

Abstract
Given the putative role of PHGDH in cancer, development of inhibitors is required to explore its function. In this context, we established and validated a straightforward enzymatic assay suitable for high-throughput screening and we identified inhibitors with similar chemical scaffolds. Through a convergent pharmacophore approach, we synthesized α-ketothioamides that exhibit interesting in vitro PHGDH inhibition and encouraging cellular results. These novel probes may be used to understand the emerging biology of this metabolic target.
Graphical Abstract
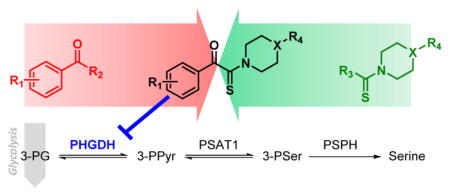
INTRODUCTION
In this past decade, important efforts have been made by scientists to characterize the metabolic alterations in tumor cells and it has been demonstrated that cancer cells exhibit an increase in the uptake of serine and glycine.1 These amino acids provide essential precursors for the synthesis of proteins, nucleic acids, and lipids that are crucial to cancer cell growth.2 Moreover, serine/glycine biosynthesis also affects cellular antioxidative capacity, thus supporting tumor homeostasis.
Serine can be imported from the extracellular compartment via amino acid transporters, but it can also be synthesized by an intracellular pathway that consists of three successive enzymatic reactions (Figure 1). Phosphoglycerate dehydrogenase (PHGDH) catalyzes the first step of this pathway and produces 3-phosphohydroxypyruvate (3-PPyr) by NAD+-coupled oxidation of 3-phosphoglycerate (3-PG). Subsequently, 3-PPyr is converted to phosphoserine by phosphoserine aminotransferase 1 (PSAT1) and then to serine by phosphoserine phosphatase (PSPH).
Figure 1.
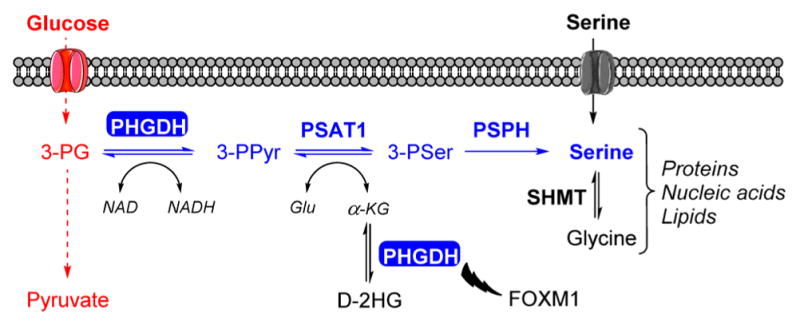
Schematic representation of the SSP. SSP (represented in blue) is a branching route of the glycolysis (represented in red) that contributes to the production of proteins, nucleic acids, and lipids. In addition to catalyzing the oxidation of 3-PG, PHGDH catalyzes the reduction of α-KG to D-2HG and stabilizes FOXM1.
In 2011, Pollari and co-workers described the first clinical results associated with serine biosynthetic elevation in cancer cells.3 More precisely, they demonstrated that the three genes involved in the serine biosynthesis were upregulated in the highly metastatic MDA-MB-231(SA) breast cancer cell line compared to the parental cell line MDA-MB-231. Subsequently, the role of the first serine synthetic pathway (SSP) enzyme, PHGDH, as a putative metabolic oncogene has emerged following two important publications near-simultaneously in 2011. Possemato et al. identified PHGDH as a metabolic gene required for tumorigenesis via an in vivo shRNA screen, while Locasale et al. found that in some cancer cells, a relatively large amount of glycolytic carbon was diverted into serine and glycine biosynthesis due to genomic amplification of PHGDH.4,5 In these two studies, RNAi-mediated suppression of PHGDH caused a decrease in survival and growth of cancer cells that harbored PHGDH amplification or PHGDH overexpression but had no effect on lines lacking PHGDH. A recent study also demonstrated the role of PHGDH in metastasis formation and stem cell phenotype of breast cancer cells.6 Since 2011, several works extended the purported role of PHGDH in a large panel of other cancers such as colon cancer, glioma, lung cancer, thyroid cancer, or leukemia.7–12
The mechanism by which PHGDH supports tumorigenesis is not yet fully understood. Indeed, it was suggested that serine production is not the only major role for PHGDH in cancer cells. This enzyme also can produce metabolites beyond serine that are essential for cell proliferation, invasion, and tumorigenicity of cancer cells.
These reports strongly implicate PHGDH as an attractive drug target in tumors that overexpress PHGDH or have PHGDH gene amplification.13 Currently, only few specific inhibitors of PHGDH have been described, severely limiting investigation into this interesting novel cancer target (Figure 2).14–16 To address this need, we have initiated a study toward the discovery of PHGDH inhibitors and herein, we describe this novel class of inhibitors.
Figure 2.
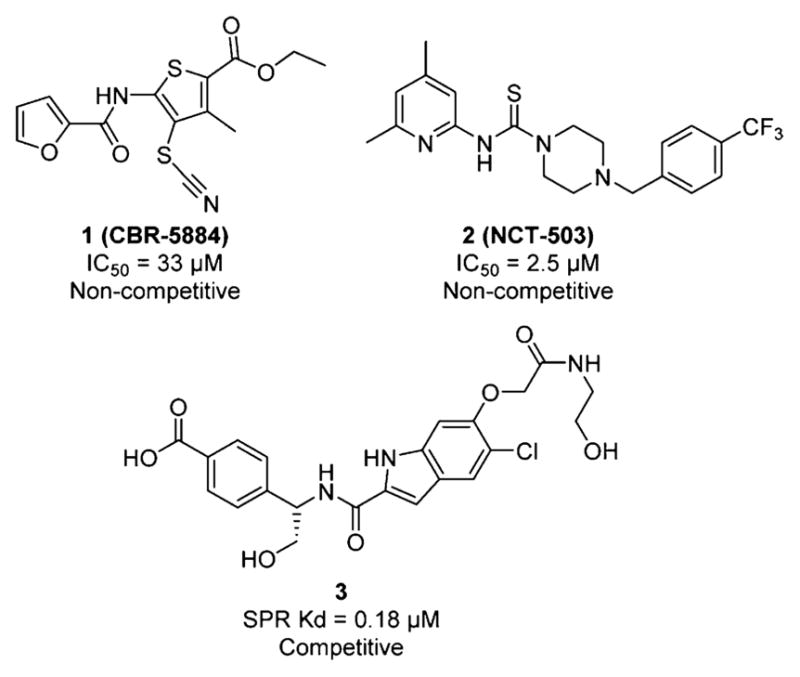
Structure of reported PHGDH inhibitors.
RESULTS AND DISCUSSION
Enzymatic Assay Development
Prior to screening the compound library, we initially optimize a robust quantitative enzymatic assay that is based on PHGDH activity. More precisely, PHGDH oxidizes 3-PG to 3-PPyr with NAD+ as the electron acceptor to yield NADH. The formation of 3-PPyr is therefore directly correlated with the NADH formation, and the enzymatic activity of PHGDH can be monitored by following the fluorescence intensity (excitation wavelength 340 nm; emission wavelength 460 nm). After optimization of the assay (Supporting Information, Figure S1), we undertook the process of hit identification.
Primary Screening
As described in Figure 3, the primary screening was carried out on a compound library of 336 molecules from a fragment library and an in-house compound collection at high concentration (100 μM). Commercial fragments (4–7) were purchased from Prestwik Chemical, and most of them are derived from actual drugs following smart fragmentation. The in-house collection was composed by original molecules (8–18) showing a large structural diversity.
Figure 3.

Process of hit identification. PHGDH inhibition percentage at 100 μM is represented under each identified hit.
Of the 336 samples initially screened, 29 compounds (8.6%) exhibited an inhibitory effect, and among them, 15 molecules (4.5%) decreased PHGDH activity by greater than 50%. Among them, three molecules were quinone derivatives (4, 16, 18). It is well-known that quinones represent a class of toxicological compounds which can promote a variety of side effects in vivo, including acute cytotoxicity, immunotoxicity, and carcinogenesis.17 For this reason, these three molecules were not selected for the optimization process. The screening also highlighted promising fragments like 6 that will be the subject of further optimization studies.
As highlighted in Figure 3, the screening also highlighted several inhibitors that present common structural groups. For example, several compounds contain a nitrogen moiety such as morpholine, piperazine, or cyclohexamine that is often attached to a thiocarbonyl (8–11,17). This observation is consistent with the recent results of Pacold et al. that highlighted PHGDH inhibitors containing a piperazine-1-carbothioamide scaffold (see compound 2).16 Three other compounds (12–14) showed an acetophenone part attached to a bulky group such as aniline, biphenylmethylamine, or aminopyrimidine.
PHGDH Inhibitors Design
On the basis of the primary screening, we have merged the arylketo moiety of 12–14 and the highlighted thioamide template to design α-ketothioamides (Figure 4). This operation of design which was undertaken is sometimes referred to as convergent pharmacophore synthesis.
Figure 4.

Process of convergent pharmacophore operation to design α-ketothioamides.
Chemistry
To warrant a proof of concept, a limited set of α-ketothioamides was synthesized (Scheme 1). First, the bromination of appropriate acetophenone (1 equiv) was carried out in the presence of dibromine (1.2 equiv) and TBAB in chloroform. The resulting product was engaged in the Willgerodt–Kindler reaction without any further purification according to a reported procedure.18 DMF, octasulfur (1.5 equiv), and morpholine (3 equiv) were introduced into the reaction mixture and the reaction proceeded to completion at room temperature.
Scheme 1. Synthetic Pathway of α-Ketothioamide Derivativesa.
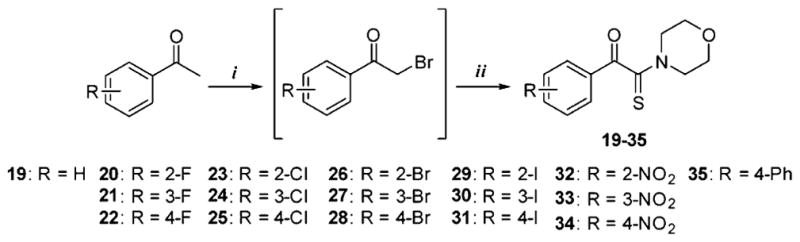
aReagents and conditions: (i) Br2, CHCl3, TBAB, rt for 2 h; (ii) S8, morpholine, DMF, rt.
Enzymatic Evaluation
All the synthesized compounds (19–35) were evaluated on PHGDH thanks to a coupled-enzyme assay (Table 1). Compound 25 is the strict fusion between hit compounds 12 and 8 identified during the primary screening. Given the IC50 values of these three compounds on PHGDH (8.7, 75.2, and 16.0 μM, respectively), we can validate the design based on the convergent pharmacophore synthesis. In addition, within this concise series of α-ketothioamides, it appears that the para-substitution in the aryl moiety is more favorable than the substitution in the ortho or meta-position, as illustrated by the fluoro (22: 68.9 μM), chloro (25: 8.7 μM), and nitro (34: 35.1 μM) derivatives. However, only the 4-chloro derivative 25 exhibits a better inhibition than the nonsubstituted α-ketothioamide 19.
Table 1.
PHGDH Inhibition and Cell Proliferation Inhibition Results of Compounds 19–35
| compd | R | PHGDH inhibition (IC50, μM)a | cell proliferation inhibition
(IC50, μM)a
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MDA-MB-231 | SiHa | HL-60 shCTRL | HL-60 shPHGDH | |||||||
|
|
|
|||||||||
| +Ser | −Ser | IC50+Ser/IC50−Ser | +Ser | −Ser | IC50+Ser/IC50−Ser | |||||
| 12 | 75.2 | >100 | >100 | 57.5 | >1.7 | >100 | 83 | >1.2 | >100 | |
| 8 | 16.0 | >100 | 19.1 | 5.6 | 3.4 | 24.3 | 7.0 | 3.5 | >100 | |
| 19 | H | 30.9 | >100 | 71.7 | 7.9 | 9,1 | >100 | 18.3 | >5.5 | >100 |
| 20 | 2-F | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
| 21 | 3-F | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
| 22 | 4-F | 68.9 | >100 | >100 | 43.3 | >2.3 | >100 | 64.4 | 1.5 | >100 |
| 23 | 2-Cl | >200 | >100 | >100 | >100 | >100 | >100 | >100 | ||
| 24 | 3-Cl | >200 | >100 | >100 | >100 | >100 | >100 | >100 | ||
| 25 | 4-Cl | 8.7 | >100 | 21.0 | 6.6 | 3.2 | 50.5 | 15.5 | 3.3 | >100 |
| 26 | 2-Br | 160.7 | nd | nd | nd | nd | nd | nd | nd | nd |
| 27 | 3-Br | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
| 28 | 4-Br | 130.7 | >100 | 20.4 | 7.9 | 2.6 | 37.2 | 16.9 | 2.2 | >100 |
| 29 | 2-I | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
| 30 | 3-I | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
| 31 | 4-I | 177.7 | >100 | 25.7 | 9.3 | 2.8 | 19.6 | 7.1 | 2.8 | >100 |
| 32 | 2-NO2 | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
| 33 | 3-NO2 | 88.1 | nd | nd | nd | nd | nd | nd | nd | nd |
| 34 | 4-NO2 | 35.1 | >100 | 22.8 | 7.6 | 3 | 17.6 | 6.0 | 2.9 | >100 |
| 35 | 4-Ph | >200 | nd | nd | nd | nd | nd | nd | nd | nd |
All experiments to determine IC50 values were performed with at least triplicates at each compound dilution, and all IC50 values were averaged when determined in two or more independent experiments. nd = not determined.
Cellular Assays
To determine if the preliminary SARs can be translated to cellular activities, the nonsubstituted compound (19), the 4-substituted derivatives (22, 25, 28, 31, and 34) as well as the 2- and 3-chloro α-ketothioamides (23 and 24) were evaluated in several cell-based assays (Table 1).
First of all, the proliferation ability of the three selected cancer cell lines (MDA-MB-231, human breast adenocarcinoma cells (PHGDH−); SiHa, human cervix carcinoma cells (PHGDH+); HL-60, human promyelocytic leukemia cells (PHGDH+); see immunoblots in Figure 5A, C) was evaluated in serine replete or deplete medium. As depicted in Figure 5B, D, extracellular depletion of serine had low effect on proliferation of high PHGDH-expressing lines (SiHa and HL-60-shScr) while the lack of serine considerably decreased the proliferation of the cell line lacking PHGDH (MDA-MB-231 and HL-60-shPHGDH). Given that the ability to proliferate in the absence of extracellular serine is indicative of a high dependence for serine synthesis, SiHa and HL-60-shScr cell lines should be sensitive to α-ketothioamides. Conversely, the identified inhibitors should have no effect on MDA-MB-231 and HL-60-shPHGDH cell lines.
Figure 5.
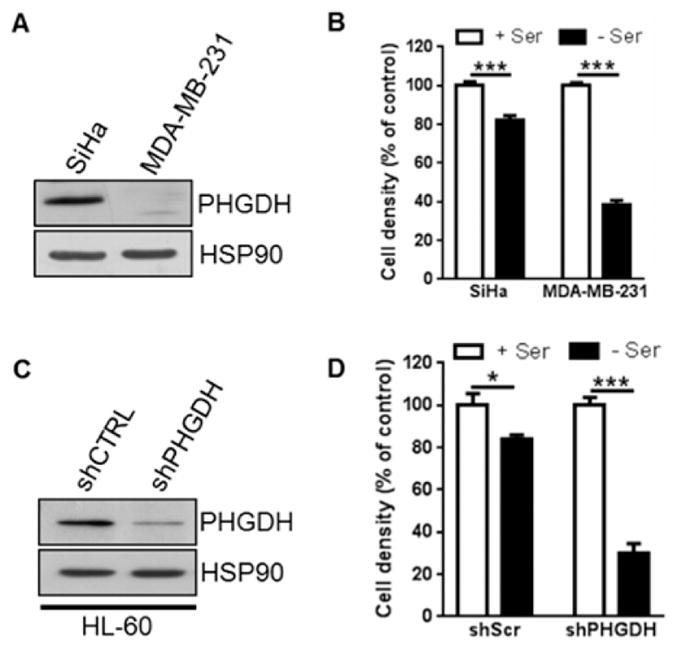
Ability of the selected cancer cell lines to proliferate in serine-replete or -deplete medium. (A, C) Representative immunobolot for PHGDH. (B, D) Cell growth extent (for 5 days) in either serine-replete (+Ser) or serine-deplete (−Ser) medium. Data are shown as mean ± SEM (n ≥ 3).
Subsequently, the selected molecules were evaluated on the following systems: (i) in MDA-MB-231 lacking PHGDH, (ii) in SiHa and HL-60 expressing PHGDH in serine-replete or serine-deplete medium, and (iii) in HL-60 with PHGDH knockdown (shPHGDH) (Table 1). Assays in deplete-serine medium were expected to enhance the de novo serine synthesis and increase the cell sensitivity toward the inhibitors.
As expected, all tested molecules had no effect on the MDA-MB-231 and HL-60-shPHGDH cell lines sensitive to serine removal. In contrast, the inhibitors (except compound 22) exhibit interesting inhibition of SiHa and HL-60 cell proliferation in serine-replete (+Ser) medium. For example, 25 shows IC50 values of 21.0 and 50.5 μM on SiHa and HL-60, respectively. According to plan, the amino acid depletion (−Ser) enhanced the efficacy of the inhibitors by a factor of 3 for SiHa line and a factor 1.5–5.5 for the HL-60 line. In these conditions, 25 had an IC50 of 6.6 and 15.5 μM on SiHa and HL-60, respectively. Finally, the preliminary SARs established were confirmed in the cellular assay. Indeed, ortho- and meta-chloro compounds (23 and 24) that show no inhibition in the in vitro assay also show no cell proliferation inhibition on all systems (IC50 > 100 μM).
Mechanism of Action of Compound 19
To characterize the mechanism by which α-ketothioamides inhibit PHGDH, we have undertaken competition and rapid dilution assays with the leader of the series (19). Inhibition constants (Ki) for 19 with respect to 3-PG and NAD+ were determined (Figure 6).
Figure 6.

Mechanism of action of 19. Inhibition constants (Ki) were determined by titrating (A) 3-PG and (B) NAD+ at four concentrations of 19 (in nanomolar). (C) Washout experiment of 19.
As highlighted in Figure 6A, B, the α-ketothioamide 19 inhibits PHGDH in a noncompetitive mode of action with respect to both substrates. The determined Ki values are 40 ± 2 and 27 ± 7 μM for 3-PG and NAD+, respectively. Moreover, the rapid dilution experiment in Figure 6C suggests that 19 may be a covalent inhibitor because enzymatic activity is not recovered after dilution.
CONCLUSION
Recent studies implicate PHGDH as an attractive drug target in tumors that overexpress PHGDH or amplify the PHGDH gene. In the present work, we explored whether this enzyme was druggable with small molecules using a screening approach. Initially, we established a straightforward fluorescence-based enzymatic assay that allowed us to screen around 350 molecules. Structure analyses of these hits highlighted common structural elements such as a thioamide or acetophenone part. On these observations, a convergent pharmacophore approach led to the synthesis of α-ketothioamides that appear as promising in vitro PHGDH inhibitors. Moreover, cellular assays confirmed that the identified inhibitors selectively abrogated the proliferation of cancer cell with elevated PHGDH expression. These encouraging results open the way to the development of a novel series of PHGDH inhibitors around the α-ketothioamide scaffold.
EXPERIMENTAL SECTION
General Chemistry
All reagents were purchased from chemical suppliers and used without purification. Thin-layer chromatography (TLC) was performed using silica gel 60 F254 plates, with observation under UV when necessary. Melting points were recorded on an Electrothermal IA9000 melting point system. 1H NMR spectra were recorded an AVANCE II 400 MHz Bruker spectrometer with CDCl3 or DMSO-d6 as the solvent. 13C NMR spectra were recorded at 100 MHz. All coupling constants are measured in hertz (Hz), and the chemical shifts (δH and δC) are quoted in parts per million (ppm) relative to TMS (δ0), which was used as the internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, and coupling constant (Hz). High-resolution mass spectroscopy (HRMS) analyses were carried out on a LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Data were acquired in positive ion mode using full-scan MS with a mass range of 100–1000 m/z. The orbitrap operated at 30000 resolutions (fwhm definition). All experimental data were acquired using daily external calibration prior to data acquisition. Appropriate tuning of the electrospray ion source was done. The following electrospray inlet conditions were applied: flow rate, 100 μL min−1; spray voltage, 5 kV; sheath gas (N2) flow rate, 20 au; auxiliary gas (N2) flow rate, 10 au; capillary temperature, 275 °C; capillary voltage, 45 V; tube lens, 80 V. High-performance liquid chromatography (HPLC) analyses were performed on a LC system using a YMC-Triart C-18 (250 mm × 4.6 mm, 5 μm) column as the stationary phase. Mobile phase contained water/CH3CN (30:70, v/v) and was maintained isocratically at the flow rate of 1 mL/min. The column temperature was maintained at room temperature. The peaks were monitored at the wavelength of 215 nm. The purity of all compounds tested was greater than 95%, as determined by HPLC and 1H NMR.
General Procedure for the Synthesis of α-Ketothioamide Analogues (19–35)
To a stirred solution of ethanone derivative (1 equiv) in chloroform was added dropwise a solution of dibromine (1.2 equiv) in chloroform. After 2 h, the solvent was evaporated in vacuo to give a crude oil consisting mainly of 2-bromo-1-(substituted)-ethanone compound along with trace amounts of 2,2-dibromo-1-(substituted)-ethanone compound. The mixture was used without purification in the next step. To the synthesized or commercial 2-bromo-1-(substituted)-ethanone derivative were added in sequence DMF, cyclooctasulfur (1.5 equiv), and morpholine (3 equiv). The mixture was then stirred at room temperature. After completion, the reaction mixture was quenched with distilled water to give a precipitate, which was further washed with distilled water. The residue was recrystallized or purified by silica gel chromatography if necessary.
1-Phenyl-2-morpholino-2-thioxoethanone (19)
Brown solid (73%). This compound was synthesized according to the general procedure described above. Acetophenone (2.00 g, 16.60 mmol) and dibromine (1.01 mL, 19.90 mmol) were mixed in chloroform (15 mL) to obtain the 2-bromo-1-phenyl-ethanone, and this intermediate was reacted in a second time with morpholine (4.34 mL, 49.80 mmol) and sulfur (0.79 g, 24.90 mmol) in DMF (10 mL). Methanol was used for recrystallization to afford the title compound; Rf 0.2 (cyclohexane/EtOAc 8:2); mp 110–112 °C. 1H NMR (400 MHz, CDCl3): δH (ppm) 3.59–3.62 (t, 2H, J = 4.8 Hz), 3.69–3.71 (t, 2H, J = 4.8 Hz), 3.90–3.92 (t, 2H, J = 4.8 Hz), 4.33–4.35 (t, 2H, J = 4.8 Hz), 7.48–7.52 (m, 2 ArH), 7.59–7.65 (m, 1 ArH), 7.99–8.01 (D, 2 ArH, J = 8.2 Hz). 13C NMR (100 MHz, CDCl3): δC (ppm) 47.13, 51.95, 66.40, 66.52, 128.99 (2C), 129.86 (2C), 133.26, 134.48, 187.90 (C═O), 195.70 (C═S). HRMS (ESI+): m/z calcd for C12H13NO2S (M + H)+ 236.0739, found 236.0737.
Compounds 20–35 were synthesized with the same conditions and procedure. The full descriptive paragraph of each compound is in the Supporting Information.
1-(2-Fluorophenyl)-2-morpholino-2-thioxoethanone (20)
Starting from the 1-(2-fluorophenyl)ethanone (1.50 g, 11.10 mmol), the title compound 20 was obtained after recrystallization in methanol as a yellow solid (39%).
1-(3-Fluorophenyl)-2-morpholino-2-thioxoethanone (21)
Starting from the 2-bromo-1-(3-fluorophenyl)ethanone (0.50 g, 2.30 mmol), the title compound 21 was obtained after recrystallization in methanol as a yellow solid (23%).
1-(4-Fluorophenyl)-2-morpholino-2-thioxoethanone (22)
Starting from the 2-bromo-1-(4-fluorophenyl)ethanone (0.50 g, 2.30 mmol), the title compound 22 was obtained after recrystallization in methanol as a beige solid (41%).
1-(2-Chlorophenyl)-2-morpholino-2-thioxoethanone (23)
Starting from the 2-bromo-1-(2-chlorophenyl)ethanone (0.50 g, 2.14 mmol), the title compound 23 was obtained after recrystallization in acetonitrile as a yellow solid (36%).
1-(3-Chlorophenyl)-2-morpholino-2-thioxoethanone (24)
Starting from the 1-(3-chlorophenyl)ethanone (2.00 g, 12.90 mmol), the title compound 24 was obtained after purification by silica gel chromatography (cyclohexane/EtOAc, 8:2) as a yellow solid (43%).
1-(4-Chlorophenyl)-2-morpholino-2-thioxoethanone (25)
Starting from the 2-bromo-1-(2-chlorophenyl)ethanone (0.50 g, 2.14 mmol), the title compound 25 was obtained after recrystallization in acetonitrile as a yellow solid (38%).
1-(2-Bromophenyl)-2-morpholino-2-thioxoethanone (26)
Starting from the 2-bromo-1-(2-bromophenyl)-ethanone (0.50 g, 1.81 mmol), the title compound 26 was obtained after recrystallization in ethanol as a white solid (52%).
1-(3-Bromophenyl)-2-morpholino-2-thioxoethanone (27)
Starting from the 2-bromo-1-(3-bromophenyl)-ethanone (0.50 g, 1.81 mmol), the title compound 27 was obtained after recrystallization in methanol as a white solid (26%).
1-(4-Bromophenyl)-2-morpholino-2-thioxoethanone (28)
Starting from the 2-bromo-1-(4-bromophenyl)-ethanone (0.50 g, 1.81 mmol), the title compound 28 was obtained after recrystallization in cyclohexane as a white solid (19%).
1-(2-Iodophenyl)-2-morpholino-2-thioxoethanone (29)
Starting from the 2-bromo-1-(2-iodophenyl)-ethanone (1.00 g, 4.00 mmol), the title compound 29 was obtained after purification by silica gel chromatography (cyclohexane/EtOAc, 8:2) as a yellow oil (47%).
1-(3-Iodophenyl)-2-morpholino-2-thioxoethanone (30)
Starting from the 2-bromo-1-(3-iodophenyl)-ethanone (1.00 g, 4.06 mmol), the title compound 30 was obtained after purification by silica gel chromatography (cyclohexane/EtOAc, 8:2) as a yellow solid (51%).
1-(4-Iodophenyl)-2-morpholino-2-thioxoethanone (31)
Starting from the 2-bromo-1-(4-iodophenyl)-ethanone (1.00 g, 4.06 mmol), the title compound 31 was obtained after purification by silica gel chromatography (cyclohexane/EtOAc, 8:2) as a yellow solid (63%).
1-(2-Nitrophenyl)-2-morpholino-2-thioxoethanone (32)
Starting from the 2-bromo-1-(2-nitrophenyl)-ethanone (0.50 g, 2.05 mmol), the title compound 32 was obtained after recrystallization in a mixture of cyclohexane/EtOAc (8:2) as a yellow solid (26%).
1-(3-Nitrophenyl)-2-morpholino-2-thioxoethanone (33)
Starting from the 2-bromo-1-(3-nitrophenyl)-ethanone (1.50 g, 6.17 mmol), the title compound 33 was obtained after recrystallization in methanol as a yellow solid (73%).
1-(4-Nitrophenyl)-2-morpholino-2-thioxoethanone (34)
Starting from the 2-bromo-1-(4-nitrophenyl)-ethanone (1.50 g, 6.17 mmol), the title compound 34 was obtained after recrystallization in methanol as a yellow solid (47%).
1-([1,1′-biphenyl]-4-yl)-2-morpholino-2-thioxoethanone (35)
Starting from the 1-([1,1′-biphenyl]-4-yl)-2-bromoethanone (1.00 g, 3.64 mmol), the title compound 35 was obtained after purification by silica gel chromatography (cyclohexane/EtOAc, 8:2) as a yellow solid (61%).
Primary PHGDH Screen
PHGDH inhibition assay was performed in black polypropylene 96-well plates (Sigma-Aldrich) using NAD (Sigma-Aldrich) as cofactor and 3-phosphoglycerate (Sigma-Aldrich) as substrate. Tested compounds were dissolved in DMSO. The final concentration of DMSO in assay solutions was 10%, which was shown to have no effect on PHGDH activity. The reaction volume per well was 50 μL. Each assay was carried out in triplicate. Briefly, a solution buffer (100 mM Tris HCl buffer (pH 8.8), 400 mM NaCl, 0.2 mM DTT, 1 mM NAD, and 0.5 mM 3-PG) was added at various concentrations of tested compounds. Then, an amount of 100 ng of PHGDH (human recombinant protein, BPS Bioscience) is added to start the reaction. Finally, the product formation, correlated with NADH formation, is instantly monitored for 7 min at 25 °C using a SpectraMax spectrophotometer at an excitation wavelength of 360 nm and emission wavelength of 460 nm.
PHGDH Assay
PHGDH activity was measured in 96-well plates (96 μL per well) at 37 °C by monitoring resorufin fluorescence (Ex 550 nM/Em 580 nM) over time with a FLUOstar Omega (BMG Labtech). PSAT1 was included to prevent product inhibition of PHGDH. Assays were performed in PHGDH assay buffer (50 mM Tris, pH 8.5, and 1 mM EDTA). Substrates and enzyme concentrations were as follows: 3-PG, 240 μM; NAD+, 120 μM; glutamate, 30 mM; resazurin, 0.1 mM; PHGDH, 12 ng/μL; PSAT1, 20 ng/μL; diaphorase 0.18 μg/μL except for Ki measurements where one of the PHGDH substrates was held constant at 3 mM whereas the other substrate was titrated. For Ki measurements, drug and enzyme were preincubated for 30 min. Initial rate plots were fit using Prism 6. Chemicals were purchased from Sigma.
Cell Models and Cytotoxicity Assay
All cell lines were acquired in the last three years from ATCC, where they are regularly authenticated by short tandem profiling. Cells were stored according to the supplier’s instructions and used within 6 months after resuscitation of frozen aliquots. SiHa cervix carcinoma cells and MDA-MB-231 breast cancer cells were routinely cultured in DMEM supplemented with 10% FBS and antibiotics. For the cytotoxicity assay, cells were seeded at low density I 96-well plates in serine containing media. Media were then aspirated, and cells were incubated in fresh serine-replete or -deplete media containing drugs or vehicle (DMSO). Cells were grown for 5 days at 37 °C with drug and media changed daily before assaying cell viability using a Presto Blue assay (Life Technologies) according to manufacturer’s instructions.
Immunoblots
Protein extraction and Western blot analysis were carried out as reported elsewhere (Polet et al.11).
Dilution Experiment
Compound 19 (160 μM) or DMSO control was incubated with the enzyme mix for 45 min at 37 °C. At the end of the incubation, the enzyme drug mix was diluted 10-fold using assay buffer and concentrated back to the original volume by centrifuging through a 10 kD molecular weight cutoff filter. Subsequently, enzyme activity was measured via the PHGDH assay. An undiluted compound (160 μM) condition was included as a positive control for inhibition.
Supplementary Material
Acknowledgments
We are grateful to Laurenne Petit, Prof. Jacques Poupaert, and Romain Marteau for their participation in this project and to Dr. Marie-France Hérent and Prof. Giulio Muccioli (UCL, BPBL) for helpful discussions and assistance with HRMS. This work was supported by a J. Maisin Foundation grant, the Fondatioun Kriibskrank Kanner (Luxembourg), a Crédit de Recherche (CDR) grant from the F.R.S.-FNRS, and an Action de Recherche Concertée (ARC 14/19-058) grant from the Fédération Wallonie-Bruxelles. Q.S. is a Télévie Research Fellow. L.C.C. was supported by NIH grants P01CA117969 and P01CA120964.
ABBREVIATIONS USED
- 3-PG
3-phosphoglycerate
- 3-PPyr
3-phosphohydroxypyruvate
- NAD
nicotinamide adenine dinucleotide
- PHGDH
3-phosphoglycerate dehydrogenase
- PSA
molecular polar surface area
- PSAT1
phosphoserine amintransferase 1
- PSPH
phosphoserine phosphatase
- TNBC
triple-negative breast cancer
Footnotes
Author Contributions
S.R. and C.C. equally contributed to this work and share first author position. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare the following competing financial interest(s): A patent application has been submitted based on the results described in this manuscript. L.C.C. owns equity in, receives compensation from, and serves on the Board of Directors and Scientific Advisory Board of Agios Pharmaceuticals. Agios Pharmaceuticals is identifying metabolic pathways of cancer cells and developing drugs to inhibit such enzymes in order to disrupt tumour cell growth and survival. The authors declare that they have no other conflicts of interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01166.
Synthetic details for 20–35, 1H NMR, 13C NMR, and enzymatic assay optimization (PDF)
Molecular formula strings (CSV)
References
- 1.Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287:19786–19791. doi: 10.1074/jbc.R112.357194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollari S, Käkönen SM, Edgren H, Wolf M, Kohonen P, Sara H, Guise T, Nees M, Kallioniemi O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat. 2011;125:421–430. doi: 10.1007/s10549-010-0848-5. [DOI] [PubMed] [Google Scholar]
- 4.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, Vokes NI, Sasaki M, Beroukhim R, Stephanopoulos G, Ligon AH, Meyerson M, Richardson AL, Chin L, Wagner G, Asara JM, Brugge JS, Cantley LC, Vander Heiden MG. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 2016;76:4430–4442. doi: 10.1158/0008-5472.CAN-16-0530. [DOI] [PubMed] [Google Scholar]
- 7.Jia XQ, Zhang S, Zhu HJ, Wang W, Zhu JH, Wang XD, Qiang JF. Increased expression of PHGDH and prognostic significance in colorectal cancer. Transl Oncol. 2016;9:191–196. doi: 10.1016/j.tranon.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Guo S, Li Q, Yang L, Xia Z, Zhang L, Huang Z, Zhang Nu. Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J Neuro-Oncol. 2013;111:245–255. doi: 10.1007/s11060-012-1018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, Wistuba II, Minna JD, DeBerardinis RJ, Cantley LC. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun WY, Kim HM, Jung WH, Koo JS. Expression of serine/glycine metabolism-related proteins is different according to the thyroid cancer subtype. J Transl Med. 2016;14:168. doi: 10.1186/s12967-016-0915-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polet F, Corbet C, Pinto A, Rubio LI, Martherus R, Bol V, Drozak X, Gregoire V, Riant O, Feron O. Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget. 2016;7:1765–1776. doi: 10.18632/oncotarget.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan J, Teng X, Liu L, Mattaini KR, Looper RE, Vander Heiden MG, Rabinowitz JD. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol. 2015;10:510–516. doi: 10.1021/cb500683c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J. Cancer’s sweet tooth for serine. Breast Cancer Res. 2011;13:317. doi: 10.1186/bcr2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullarky E, Lucki NC, BeheshtiZavareh R, Anglin JL, Gomes AP, Nicolay BN, Wong JC, Christen S, Takahashi H, Singh PK, Blenis J, Warren JD, Fendt SM, Asara JM, DeNicola GM, Lyssiotis CA, Lairson LL, Cantley LC. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A. 2016;113:1778–1783. doi: 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuller N, Spadola L, Cowen S, Patel J, Schönherr H, Cao Q, McKenzie A, Edfeldt F, Rabow A, Goodnow R. An improved model for fragment-based lead generation at AstraZeneca. Drug Discovery Today. 2016;21:1272–1283. doi: 10.1016/j.drudis.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 16.Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, Possemato R, Chen WW, Sullivan LB, Fiske BP, Cho S, Freinkman E, Birsoy K, Abu-Remaileh M, Shaul YD, Liu CM, Zhou M, Koh MJ, Chung H, Davidson SM, Luengo A, Wang AQ, Xu X, Yasgar A, Liu L, Rai G, Westover KD, Vander Heiden MG, Shen M, Gray NS, Boxer MB, Sabatini DM. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452–458. doi: 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 18.Kassehin U, Gbaguidi FA, Kapanda CN, McCurdy CR, Poupaert JH. Insight into the Willgerodt–Kindler reaction of ω-haloacetophenone derivatives: mechanistic implication. Org Chem Int. 2014;2014:486540. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.