

Abstract
Background
The Fusarium oxysporum species complex (FOSC) contains several phylogenetic lineages. Phylogenetic studies identified two to three major clades within the FOSC. The mitochondrial sequences are highly informative phylogenetic markers, but have been mostly neglected due to technical difficulties.
Results
A total of 61 complete mitogenomes of FOSC strains were de novo assembled and annotated. Length variations and intron patterns support the separation of three phylogenetic species. The variable region of the mitogenome that is typical for the genus Fusarium shows two new variants in the FOSC. The variant typical for Fusarium is found in members of all three clades, while variant 2 is found in clades 2 and 3 and variant 3 only in clade 2. The extended set of loci analyzed using a new implementation of the genealogical concordance species recognition method support the identification of three phylogenetic species within the FOSC. Comparative analysis of the mitogenomes in the FOSC revealed ongoing mitochondrial recombination within, but not between phylogenetic species.
Conclusions
The recombination indicates the presence of a parasexual cycle in F. oxysporum. The obstacles hindering the usage of the mitogenomes are resolved by using next generation sequencing and selective genome assemblers, such as GRAbB. Complete mitogenome sequences offer a stable basis and reference point for phylogenetic and population genetic studies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4116-5) contains supplementary material, which is available to authorized users.
Keywords: Comparative genomics, Mitochondrial genome, Mitochondrial recombination, Phylogenomics
Background
Members of the Fusarium oxysporum species complex (FOSC) are important plant pathogens causing vascular wilts, rots and damping-off on a broad range of agronomically and horticulturally important crops [1, 2]. In addition, members of this species complex are also clinically important, causing infections in both human and animal hosts [3, 4]. Furthermore, despite the pathogenic potential of these fungi, not all strains are virulent. Putative non-pathogenic strains belonging to this group are used as biocontrol agents against pathogens [5].
The taxonomy of Fusarium has historically been based on the morphology of the asexual reproductive structures, leading to a broad definition of F. oxysporum. To capture intraspecific variability within the morphospecies, formae speciales (ff. spp.) were introduced based on the pathogenicity of the strains towards particular plant hosts [6]. F. oxysporum is closely related to the F. fujikuroi species complex that contains several heterothallic species. The genome of F. oxysporum resembles the genome of heterothallic species, both mating types can be found in populations, the mating type genes are expressed and introns are correctly spliced from the transcript [7]. However, since no sexual stage has been found for this species nor could it be induced under laboratory conditions, F. oxysporum is considered to be asexual [8, 9].
The use of molecular markers revealed that multiple ff. spp. have evolved polyphyletically [1, 10, 11]. Phylogenetic studies by O’Donnell et al. [8, 10] identified three major clades within the FOSC. However, later phylogenetic analysis conducted using genealogical concordance phylogenetic species recognition (GCPSR) based on eight loci supported the separation of only two phylogenetic species within the complex: one species corresponding to clade 1 and the other to the remaining clades [12].
The operational criteria for GCPSR for fungi were introduced in the theoretical paper of Taylor et al. [13], and first implemented as an operational framework using a two-step methodology by Dettman et al. [14]. After genetic isolation, two populations (or species) undergo the following stages: shared polymorphism (apparent polyphyly), loss of shared polymorphism (after fixation in one of the species) and reciprocal monophyly (after fixation in both species). According to GCPSR, the genetic isolation between populations (phylogenetic species) can be detected by a combination of genealogical concordance and non-discordance. Genealogical concordance is meant by the concordant reciprocal monophyly of multiple gene genealogies. Genealogical non-discordance means that no grouping supported by high support values for one of the genes is contradicted by another gene with the same level of support [13, 14].
Mitogenome sequences were used for resolving phylogenetic and evolutionary relationships between fungi at all taxonomic levels [15–17]. The advances in sequencing technologies and the reduction of costs involved, as well as, the development of tools to selectively assemble target genomic regions, like GRAbB, made it feasible to conduct complete mitochondrial genome (mitogenome) sequence analysis of a large number of strains [18].
The mitochondrial genomes of Fusarium spp. contain a set of fourteen “standard” mitochondrial polypeptide-encoding genes, two rRNA-encoding genes, rnl (mtLSU) and rns (mtSSU), and more than twenty tRNA-encoding genes [19]. The orientation and order of the genes are conserved within the genus, with the exception of some of the tRNA-encoding genes [17]. The mitochondrial genomes of Fusarium spp. contain variable numbers of introns, which causes significant size variation between the different species [17, 19]. One of the introns is located in the rnl gene and encodes a small ribosomal protein Rps3. This intron is conserved in the Pezizomycotina [20].
In addition to the genes with functional predictions, a large open reading frame (ORF) was found in all Fusarium spp. except in F. oxysporum (represented by strain F11) [17, 19, 21]. This large ORF was found in a region that is variable and contains several tRNA genes. For these reasons the region was referred to as large variable (LV) region and the large ORF with unknown function as LV-uORF [19] (orf2229 in Fig. 1). The mitogenome of the representative strain for F. oxysporum was sequenced using Sanger sequencing and primer walking [21]. This sequence did not contained the LV-uORF, and this is the only F. oxysporum mitogenome that was used for comparative studies so far [17, 19, 21]. Although several F. oxysporum strains have been sequenced using next generation sequencing (NGS) methods, there are only two strains for which the complete mitogenome was assembled. Both of these mitogenome sequences, GenBank accession no. KR952337 (unpublished) and LT571433 [18] (Fig. 1), contain the large variable region with the large variable ORF.
Fig. 1.
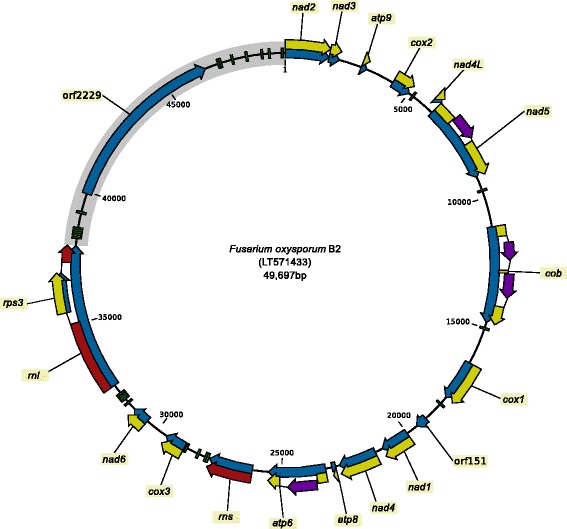
Mitochondrial genome of Fusarium oxysporum f. sp. cubense race 4 (strain B2; LT571433). Green blocks: tRNA coding genes, blue arrows: genes, yellow arrows: protein coding sequences, red arrows: rDNA coding sequence, purple arrows: intron encoded homing endonuclease genes, gray segment: large variable (LV) region with orf2229 (LV-uORF)
The goals of this study were (i) to prove that the mitochondrial genomes of a large number of strains can be analyzed, as we suggested in an earlier study [18], (ii) to present a detailed analysis of these mitochondrial genomes and (iii) to demonstrate how a detailed analysis of these genomes can contribute to our understanding of the biology of the given organism. To achieve these goals we present the following in this study. First, the phylogenetic relationships between 61 strains of the F. oxysporum species complex is analyzed. For this, we used a revised implementation of the GCPSR. Second, a detailed analysis of the genetic diversity of the mitochondrial genomes within the complex is given. Third, an interpretation of the mitogenome diversity in the light of the phylogeny is provided. Lastly, the biological implications of our results are discussed.
Methods
Fungal strains used and whole genome sequencing of the strains
Sixty-one F. oxysporum strains, two F. proliferatum strains and one F. commune strain were analyzed in this study (see Additional file 1). The F. commune strain (JCM 11502) was previously identified as F. oxysporum, but BLAST and Fusarium-MLST (http://www.westerdijkinstitute.nl/fusarium/) results showed that this strain belongs to F. commune.
The F. oxysporum f. sp. cumini strain F11, kindly provided by Dimitrios Tsirogiannis (Benaki Phytopathological Institute, Kifissia, Greece), was used for re-sequencing.
For the two F. proliferatum strains (ITEM2287 and ITEM2400), the two F. oxysporum f. sp. dianthi strains (Fod001 and Fod008) and F. oxysporum f. sp. cumini strain F11 shotgun libraries were made using the Illumina TruSeq nano DNA library prep kit, according to manufacturer’s protocols (Illumina). Each of the libraries was loaded as (part of) one lane of an Illumina paired-end flowcell for cluster generation using a cBot. Sequencing was done on an Illumina HiSeq2000 instrument using 101, 7, 101 flow cycles for forward, index and reverse reads respectively. De-multiplexing of resulting data was done using Casava 1.8 software. The sequencing data has been deposited into the European Nucleotide Archive (ENA) with the following accession numbers: PRJEB18591 (ITEM2287 and ITEM2400), PRJEB18594 (F11) and PRJEB18595 (Fod001 and Fod008) (see Additional file 1).
The remaining strains were sequenced by other research groups or as part of previous studies [22–24]. The sequencing reads for these strains were retrieved through NCBI’s Sequence Read Archive (see Additional file 1).
Assembling sequences
The following nuclear protein coding genes were assembled from NGS data for all 64 strains: γ-actin (act), ATP citrate lyase (acl1), β-tubulin II (tub2), calmodulin (cal), nitrate reductase (NIR), phosphate permease (PHO), 60S ribosomal protein L10 (rpl10a), the largest and second largest subunit of DNA-dependent RNA polymerase II (RPB1 and RPB2, respectively), translation elongation factor 1 α (tef1a), translation elongation factor 3 (tef3) and topoisomerase I (top1). Besides the nuclear protein coding genes, part of the mitochondrial SSU rRNA gene (mtSSU or rns), the complete nuclear rDNA repeat region (18S rRNA - ITS1 - 5.8S rRNA - ITS2 - 28S rRNA - IGS) and the complete mitochondrial genome were also assembled for all 64 strains.
The eight loci used by Laurence et al. [12] for genealogical concordance phylogenetic species recognition were only barcoding regions of the following genes: acl1, tub2, cal, NIR, PHO, RPB1, RPB2 and tef1a. A set of eight complete protein coding genes were used in this study, which have been traditionally used as barcoding genes or have recently been suggested as such [25]: act, tub2, cal, rpl10a, RPB2, tef1a, tef3 and top1.
All of the regions listed above were assembled from NGS reads using GRAbB (Genomic Region Assembly by Baiting; [18]) by specifying the appropriate reference sequence and employing SPAdes 3.6 [26, 27] as assembler. The assembled sequences have been uploaded to the European Nucleotide Archive under the following accession numbers: LT841199-LT841268 and LT905535-LT906358.
Sequence annotation
The initial mitogenome annotations were done using MFannot (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl) and were manually curated: annotation of tRNA genes was performed using tRNAscan-SE [28], annotation of protein-coding genes and the rnl gene was corrected by aligning intronless homologs to the genome. Intron encoded proteins were identified using NCBI’s ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) and annotated using InterPro [29] and CD-Search [30]. The visualization of the annotated sequences was produced using CLC Sequence Viewer 7.7.1 (https://www.qiagenbioinformatics.com/products/clc-sequence-viewer/), the graphical output was subsequently manually adjusted.
Sequence analysis
All sequence alignments were done using MUSCLE [31, 32]. The barcoding regions and genes were each aligned per locus. The alignment of the mitochondrial genome required a different approach. The mitochondrial genome sequences were split into two regions: the large variable region (between the trnT(tgt) and the nad2 genes) and remaining part of the mitochondrial genome, which we refer to as the conserved part of the mitochondrial genome. Since the conserved part of the mitogenome and the variants of the large variable region were too long for MUSCLE to handle in a single run, we used the following approach. First, the given sequences were divided into non-overlapping homologous blocks (5000–7000 bp). Second, each of these blocks were aligned using MUSCLE. Finally, the individual aligned blocks were concatenated in the original order.
Sequence variability of each region was calculated by aligning the sequences, then the number of characters with multiple character states was calculated and divided by the total number of characters in the alignment. This step was done using fasta_variability from the fasta_tools package (https://github.com/b-brankovics/fasta_tools).
Phylogenetic analysis
The most appropriate substitution evolution model was determined using jModelTest 2 [33] for each of the single locus data sets. Phylogenetic reconstruction has been conducted using MrBayes 3.2.5 [34]. The MCMC algorithm was run for 4,000,000 generations with four incrementally-heated chains, starting from random trees and sampling one out every 400 generations. Burn-in was set to a relative value, 0.25.
Majority-rule consensus (MRC) trees were calculated based on the trees generated by the MrBayes run using PAUP* 4.0a147 for Unix/Linux with “percent” set to 95 (corresponding to 0.95 posterior probability), using the two F. proliferatum strains and the F. commune strain as outgroups and rooting was set to “monophyl”.
Genealogical concordance phylogenetic species recognition
Genealogical concordance phylogenetic species recognition (GCPSR) was implemented in two steps: (i) identifying independent evolutionary lineages (IELs) and (ii) exhaustive subdivision of strains into phylogenetic species. These were implemented using Perl scripts developed in house that are available at GitHub (https://github.com/b-brankovics/GCPSR). The GCPSR method applied in this study is a modified implementation of GCPSR sensu Dettman et al. [14]. The revised implementation of the GCPSR method is briefly described below (for a detailed description as well as recommendations see Additional file 2).
Identifying independent evolutionary lineages
Independent evolutionary lineages (IELs) have two criteria: concordance and non-discordance. In our analysis, a clade was considered concordant when it was supported by at least two single locus MRC phylogenies (for explanation on alternative settings, see Additional file 2). Clades “A” and “B” are discordant if A∩B≠∅ (they have common elements) and neither one is a subset of the other. In our implementation, we used these two criteria in a sequential order: First, identifying concordant clades, then comparing concordant clades and removing those that were discordant with each other. The concordant clades obtained in this manner define an unambiguous tree topology. This tree can be visualized and the number of loci supporting each clade can be used as a support value for the given clade. This tree can be used for exhaustive subdivision.
Exhaustive subdivision and phylogenetic species recognition
After identifying IELs, the IELs that are supported by a large number of single locus genealogies (e.g. half of the loci), are considered as putative phylogenetic species, the rest of the IELs is removed (see Figure S2b in Additional file 2). Each isolate must be classified within a putative phylogenetic species. When an isolate is grouped within a given clade (putative phylogenetic species), then all subclades of the given clade are removed (see Figure S2c in Additional file 2). This is referred to as exhaustive subdivision, which ensures that all phylogenetic species are monophyletic and there are no paraphyletic species. The clades that are kept after this step are recognized as phylogenetic species.
Results
Phylogenetic analysis and GCPSR
Sixty-one strains belonging to Fusarium oxysporum species complex, one F. commune strain and two F. proliferatum strains have been analyzed in this study. Applying genealogical concordance phylogenetic species recognition (GCPSR) on the eight single copy nuclear (complete) protein coding genes (act, cal, RPB2, rpl10a, tef1a, tef3, top1 and tub2) resulted in the recognition of three phylogenetic species within the FOSC. All three clades were present in at least half of the single locus phylogenies with high support (BPP ). Half of the single locus phylogenies still supported the three species when the rDNA repeat region and the conserved part of the mitogenome were added besides the eight genes (Fig. 2). The five loci that supported the recognition of all three clades with high support were among the six most variable loci included in the analysis (Table 2). The phylogeny of the conserved part of the mitogenome also supported the recognition of the three phylogenetic species that correspond to clades 1, 2 and 3 sensu O’Donnell et al. [10, 35].
Fig. 2.
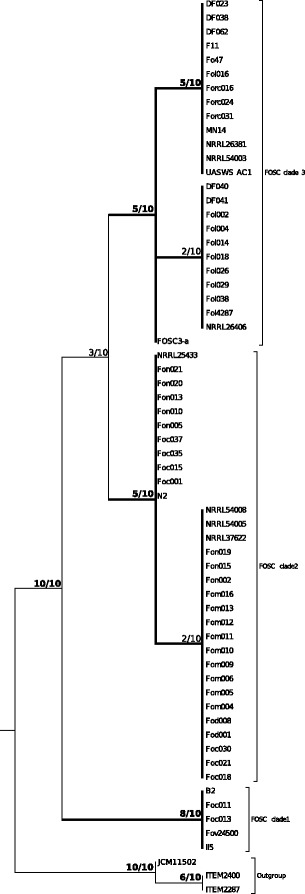
Genealogical concordance phylogenetic species recognition based on the 10 loci data set. The 10 loci used in the analysis were: act, tub2, cal, tef1a, tef3, rpl10a, rpb2, top1, rDNA repeat and the conserved part of the mitogenome. Only clades that were highly supported (BPP ) in at least two single locus phylogenies were included in the analysis. The three clades within the FOSC were recognized as phylogenetic species and shown in the tree. The support values indicate how many single locus phylogenies supported the given clade with BPP out of the 10 loci
Table 2.
The variability of the individual loci used for the GCPSR based on the 10 loci data set. The loci are ordered based on the number of variable sites
| Locus | Length | Conserved | PIa | The three clades | ||
|---|---|---|---|---|---|---|
| Interb | Intrac | Interb | Intrac | were monophyletic | ||
| rpl10a | 795 | 762 | 783 | 28 | 7 | no |
| cal | 979 | 920 | 963 | 52 | 8 | no |
| act | 1634 | 1572 | 1605 | 54 | 21 | no |
| tub2 | 1671 | 1586 | 1634 | 78 | 27 | no |
| tef1a | 1776 | 1650 | 1711 | 99 | 43 | yes |
| tef3 | 3588 | 3456 | 3532 | 111 | 43 | yes |
| top1 | 3170 | 2981 | 3114 | 177 | 45 | yes |
| rpb2 | 3907 | 3653 | 3812 | 213 | 61 | yes |
| rDNA | 7990 | 7437 | 7689 | 483 | 223 | no |
| mt | 40841 | 39121 | 40153 | 1501 | 464 | yes |
aParsimony informative sites
bOutgroup + FOSC (clades 1, 2, 3)
cFOSC (clades 1, 2, 3)
In comparison, GCPSR based on the eight partial loci (acl1, cal, NIR, PHO, RPB1, RPB2, tef1a and tub2) used by Laurence et al. [12] had insufficient support to recognize the three phylogenetic species within the FOSC. Only tef1a showed high support for all three clades (data not shown).
Mitogenomes
The mitogenomes of all 64 strains (61 FOSC and the 3 outgroup strains) were successfully assembled into single contigs each showing circular topology (Fig. 3). Some mitogenomes had identical sequences, in total there were 42 unique haplotypes identified for mitochondrial genome in this data set of 64 strains (Table 1).
Fig. 3.
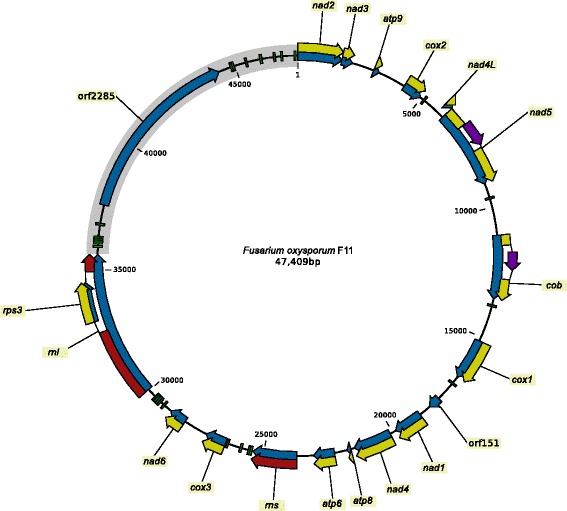
Mitochondrial genome of Fusarium oxysporum f. sp. cumini strain F11. Green blocks: tRNA coding genes, blue arrows: genes, yellow arrows: protein coding sequences, red arrows: rDNA coding sequence, purple arrows: intron encoded homing endonuclease genes, gray segment: large variable (LV) region with orf2285 (LV-uORF)
Table 1.
Mitochondrial genome lengths and intron presence. The strains of clades 2 and 3 are ordered based on the corrected mitochondrial genome length
| Strain | Species | forma specialis | Clade | Introns | LV | mt length | Correcteda | |||
|---|---|---|---|---|---|---|---|---|---|---|
| nad5 | cob(1) | cob(2) | atp6 | variant | (bp) | (bp) | ||||
| Foc011 | F. oxysporum | f.sp. cucumerinum | 1 | yes | yes | - | - | 1 | 46476 | 33615 |
| Foc013 | F. oxysporum | f.sp. cucumerinum | 1 | yes | yes | - | - | 1 | 46476 | 33615 |
| Fov24500 | F. oxysporum | f.sp. vasinfectum | 1 | yes | yes | yes | yes | 1 | 48054 | 33727 |
| B2 | F. oxysporum | f.sp. cubense | 1 | yes | yes | yes | yes | 1 | 49697 | 33419 |
| II5 | F. oxysporum | f.sp. cubense | 1 | yes | yes | yes | yes | 1 | 49692 | 33414 |
| Foc021 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 2 | 50783 | 33449 |
| Foc018 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 2 | 50783 | 33449 |
| Foc030 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 2 | 50783 | 33449 |
| NRRL25433 | F. oxysporum | f.sp. vasinfectum | 2 | yes | - | - | - | 1 | 44941 | 33458 |
| N2 | F. oxysporum | f.sp. cubense | 2 | yes | - | - | - | 1 | 45639 | 33465 |
| Foc035 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 1 | 45706 | 33477 |
| Fon002 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 1 | 45677 | 33477 |
| Foc037 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 1 | 45705 | 33479 |
| Foc015 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 1 | 45712 | 33486 |
| Fom016 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 1 | 45538 | 33487 |
| Fom013 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 1 | 45538 | 33487 |
| Fom005 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 1 | 45538 | 33487 |
| Fom012 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 1 | 45538 | 33487 |
| NRRL54005 | F. oxysporum | f.sp. raphani | 2 | yes | - | - | - | 1 | 45536 | 33487 |
| Fom004 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 1 | 45538 | 33487 |
| Fom006 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 1 | 45538 | 33487 |
| NRRL37622 | F. oxysporum | f.sp. pisi | 2 | yes | - | - | - | 3 | 39941 | 33490 |
| Fom009 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 3 | 38736 | 33492 |
| Fom011 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 3 | 38736 | 33492 |
| Fom010 | F. oxysporum | f.sp. melonis | 2 | yes | - | - | - | 3 | 38736 | 33492 |
| Fod008 | F. oxysporum | f.sp. dianthi | 2 | yes | - | - | - | 1 | 45694 | 33498 |
| Fod001 | F. oxysporum | f.sp. dianthi | 2 | yes | - | - | - | 1 | 44947 | 33498 |
| Foc001 | F. oxysporum | f.sp. cucumerinum | 2 | yes | - | - | - | 1 | 45741 | 33509 |
| Fon015 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 1 | 45708 | 33514 |
| Fon019 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 2 | 50087 | 33514 |
| Fon020 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 1 | 45752 | 33526 |
| Fon005 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 1 | 45752 | 33526 |
| Fon010 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 1 | 45752 | 33526 |
| Fon021 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 1 | 45752 | 33526 |
| Fon013 | F. oxysporum | f.sp. niveum | 2 | yes | - | - | - | 2 | 50795 | 33527 |
| NRRL54008 | F. oxysporum | f.sp. conglutinans | 2 | yes | - | - | - | 2 | 50023 | 33534 |
| Forc031 | F. oxysporum | f.sp. radicis-cucumerinum | 3 | yes | yes | - | - | 1 | 47541 | 33667 |
| DF023 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52324 | 33667 |
| Fo47 | F. oxysporum | 3 | yes | yes | - | - | 1 | 47547 | 33667 | |
| Forc024 | F. oxysporum | f.sp. radicis-cucumerinum | 3 | yes | yes | - | - | 1 | 47541 | 33667 |
| Forc016 | F. oxysporum | f.sp. radicis-cucumerinum | 3 | yes | yes | - | - | 1 | 47541 | 33667 |
| NRRL54003 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 1 | 47588 | 33674 |
| UASWS AC1 | F. oxysporum | 3 | yes | yes | - | - | 1 | 47598 | 33682 | |
| DF038 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 1 | 47562 | 33688 |
| DF062 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 1 | 47562 | 33688 |
| Fol016 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 1 | 47562 | 33688 |
| NRRL26406 | F. oxysporum | f.sp. melonis | 3 | yes | yes | - | - | 1 | 47444 | 33688 |
| Fol004 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol014 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol4287 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol038 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| DF041 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol018 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol002 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol026 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| DF040 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| Fol029 | F. oxysporum | f.sp. lycopersici | 3 | yes | yes | - | - | 2 | 52352 | 33694 |
| MN14 | F. oxysporum | 3 | yes | yes | - | - | 1 | 47573 | 33696 | |
| F11 | F. oxysporum | f.sp. cumini | 3 | yes | yes | - | - | 1 | 47409 | 33704 |
| NRRL26381 | F. oxysporum | f.sp. radicis-lycopersici | 3 | yes | yes | - | - | 1 | 47263 | 33707 |
| FOSC3-a | F. oxysporum | 3 | yes | yes | yes | - | 2 | 53639 | 33727 | |
| JCM11502 | F. commune | - | yes | - | - | - | 1 | 47526 | 33565 | |
| ITEM2287 | F. proliferatum | - | - | - | - | - | 1 | 46555 | 33558 | |
| ITEM2400 | F. proliferatum | - | - | - | - | - | 1 | 46549 | 33549 | |
aThe length of the conserved part of the mitogenome after excluding the introns of protein coding genes
Intron presence
All of the strains analyzed contained an intron in the rnl gene (mtLSU) that is conserved in the Pezizomycotina and contains a gene coding for a ribosomal protein, Rps3. The two F. proliferatum mitogenomes contained no further introns. The F. commune mitogenome contained an intron in the nad1 gene, which was not found in the other strains. The mitogenomes of the FOSC strains contained intron positions in the following protein coding genes: atp6, cob (this gene had two intron positions) and nad5 (Table 1). The members of clade 2 could be differentiated from all other strains based on their intron patterns. Intron patterns showed almost no variation within clades 2 and 3, the only exception being strain FOSC3-a in clade 3. Clade 1 showed marked variation in intron pattern.
Genetic diversity of the conserved part of the mitogenome
The mitogenomes of Fusarium spp. contain a region that shows higher levels of variation than other parts of the mitogenome [19]. This region is referred to as the large variable (LV) region and it is located between rnl (mitochondrial LSU rRNA gene) and nad2 (gray area in Fig. 3). In this paragraph, we present the analysis of the genetic diversity of the mitochondrial sequences located outside the LV region, the analysis of the LV region can be found in the next subsection.
Strains that belong to clades 2 and 3 had clearly different lengths of the conserved region of the mitogenome. This is partially due to the difference in intron patterns, but even after these introns were excluded, the two populations, clades 2 and 3, had significantly different lengths, 33492.32 ± 23.95 and 33688.68 ± 14.42, respectively (p-value <2.2∗10−16 according to two-sample t-test; Table 1 and Additional file 3). The significant difference between lengths of the conserved part of mitogenome suggests that these two populations have been genetically isolated. The variation observed in clade 1 was larger than in the two other clades. There were only 5 strains within clade 1, thus, further grouping of the strains based on mitogenome length within this clade would not produce statistically supported results.
Large variable region
Re-sequencing of F. oxysporum f. sp. cumini strain F11 revealed that the LV-uORF (orf2285) gene, typical for Fusarium spp., is present in the mitogenome of this strain at the expected position, within the large variable (LV) region located between rnl and nad2 (Fig. 3).
The LV region has three variants in the FOSC, which do not appear to be homologous (Fig. 4). Although the three variants contained tRNA genes with identical anticodon sequences in a similar order (Fig. 4), there is a high level of sequence variation between the three variants. BLAST was unable to identify homology between the tRNA genes of the different variants of the LV region. The variant that is typical for Fusarium spp., variant 1, was 13424 or 13428 bp long in the F. proliferatum strains, 12422 bp in the F. commune strain and 9833-12003 bp long in FOSC. This variant was present in all three major clades of the FOSC. Variant 2 of the LV region was 15816-16749 bp long and it was present only in clades 2 and 3 within the FOSC. Finally, variant 3 was 4570 or 5777 bp long and it was found only in clade 2.
Fig. 4.
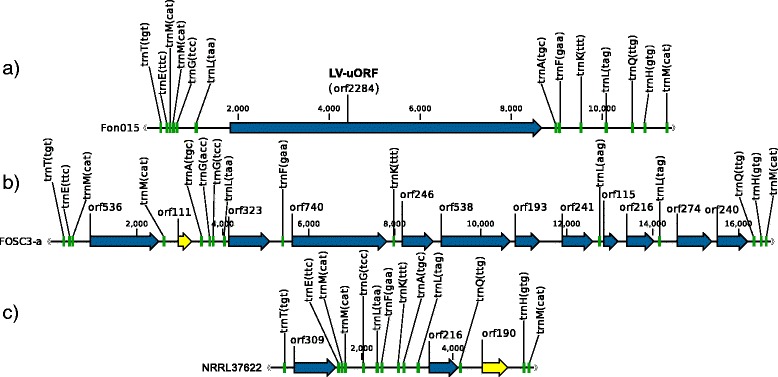
The three variants of the large variable region. a Variant 1 represented by F. oxysporum strain Fon015, b variant 2 represented by F. oxysporum strain FOSC3-a and c variant 3 represented by F. oxysporum strain NRRL37622. Green blocks: tRNA coding genes, blue arrows: ORFs, yellow arrows: ORFs that are not present in all representatives of the given variant
Variant 1 of the LV region
This variant contained thirteen tRNA genes and the LV-uORF located between trnL(taa) and trnA(tgc) (Fig. 4). LV-uORF has no known function. Domain predictions returned low quality hits to a limited part of the protein sequence (see Additional file 4). Blast searches against the NCBI and UniProt databases returned hits against only Fusarium LV-uORF sequences. The GC-content of LV-uORF was higher than the average of that of conserved protein coding genes (both exonic and intronic regions). This higher GC-content was similar to that of the intergenic regions (see Additional file 4).
Variant 2 of the LV region
This variant contained fifteen tRNA genes, thirteen of which are also found in variant 1 and two additional tRNAs: trnG(acc) and trnL(aag). This region contains eleven or twelve ORFs interleaved with the tRNA genes (Fig. 4). Most of the variant 2 sequences contain twelve ORFs, but strains Fon019 and NRRL54008 have lost the ORF found between the second copy of trnM(cat) and trnA(tgc), due to a deletion.
Most of the ORFs have no functional prediction or returned no hits using BLAST. Four ORFs appeared to be homing endonuclease genes (HEGs) based on conserved domain matches: two ORFs, the one between the first and second trnM(cat) (orf536 in Fig. 4) and the one between trnL(taa) and trnF(gaa) (orf323 in Fig. 4), matched to LAGLIDADG endonucleases; the two ORFs between trnL(tag) and trnQ(ttg) gene (orf274 and orf240 in Fig. 4) matched to the GIY-YIG endonucleases.
Variant 3 of the LV region
This variant contained a set of thirteen tRNA genes that were also present in the other two variants. Strains Fom009, Fom010 and Fom011 contained an additional trnQ(ttg) gene. Besides the tRNA genes there were two ORFs present in all four strains containing this variant, while strain NRRL37622 contained an additional ORF (Fig. 4).
The two ORFs present in all strains with variant 3 of the LV region showed similarity to HEGs based on conserved domain search results, one belonging to the LAGLIDADG family and one to the GIY-YIG family. The putative LAGLIDADG HEG was located between trnT(tgt) and trnE(ttc) gene. The second ORF, between trnL(tag) and trnQ(ttg) gene, was a putative homolog of the GIY-YIG endonuclease gene upstream of trnQ(ttg) in variant 2, based on BLASTp results.
F. oxysporum f.sp. pisi strain NRRL37622 contains a third ORF, which returned partial hits to a hypothetical protein in Rhizophagus irregularis using BLASTp, and CD-Search (Conserved Domain Search [30]) showed similarity to a zinc-binding domain (zf-3CxxC). This ORF is located at a homologous position to the second trnQ(ttg) gene present in the other three strains possessing variant 3 of the large variable region (Fig. 4).
LV region phylogeny and conserved mitogenome phylogeny
In the phylogenetic tree based on variant 1 of the large variable region, strain Fov24500 grouped with members of clade 3, although this strain belonged to clade 1 based on other markers (data not shown). Sequence comparison between the LV-uORF of strain Fov24500 and other strains possessing the homologous region revealed that there is a large deletion in the copy found in Fov24500 (see Additional file 4). Since this deletion was unique to this strain, the phylogenetic reconstruction was also done after removing the LV-uORF region from the alignment of variant 1 sequences. The resulting tree and the phylogenetic tree of variant 2 are congruent with the phylogenetic tree based on the conserved part of the mitochondrial genome (presented as a tanglegram in Fig. 5 to highlight the co-evolution of these sequences). The topological incongruence is due to the differences in the resolution power of the regions. The only topological incongruence beyond the difference in resolution is the location of strain Fon015 within clade 2 of the FOSC. Fon015 was grouped with strains Fon019, Fom009, Fom010, Fom011 and NRRL54008 in the tree based on the conserved part of the mitogenome, while it appeared separate from all other clade 2 isolates in the tree based on variant 1 of the LV region.
Fig. 5.
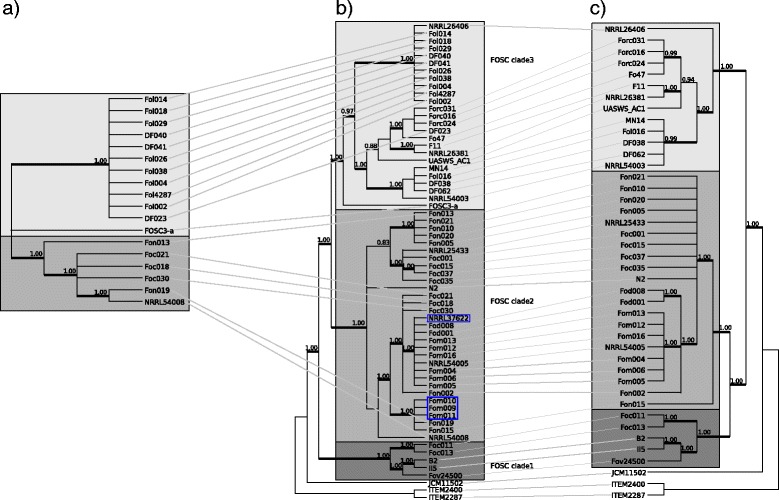
Tanglegram of the trees based on a the large variable region variant 2, b the conserved part of the mitogenome and c the variant 1 of the LV region, respectively. Clades with high Bayesian posterior probability (BPP) support are displayed with thicker branches. The support values are BPP values. The three phylogenetic clades identified within the FOSC are highlighted in different shades of gray. The strains that contain variant 3 are highlighted by blue boxes
Highly similar conserved region irrespective of different variants of the LV region
Sequence similarity of the conserved part of the mitochondrial genome was greater between members of the same clade than between strains that shared the same variant of the LV region, but belonging to different clades. The sequences of the conserved part of the mitogenome of F. oxysporum f. sp. niveum strains Fon015 and Fon019 from clade 2 were completely identical, despite the fact that they contain variants 1 and 2, respectively. A similar example was found in clade 3, where a high sequence similarity was observed between the conserved part of the mitogenome of the F. oxysporum f. sp. melonis strain NRRL 26406 and F. oxysporum f. sp. lycopersici strain FOL4287 (99.85%), despite the fact that they contain variants 1 and 2, respectively.
Discussion
The theory of genealogical concordance phylogenetic species recognition (GCPSR) for fungi was introduced by Taylor et al. [13]. GCPSR consists of two steps: (i) identifying independent evolutionary lineages (IELs) based on genetic isolation estimated from multiple single locus phylogenies and (ii) exhaustive subdivision (classifying each isolate into clades substantiated by IELs). Previous implementations of GCPSR [12, 14] presented no programmatic tools for performing the analysis, required that balancing selection should not be influencing any of the loci in the analysis, identified IELs based on being at least concordant or non-discordant, and preformed exhaustive subdivision after superimposing the IELs on a maximum likelihood tree based on concatenated sequences. The last two characteristics make the method difficult to automate, which means that it is challenging to scale the method to a large number of loci. The scaling is further hindered by the requirement that loci should not be under balancing selection, which is difficult to detect before conducting phylogenetic comparisons.
We have employed a new strategy for GCPSR that uses sequential selection of clades: first identifying concordant clades, and subsequently removing the discordant clades from the selection. The programmatic implementation allows the user to set the minimum number of phylogenies required to recognize a given clade as concordant, which in combination with applying the two criteria sequentially makes the detection indifferent to some loci being under balancing selection. Our implementation of the GCPSR is scalable to any number of loci. Since the criteria for clade selection is applied sequentially and not in parallel, there can be no conflict left between the clades that are kept, and these clades already define a tree topology. The number of loci supporting a given clade can be used to display the support in the constructed tree. As a final selection, exhaustive subdivision can be used, by removing all subclades that would make a clade paraphylic. Employing exhaustive subdivision also ensures that no single strain can be recognized as unique species, thereby adhering to the genetic differentiation criterion.
Phylogenetic studies by O’Donnell et al. [8, 10] have identified three major clades within the Fusarium oxysporum species complex. Further phylogenetic analysis conducted using genealogical concordance phylogenetic species recognition based on eight loci supported the separation of two phylogenetic species within the complex: one species corresponding to clade 1 and the other to the remaining three clades [12].
In the current study, the eight loci used by Laurence et al. [12] were tested with our GCPSR approach. Only one locus, tef1a, contained sufficient phylogenetic information to support the grouping with sufficient Bayesian posterior support (BPP ) within the FOSC. In contrast, when the eight complete protein coding genes, the rDNA repeat region and the conserved part of the mitochondrial genome were used for the GCPSR analysis, three phylogenetic species were identified. All three species were highly supported in half of the single locus phylogenies. The three phylogenetic species correspond to clades 1, 2 and 3 sensu O’Donnell within the FOSC, and they were identified with low support (supported only by tef1a) by the eight loci analysis. The eight loci set did not contain sufficient phylogenetic information to separate the three species. The ten loci used for GCPSR contained more phylogenetic information, five out of the six most informative loci could resolve the three species with high support (BPP , Table 2), the exception being the rDNA repeat region. Most of the variable sites in the rDNA repeat region were located in the intergenic spacer region (IGS), which is frequently the target of indel mutations [36]. Indel mutations make it difficult to estimate correctly which nucleotide characters are homologous, and this can lead to incorrect phylogenetic tree estimations. This could explain why the rDNA repeat sequence did not support the clustering supported by the other variable markers, despite the large number of parsimony informative sites.
The recognition of two phylogenetic species corresponding to clades 2 and 3 was further supported by the fact that the conserved part of their mitogenomes had significantly different lengths and their mitochondrial genomes had distinctly different intron patterns. This length difference was statistically significant even after excluding the introns of protein coding genes from the analysis. This marked difference suggests that the two populations have been genetically isolated. This genetic isolation could not be explained by the geographic origin of the strains.
The distribution of the variants of the large variable (LV) region of the mitogenome across the clades seems to contradict the recognition of the three phylogenetic species. However, the trees based on the variant regions and on the conserved part of the mitogenome are congruent. This demonstrates that three phylogenetic species are genetically isolated and differentiated from each other. It is likely that variants 1 and 2 of the LV region appeared in the common ancestor of clades 2 and 3, and they were maintained by recombination during the separation of the two lineages. This is supported by the fact that the two variants are present in both clades 2 and 3, and that the separation of clades 2 and 3 is supported by the phylogeny of both the LV region as well as the conserved part of the mitogenome. This hypothesis is also supported by the complete sequence identity of the conserved part of the mitogenome of F. oxysporum f. sp. niveum strains Fon015 and Fon019 from clade 2, despite the fact that these strains contain variants 1 and 2, respectively. A similar example in clade 3 is the high sequence similarity between the conserved part of the mitogenome of the F. oxysporum f. sp. melonis strain NRRL26406 and F. oxysporum f. sp. lycopersici strain FOL4287 (99.85%), while they contain variants 1 and 2 of the LV region, respectively.
In this study strain F11, which was used for previous comparative mitogenome studies [17, 19], was re-sequenced using next generation sequencing, and its mitogenome was assembled from the sequencing reads. The curated sequence contains the LV region, which is typical for Fusarium spp. [17, 19]. In addition, two introns were found that were absent from the original assembly [37].
Al-Reedy et al. [19] reported that the LV-uORF may represent a gene with unknown function and it is under purifying selection. Because the LV-uORF region has a higher GC content than the conserved protein encoding genes or the intron encoded genes and because its codon usage differs significantly from other mitochondrial genes, they suggested that the LV-uORF was acquired via horizontal gene transfer. However, examining the mitogenome sequence presented by Al-Reedy et al. [19] reveals that the GC contents of the LV-uORF and the intergenic regions are similar. So the GC content does not necessarily suggest horizontal gene transfer. Comparative genome analysis conducted in this study revealed that within the FOSC this region contains multiple indels as well as point mutations, which in some strains lead to fragmentation of the ORF into smaller ORFs (Additional file 4). The variability of this region as well as its GC content is similar to intergenic regions of the mitochondrial genome. All these facts combined suggest that the LV-uORF has lost its function within the FOSC or it never had a function within Fusarium. To understand whether the LV-uORF does have a function in other species complexes within Fusarium a study of the mitogenomes of these groups should be undertaken.
The analysis of the 61 strain data set revealed two additional variants of the LV region, both of which had no LV-uORF. Both contain at least thirteen tRNA genes containing the same anticodons that are present in variant 1 which is present in F. proliferatum as well. The different variants share only low sequence similarity and the order of the tRNA genes is not completely syntenic. Variant 1 is present in all three clades of the FOSC and also in other Fusarium spp. [17, 19], variant 2 is found in clades 2 and 3 within the FOSC, and variant 3 is confined to clade 2.
The origin of the two new variants of the LV region is unclear. They may have evolved from variant 1 by the insertion of mobile elements similar to mitochondrial introns, because the newly discovered variants contain ORFs that have domains matching those present in homing endonuclease genes (HEGs). HEGs are commonly associated with mitochondrial introns [38]. These may have initiated double strand breaks inside this region, which through homologous recombination repair could have led to the rearrangement of the tRNA genes, while still preserving some blocks of synteny.
F. oxysporum has been considered to be an asexual species. In asexual species, genetic exchange between strains is possible only between members of the same vegetative compatibility group (VCG). We found recombination of the mitochondrial genome, which indicates the presence of either a sexual or a parasexual cycle. The sexual cycle in haploid fungi involves the fusion of two haploid nuclei leading to a diploid nucleus which through meiosis results in haploid offspring. The parasexual cycle also begins by the fusion of two haploid nuclei, but recombination occurs by mitotic crossing over during multiplication of the diploid nucleus, and haploid cells emerge through vegetative haploidization instead of meiosis [39]. No sexual cycle could be induced in F. oxysporum, but the fusion of the nuclei of strains that were not members of the same VCG was shown [23, 40]. Genome analysis of F. oxysporum f. sp. lycopersici strain FOL4287 revealed that pathogenicity factors are located on accessory chromosomes and that transfer of these chromosomes to a non-pathogenic recipient conveyed pathogenicity to the recipient strain [23]. The proposed underlying mechanism for this chromosome transfer is that the nuclei of the two strains fuse, followed by selective loss of chromosomes from one of the fusion partners [40]. Genetic exchange may be restricted to members of the same phylogenetic species since anastomosis was observed between F. oxysporum f.sp. lycopersici Fol007 (clade 3) and F. oxysporum Fo47 (clade 3), between F. oxysporum f.sp. lycopersici Fol007 (clade 3) and F. oxysporum f.sp. melonis NRRL26406 (clade 3), but anastomosis could not be induced between F. oxysporum f.sp. lycopersici Fol007 (clade 3) and F. oxysporum f.sp. cubense NRRL25603 (clade 1) [23, 35]. These results demonstrates that recombination is possible between members of the same clade, but there is genetic isolation between the different clades.
Conclusions
In this study a programmatic implementation of genealogical concordance phylogenetic species recognition (GCPSR) strategy was introduced, which allows the method to scale even to a large set of loci. It was shown to be robust in recognizing phylogenetic species. Our new GCPSR approach revealed three phylogenetic species within the Fusarium oxysporum species complex, which correspond to clades 1, 2 and 3 sensu O’Donnell et al. [10, 35]. We conclude that there has been genetic isolation between the different phylogenetic species leading to reciprocal monophyly of multiple loci. Thus, the phylogenetic species appear to define the limit of potential genetic exchange between members of the FOSC.
Both the recombination identified in the mitochondrial genome of the FOSC in our study and previous chromosome transfer studies [23, 40] indicate that there is a parasexual cycle in F. oxysporum and recombination is possible between members of the same phylogenetic species.
Mitochondrial genomes are highly informative for resolving phylogenetic relationships even between closely related species and populations. Complete mitochondrial genome sequences offer a stable basis and reference point for phylogenetic and population genetic studies. Our study demonstrates that a detailed comparative analysis of the mitogenome may offer new insights into the biology of the studied organism.
F. oxysporum strains contain the large variable region containing the LV-uORF that is not a functional gene. Furthermore, two new variants of the LV region were discovered within the FOSC. The distribution pattern and the sequence comparisons demonstrates that there has been mitochondrial recombination during the separation of clades 2 and 3. Clade 1 may contain more phylogenetic species. Extended sampling of this group may shed more light on the composition of the Fusarium oxysporum species complex.
Additional files
Strains analyzed and accession numbers of the NGS data used. (XLSX 9 kb)
Scalable Genealogical Concordance Phylogenetic Species Recognition: Detailed description of the new implementation of the GCPSR method, including algorithm overview and recommended workflow. (PDF 420 kb)
Boxplot of the length of the conserved part of the mitogenome of the three clades of the FOSC. (PDF 111 kb)
Analysis of the LV-uORF region In depth comparative analysis of the LV-uORF region. (PDF 434 kb)
Acknowledgments
We would like to thank Dimitrios Tsirogiannis from the Benaki Phytopathological Institute (Kifissia, Greece) for providing F. oxysporum f. sp. cumini strain F11 for re-sequencing.
Funding
The investigations were supported by the Division for Earth and Life Sciences (ALW) with financial aid from the Netherlands Organization for Scientific Research (NWO, http://www.nwo.nl/) under grant number 833.13.006. This work was further supported by the Horizon programme (project 93512007) of the Netherlands Genomics Initiative (NGI) through a grant to MR. CW was supported from the MycoKey project (Horizon2020, nr. 678781). The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The sequencing data has been deposited into the European Nucleotide Archive (ENA) with the following accession numbers: PRJEB18591, PRJEB18594 and PRJEB18595. The assembled sequences have been uploaded to the European Nucleotide Archive under the following accession numbers: LT841199-LT841268 and LT905535-LT906358. Perl script, fasta_variability, is available from the fasta_tools package (https://github.com/b-brankovics/fasta_tools). GCPSR Perl scripts are available at GitHub (https://github.com/b-brankovics/GCPSR).
Abbreviations
- acl1
ATP citrate lyase
- act
γ-actin
- atp6
Mitochondrially encoded ATP synthase 6
- atp8
Mitochondrially encoded ATP synthase 8
- atp9
Mitochondrially encoded ATP synthase 9
- BLAST
Basic Local Alignment Search Tool
- BPP
Bayesian posterior probability
- cal
Calmodulin
- CD-Search
Conserved domain search
- cob
Mitochondrially encoded cytochrome b
- cox1
Mitochondrially encoded cytochrome c oxidase I
- cox2
Mitochondrially encoded cytochrome c oxidase II
- cox3
Mitochondrially encoded cytochrome c oxidase III
- DNA
Deoxyribonucleic acid
- ENA
European Nucleotide Archive
- F
Fusarium
- f. sp
forma specialis
- ff. spp
formae speciales
- FOSC
Fusarium oxysporum species complex
- GCPSR
Genealogical concordance phylogenetic species recognition
- GRAbB
Genomic Region Assembly by Baiting
- GTR
General time reversible
- HEG
Homing endonuclease
- IEL
Independent evolutionary lineages
- IGS
Intergenic spacer
- ITS
Internal transcribed spacer
- LV
Large variable
- LV-uORF
Large variable open reading frame with unknown function
- MCMC
Markov chain Monte Carlo
- MRC
Majority-rule consensus
- mt
Mitochondrial genome
- nad1
Mitochondrially encoded NADH dehydrogenase 1
- nad2
Mitochondrially encoded NADH dehydrogenase 2
- nad3
Mitochondrially encoded NADH dehydrogenase 3
- nad4
Mitochondrially encoded NADH dehydrogenase 4
- nad4L
Mitochondrially encoded NADH dehydrogenase 4L
- nad5
Mitochondrially encoded NADH dehydrogenase 5
- nad6
Mitochondrially encoded NADH dehydrogenase 6
- NCBI
National Center for Biotechnology Information
- NGS
Next generation sequencing
- nir
Nitrate reductase
- ORF
Open reading frame
- pho
Phosphate permease
- rDNA
Ribosomal DNA
- rnl
Mitochondrially encoded 16S RNA
- rns
Mitochondrially encoded 12S RNA
- rpb1
Largest subunit of DNA-dependent RNA polymerase II
- rpb2
Second largest subunit of DNA-dependent RNA polymerase II
- rpl10a
60S ribosomal protein L10
- rps3
Ribosomal protein S3
- sp
Species (singular form)
- spp
Species (plural form)
- tef1a
Translation elongation factor 1 α
- tef3
Translation elongation factor 3
- top1
Topoisomerase I
- tRNA
Transfer ribonucleic acid
- tub2
β-tubulin
- VCG
Vegetative compatibility group
Authors’ contributions
BB and AD designed the study. PD, MR, TAJL and CW sequenced large part of the strains used for this study. BB wrote the scripts and carried out the analysis. All authors (BB, PD, MR, GSH, TAJL, CW and AD) helped shaping the manuscript. All authors approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4116-5) contains supplementary material, which is available to authorized users.
Contributor Information
Balázs Brankovics, Email: b.brankovics@westerdijkinstitute.nl.
Peter van Dam, Email: p.vandam@uva.nl.
Martijn Rep, Email: m.rep@uva.nl.
G. Sybren de Hoog, Email: s.hoog@westerdijkinstitute.nl.
Theo A. J. van der Lee, Email: theo.vanderlee@wur.nl
Cees Waalwijk, Email: cees.waalwijk@wur.nl.
Anne D. van Diepeningen, Email: anne.vandiepeningen@wur.nl
References
- 1.Baayen RP, O’Donnell K, Bonants PJM, Cigelnik E, Kroon LPNM, Roebroeck EJA, Waalwijk C. Gene genealogies and AFLP analyses in the Fusarium oxysporum complex identify monophyletic and nonmonophyletic formae speciales causing wilt and rot disease. Phytopathology. 2000;90(8):891–900. doi: 10.1094/PHYTO.2000.90.8.891. [DOI] [PubMed] [Google Scholar]
- 2.Michielse CB, Rep M. Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol. 2009;10(3):311–24. doi: 10.1111/j.1364-3703.2009.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Donnell K, Sutton DA, Wiederhold N, Robert VARG, Crous PW, Geiser DM. Veterinary Fusarioses within the United States. J Clin Microbiol (Epub ahead of print). 2016. doi:10.1128/JCM.01607-16. [DOI] [PMC free article] [PubMed]
- 4.van Diepeningen AD, Al-Hatmi AMS, Brankovics B, de Hoog GS. Taxonomy and Clinical Spectra of Fusarium Species: Where Do We Stand in 2014? Curr Clin Microbiol Rep. 2014;1(1-2):10–18. doi: 10.1007/s40588-014-0003-x. [DOI] [Google Scholar]
- 5.Fuchs JG, Moënne-Loccoz Y, Défago G. Nonpathogenic Fusarium oxysporum strain Fo47 induces resistance to Fusarium wilt in tomato, Plant Dis. 1997;81(5):492–6. doi: 10.1094/PDIS.1997.81.5.492. [DOI] [PubMed] [Google Scholar]
- 6.Snyder WC, Hansen HN. The species concept in Fusarium. Am J Bot. 1940;27(2):64–7. doi: 10.2307/2436688. [DOI] [Google Scholar]
- 7.Yun SH, Arie T, Kaneko I, Yoder OC, Turgeon BG. Molecular organization of mating type loci in heterothallic, homothallic, and asexual Gibberella/Fusarium species. Fungal Genet Biol. 2000;31(1):7–20. doi: 10.1006/fgbi.2000.1226. [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell K, Sutton DA, Rinaldi MG, Magnon KC, Cox PA, Revankar SG, Sanche S, Geiser DM, Juba JH, van Burik J-AH, Padhye A, Anaissie EJ, Francesconi A, Walsh TJ, Robinson JS. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses. J Clin Microbiol. 2004;42(11):5109–120. doi: 10.1128/JCM.42.11.5109-5120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fourie G, Steenkamp ET, Gordon TR, Viljoen A. Evolutionary relationships among the Fusarium oxysporum f. sp. cubense vegetative compatibility groups. Appl Environ Microbiol. 2009;75(14):4770–781. doi: 10.1128/AEM.00370-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Donnell K, Kistler HC, Cigelnik E, Ploetz RC. Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc Natl Acad Sci U S A. 1998;95:2044–049. doi: 10.1073/pnas.95.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skovgaard K, Nirenberg HI, O’Donnell K, Rosendahl S. Evolution of Fusarium oxysporum f. sp. vasinfectum races inferred from multigene genealogies. Phytopathology. 2001;91(12):1231–1237. doi: 10.1094/PHYTO.2001.91.12.1231. [DOI] [PubMed] [Google Scholar]
- 12.Laurence MH, Summerell BA, Burgess LW, Liew ECY. Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biology. 2014;118(4):374–84. doi: 10.1016/j.funbio.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC. Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol. 2000;31(1):21–32. doi: 10.1006/fgbi.2000.1228. [DOI] [PubMed] [Google Scholar]
- 14.Dettman JR, Jacobson DJ, Taylor JW. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution. 2003;57(12):2703–720. doi: 10.1111/j.0014-3820.2003.tb01514.x. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Steenkamp ET, Brinkmann H, Forget L, Philippe H, Lang BF. Phylogenomic analyses predict sistergroup relationship of nucleariids and Fungi and paraphyly of zygomycetes with significant support. BMC Evol Biol. 2009;9:272. doi: 10.1186/1471-2148-9-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avila-Adame C, Gómez-Alpizar L, Zismann V, Jones KM, Buell CR, Ristaino JB. Mitochondrial genome sequences and molecular evolution of the Irish potato famine pathogen, Phytophthora infestans. Curr Genet. 2006;49(1):39–46. doi: 10.1007/s00294-005-0016-3. [DOI] [PubMed] [Google Scholar]
- 17.Fourie G, van der Merwe NA, Wingfield BD, Bogale M, Tudzynski B, Wingfield MJ, Steenkamp ET. Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genomics. 2013;14(1):605. doi: 10.1186/1471-2164-14-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brankovics B, Zhang H, van Diepeningen AD, van der Lee TAJ, Waalwijk C, de Hoog GS. GRAbB: Selective assembly of genomic regions, a new niche for genomic research. PLoS Comput Biol. 2016;12(6):1004753. doi: 10.1371/journal.pcbi.1004753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Reedy RM, Malireddy R, Dillman CB, Kennell JC. Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet Biol. 2012;49(1):2–14. doi: 10.1016/j.fgb.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Ghikas DV, Kouvelis VN, Typas MA. The complete mitochondrial genome of the entomopathogenic fungus Metarhizium anisopliae var. anisopliae: gene order and trn gene clusters reveal a common evolutionary course for all Sordariomycetes, while intergenic regions show variatio. Arch Microbiol. 2006;185(5):393–401. doi: 10.1007/s00203-006-0104-x. [DOI] [PubMed] [Google Scholar]
- 21.Cunnington JH. Organization of the mitochondrial genome of Fusarium oxysporum (anamorphic Hypocreales) Mycoscience. 2007;48(6):403–6. doi: 10.1007/S10267-007-0379-Z. [DOI] [Google Scholar]
- 22.Guo L, Han L, Yang L, Zeng H, Fan D, Zhu Y, Feng Y, Wang G, Peng C, Jiang X, Zhou D, Ni P, Liang C, Liu L, Wang J, Mao C, Fang X, Peng M, Huang J. Genome and transcriptome analysis of the fungal pathogen Fusarium oxysporum f. sp. cubense causing banana vascular wilt disease. PLoS ONE. 2014;9(4):95543. doi: 10.1371/journal.pone.0095543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman PM, Kang S, Shim WB, Woloshuk C, Xie X, Xu JR, Antoniw J, Baker SE, Bluhm BH, Breakspear A, Brown DW, Butchko RAE, Chapman S, Coulson R, Coutinho PM, Danchin EGJ, Diener A, Gale LR, Gardiner DM, Goff S, Hammond-Kosack KE, Hilburn K, Hua-Van A, Jonkers W, Kazan K, Kodira CD, Koehrsen M, Kumar L, Lee YH, Li L, Manners JM, Miranda-Saavedra D, Mukherjee M, Park G, Park J, Park SY, Proctor RH, Regev A, Ruiz-Roldan MC, Sain D, Sakthikumar S, Sykes S, Schwartz DC, Turgeon BG, Wapinski I, Yoder O, Young S, Zeng Q, Zhou S, Galagan J, Cuomo CA, Kistler HC, Rep M. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464(7287):367–73. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dam P, Fokkens L, Schmidt SM, Linmans JHJ, Kistler HC, Ma LJ, Rep M. Effector profiles distinguish formae speciales of Fusarium oxysporum. Environ Microbiol. 2016;18(11):4087–102. doi: 10.1111/1462-2920.13445. [DOI] [PubMed] [Google Scholar]
- 25.Stielow JB, Lévesque CA, Seifert KA, Meyer W, Irinyi L, Smits D, Renfurm R, Verkley GJM, Groenewald M, Chaduli D, Lomascolo A, Welti S, Lesage-Meessen L, Favel A, Al-Hatmi AMS, Damm U, Yilmaz N, Houbraken J, Lombard L, Quaedvlieg W, Binder M, Vaas LAI, Vu TD, Yurkov AM, Begerow D, Roehl O, Guerreiro MA, Fonseca A, Samerpitak K, van Diepeningen AD, Dolatabadi S, Moreno LF, Casaregola S, Mallet S, Jacques N, Roscini L, Egidi E, Bizet C, Garcia-Hermoso D, Martín MP, Deng S, Groenewald JZ, Boekhout T, de Beer ZW, Barnes I, Duong TA, Wingfield MJ, de Hoog GS, Crous PW, Lewis CT, Hambleton S, Moussa TAA, Al-Zahrani HS, Almaghrabi OA, Louis-Seize G, Assabgui R, McCormick W, Omer G, Dukik K, Cardinali G, Eberhardt U, de Vries M, Robert V. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia. 2015;35:242–63. doi: 10.3767/003158515X689135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 2013;20(10):714–37. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavesi A, Conterio F, Bolchi A, Dieci G, Ottonello S. Identification of new eukaryotic tRNA genes in genomic DNA databases by a multistep weight matrix analysis of transcriptional control regions. Nucleic Acids Res. 1994;22(7):1247–1256. doi: 10.1093/nar/22.7.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, Yong SY, Bateman A, Punta M, Attwood TK, Sigrist CJA, Redaschi N, Rivoire C, Xenarios I, Kahn D, Guyot D, Bork P, Letunic I, Gough J, Oates M, Haft D, Huang H, Natale DA, Wu CH, Orengo C, Sillitoe I, Mi H, Thomas PD, Finn RD. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015;43(Database issue):213–21. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchler-Bauer A, Bryant SH. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32(Web Server issue):327–31. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinforma. 2004;19(5):113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772–2. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Donnell K, Gueidan C, Sink S, Johnston PR, Crous PW, Glenn A, Riley R, Zitomer NC, Colyer P, Waalwijk C, van der Lee TAJ, Moretti A, Kang S, Kim HS, Geiser DM, Juba JH, Baayen RP, Cromey MG, Bithell S, Sutton DA, Skovgaard K, Ploetz R, Kistler HC, Elliott M, Davis M, Sarver BAJ. A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genet Biol. 2009;46(12):936–48. doi: 10.1016/j.fgb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Appel DJ, Gordon TR. Relationships among pathogenic and nonpathogenic isolates of Fusarium oxysporum based on the partial sequence of the intergenic spacer region of the ribosomal DNA. Mol Plant Microbe Int. 1996;9(2):125–38. doi: 10.1094/MPMI-9-0125. [DOI] [PubMed] [Google Scholar]
- 37.Pantou MP, Kouvelis VN, Typas MA. The complete mitochondrial genome of Fusarium oxysporum: Insights into fungal mitochondrial evolution. Gene. 2008;419:7–15. doi: 10.1016/j.gene.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Burt A, Koufopanou V. Homing endonuclease genes: The rise and fall and rise again of a selfish element. Curr Opin Genet Dev. 2004;14(6):609–15. doi: 10.1016/j.gde.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Pontecorvo G. The parasexual cycle in Fungi. Annu Rev Microbiol. 1956;10(1):393–400. doi: 10.1146/annurev.mi.10.100156.002141. [DOI] [PubMed] [Google Scholar]
- 40.Vlaardingerbroek I, Beerens B, Rose L, Fokkens L, Cornelissen BJC, Rep M. Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ Microbiol. 2016;18(11):3702–13. doi: 10.1111/1462-2920.13281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains analyzed and accession numbers of the NGS data used. (XLSX 9 kb)
Scalable Genealogical Concordance Phylogenetic Species Recognition: Detailed description of the new implementation of the GCPSR method, including algorithm overview and recommended workflow. (PDF 420 kb)
Boxplot of the length of the conserved part of the mitogenome of the three clades of the FOSC. (PDF 111 kb)
Analysis of the LV-uORF region In depth comparative analysis of the LV-uORF region. (PDF 434 kb)
Data Availability Statement
The sequencing data has been deposited into the European Nucleotide Archive (ENA) with the following accession numbers: PRJEB18591, PRJEB18594 and PRJEB18595. The assembled sequences have been uploaded to the European Nucleotide Archive under the following accession numbers: LT841199-LT841268 and LT905535-LT906358. Perl script, fasta_variability, is available from the fasta_tools package (https://github.com/b-brankovics/fasta_tools). GCPSR Perl scripts are available at GitHub (https://github.com/b-brankovics/GCPSR).