

Abstract
Src Homology 2 domain–containing phosphotyrosine phosphatase 2 (Shp2) functions in synaptic plasticity, learning, and memory. However, the precise mechanisms by which this multifunctional protein contributes to synaptic function remains largely unknown. Homeostatic plasticity may be viewed as a process of bidirectional synaptic scaling, up or down. Through this process, neuronal circuitry stability is maintained so that changes in synaptic strength may be preserved under changing conditions. A better understanding of these processes is needed. In this regard, we report that phosphorylation of Shp2 at tyrosine 542 and its translocation to the postsynaptic compartment are integral processes in synaptic scaling. Furthermore, we show, using both pharmacological and genetic approaches, that Shp2 phosphatase activity is critical to the regulation of Ser(P)845 GluA1 and surface expression of this AMPA receptor subunit during synaptic scaling. Thus, Shp2 may contribute meaningfully to synaptic homeostasis.
Keywords: AMPA receptor (AMPAR), phosphorylation, Src homology 2 domain (SH2 domain), trafficking, protein–tyrosine phosphatase (tyrosine phosphatase), GluA1, S845, Shp2, Tyr542, synaptic scaling
Introduction
Different types of stimuli elicit synapses of neurons to change their strength in a bidirectional manner, most studied as long-term potentiation (LTP)3 and long-term depression (LTD), the phenomenon well known as Hebbian-type synaptic plasticity (1). It is widely believed that the changes in relative synaptic strength via synaptic plasticity underpin the molecular and cellular basis of learning and memory (2, 3). However, LTP and LTD exert a vigorous long-term impact on neuronal circuits. Homeostatic plasticity, by synaptic scaling either up or down, maintains a stable neuronal circuit by adjusting synaptic properties to keep activity close to its optimal operating range, preserving network stability and, in turn, promoting learning and memory by offsetting deleterious effects induced by continuous LTP or LTD (4).
There is considerable evidence that changes in synaptic strength associated with synaptic plasticity or homeostatic plasticity are mediated by changes in the levels of AMPA receptors (1, 4). The majority of AMPA receptors in the hippocampus of rodents consists of GluA1/GluA2 or GluA2/GluA3 heterodimers, and homeostatic plasticity is tightly associated with changes in surface GluA1-containing AMPA receptors (1, 5). To bidirectionally regulate surface delivery of GluA1, phosphorylation of the GluA1 cytoplasmic C-terminal tail has been adopted in hippocampal neurons (5, 6). In particular, elevated phosphorylation of the PKA GluA1 Ser845 target site is widely speculated to facilitate forward surface delivery of GluA1 after LTP or synaptic upscaling (5, 7, 8). Recently we demonstrated that Shp2 contributes to the regulation of Ser(P)845 GluA1 during LTP. Specifically, inhibition of Shp2 phosphatase activity via pharmacological or genetic means impaired regulation of GluA1 phosphorylation during LTP induction in hippocampal neurons (9).
The ubiquitous tyrosine phosphatase Shp2 (encoded by PTPN11) has emerged as a critical regulator of synaptic plasticity, learning, and memory (9–13). In light of its structure, Shp2 contains two tandem N-terminal Src homology 2 (SH2) domains, a catalytic protein–tyrosine phosphatase (PTP) domain, and a C-terminal tail with tyrosyl phosphorylation sites and a proline-rich motif (14, 15). The N-terminal SH2 domain binds to the PTP domain, resulting in Shp2 autoinhibition, which can be relieved by phosphorylation of Tyr542 to disrupt the association of the SH2 and PTP domains (14, 16, 17). Hence, phosphorylation of Tyr542 correlates with activity of Shp2, which can be utilized as an indicative marker for the activity of Shp2. Germ line mutations in Shp2 cause Noonan syndrome, which may cause mental retardation and intellectual disability (18). Additionally, deregulation of Shp2 has been reported in various diseases, including juvenile myelomonocytic leukemia, breast cancer, and liver cancer (18–20). Thus, considerable effort has been directed toward the development of Shp2-specific inhibitors as potential drug therapies. Given this clinical relevance and the emerging role of Shp2 in synaptic plasticity, studies are warranted to further understand the role Shp2 plays in synaptic function and homeostasis.
In this study, we demonstrate that phosphorylation of Shp2, particularly at Tyr542, is regulated by bidirectional homeostatic plasticity. Additionally, we provide convincing evidence that Shp2 is critical for elevated surface GluA1 expression in homeostatic plasticity. We show that inhibiting Shp2 phosphatase activity with its inhibitors, expressing an Shp2 phosphatase-inactive mutant, and adeno-associated virus (AAV)-mediated KO of Shp2 in hippocampal neurons prevented an increase in pS845 levels and surface GluA1. Therefore, our results reveal an undefined role of Shp2 in regulating GluA1 surface expression in homeostatic plasticity.
Results
The phosphorylation level of Shp2 Tyr542 is regulated by synaptic scaling
To explore whether the phosphorylation level of Tyr542 in Shp2 (hereafter referred to as the Tyr(P)542 level) is regulated during synaptic scaling, we treated cultured rat hippocampal neurons with bicuculline (Bic) or tetrodotoxin (TTX) for 48 h to induce synaptic scaling down or scaling up, as reported previously (21). The phosphorylation level of Ser845 (hereinafter referred to as the Ser(P)845 level) in GluA1 and the total GluA1 level were simultaneously monitored by Western blotting to ascertain the success of synaptic scaling. As expected, TTX treatment elevated GluA1 and Ser(P)845 levels, whereas bicuculline treatment reduced GluA1 and Ser(P)845 levels (Fig. 1, A and B; for GluA1, TTX, 1.494 ± 0.069 and Bic, 0.664 ± 0.075; for Ser(P)845, TTX, 1.760 ± 0.097 and Bic, 0.544 ± 0.040; n = 4), which is consistent with others reports (21, 22). Western blot analysis also showed an increased Tyr(P)542 level in synaptic upscaling and a decreased Tyr(P)542 level in synaptic downscaling (Fig. 1, A and B; TTX, 2.051 ± 0.094 and Bic, 0.369 ± 0.135), respectively. Interestingly, the total Shp2 level exhibited no significant difference among the control, the upscaling (TTX), and the downscaling (Bic) groups (Fig. 1, A and B). These results reveal that the activity of Shp2 is bidirectionally regulated during synaptic scaling.
Figure 1.

Phosphorylation of Shp2 at Tyr542 is regulated bidirectionally in synaptic scaling. A, hippocampal neurons were treated for 48 h with TTX or Bic, followed by Western blot analysis. MW, molecular weight. B, quantification of Ser(P)845, GluA1, Tyr(P)542, and Shp2 levels. Error bars indicate S.E. n = 4 independent experiments. **, p ≤ 0.01; #, p ≤ 0.001 against control; one-way ANOVA followed by Tukey's post hoc test.
Increased colocalization of Shp2 and PSD95 after synaptic upscaling
Our previous study suggested increased incorporation of Shp2 into excitatory postsynaptic sites after acute phase of chemical LTP (9). We wondered whether there was a similar process during synaptic upscaling. Therefore, we set out to examine the colocalization of endogenous Shp2 and PSD95 after 48-h TTX treatment. When the neurons were labeled with the secondary antibody but not the primary antibody against Shp2, no signal was detected under microscopy (supplemental Fig. S1). Afterward, colocalization analysis of Shp2 and PSD95 in dendrites was assessed by immunocytochemistry with or without 48-h TTX treatment in cultured rat hippocampal neurons (Fig. 2, A and B). We found that the proportion of Shp2 clusters merged with PSD95 clusters was increased after upscaling (Fig. 2, A and B). This result suggests that Shp2 translocates to postsynaptic sites after synaptic upscaling. To further confirm this result, the 48-h TTX-treated group and the control group were collected, followed by extraction of the postsynaptic density (PSD) fraction. Western blot analysis showed an increased Shp2 proportion in TTX-mediated synaptic upscaling against the control group in the PSD fraction (Fig. 2, C and D; GluA1, 2.088 ± 0.189, TTX versus control, n = 4; Shp2, 2.677 ± 0.237, TTX versus control, n = 4), indicating a translocation of Shp2 to postsynaptic sites after TTX-mediated upscaling. The results from confocal imaging and biochemistry suggest that Shp2 is subject to fine regulation by synaptic scaling in hippocampal neurons.
Figure 2.
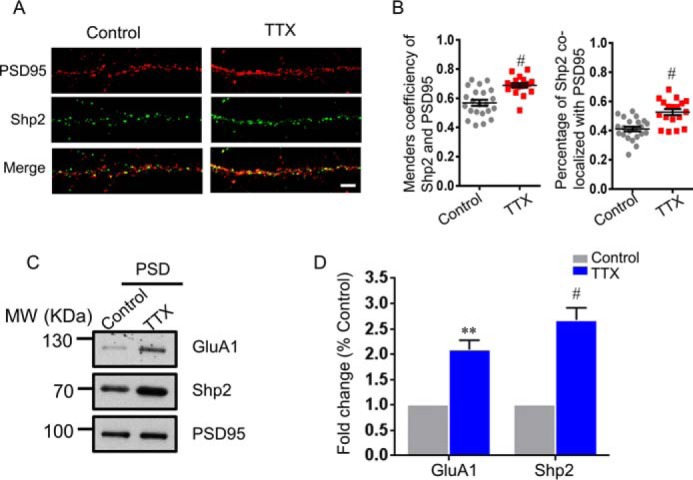
Shp2 is accumulated at the postsynaptic site after TTX-mediated synaptic upscaling. A, hippocampal neurons were treated for 48 h with TTX, and colocalization of Shp2 (green) and PSD95 (red) was analyzed by immunocytochemistry. Scale bar = 5 μm. B, Manders coefficiency for colocalization of Shp2 and PSD95 and percentage of Shp2 colocalized with PSD95 (n = 22 or 18 neurons for the control condition or TTX treatment, respectively). #, p ≤ 0.001. C, hippocampal neurons were treated for 48 h with TTX, and the PSD fraction was analyzed. MW, molecular weight. D, quantification of GluA1 and Shp2 levels in the PSD fraction (n = 4 independent experiments). **, p ≤ 0.01; #, p ≤ 0.001 versus control.
Inhibition of Shp2 phosphatase activity by specific inhibitors dampens surface GluA1 up-regulation in synaptic upscaling
Given our demonstration of the regulation of the Tyr(P)542 level in synaptic scaling, we wondered whether activity of Shp2 was required for enhanced surface GluA1 after TTX-induced upscaling. To address this issue, we utilized two widely used inhibitors that block Shp2 activity. Our recent study revealed that 10 μm NSC87877 blocked chemical LTP-induced Tyr(P)542 level elevation in cultured hippocampal neurons (9). Based on our previous data, we co-incubated 10 μm NSC87877 with TTX for 48 h. A Western blot analysis of cultured hippocampal neurons demonstrated that the level of Tyr(P)542 was dramatically decreased in response to NSC87877 + TTX treatment compared with TTX alone, without altering the total Shp2 level (Fig. 3, A and B; Tyr(P)542 level, TTX, 1.984 ± 0.216; NSC87877 + TTX, 1.028 ± 0.178; NSC87877, 0.974 ± 0.065; n = 3). Additionally, NSC87877 + TTX treatment blunted elevation of the Ser(P)845 level (Fig. 3, A and B; TTX, 1.887 ± 0.144; NSC87877 + TTX, 1.094 ± 0.028; NSC87877, 1.012 ± 0.067; n = 3). Interestingly, NSC87877 also blocked the increase in total GluA1 level induced by TTX treatment (Fig. 3, A and B; TTX, 1.519 ± 0.092; NSC87877 + TTX, 1.072 ± 0.028; NSC87877, 0.905 ± 0.097; n = 3). Notably, the total GluA1 level remained stable under 48-h NSC87877 treatment, suggesting that activity of Shp2 is required for the increased levels of Ser(P)845 and GluA1 during synaptic upscaling. Because phosphorylation of Ser845 is well correlated with surface GluA1 (5, 7, 8), we decided to measure surface GluA1 in the next step. To evaluate surface GluA1 in this process, surface staining of GluA1 was assessed with and without NSC87877 incubation during 48-h TTX treatment. We found that surface GluA1 significantly increased in the 48-h TTX-treated neurons (Fig. 3, C and D). However, surface GluA1 in the TTX and NSC87877 co-incubation group was comparable with the control group or the NSC87877 alone group (Fig. 3, C and D; TTX, 1.406 ± 0.028; NSC87877 + TTX, 0.981 ± 0.063; NSC87877, 0.985 ± 0.054). Collectively, our results suggest that activity of Shp2 is critical for activity-dependent forward trafficking of GluA1 after 48-h TTX-induced synaptic scaling.
Figure 3.

Inhibition of Shp2 prevents elevation of the Ser(P)845 level and surface GluA1 after synaptic upscaling. A, hippocampal neurons were treated for 48 h with TTX, TTX + NSC87877 (NSC), or NSC alone, followed by Western blot analysis. MW, molecular weight. B, summary data of A (n = 3 independent experiments). **, p ≤ 0.01; #, p ≤ 0.001 versus control. C, hippocampal neurons were treated for 48 h with TTX, TTX + NSC, or NSC alone, followed by surface GluA1 and MAP2 staining. Scale bar = 5 μm. D, statistical analysis of surface GluA1 (n = 19 neurons for each condition from three independent experiments). #, p ≤ 0.001 versus control. E, hippocampal neurons were treated for 48 h with TTX, TTX + PHPS1, or PHPS1 alone, followed by Western blot analysis with the indicated antibodies. F, quantification of E (n = 5 independent experiments). **, p ≤ 0.01; #, p ≤ 0.001 versus control. G, hippocampal neurons were treated for 48 h with TTX, TTX + PHPS1, or PHPS1 alone, followed by surface GluA1 and MAP2 staining. Scale bar = 5 μm. H, statistical analysis of surface GluA1 (n = 15 neurons for each condition from three independent experiments). #, p ≤ 0.001 against control; one-way ANOVA followed by Tukey's post hoc test. I, hippocampal neurons were treated for 48 h with TTX, TTX + PHPS1, TTX + NSC, NSC87877, or PHPS1. Changes in GluA1 and Shp2 levels in the PSD fraction were assessed. J, Summary data of I (n = 4 independent experiments). #, p ≤ 0.001 versus control.
To strengthen the conclusion from the NSC87877 experiment, we sought to find out whether PHPS1, which interacts with the PTP signature loop of Shp2 to restrain its enzymatic activity (23), exerted a similar effect on the Ser(P)845 level and surface GluA1 level during synaptic upscaling. A previous study indicates that a dose range of 5–20 μm PHPS1 exerts effects on Shp2-dependent downstream signaling, i.e. hepatocyte growth factor/scatter factor–induced sustained phosphorylation of the ERK1/2 in epithelial cells (23). Accordingly, we applied 5 μm PHPS1 during a 48-h TTX treatment. Similar to the results observed with NSC87877, the Western blots showed that the Tyr(P)542 level was dramatically reduced in the PHPS1 + TTX group compared with the TTX alone group (Fig. 3, E and F; TTX, 1.857 ± 0.050; PHPS1 + TTX, 0.941 ± 0.159; PHPS1, 0.906 ± 0.135; n = 5), whereas total Shp2 levels were nearly comparable among the four groups. Additionally, PHPS1 blocked elevation of the Ser(P)845 level (Fig. 3, E and F; TTX, 1.920 ± 0.227; PHPS1 + TTX, 0.863 ± 0.164; PHPS1, 0.878 ± 0.156; n = 5). Notably, we observed that the increment of total GluA1 level induced by 48-h TTX treatment was negated by co-application of PHPS1 (Fig. 3, E and F; TTX, 1.615 ± 0.168; PHPS1 + TTX, 0.975 ± 0.065; PHPS1, 0.960 ± 0.048; n = 5). Surface staining of GluA1 in hippocampal neurons was also accessed in PHPS1 experiments. The increase in surface GluA1 was detected in the TTX-treated group but not in the TTX + PHPS1–treated group, which was consistent with changes in the Ser(P)845 level in biochemistry experiments (Fig. 3, G and H; TTX, 1.357 ± 0.047; PHPS1 + TTX, 0.939 ± 0.039; PHPS1, 1.019 ± 0.025). In addition, we evaluated GluA1 and Shp2 levels in the PSD fraction after co-application of TTX and Shp2 inhibitors. We found that NSC87877 and PHPS1 blocked the increase in GluA1 in the PSD fraction after 48-h TTX treatment, whereas these two inhibitors failed to affect the Shp2 level after TTX treatment (Fig. 3, I and J), suggesting that the inhibitors of Shp2 have little effect on the translocation of Shp2 to the PSD fraction after synaptic upscaling. Together, our data suggest that inhibition of Shp2 activity by specific inhibitors dampens Ser(P)845 and GluA1 up-regulation in synaptic upscaling.
Shp2 inactive mutation negates surface GluA1 up-regulation in synaptic upscaling
Accumulating evidence illustrates that Shp2 is a protein–tyrosine phosphatase implicated in activation of cell signaling and that mutation of C459S of Shp2 results in diminished Shp2 enzymatic activity (24, 25). Based on the above results, we predicted that the phosphatase-inactive Shp2 mutant, in which Cys459 was substituted with serine, would lead to the same phenomenon that was observed in Shp2 inhibitor experiments. To investigate this hypothesis, we transfected the Shp2 WT or C459S mutation together with GFP in DIV8 hippocampal neurons. The effect of TTX treatment (48 h) on surface expression of GluA1 in GFP-positive hippocampal neurons (DIV16) was assessed by immunostaining of cell surface receptors. As expected, hippocampal neurons transfected with Shp2 WT showed increased surface GluA1 expression after TTX treatment, whereas the increase was not observed in neurons transfected with the Shp2 C459S mutation after 48 h of TTX (Fig. 4, A and B; TTX for Shp2 WT, 1.429 ± 0.070; control for Shp2 C459S, 1.052 ± 0.029; TTX for Shp2 C459S, 1.028 ± 0.031). These data suggest that constitutive inactivation of Shp2 impairs surface GluA1 up-regulation in synaptic upscaling.
Figure 4.

Loss of functional Shp2 blocks elevation of the Ser(P)845 level and surface GluA1 after synaptic upscaling. A, hippocampal neurons were transfected with the Shp2 WT or Shp2 C459S mutant (phosphatase-inactive Shp2). After 48-h TTX treatment, surface GluA1 was analyzed. Scale bar = 5 μm. B, quantification of surface GluA1 levels (n = 15 neurons for each condition from three independent experiments). #, p ≤ 0.001 against control. C, hippocampal neurons from Shp2flox/flox mice were infected with AAV-GFP as a control or AAV-Cre to knock out Shp2. Seven days post-infection, 48-h TTX treatment was performed, followed by Western blot analysis with the indicated antibodies. MW, molecular weight. D, quantification of Ser(P)845, GluA1, Tyr(P)542, and Shp2 levels (n = 4 independent experiments). **, p ≤ 0.01; #, p ≤ 0.001 against the control; one-way ANOVA followed by Tukey's post hoc test. E, hippocampal neurons were treated as in C. Surface GluA1 and MAP2 staining was performed after 48-h TTX treatment. Scale bar = 5 μm. F, quantification of surface GluA1 levels (n = 20 neurons for each condition from three independent experiments). #, p ≤ 0.001; one-way ANOVA followed by Tukey's post hoc test.
AAV-mediated KO of Shp2 blocks surface GluA1 up-regulation in synaptic upscaling
To rule out possible off-target effects of the Shp2 inhibitor, we utilized AAV-Cre–mediated KO of Shp2 in hippocampal neurons to evaluate the role of Shp2 in synaptic upscaling. As described in our previous study (9), we infected hippocampal neurons on DIV6–7. Seven days post-infection, TTX was applied to the neurons for 48 h. Shp2, Tyr(P)542, GluA1, and Ser(P)845 levels were measured by Western blot analysis afterward. Genetic ablation of Shp2 prevented an increase in GluA1 and Ser(P)845 levels elicited by 48-h TTX treatment (Fig. 4, C and D; GluA1: TTX for AAV-GFP, 1.632 ± 0.108; control for AAV-Cre, 1.187 ± 0.102; TTX for AAV-Cre, 1.077 ± 0.110; Ser(P)845: TTX for AAV-GFP, 1.686 ± 0.083; control for AAV-Cre, 1.025 ± 0.110; TTX for AAV-Cre, 1.061 ± 0.107; n = 4). In addition, surface staining of GluA1 was accessed on hippocampal neurons in the absence or presence of 48-h TTX treatment in WT or Shp2 KO hippocampal neurons. In line with our results from Shp2 inhibitors and the Shp2-inactive mutant, hippocampal neurons infected with AAV-GFP displayed increased surface GluA1 expression after 48-h TTX treatment, whereas surface GluA1 expression failed to elevate in neurons infected with AAV-Cre after 48 h of TTX (Fig. 4, E and F; TTX for AAV-GFP, 1.334 ± 0.039; control for AAV-Cre, 0.990 ± 0.038; TTX for AAV-Cre, 1.093 ± 0.046). These data suggest that loss of Shp2 function occludes surface GluA1 up-regulation during synaptic upscaling.
Discussion
Previously, we have shown that activity of Shp2 is critical for the increase in Ser(P)845 level in GluA1 as well as surface GluA1 after LTP (9). In this study, we focused on the molecular role of Shp2 in homeostatic plasticity and uncovered a novel role of Shp2 in synaptic upscaling-mediated changes of surface GluA1 by pharmacological and genetic approaches. We found that the phosphorylation level of Shp2 at Tyr542 was regulated by synaptic scaling. Additionally, increased colocalization of Shp2 and PSD95 and accumulated Shp2 sorting into the PSD fraction were observed after synaptic upscaling. The results suggest Shp2 translocation into the postsynaptic site to facilitate its function in the synapse during synaptic upscaling. Recently, we showed that Shp2 rapidly accumulated at the postsynaptic site after LTP stimuli (9). Given both synaptic upscaling and LTP-induced Shp2 translocation into the postsynaptic site, the shared phenomenon of accumulation of Shp2 at the postsynaptic site may contribute to modulating the phosphorylation of GluA1 in the two processes.
We provide evidence that phosphatase activity of Shp2 is critical for regulating GluA1 in synaptic upscaling. Considering that Shp2 constitutes two tandem SH2 domains (14), it is rational to hypothesize that Shp2 may also serve as a scaffolding protein to recruit other signaling proteins during synaptic upscaling for coordinated regulation of GluA1. However, expression of the phosphatase-inactive form of Shp2 in hippocampal neurons sufficed to negate increased surface expression of GluA1 after synaptic upscaling, implying that the phosphatase activity of Shp2 was critical for the process, whereas its scaffolding function exerted minimal effects on synaptic scaling up.
It is worth noting that both acute glycine-induced chemical LTP and chronic TTX treatment drive the accumulation of Shp2 at postsynaptic sites and elevate the Ser(P)845 level, which is Shp2-dependent. The signaling pathways are substantially complex with respect to synaptic plasticity (LTP) and synaptic upscaling, consisting of overlapping and separated signaling nodes for these two processes. Nevertheless, how these two distinct stimuli (chemical LTP and chronic TTX treatment) elicit similar phosphorylation outcomes remains elusive. Our results, demonstrating that Shp2 is activated after synaptic upscaling and that phosphatase activity of Shp2 is vital for elevated surface expression of GluA1, suggest that Shp2 is a common element in the regulation of GluA1 during the two processes. Recently, it has been suggested that Shp2 contributes to ERK1/2 pathway activation during LTP, which would agree with the results presented by us and others (9, 12). However, there is evidence showing that the activity of ERK1/2 was decreased at a time scale of dozens of minutes following blockade of neuronal activity by TTX and that ERK1/2 activity reached a stable level after 6–24 h of TTX treatment (26). The contradiction of activated Shp2 and inactivated ERK1/2 suggests dissociated roles of Shp2 and ERK1/2 during synaptic upscaling, adding complexity to the signaling pathway involved in synaptic scaling.
The downstream signaling mechanisms by which Shp2-dependent regulation of GluA1 is mediated during synaptic scaling remain unclear. Shp2 functions both as a protein phosphatase and scaffolding protein promoting assembly of protein complexes (14–16). Thus, depending on different circumstances, Shp2 may contribute to regulating the ERK1/2, Src family kinase, PI3K—Akt, and NF-κB pathways (27). More study is required to understand by which of the multiple functions and downstream pathways Shp2 regulates phosphorylation of GluA1 at Ser845 during synaptic scaling and the broader processes of synaptic plasticity.
Experimental procedures
DNA constructs
The cDNAs encoding WT mouse Shp2 were subcloned into the pMSCV-FLAG vector between the BamHI and KpnI restriction sites. This was a gift from Dr. Yuehai Ke (Zhejiang University) and has been described previously (28). The Shp2 C459S mutant was generated by site-directed mutagenesis using the QuikChange mutagenesis system (Stratagene, La Jolla, CA). EGFP-N1 was from Clontech. All cDNA constructs were verified by DNA sequencing before use.
Hippocampal neuron culture and transfection
The use and care of animals in this study were performed in strict accordance with Hangzhou Normal University Animal Experimentation Committee guidelines and completely followed the guidelines of the National Institutes of Health Animal Research Advisory Committee. We made maximal efforts to minimize animal numbers and to reduce animal suffering. Hippocampal primary neuronal cultures were harvested from embryonic day 18 Sprague-Dawley rats or newborn (postnatal day 1–2) Shp2flox/flox mice of either sex, as described previously (9, 29). Hippocampal tissue was quickly dissected in ice-cold Hanks' balanced salt solution (HBSS) buffer and then digested in 0.5% trypsin for 15 min at 37 °C. Dissociated neurons were seeded on poly-l-lysine-coated (Sigma-Aldrich, St. Louis, MO) coverslips (Deckgläser, Carolina Biological, Burlington, NC) or dishes (Corning, Rochester, NY) at a proper density in Neurobasal medium containing 10% horse serum at 37 °C under 5% CO2. Four hours later, the primary medium was substituted with Neurobasal medium containing 0.5 mm GlutaMax supplemented with 2% B27 and 1% penicillin/streptomycin. Subsequently, the culture medium was replaced every 4 days. At days in vitro 4 (DIV4), cytosine arabinoside was added into the culture medium at a final concentration of 2.5 μm. All buffers were purchased from Gibco (Life Technologies) unless otherwise stated.
EGFP-N1 was co-transfected with Shp2 WT or mutant C459S at a mass ratio of 1:10 at DIV8 using the CalPhosTM mammalian transfection kit (Clontech). Shp2flox/flox neurons were infected with adeno-associated viruses at DIV7. All cell culture reagents were acquired from Gibco (Life Technologies) unless otherwise stated.
Synaptic scaling induction and drug treatment
To induce homeostatic scaling, hippocampal neurons at DIV14–16 were incubated for 48 h with bicuculline or TTX at a final concentration of 40 μm or 1 μm, respectively. For some experiments, TTX was used in combination with PHPS1 (5 μm) or NSC87877 (10 μm). Neurons treated with DMSO were considered the control group.
Immunocytochemistry
We followed a protocol described previously for surface staining of GluA1 (9). Briefly, hippocampal neurons were washed twice with 37 °C PBS. Immediately after washing, surface GluA1 was labeled with mouse anti-GluA1 (MAB2263, Millipore, 1:50) for 15 min at 37 °C. Neurons were washed and fixed and then incubated with the respective secondary antibody for 15 min at 37 °C to visualize surface GluA1.
For staining of endogenous Shp2 and PSD95, neurons were fixed with 4% paraformaldehyde for 10 min and permeabilized and blocked simultaneously in PBS buffer containing 0.2% Triton X-100 and 5% BSA for 0.5 h. Then Shp2 was labeled with rabbit anti-Shp2 (SC-280, Santa Cruz Biotechnology, 1:50) and PSD95 was labeled with mouse anti-PSD95 (ab2723, Abcam, 1:500) primary antibodies for 1 h at room temperature. Neurons were washed and incubated with the respective secondary antibody for 1 h at room temperature or overnight at 4 °C. For colocalization of Shp2 and PSD95, donkey anti-rabbit Alexa 488–conjugated (A21206, Invitrogen, 1:500) and goat anti-mouse Alexa 546–conjugated (A11030, Invitrogen, 1:500) secondary antibodies were used. For the PHPS1 and NSC87877 treatment experiment, donkey anti-mouse Alexa 488–conjugated (A21202, Invitrogen, 1:500) antibody was used for surface GluA1 and goat anti-rabbit Alexa 633–conjugated (A21082, Invitrogen, 1:500) antibody was used for MAP2. For AAV-mediated Shp2 KO neurons or neurons co-transfected with EGFP-N1 and Shp2 or C459S, surface GluA1 was labeled with goat anti-mouse Alexa 546–conjugated (A11030, Invitrogen, 1:500) secondary antibody, and MAP2 was labeled with goat anti-rabbit Alexa 633–conjugated (A21082, Invitrogen, 1: 500) secondary antibody.
Cells were imaged on a confocal microscope (Fluoview FV1000, Olympus). Manders coefficiency was analyzed using the ImageJ plugin JACoP, and the percentage of Shp2 colocalized with PSD95 was analyzed by manually counting the overlapping cluster number of Shp2 and PSD95, which was divided by the total cluster number of PSD95. For surface AMPA receptor intensity, the integrated intensity of individual puncta of endogenous surface GluA1 per unit length of dendrite was measured, and the ratio of surface GluA1/MAP2 in other groups was normalized to the ratio of the control group. Experiments were repeated at least three times independently, and significance was analyzed using one-way ANOVA followed by Tukey's post hoc test (n = number of cells).
PSD fractionation of cultured neurons
PSD fractionation was performed as described previously (9, 30).
Immunoblotting
Western blotting was adopted from previous publications with minor modifications (9, 30). After pharmacological treatments, neurons were washed with ice-cold PBS and lysed by radioimmune precipitation assay buffer containing 10 mm Tris-HCl (pH 7.4), 0.15 m NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, and 1% sodium deoxycholate (Beyotime, Shanghai, China). Then the cell lysate was clarified by centrifugation at 16,000 × g for 30 min. Subsequently, the supernatant was quantified using a BCA protein assay kit (Thermo Fisher Scientific, Pittsburgh, PA). Samples containing equal amounts of protein were denatured in loading buffer by boiling for 5 min, subjected to SDS-PAGE, transferred onto nitrocellulose membranes (Whatman, GE Healthcare), and subjected to immunoblot analysis. Blots were probed with ECL reagent (Thermo Fisher Scientific), and chemiluminescence was detected by photographic film. Protein bands were analyzed with Quantity One software (Bio-Rad).
Antibodies and reagents
The primary antibodies used in immunoblotting were rabbit anti-Shp2 (SC-280, Santa Cruz Biotechnology, 1:5000), rabbit anti-pShp2-Tyr542 (ab62322, Abcam, 1:5000), mouse anti-GluA1 (MAB2263, Millipore, 1:2000), rabbit anti-pGluA1-Ser845 (04-1073, Millipore, 1:1000), and mouse anti-β-actin (A5316, Sigma-Aldrich, 1:10,000). Rabbit anti-MAP2 antibody (4542, Cell Signaling Technology, 1:200) was used for immunocytochemistry. The secondary antibodies for immunoblotting were goat anti-rabbit IgG-HRP–conjugated secondary antibody (31460, Pierce, 1:10,000) and goat anti-mouse IgG-HRP–conjugated secondary antibody (31460, Pierce, 1:10,000).
TTX (1078), Bic (2503), and NSC87877 (2613) were acquired from Tocris Bioscience (Minneapolis, MN), and PHPS1 (sc-253272) was purchased from Santa Cruz Biotechnology. AAVs, both AAV-CaMKII-GFP-Cre and AAV-CaMKII-GFP, were packaged by Obio Technology (Shanghai, China) and have been described previously (9).
Statistical analysis
All data were analyzed using GraphPad prism 6.0 software (GraphPad Software, La Jolla, CA) and are represented as mean ± S.E. Comparisons of two groups were done by Student's t test. Data of multiple groups (three or more groups) were tested using one-way ANOVA followed by Tukey's post hoc test. p ≤ 0.05 was considered significant. No significance (NS) indicates p > 0.05.
Author contributions
B. Z. designed and performed the biochemical and immunostaining experiments. W. L. designed and performed some biochemical experiments, analyzed the data, and conceived and coordinated the study. B. Z. and W. L. wrote the paper. Both authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. Yuehai Ke for providing the Shp2flox/flox mice and Shp2 WT plasmid. We also thank Prof. Yang Liu for critical reading of the manuscript.
This work was supported by National Natural Science Foundation of China Grant 31600863 (to W. L.) and a start-up fund of Hainan Medical University (to W. L.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Fig. S1.
- LTP
- long-term potentiation
- LTD
- long-term depression
- SH
- Src homology
- PTP
- protein–tyrosine phosphatase
- AAV
- adeno-associated virus
- Bic
- bicuculline
- TTX
- tetrodotoxin
- PSD
- postsynaptic density
- DIV
- days in vitro
- ANOVA
- analysis of variance
- NS
- no significance
- NSC
- NSC87877.
References
- 1. Pozo K., and Goda Y. (2010) Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Citri A., and Malenka R. C. (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41 [DOI] [PubMed] [Google Scholar]
- 3. Kessels H. W., and Malinow R. (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Turrigiano G. G. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anggono V., and Huganir R. L. (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 22, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shepherd J. D., and Huganir R. L. (2007) The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643 [DOI] [PubMed] [Google Scholar]
- 7. Oh M. C., Derkach V. A., Guire E. S., and Soderling T. R. (2006) Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 281, 752–758 [DOI] [PubMed] [Google Scholar]
- 8. Lee H. K., Barbarosie M., Kameyama K., Bear M. F., and Huganir R. L. (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959 [DOI] [PubMed] [Google Scholar]
- 9. Zhang B., Du Y. L., Lu W., Yan X. Y., Yang Q., Yang W., and Luo J. H. (2016) Increased activity of Src homology 2 domain containing phosphotyrosine phosphatase 2 (Shp2) regulates activity-dependent AMPA receptor trafficking. J. Biol. Chem. 291, 18856–18866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pagani M. R., Oishi K., Gelb B. D., and Zhong Y. (2009) The phosphatase SHP2 regulates the spacing effect for long-term memory induction. Cell 139, 186–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kusakari S., Saitow F., Ago Y., Shibasaki K., Sato-Hashimoto M., Matsuzaki Y., Kotani T., Murata Y., Hirai H., Matsuda T., Suzuki H., Matozaki T., and Ohnishi H. (2015) Shp2 in forebrain neurons regulates synaptic plasticity, locomotion, and memory formation in mice. Mol. Cell. Biol. 35, 1557–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee Y. S., Ehninger D., Zhou M., Oh J. Y., Kang M., Kwak C., Ryu H. H., Butz D., Araki T., Cai Y., Balaji J., Sano Y., Nam C. I., Kim H. K., Kaang B. K., et al. (2014) Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat. Neurosci. 17, 1736–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan X., Zhang B., Lu W., Peng L., Yang Q., Cao W., Lin S., Yu W., Li X., Ke Y., Li S., Yang W., and Luo J. (2016) Increased Src family kinase activity disrupts excitatory synaptic transmission and impairs remote fear memory in forebrain Shp2-deficient mice. Mol. Neurobiol. 10.1007/s12035-016-0222-7 [DOI] [PubMed] [Google Scholar]
- 14. Dance M., Montagner A., Salles J. P., Yart A., and Raynal P. (2008) The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 20, 453–459 [DOI] [PubMed] [Google Scholar]
- 15. Neel B. G., Gu H., and Pao L. (2003) The “Shp”ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 [DOI] [PubMed] [Google Scholar]
- 16. Poole A. W., and Jones M. L. (2005) A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell. Signal. 17, 1323–1332 [DOI] [PubMed] [Google Scholar]
- 17. Prahallad A., Heynen G. J., Germano G., Willems S. M., Evers B., Vecchione L., Gambino V., Lieftink C., Beijersbergen R. L., Di Nicolantonio F., Bardelli A., and Bernards R. (2015) PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep. 12, 1978–1985 [DOI] [PubMed] [Google Scholar]
- 18. Bentires-Alj M., Paez J. G., David F. S., Keilhack H., Halmos B., Naoki K., Maris J. M., Richardson A., Bardelli A., Sugarbaker D. J., Richards W. G., Du J., Girard L., Minna J. D., Loh M. L., et al. (2004) Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 64, 8816–8820 [DOI] [PubMed] [Google Scholar]
- 19. Aceto N., Sausgruber N., Brinkhaus H., Gaidatzis D., Martiny-Baron G., Mazzarol G., Confalonieri S., Quarto M., Hu G., Balwierz P. J., Pachkov M., Elledge S. J., van Nimwegen E., Stadler M. B., and Bentires-Alj M. (2012) Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat. Med. 18, 529–537 [DOI] [PubMed] [Google Scholar]
- 20. Han T., Xiang D. M., Sun W., Liu N., Sun H. L., Wen W., Shen W. F., Wang R. Y., Chen C., Wang X., Cheng Z., Li H. Y., Wu M. C., Cong W. M., Feng G. S., et al. (2015) PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J. Hepatol. 63, 651–660 [DOI] [PubMed] [Google Scholar]
- 21. Diering G. H., Gustina A. S., and Huganir R. L. (2014) PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anggono V., Clem R. L., and Huganir R. L. (2011) PICK1 loss of function occludes homeostatic synaptic scaling. J. Neurosci. 31, 2188–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hellmuth K., Grosskopf S., Lum C. T., Würtele M., Röder N., von Kries J. P., Rosario M., Rademann J., and Birchmeier W. (2008) Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc. Natl. Acad. Sci. U.S.A. 105, 7275–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanafusa H., Torii S., Yasunaga T., Matsumoto K., and Nishida E. (2004) Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J. Biol. Chem. 279, 22992–22995 [DOI] [PubMed] [Google Scholar]
- 25. Park C. Y., and Hayman M. J. (1999) The tyrosines in the bidentate motif of the env-sea oncoprotein are essential for cell transformation and are binding sites for Grb2 and the tyrosine phosphatase SHP-2. J. Biol. Chem. 274, 7583–7590 [DOI] [PubMed] [Google Scholar]
- 26. Bateup H. S., Denefrio C. L., Johnson C. A., Saulnier J. L., and Sabatini B. L. (2013) Temporal dynamics of a homeostatic pathway controlling neural network activity. Front. Mol. Neurosci. 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan G., Kalaitzidis D., and Neel B. G. (2008) The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 27, 179–192 [DOI] [PubMed] [Google Scholar]
- 28. Meng S., Gui Q., Xu Q., Lu K., Jiao X., Fan J., Ge B., Ke Y., Zhang S., Wu J., and Wang C. (2010) Association of Shp2 with phosphorylated IL-22R1 is required for interleukin-22-induced MAP kinase activation. J. Mol. Cell Biol. 2, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X. M., Yan X. Y., Zhang B., Yang Q., Ye M., Cao W., Qiang W. B., Zhu L. J., Du Y. L., Xu X. X., Wang J. S., Xu F., Lu W., Qiu S., Yang W., and Luo J. H. (2015) Activity-induced synaptic delivery of the GluN2A-containing NMDA receptor is dependent on endoplasmic reticulum chaperone Bip and involved in fear memory. Cell Res. 25, 818–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu W., Fang W., Li J., Zhang B., Yang Q., Yan X., Peng L., Ai H., Wang J. J., Liu X., Luo J., and Yang W. (2015) Phosphorylation of tyrosine 1070 at the GluN2B subunit is regulated by synaptic activity and critical for surface expression of N-methyl-d-aspartate (NMDA) receptors. J. Biol. Chem. 290, 22945–22954 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.