

Abstract
The genus Conus comprises approximately 700 species of venomous marine cone snails that are highly efficient predators of worms, snails, and fish. In evolutionary terms, cone snails are relatively young with the earliest fossil records occurring in the Lower Eocene, 55 Ma. The rapid radiation of cone snail species has been accompanied by remarkably high rates of toxin diversification. To shed light on the molecular mechanisms that accompany speciation, we investigated the toxin repertoire of two sister species, Conus andremenezi and Conus praecellens, that were until recently considered a single variable species. A total of 196 and 250 toxin sequences were identified in the venom gland transcriptomes of C. andremenezi and C. praecellens belonging to 25 and 29 putative toxin gene superfamilies, respectively. Comparative analysis with closely (Conus tribblei and Conus lenavati) and more distantly related species (Conus geographus) suggests that speciation is associated with significant diversification of individual toxin genes (exogenes) whereas the expression pattern of toxin gene superfamilies within lineages remains largely conserved. Thus, changes within individual toxin sequences can serve as a sensitive indicator for recent speciation whereas changes in the expression pattern of gene superfamilies are likely to reflect more dramatic differences in a species’ interaction with its prey, predators, and competitors.
Keywords: venom evolution, speciation, conotoxins, exogenes
Introduction
Exogenes are the subset of genes that directly mediate the biotic interactions between organisms (Olivera 2006). They are sensitively tuned to the particular ecological niche for which a given species is maximally fit. The genes that encode the components of animal venoms are an example of exogenes. When, for example, speciation is associated with a change in prey preference, genes encoding venom components may experience strong selection pressure for optimal efficacy in the new prey (Olivera 2006; Chang and Duda 2016). A recent study of venom insulins has highlighted how exogene products, targeted at other organisms, appear to evolve very differently from their endogenous counterparts (Safavi-Hemami etal. 2016). In this case, exogenes were characterized by high diversity of sequence variants, thought to reflect duplication events coupled with strong positive selection. In contrast, their endogenous counterparts, due to strong purifying selection, were essentially identical between distantly related species. Thus, exogenes can be sensitive indicators of hidden biological differences that are not obvious from morphology or endogene sequence analyses, and offer insight into the molecular events that accompany speciation.
The venom components of cone snails (genus Conus) are one of the best-studied examples of exogenes. Conus is a large genus (∼730 extant species according to WoRMS—accessed on September 28, 2016) of predatory marine mollusks that use venom for prey capture, defense, and competitive interactions (Rockel etal. 1995; Puillandre etal. 2014). According to the recent dated phylogeny (Uribe etal. 2017), the first rapid diversification of the genus Conus started in Upper Oligocene about 30–25 Ma, and was followed by a mass extinction at the Miocene–Pliocene boundary. Thus, Conus is a relatively young genus, with most extant species originated less than 10 Ma (Duda etal. 2001; Duda and Kohn 2005; Uribe etal. 2017). Several major shifts in prey preference occurred in the evolution of this genus and living cone snail species can typically be divided into those that prey on vermiform invertebrates, gastropods, or fish (Duda etal. 2001).
The venom of each species is a complex mixture of approximately 70–400 peptide toxins (Hu etal. 2012; Barghi etal. 2015a, 2015b; Phuong etal. 2016) (named conotoxins or conopeptides). Conotoxins are initially translated as precursor peptides in the epithelial cells of a specialized venom gland. Most conotoxins precursors have a well-defined primary structure: At the N-terminus, a highly conserved signal sequence, at the C-terminus, a variable mature toxin region and in between, a propeptide region (Woodward etal. 1990). The mature toxin region encodes the actual bioactive peptide injected by the cone snail into another animal. According to signal sequence conotoxins can be grouped into probably 40–50 gene superfamilies (Robinson and Norton 2014). Several of these are found in nearly every species examined and exhibit the characteristics typical of many other venom exogenes: Extraordinarily high diversity within and between species. Consistent with this observation, considerable prior work has demonstrated that conotoxins are hyperdiverse, with each species of Conus suggested to have its own distinct venom exogene repertoire, presumably shaped by species-specific interactions with prey, predators, and competitors (Olivera etal. 2014).
Conus andremenezi is a recently described cone snail (Biggs etal. 2010), and was chosen as one of “100 of our Planets Most Amazing New Species” (Wheeler etal. 2013). Conus andremenezi belongs to a group of small forms of Conus that were conventionally regarded by taxonomists as a single variable species Conus praecellens, which has recently been demonstrated to constitute a species complex (Biggs etal. 2010) (fig. 1A). Conus andremenezi and C. praecellens are distinguishable by several morphological criteria, as well as by primary sequence of mitochondrial DNA (Biggs etal. 2010). A rare aquarium recording of C. andremenezi and C. praecellens shows differences in foot and siphon coloration, potentially pointing toward different camouflage strategies (fig. 1B).
Fig. 1.

—(A) Selected species of the subgenus Turriconus. (i) Conus andremenezi, (ii) Conus praecellens, (iii) Conus excelsus, a famous rarity known from only a few specimens for many centuries, and still a desirable shell collectors treasure, (iv) Conus miniexcelsus, and (v) Conus acutangulus. The shell of C. andremenezi can be distinguished from that of similar closely related species by a generally broader shape, characteristic purplish-brown maculations, undulating spiral ribs, widely spaced ribs on the spire, and a distinct protoconch. (B) Aquarium images of living C. andremenezi (top) and C. praecellens (bottom). Note color difference in siphon (blue arrows) and foot (red arrows). Conus andremenezi has a darker foot but whiter siphon than C. praecellens.
The distributions of the two species overlap, in the Central Indo Pacific (Philippines, Papua New Guinea, and Solomon Islands); however, C. andremenezi is sometimes collected at greater depths than C. praecellens, based on material from several field expeditions carried out by the Museum of Natural History (Paris) (Olivera B, Bouchet P, unpublished observations).
According to a recently published classification of cone snails (Puillandre etal. 2014), C. praecellens and C. andremenezi belong to the subgenus Turriconus, one of the early diverging lineages of the genus Conus (fig. 1A). As most of the Turriconus species, including C. andremenezi and C. praecellens, occur only in relatively deep water, direct behavioral observations are difficult to obtain, and little is known about their biology, although all early diverging clades of Conus are believed to be worm-hunting (Duda etal. 2001) and phylogenetic analysis strongly suggests that fish- and mollusk-hunting behaviors only evolved in certain more recently diverging groups of Conus (Uribe etal. 2017). Additionally, radular teeth of Turriconus bear strong barbs with no accessory process (Tucker and Tenorio 2009); such morphology is common for vermivorous Conus species, but was not observed in either fish or mollusk hunters (Olivera etal. 2015). Thus, although the possibility that members of the Turriconus clade are specialists on other prey cannot be eliminated it appears highly likely that these species are vermivorous.
In this study, we analyzed the venom composition of C. andremenezi and C. praecellens, neither of which has previously been comprehensively investigated at a molecular level. Our results provide new insight into the biology of these species and of the subgenus Turriconus in general. Moreover, comparison of the venom exogene repertoires of C. praecellens and C. andremenezi has provided insights into the genetic mechanisms associated with recent speciation. Our findings show that the expression pattern of toxin gene families remains largely conserved between these sister species, but that speciation is associated with significant diversification of individual toxin genes.
Materials and Methods
Specimen Collection, RNA Extraction, and Sequencing
All studied specimens were collected in the central Philippines. Adult specimens of C. andremenezi and C. praecellens were collected at depth about 180–250 m, off Sogod, North of Cebu Island, and the specimens of C. geographus—from the shallow water (10–25 m) off Caw-Oy, Olango Island. Specimen identification was initially performed by morphological examination and later verified by sequence analysis of the cytochrome oxidase c subunit 1 (COI) gene (as described under phylogenetic analysis below). Venom glands of live specimens were dissected and stored in RNAlater at −80 °C. Total RNA was isolated from the homogenized venom glands of each species using TRIzol reagent following the manufacturer’s protocol (Life Technologies Corporation). Integrity of RNA was verified on a Bioanalyzer instrument (Agilent Technologies), and Illumina libraries were prepared with a mean insert size of 170 bp at Cofactor Genomics (St. Louis, MO) for C. andremenezi (specimen 1), C. praecellens (specimen 1), and C. geographus. For C. praecellens (specimen 2), strand-specific Illumina libraries with a mean insert size of 220 bp were prepared at the University of Utah High-Throughput and Genomics Core Facility. For C. andremenezi (specimen 2), mRNA was extracted from the venom gland using Dynabeads mRNA DIRECT kit (Invitrogen Dynal AS, Oslo, Norway) and a cDNA library was constructed with an average insert size of 200 bp. All libraries were sequenced on an Illumina HiSeq 2000 platform. Prior to assembly, quality filtering and adaptor trimming were performed using Fqtrim software (version 0.9.4, http://ccb.jhu.edu/software/fqtrim/) and Prinseq (version 0.20.4) (Schmieder and Edwards 2011). After preprocessing, reads shorter than 70 bp and those containing more than 5% ambiguous bases (Ns) were discarded. 58, 834, 536 (specimen 1) and 54, 177, 324 (specimen 2) reads for C. andremenezi; 70, 826, 570 (specimen 1) and 32, 695, 570 (specimen 2) reads for C. praecellens and 146, 630, 264 reads for C. geographus were used for each transcriptome assembly.
Phylogenetic Analysis
Phylogenetic analysis was performed using a partial sequence of the COI gene segment of mitochondrial DNA, corresponding to the standard barcode region. COI nucleotide sequences for specimens used in this study were retrieved from the assembled venom gland transcriptomes, and those from additional members of the Turriconus subgenus were obtained from GenBank (KJ550384, KJ550243, KJ550048, KJ549906, KJ549742, KJ550110, KJ550109, KJ550108, KJ550107, KJ550106, KJ550105, KJ550104, KJ550103, KJ550102, KJ550101, KJ549855, KJ550425, KJ549743, KJ550125, KJ549858, KJ550424, KJ550423, KJ550124, KJ550123).
Phylogenetic analyses were performed using MrBayes v.3.2.6 (Huelsenbeck and Ronquist 2001), running two parallel analyses, consisting each of six Markov chains of 15,000,000 generations with a sampling frequency of one tree each 1,000 generations, and chain temperature set to 0.02. Parameters of the substitution model were estimated during the analysis (six substitution categories, a gamma-distributed rate variation across sites, and a proportion of invariable sites approximated in four discrete categories). Each codon position of the COI gene was treated as a separate partition. The first 25% of trees were omitted as burn in prior to the construction of consensus trees. Convergence of the analysis was evaluated using Tracer 1.4.1 (http://tree.bio.ed.ac.uk/software/tracer/) to ensure that effective sample size values were all greater than 200 (default burning).
Bootstrap branch support values were obtained for an equivalent maximum-likelihood tree estimated by RaxML (Stamatakis 2006) with each codon position treated as an independent partition. Nodal support of the obtained tree was estimated using the thorough bootstrapping algorithm (Felsenstein 1985) with 1,000 iterations. All phylogenetic analyses were carried out at Cipres Science Gateway (Miller etal. 2010) Kimura 2-parameter (K2P) genetic distances were calculated with MEGA 5.2.1 (Tamura etal. 2011). Conus distans, the species recovered in a sister position to all other Conus lineages by Puillandre etal. (2014), was used as an outgroup in all phylogenetic analyses.
Transcriptome Assembly, Annotation, and Postprocessing
Venom gland transcriptomes for C. andremenezi (both specimens) and C. praecellens (specimen 1) and C. geographus were assembled with Trinity (version 2.0.5) using the paired-end mode, wherein kmer size used to build the De Bruijn Graphs was 32, minimum percent identity for two paths to be merged into single paths was 99, maximum allowed difference encountered between path sequences to combine them was 1, maximum internal gap length allowed between path sequences to combine them was 3, and the minimum output contig length was 75 (Grabherr etal. 2011). Trinity settings were optimized for sequence read length and library type according to simulation results; RNAseq reads were simulated using the published Conus victoriae venom gland transcriptome (Robinson etal. 2014) with the same read length and quality obtained for RNAseq data sets sequenced in this study. Systematic, serial modifications of the assembly parameters and batch assemblies were performed to optimize assemblies. The true positive rate, defined as the percentage of C. victoriae toxins that were correctly assembled, was used as the criteria of optimality. Assembly parameters resulting in the highest true positive rate were used. A bash script for this procedure is available online (git@github.com/qingl0331/turriconus.git). Because the second specimen of C. praecellens (specimen 2) was sequenced with longer (125 nt instead of 101 nt) and stranded reads, different assembly parameters were used in Trinity (version 2.0.5): Paired-end mode, wherein the kmer size used to build De Bruijn Graphs was 31, sequence library type was set to reverse forward, minimum kmer coverage was 10, and the minimum glue was 10. Transcriptome assemblies were annotated using NCBI-BLASTX against a combined ConoServer (Kaas etal. 2012) and UniProtKB database (release April 2015). Recently published conotoxin sequences that were not yet uploaded into Uniprot at the time of analysis (Hu etal. 2012; Barghi etal. 2015a, 2015b; Phuong etal. 2016) were also added to our in-house database. Contigs with significant similarity (e-value 1e−4) to known conotoxins were designated as putative conotoxin sequences. Contigs with no blast annotation were translated into six frames and predicted open reading frames that contained a start and stop codon and a total length of 50–200 amino acids were selected for further analysis. Sequences that did not have a BlastX hit but fulfilled all of the following criteria were designated as putative conotoxins: Contained an N-terminal signal sequence (predicted using SignalP [Petersen etal. 2011]), were abundantly expressed in at least one specimen examined in this study (>10,000 mapped reads), and shared high similarity with a sequence expressed in another specimen examined in this study (BlastP e-value <1e−10). The rational for implementing these filtering criteria is that a putative new toxin gene family should not be described based on a single putative toxin sequence. Identification of a similar but distinct sequence in another specimen and/or species increases the likelihood of a sequence to be indeed a member of a novel toxin gene family.
All specimens sequenced in this study were multiplexed with other cone snail species on the same Illumina flowcell (see supplementary table 1, Supplementary Material online, for number of species multiplexed per lane) and examined for cross-contamination resulting from index misassignment (Kircher etal. 2012; Sloan etal. 2013). Conotoxin sequences with low abundance in one species (ratio of single read count/total read count < 10−6) but highly similar (>95% sequence identity) to a high abundant one in another species (200-fold higher) were considered as cross-contamination resulting from index misassignment. This ensured correct sequence assignments, however, could have resulted in the removal of a small number of genuine sequences that are present at very low expression levels (and are therefore unlikely to play a significant role in envenomation). Next, any erroneous duplicate transcripts resulting from the assembly process were removed. Finally, a subset of raw reads was remapped to the conotoxin data set using the map-to-reference tool in the Geneious package (version 8.1.7 [Kearse etal. 2012]), in order to identify variants overlooked by the assembler and to remove misassembled contigs.
Read Counts for Expression Analysis
To determine approximate relative expression levels, reads were mapped back to the final conotoxin data set with Bowtie2 (Langmead and Salzberg 2012). Mapped reads that were shared between > 2 sequences were divided equally. For comparative analysis with members of the Splinoconus clade, Illumina reads for the venom glands of Conus tribblei and Conus lenavati were obtained from GenBank (SRR1803937, SRR1803938, SRR1803939, SRR1803940, SRR1803941, SRR1803942) and mapped back to conotoxin sequences expressed in these two species (Barghi etal. 2015a) with Bowtie2 as described above.
Comparative Analysis
All-by-all NCBI-BLASTN searches were carried out to identify reciprocal best-hit (rbh) pairs among the inter-/intraspecific conotoxin nucleotide sequences. The rbh pairs were compared using NCBI-BLASTX to our in-house conotoxin database in order to classify rbh pairs as either conotoxin or nonconotoxin rbhs. ClustalW alignment was then used to globally align the rbh pairs (Larkin etal. 2007). The alignment results were parsed to obtain the percent identity of each aligned rbh pair. The program codeml from PAML 4.7 (Yang 2007) was used to estimate synonymous and nonsynonymous substitutions with model = 2 (fix omega), over all codon positions in an alignment of rbh pairs of both the conotoxin and nonconotoxin sequences. Analyses were conducted in runmode = -2. Rbh pairs with branches showing dS < 0.01 were discarded as inaccurate dN/dS estimation. Next, the average dN/dS ratio was calculated by averaging the values from all pairwise comparisons of toxin and nontoxin sequences.
Principal Component Analysis
Principal component analysis (PCA) was performed on relative expression levels of conotoxin superfamilies (as determined with Bowtie2 [Langmead and Salzberg 2012]) in each of the specimens analyzed. Expression levels were normalized to total read counts after trimming. Principal components were calculated using the prcomp package in R (R Core Team 2013).
Naming of Conotoxin Precursors
Conotoxin precursor sequences identified in C. andremenezi (prefix Amz) and C. praecellens (prefix Ps) were named according to the conventional conotoxin nomenclature (Walker etal. 1999) (where the species is represented by one to three letters, cysteine framework by an Arabic numeral and, following a decimal, order of discovery by a second numeral), with slight modifications. The gene superfamily was provided as a prefix (Robinson etal. 2014), and sequences that likely represent allelic variants rather than being encoded at distinct loci (i.e., only differ by 1–3 nucleotides) were given the same name but a small numeral was added as a suffix. Putative new superfamilies were named by the first five amino acids of their predicted signal peptides (e.g., putative MWSGK or put.MWSGK).
Data Availability
All C. andremenezi and C. praecellens conotoxin prepropeptide sequences from this study are provided in the Supplementary Material online and have been deposited at GenBank (accession numbers MF576542–MF576988).
Results
Phylogenetic Analysis of Turriconus Resolves C. andremenezi and C. praecellens as Two Sister Species
The Turriconus segment of the COI tree (highlighted in red) is characterized by the overall high posterior probability (PP) and bootstrap (B) support values, and distinctive well-supported terminal clades (PP value 0.99 or 1; fig. 2) corresponding to Conus acutangulus, Conus excelsus, C. praecellens, and C. andremenezi; Conus miniexcelsus is represented by a single sequence. Conus praecellens and C. andremenezi show sister relationship, with the grouping of C. andremenezi–C. praecellens being highly supported in both Bayesian analysis (PP = 0.99) and RaxML (B = 92). Conus miniexcelsus forms a sister group to C. andremenezi–C. praecellens, and the resulting three-species clade is also well supported (PP = 1, B = 99). In the Splinoconus subtree (PP = 1, B = 100; shown in green), the well-supported clade corresponding to C. tribblei shows sister relationship to the C. queenslandis with a moderate support (PP = 0.96, B = 88), and the highly supported C. lenavati clade is a sister group to the C. tribblei–C. queenslandis clade.
Fig. 2.

—Bayesian phylogenetic tree of some Turriconus and Splinoconus species based on the analysis of the COI fragment. Support values at each node correspond to RaxML bootstrap (B)/Bayesian posterior probability (PP). The specimens for which venom gland transcriptomes were generated are labeled as C. andremenezi Spm1, C. andremenezi Spm2, C. praecellens Spm1, and C. praecellens Spm2. Shells of analyzed Turriconus and Splinoconus species and of C. geographus are shown right to the respective species clade.
The mean K2P genetic distance among specimens of C. andremenezi is 1.9% and among specimens of C. praecellens is 0.9%. The mean K2P genetic distance between specimens of C. praecellens and C. andremenezi is 5.4%. Within-group K2P genetic distance in the analyzed data set ranges from 0% to 3.4%, whereas the minimal K2P value for between-group comparison is 4.8%. This difference corresponds to the so-called “barcode gap” marking the difference in within- and among-species comparisons, and is overall consistent with within- and among-species genetic distances in other groups of Conoidea (Puillandre etal. 2009; Fedosov and Puillandre 2012).
Similarities in Toxin Number and Superfamily Expression Patterns within the Turriconus Clade
We analyzed venom component transcripts from five venom gland transcriptomes: Two specimens of C. andremenezi and C. praecellens, from the Turriconus subgenus, and one specimen of C. geographus from the distantly related subgenus Gastridium that served as a reference. An in-depth analysis of the venom components of C. geographus has been provided elsewhere (Hu etal. 2012; Dutertre etal. 2014; Safavi-Hemami etal. 2014). Information on RNA sequencing and assembly statistics for these transcriptomes is provided in supplementary table 1, Supplementary Material online. Additionally, published data for two species of the Splinoconus clade (C. tribblei and C. lenavati) were interrogated. Comparison between the two specimens of the same species within Turriconus allowed us to estimate intraspecific variation in venom composition, whereas comparison between C. andremenezi and C. praecellens made it possible to examine interspecific variation between these two sister species. Finally, comparison of C. andremenezi and C. praecellens to C. tribblei and C. lenavati from the closely related subgenus Splinoconus (Barghi etal. 2015a) and C. geographus, a distantly related cone snail of the Gastridium clade with distinct prey preferences (C. geographus feeds on fish), provided relative scale by which to measure the magnitude of interspecific variation in Turriconus.
A number of general similarities exist across all transcriptomes examined (fig. 3): Each species’ venom repertoire is dominated by less than five conotoxin superfamilies, which typically account for over 70% of all of the sequences found. Multiple members of some superfamilies were observed, whereas other superfamilies were represented by a single toxin in the transcriptome of a species (table 1, supplementary table 2, Supplementary Material online).
Fig. 3.

—Comparison of relative toxin gene superfamily expression in C. andremenezi and C. praecellens with two members of the Splinoconus clades (C. tribblei and C. lenavati) and one member of the Gastridium clade (C. geographus). (A) Donut charts of expression levels. (B) Heat map showing a select number of superfamilies, including all that were highly expressed in the five species compared here. Plotted numbers represented normalized superfamily read counts. Amz1, C. andremenezi specimen 1; Amz2, C. andremenezi specimen 2; Ps1, C. praecellens specimen 1; Ps2, C. praecellens specimen 2; Trb, C. tribblei (pooled data from three individuals); Len, C. lenavati (pooled data from three individuals); Geo, C. geographus.
Table 1.
Venom Composition of the Two Species of Turriconus
|
Conus andremenezi 1 |
Conus andremenezi 2 |
Conus praecellens 1 |
Conus praecellens 2 |
||||
|---|---|---|---|---|---|---|---|
| Superfamily | Relative Expression Level | Superfamily | Relative Expression Level | Superfamily | Relative Expression Level | Superfamily | Relative Expression Level |
| O1d | 22.8% (30) | O1d | 36.8% (3) | P | 44.3% (16) | P | 41.2 (27) |
| T | 19.8% (4) | T | 13.9% (18) | T | 19% (28) | O1d | 12.9 (7) |
| M | 14.3% (18) | P | 11.6% (9) | M | 11.9% (24) | T | 11.5 (17) |
| P | 9.9% (18) | M | 8.1% (14) | O1d | 6.4% (7) | S | 8.5 (2) |
| O1 | 8% (11) | O1 | 8.1% (13) | S | 4.5% (2) | O1 | 3.9 (19) |
| I3 | 4.9% (4) | S | 4.9% (2) | O1 | 3.3% (24) | M | 3.7 (18) |
| S | 3.9% (1) | I3 | 4% (2) | O2 | 1.2% (7) | I2 | 3.0 (5) |
| O2 | 3.4% (10) | O2 | 3.2% (10) | SF-06 | 1.2% (1) | O2 | 2.9 (13) |
| I1 | 2.9% (2) | I2 | 2.8% (2) | J | 1% (6) | I1 | 2.3 (2) |
| Sf-mi1 | 2.7% (2) | I1 | 1.7% (2) | I3 | 1% (4) | SF-mi2 | 1.5 (1) |
| SF-01 | 2.6% (2) | V | 1.3% (1) | Con-ikot-ikot | 0.8% (3) | SF-06 | 1.4 (1) |
| put.MGGRF | 2% (4) | put.MKAVA | 0.5% (1) | put.MSGLR | 0.8% (1) | DivMKFPLLFISL | 1.3 (1) |
| I2 | 1.7% (1) | SF-mi1 | 0.3% (1) | I1 | 0.7% (1) | I3 | 1.1 (3) |
| V | 1.5% (2) | put.MSGLR | 0.3% (1) | DivMKFPLLFISL | 0.6% (2) | SF-01 | 1.0 (3) |
| PH4a | 0.7% (1) | put.MWSGK | 0.3% (1) | B2 | 0.6% (2) | J | 1.0 (5) |
| put.MWSGK | 0.7% (1) | put.MGGRF | 0.2% (1) | SF-mi1 | 0.5% (4) | put.MWSGK | 1.0 (2) |
| put.MKAVA | 0.6% (2) | PH4a | 0.1% (1) | PH4 | 0.5% (1) | L | 0.51 (2) |
| B2 | 0.3% (2) | B2 | 0.03% (1) | put.MWSGK | 0.4% (1) | put.MSGLR | 0.39 (1) |
| I4 | 0.2(3) | SF-01 | 0.01% (1) | I4 | 0.4% (3) | Con-ikot-ikot | 0.29 (3) |
| put.MSGLR | 0.2% (2) | Con-ikot-ikot | 0.01% (2) | SF-01 | 0.2% (2) | U | 0.20 (2) |
| put.MMLFM | 0.2% (3) | I4 | 0.01% (2) | SF-mi2 | 0.2% (1) | H | 0.07 (3) |
| Conopressina | 0.2% (1) | Conopressina | 0.009% (1) | Conopressina | 0.2% (2) | D | 0.04 (1) |
| B4 | 0.02% (1) | H | 0.008% (1) | D | 0.1% (2) | Cono-NPYa | 0.03 (1) |
| Conoporina | 0.02% (1) | A2 | 0.0004% (2) | Cono-NPYa | 0.1% (3) | Conopressina | 0.02 (2) |
| A2 | 0.01% (3) | L | 0.03% (2) | I4 | 0.02 (3) | ||
| I2 | 0.02% (2) | N | 0.01 (1) | ||||
| A2 | 0.02% (2) | SF-mi1 | 0.01 (1) | ||||
| V | 0.02% (1) | B2 | 0.004 (2) | ||||
| H | 0.02% (1) | V | 0.002 (1) | ||||
Note.—Relative expression levels are provided for each superfamily, from highest to lowest expression. The number of individual sequences detected per superfamily is shown in parentheses.
Sequences that may have derived from housekeeping proteins or serve in endogenous processes in the venom gland rather than in envenomation.
In the venom gland transcriptome of C. andremenezi (specimen 1), a total of 128 distinct conotoxin transcripts were identified. These could be grouped into 24 conotoxin gene superfamilies. Nevertheless, the transcriptome was dominated by only a few conotoxin gene superfamilies, namely the O1∂-like- (O1d), T-, M-, and P-superfamilies (in descending order of relative expression level) in terms of both expression and number of isoforms (fig. 3, table 1). Analysis of O1-superfamily sequences revealed two distinct gene classes within the O1 family, one of which shares high sequence similarity with so-called ∂-conotoxins (Heinemann and Leipold 2007). Thus, this class was named the O1d superfamily whereas all other O1 sequences were grouped into the O1 superfamily.
The venom gland transcriptome of a second specimen of C. andremenezi closely mirrored that of the first specimen: A total of 107 distinct conotoxin transcripts were identified belonging to 23 conotoxin superfamilies. High similarities were also observed in the expression of the most abundant toxin superfamilies: The O1d was the most highly expressed superfamily followed by the T-, P-, and M-superfamily (in descending order of relative expression level) (fig. 3, table 1). Together the two specimens of C. andremenezi expressed 196 distinct sequences of which 77 were likely to be allelic variants (defined as differing by 1–3 nucleotides).
A total of 155 and 149 distinct conotoxin transcripts representative of 28 and 29 conotoxin gene superfamilies were identified in the venom gland transcriptomes of C. praecellens specimens 1 and 2, respectively. The majority of the identified transcripts represented the same conotoxin superfamilies expressed in C. andremenezi: O1d-, T-, P-, and M- (fig. 3, table 1). Notably, however, the relative expression ranks of these superfamilies are different: Although the P-superfamily is most highly expressed in C. praecellens, the O1d superfamily predominates in both C. andremenezi specimens. Conotoxin transcripts from ten gene superfamilies were not detected in either C. andremenezi specimen but were identified in C. praecellens, albeit at very low expression levels (table 1). Together the two specimens of C. praecellens expressed 250 distinct sequences of which 81 were likely to be allelic variants.
Consistent with previous observations, conotoxin-encoding genes are under strong positive selection (average ω of 1.243) (Chang and Duda 2012; Sunagar etal. 2015) whereas other venom gland proteins remain largely conserved (average ω of 0.182). Pairwise dN/dS values are provided in supplementary table 3, Supplementary Material online. dN/dS ratios greatly vary between individual pairs of sequences within each superfamily, and almost all superfamilies contain sequences that appear to be under both, positive and purifying selection.
A side-by-side comparison with the venom composition of C. tribblei and C. lenavati (subgenus Splinoconus), and with a distantly related fish-hunting species, C. geographus (subgenus Gastridium) (fig. 3), lends scale to these differences. The venom composition of these three species differs significantly from the two Turriconus members and from each other, with little overlap in the expression pattern of the most highly expressed toxin gene superfamilies (fig. 3). The total of 39 toxin gene superfamilies identified in C. tribblei and 40 in C. lenavati exceed the gene superfamily diversity in both C. andremenezi and C. praecellens. The two Splinoconus species are characterized by very high expression of the con-ikot-ikot and B2 superfamilies (fig. 3), which together account for 63% and 55% of the expressed venom toxins in C. tribblei and C. lenavati, respectively (supplementary table 2, Supplementary Material online). At the same time, only about 0.3% of identified toxins in C. andremenezi and C. praecellens represent the B2 superfamily, and the con-ikot-ikot superfamily was not detected in either Turriconus species. Similarly, the O1d superfamily, which is highly expressed in Turriconus species, was not identified in either, C. tribblei or C. lenavati. Nevertheless, the toxin gene superfamilies P-, M-, O1, and O2 are detected in venom gland transcriptomes of all four species, and although their relative expression levels differ notably between Turriconus and Splinoconus, the P-, M-, O1, and O2 are among the ten most highly expressed toxin gene superfamilies in all four species. Furthermore, the T-superfamily toxins that are not detected in the venom of C. tribblei are highly expressed in the venom glands of C. andremenezi and C. praecellens, as well, as in C. lenavati. The total contribution of the P-, M-, O1-, O2-, and T-toxin gene superfamilies ranges from 24% of all putative toxin transcripts in C. lenavati to as much as 71% in C. praecellens.
The venom of C. geographus is constituted by relatively few (21) toxin gene superfamilies (Hu etal. 2012), with very high relative expression of the A-, B-, and O1 superfamilies toxins, followed by the S-, B2-, M- and T-superfamilies. Both A- and B-gene superfamilies are absent from the venom gland transcriptomes of Turriconus and Splinoconus species. However, the O1- and M-superfamilies are expressed by all five species, although with varying expression levels (fig. 3), the T-superfamily toxins show relatively high expression in all species with the exception of C. tribblei, and S-superfamily toxins are expressed at similar levels in Turriconus spp. and C. geographus, while showing very low expression levels in Splinoconus. The B2- and Con-ikot-ikot superfamilies that are characterized by very high expression in Splinoconus spp. and on the contrary, low in Turriconus, display intermediate expression levels in C. geographus. Expectedly, several gene families expressed in the two Turriconus species are absent (or of negligible expression) in C. geographus: P, I1, I2, V, I4, and A2.
Collectively, these data suggest rather minor differences in venom composition between the two closely related Turriconus species but pronounced differences between Turriconus spp. and members of other Conus lineages. Indeed, at least at the gene superfamily level, it appears that the notable difference between the C. andremenezi and C. praecellens venom repertoires is limited to differences in the relative expression levels of ∂-like conotoxins (O1 superfamily) (Heinemann and Leipold 2007) and P-superfamily peptides (table 1). This is further illustrated by PCA based on the relative expression of conotoxin superfamilies (fig. 4): The two Turriconus species are projected closely together in Eigen PC1 and PC2 space (relative to the other three species), and the slight separation in projection is largely due to differences in the relative expression of the O1d- and P-superfamilies. The two members of the Splinoconus clade group closely together in PC1 and PC2 space and are well separated from the other three species.
Fig. 4.
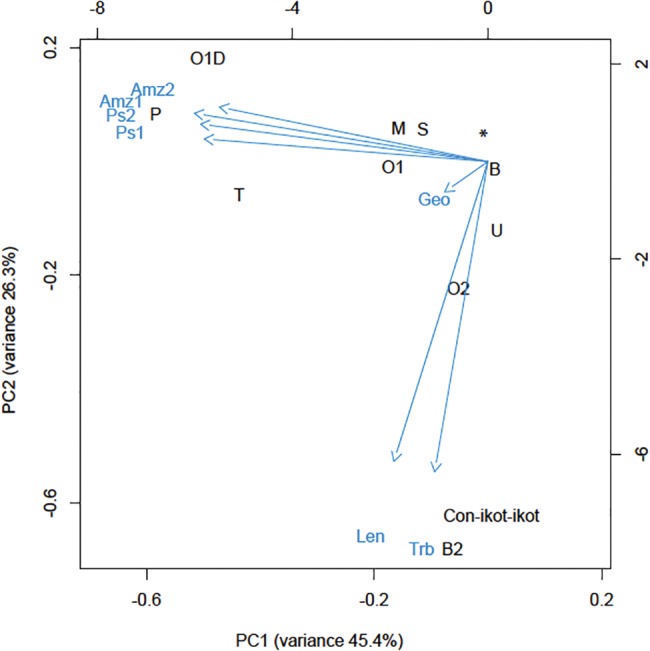
—PCA of expression of individual superfamilies. Relative expression levels of conotoxin superfamilies were determined by mapping reads back to contigs with Bowtie2 (Langmead and Salzberg 2012). Expression levels were normalized to total read counts after trimming. Principal components were calculated using the prcomp package in R (R Core Team 2013). O1∂: ∂-like O1 superfamily; *Superfamilies that did not contribute to separation on PC1 and PC2.
Interspecific Differences within Turriconus: The P- and O1d-Superfamilies
In all four Turriconus individuals analyzed, P-superfamily conotoxins were both diverse and highly expressed. In contrast, P-superfamily conotoxin expression was lower in the two species of Splinoconus and not detected in the venom gland transcriptome of the fish-hunter C. geographus. In C. praecellens, the P-superfamily is by far the most highly expressed conotoxin superfamily (41–44%), with 16–27 variants identified. In both specimens of C. andremenezi P-superfamily expression levels, although generally high (9.9% and 11.6%), are still substantially lower than in C. praecellens.
Interestingly, much of the difference in the total change in frequency for the P-superfamily between the two Turriconus species can be accounted for by the extremely high relative expression of two unusual peptide isoforms in C. praecellens, Ps9.1 and Ps9.2 (∼14% and 24%, respectively). These peptides share the cysteine framework typical of the P-superfamily, but are substantially longer than most previously reported sequences and exhibit a remarkably high frequency of histidine residues (12 of 45 residues in Ps9.2). A putative ortholog of Ps9.2 was also detected in C. andremenezi, where it also shows relatively high expression (1.9%), but no ortholog was detected for Ps9.1 (fig. 5).
Fig. 5.

—Inter- and intraspecific variation in Conus venom composition. (A) Sequence alignments of a selection of highly expressed conotoxins, where putative orthologs were identified in all four Turriconus specimens (predicted mature peptides and their names are shown); cysteines are highlighted in yellow and differences in amino acids are shown in red. *Predicted C-terminal amidation. (B) Histograms of rbh pair counts versus percent sequence identity for all species analyzed in this study. The total number of rbh pairs is depicted on graphs.
Another major difference between these closely related species is the high expression of the O1d superfamily in both specimens of C. andremenezi (∼20% and 36%). In vertebrate prey ∂-conotoxins block deactivation of sodium channels, which can lead to tetanic paralysis producing prey immobilization (Heinemann and Leipold 2007). Although this superfamily is also relatively highly expressed in C. praecellens (7–13%), its expression is still considerably lower than in C. andremenezi.
A Fine-Grained Comparison—Interspecific Difference at the Sequence Level
To investigate interspecific differences at sequence level, we performed comparative sequence analyses between the venom repertoires of each species. All-by-all NCBI-BLASTN searches identified 67 and 90 rbh pairs between the two specimens of each Turriconus species (C. andremenezi and C. praecellens, respectively) and between 53 and 57 rbh pairs when comparing the two species to one another (fig. 5B). In contrast, only between 9 and 16 rbh pairs were identified between the two Turriconus species and C. tribblei, C. lenavati, and C. geographus (fig. 5B).
Comparative analysis of conotoxin sequences between the two C. andremenezi specimens revealed that 98.5% of rbh pairs were greater than 95% identical, and of these, 46% of sequences were 100% identical (fig. 5B). Comparative analysis of conotoxin sequences between the two specimens of C. praecellens showed that 89% of rbh pairs were greater than 95% identical, and of these, 52% of sequences were 100% identical. These data are consistent with low variation between individuals of the same species of Conus.
Comparative analysis of C. praecellens conotoxin sequences to those of the two C. andremenezi specimens revealed that 68/60% of conotoxin precursors shared >95% identity, but of these, only 9/2% of sequences shared 100% identity. These data indicate that although there is overall similarity between the two species of Turriconus, considerable variation is present at individual sequence level. The low number of identical conotoxin sequences between C. andremenezi and C. praecellens suggests that despite being closely related these species have distinct venom repertoires. Comparison of conotoxin sequences from the two Splinoconus species and C. geographus to those of C. andremenezi and C. praecellens revealed that no toxin precursor shared >87% identity, highlighting the drastic differences between conotoxins sequences belonging to species from different Conus lineages.
Sequences of highly expressed conotoxins, for which similar sequences were identified in all four Turriconus specimens, are shown in figure 5B. This comparison provides concrete examples of sequences from two individuals of the same species, with these compared with the most similar peptide from a sister species. The transcripts of the first set shown (T-superfamily, fig. 5Bi) have identical mature toxin sequences in the four specimens analyzed. This identity appears atypical; more common are the sets of peptides in the same superfamily (T-superfamily fig. 5Bii) where the two C. andremenezi specimens are identical, but a small number of amino acid substitutions are found in the putative C. praecellens homolog. An interesting variation of this is shown in the next group of peptides compared (O1- and P-superfamily, fig. 5Biii and iv) for which the two C. andremenezi specimens have identical sequences, and C. praecellens expresses peptides with a few amino acid substitutions. In all of these cases however, the sequences align with few gaps and identical cysteine arrangement, and the differences between C. andremenezi and C. praecellens are predominantly accounted for by amino acid substitutions. The I1-superfamily of peptides provides an example of identical sequences in C. andremenezi, with a divergent sequence in C. praecellens that involves indels (fig. 5Bv). Thus, there is considerable divergence in this example between the two species, but no amino acid substitutions between the two specimens in the same species.
The A2-Superfamily in Turriconus
The A-superfamily, a prominent and well-represented group of venom components in most Conus species investigated to date, is not detected in the four Turriconus specimens analyzed and also absent (or of negligible expression) in the two members of Splinoconus. In C. geographus these comprise over 10% of all transcripts. A related unexpected discovery from the analysis of the Turriconus transcriptomes was that although the A-superfamily is not detected, a distinct class of A-like sequences is present, albeit at low expression levels (0.0004–0.02%, table 1). As discussed later, similar sequences have recently been reported in other cone snail species (Biggs etal. 2008; Barghi etal. 2015a). Although these were named A-like, based on distinct sequence characteristics, we have now designated these peptides as the A2-superfamily. A2 conotoxins do not share the signal peptide that defines the A-superfamily (see fig. 6, examples of A-superfamily toxins from C. geographus); however there is some weak similarity in the propeptide region, and the encoded mature peptides exhibit the structural features that define the A-superfamily (i.e., cysteine framework I, similar intercysteine loop spacing, as well as other key residues).
Fig. 6.

—The A2-superfamily sequences identified in the venom gland transcriptomes of C. praecellens and C. andremenezi. For comparison, another A2-superfamily sequence identified in the salivary gland of C. pulicarius PuSG1.1 (Uniprot: P0C8U6), and two A-superfamily sequences, GID and (Uniprot: P60274) GI (Uniprot: P01519) from C. geographus are shown. Signal peptides are underlined in purple, predicted mature peptides are underlined in black.
Discussion
We have provided the first comprehensive portrait of the venom repertoire of two sister species in the Turriconus subgenus of Conus and insights into the venom divergence upon speciation.
The Turriconus Venom Repertoire
In general, with between 107 and 155 conotoxins derived from 21 to 29 superfamilies, the venom diversity of the two species from the Turriconus clade is consistent with what has been reported for other species of Conus including studies addressing intraspecific variations in Conus venom gland transcriptomes (Hu etal. 2012; Robinson etal. 2014, 2017; Barghi etal. 2015a, 2015b; Phuong etal. 2016). Nevertheless, these numbers conflict with recent reports of as many as 3,303 conotoxins in Conus episcopatus that led authors to hypothesize that “transcriptomic messiness” accounts for the rapid evolution of conotoxin encoding genes (Jin etal. 2013; Lavergne etal. 2015). We believe that these are overestimates that result from inadequate processing of next-generation sequencing data sets, as recently suggested by others (Phuong etal. 2016).
Although the overall number of conotoxins is comparable to other cone snail species, the two members of the Turriconus clade exhibit clear differences in toxin superfamily expression. Relative to other species of Conus, Turriconus appears to be more specialized in P-superfamily conotoxins. P-conotoxins account for almost half of total toxin number expressed in C. praecellens and approximately 10% in C. andremenezi. The specific biological activity of P-superfamily conotoxins is not well established: Very few representatives have been characterized to-date. Of these, TxIXA from Conus textile produces a “spasmodic phenotype” in mice when injected intracranially (Lirazan etal. 2000), nevertheless, the molecular target of TxIXA remains to be elucidated. Very high expression levels suggest a key functional role for these toxins in Turriconus, so future work will be directed at investigating the biological activity of these peptides and correlating findings to the biology of Turriconus.
Another difference is the absence (or very low expression) of A-superfamily toxins that are functionally important components of several other Conus venoms characterized to date, especially in species that prey on fish (Santos etal. 2004; Terrat etal. 2012), including C. geographus. α-conotoxins comprise the largest and most-studied group of A-superfamily toxins. These peptides potently and selectively block neuronal and muscular nicotinic acetylcholine receptors (Santos etal. 2004). α-conotoxins have been identified from diverse phylogenetic lineages of Conus, including the early-diverging worm-hunting Stephanoconus clade (Ellison etal. 2003). Thus, the absence (or very low expression) of A-superfamily toxins in Turriconus suggests that these peptides ceased to serve a functional role in envenomation. As noted earlier, we instead detected expression of a genetically distinct A2-superfamily that would be predicted to play a similar functional role, but is expressed at relatively low levels.
Although the A2-superfamily sequences display all of the hallmarks of typical conotoxins, one puzzling finding is that they appear to be expressed at only very low relative levels. It is notable that structurally similar conotoxins had previously been characterized from Conus pulicarius (Biggs etal. 2008), C. tribblei, and C. lenavati (Barghi etal. 2015a), all presumably worm-hunting species. In C. tribblei and C. lenavati, A2 conotoxins are similarly expressed at low levels, whereas in C. pulicarius, which belongs to the more recently diverged subgenus Puncticulus, this gene superfamily was expressed in the salivary glands and not the venom gland, where conventional A-superfamily conotoxins were expressed. It will be of interest to further investigate whether the absence (or very low expression) of the A-superfamily correlates with presence of the A2-superfamily in other Conus species.
Besides these apparent differences between Turriconus and other Conus clades, several similarities could be observed. Highly expressed toxin superfamilies in Turriconus included the O1d-, M-, and T-superfamilies, which are found in almost all Conus venoms characterized to date. Thus, these gene superfamilies must play a fundamental role in envenomation and can be considered hallmarks of Conus venoms.
Speciation and Rapid Divergence of Conotoxin Exogenes
Conotoxins are among the fastest evolving gene products known in nature (Chang and Duda 2012; Sunagar etal. 2015). Considerable prior work has demonstrated that at the sequence level, each species of Conus appears to have its own distinct repertoire of conotoxins. Individuals of the same species would be expected to share a near identical venom repertoire, with only allelic differences among genes evident (Duda etal. 2009; Chang etal. 2015). The comparison of the venom repertoires of two individuals of the same species is also clearly consistent with expectations—approximately half of the identified rbh pairs matched with 100% identity, whereas the other half demonstrated >90% identity, suggesting reasonable levels of allelic polymorphism of conotoxin genes within the species population (Chang etal. 2015). Our findings on intraspecific variation of venom repertoire are largely consistent with the recent studies on multiple individuals of two closely related species C. tribblei and C. lenavati (Barghi etal. 2015a). The expression level of conotoxin gene superfamilies was similar across all three specimens of C. tribblei analyzed, and two of the three specimens of C. lenavati also showed low levels of intraspecific variation in conotoxin gene superfamily expression. Interestingly, the third C. lenavati specimen showed some substantial differences. Numerous factors might contribute to this observed difference, and the authors noted that this specimen was smaller than the two others examined, suggesting that age may be a factor contributing to venom composition as reported by others (Safavi-Hemami etal. 2011).
Consistent with published data, closely related species often share similar, but very rarely identical conotoxin sequences. Nevertheless, interpretation of the differences in toxin expression patterns between two species of Conus cannot be solely addressed by the degree of phylogenetic relatedness between these two species (Phuong etal. 2016). Differences in the ecology between these species should ultimately be taken in consideration, as this provides insights into the different ecological niches’ requirements that determined the adaptation of these species upon speciation. In this respect, syntopic species constitute a perfect model for addressing divergence of toxin expression patterns.
In addition to the two Turriconus species analyzed in this study, the Splinoconus species, C. tribblei and C. lenavati (Barghi etal. 2015a), were sampled from the same geographic locality off North Cebu in the Philippines. As all four species occur in (and were sampled from) the same locality and habitat, it is reasonable to expect that the venom gland transcriptomes of species in these two pairs are not greatly affected by adaptations to different environmental variables. Furthermore, as muddy bottom communities at depths of 180–250 m are more homogeneous at small spatial scale, and feature very limited diversity of microhabitats comparing to shallow water coral reef ecosystems (Gray 2002), we hypothesize that the main evolutionary force driving diversification in both species pairs were differential biotic interactions, primarily adaptation to different prey and predator species. This is why exogenes that are presumed to be sensitively tuned to mediate particular biological interactions following speciation may be expected to diversify rapidly in the case of both Turriconus and Splinoconus.
Indeed, we have shown that less than 10% of the venom exogene repertoire is identical between C. praecellens and C. andremenezi. Only a very small fraction of rbh pairs were 100% identical (2–9%). On the other hand, over half (68–78%) shared >90% identity, reflecting the close relationship between these two species. Although at conotoxin superfamily level, the venoms of C. andremenezi and C. praecellens are highly similar, there are two apparent differences: The much higher relative expression of several O1d conotoxins in C. andremenezi and very high relative expression of P-superfamily peptides in C. praecellens. In additional to these differences, it should be noted that within each superfamily there exists a large repertoire of species-specific sequences and structural scaffolds. Together, these data demonstrate that beyond the relatively subtle differences evident at the level of conotoxin gene superfamily expression, there are substantial differences present at the sequence level between the two sister species. The demonstrated notable divergence of the venom composition between C. andremenezi and C. praecellens further highlights that exogenes are a sensitive indicator of hidden biological differences.
Our comparison of the two species of Turriconus and C. tribblei, C. lenavati revealed no sequence similarity exceeding 87% across the entire conotoxin repertoire. Nevertheless, certain similarity can be noted at the gene superfamily level, with a set of 4–5 toxin gene superfamilies showing comparable and generally high relative expression levels in the four species. Therefore, at the gene superfamily level, our results highlight the role of the species-specific toxin expression patterns that are closely comparable among closely related species, but retain some recognizable features even in comparison of distantly related lineages.
Several studies have now demonstrated that diet is a major contributor to venom evolution in cone snails. Conotoxin gene diversity appears to be highest in species that prey on the widest diversity of prey (Phuong etal. 2016; Phuong and Mahardika 2017). Our results reveal that the overall diversity of conotoxin transcripts and gene superfamilies does not significantly change upon speciation in the two species of Turriconus but demonstrate variations of expression levels of individual gene families (i.e., the P- and O1d-superfamilies). This finding indicates that changes in dietary breadth may not have contributed to speciation in Turriconus. Our recent study on weaponized insulins expressed in the venoms of several cone snail species suggests that toxin gene expression may correlate with prey preference and hunting strategies (Safavi-Hemami etal. 2016). In this scenario, speciation in Turriconus may have been accompanied by changes in the expression levels of toxin genes evolved to target different types of prey and/or to induce different physiological endpoints in their prey. Although this is an intriguing hypothesis, more dense taxon sampling combined with behavioral studies is required to correlate the role of prey preference and predation strategy in shaping toxin expression and to disentangle its contribution to speciation in this genus.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The specimens used in this study were obtained in conjunction with a collection trip supported in part by an ICBG Grant No. 1U01TW008163. We would like to thank Noel Saguil for help with sample collection. This work was supported in part by National Institutes of Health Grants GM 48677 (to B.M.O.) and GM 099939 (to M.Y. and P.K.B.), an International Outgoing Fellowship Grant from the European Commission (CONBIOS 330486 to H.S.-H.), and the National Natural Science Foundation of China (Grant No. 31500626 to A.L.).
Literature Cited
- Barghi N, Concepcion GP, Olivera BM, Lluisma AO.. 2015a. Comparison of the venom peptides and their expression in closely related Conus species: insights into adaptive post-speciation evolution of Conus exogenomes. Genome Biol Evol. 7(6): 1797–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghi N, Concepcion GP, Olivera BM, Lluisma AO.. 2015b. High conopeptide diversity in Conus tribblei revealed through analysis of venom duct transcriptome using two high-throughput sequencing platforms. Mar Biotechnol (NY). 17(1): 81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs JS, Olivera BM, Kantor YI.. 2008. Alpha-conopeptides specifically expressed in the salivary gland of Conus pulicarius. Toxicon 52(1): 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs JS, Watkins M, Corneli PS, Olivera BM.. 2010. Defining a clade by morphological, molecular and toxinological criteria: distinctive forms related to Conus praecellens A. Adams, 1854. Nautilus (Philadelphia) 124: 1–19. [PMC free article] [PubMed] [Google Scholar]
- Chang D, Duda TF Jr.. 2016. Age-related association of venom gene expression and diet of predatory gastropods. BMC Evol Biol. 16: 27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Duda TFJ.. 2012. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol Biol Evol. 29(8): 2019–2029. [DOI] [PubMed] [Google Scholar]
- Chang D, Olenzek AM, Duda TF Jr.. 2015. Effects of geographical heterogeneity in species interactions on the evolution of venom genes. Proc Biol Sci. 282(1805): 20141984.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda TF, Chang D, Lewis BD, Lee T, DeSalle R.. 2009. Geographic variation in venom allelic composition and diets of the widespread predatory marine gastropod Conus ebraeus. PLoS One 4(7): e6245.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda TF Jr, Kohn AJ.. 2005. Species-level phylogeography and evolutionary history of the hyperdiverse marine gastropod genus Conus. Mol Phylogenet Evol. 34(2): 257–272. [DOI] [PubMed] [Google Scholar]
- Duda TF Jr, Kohn AJ, Palumbi S.. 2001. Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol J Linn Soc. 73(4): 391–409. [Google Scholar]
- Dutertre S, et al. 2014. Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat Commun. 5: 3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison M, McIntosh JM, Olivera BM.. 2003. Alpha-conotoxins ImI and ImII. Similar alpha 7 nicotinic receptor antagonists act at different sites. J Biol Chem. 278(2): 757–764. [DOI] [PubMed] [Google Scholar]
- Fedosov A, Puillandre N.. 2012. Phylogeny and taxonomy of the Kermia–Pseudodaphnella (Mollusca: Gastropoda: Raphitomidae) genus complex: a remarkable radiation via diversification of larval development. Syst Biodivers. 10(4): 447–477. [Google Scholar]
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evol Theory. 39(4): 783–791. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7): 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JS. 2002. Species richness of marine soft sediments. Mar Ecol Prog Ser. 244: 285–297. [Google Scholar]
- Heinemann SH, Leipold E.. 2007. Conotoxins of the O-superfamily affecting voltage-gated sodium channels. Cell Mol Life Sci. 64(11): 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Bandyopadhyay PK, Olivera BM, Yandell M.. 2012. Elucidation of the molecular envenomation strategy of the cone snail Conus geographus through transcriptome sequencing of its venom duct. BMC Genomics 13(1): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F.. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17(8): 754–755. [DOI] [PubMed] [Google Scholar]
- Jin AH, et al. 2013. Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity. Mol Cell Proteomics. 12(12): 3824–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas Q, Yu R, Jin AH, Dutertre S, Craik DJ.. 2012. ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40(Database issue): D325–D330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12): 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Sawyer S, Meyer M.. 2012. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40(1): e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4): 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23(21): 2947–2948. [DOI] [PubMed] [Google Scholar]
- Lavergne V, et al. 2015. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc Natl Acad Sci U S A, 112: E3782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lirazan MB, et al. 2000. The spasmodic peptide defines a new conotoxin superfamily. Biochemistry 39(7): 1583–1588. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov., New Orleans, LA, 1–8. doi: 10.1109/gce.2010.5676129.
- Olivera BM. 2006. Conus peptides: biodiversity-based discovery and exogenomics. J Biol Chem. 281(42): 31173–31177. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Seger J, Horvath MP, Fedosov AE.. 2015. Prey-Capture Strategies of Fish-Hunting Cone Snails: Behavior, Neurobiology and Evolution. Brain Behav Evol. 86: 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera BM, Showers Corneli P, Watkins M, Fedosov A.. 2014. Biodiversity of cone snails and other venomous marine gastropods: evolutionary success through neuropharmacology. Annu Rev Anim Biosci. 2: 487–513. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H.. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- Phuong MA, Mahardika GN.. 2017. Targeted sequencing of venom genes from cone snail genomes reveals coupling between dietary breadth and conotoxin diversity. bioRxiv 107672. Available from: https://doi.org/10.1101/107672 [Google Scholar]
- Phuong MA, Mahardika GN, Alfaro ME.. 2016. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genomics 17: 401.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puillandre N, Baylac M, Boisselier MC, Cruaud C, Samadi S.. 2009. An integrative approach to species delimitation in Benthomangelia (Mollusca: Conoidea). Biol J Linn Soc. 96(3): 696–708. [Google Scholar]
- Puillandre N, et al. 2014. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol Phylogenet Evol. 78: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2013. R: a language and environment for statistical computing. Vienna (Austria: ): R Core Team. [Google Scholar]
- Robinson SD, et al. 2014. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS One 9(2): e87648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SD, et al. 2017. The venom repertoire of Conus gloriamaris (Chemnitz, 1777), the glory of the sea. Mar Drugs. 15. doi:10.3390/md15050145 [DOI] [PMC free article] [PubMed]
- Robinson SD, Norton RS.. 2014. Conotoxin gene superfamilies. Mar Drugs. 12(12): 6058–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockel D, Korn W, Kohn A.. 1995. Manual of the living Conidae. Wiesbaden, Germany: Hemmen Verlag. [Google Scholar]
- Safavi-Hemami H, et al. 2014. Combined proteomic and transcriptomic interrogation of the venom gland of Conus geographus uncovers novel components and functional compartmentalization. Mol Cell Proteomics. 13(4): 938–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavi-Hemami H, et al. 2016. Venom insulins of cone snails diversify rapidly and track prey taxa. Mol Biol Evol. 33(11): 2924–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavi-Hemami H, et al. 2011. Embryonic toxin expression in the cone snail Conus victoriae: primed to kill or divergent function? J Biol Chem. 286(25): 22546–22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AD, McIntosh JM, Hillyard DR, Cruz LJ, Olivera BM.. 2004. The A-superfamily of conotoxins. J Biol Chem. 279(17): 17596–17606. [DOI] [PubMed] [Google Scholar]
- Schmieder R, Edwards R.. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Bennett GM, Engel P, Williams DJ, Ochman H.. 2013. Disentangling associated genomes In: FD E, editor. Methods enzymol. Academic Press; p. 445–464. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21): 2688–2690. [DOI] [PubMed] [Google Scholar]
- Sunagar K, Moran Y, Hoekstra HE.. 2015. The rise and fall of an evolutionary innovation: contrasting strategies of venom evolution in ancient and young animals. PLoS Genet. 11(10): e1005596.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28(10): 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrat Y, et al. 2012. High-resolution picture of a venom gland transcriptome: case study with the marine snail Conus consors. Toxicon 59(1): 34–46. [DOI] [PubMed] [Google Scholar]
- Tucker JK, Tenorio MJ.. 2009. Systematic classification of recent and fossil conoidean gastropods. Hackenheim (Germany: ): Conchbooks. [Google Scholar]
- Uribe JE, Puillandre N, Zardoya R.. 2017. Beyond Conus: phylogenetic relationships of Conidae based on complete mitochondrial genomes. Mol Phylogenet Evol. 107: 142–151. [DOI] [PubMed] [Google Scholar]
- Walker CS, et al. 1999. The T-superfamily of conotoxins. J Biol Chem. 274(43): 36030–36030. [DOI] [PubMed] [Google Scholar]
- Wheeler QD, Wheeler Q, Pennak S.. 2013. What on earth? 100 of our planet’s most amazing new species. New York: Plume. [Google Scholar]
- Woodward SR, Cruz LJ, Olivera BM, Hillyard DR.. 1990. Constant and hypervariable regions in conotoxin propeptides. EMBO J. 9: 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8): 1586–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All C. andremenezi and C. praecellens conotoxin prepropeptide sequences from this study are provided in the Supplementary Material online and have been deposited at GenBank (accession numbers MF576542–MF576988).