

Abstract
Fluorescence of Reduced Nicotinamide Adenine Dinucleotide (NADH) is extensively employed in studies of oxidoreductases. A substantial amount of static and kinetic work has focused on the binding of pyruvate or substrate mimic oxamate to LDH•NADH where substantial fluorescence quenching is typically observed. However, the quenching mechanism is not well understood limiting structural interpretation. Based on time-dependent density functional theory (TDDFT) computations with cam-B3LYP functional in conjunction with the analysis of previous experimental results, we propose that bound oxamate acts as an electron acceptor in the quenching of fluorescence of NADH in the ternary complex, where a charge transfer (CT) state characterized by excitation from the highest occupied molecular orbital (HOMO) of the nicotinamide moiety of NADH to the lowest unoccupied molecular orbital (LUMO) of oxamate exists close to the locally excited (LE) state involving only the nicotinamide moiety. Efficient quenching in the encounter complex like in pig heart LDH requires that oxamate forms a salt bridge with Arg-171 and hydrogen bonds with His-195, Thr-246 and Asn-140. Further structural rearrangement and loop closure, which also brings about another hydrogen bond between oxamate and Arg-109, will increase the rate of fluorescence quenching as well.
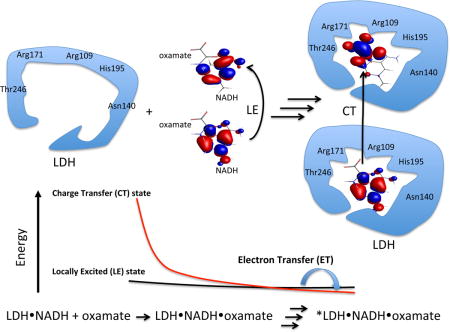
Graphical abstract
Oxamate efficiently quenches the fluorescence of NADH in the lactate dehydrogenase (LDH) ternary complex, but not in water, through electron transfer (ET) mechanism. Oxamate interacts with LDH by forming a salt bridge with Arg-171 and hydrogen bonds with His-195, Thr-246, and Asn-140 in the encounter complex. It adds another hydrogen bond with Arg-109 after loop is closed. Such interactions decrease the energy of a charge transfer (CT) state involving the nicotinamide moiety and oxamate. Then electron transfer from excited states of NADH to oxamate, and consequent fluorescence quenching take place.
INTRODUCTION
Monitoring the fluorescence of Reduced Nicotinamide Adenine Dinucleotide (NADH) bound to oxidoreductases has been and is a significant method for in vivo or in vitro studies because of its essential role as a coenzyme in metabolism (1–5). NADH has been a subject of research for many years (6–8). In general, the molecular factors that regulate NADH emission are not firmly established. NADH has two strong UV-vis absorption bands in water. The stronger one at 260 nm is assigned to transition within the adenylyl moiety. The absorption band at 340 nm is due to the dihydronicotinamide moiety, which is the origin of NADH fluorescence. In aqueous solution, it is believed that NADH exists in equilibrium between folded and extended conformations (9–14). In folded state, the adenylyl moiety, less than 6 Å away from the dihydronicotinamide ring, quenches the fluorescence from the dihydronicotinamide moiety. Hence, NADH has a lower quantum yield in water than in alcohol solution (15). The absolute emission quantum yields (QY) of NADH was reported to be 0.019 versus 0.076 (or 0.066) in water versus 1,2-propanediol; for comparison, the QY of NADH in the LDH•NADH binary complex, where the molecule is extended so that the intra-molecular interaction is absent, is 0.099 (15). The quenching in these nucleotides is believed to be dynamic (15). As the QY of NADH is low, the non-radiative decays are major processes for the excited NADH. Some studies indicated that two-photon absorption could result in photoionization when the excitations fall into the absorption band of dihydronicotinamide (16–20). However, excitation at the adenylyl moiety, for example at 266 nm, only gives rise to a short lived excited state with a lifetime of ~3 ps, which decays through energy transfer to the excited dihydronicotinamide moiety and other internal conversion processes (21). In summary, the photodynamics of NADH could be presented by a simple Jablonski diagram (Fig. 1). However, with the interaction between the adenylyl and the dihydronicotinamide moieties, the photodynamics is more complex (22) and may need more studies to fully be resolved, especially with an excitation of the dihydronicotinamide.
Figure 1.
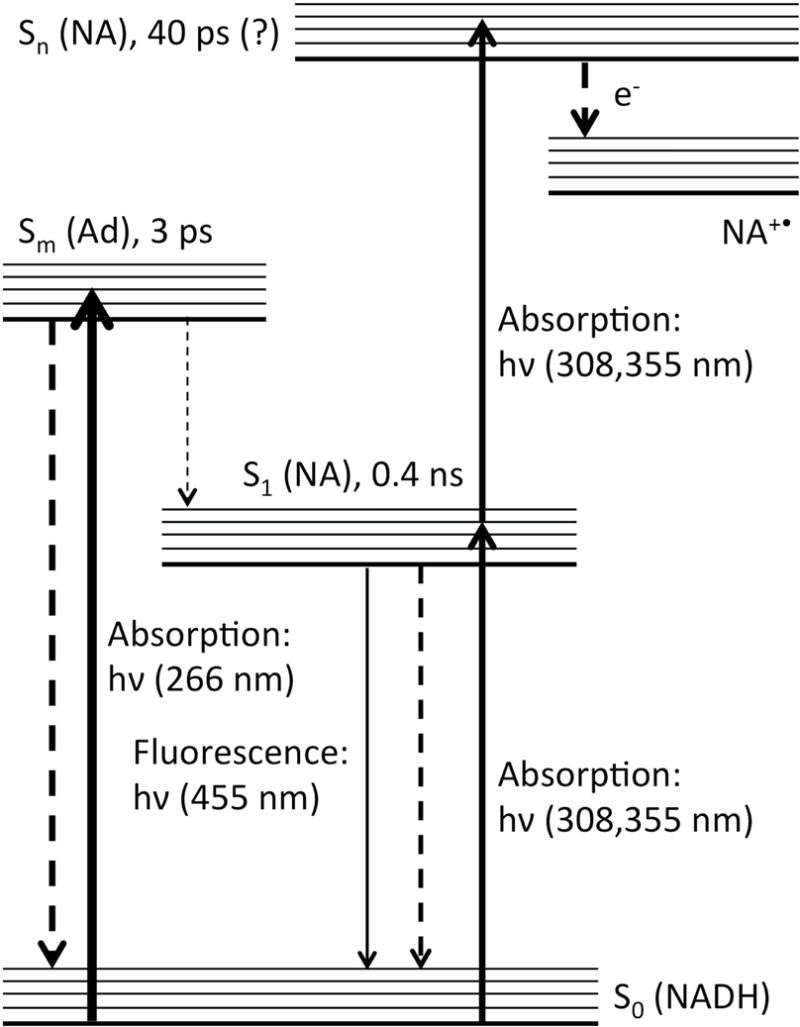
Jablonski diagram of the photodynamics of NADH. Some reported lifetimes are shown for selected excited states. Ad = adenylyl group; NA = dihydronicotinamide group.
The emission from NADH has been a broadly useful tool in examining the dynamical nature of certain enzymes, in particular the NADH and NADPH linked enzymes (23–25). In our work, NADH emission is used to monitor the dynamics of binding of NADH and substrate to enzymes employing various mixing approaches and also laser induced T-jump studies, where resolution of specific events can approach the nanosecond time scale (26, 27). The fluorescence intensity of a molecule like NADH generally increases accompanied by a small blue shift when it binds to a hydrophobic like protein pocket. This is what is observed when NADH binds to lactate dehydrogenase (LDH), an enzyme that catalyzes the interconversion of lactate and pyruvate. There is a substantial and unusual drop in intensity when substrate binds to the LDH•NADH complex. Moreover, the events in the pathway of substrate binding show varying emission quantum yields. Fluorescence T-jump studies indicate that NADH fluorescence is quenched by forming an encounter complex in pig heart lactate dehydrogenase phLDH•NADH•substrate system (26). Specific LDH isozymes included in this study are pig heart, phLDH, and Bacillus Stearothermophilus, bsLDH. The relative quantum yields of NADH in free form, binary complex, encounter complex and two down stream structure-rearranged complexes in the pig heart system are estimated to be 1, 2.5, 0.59, 0.29 and 0.23 respectively. There is clearly structural information contained in these numbers. It would be useful and highly important, given that it is generally very difficult to determine the structure of kinetics intermediates, to develop molecular mechanisms that bear on quantum yields so as to be able to interpret these data in structural terms. Dihydronicotinamide has shown to be an electron donor in some fluorescence quenching studies(28–30). We thus explore a photoinduced electron transfer (PET) mechanism responsible for the quenching in enzymes. We propose, by dissecting the mechanism under the fluorescence changes along the dynamic processes of enzymes, structural information about enzyme intermediates or sub-states can be determined. If the quantum yields (QY), for example, of each protein species can be estimated experimentally and calculated theoretically (31–33) from possible structures, important structural information is derived.
For example, aspects of the dynamics of substrate binding to lactate dehydrogenases (LDH) are resolved by monitoring the fluorescence changes in the systems of LDH•NADH•pyruvate and LDH•NADH•oxamate (26, 27, 34–36). The active sites of LDHs are well conserved, and it is reasonably clear that interactions between active site residues and substrate (or substrate mimic) modulate the emission of bound NADH. LDHs are characterized by an essential mobile surface loop, residues 98–110 in the pig heart protein. It is generally thought that the protein is only fully active when this loop has properly closed over the active site. One study has shown directly that loop closure, which brings Arg-109 in close contact with bound substrate has a direct and sizable effect on the steady state NADH emission(37). Moreover, several fluorescence T-jump studies of NADH indicate that binding involves several structural rearrangement steps affecting NADH emission differentially while overall values of kcat and Km are close under their physiological conditions(38, 39). For example, in phLDH NADH fluorescence is mostly quenched in the bimolecular step of substrate mimic binding to LDH•NADH (26). For bsLDH, the fluorescence is largely quenched in subsequent unimolecular steps (40). These differences are most likely due to that different binding arrangements of substrate (or substrate mimic) with several key active sites residues such as Arg-171, His-195, Asn-140, and Arg-109. Among these, an important interaction is the salt bridge formed between the guanidinium group of Arg-171 and the carboxylate group of oxamate. Isotope edited FTIR studies have demonstrated that the salt bridge is stronger in phLDH than that in bsLDH (41, 42). In bsLDH, the band assigned to the salt bridge has a frequency of 1581 cm−1, while the frequency is 1589 cm−1 in phLDH. Higher frequency means a stronger salt bridge. X-ray structures also indicated that a shorter salt bridge in phLDH than that in bsLDH (Fig. 2) (43, 44). It is obvious that kinetic studies using NADH emission report on kinetic steps as well as, in principle, accompanying structural changes. However, it is necessary to have a mechanistic understanding of the quenching mechanism to unravel specific structural changes.
Figure 2.
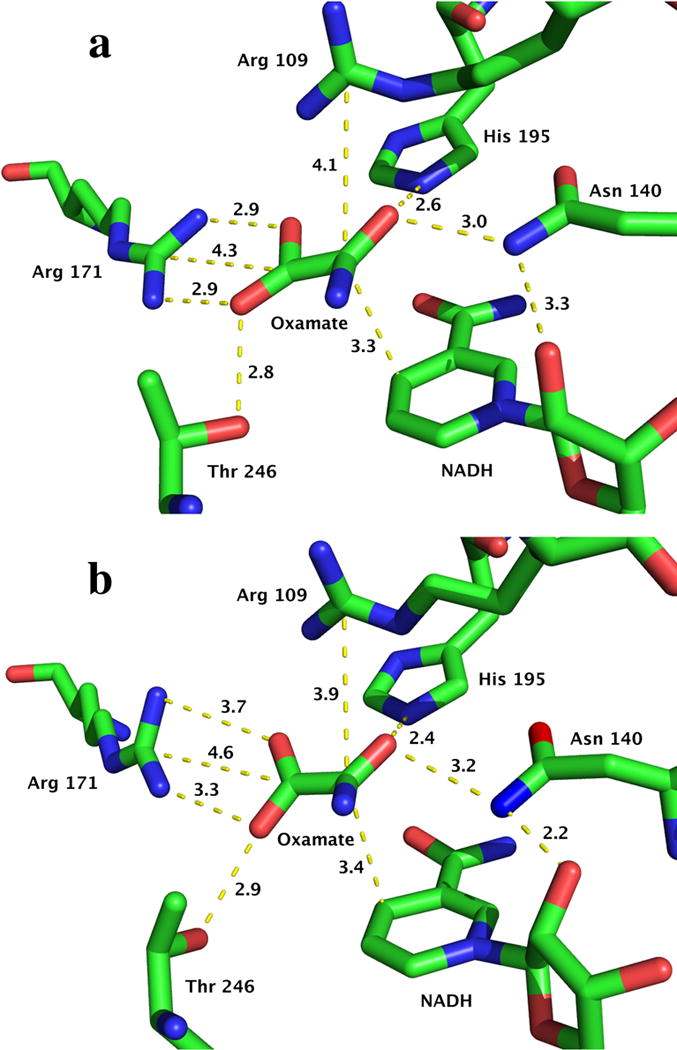
X-ray crystal structural information of the active sites for phLDH (top, PDB ID: 9LDB) and bsLDH (bottom, PDB ID: 1LDN). Distance unit: Å. Here the structure of pig muscle LDH is used since pig heart LDH structure (PDB ID: 5LDH) does not have oxamate. It should be noted that the structure of LDH-NADH-oxamate of pig muscle is very similary with that of pig heart (unpublished results).
Here we examine deeply the changes of NADH fluorescence in binary LDH•NADH and ternary complexes such as LDH•NADH•substrate. We and others have performed a number of studies of the reaction pathway of LDH•NADH•pyruvate conversion to LDH•NAD+•lactate(27, 34, 35, 45, 46). The pathway of this system is arguably better known than other systems having a simple chemical event. In a number of studies, oxamate is employed as non-reactive substrate mimic (for pyruvate) (26, 41, 42, 47, 48). For this reason, many crystal structures of LDH•NADH•oxamate can be obtained from the PDB database. In fact, more studies on the ternary complexes of oxamate have come out and understanding in this system is much better than in the ‘live’ system of the pyruvate complex. The reaction pathway is very complex with a number of clearly identifiable protein LDH•NADH•pyruvate type species with varying degrees of NADH emission (26, 49, 50). In this regard, we apply quantum mechanical computations on some oxamate complexes, which would mimic the structures along the substrate binding. By comparing those results with some experimental and theoretical findings on small molecular quenchers, in the final, we proposed an operative mechanism of PET enabling a structural analysis.
MATERIALS AND METHODS
Experimental
Chemicals used in this study, including sodium pyruvate and imidazole were purchased from Sigma or Aldrich. Acrylamide were purchased from Fisher Scientific (Alfa Aesar). NADH was purchased from Roche Diagnostics. Sodium oxamate and other chemicals were from Fisher Scientific (Acros). Those chemicals, generally having 97%, 98% or greater purity, were used as purchased except oxamate was recrystallized in 3:1 (v:v) water/ethanol.
Fluorescence spectra were measured on a FluoroMax-2 spectrometer. The solutions were freshly made right before measurements in 100 mM phosphate buffer at pH 7; final pH was adjusted by adding HCl or NaOH. Excitation wavelength was set at 385 nm for NADH in the quenching measurements. The quencher concentrations were from 0–0.5 M. The absorbance, measured on a Beckman DU-7400 spectrometer, at the excitation wavelength was about 0.1 OD or below. 1 cm quartz cell was used in the experiments and inner filter correction and subtraction of buffer background were performed to obtain final fluorescence intensities for Stern-Volmer plots.
Computational details
GAMESS (version Dec 5, 2014 R1) was used in the quantum mechanical computations (51, 52). Density functional B3LYP (53, 54) with basis sets 6–31g(d) were employed in ground state geometry optimization. The excitation energies of single molecules were obtained from time-dependent density function theory (TDDFT) computations with B3LYP functional. As TDDFT with B3LYP tends to underestimate the charge transfer (CT) states due to lacking the correct 1/R asymptotic behavior (55–57), Coulomb attenuated method (CAM) was applied to oxamate complexes for a better description of CT states (58, 59). In the CAM approach, the contribution of Hartree-Fock exchange integral is varied along the distance R to produce the correct long-range behavior. Solvent effect was considered by employing polarized continuum model (PCM) to the systems in water (60). For the models of oxamate complexes, pruned 3-D structures (see Fig. 3) were obtained from the protein data bank (PDB ID: 9LDB (44)) by only keeping atoms in the functional group from x-ray crystallographic structure. Constrained geometry optimizations were then performed on the structures with heavy atoms fixed except terminal methyl carbon (at the cutting position during model structure building).
Figure 3.
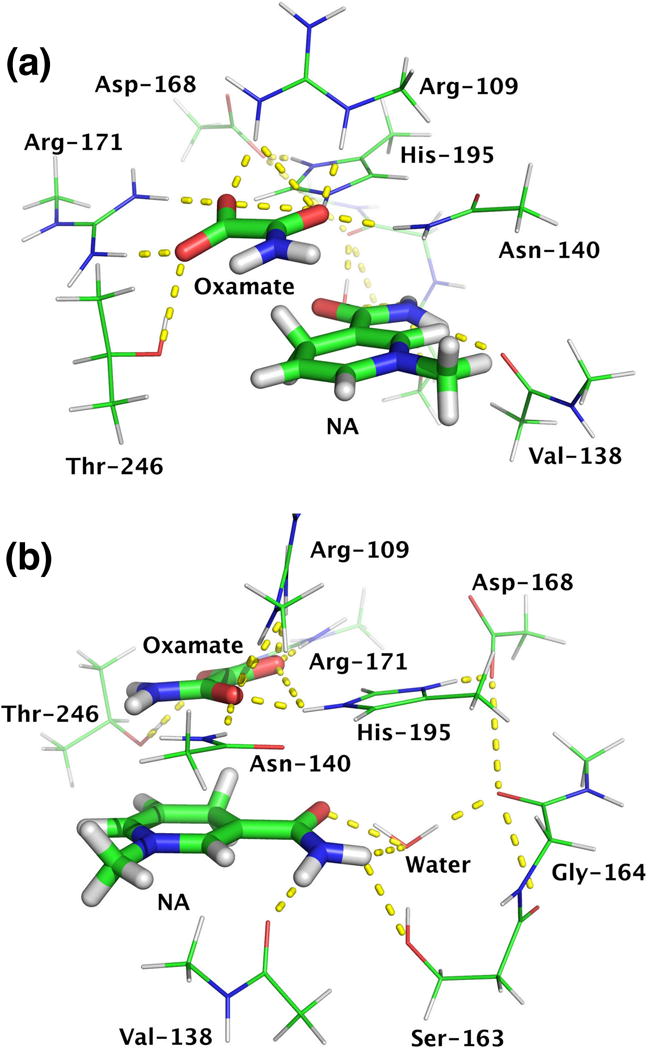
Different views of pruned structures from x-ray crystallographic structure (PDB ID: 9LDB). Residues form hydrogen bonds with oxamate include Arg-171, Thr-246, Asn-140, His-195, and Arg-109. Residues involve hydrogen bonds with dihydronicotinamide are Val-138, Ser-163, Gly-164, and a water molecule. Asp-168 is also included because it interacts with His-195 and neutralizes the positive charge on it. NA = 1-methyl-1,4-dihydronicotinamide.
Electrostatic potential surface of phLDH (PDB ID: 9LDB) was generated by Apbs (61) and pdb2pqr (62) in Pymol (63). Orbitals were generated using MacMolPlt (64). Gabedit (65) was used to generate GAMESS input files and monitor computations.
RESULTS AND DISSCUSSION
Experimental findings
In solution, quenching of NADH fluorescence by oxamate is quite inefficient. Especially, Stern-Volmer constant, KSV for oxamate is only 0.1 M−1, much smaller than those of pyruvate and acrylamide (Fig. 4 and Table 1), where pyruvate (66) and acrylamide (67–72) were used as electron acceptors in fluorescence quenching studies. In general, amides like acetamide and carboxylate groups are not good electron acceptors in fluorescence quenching through a PET quenching mechanism (73). However, ternary complexes with LDH•NADH, oxamate or pyruvate both efficiently quench fluorescence of NADH. By binding oxamate to active site, the enzyme not only brings oxamate (quencher) and NADH (fluorophore) close to each other (important in quenching because the rate of electron transfer shows exponential distance dependence (74)) but also brings about formation of salt bridges or hydrogen bonds that greatly increase the electrophilicity of oxamate. After binding, the guanidinium group of the Arg-171 salt bridge can behave as an electron-withdrawing group. The oxamate amide group also becomes electrophilic due to strong hydrogen bonds between the amide and His-195, Asn-140, and Arg-109. Additionally, the positively charged environment at active site (as shown in Fig. 5), especially when closed, will tend to increase electron transfer rate. With a contribution from PET quenching, the quantum yield (Φf) of fluorescence is determined by the eqn.1:
| (1) |
Figure 4.
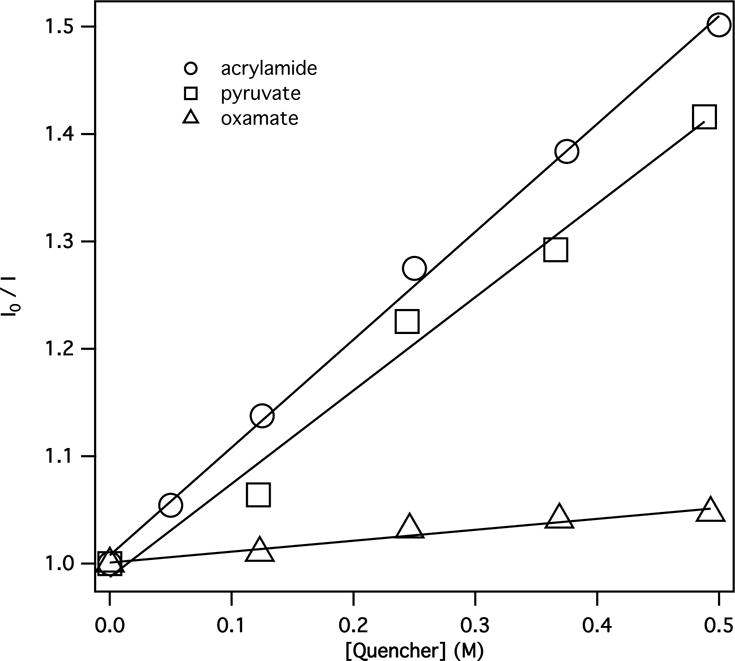
Stern-Volmer plots of fluorescence quenching of NADH by acrylamide, pyruvate, and oxamate.
Table 1.
Stern-Volmer constants for fluorescence quenching of NADH by selected quenchers in 100 mM phosphate buffer (pH 7).
| Molecule | KSV (M−1) | LUMO energy (eV) |
|---|---|---|
| Methyl viologen (MV2+) | 3a | −3.72 |
| Acrylamide | 1.0 | −0.96 |
| Pyruvate | 0.87 | −0.48 |
| Oxamate | 0.1 | 0.27 |
From ref. (28)
Figure 5.
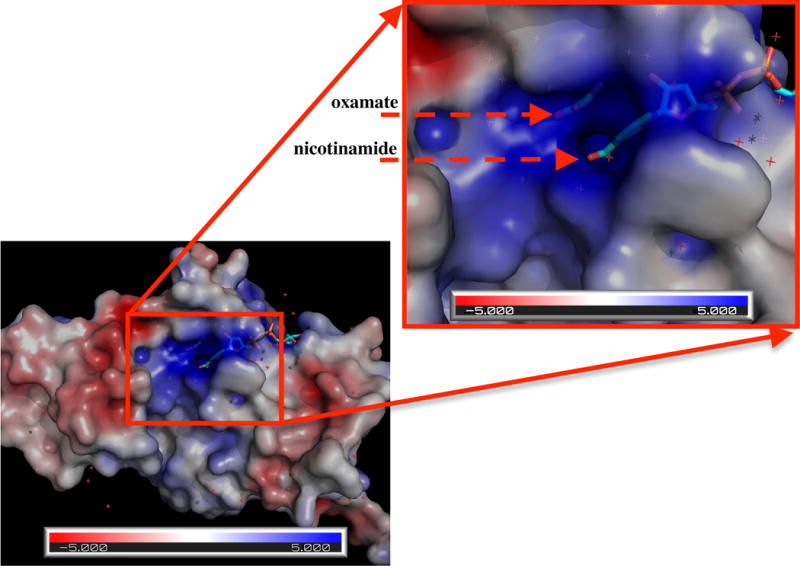
Electrostatic potential surface of phLDH(PDB ID: 9LDB). It is clearly seen that the cavity around oxamate and the nicotinamide moiety of NADH is positively charged.
where, kf, ket and knr are radiative, electron transfer and other non-radiative rate constants respectively. Factors that enhance electron transfer will decrease the quantum yield. Since the interactions mostly through hydrogen bonds and salt bridges between the dihydronicotinamide moiety and protein residues become stronger along the process of the ternary complex formation, the quantum yield is expected to be lower and lower, which is constant with the finding in the t-jump study (26).
It has been shown that some compounds such as oxalate, malonate, and L-malate (shown in Fig. 6, right) cause an increase of NADH fluorescence while others are quenchers like oxamate, nicotinate, and pyrimidine-4-carboxylate (shown in Fig. 6, left) (75). One important feature among these compounds is that they can bind to Arg-171 to form a salt bridge with their carboxylate or similar moiety, as in the case of oxamate. The fluorescence enhancers have neutral (lactate) or a negative charged carboxylate group on the top of the dihydronicotinamide moiety (Fig. 7), while the quenchers all have a group that can be an electron acceptor.
Figure 6.
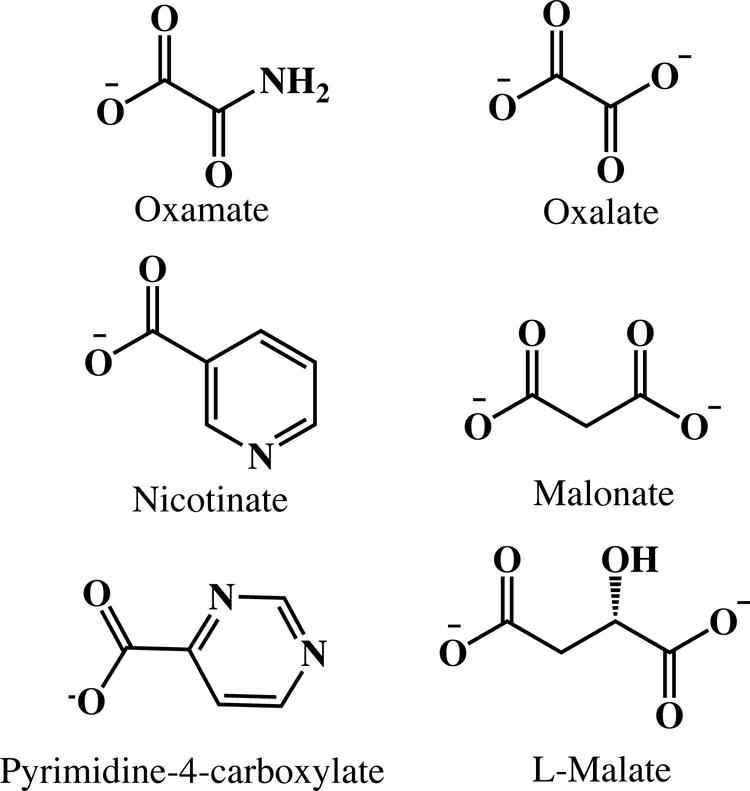
Structures of selected known NADH fluorescence quenchers (left) and enhancers (right).
Figure 7.
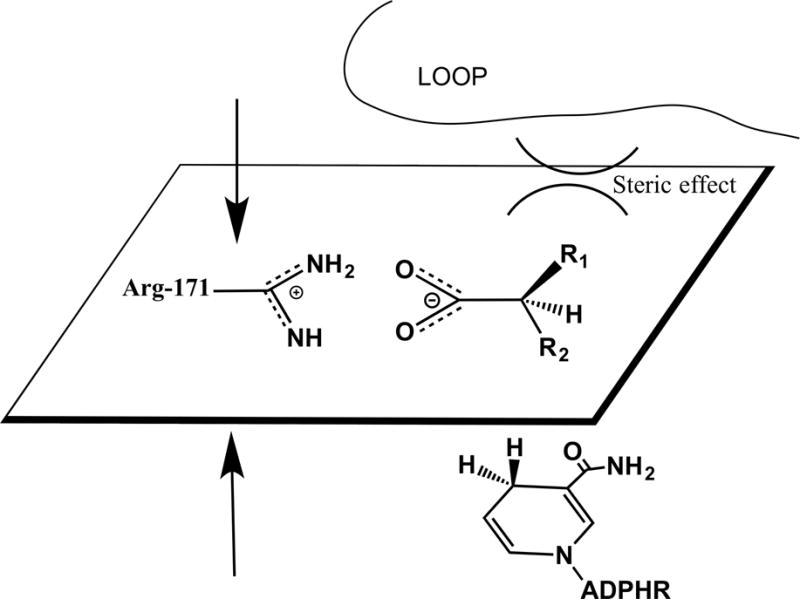
Structural description of steric effect induced by substrates.
In the case of NADH, the dihydronicotinamide group is the fluorophore and the electron donor in the fluorescence quenching process. When lactate or a negative ion like oxalate is bound, without an electron acceptor, water molecules are excluded from the active site and a cavity with more hydrophobic character is formed. Negative charge will also neutralize the positively charged environment. Thus, a decrease of ket and knr is expected along with a concomitant increase of NADH fluorescence.
Steric effects are another important factor. As shown in Fig. 7, where the volume of -R1 is larger than that of -R2 along with proper binding of the substrates, there should be no steric interaction between the dihydronicotinamide group and the substrates as previously suggested(75). However, steric hindrances from -R1 group can keep the loop from closing. Such situation can be found in the case of D-malate. In contrast to that, in the L-malate complex, -R1 is smaller than that of -R2, allowing the loop to close and induce a tighter active site. As a result, fluorescence increases in the ternary L-malate/pig heart LDH•NADH complex. In other words, enzyme backbones and side chains around dihydronicotinamide are not NADH fluorescence quencher. However, structural changes could affect non-radiative rate knr and thus QY of NADH.
It is known that, the protonated imidazole, imidazole-H+ could be fluorescence quencher in solution through PET quenching mechanism(76–78). In the binary and ternary complexes, His-195 is just c.a. 3 Å away from the dihydronicotinamide moiety. Thus it is possible that imidazole-H+ quenches NADH fluorescence in complexes despite that the quenching of NADH fluorescence was not observed in 0.5 M imidazole solution at pH 6 or pH 8 from our experiments. Reported pKas of His-195 are about 6.8 respectively in apo and binary pig heart LDH (79, 80). But NADH fluorescence still increases after formation of binary complexes at pH 6, where around 90% of imidazole is protonated (Fig. 8). As a consequence, quenching of NADH fluorescence by His-195 is unlikely to be operative. So the only quencher candidate in the ternary complex would be bound oxamate (or pyruvate).
Figure 8.
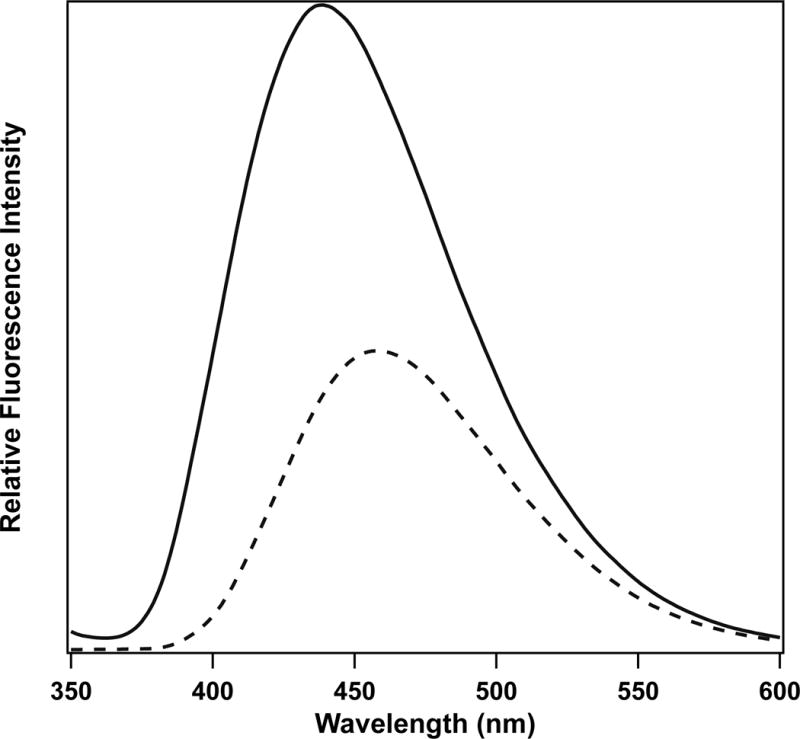
Fluorescence of NADH in solution (dashed line) and binary complex (phLDH/NADH) (solid line) at pH 6 buffer. Excited at 340 nm. [NADH] = 10 μM, [phLDH] = 22 μM.
Computational results
Since photoinduced electron transfer from excited dihydronicotinamide moiety to bound oxamate is a prime suspect in the quenching mechanism of bound NADH as outlined above, we examine the complex for the formation of charge transfer states; this is a minimal requirement to assess the conjecture. We first look into molecular characteristics of NADH that affect its optical properties. Then we construct a series of structural models of the protein-cofactor that simulate putative structures involved with the binding pathway of substrate mimic with LDH•NADH; each is evaluated for the formation of a CT state using ab-initio calculations at the cam-B3LYP level.
NADH optical transitions
The variance of QY is not only due to the conformation of NADH, but is also affected by interactions within the dihydronicotinamide group. Calculations (Table 2) indicate that, in the fully optimized 1-methyl-dihydronicotinamide at B3LYP/6-31g(d) level, dihydropyridine and carboxamide are not coplanar because of steric interactions. However the dihedral angle C2-C3-C-O is small, about 160 degrees, and will have little affect on the conjugation. Good conjugation between dihydropyridine and carboxamide is necessary for the transition with π-π* character. When dihydropyridine is twisted away from planar conformation, as in the case of NADH in liver alcohol dehydrogenase (PDB ID: 1HET) (81), the transition is weakened (smaller oscillator strength). Dihydropyridine without carboxamide group does not have 340 nm band (82–84), The S1 (π-π*) transition is found to be at around 280 nm. With less contribution from conjugation, the π-π* transition becomes a higher energy transition (S2) and 1,4,5,6-tetrahydronicotinamides do not absorb above 300 nm (85–88). On the other hand, acetyldihydropyridine absorbs at around 350 nm due to similar conjugation as found in dihydronicotinamide (82, 89–91). In short, TDDFT computations presented here match the experimental results very well (See Table S1), although the computations normally give higher transition energies; the results could be improved with a higher level calculation. Nonetheless, the key observation is the carboxamide group, the dihydropyridine group, and their conjugation are necessary for the electronic transitions and thus the fluorescence of NADH. Any factors including hydrogen bonds and steric effects involving these groups could induce a fluorescence change in bound NADH.
Table 2.
Electronic transition energies of dihydronicotinamide and related compounds at td-B3LYP/6-31g(d) level. Structures of these compounds can be found in the Supporting Information Figure S1.
| Compound | Electronic State | Energy, eV (nm) | f |
|---|---|---|---|
| 1-methyl-1,4-dihydronicotinamide | S1 (π-π*) | 3.747 (331) | 0.113 |
| S2 (n-π*) | 4.514 (275) | 0.001 | |
| Twisted 1-methyl-1,4-dihydronicotinamide | S1 (π-π*) | 3.652 (340) | 0.07 |
| S2 (n-π*) | 4.489 (276) | 0.002 | |
| 1-methyl-1,4-dihydropyridine | S1 (π-π*) | 4.417 (281) | 0.034 |
| 1-methyl-3-acetyl-1,4-dihydropyridine | S1 (π-π*) | 3.671 (338) | 0.138 |
| 1-methyl-1,4,5,6-tetrahydronicotinamide | S1 (n-π*) | 4.701 (264) | 0 |
| S2 (π-π*) | 4.883 (254) | 0.365 |
Electronic transitions in selected complexes
As oxamate binds to the LDH•NADH binary complex, it interacts with various residues. The tightest interactions are with the active site residues. We can assess the effect that these core residues have on quenching by adding different residues to the core structure, which is composed of dihydronicotinamide (fluorophore) and oxamate (quencher), a series of computations were performed to simulate the possible intermediate structures. His-195 is a key residue in LDH catalysis, and protonation is necessary for the binding of oxamate (or pyruvate; refs. (79, 80)). In Case 1, only protonated His-195 was added; see Table 3. The lowest electronic transition S1 state of dihydronicotinamide π-π* (LE) lies at 3.087 eV with an oscillator strength of 0.067. (It is of interest that the energy of the lowest electronic state does not vary too much in different models in our calculations). The lowest oxamate state is S2, a forbidden n-π* transition, with a much higher energy at 4.102 eV. The Kohn-Sham orbitals involving the transitions are shown in Fig. 9. The charge transfer (CT) state from π of dihydronicotinamide to π* of oxamate was found to be S14 at an energy level of 5.658 eV. A transition of mostly CT character from dihydronicotinamide to imidazole of His-195 was found to be S10 with 5.156 eV of energy. Thus the quenching of NADH fluorescence by photoinduced electron transfer (PET) is close to zero. However, if the carboxylate group is protonated, as in Case 2, a charge transfer (CT) state was found to be S1 state. The energy of 3.094 eV of the CT state is just below 3.209 eV for the lowest energy state of dihydronicotinamide. Then it is highly possible that PET could account for the fluorescence quenching here. It is also in line with previous findings that carboxylic acid could be an electron acceptor in PET (77). We should note the CT state energy from TDDFT calculation at B3LYP level functional is about 0.9 eV lower than that from CAM-B3LYP calculation in this case.
Table 3.
Electron transition energies of selected models at cam-B3LYP/6-31g(d) level. CT state energies are emphasized with a bold font.
| Case | Added Residues | Electronic State | Energy, eV (nm) | ELUMO (eV) | f |
|---|---|---|---|---|---|
| 1 | His-195 | S1 (π-π*, nicotinamide) | 3.087 (402) | −0.139 | 0.067 |
| S2 (n-π*, oxamate) | 4.102 (302) | 0 | |||
| 2 | H+(oxamic acid) + His-195 | S1 (π-π*, CT) | 3.094 (401) | −1.804 | 0.080 |
| S2 ((π-π*, nicotinamide) | 3.209 (386) | 0.059 | |||
| 3 | Arg-171 | S1 (π-π*, nicotinamide) | 3.271 (379) | −0.384 | 0.053 |
| S2 (n-π*, oxamate) | 4.238 (293) | 0 | |||
| S5 (π-π*, CT) | 5.297 (234) | 0.101 | |||
| 4 | Arg-171 + Thr246 | S1 (π-π*, nicotinamide) | 3.261 (380) | −0.631 | 0.057 |
| S2 (n-π*, oxamate) | 4.227 (293) | 0 | |||
| S5 (π-π*, CT) | 4.827 (257) | 0.058 | |||
| 5 | Arg-171 + His-195 | S1 (π-π*, nicotinamide) | 3.186 (389) | −0.816 | 0.068 |
| S2 (π-π*, CT) | 4.333 (286) | 0.060 | |||
| 6 | Arg-171 + His-195 + Thr-246 | S1 (π-π*, nicotinamide) | 3.180 (390) | −0.985 | 0.075 |
| S2 (π-π*, CT) | 4.008 (309) | 0.056 | |||
| 7 | Arg-171 + His-195 + Thr-246 + Asn-140 | S1 (π-π*, nicotinamide) | 3.276 (380) | −1.222 | 0.062 |
| S2 (π-π*, CT) | 3.740 (332) | 0.055 | |||
| 8 | Full binding | S1 (π-π*, CT) | 3.167 (391) | −1.859 | 0.085 |
| S2 (π-π*, nicotinamide) | 3.265 (380) | 0.090 |
Figure 9.
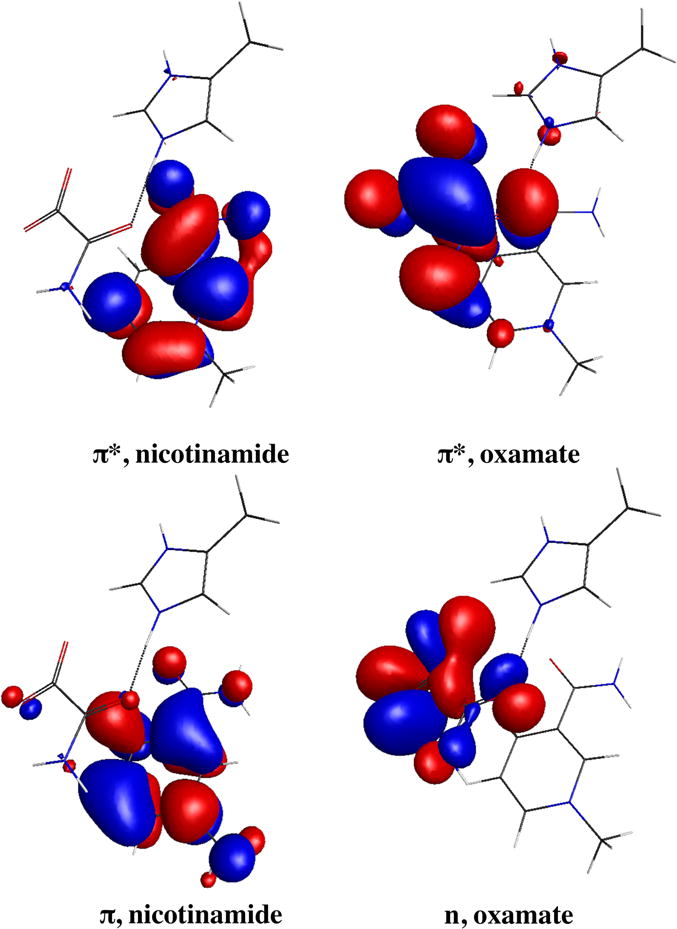
Sample frontier orbitals involved in the locally excited (LE) states and charge transfer (CT) states. The LE state will refer to π-π* transition of nicotinamide. The CT state will refer to π, nicotinamide-π*, oxamate transition.
Arg-171 is a key residue involving substrate binding. When it is the only residue added to the core structure in Case 3, a CT state was found at 5.297 eV, well above the lowest energy state of dihydronicotinamide (calculated to be at 3.271 eV). Adding Thr-246 to form a hydrogen bond with the carboxylate group of oxamate (Case 4) decreases the CT energy to 4.827 eV, about 0.5 eV difference. His-195 has a stronger effect on the CT transition as shown in Case 5, where the CT state is decrease by about 1 eV. Including His-195 and Thr-246 together with Arg-171, the energy of the CT state is 4.008 eV and the decrease from Arg-171 only complex is roughly a sum of contributions from His-195 and Thr-246. In this case, the frontier LUMOs (lowest unoccupied molecular orbitals), π* orbitals of dihydronicotinamide, oxamate, and imidazole of His-195, become near-degenerated. The degeneracy is removed when Asn-140 is added to the model. Asn-140 is another important residue involving substrate binding in LDH (92). In Case 7, with His-195, Arg-171, Thr-246, and Asn-140, the structure is close to the active site structure with loop open. The energy of the CT state is 3.740 eV, less than 0.5 eV above the lowest energy state of dihydronicotinamide. The contribution from Asn-140 is less than that of other three residues, which is reasonable considering a weaker hydrogen bond or longer distance between oxamate and Asn-140. According to above results, especially shown in Fig. 10, it is obvious that interactions between residues and oxamate could lead to electron transfer, as CT and LE states become close in energy.
Figure 10.
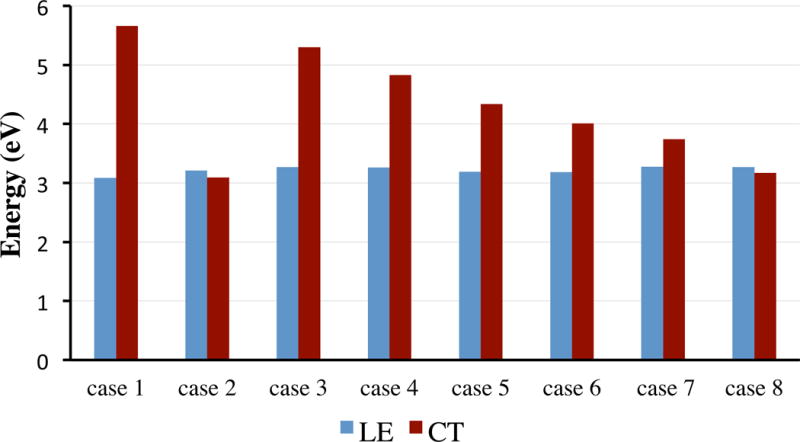
Energetic comparison of LE and CT states in selected cases in vacuum.
Including most interactions involving oxamate and dihydronicotinamide simulates the loop closed ternary complex (Case 8). Optimization of this complex in vacuum will result that Asp-168 abstracts the δ-H from His-195. So the polarizable continuum model (PCM) was applied to the computation to simulate media polarization on the transitions. The dielectric constant was set to 4 although this value may underestimate the polarity of LDH active site. The possible effect will be discussed below. In this case, the CT state is just below the lowest energy state with difference of only about 0.1 eV. The mechanism of photoinduced electron transfer (PET), involving transferring an electron from excited NADH (or dihydronicotinamide) to oxamate, does quite well to account for the fluorescence quenching of NADH in ternary complex.
As electron transfers (ET) can be strongly affected by media polarity, solvent effect needs to be considered, which in some cases is necessary in order to obtain right structure. A strong effect was found when a model is neutral (Table S2). In cases where the dielectric constant was set to 4, there is about 0.3–0.6 eV decrease of the CT state. Further increasing the constant to 78 yields another 0.2 eV stabilization. Positively charged models don’t show such effect; this is also true for locally excited states. As substrate binding may change the polarity and charge in the active site, which in turn affects fluorescence quenching by PET, a more careful selection of the dielectric constant is required to model the solvent effect. Notwithstanding this, the results here show the trend of the charge transfer state is not changed much by varying dielectric (see Supporting Information Figures S2 and S3).
From the cam-B3LYP computational results summarized in Table 3, one may see the contribution to the ET efficiency from LDH residues roughly add up. The CT state only can have an energy level close to or below the LE state (a necessary requirement for an efficient electron transfer) when the hydrogen bonds between oxamate and Arg-171 and His-195 are formed. Hence our results suggest that those bonds must be formed for the efficient quenching of NADH fluorescence in the formation of the encounter complex for phLDH. Thr-246, whose contribution is important, is not a conserved residue; its hydrogen bonding with oxamate is not indispensable in the formation of the encounter complex. Asn-140 might be helping in the step. But due to its weaker interaction, the contribution would be less important. Recently molecular dynamics study on pyruvate ternary complex also suggested that the hydrogen bonds between these residues and pyruvate form in the encounter complex(93). Following structural rearrangement and loop closure, the active site becomes tighter and hydrogen bonding, electrostatic interactions, and the hydrophobic effect all become stronger. Thus, quenching via PET become more efficient.
LUMO energies of oxamate in selected complexes
Electron transfer rates (ket) have been shown good correlations with reduction potentials of electron donors and acceptors according to Marcus theory (eqn. 2) (94) or diffusion controlled Sandros-Boltzmann dependence (eqn. 3) (95–98).
| (2) |
| (3) |
In eqn 2., |HAB| is the electronic coupling between initial and final states, λ is the reorganization energy, , and T are the reduced Planck constant, the Boltzmann constant, and the absolute temperature respectively. klim is the rate limit and R is the gas constant in eqn. 3. The free energy change, is given by Rehm-Weller equation (eqn. 4) (99):
| (4) |
where Ered(D+/D) and Ered(A/A−) are the reduction potentials of electron donor and acceptor. E00 is the energy of the excited reactant. C is the Coulombic energy term and generally is small. However, the potentials may not be experimentally available, especially in the case of one-electron transfer or for those transient species in our cases. We thus look into how the binding could affect the energies of LUMO of oxamate since the reduction potentials of electron acceptors have been well proved to be linearly related to their LUMO energies (100, 101). As the Stern-Volmer constant is given by KSV = kq τ0, the relationship between KSV and LUMO energies could be established. In fact, with 1-benzyl-dihydronicotinamide (BNAH) as an electron donor, we demonstrated that LUMO energies of several quenchers are linearly correlated to their reduction potentials (Ered) and then the relationship between KSV (or kq) and LUMO is similar with that between KSV (or kq) and Ered (see Fig. S4).
Back to LDH complexes, single point energy calculations at 6–31g(d) level in water were carried out and thus their oxamate LUMO (π*) energies (Table 2) are compared to those of some quenchers in solution (Table 1). As can be seen, the energy in case 6 is just greater than that of acrylamide, which in solution has KSV of 1.0 M−1. If we apply Sandros-Boltzmann equation to our quenching results, KSV of oxamate in the full complex where the LUMO energy is −1.859 eV, could be roughly estimated to be about 1.8 M−1. Considering the close proximity between oxamate and the dihydronicotinamide moiety, the apparent KSV could be much higher. Let’s assume it is 50 M−1. This estimation should be justified since it has shown the quenching of BNAH by methyl viologen in micelle is more than 30 times of efficiency than in solution (28). On the other hand, in a similar complex LDH•NADH•oxalate, the measured lifetime and the quantum yield (QY) is 6.53 ns and 0.45 respectively (15). According to these results, by using eqn. 1, we find the QY in the fully binding complex is about 0.02, which is 1/5 of that in binary complex and is fairly consistent with the results from the T-jump study (26). It should be noted that a larger basis set in quantum mechanical calculations might be needed. Like in pyruvate, the LUMO energy is strongly affected (not shown here) by applying 6–311++g(d,p) basis set, which is however computationally impractical for large complexes. Anyhow, for a more accurate estimation, the process needs more careful optimization.
CONCLUSIONS
In summary, we have discussed how is the fluorescence of NADH is quenched in ternary LDH complexes and propose photoinduced electron transfer (PET) as the major mechanism accounting for the fluorescence quenching. Oxamate is transformed into an effective electron acceptor by a series of interactions upon binding and thus becomes a good fluorescence quencher for NADH. The quenching ability is modulated by different substrate-protein interactions. Arg-171 and His-195 are most important among residues around oxamate. The models in the computations are based on an X-ray structure with a loop-closed form. These structures are likely tighter than for an encounter complex. We have observed strong quenching in the encounter complex of phLDH ternary complex; hence, those hydrogen bonding patterns must be formed even in the encounter complex. With a weaker interaction in bsLDH encounter complex, we expect to observe weaker quenching in the analogous structure, as observed by experiment (41).
Monitoring the fluorescence of NAD(P)H is a major means to study oxidoreductases and its physiological significance has been well known. Due to the complexity of proteins, understanding the quenching process NAD(P)H emission is a necessary step for a proper explanation of fluorescence experimental results. Unfortunately, most studies using fluorescence or absorption stopped-flow or T-jump techniques do not provide much structural information along the dynamic processes. In the present study, we demonstrated a correlation between the fluorescence quenching properties and the frontier orbital, LUMO energies, which are determined by the structural interactions along the enzyme dynamics. The relationships among those properties allowed us to envision the transient enzyme structures. All in all, by combining the theoretical computational methods, we can resolve important aspects of those intermediate structures.
Supplementary Material
Table S1. Electronic transition energies of dihydronicotinamide related compounds obtained by computations at td-B3LYP/6-31g(d) level and from experimental. Numbers in parentheses are oscillator strength and extinction coefficient respectively.
Table S2. Energies (eV) of locally excited (LE) sates and charge transfer (CT) states in vacuum and in water with polarizable continuum model (PCM) at selected dielectric constant (ε).
Figure S1. Structures of dihydronicotinamide and related compounds.
Figure S2. Energetic comparison of LE and CT states in selected cases in water (PCM, ε=4).
Figure S3. Comparison of Energies of CT states in selected cases in vacuum and water (PCM, ε=4).
Figure S4. Correlation between LUMO energies and the reduction potentials (b) and the quenching constants (a) of selected quenchers. LUMO energies were calculated at B3LYP/6-31g(d) level. The reduction potentials and the quenching constants were from the reference (7). Quenchers in the figure are given in order of increasing LUMO energy: 1. Trifluoroacetophanone, 2. Diethylfumarate, 3. Dimethylterephthalate, 4. 1-Cyanonaphthalene, 5. Benzophenone, 6. Acetophenone, 7. Cyanobenzene, 8. E-stilbene, 9. Methylbenzoate, 10. Styrene, and 11. 1-methylnaphthalene.
Acknowledgments
This work supported by the Institute of General Medicine of the National Institutes of Health, program project grant number 5P01GM068036. The authors also thank Dr. Deng, Hua for reviewing the manuscript and suggestions.
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article:
References
- 1.Wu Y, Zheng W, Qu JY. Sensing cell metabolism by time-resolved autofluorescence. Opt Lett. 2006;31:3122. doi: 10.1364/ol.31.003122. [DOI] [PubMed] [Google Scholar]
- 2.Wang H-W, Wei Y-H, Guo H-W. Reduced Nicotinamide Adenine Dinucleotide (NADH) Fluorescence for the Detection of Cell Death. Anticancer Agents Med Chem. 2009;9:1012–1017. doi: 10.2174/187152009789377718. [DOI] [PubMed] [Google Scholar]
- 3.Heikal AA. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 2010;4:241–263. doi: 10.2217/bmm.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, Duchen MR. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun. 2014;5:3936. doi: 10.1038/ncomms4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartolome F, Abramov AY. Measurement of mitochondrial NADH and FAD autofluorescence in live cells. In: Weissig V, Edeas M, editors. Mitochondrial Medicine: Volume I, Probing Mitochondrial function 1264. Springer; New York: 2015. pp. 263–270. [DOI] [PubMed] [Google Scholar]
- 6.Dolphin D, Poulson R, Avramovic O. Pyridine nucleotide coenzymes: Chemical, biological, and medical aspects, Pt. A. Wiley-Interscience; United States: 1987. [Google Scholar]
- 7.Dolphin D, Poulson R, Avramovic O. Pyridine nucleotide coenzymes: Coenzymes and Cofactors, Pt. B. Wiley-Interscience; United States: 1987. [Google Scholar]
- 8.Everse J. The Pyridine Nucleotide Coenzymes. Academic Press; 1982. [Google Scholar]
- 9.Sarma RH, Ross V, Kaplan NO. Investigation of the conformation of beta-diphosphopyridine nucleotide (beta-nicotinamide-adenine dinucleotide) and pyridine dinucleotide analogs by proton magnetic resonance. Biochemistry. 1968;7:3052–3062. doi: 10.1021/bi00849a005. [DOI] [PubMed] [Google Scholar]
- 10.Oppenheimer NJ, Arnold LJ, Kaplan NO. A structure of pyridine nucleotides in solution. Proc Natl Acad Sci USA. 1971;68:3200–3205. doi: 10.1073/pnas.68.12.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald G, Brown B, Hollis D, Walter C. Effects of environment on the folding of nicotinamide-adenine dinucleotides in aqueous solutions. Biochemistry. 1972;11:1920–1930. doi: 10.1021/bi00760a029. [DOI] [PubMed] [Google Scholar]
- 12.Sovago I, Bruce Martin R. Nicotinamide adenine dinucleotide is about 44% folded in neutral aqueous solutions. FEBS Lett. 1979;106:132–134. doi: 10.1016/0014-5793(79)80710-3. [DOI] [PubMed] [Google Scholar]
- 13.Smith PE, Tanner JJ. Conformations of nicotinamide adenine dinucleotide (NAD(+)) in various environments. J Mol Recogn. 2000;13:27–34. doi: 10.1002/(SICI)1099-1352(200001/02)13:1<27::AID-JMR483>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 14.Hull RV, Conger PS, Hoobler RJ. Conformation of NADH studied by fluorescence excitation transfer spectroscopy. Biophys Chem. 2001;90:9–16. doi: 10.1016/s0301-4622(00)00239-8. [DOI] [PubMed] [Google Scholar]
- 15.Scott TG, Spencer RD, Leonard NJ, Weber G. Emission properties of NADH. Studies of fluorescence lifetimes and quantum efficiencies of NADH, AcPyADH, and simplified synthetic models. J Am Chem Soc. 1970;92:687–695. [Google Scholar]
- 16.Czochralska B, Lindqvist L. Diphotonic one-electron oxidation of NADH on laser excitation at 353 nm. Chem Phys Lett. 1983;101:297–299. [Google Scholar]
- 17.Boldridge DW, Morton TH, Scott GW. Formation kinetics and quantum yield of photon-induced electron ejection from NADH in aqueous solution. Chem Phys Lett. 1984;108:461–465. [Google Scholar]
- 18.Lindqvist L, Czochralska B, Grigorov I. Determination of the mechanism of photo-ionization of NADH in aqueous solution on laser excitation at 355 nm. Chem Phys Lett. 1985;119:494–498. [Google Scholar]
- 19.Noguchi N, Tachikawa M, Takahashi H. One-Electron Photooxidation Of Nadh: Laser Photolysis, Time-Resolved Raman And Ab Initio Mo Calculation Study. In: Carmona P, Navarro R, Hernanz A, editors. Spectroscopy of Biological Molecules: Modern Trends. Springer; Netherlands, Dordrecht: 1997. pp. 161–162. [Google Scholar]
- 20.Pakalnis S, Sitas V, Schneckenburger H, Rotomskis R. Picosecond absorption spectroscopy of biologically active pigments NADH, FMN and fluorescence marker Rhodamine-123. The Third Internet Photochemistry and Photobiology Conference.2000. [Google Scholar]
- 21.Chergui M, Heiner Z, Roland T, Léonard J, Haacke S, Groma GI, Taylor A, Cundiff S, de Vivie-Riedle R, Yamagouchi K. Ultrafast Absorption Kinetics of NADH in Folded and Unfolded Conformations. EPJ Web of Conferences. 2013;41:07003. [Google Scholar]
- 22.Ross JBA, Subramanian S, Brand L. Spectroscopic Studies of the Pyridine Nucleotide Coenzymes and Their Complexes with Dehydrogenases. In: Everse J, editor. The Pyridine Nucleotide Coenzymes. Academic Press; 1982. pp. 19–49. [Google Scholar]
- 23.Fierke CA, Hammes GG. Transient kinetic approaches to enzyme mechanisms. In: Purich D, editor. Enzyme Kinetics and Mechanism Part D: Developments in Enzyme Dynamics. Vol. 249. Academic Press; 1995. pp. 3–37. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Ariola FS, Kim H-R, Yu Q, Ronn W, Ahmed H. Two-photon excited-state and conformation dynamics of NADH binding with dehydrogenases. In: Castleman AW, Kimble ML, editors. Femtochemistry VII. Elsevier; Amsterdam: 2006. pp. 396–401. [Google Scholar]
- 25.Dalziel K. Kinetics of Oxidative Decarboxylases. In: Ricard J, Cornish-Bowden A, editors. Dynamics of Biochemical Systems. Springer; US: 1984. pp. 65–81. [Google Scholar]
- 26.McClendon S, Zhadin N, Callender R. The approach to the Michaelis complex in lactate dehydrogenase: the substrate binding pathway. Biophys J. 2005;89:2024–2032. doi: 10.1529/biophysj.105.062604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhadin N, Gulotta M, Callender R. Probing the role of dynamics in hydride transfer catalyzed by lactate dehydrogenase. Biophys J. 2008;95:1974–1984. doi: 10.1529/biophysj.108.132464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martens FM, Verhoeven JW. Photo-induced electron transfer from NADH and other 1,4-dihydronicotinamides to methyl viologen. Recl Trav Chim Pays-Bas. 1981;100:228–236. [Google Scholar]
- 29.Martens FM, Verhoeven JW, Gase RA, Pandit UK, de Boer TJ. On the question of one-electron transfer in the mechanism of reduction by nadh-models. Tetrahedron. 1978;34:443–446. [Google Scholar]
- 30.Martens FM, Verhoeven JW, Varma CAGO, Bergwerf P. Photo-oxidation of 1,4-dihydropyridines by various electron acceptors: a laser flash photolysis study. J Photochem. 1983;22:99–113. [Google Scholar]
- 31.Callis PR, Vivian JT. Understanding the variable fluorescence quantum yield of tryptophan in proteins using QM-MM simulations. Quenching by charge transfer to the peptide backbone. Chem Phys Lett. 2003;369:409–414. [Google Scholar]
- 32.Callis PR, Liu TQ. Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J Phys Chem B. 2004;108:4248–4259. [Google Scholar]
- 33.Peng Q, Yi Y, Shuai Z, Shao J. Toward quantitative prediction of molecular fluorescence quantum efficiency: role of duschinsky rotation. J Am Chem Soc. 2007;129:9333–9339. doi: 10.1021/ja067946e. [DOI] [PubMed] [Google Scholar]
- 34.Stinson RA, Gutfreund H. Transient-kinetic studies of pig muscle lactate dehydrogenase. Biochem J. 1971;121:235–240. doi: 10.1042/bj1210235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke AR, Waldman ADB, Hart KW, John Holbrook J. The rates of defined changes in protein structure during the catalytic cycle of lactate dehydrogenase. Biochim Biophys Acta. 1985;829:397–407. doi: 10.1016/0167-4838(85)90250-x. [DOI] [PubMed] [Google Scholar]
- 36.Holbrook JJ, Stinson RA. The use of ternary complexes to study ionizations and isomerizations during catalysis by lactate dehydrogenase. Biochem J. 1973;131:739–748. doi: 10.1042/bj1310739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clarke AR, Wigley DB, Chia WN, Barstow D, Atkinson T, Holbrook JJ. Site-directed mutagenesis reveals role of mobile arginine residue in lactate dehydrogenase catalysis. Nature. 1986;324:699–702. doi: 10.1038/324699a0. [DOI] [PubMed] [Google Scholar]
- 38.Boland MJ, Gutfreund H. Pig heart lactate dehydrogenase. Binding of pyruvate and the interconversion of pyruvate-containing ternary complexes. Biochem J. 1975;151:715–727. doi: 10.1042/bj1510715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luyten MA, Bur D, Wynn H, Parris W, Gold M, Friesen JD, Jones JB. An Evaluation of the Substrate-Specificity, and of Its Modification by Site-Directed Mutagenesis, of the Cloned L-Lactate Dehydrogenase from Bacillus-Stearothermophilus. J Am Chem Soc. 1989;111:6800–6804. [Google Scholar]
- 40.Nie B, Deng H, Desamero R, Callender R. Large scale dynamics of the Michaelis complex in Bacillus stearothermophilus lactate dehydrogenase revealed by a single-tryptophan mutant study. Biochemistry. 2013;52:1886–1892. doi: 10.1021/bi3017125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng H, Brewer S, Vu DM, Clinch K, Callender R, Dyer RB. On the pathway of forming enzymatically productive ligand-protein complexes in lactate dehydrogenase. Biophys J. 2008;95:804–813. doi: 10.1529/biophysj.108.128884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng H, Vu DV, Clinch K, Desamero R, Dyer RB, Callender R. Conformational heterogeneity within the Michaelis complex of lactate dehydrogenase. J Phys Chem B. 2011;115:7670–7678. doi: 10.1021/jp2015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wigley DB, Gamblin SJ, Turkenburg JP, Dodson EJ, Piontek K, Muirhead H, Holbrook JJ. Structure of a Ternary Complex of an Allosteric Lactate-Dehydrogenase from Bacillus-Stearothermophilus at 2.5 a Resolution. J Mol Biol. 1992;223:317–335. doi: 10.1016/0022-2836(92)90733-z. [DOI] [PubMed] [Google Scholar]
- 44.Dunn CR, Wilks HM, Halsall DJ, Atkinson T, Clarke AR, Muirhead H, Holbrook JJ. Design and Synthesis of New Enzymes Based on the Lactate Dehydrogenase Framework. Philos Trans R Soc Lond, Ser B: Biol Sci. 1991;332:177–184. doi: 10.1098/rstb.1991.0047. [DOI] [PubMed] [Google Scholar]
- 45.Borgmann U, Moon TW, Laidler KJ. Molecular kinetics of beef heart lactate dehydrogenase. Biochemistry. 1974;13:5152–5158. doi: 10.1021/bi00722a016. [DOI] [PubMed] [Google Scholar]
- 46.Heck HD, McMurray CH, Gutfreund H. The resolution of some steps of the reactions of lactate dehydrogenase with its substrates. Biochem J. 1968;108:793–796. doi: 10.1042/bj1080793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parker DM, Jeckel D, Holbrook JJ. Slow structural changes shown by the 3-nitrotyrosine-237 residue in pig heart [Tyr(3NO2)237] lactate dehydrogenase. Biochem J. 1982;201:465–471. doi: 10.1042/bj2010465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gulotta M, Deng H, Deng H, Dyer RB, Callender RH. Toward an understanding of the role of dynamics on enzymatic catalysis in lactate dehydrogenase. Biochemistry. 2002;41:3353–3363. doi: 10.1021/bi016009a. [DOI] [PubMed] [Google Scholar]
- 49.Reddish M, Peng H-L, Deng H, Panwar KS, Callender R, Dyer RB. Direct Evidence of Catalytic Heterogeneity in Lactate Dehydrogenase by Temperature Jump Infrared Spectroscopy. J Phys Chem B. 2014;118:10854–10862. doi: 10.1021/jp5050546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Callender R, Dyer RB. The dynamical nature of enzymatic catalysis. Acc Chem Res. 2015;48:407–413. doi: 10.1021/ar5002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA. General atomic and molecular electronic structure system. J Comput Chem. 1993;14:1347–1363. [Google Scholar]
- 52.Gordon MS, Schmidt MW. Advances in electronic structure theory: GAMESS a decade later. In: Dykstra CE, Frenking G, Kim KS, Scuseria GE, editors. Theory and Applications of Computational Chemistry: the first forty years. Elsevier; 2005. pp. 1167–1189. [Google Scholar]
- 53.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 54.Hertwig RH, Koch W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem Phys Lett. 1997;268:345–351. [Google Scholar]
- 55.Dreuw A, Head-Gordon M. Failure of time-dependent density functional theory for long-range charge-transfer excited states: the zincbacteriochlorin-bacteriochlorin and bacteriochlorophyll-spheroidene complexes. J Am Chem Soc. 2004;126:4007–4016. doi: 10.1021/ja039556n. [DOI] [PubMed] [Google Scholar]
- 56.Magyar RJ, Tretiak S. Dependence of Spurious Charge-Transfer Excited States on Orbital Exchange in TDDFT: Large Molecules and Clusters. J Chem Theory Comput. 2007;3:976–987. doi: 10.1021/ct600282k. [DOI] [PubMed] [Google Scholar]
- 57.Lange AW, Herbert JM. Both intra- and interstrand charge-transfer excited states in aqueous B-DNA are present at energies comparable to, or just above, the (1)pipi* excitonic bright states. J Am Chem Soc. 2009;131:3913–3922. doi: 10.1021/ja808998q. [DOI] [PubMed] [Google Scholar]
- 58.Yanai T, Tew DP, Handy NC. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem Phys Lett. 2004;393:51–57. [Google Scholar]
- 59.Kobayashi R, Amos RD. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin–bacteriochlorin complex. Chem Phys Lett. 2006;420:106–109. [Google Scholar]
- 60.Cossi M, Scalmani G, Rega N, Barone V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J Chem Phys. 2002;117:43–54. [Google Scholar]
- 61.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schrodinger LLC. The PyMOL Molecular Graphics System. 2015 Version 1.8. [Google Scholar]
- 64.Bode BM, Gordon MS. Macmolplt: a graphical user interface for GAMESS. J Mol Graphics Model. 1998;16:133–138. doi: 10.1016/s1093-3263(99)00002-9. [DOI] [PubMed] [Google Scholar]
- 65.Allouche AR. Gabedit–a graphical user interface for computational chemistry softwares. J Comput Chem. 2011;32:174–182. doi: 10.1002/jcc.21600. [DOI] [PubMed] [Google Scholar]
- 66.Ando T, Miyata H. Pyruvate as a fluorescence quencher: A new spectroscopic assay for pyruvate reactions. Anal Biochem. 1983;129:170–175. doi: 10.1016/0003-2697(83)90065-9. [DOI] [PubMed] [Google Scholar]
- 67.Tallmadge DH, Huebner JS, Borkman RF. Acrylamide quenching of tryptophan photochemistry and photophysics. Photochem Photobiol. 1989;49:381–386. doi: 10.1111/j.1751-1097.1989.tb09183.x. [DOI] [PubMed] [Google Scholar]
- 68.Ilich P, Prendergast FG. Electronic states of the indole-acrylamide molecular pair. Photochem Photobiol. 1991;53:445–453. doi: 10.1111/j.1751-1097.1991.tb03655.x. [DOI] [PubMed] [Google Scholar]
- 69.Zelent B, Kusba J, Gryczynski I, Johnson ML, Lakowicz JR. Time-resolved and steady-state fluorescence quenching of N-acetyl-L-tryptophanamide by acrylamide and iodide. Biophys Chem. 1998;73:53–75. doi: 10.1016/s0301-4622(98)00137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eftink MR, Selva TJ, Wasylewski Z. Studies of the Efficiency and Mechanism of Fluorescence Quenching Reactions Using Acrylamide and Succinimide as Quenchers. Photochem Photobiol. 1987;46:23–30. [Google Scholar]
- 71.Bastyns K, Engelborghs Y. Acrylamide quenching of the fluorescence of glyceraldehyde-3-phosphate dehydrogenase: reversible and irreversible effects. Photochem Photobiol. 1992;55:9–16. doi: 10.1111/j.1751-1097.1992.tb04203.x. [DOI] [PubMed] [Google Scholar]
- 72.Eftink MR, Selvidge LA. Fluorescence quenching of liver alcohol dehydrogenase by acrylamide. Biochemistry. 1982;21:117–125. doi: 10.1021/bi00530a021. [DOI] [PubMed] [Google Scholar]
- 73.Chen Y, Liu B, Yu H-T, Barkley MD. The Peptide Bond Quenches Indole Fluorescence. J Am Chem Soc. 1996;118:9271–9278. [Google Scholar]
- 74.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; US: 2006. Quenching of Fluorescence; pp. 277–330. [Google Scholar]
- 75.Winer AD, Schwert GW. Lactic dehydrogenase. VII. Fluorescence spectra of ternary complexes of lactic dehydrogenase, reduced diphosphopyridine nucleotide, and carboxylic acids. J Biol Chem. 1959;234:1155–1161. [PubMed] [Google Scholar]
- 76.Willaert K, Engelborghs Y. The Quenching of Tryptophan Fluorescence by Protonated and Unprotonated Imidazole. Eur Biophys J. 1991;20:177–182. [Google Scholar]
- 77.Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 78.Vos R, Engelborghs Y. A fluorescence study of tryptophan-histidine interactions in the peptide anantin and in solution. Photochem Photobiol. 1994;60:24–32. doi: 10.1111/j.1751-1097.1994.tb03938.x. [DOI] [PubMed] [Google Scholar]
- 79.Holbrook JJ, Ingram VA. Ionic properties of an essential histidine residue in pig heart lactate dehydrogenase. Biochem J. 1973;131:729–738. doi: 10.1042/bj1310729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clarke AR, Wilks HM, Barstow DA, Atkinson T, Chia WN, Holbrook JJ. An investigation of the contribution made by the carboxylate group of an active site histidine-aspartate couple to binding and catalysis in lactate dehydrogenase. Biochemistry. 1988;27:1617–1622. doi: 10.1021/bi00405a034. [DOI] [PubMed] [Google Scholar]
- 81.Meijers R, Adolph HW, Dauter Z, Wilson KS, Lamzin VS, Cedergren-Zeppezauer ES. Structural evidence for a ligand coordination switch in liver alcohol dehydrogenase. Biochemistry. 2007;46:5446–5454. doi: 10.1021/bi6023594. [DOI] [PubMed] [Google Scholar]
- 82.Kosower EM, Sorensen TS. The Synthesis and Properties of Some Simple 1,4-Dihydropyridines. J Org Chem. 1962;27:3764–3771. [Google Scholar]
- 83.Cook NC, Lyons JE. 1,4-Dihydropyridine. J Am Chem Soc. 1965;87:3283–3284. [Google Scholar]
- 84.Eisner U, Kuthan J. Chemistry of dihydropyridines. Chem Rev. 1972;72:1–42. [Google Scholar]
- 85.Quan PM, Quin LD. 1,4,5,6-Tetrahydropyridines from Catalytic Reduction of Nicotinoyl Derivatives and Their Ring Opening with Hydrazine. J Org Chem. 1966;31:2487–2490. [Google Scholar]
- 86.Dave KG, Dunlap RB, Jain MK, Cordes EH, Wenkert E. Highly reduced analogues of the pyridine nucleotide coenzymes. Synthesis of tetrahydro- and hexahydronicotinamide adenine dinucleotides and certain of their derivatives. J Biol Chem. 1968;243:1073–1074. [PubMed] [Google Scholar]
- 87.Quin LD, Pinion DO. Reaction of 1,4,5,6-tetrahydronicotinamide with hydroxylamine [isoxazolinones and aminocyanopentanoic acid] J Org Chem. 1970;35:3130–3134. [Google Scholar]
- 88.Branlant G, Eiler B, Biellmann JF. A word of caution: 1,4,5,6-tetrahydronicotinamide adenine dinucleotide (phosphate) should be used with care in acidic and neutral media. Anal Biochem. 1982;125:264–268. doi: 10.1016/0003-2697(82)90005-7. [DOI] [PubMed] [Google Scholar]
- 89.Anderson AG, Berkelhammer G. A Study of the Primary Acid Reaction on Model Compounds of Reduced Diphosphopyridine Nucleotide1,2. J Am Chem Soc. 1958;80:992–999. [Google Scholar]
- 90.Evleth EM. The electronic structures of the dihydronicotinamides and related materials. J Am Chem Soc. 1967;89:6445–6453. doi: 10.1021/ja01001a012. [DOI] [PubMed] [Google Scholar]
- 91.Brewster ME, Simay A, Czako K, Winwood D, Farag H, Bodor N. Reactivity of biologically important reduced pyridines. IV. Effect of substitution on ferricyanide-mediated oxidation rates of various 1,4-dihydropyridines. J Org Chem. 1989;54:3721–3726. [Google Scholar]
- 92.Cortes A, Emery DC, Halsall DJ, Jackson RM, Clarke AR, Holbrook JJ. Charge balance in the alpha-hydroxyacid dehydrogenase vacuole: an acid test. Protein Sci. 1992;1:892–901. doi: 10.1002/pro.5560010707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pan X, Schwartz SD. Free energy surface of the michaelis complex of lactate dehydrogenase: a network analysis of microsecond simulations. J Phys Chem B. 2015;119:5430–5436. doi: 10.1021/acs.jpcb.5b01840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mattay J. Electron Transfer I. Springer; Berlin Heidelberg: 1994. [Google Scholar]
- 95.Farid S, Dinnocenzo JP, Merkel PB, Young RH, Shukla D. Bimolecular electron transfers that follow a Sandros-Boltzmann dependence on free energy. J Am Chem Soc. 2011;133:4791–4801. doi: 10.1021/ja104536j. [DOI] [PubMed] [Google Scholar]
- 96.Luo P, Dinnocenzo JP, Merkel PB, Young RH, Farid S. Bimolecular electron transfers that deviate from the Sandros-Boltzmann dependence on free energy: steric effect. J Org Chem. 2012;77:1632–1639. doi: 10.1021/jo202385f. [DOI] [PubMed] [Google Scholar]
- 97.Rosspeintner A, Angulo G, Vauthey E. Bimolecular photoinduced electron transfer beyond the diffusion limit: the Rehm-Weller experiment revisited with femtosecond time resolution. J Am Chem Soc. 2014;136:2026–2032. doi: 10.1021/ja4118279. [DOI] [PubMed] [Google Scholar]
- 98.Farid S, Dinnocenzo JP, Merkel PB, Young RH, Shukla D, Guirado G. Reexamination of the Rehm-Weller data set reveals electron transfer quenching that follows a Sandros-Boltzmann dependence on free energy. J Am Chem Soc. 2011;133:11580–11587. doi: 10.1021/ja2024367. [DOI] [PubMed] [Google Scholar]
- 99.Rehm D, Weller A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr J Chem. 1970;8:259–271. [Google Scholar]
- 100.Seto K, Nakayama T, Uno B. Formal redox potentials of organic molecules in ionic liquids on the basis of quaternary nitrogen cations as adiabatic electron affinities. J Phys Chem B. 2013;117:10834–10845. doi: 10.1021/jp402457k. [DOI] [PubMed] [Google Scholar]
- 101.Mendez-Hernandez DD, Tarakeshwar P, Gust D, Moore TA, Moore AL, Mujica V. Simple and accurate correlation of experimental redox potentials and DFT-calculated HOMO/LUMO energies of polycyclic aromatic hydrocarbons. J Mol Model. 2013;19:2845–2848. doi: 10.1007/s00894-012-1694-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Electronic transition energies of dihydronicotinamide related compounds obtained by computations at td-B3LYP/6-31g(d) level and from experimental. Numbers in parentheses are oscillator strength and extinction coefficient respectively.
Table S2. Energies (eV) of locally excited (LE) sates and charge transfer (CT) states in vacuum and in water with polarizable continuum model (PCM) at selected dielectric constant (ε).
Figure S1. Structures of dihydronicotinamide and related compounds.
Figure S2. Energetic comparison of LE and CT states in selected cases in water (PCM, ε=4).
Figure S3. Comparison of Energies of CT states in selected cases in vacuum and water (PCM, ε=4).
Figure S4. Correlation between LUMO energies and the reduction potentials (b) and the quenching constants (a) of selected quenchers. LUMO energies were calculated at B3LYP/6-31g(d) level. The reduction potentials and the quenching constants were from the reference (7). Quenchers in the figure are given in order of increasing LUMO energy: 1. Trifluoroacetophanone, 2. Diethylfumarate, 3. Dimethylterephthalate, 4. 1-Cyanonaphthalene, 5. Benzophenone, 6. Acetophenone, 7. Cyanobenzene, 8. E-stilbene, 9. Methylbenzoate, 10. Styrene, and 11. 1-methylnaphthalene.