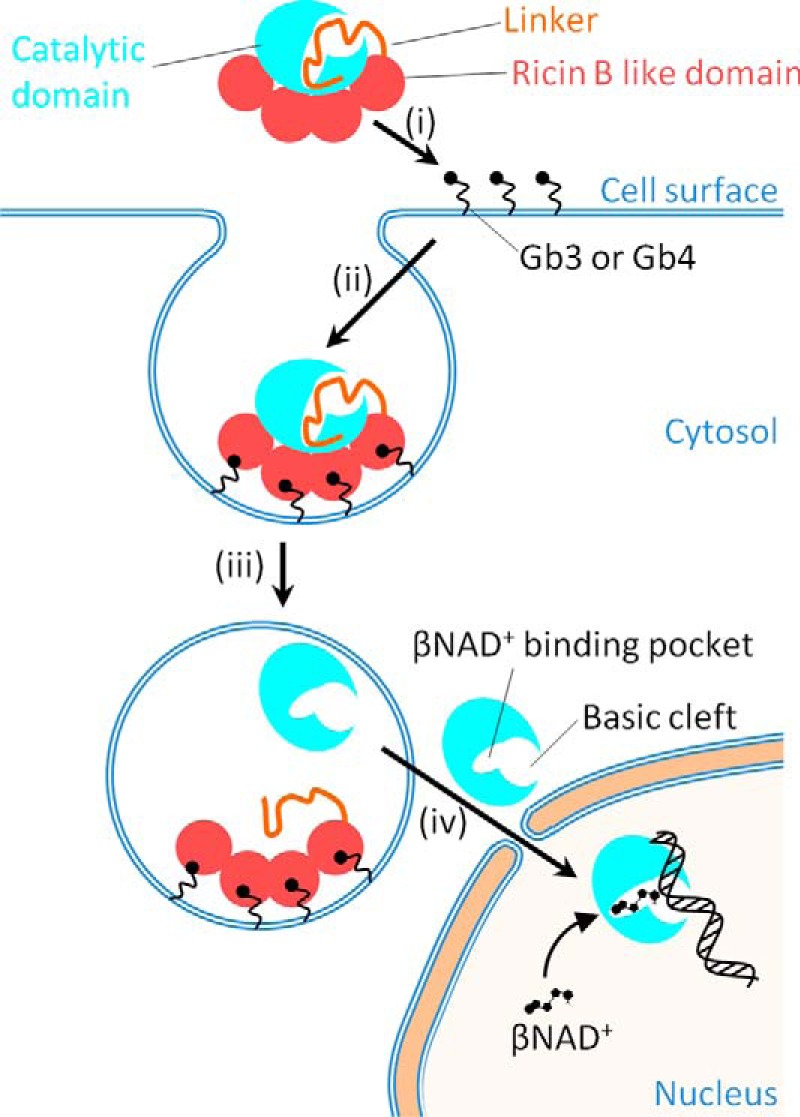
A model for the activation and DNA binding of DNA ADP-ribosylating pierisin-1. Pierisin-1 consists of an N-terminal catalytic domain colored light blue, a linker shown as an orange thread, and a C-terminal ricin B–like domain colored red and autoinhibits a DNA ADP-ribosylating activity by occupying its catalytic cleft with its linker region. i and ii, pierisin binds to cell surface glycosphingolipid receptors, such as globotriaosylceramide (Gb3) and globotetraosylceramide (Gb4) through its four ricin B–like domains (13) and is internalized into cells. iii, the pierisin incorporated into lysozomes is cleaved at a C-terminal site of the catalytic domain and then followed by the catalytic domain dissociation from the rest of the linker and the ricin B–like domains, resulting in the activation of its catalytic domain. Thus, the catalytic domain is exposed at a catalytic site and freely accessible by a substrate βNAD+ and an acceptor DNA. iv, the active catalytic domain (residues 1–233) released into the cytosol migrates into the nucleus and binds to DNA, where it transfers the ADP-ribose moiety of βNAD+ to the N2 amino group of guanine.
Discussion
In this study, we determined the crystal structures of pierisin(1–233)E165Q and a βNAD+-bound pierisin(1–233)E165Q. These structures share a common fold with such mono-ADP-ribosylating toxins as cholera toxin, diphtheria toxin, MTX, and others, despite high sequence diversity. βNAD+ is also recognized in a similar manner to the mono-ADP-ribosylating toxins. The mutation analysis based on the 3D structure of the catalytic domain of pierisin-1 shows that the PN loop and the basic cleft are important for dsDNA binding. Modeling dsDNA onto pierisin(1–233)E165Q shows that dsDNA fits well between the PN loop and the basic cleft (Fig. 4A). The width of the basic cleft (the distance between Cα atoms of Lys123 and Arg187) was 19.2 Å, which is suitable for dsDNA but too large for ssDNA. Our results from DNA binding assays substantiated the previous finding that as an acceptor molecule for ADP-ribosylation, dsDNA is much better than ssDNA. The PN loop exhibits structural flexibility (Fig. 1D). It is possible that this flexibility is necessary for dsDNA recognition to fit the DNA into the binding surface. The basic residues of the PN loop and the basic cleft nearby are conserved between pierisin-1, -1b, -2, -3, -4, and -5, all of which ADP-ribosylate DNA (supplemental Fig. S5), suggesting a common feature of all pierisins. Despite the high similarity in 3D structure between pierisin-1 and MTX, their electrostatic surface potentials are distinctively different (Fig. 4A). This difference may reflect acceptor specificity of the enzymes. In fact, the surface that corresponds to the basic cleft of pierisin-1, is also masked by the linker region in MTX, although the substrate binding region of MTX has not been identified.
Recently, Lyons et al. (34) determined the crystal structure of scabin, a DNA-targeting ADP-ribosyltransferase from Streptomyces scabies. They showed that scabin has a positively charged surface suggested to be a DNA-binding surface (supplemental Fig. S7A). The core structure is similar to those of pierisin-1 (Z score = 16.7) as well as MTX and other ADP-ribosyltransferases (supplemental Fig. S7). However, scabin has neither a ricin B–like domain nor an autoinhibitory linker, indicating that activity control is different between scabin and pierisin-1. Furthermore, arrangement of α-helices and loop regions around the core structure are different, and the PN loop of scabin is significantly shorter than that of pierisin-1 (supplemental Figs. S5 and S7B). The key residues for DNA binding are also not conserved between pierisin-1 and scabin (supplemental Fig. S5). These observations indicate that the binding mode for dsDNA and preference for dsDNA, ssDNA, and mononucleotide differ between scabin and pierisin-1. Thus, the DNA-targeting mechanisms seem to be divergent among ADP-ribosyltransferases. We are currently determining the crystal structure of pierisin(1–233)E165Q in complex with dsDNA and with dsDNA and βNAD+ to obtain further structural insight into the ADP-ribosyltransferase DNA-targeting mechanism.
Experimental procedures
Expression and purification of pierisin(1–233)E165Q and pierisin(1–267)E165Q
The plasmid vector pET-32a containing a cDNA encoding pierisin(1–233)E165Q was transformed into E. coli strain BL21(DE3) pLysS. Pierisin(1–233)E165Q was expressed as an N-terminal thioredoxin and His tag fusion protein. The cells were grown at 37 °C in LB medium and induced with 1 mm isopropyl-thio-β-d-galactopyranoside when they reached an optical density of 0.4–0.6 at 660 nm and were further incubated at 16 °C for 3 h. The cells were harvested; resuspended in a lysis buffer containing 50 mm Tris-HCl, pH 7.5, 0.5 m NaCl, and 1 mm PMSF; and disrupted by sonication on ice. After centrifugation, the supernatant was applied to a nickel-Sepharose column (GE Healthcare). Thioredoxin and His tag fusion protein was eluted with 50 mm Tris-HCl, pH 7.5, containing 0.5 m NaCl, 200 mm imidazole. After digestion of the fusion protein with thrombin, samples were diluted with 50 mm Tris-HCl, pH 7.5, and loaded onto a HiTrapSP HP column (GE Healthcare). The protein was eluted by a linear gradient of 0–1 m NaCl. The eluted protein was concentrated to 1.5–2.0 mg/ml, and the buffer solution was exchanged with 20 mm HEPES-NaOH, pH 7.0, containing 400 mm NaCl. The purified protein was stored at 4 °C. All of the mutant proteins of pierisin(1–233)E165Q used for EMSAs were purified as described above.
The plasmid vector pGEX6p-1 containing a cDNA encoding pierisin(1–267)E165Q was transformed into E. coli BL21(DE3) codon plus RIPL. Pierisin(1–267)E165Q was expressed as an N-terminal GST tag fusion protein. The cells were grown at 37 °C in LB medium and induced for protein expression with 1 mm isopropyl-thio-β-d-galactopyranoside when they reached an optical density of 0.4–0.6 at 660 nm and incubated at 18 °C for 3 h. The cells were harvested and stored at −80 °C. The thawed cells were resuspended in lysis buffer (50 mm Tris-HCl, pH 7.5, 0.5 m NaCl, 1 mm PMSF) and disrupted by sonication on ice. After centrifugation, the supernatant was applied to a glutathione-Sepharose 4B resin (GE Healthcare), and unbound protein was washed out by 50 mm Tris-HCl, pH 7.5, containing 50 mm NaCl. The GST tag was removed by GST-HRV3C protease on resin, and pierisin(1–267)E165Q was eluted. The pierisin(1–267)E165Q was loaded onto a HiTrapQ HP column (GE Healthcare) and eluted with a stepwise gradient of 0.1, 0.2, and 1.0 m NaCl. Further purification was performed on a HiLoad 26/60 Superdex 75 pg column (GE Healthcare) with a running buffer of 50 mm Tris-HCl, pH 7.5, 100 mm NaCl. The eluted pierisin(1–267)E165Q was concentrated at 2 mg/ml and stored at 4 °C. Pierisin (1–267)E165Q, pierisin(1–233)E165Q, and all other mutant proteins were checked for purity using SDS-PAGE (supplemental Fig. S4).
Crystallization and structure determination of pierisin(1–267)E165Q, pierisin(1–233)E165Q, and pierisin(1–233)E165Q/βNAD+
All crystallization was performed by vapor diffusion at 20 °C by using 1.5–2.0 mg/ml protein solution. All crystals were obtained after 1–3 weeks. Crystals of pierisin(1–233)E165Q (form 1) were obtained using a sample containing 50 μm pierisin(1–233)E165Q (1.6 mg/ml) and 50 μm 14-bp dsDNA. The 14-bp dsDNA was the same as that used for EMSA. The reservoir solution used for crystallization was 0.1 m Tris-HCl, pH 8.0, 20% (v/v) 2-methyl-2,4-pentanediol (MPD). As a result, this crystal did not contain DNA. The crystal was transferred to a reservoir solution containing 5–25% (v/v) ethylene glycol stepwise and flash-frozen at −180 °C. For preparation of the βNAD+-bound form, the crystal was soaked in a reservoir solution containing 10 mm βNAD+ for 18 h. Then the crystal was transferred to a reservoir solution containing 10 mm βNAD+ and 5–25% (v/v) ethylene glycol stepwise and flash-frozen at −180 °C.
Crystals of pierisin(1–233)E165Q (form 2) were obtained using a reservoir solution containing 0.1 m NaF and 6–10% (w/v) PEG 3350. The crystal was transferred to a reservoir solution containing 5–25% (v/v) ethylene glycol stepwise and flash-frozen at −180 °C. For the preparation of the βNAD+-bound form, the crystal was soaked in a reservoir solution containing 5 mm βNAD+ without NaF for 10 min. Then the crystal was transferred to 5–25% (v/v) ethylene glycol containing 10 mm βNAD+, 6% (w/v) PEG 3350 stepwise and flash-frozen at −180 °C.
Crystals of pierisin(1–267)E165Q were obtained by using a reservoir solution containing 22% (v/v) t-butanol and 0.1 m Tris-HCl, pH 8.0. The crystal was transferred to a reservoir solution containing 22.5% (v/v) glycerol and flash-frozen at −180 °C.
X-ray diffraction data were collected on a Quantum315 CCD detector (ADSC) in beamline BL5A or BL17A at PF (Tsukuba, Japan) and processed using the HKL-2000 program suite (35). The structure of pierisin(1–267)E165Q was solved by molecular replacement with the program MOLREP (36) using the MTX-1 catalytic domain (PDB code 2CB4) as a search model. In the same way, the structure of pierisin(1–233)E165Q was solved using the structure of pierisin(1–267)E165Q. The structures were manually improved with the program COOT (37) and refined with the program REFMAC (38) and PHENIX (39). Geometries of the final structures were validated with molprobity (40, 41). Data collection and refinement are summarized in Table 1. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB entries 5H6K, 5H6J, 5H6M, 5H6L, and 5H6N).
EMSA
The binding activity of pierisin to various DNAs was assessed by EMSA. 6-Carboxyfluorescein (6-FAM)-labeled 15-bp dsDNA (5′-CTACAGTCGTCAGGA-3′ labeled with 6-FAM at 5′-end and unlabeled complementary sequence) or its labeled strand as ssDNA were used. The 15-bp dsDNA was prepared by annealing in 0.1 m KCl, 0.1 mm EDTA. Sample solutions (20 μl) containing 3.1 pmol of dsDNA or ssDNA and varying amounts (0, 3.1, 6.2, 12.4, 24.8, and 49.6 pmol) of pierisin(1–233)E165Q in a buffer solution (2 mm HEPES-NaOH, 40 mm NaCl, 40% (v/v) glycerol, 40 mm Tris, 40 mm acetate, and 1 mm EDTA with a final pH value of 8.3) were incubated for 30 min at 4 °C. Then the sample solutions were subject to electrophoresis on a native 5–20% gradient polyacrylamide gel with a conventional Tris acetate-EDTA buffer (TAE: 40 mm Tris, 40 mm acetate, 1 mm EDTA, final pH 8.3) at 4 °C. The bands of DNA were detected by fluorescence of 6-FAM using a ChemiDocTM MP system (Bio-Rad).
For analysis of DNA-binding activity of pierisin(1–267)E165Q and pierisin(1–233)E165Q, 14-bp dsDNA (5′-CTTCCACGTGGCAT-3′ and complementary sequence) was used. Sample solutions (15 μl) containing 100 pmol of 14-bp dsDNA, 300 pmol of pierisin, and 60 nmol of βNAD+ in a buffer solution (8 mm Tris-HCl, pH 7.5, 160 mm NaCl, 13 mm KCl, and 10% (v/v) glycerol) were incubated for 30 min at 4 °C. Then the sample solutions were subjected to electrophoresis on a native 6% polyacrylamide gel with a TAE buffer at 4 °C. The gels were stained with ethidium bromide.
For analysis of DNA-binding activity of pierisin(1–233)E165Q and other mutants, 201-bp dsDNA (a sequence between the T7 promoter and T7 terminator of pET-32a plasmid) was used. The 201-bp dsDNA was amplified by PCR using KOD plus DNA polymerase (TOYOBO). The amplified 201-bp dsDNA was purified by electrophoresis on a 1.5% agarose gel with a TAE buffer and extracted from the gel using the Wizard SV Gel and PCR Clean-Up System (Promega). Sample solutions (10 μl) containing 0.1 pmol of 201-bp dsDNA and varying amounts (0, 0.25, 0.5, 1.0, 2.0, and 4.0 pmol) of pierisin(1–233) mutants in a buffer solution (2 mm HEPES-NaOH, 40 mm NaCl, 40% (v/v) glycerol, 40 mm Tris, 40 mm acetate, and 1 mm EDTA with a final pH value of 8.3) were incubated for 30 min at 4 °C. Then the sample solutions were subjected to electrophoresis on a 1.5% (w/v) agarose gel with TAE buffer at 4 °C. The gel was stained with SYBR Gold (Invitrogen).
Author contributions
T. O., M. Y., K. W., T. S., and M. S. designed the research; T. O., H. Hirabayashi, G. S., R. T., K. H., and H. M. performed the research; T. O., H. Hirabayashi, H. Hashimoto, and T. S. analyzed the data; and T. O., H. Hashimoto, K. W., T. S., and M. S. wrote the paper.
Supplementary Material
Acknowledgments
We are deeply grateful to President of the Japan Academy, Takashi Sugimura, for his great interest in this work and for critical reading of the manuscript. We thank the staff of the Photon Factory for assistance with data collection. We also thank Terukazu Nogi and Kyouhei Arita for support of structural analysis. We also thank Michiyuki Yamada for support of writing and critical reading of the manuscript.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S7.
- MTX
- mosquitocidal toxin
- PDB
- Protein Data Bank
- PN loop
- phosphate–nicotinamide loop
- ARTT loop
- ADP-ribosylating turn-turn loop
- 6-FAM
- 6-carboxyfluorescein
- TAE
- Tris acetate-EDTA buffer.
References
- 1. Corda D., and Di Girolamo M. (2003) Functional aspects of protein mono-ADP-ribosylation. EMBO J. 22, 1953–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Holbourn K. P., Shone C. C., and Acharya K. R. (2006) A family of killer toxins: exploring the mechanism of ADP-ribosylating toxins. FEBS J. 273, 4579–4593 [DOI] [PubMed] [Google Scholar]
- 3. Hottiger M. O., Hassa P. O., Lüscher B., Schüler H., and Koch-Nolte F. (2010) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 35, 208–219 [DOI] [PubMed] [Google Scholar]
- 4. Jørgensen R., Merrill A. R., Yates S. P., Marquez V. E., Schwan A. L., Boesen T., and Andersen G. R. (2005) Exotoxin A–eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature 436, 979–984 [DOI] [PubMed] [Google Scholar]
- 5. Jørgensen R., Wang Y., Visschedyk D., and Merrill A. R. (2008) The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 9, 802–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsuge H., Nagahama M., Oda M., Iwamoto S., Utsunomiya H., Marquez V. E., Katunuma N., Nishizawa M., and Sakurai J. (2008) Structural basis of actin recognition and arginine ADP-ribosylation by Clostridium perfringens ι-toxin. Proc. Natl. Acad. Sci. U.S.A. 105, 7399–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koyama K., Wakabayashi K., Masutani M., Koiwai K., Watanabe M., Yamazaki S., Kono T., Miki K., and Sugimura T. (1996) Presence in Pieris rapae of cytotoxic activity against human carcinoma cells. Jpn. J. Cancer Res. 87, 1259–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Watanabe M., Kono T., Koyama K., Sugimura T., and Wakabayashi K. (1998) Purification of pierisin, an inducer of apoptosis in human gastric carcinoma cells, from cabbage butterfly, Pieris rapae. Jpn. J. Cancer Res. 89, 556–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kono T., Watanabe M., Koyama K., Kishimoto T., Fukushima S., Sugimura T., and Wakabayashi K. (1999) Cytotoxic activity of pierisin, from the cabbage butterfly, Pieris rapae, in various human cancer cell lines. Cancer Lett. 137, 75–81 [DOI] [PubMed] [Google Scholar]
- 10. Watanabe M., Nakano T., Shiotani B., Matsushima-Hibiya Y., Kiuchi M., Yukuhiro F., Kanazawa T., Koyama K., Sugimura T., and Wakabayashi K. (2004) Developmental stage-specific expression and tissue distribution of pierisin-1, a guanine-specific ADP-ribosylating toxin, in Pieris rapae. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 139, 125–131 [DOI] [PubMed] [Google Scholar]
- 11. Watanabe M., Kono T., Matsushima-Hibiya Y., Kanazawa T., Nishisaka N., Kishimoto T., Koyama K., Sugimura T., and Wakabayashi K. (1999) Molecular cloning of an apoptosis-inducing protein, pierisin, from cabbage butterfly: possible involvement of ADP-ribosylation in its activity. Proc. Natl. Acad. Sci. U.S.A. 96, 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanazawa T., Watanabe M., Matsushima-Hibiya Y., Kono T., Tanaka N., Koyama K., Sugimura T., and Wakabayashi K. (2001) Distinct roles for the N- and C-terminal regions in the cytotoxicity of pierisin-1, a putative ADP-ribosylating toxin from cabbage butterfly, against mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 98, 2226–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsushima-Hibiya Y., Watanabe M., Hidari K. I., Miyamoto D., Suzuki Y., Kasama T., Kanazawa T., Koyama K., Sugimura T., and Wakabayashi K. (2003) Identification of glycosphingolipid receptors for pierisin-1, a guanine-specific ADP-ribosylating toxin from the cabbage butterfly. J. Biol. Chem. 278, 9972–9978 [DOI] [PubMed] [Google Scholar]
- 14. Takamura-Enya T., Watanabe M., Totsuka Y., Kanazawa T., Matsushima-Hibiya Y., Koyama K., Sugimura T., and Wakabayashi K. (2001) Mono(ADP-ribosyl)ation of 2′-deoxyguanosine residue in DNA by an apoptosis-inducing protein, pierisin-1, from cabbage butterfly. Proc. Natl. Acad. Sci. U.S.A. 98, 12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reinert D. J., Carpusca I., Aktories K., and Schulz G. E. (2006) Structure of the mosquitocidal toxin from Bacillus sphaericus. J. Mol. Biol. 357, 1226–1236 [DOI] [PubMed] [Google Scholar]
- 16. Carpusca I., Jank T., and Aktories K. (2006) Bacillus sphaericus mosquitocidal toxin (MTX) and pierisin: the enigmatic offspring from the family of ADP-ribosyltransferases. Mol. Microbiol. 62, 621–630 [DOI] [PubMed] [Google Scholar]
- 17. Schirmer J., Wieden H. J., Rodnina M. V., and Aktories K. (2002) Inactivation of the elongation factor Tu by mosquitocidal toxin-catalyzed mono-ADP-ribosylation. Appl. Environ. Microbiol. 68, 4894–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Orth J. H. C., Schorch B., Boundy S., Ffrench-Constant R., Kubick S., and Aktories K. (2011) Cell-free synthesis and characterization of a novel cytotoxic pierisin-like protein from the cabbage butterfly Pieris rapae. Toxicon 57, 199–207 [DOI] [PubMed] [Google Scholar]
- 19. Matsushima-Hibiya Y., Watanabe M., Kono T., Kanazawa T., Koyama K., Sugimura T., and Wakabayashi K. (2000) Purification and cloning of pierisin-2, an apoptosis-inducing protein from the cabbage butterfly, Pieris brassicae. Eur. J. Biochem. 267, 5742–5750 [DOI] [PubMed] [Google Scholar]
- 20. Takamura-Enya T., Watanabe M., Koyama K., Sugimura T., and Wakabayashi K. (2004) Mono(ADP-ribosyl)ation of the N2 amino groups of guanine residues in DNA by pierisin-2, from the cabbage butterfly, Pieris brassicae. Biochem. Biophys. Res. Commun. 323, 579–582 [DOI] [PubMed] [Google Scholar]
- 21. Matsumoto Y., Nakano T., Yamamoto M., Matsushima-Hibiya Y., Odagiri K., Yata O., Koyama K., Sugimura T., and Wakabayashi K. (2008) Distribution of cytotoxic and DNA ADP-ribosylating activity in crude extracts from butterflies among the family Pieridae. Proc. Natl. Acad. Sci. U.S.A. 105, 2516–2520; Erratum (2008) Proc. Natl. Acad. Sci. U.S.A.105, 5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamamoto M., Nakano T., Matsushima-Hibiya Y., Totsuka Y., Takahashi-Nakaguchi A., Matsumoto Y., Sugimura T., and Wakabayashi K. (2009) Molecular cloning of apoptosis-inducing Pierisin-like proteins, from two species of white butterfly, Pieris melete and Aporia crataegi. Comp. Biochem. Physiol. B 154, 326–333 [DOI] [PubMed] [Google Scholar]
- 23. Subbarayan S., Marimuthu S. K., Nachimuthu S. K., Zhang W., and Subramanian S. (2016) Characterization and cytotoxic activity of apoptosis-inducing pierisin-5 protein from white cabbage butterfly. Int. J. Biol. Macromol. 87, 16–27 [DOI] [PubMed] [Google Scholar]
- 24. Nakano T., Matsushima-Hibiya Y., Yamamoto M., Enomoto S., Matsumoto Y., Totsuka Y., Watanabe M., Sugimura T., and Wakabayashi K. (2006) Purification and molecular cloning of a DNA ADP-ribosylating protein, CARP-1, from the edible clam Meretrix lamarckii. Proc. Natl. Acad. Sci. U.S.A. 103, 13652–13657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D, Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Otto H., Reche P. A., Bazan F., Dittmar K., Haag F., and Koch-Nolte F. (2005) In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs) BMC Genomics 6, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han S., Arvai A. S., Clancy S. B., and Tainer J. A. (2001) Crystal structure and novel recognition motif of Rho ADP-ribosylating C3 exoenzyme from Clostridium botulinum: structural insights for recognition specificity and catalysis. J. Mol. Biol. 305, 95–107 [DOI] [PubMed] [Google Scholar]
- 28. O'Neal C. J., Jobling M. G., Holmes R. K., and Hol W. G. J. (2005) Structural basis for the activation of cholera toxin by human ARF6-GTP. Science 309, 1093–1096 [DOI] [PubMed] [Google Scholar]
- 29. Watanabe M., Enomoto S., Takamura-Enya T., Nakano T., Koyama K., Sugimura T., and Wakabayashi K. (2004) Enzymatic properties of pierisin-1 and its N-terminal domain, a guanine-specific ADP-ribosyltransferase from the cabbage butterfly. J. Biochem. 135, 471–477 [DOI] [PubMed] [Google Scholar]
- 30. Han S., and Tainer J. A. (2002) The ARTT motif and a unified structural understanding of substrate recognition in ADP-ribosylating bacterial toxins and eukaryotic ADP-ribosyltransferases. Int. J. Med. Microbiol. 291, 523–529 [DOI] [PubMed] [Google Scholar]
- 31. Treiber N., Reinert D. J., Carpusca I., Aktories K., and Schulz G. E. (2008) Structure and mode of action of a mosquitocidal holotoxin. J. Mol. Biol. 381, 150–159 [DOI] [PubMed] [Google Scholar]
- 32. Thanabalu T., Hindley J., and Berry C. (1992) Proteolytic processing of the mosquitocidal toxin from Bacillus sphaericus SSII-1. J. Bacteriol. 174, 5051–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schirmer J., Just I., and Aktories K. (2002) The ADP-ribosylating mosquitocidal toxin from Bacillus sphaericus: proteolytic activation, enzyme activity, and cytotoxic effects. J. Biol. Chem. 277, 11941–11948 [DOI] [PubMed] [Google Scholar]
- 34. Lyons B., Ravulapalli R., Lanoue J., Lugo M. R., Dutta D., Carlin S., and Merrill A. R. (2016) Scabin, a novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem. 291, 11198–11215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collection in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 36. Vagin A. A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 37. Emsley P., and Cowtan K. (2004) Coot: model building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 39. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.