

Abstract
Ten antifouling 14-membered resorcylic acid lactones 1–10 were isolated previously with low or trace natural abundance from the zoanthid-derived Cochliobolus lunatus fungus. Further optimization of fermentation conditions led to the isolation of two major natural compounds 7 and 8 with multi-gram quantities. By one or two steps, we semisynthesized the six trace natural compounds 1–6 and a series of derivatives 11–27 of compounds 7 and 8 with high yields (65–95%). Compounds 11–13 showed strong antiplasmodial activity against Plasmodium falciparum with IC50 values of 1.84, 8.36, and 6.95 μM, respectively. Very importantly, 11 and 12 were non-toxic with very safety and high therapeutic indices (CC50/IC50 > 180), and thus representing potential promising leads for antiplasmodial drug discovery. Furthermore, 11 was the only compound showed obvious antileishmanial activity against Leishmania donovani with an IC50 value of 9.22 μM. Compounds 11 and 12 showed the values of IC50 at 11.9 and 17.2 μM against neglected Chagas’ disease causing Trypanosoma cruzi, respectively.
Introduction
Protozoan infections such as malaria, leishmaniasis, and Chagas’ disease remain a major threat to public health especially in the tropical regions of the world. Malaria is a life-threatening mosquito-borne infectious disease caused by a parasite of the genus Plasmodium. According to the World Health Organization, there are 212 million new cases of malaria with 429,000 deaths in 2015. Malaria is a serious problem especially in Africa, where it is responsible for most global malaria cases (90%)1. Leishmaniasis is a debilitating and devastating disease caused by protozoan parasites of the genus Leishmania, which is endemic in 98 countries, with 20,000–30,000 deaths and 0.7–1.0 million new cases annually2. Chagas’ disease or American trypanosomiasis is caused by the parasite Trypanosoma cruzi and is responsible for about 5.7 million people infected with the parasite and 7,000 deaths per year3,4. Current drugs used for the treatment of these diseases are effective, but resistance and side effects, in some cases serious, have been reported for all the current therapeutic agents5–8. There is therefore an urgent need to develop new antimalarial, antileishmanial, and antitrypanosomal agents that act on new targets with novel modes of action.
Fungi have been proven to be a rich source of structurally diverse and biologically active secondary metabolites that are of considerable synthetic interest and dramatic significance as promising leads for drug discovery and development9,10. 14-Membered resorcylic acid lactones (RALs) constitute a class of fungal polyketide metabolites which exhibit a wide range of biological properties including antimalarial11,12, cytotoxic13, antiparasitic, antiviral14, and kinase inhibitory15,16 activities. Paecilomycins E and F, with potent antiplasmodial activity against P. falciparum lines 3D7 (IC50: 20 nM and 1.1 μM) and Dd2 (IC50: 8.8 μM and 1.7 μM, respectively), were isolated from a Paecilomyces fungus11,12. Hypothemycin and aigialomycin D that exhibited antimalarial activity against P. falciparum lines K1 with IC50 values of 2.2 and 6.6 μg/mL respectively and cytotoxicity, were isolated from Aigialus parvus 13. In our previous studies, cochliomycins A, C–F (1–5), and several analogues (6–10) have been isolated with low or trace yields from the fungus C. lunatus (M351)17 and C. lunatus (TA26-46)18. In view of their potentially biological applications, cochliomycins have attracted the attention of academic groups to synthesize these compounds. The most challenging part of their syntheses was the assembly of the macrolide ring and a significant range of techniques with complicated procedures have been tried and improved19–25. For example, cochliomycin A was firstly synthesized with 22 steps (6.5% overall yields) employing stereo-selective Keck allylation, Juliae–Kocienski olefination and a late-stage RCM reaction as the key steps19. Meanwhile, the scarce availability of the natural products including time-consuming isolation protocols has also prompted our study of alternative sources of these materials for additional studies.
Up to now, total syntheses of cochliomycins A–C have been accomplished extensively by Nanda’s group19,24, Du’s group20,21, Srihari’s group23, and Banwell’s group22,25. Cochliomycin A (1) was firstly synthesized in overall yield of 6.5% in 201219, and then was stereo-selectively synthesized by Wang et al. with 16 steps and only mg scale (23.0 mg)20. Cochliomycin B has been synthesized in 4.8% overall yields21 and the latest progress in the modular total syntheses of 1 (36.8 mg) and cochliomycin B (58.0 mg) were reported with 18 steps22. Cochliomycins A–C were in close resemblance to antiplasmodial paecilomycins E and F structurally, and cochliomycin C (2) is a chlorinated derivative of paecilomycin F on aromatic ring at C5 carbon. Total syntheses of paecilomycins E and F were also accomplished and reported during the period 2012–201626–30. Very recently, cochliomycin C (2) and paecilomycin F were synthesized successively by Srihari’s group23 with 16 steps, Nanda’s group24 with 12 steps in overall yield of 14.1%, and Banwell’s group25 with 11 steps. There is no report on total syntheses of compounds 3–6. Alternatively, semisynthetic strategy could provide an efficient and economical route to obtain natural products for further investigation of their pharmacological activities.
In this study, further optimization of the fermentation conditions of C. lunatus resulted in multi-gram quantities of compounds 7 and 8, and the one or two-step semisyntheses of natural compounds 1–6 and a series of derivatives 11–27 with high yields were reported (Fig. 1). The antiplasmodial, antileishmanial, antitrypanosomal, and cytotoxic activities of these compounds were evaluated in vitro. The preliminary structure-activity relationships were discussed.
Figure 1.

Chemical structures of natural compounds 1–10 and their derivatives 11–27.
Results and Discussion
Progress of the optimization of fermentation
The fungal strain C. lunatus was cultured in a 500 mL flask containing 200 mL liquid medium and incubated at 28 °C for 7 days on a rotary shaker at 120 rpm. The result of analysis showed that 7 and 8 are two predominant products in oligotrophic liquid medium (soluble starch 10 g/L, tryptone 1 g/L, 3% salinity). Then, the medium was selected as the basic medium for further optimization. The level of all the single factors including carbon source, nitrogen source, salinity, pH, inoculum size, and cultural time were studied. Subsequently, orthogonal test was carried out to approach the optimum point. All experiments were carried out in triplicate and the yields of 7 and 8 were calculated as the average values of three independent experiments. The fermented liquid medium was extracted with 200 mL ethyl acetate for each flask three times. The yield of 8 was 155.4 mg/L (soluble starch 10 g/L, NaNO3 5 g/L, NaOAc 1 g/L, 1% salinity, 10% inoculum size, adjusting initial pH value to 6 with 10% HCl/NaOH, Medium I). The yield of 7 reached 137.8 mg/L (soluble starch 10 g/L, NaNO3 5 g/L, 0.1% salinity, 15% inoculum size, adjusting initial pH value to 6 with 10% HCl/NaOH, Medium II) (Fig. 2). This supplied with a feasible method to obtain multi-gram quantities of 7 and 8 by fermentation in the laboratory. The advantages of the fermentation method were low cost, time saving, and easy separation by recrystallization. The structures of 7 and 8 were determined by comparing the NMR and MS spectroscopic data with that reported in literature31,32.
Figure 2.

HPLC profiles for the EtOAc extract of metabolites in the media Medium I and Medium II.
Semisynthesis
Compounds 1–6 and 11–27 were designed and synthesized through one or two-step semisynthetic reaction with high yields ranging from 65–95% from 7 and 8 (Fig. 1). The structures of new derivatives 12, 14–19, 21–22, 24, and 27 were identified by extensive spectroscopic methods including HRESIMS, 1H NMR, and 13C NMR. Compound 1 was prepared in yield of 95% through one-step acetonide reaction from 8. Other acetonide derivatives (11, 17, 18, 20, 23, 25, and 26) were also prepared with the same yields. Chlorination of compound 8 with sulfuryl chloride33 followed by selective reduction with 10% Pd-C under H2 atmosphere led to the natural compound 2 (68%, yield). Acylation reaction was carried out by reaction of anhydride with the corresponding materials (1–3, and 6–8) in high yields (85–95%). In order to prepare 6, oxidation of the allyl alcohol 8 was examined under various oxidation conditions (PCC, PDC, MnO2, DMP, and IBX). However, only DMP and IBX enabled the selective oxidation of the allyl alcohol to give 6 in 65% and 35% yields, respectively (Fig. 3). Interestingly, a fortuitous observation revealed that 6 can slowly proceed in chemical conversion to 3–5 in the process of silica gel column separation. Compounds 3–6 are diastereomers differing from each other by the absolute configurations of the 4′, 5′-diol chiral centers. The proportions of compounds 3–6 were in dynamic equilibrium, and the final ratios of 3–6 were approximately 8: 2: 5: 6 (Fig. 4). Further study revealed that a subtle chemical conversion of 6 to 3–5 was observed in protic solvents (MeOH, EtOH, and MeOH-d 4, except for H2O) in the stationary state within a week, and similar interconversion of 4 and 5 were also observed under the same conditions, while compound 3 was found to be quite stable (Fig. 5). However, compounds 4–6 were found to be stable in aprotic solvents (EtOAc, CH3CN, acetone, and CH2Cl2). Compounds 4–6 were unstable because of the existence of trans-enone moieties and the absolute configurations of the 4′, 5′-diol groups, causing chemical conversions under the condition of protic solvent, such as intramolecular hetero Michael addition34 and carbon migration of hemiacetal35. This conversion played a vital role in obtaining compounds 3–5 for further biological study.
Figure 3.

Example of one or two-step semisyntheses of compounds 1, 2, 6, 13, and 16.
Figure 4.

Semisynthesis of compound 6 and chemical conversion of 6 to 3–5.
Figure 5.

Chemical interconversion of compounds 3–6 in MeOH.
Antiplasmodial activity
Results for the in vitro antiplasmodial activity of all of the compounds were shown in Table 1. Compounds 11–13 and 26 exhibited strong antiplasmodial activity with the IC50 values of 1.84, 8.36, 6.95, and 8.95 μM, respectively. It should be pointed that the introduction of the acetoxy groups in compounds 1 and 8 appreciably change the activity, indicating that adding the acetoxy groups has a positive effect on the antiplasmodial activity. The acetonide functionality in 11 improved the IC50 value approximately 4-fold over that of 13 with the acetoxy groups at C5′-C6′. This suggests that the acetonide functionality might contribute to the antiplasmodial activity. However, the introduction of the chlorine atom at C5 in 2 and 16–19 was all found to be inactive, indicating that the chlorine atom has a negative effect on the antiplasmodial activity (Fig. 6).
Table 1.
Antiplasmodial, antileishmanial, antitrypanosomal, and toxic activities of compounds 1–27.
| No. | Plasmodium falciparum | Leishmania donovani | Trypanosoma cruzi | Vero cells | |||
|---|---|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | CC50 (μM) | |
| 1 | 30.7 | 3 | I | — | I | — | 96.0 |
| 2 | I | — | I | — | I | — | nt |
| 3 | I | — | 9.11 | — | I | — | nt |
| 4 | I | — | 15.2 | — | I | — | nt |
| 5 | I | — | I | — | I | — | nt |
| 6 | 14.9 | <1 | 1.93 | 2 | nt | — | 3.20 |
| 7 | 11.0 | <1 | 2.21 | <1 | nt | — | <1 |
| 8 | I | — | I | — | I | — | 227.1 |
| 9 | I | — | I | — | I | — | 131.0 |
| 10 | I | — | I | — | I | — | 273.8 |
| 11 | 1.84 | 184 | 9.22 | 37 | 11.9 | 28 | 338.7 |
| 12 | 8.36 | 182 | 15.7 | 97 | 17.2 | 88 | 1520.5 |
| 13 | 6.95 | 61 | I | — | I | — | 426.6 |
| 14 | I | — | I | — | I | — | nt |
| 15 | 22.2 | 71 | I | — | I | — | 1567.8 |
| 16 | I | — | I | — | I | — | 260.0 |
| 17 | I | — | I | — | I | — | 143.1 |
| 18 | I | — | I | — | I | — | 101.9 |
| 19 | I | — | I | — | I | — | nt |
| 20 | I | — | 6.46 | <1 | nt | — | <1 |
| 21 | I | — | 3.69 | 2 | nt | — | 5.80 |
| 22 | 20.1 | <1 | 3.03 | 4 | 6.31 | 2 | 11.5 |
| 23 | I | — | I | — | I | — | nt |
| 24 | I | — | 12.1 | <1 | nt | — | 3.20 |
| 25 | I | — | I | — | I | — | nt |
| 26 | 8.95 | <1 | 1.24 | <1 | nt | — | <1 |
| 27 | I | — | 3.14 | <1 | nt | — | <1 |
SI: Selectivity Index (CC50/IC50); I: Inactive; nt: Not tested.
Figure 6.
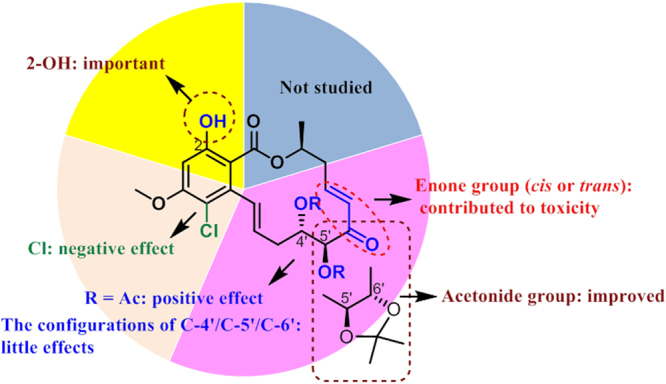
The structure-activity relationships of RALs.
Antileishmanial activity
Antileishmanial activity of all of the compounds was tested in vitro against L. donovani and were also shown in Table 1. For the enone RALs, compounds 3, 6, 7, 20–22, 26, and 27 displayed obvious antileishmanial activity with IC50 values ranging from 1.24 μM to 9.11 μM. Additionally, antiplasmodial compound 11 also showed antileishmanial activity with an IC50 value of 9.22 μM.
Antitrypanosomal activity
The bioassays against T. cruzi of all 27 molecules showed little activity against this parasite in initial screenings at 10 μg/mL, except in the cases when the compound was also discovered to be toxic (data not shown). However, compounds 11 and 12 showed IC50 values of 11.9 and 17.2 μM, respectively, with selectivity indexes of 28 and 88, making 12 a good candidate for in vivo studies.
Toxicity
It should be emphasized that toxicity is a major concern of drug discovery and development. The above active compounds were evaluated for in vitro toxicity against a mammalian kidney cell line (Vero). The selectivity index (SI) was used as the evaluation parameter of drug potential of the test samples by comparing the toxicity on the Vero cell line (CC50) and the selective inhibitory effect on P. falciparum, L. donovani or T. cruzi (IC50) calculated here as CC50/IC50. Compounds 11–13 and 15 were found to be non-toxic and showed encouraging therapeutic indices, which were far greater than the value requested by the Medicine for Malaria Venture (SI > 10). Specially, antiplasmodial compounds 11 and 12 showed pronounced selectivity indices (SI > 180), and thus represent potentially promising antiplasmodial leads for further development. Compounds 12 and 13, where 12, with a 2-OH group, displayed 3-fold higher therapeutic ratio and equivalent or stronger activities than compound 13, indicating that 2-OH is an important functionality for reducing the toxicity. For the enone RALs, most of them had high antileishmanial efficacy but poor therapeutic indices, indicating that the cis or trans-functionality has a positive contribution to toxicity, and that the antileishmanial activity of these compounds against L. donovani may well be related to their toxicity (Fig. 6).
Cytotoxic activity
All of the compounds were also evaluated for their cytotoxic properties against six human cancer cell lines (lung carcinoma epithelian (A549), colon carcinoma (HCT-116), pancreatic carcinoma (BXPC-3), breast cancer (MCF-7), cervical epithelium (HeLa), and erythroleukemic (K562)) and a human umbilical vein endothelial cell (HUVEC) line (Table 2). Compounds 3, 6, 7, 20, 21, 23, 26, and 27 with the enone functionality exhibited selectively cytotoxicity against the above cell lines with IC50 values less than 10 μM. This further confirmed that the enone functionality was related to the toxicity.
Table 2.
Cytotoxic activity of compounds 1–27.
| No. | Cytotoxicity IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| HCT-116 | BXPC-3 | HeLa | MCF-7 | A-549 | K562 | HUVEC | |
| 1 | I | I | I | I | I | I | nt |
| 2 | I | I | I | I | I | I | nt |
| 3 | I | I | 7.18 | I | I | I | nt |
| 4 | I | I | I | I | I | I | nt |
| 5 | I | I | I | I | I | I | nt |
| 6 | I | 8.47 | I | I | I | I | 8.59 |
| 7 | 1.09 | 5.07 | I | I | I | I | I |
| 8 | I | I | I | I | I | I | I |
| 9 | I | I | I | I | I | I | nt |
| 10 | I | I | I | I | I | I | nt |
| 11 | I | I | I | I | I | I | I |
| 12 | I | I | I | I | I | I | I |
| 13 | I | I | I | I | I | I | I |
| 14 | I | I | I | I | I | I | nt |
| 15 | nt | nt | nt | nt | nt | nt | nt |
| 16 | I | I | I | I | I | I | nt |
| 17 | I | I | I | I | I | I | no |
| 18 | I | I | I | I | I | I | no |
| 19 | I | I | I | I | I | I | no |
| 20 | I | I | 7.21 | I | I | I | 9.78 |
| 21 | I | I | 6.95 | I | I | I | 4.52 |
| 22 | nt | nt | nt | nt | nt | nt | nt |
| 23 | 3.51 | nt | 6.59 | nt | 9.75 | nt | 7.51 |
| 24 | I | nt | I | nt | I | nt | I |
| 25 | I | I | I | I | I | I | I |
| 26 | 9.16 | 7.22 | 4.02 | I | I | I | 4.81 |
| 27 | 2.83 | nt | 3.85 | nt | 7.30 | nt | 5.90 |
| Adriamycin | 0.21 | 0.033 | 0.60 | 0.86 | 0.16 | 0.25 | — |
I: Inactive; nt: Not tested.
Conclusion
In summary, the natural products 1–6 and a group of derivatives 11–27 were semisynthesized with multi-gram scales of 7 and 8 by one or two steps with high yields. This semisynthetic route provided a convenient source of these RALs for further potentially biological applications. Compounds 11–13 showed strong antiplasmodial activity against P. falciparum, of which 11 and 12 were discovered to be promising non-toxic antiparasitic candidates. The structure-activity relationship analysis indicated that the acetoxy and acetonide groups are required for antiplasmodial activity, while the introduction of chlorine atom at C-5 is not necessary to the activity. Additionally, enone group can lead to an obvious toxicity and a comparatively lower therapeutic ratio, but 2-OH is critical for reducing toxicity. Further studies of selected compounds 11–13 on assessing their in vivo activities and a systematic optimization based upon these identified promising chemical leads are in progress and will be reported in due course.
Methods
General experimental procedures
NMR spectra were recorded with TMS as internal standard on an Agilent DD2 NMR spectrometer (500 and 125 MHz for 1H and 13C NMR, respectively). HRESIMS and ESIMS spectra were obtained from a Micromass Q-TOF spectrometer. Optical rotations were measured on a JASCO P-1020 digital polarimeter. Silica gel (Qing Dao Hai Yang Chemical Group Co.; 200–300 mesh), and Sephadex LH-20 (Amersham Biosciences) were used for column chromatography (CC). TLC silica gel plates (Yan Tai Zi Fu Chemical Group Co.; G60, F-254) were used for thin-layer chromatography. Semipreparative HPLC was carried out on a Waters 1525 system using a semipreparative C18 (Kromasil, 5 μm, 10 × 250 mm) column equipped with a Waters 2996 photodiode array detector, at a flow rate of 2.0 mL/min.
Fungal material
The fungal strain C. lunatus (CHNSCLM-0009) was isolated from the inner part of the zoanthid Palythoa haddoni, collected by hand from the South China Sea in April 2010. The fungus was identified as C. lunatus on the basis of 16 S rRNA gene analysis with the access code JF819163.
Fermentation, extraction, and isolation
The fungal strain was cultivated in 10 L of optimal liquid medium (Medium I and Medium II, respectively) at 28 °C for 7 days on a rotary shaker at 120 rpm. Then the culture was filtered to separate the culture broth from the mycelia. The culture broth was extracted with an equal volume of EtOAc. The combined EtOAc solution was evaporated to dryness under a vacuum to give an EtOAc extract. The EtOAc extract (4.22 g) was subjected to Sephadex LH-20 column chromatography (120.0 cm × 3.0 cm i.d.) with 1.5 L petroleum ether/CH2Cl2/MeOH (2:1:1, v/v/v), and then recrystallization with MeOH/CH2Cl2 to obtain 8 (1.71 g) and 7 (1.35 g).
Semisynthesis of cochliomycin C (2)
SO2Cl2 (0.045 mL, 0.36 mmol) in CH2Cl2 (5.0 mL) was added to the solution of zeaenol (8) (90.0 mg, 0.25 mmol) in dry CH2Cl2 (25.0 mL) at 0 °C. After 1 h the reaction was quenched by the addition of 25.0 mL of 5% aqueous NH4Cl solution and diluted with CH2Cl2 (30.0 mL). The organic layer was separated and the organic solvents were removed under a vacuum. The residue as purified by silica gel column chromatography (EtOAc/petroleum, 1:1, v/v) to give 16 as a white solid (87.5 mg, 90%). The resulting 16 (50.0 mg, 0.13 mmol) was then treated with 10% Pd-C (5.0 mg) in MeOH (35.0 mL) under H2 atmosphere, and the solution was stirred for 1.5 h at room temperature. The mixture was filtered and concentrated under a vacuum. The residue was purified by semipreparative HPLC to give 2 as a white solid (37.4 mg, 75%), the spectroscopic data of which were consistent with that reported in literature17.
Allyl alcohol-oxidation of 8 to yield 3–6
Pyridine (24 µL, 0.25 mmol) was added to a solution of 8 (30 mg, 0.082 mmol) in CH2Cl2 (20 mL). After the solution was cooled to 0 °C, the Dess-Martin periodinane (45 mg, 0.11 mmol) and NaHCO3 (14 mg, 0.16 mmol) were added as a solid. The mixture was stirred at 0 °C for 30 min, and then was allowed to stir at room temperature for 2 h. The mixture was diluted with Et2O (30 mL), 1 M aqueous Na2S2O3 (15 mL), and 5% aqueous NaHCO3 (15 mL) were added. This biphasic mixture was stirred for 0.5 h and the organic layer was separated and concentrated to dryness in vacuo. The crude mixture was immediately purified by Sephadex LH-20 CC eluted with petroleum ether/CH2Cl2/MeOH (2:1:1, v/v/v) to obtain pure compound 6 (19.5 mg). The obtained crude mixture was purified by silica gel CC eluted with EtOAc-petroleum (1:1), and then subjected to semipreparative HPLC to give the compounds 3 (6.8 mg), 4 (1.7 mg), 5 (4.3 mg), and 6 (5.2 mg), respectively. The structures of 3–6 were determined by comparing NMR data and MS spectrum with the literature18.
Antiplasmodial assay
Activity against the causative agent of malaria was performed by culturing human erythrocytes and infecting them with P. falciparum, as described by Trager and Jensen36. Briefly, the W2 (Chloroquine resistant) and 3D7 (Chloroquine sensitive) strains of P. falciparum were cultured in RPMI 1640 medium (Sigma-Aldrich, USA) supplemented with 10% human serum (from O+ blood) at a hematocrit of 2% erythrocytes (O+) at 37 °C in a gas mixture of 5% CO2, 5% O2, and 90% N2. Parasites were synchronized by a temperature cycling technique as described by Almanza et al.37 Malaria bioassays were performed following the procedure of Corbett et al., which used Pico-Green to assess parasite growth inhibition by drugs and used chloroquine as positive control, with an IC50 of 32.9 nM38.
Antileishmanial assay
The anti-leishmania activity was evaluated following the protocol described by Calderon et al.39, using the fluorescent DNA intercalator PicoGreen (Invitrogen, USA). The species responsible for visceral leishmaniasis, L. donovani, was used for the assays. Amphotericin B was used as the positive control and had an IC50 of 76.3 nM40.
Antitrypanosomal assay
The antitrypanosomal test used the methods recommended in Romanha et al. for screening against this parasite with benznidazole as the control drug, with an IC50 of 3.85 μM41. Briefly, the expression of the reporter gene for beta-galactosidase (β-Gal) in the Tulahuen clone C4 of T. cruzi is assessed by colorimetry at 570 nm, which correlates with the parasite growth42. Assays were performed on the intracellular amastigote form of the parasite infecting African green monkey kidney (Vero) cells, incubated for 120 h at 37 °C with 5% CO2.
SRB and MTT assays
The cytotoxicity against HCT-116, BXPC-3, HeLa, MCF-7, A-549, and HUVEC cell lines was evaluated using the SRB method43. The cytotoxicity against K562 and Vero cell lines was evaluated using the MTT method44. Adriamycin was used as a positive control. All cells were cultivated in T-75 flasks containing 10% Fetal Bovine Serum (FBS), Dulbeco’s Modified Eagle Medium (DMEM) with 2 mM L-glutamine, and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere with 5% CO2. For SRB assay, cells in their log phase of growth were seeded into 96 well micro plates (4 × 104 cells per well) and followed by treating with test samples at 10 μM or DMSO. After incubation at 37 °C for 72 h, the cells were fixed with the cold 20% (w/v) TCA for 2 h and stained with Sulforhodamine B (SRB) dye for 0.5 h. The protein-bound dye is dissolved in Tris base solution for OD determination at 540 nm on an ELISA Plate Reader. For MTT assay, cells (5 × 103 cells per well) were plated in 96 well micro plates. After cell attachment overnight, new medium containing test compounds or DMSO was added. The plate was incubated for 48 h at 37 °C and incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for additional 2–4 hours. The assay plate was then read at 490 nm on an ELISA Plate Reader. The data were obtained from experiments carried out in triplicate.
Electronic supplementary material
Acknowledgements
This work was supported by the Program of Natural Science Foundation of Shandong Province of China (No. JQ201510), the Program of National Natural Science Foundation of China (Nos 41376145, 41322037 and 41606172), AoShan Talents Program Supported by Qingdao National Laboratory for Marine Science and Technology (No. 2015ASTP-ES11), and the Taishan Scholars Program, China. L. P. and M. N. were funded by a GEF grant to Panama (81860) through C. S., who was partially supported by the SNI Panama.
Author Contributions
X.Q.Z. contributed to semisynthesis, isolation, identification, and manuscript preparation. J.H.S. contributed to optimization of media composition of C. lunatus. C.S., L.P., M.N. and W.W. contributed to bioactivity tests. C.Y.W., Y.C.G. and C.L.S. conceived of and proposed the idea and prepared the manuscript. All authors read, revised and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-12336-0.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World malaria report 2016; World Health Organization (2016).
- 2.WHO | Leishmaniasis. Available from: http://www.who.int/mediacentre/factsheets/fs375/en/.
- 3.WHO Weekly Epidemiological Record (WER) 6 February, No. 6, 90, 33–44 (2015).
- 4.Third WHO report on NTDs, WHO (2015).
- 5.WHO. Guidelines for the treatment of malaria, 2nd edition. World Health Organization: Geneva, ISBN 9789241547925 (2010).
- 6.Dondorp AM, et al. Artemisinin resistance: current status and scenarios for containment. Nat. Rev. Microbiol. 2010;8:272–280. doi: 10.1038/nrmicro2385. [DOI] [PubMed] [Google Scholar]
- 7.Minodier P, Retornaz K, Horelt A, Garnier JM. Liposomal amphotericin B in the treatment of visceral leishmaniasis in immunocompetent patients. Fundam. Clin. Pharmacol. 2003;17:183–188. doi: 10.1046/j.1472-8206.2003.00168.x. [DOI] [PubMed] [Google Scholar]
- 8.Walker J, et al. Discovery of factors linked to antimony resistance in Leishmania panamensis through differential proteome analysis. Mol. Biochem. Parasitol. 2012;183:166–176. doi: 10.1016/j.molbiopara.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Blunt JW, et al. Marine natural products. Nat. Prod. Rep. 2016;33:382–431. doi: 10.1039/C5NP00156K. [DOI] [PubMed] [Google Scholar]
- 10.Jia YL, et al. (+)- and (−)- Pestaloxazine A, a pair of antiviral enantiomeric alkaloid dimers with a symmetric spiro[oxazinane-piperazinedione] skeleton from Pestalotiopsis sp. Org. Lett. 2015;17:4216–4219. doi: 10.1021/acs.orglett.5b01995. [DOI] [PubMed] [Google Scholar]
- 11.Xu LX, He ZX, Xue JH, Chen XP, Wei XY. β-Resorcylic acid lactones from a Paecilomyces fungus. J. Nat. Prod. 2010;73:885–889. doi: 10.1021/np900853n. [DOI] [PubMed] [Google Scholar]
- 12.Xu LX, He ZX, Xue JH, Chen XP, Wei XY. Correction to β-resorcylic acid lactones from a Paecilomyces fungus. J. Nat. Prod. 2012;75:1006–1006. doi: 10.1021/np300293b. [DOI] [PubMed] [Google Scholar]
- 13.Isaka M, Suyarnsestakorn C, Tanticharoen M, Kongsaeree P, Thebtaranonth Y. Aigialomycins A–E, new resorcylic macrolides from the marine mangrove fungus Aigialus parvus. J. Org. Chem. 2002;67:1561–1566. doi: 10.1021/jo010930g. [DOI] [PubMed] [Google Scholar]
- 14.Hellwig V, et al. Pochonins A–F, new antiviral and antiparasitic resorcylic acid lactones from Pochonia chlamydosporia var. catenulate. J. Nat. Prod. 2003;66:829–837. doi: 10.1021/np020556v. [DOI] [PubMed] [Google Scholar]
- 15.Dutton BL, et al. Synthesis of macrolactam analogues of radicicol and their binding to heat shock protein Hsp90. J. Org. Biomol. Chem. 2014;12:1328–1330. doi: 10.1039/c3ob42211a. [DOI] [PubMed] [Google Scholar]
- 16.Barluenga S, Dakas PY, Boulifa M, Moulin E, Winssinger N. Resorcylic acid lactones: a pluripotent scaffold with therapeutic potential. C. R. Chimie. 2008;11:1306–1317. doi: 10.1016/j.crci.2008.01.020. [DOI] [Google Scholar]
- 17.Shao, C. L. et al. Potent antifouling resorcylic acid lactones from the gorgonian-derived fungus Cochliobolus lunatus. J. Nat. Prod.74, 629–633 (2011). [DOI] [PubMed]
- 18.Liu QA, et al. Antifouling and fungicidal resorcylic acid lactones from the sea anemone-derived fungus Cochliobolus lunatus. J. Agric. Food. Chem. 2014;62:3183–3191. doi: 10.1021/jf500248z. [DOI] [PubMed] [Google Scholar]
- 19.Jana N, Nanda S. Asymmetric total syntheses of cochliomycin A and zeaenol. Eur. J. Org. Chem. 2012;23:4313–4320. doi: 10.1002/ejoc.201200241. [DOI] [Google Scholar]
- 20.Wang LL, Gao YG, Liu J, Cai C, Du YG. Stereoselective total synthesis of cochliomycin A. Tetrahedron. 2014;70:2616–2620. doi: 10.1016/j.tet.2014.03.001. [DOI] [Google Scholar]
- 21.Gao YG, Liu J, Wang LL, Xiao M, Du YG. Total syntheses of cochliomycin B and zeaenol. Eur. J. Org. Chem. 2014;10:2092–2098. doi: 10.1002/ejoc.201301613. [DOI] [Google Scholar]
- 22.Bolte B, et al. Modular total syntheses of the marine-derived resorcylic acid lactones cochliomycins A and B using a late-stage Nozaki-Hiyama-Kishi macrocyclization reaction. J. Org. Chem. 2015;80:460–470. doi: 10.1021/jo5024602. [DOI] [PubMed] [Google Scholar]
- 23.Mahankali B, Srihari P. A carbohydrate approach for the first total synthesis of cochliomycin C: stereoselective total synthesis of paecilomycin E, paecilomycin F and 6′-epi-cochliomycin C. Eur. J. Org. Chem. 2015;18:3983–3993. doi: 10.1002/ejoc.201500395. [DOI] [Google Scholar]
- 24.Pal P, Chakraborty J, Mali A, Nanda S. Asymmetric total synthesis of paecilomycin F, cochliomycin C, zeaenol, 5-bromo-zeaenol and 3, 5-dibromo-zeaenol by Heck coupling and late stage macrolactonization approach. Tetrahedron. 2016;72:2336–2348. doi: 10.1016/j.tet.2016.03.054. [DOI] [Google Scholar]
- 25.Ma X, Bolte B, Banwell MG, Willis AC. Total syntheses of the resorcylic acid lactones paecilomycin F and cochliomycin C using an intramolecular Loh-type α-allylation reaction for macrolide formation. Org. Lett. 2016;18:4226–4229. doi: 10.1021/acs.orglett.6b01963. [DOI] [PubMed] [Google Scholar]
- 26.Srihari P, Mahankali B, Rajendraprasad K. Stereoselective total synthesis of paecilomycin E. Tetrahedron Lett. 2012;53:56–58. doi: 10.1016/j.tetlet.2011.10.137. [DOI] [Google Scholar]
- 27.Pal, P., Jana, N. & Nanda, S. Asymmetric total synthesis of paecilomycin E, 10′-epi-paecilomycin E and 6′-epi-cochliomycin C. Org. Biomol. Chem.12, 8257–8274 (2014). [DOI] [PubMed]
- 28.Mohapatra DK, Reddy DS, Mallampudi NA, Yadav JS. Stereoselective total syntheses of paecilomycins E and F through a protecting group directed diastereoselective intermolecular Nozaki-Hiyama-Kishi (NHK) reaction. Eur. J. Org. Chem. 2014;23:5023–5032. doi: 10.1002/ejoc.201402133. [DOI] [PubMed] [Google Scholar]
- 29.Bhunia N, Das B. Stereoselective total synthesis of paecilomycins E and F. Synthesis. 2015;47:1499–1509. doi: 10.1055/s-0034-1380400. [DOI] [Google Scholar]
- 30.Bakkolla M, Pabbaraja S. Stereoselective total synthesis of antiplasmodial resorcylic acid lactone paecilomycin F. ARKIVOC. 2016;2:123–136. [Google Scholar]
- 31.Sugawara F, et al. Zearalenone derivatives produced by the fungus Drechslera portulacae. Phytochemistry. 1992;31:1987–1990. doi: 10.1016/0031-9422(92)80346-G. [DOI] [Google Scholar]
- 32.Ellestad GA, Lovell FM, Perkinson NA, Hargreaves RT, McGahren WJ. New zearalenone related macrolides and isocoumarins from an unidentified fungus. J. Org. Chem. 1978;43:2339–2343. doi: 10.1021/jo00406a007. [DOI] [Google Scholar]
- 33.Danishefsky SJ, et al. New efficient synthesis of resorcinylic macrolides via ynolides: establishment of cycloproparadicicol as synthetically feasible preclinical anticancer agent based on Hsp90 as the target. J. Am. Chem. Soc. 2004;126:7881–7889. doi: 10.1021/ja0484348. [DOI] [PubMed] [Google Scholar]
- 34.Isaka M, Chinthanom P, Veeranondha S, Supothina S. Novel cyclopropyl diketones and 14-membered macrolides from the soil fungus Hamigera avellanea BCC 17816. Tetrahedron. 2008;64:11028–11033. doi: 10.1016/j.tet.2008.09.077. [DOI] [Google Scholar]
- 35.Isaka M, et al. Aigialomycins and related polyketide metabolites from the mangrove fungus Aigialus parvus BCC 5311. Tetrahedron. 2009;65:4396–4403. doi: 10.1016/j.tet.2009.03.050. [DOI] [Google Scholar]
- 36.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 37.Almanza A, Coronado L, Tayler N, Herrera L, Spadafora C. Automated synchronization of P. falciparum using a temperature cycling incubator. Curr. Trends Biotechnol. Pharm. 2011;5:1130–1133. [Google Scholar]
- 38.Corbett Y, et al. A novel DNA-based microfluorimetric method to evaluate antimalarial drug activity. Am. J. Trop. Med. Hyg. 2004;70:119–124. [PubMed] [Google Scholar]
- 39.Calderón A, et al. Evaluation of larvicidal and in vitro antiparasitic activities of plants in a biodiversity plot in the Altos de Campana National Park, Panama. Pharm. Biol. 2006;44:487–498. doi: 10.1080/13880200600878361. [DOI] [Google Scholar]
- 40.Panosian CB, Barza M, Szoka F, Wyler DJ. Treatment of experimental cutaneous leishmaniasis with liposome-intercalated amphotericin B. Antimicrob. Agents Chemother. 1984;25:655–656. doi: 10.1128/AAC.25.5.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romanha AJ, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem. Inst. Oswaldo Cruz. 2010;105:233–238. doi: 10.1590/S0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 42.Buckner FS, Verlinde CL, La Flamme AC, Van Voorhis WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob. Agents Chemother. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skehan P, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 44.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.