

Aldosterone is the most important mineralocorticoid for electrolyte and fluid homeostasis in mammals, birds, and some members of other orders. While critical for life, even subtle inappropriate increases in aldosterone contribute to the development of hypertension and cardiovascular damage. Primary aldosteronism (PA), the autonomous and excessive secretion of aldosterone, is the most common form of secondary hypertension and is associated with more pathological cardiovascular remodeling than essential hypertension of a similar duration and severity. Aldosterone-producing adenomas and idiopathic hyperaldosteronism are responsible for most primary aldosteronism. Despite the importance of PA in cardiovascular and renal disease, its pathogenesis is only partially clarified in aldosterone-producing adenomas and remains unknown in idiopathic hyperaldosteronism. A report in this issue by Yao et al1 provides crucial new information on the normal control of aldosterone synthesis that could elucidate mechanisms for its dysregulation in PA. They used elegant technology to demonstrate that TASK-3 K+ channels locates in the mitochondria and interact with proteins of the electron transport chain of the mitochondrial inner membrane and regulate mitochondrial membrane potential and morphology. They also showed that TASK-3 channels interact with the P450 side-chain cleavage enzyme that catalyzes the first step in the synthesis of all steroids from cholesterol, as well as with CYP11B2, or aldosterone synthase, the last and unique enzyme in a series of enzymatic reactions within the mitochondria.
Synthesis of aldosterone is regulated primarily by angiotensin II (A-II) and plasma potassium (K+), with lesser influence by ACTH and several paracrine factors2 at two points in its synthetic pathway. Within minutes after stimulation by A-II or K+ there is a rapid mobilization of cholesterol from lipid droplets and its transport from the outer to the inner mitochondrial membrane, a process requiring the phosphorylation and activation of StAR protein2. Cholesterol is hydroxylated and its side-chain cleaved by the P450 side chain cleavage enzyme on the inner mitochondrial membrane, generating pregnenolone, which is then acted upon by the 3β–hydroxysteroid dehydrogenase-Δ-4-5 isomerase in both the mitochondria and endoplasmic reticulum (ER) to generate progesterone. Progesterone is then hydroxylated by the 21-hydroxylase enzyme within the ER to generate deoxycorticosterone (DOC), which is then transferred to the mitochondria, where the CYP11B2 converts it successively to corticosterone, 18-hydroxycorticosterone, then finally aldosterone2. The conversion of DOC to aldosterone is the second regulatory point for aldosterone synthesis3. It was presumed to involve stimulation of CYP11B2 transcription, however, increased conversion of DOC into aldosterone in freshly isolated rat or dog zona glomerulosa cells occurs before a significant increase in transcription and translation of the CYP11B2 is likely to occur3. The membrane potential across both the plasma and mitochondrial membranes of zona glomerulosa cells regulates Ca++ transport into both the cell and mitochondria, contributing to the regulation of steroid production. A-II mediates calcium (Ca++) release from intracellular stores through store-operated and voltage-dependent Ca++ channels, whereas K+ depolarizes the membrane, thereby activating T-type voltage-dependent Ca++ Channels4. The mitochondrial Ca++ signal enhances pyridine nucleotide reduction within the mitochondrial matrix, resulting in increased ATP production and increased steroid production through NADH and NADPH generation, respectively4.
Yao et al show that mitochondria not only express the K+ selective channels mito KATP (K-ir 6.1/6.2), mito BKCa (slo 1) and mito Kv1.3, but also TASK-3, that reduce mitochondrial membrane potential and Ca++ uptake, and thus protecting the cell from apoptosis1, and also abrogating the stimulus for aldosterone synthesis. The current studies by Yao et al1 raise the possibility that rapid changes in the mitochondrial membrane potential mediated by A-II or K+ lead to an increase in the efficiency of CYP11B2 synthesis of aldosterone. As a corollary, dysregulation of the mitochondrial membrane potential may be involved in the genesis of autonomous aldosterone production in PA. The TASK-3 antagonist, C23, stimulated aldosterone secretion in the human adrenocortical carcinoma cell H295R modestly even in cells stimulated by A-II, however the modest stimulation may be due to the very low expression of TASK 3 in the H295R cells1.
Among several animal models developed to elucidate etiologies of hyperaldosteronism is a mouse in which the TASK-3 gene has been deleted, resulting in low renin high aldosterone hypertension5 that resembles human low renin hypertension. Deletion of the TASK-1 gene produced mice with a more complicated phenotype6, 7. The pattern of potassium channel expression in rodents and human differ significantly, TASK-1 is expressed both in the human, mice and rat, but TASK-3 is expressed in high levels in the mouse zona glomerulosa, but in very low levels in the human. Conversely, KCNJ5 that is highly expressed in the zona glomerulosa of the human but is not expressed in the rat8. Notwithstanding, the pattern of increased Ca++ mobilization involving the mitochondria and/or the plasma membrane is common to all species so far studied.
In 2011, the Lifton laboratory made the seminal discovery that somatic mutations of the G-protein activated inward rectifier potassium channel KCNJ5, also named GIRK4 or KIR3.4, occurs in a significant number of aldosterone-producing adenomas9. Since then, several laboratories have shown that mutations in the calcium channel genes CACNA1D and CACNA1H, the alpha subunit of the sodium potassium ATPase ATP1A1 gene, and the membrane calcium ATPase, ATP2B3 gene are responsible for increased aldosterone synthesis in aldosterone producing adenomas (reviewed in10). These mutations increase cytoplasmic Ca++ directly or indirectly through membrane depolarization, which activates Ca++ -dependent signal transduction pathways, including that of calmodulin kinases2. Somatic mutations similar to the ones found in aldosterone-producing adenomas, most commonly in the CACNA1D gene, have been found in about a third of aldosterone-producing cell clusters in human adrenals with normal aldosterone levels11. The significance of this is not yet clear.
The mechanisms responsible for the loss of regulated aldosterone synthesis in idiopathic hyperaldosteronism are unknown. To date, all genetic modifications leading to hyperaldosteronism in mice are gene deletions, while all mutations found so far in aldosterone-producing adenomas have been gain of function mutations. Information from mouse models prompted searches for mutations or polymorphisms producing loss of function of TASK-1 or analogous genes correlating idiopathic hyperaldosteronism. Genome-wide association studies identified KCNK3 (TASK-1) SNPs variants associated with mean blood pressure in humans12. A statistically significant association between baseline blood pressure measurements and the KCNK3 SNP (rs1275988) in African Americans and a nearby SNP (rs13394970) in Hispanics was found in the Multi-Ethnic Study of Atherosclerosis (MESA) comprising 7840 participants. Furthermore, aldosterone levels and plasma renin activity in a subset of 1653 participants in which they were measured also associated significantly with KCNK3 rs2586886. The effects of these SNPs on gene expression or ion channel function are yet unknown. Information from existing gene-deleted animal models has not translated into mechanisms of human hyperaldosteronism; better models are needed.
The new findings by Yao et al1 bring up another issue that has been ignored since the kinetics of CYP11B2 was first described and confirmed. CYP11B2 is a relatively inefficient partial processing enzyme which successively hydroxylates DOC to corticosterone, 18-hydroxycorticosterone, and then aldosterone. Corticosterone and 18-hydroxycorticosterone are produced in much higher quantities than aldosterone. Does the increase in K+ within the mitochondria alter the function of the CYP11B2 increasing the efficient conversion of DOC to aldosterone? Or does it facilitate the transfer of deoxycorticosterone from outside to inside the mitochondria? (Figure 1).
Figure 1.
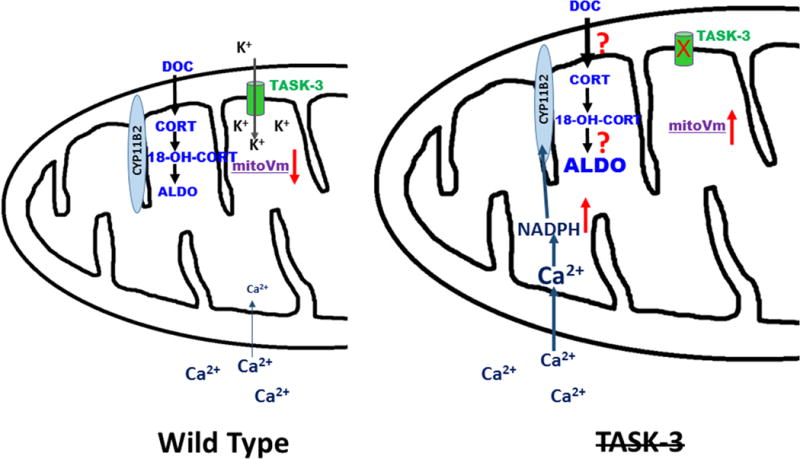
Hypothetical view of the action of TASK-3 in zona glomerulosa cell mitochondria. Normally TASK-3 contributes to the negative membrane potential in mitochondria. Deletion or inhibition of TASK-3 is postulated to depolarize the mitochondria, leading to increased intramitochondrial Ca2+, increasing the generation of NADPH, which increases the activity of the CYP11B2 enzyme. It may also increase the transport of DOC into the mitochondria by an unknown mechanism.
Acknowledgments
Sources of Funding:
CEGS and MK were supported by National Heart, Lung and Blood Institute grant R01 HL27255 and the National Institute of General Medical Sciences under Award Number 1U54GM115428. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosures: None.
References
- 1.Yao J, McHedlishvili D, McIntire WE, Guagliardo NA, Erisir A, Coburn CA, Santarelli VP, Bayliss DA, Barrett PQ. Functional task-3-like channels in mitochondria of aldosterone producing zona glomerulosa cells. Hypertension. 2017 doi: 10.1161/HYPERTENSIONAHA.116.08871. “in press”. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Molecular and Cellular Endocrinology. 2012;350:151–162. doi: 10.1016/j.mce.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilera G, Catt KJ. Loci of action of regulators of aldosterone biosynthesis in isolated glomerulosa cells. Endocrinology. 1979;104:1046–1052. doi: 10.1210/endo-104-4-1046. [DOI] [PubMed] [Google Scholar]
- 4.Spat A, Hunyady L, Szanda G. Signaling interactions in the adrenal cortex. Frontiers in Endocrinology. 2016;7:17. doi: 10.3389/fendo.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guagliardo NA, Yao J, Hu C, Schertz EM, Tyson DA, Carey RM, Bayliss DA, Barrett PQ. Task-3 channel deletion in mice recapitulates low-renin essential hypertension. Hypertension. 2012;59:999–1005. doi: 10.1161/HYPERTENSIONAHA.111.189662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heitzmann D, Derand R, Jungbauer S, et al. Invalidation of task1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. Embo J. 2008;27:179–187. doi: 10.1038/sj.emboj.7601934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, Carey RM, Bayliss DA, Barrett PQ. Task channel deletion in mice causes primary hyperaldosteronism. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2203–2208. doi: 10.1073/pnas.0712000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen AX, Nishimoto K, Nanba K, Rainey WE. Potassium channels related to primary aldosteronism: Expression similarities and differences between human and rat adrenals. Molecular and Cellular Endocrinology. 2015;417:141–148. doi: 10.1016/j.mce.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seidel E, Scholl UI. Intracellular molecular differences in aldosterone- compared to cortisol-secreting adrenal cortical adenomas. Frontiers in Endocrinology. 2016;7:75. doi: 10.3389/fendo.2016.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez-Sanchez CE, Nanba K, Rainey WE. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E4591–4599. doi: 10.1073/pnas.1505529112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung J, Barrett PQ, Eckert GJ, Edenberg HJ, Xuei X, Tu W, Pratt JH. Variations in the potassium channel genes kcnk3 and kcnk9 in relation to blood pressure and aldosterone production: An exploratory study. The Journal of Clinical Endocrinology and Metabolism. 2012;97:E2160–2167. doi: 10.1210/jc.2012-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]