

Abstract
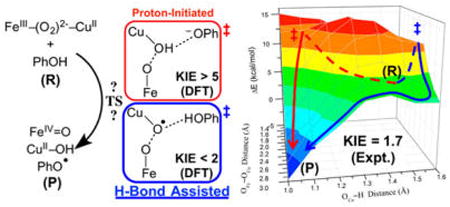
This study evaluates the reaction of a biomimetic heme–peroxo–copper complex, {[(DCHIm)(F8)-FeIII]–(O22−)–[CuII(AN)]}+ (1), with a phenolic substrate, involving a net H-atom abstraction to cleave the bridging peroxo O–O bond that produces FeIV=O, CuII—OH, and phenoxyl radical moieties, analogous to the chemistry carried out in heme–copper oxidases (HCOs). A 3D potential energy surface generated for this reaction reveals two possible reaction pathways: one involves nearly complete proton transfer (PT) from the phenol to the peroxo ligand before the barrier; the other involves O–O homolysis, where the phenol remains H-bonding to the peroxo OCu in the transition state (TS) and transfers the H+ after the barrier. In both mechanisms, electron transfer (ET) from phenol occurs after the PT (and after the barrier); therefore, only the interaction with the H+ is involved in lowering the O–O cleavage barrier. The relative barriers depend on covalency (which governs ET from Fe), and therefore vary with DFT functional. However, as these mechanisms differ by the amount of PT at the TS, kinetic isotope experiments were conducted to determine which mechanism is active. It is found that the phenolic proton exhibits a secondary kinetic isotope effect, consistent with the calculations for the H-bonded O–O homolysis mechanism. The consequences of these findings are discussed in relation to O–O cleavage in HCOs, supporting a model in which a peroxo intermediate serves as the active H+ acceptor, and both the H+ and e− required for O–O cleavage derive from the cross-linked Tyr residue present at the active site.
Graphical abstract
1. Introduction
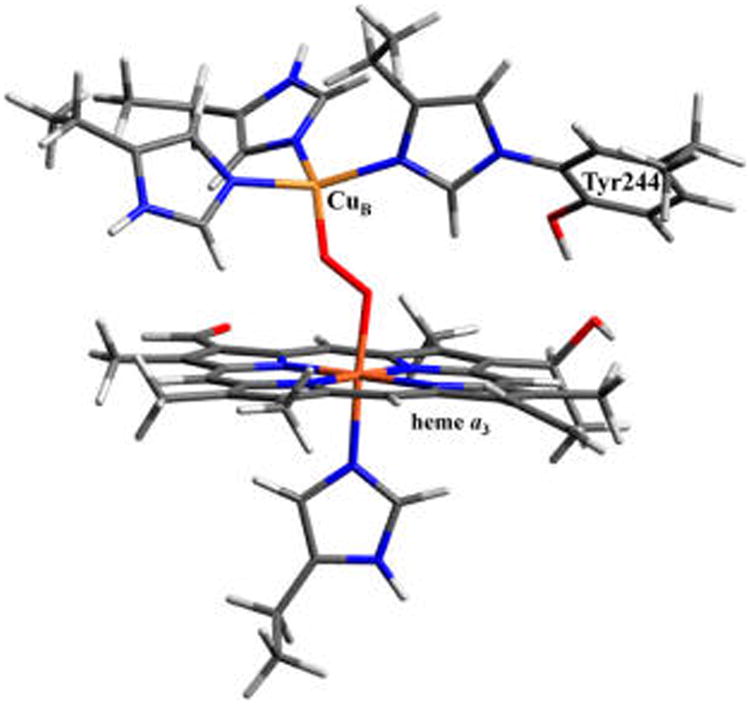
Heme–copper oxidases (HCOs) are terminal respiratory enzymes prevalent throughout biology that are responsible for the four-electron reduction of dioxygen to water.1,2 This reaction and its reverse have considerable global implications in areas ranging from biological energy conversion and mitochondrial disease to aerobic fuel cells and solar energy conversion.3 Cytochrome c oxidase (CcO), the most widely studied member of the HCO superfamily, is found in the mitochondrial membrane of eukaryotes and the bacterial membrane of aerobic prokaryotes. The biological source of electrons in CcO is cytochrome c, which results in a net free energy gain of ∼46 kcal/mol of dioxygen reduced. This energy is efficiently used to pump protons across the inner mitochondrial or bacterial membrane to generate a chemiosmotic potential used downstream to drive ATP synthesis.4 Dioxygen is reduced at an active site (termed the binuclear center (BNC)) composed of a heme (heme a3 in CcO) and a copper ion (CuB),5–7 where the heme is tethered to the protein by an axial histidine residue, and CuB is ligated by three histidine residues. One of the copper-bound histidines is covalently cross-linked to a nearby tyrosine (Tyr244 in Figure 1; note that all heme and protein residue indices referenced herein correspond to the bovine heart aa3-type CcO). Electrons are delivered to the active site through a low-spin, bis-His-ligated heme cofactor (heme a) found in all HCOs, and CcO additionally contains a binuclear Cu center (CuA) responsible for mediating electron transfer (ET) from cytochrome c to heme a. While the presence of these additional metal centers complicates data from many spectroscopic methods, the use of model chemistry (including generation of key species whose interrogation can shed light on questions of particular and important concern),8 site-selective spectroscopies, and computational studies provide promising means of studying the 4H+/4e− reduction of O2 to H2O by the BNC.
Figure 1.
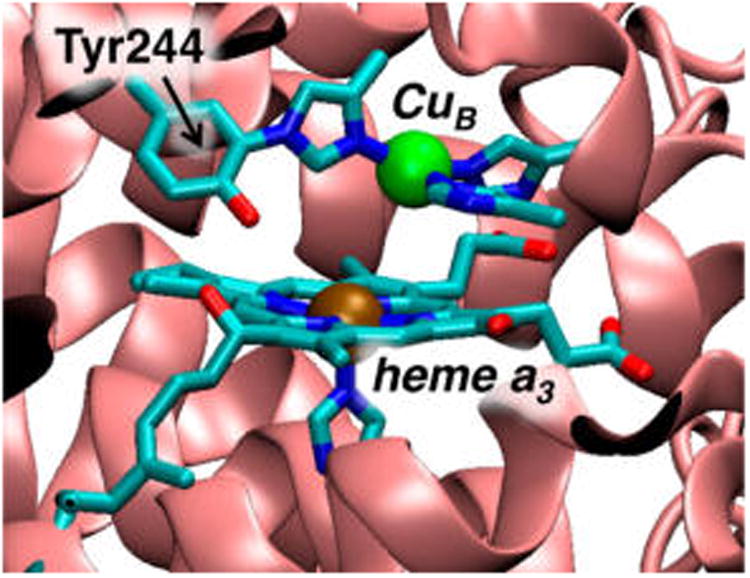
Active-site structure of the heme–copper oxidase CcO from bovine heart cytochrome aa3 (PDB deposition 1V54).
Given the global significance of the reaction, much recent emphasis has focused on developing a detailed understanding of the factors essential to O–O bond cleavage and how breaking the O–O bond drives proton pumping. An extensive set of data from spectroscopic and computational studies has evolved a likely mechanism for breaking the O–O bond in CcO.9 In the consensus mechanism, dioxygen binds to the active-site heme in the reduced state (R, in which the BNC is in the FeII/CuBI state) to generate a superoxo-level intermediate (A),10,11 which rapidly cleaves the O–O bond to generate a highly oxidized state PM consisting of FeIV=O/CuII—OH/Y• moieties (Figure 2). The active-site cross-linked tyrosine is thought to play a critical role by contributing the fourth electron required for reductive cleavage of dioxygen.12 There is, however, little direct evidence as the Y• would be antiferromagnetically (AF)-coupled to the Cu(II) and not detectable.13–16
Figure 2.
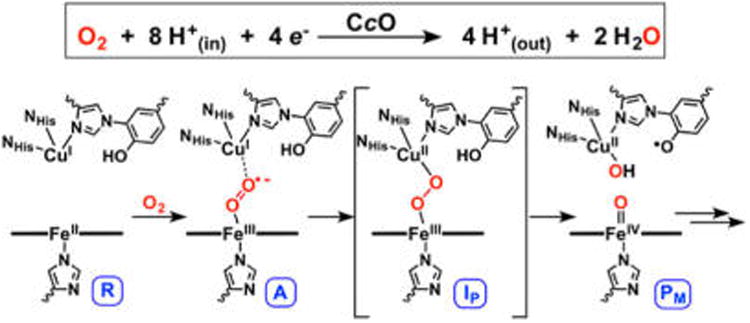
Proposed O–O cleavage mechanism in HCOs, involving O2 reduction via a net H-atom transfer from tyrosine to a peroxo intermediate (IP).
A key matter that remains unsolved is the reaction coordinate connecting intermediates A and PM. While it has not been observed experimentally, the involvement of a peroxo level intermediate (IP in Figure 2) in CcO has been a source of much discussion. Kinetic studies have indicated that if a peroxy intermediate forms in CcO, it must have an O–O cleavage barrier of <12.4 kcal/mol in order to not be observed. Nevertheless, studies of other O2-activating heme enzymes17 and computational studies calibrated to the overall thermodynamics of CcO18,19 support a peroxo level intermediate. In particular, it has been proposed that such a species serves as the active proton acceptor in CcO (rather than the superoxo intermediate, A) during turnover. Given that the terminal (i.e., distal) O-atom of an Fe(III)–superoxo in A would lie close to CuB(I), the putative peroxo species has been formulated as having a bridging FeIII–(μ-1,2-O22−)–CuII structure (IP in Figure 2). As such, a number of heme–peroxo–copper complexes have been synthesized to model this species and understand its reactivity,8,20–26 and many computational studies addressing O–O cleavage in CcO have invoked a bridging peroxo as an energetic local minimum.19,27,28
Throughout the literature, it has generally been regarded that O–O cleavage in CcO involves protonation of the peroxo early in O–O elongation and before the barrier, although the nature of the initial proton source has been disputed.19,29,30 Nevertheless, these studies agree on a reaction coordinate that proceeds through a tyrosyl radical-containing intermediate PM. Thus, while many other details remain contested, the current literature affords a consensus O–O cleavage mechanism that comprises a net H-atom transfer from tyrosine, where the barrier effectively involves cleavage of an Fe–O–O(H)–Cu moiety.
We have recently reported the first example of a reactive heme–peroxo–copper model complex, {[(DCHIm)(F8)-FeIII]–(O22−)–[CuII(AN)]}+ (1, Figure 3, middle; DCHIm = 1,5-dicyclohexylimidazole; F8 = 5,10,15,20-tetrakis(2,6-difluorophenyl)porphyrinate; AN = bis[(3-(dimethylamino)propyl)-amine]),31 which is competent for the biomimetic net H-atom abstraction from exogenous phenol to generate a high-valent (DCHIm)(F8)FeIV=O, CuII(AN)—OH, and phenoxyl radical species.32 Herein we report on the observed reactivity of an additional phenol (4-methoxy-phenol) capable of effecting O–O cleavage of 1, which has afforded valuable product characterization and kinetic data. Considering that the reaction occurs at low temperatures (around −80 °C) and yields products analogous to those proposed for PM, these results support the feasibility of a transient peroxo level intermediate in CcO. Akin to the active-site heme in CcO, 1 is axially coordinated by an imidazole (DCHIm), which generates a low-spin (S = 1/2) ferric heme that is bridged by an end-on peroxide to a cupric (S = 1/2) fragment. Owing to the low-spin nature of the heme, the overall molecule is diamagnetic due to AF coupling between the Fe and Cu through the peroxide bridge.33 In the absence of the strong axial base (DCHIm), the complex has a high-spin (S = 5/2) ferric heme and a cupric fragment bridged by a “side-on” peroxide, and no reaction with phenol occurs (Figure 3).
Figure 3.
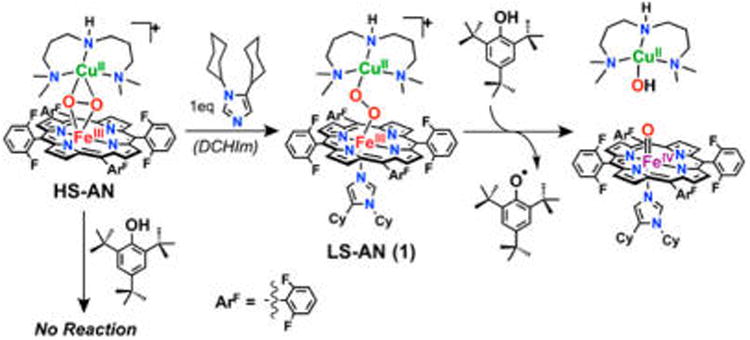
Heme–peroxo–copper complex 1 performs a net H• abstraction from phenol to generate a phenoxyl radical, analogous to the net H• thought to occur in CcO.
We have computationally evaluated the reaction of 1 with phenol using density functional theory (DFT) in order to understand how 1 is activated for reductive O–O bond cleavage and to correlate this to the analogous reaction in CcO. Since the phenols found to accomplish this reaction with 1 to date exhibit similar pKa's and bond dissociation energies, but differ in sterics,32 these calculations employed a simple, unsubstituted phenol. Investigation of the overall reaction surface reveals two possible mechanisms by which the phenol could induce O–O cleavage, providing both a H+ and an e− to fully cleave the bond and generate a phenoxyl radical. The first mechanism, comparable to that of current CcO literature (described above), involves a reaction coordinate wherein the phenolic proton transfers to the peroxo early in O–O cleavage, therefore yielding a transition state (TS) with the proton almost fully transferred to the peroxo (upper path in Figure 4). In the second reaction pathway, proton transfer (PT) occurs primarily after the TS, thus the reaction barrier is best described as O–O homolysis with the phenol remaining H-bonded to the cleaving peroxo (lower path in Figure 4).
Figure 4.
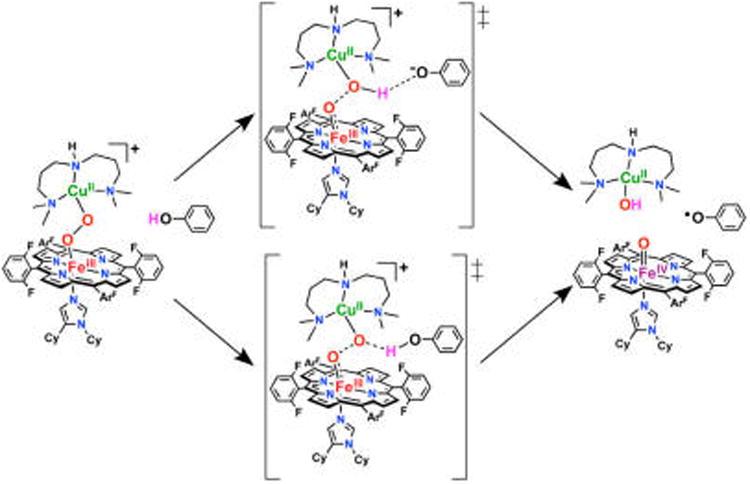
Two possible reaction mechanisms for phenol-induced O–O cleavage of 1, derived from the calculations presented below.
Upon comparing the reaction barriers for these two mechanisms to determine which is kinetically favored by DFT, we found that the relative barrier heights were highly dependent on the density functional employed. As demonstrated herein, this variability arises from differences in covalency given by different DFT methods. In order to resolve which mechanism occurs in the reaction of 1 with phenol ({1+PhOH}), we have carried out a series of kinetic experiments that has enabled us to differentiate between the two mechanisms based on the amount of PT in the TS.
Overall, the aim of the present study is to evaluate via computation and experiment the reaction of 1 with phenol, understand how 1 is activated for reductive O–O bond cleavage, and correlate this reactivity to the analogous reaction in CcO. Identifying two possible mechanisms by which a phenol can induce O–O cleavage in a heme–peroxo–Cu species, we examine the factors governing which mechanism is favored, and consider the implications of these findings. Furthermore, as this reaction involves transfer of both a H+ and an e−, two important components of this study are (1) to investigate their relative timing and (2) to evaluate their respective roles in impacting the barrier and overall thermodynamics. Given the wide variety of H+ and e− donors that are available in biology, we consider possible alternative donors in CcO to understand the importance of the cross-linked Tyr residue for O–O cleavage. A key distinction from the biological systems is that in the reaction of 1 with phenol, the phenol is the only possible source of a proton, providing a well-defined system for the calculation of the overall thermodynamics and the O–O cleavage barrier, thus allowing this system to serve as an effective benchmark comparison for O2 reduction in HCOs.
2. Methods
DFT Calculations
Density functional theory calculations were performed with the electronic structure package Gaussian09, D.01.34 Unless otherwise noted, these calculations were performed using the BP86 and B3LYP functionals within the spin unrestricted formalism, using a THF solvent polarized continuum model. Basis sets employed for geometry optimizations were as follows: 6-311g* on the metals and oxygen, 6-31g* on the metal-bound N-atoms, and 6-31g on all remaining atoms. Single point energies for the docked reactants and TSs were obtained employing 6-311g* on all metals and coordinating heteroatoms, and 6-31g on all remaining (C, F, H) atoms; these yielded nearly identical results to those obtained with the smaller basis set used for optimizations. Density fitting was employed, and the SCF used tight convergence criteria on an ultrafine integration grid. Visualization of orbitals was performed with Lumo.35 Mayer bond orders and Mulliken populations were obtained using QMForge.36 Fe and N atomic orbital populations were obtained from Natural Bonding Orbital analyses (NBO version 3.1)37 contained in the Gaussian09 package.
The previously reported DFT structure of 1 was used as a starting point for these calculations.33 To evaluate the reaction of 1 with phenol, a model containing both reactants was constructed (1·PhOH). An energetic minimum corresponding to a docked reactant was found from optimization of a series of structural guesses with phenol in various positions around the peroxo moiety, and used as the starting point for computational evaluation of the reaction. For both the separated and docked species, the singlet state for 1·PhOH is lower in energy (and remains such over the reaction surface), although the interaction of phenol with the peroxo core weakens the AF coupling between FeIII and CuII, such that the singlet and triplet states become nearly isoenergetic (within 0.5 kcal/mol in ΔE and ΔG). This reaction is therefore presented on the singlet surface. A 3D relaxed potential energy surface (PES) was calculated with axes consisting of the O–O, O⋯H(OPh), and O⋯OPh(H) coordinates, from which successive TS guesses were made by manually perturbing previous guesses along their imaginary frequencies from analytical frequency calculations until a single imaginary frequency remained that involved donating H motion and elongating O–O motion. This procedure allowed convergence to two different TSs, found by TS optimizations of the structural guesses obtained from the 3D PES. Intrinsic reaction coordinate calculations (which require unconstrained TS structures) were performed to connect the TS structures to the docked reactant, and survey the remaining reaction coordinates. All TS structures contained one imaginary frequency, and all reactants and products contained only real frequencies. To correlate with the experimental conditions,32 free energy values for the reaction of {1+PhOH} were calculated at −80 °C. In order to effectively present our results and convey our overall findings, many of the computational details are given in the Supporting Information (SI).
For comparison to the kinetic experiments, the docked reactant and TS structures were reoptimized using 4-methoxyphenol. To improve the accuracy of calculated ΔGǂ and kinetic isotope effects (KIEs), diffuse and polarization functions were applied to O-atoms and the phenolic proton (6-311+g* for O, 6-31++g** for H(OPh), all other atoms were the same as above); qualitatively similar energetics and geometric/electronic structures were obtained without the additional basis functions. Additional details are given in the SI.
General
All reagents and solvents used were of commercially available quality and used without further purification except as noted. Inhibitor-free 2-methyltetrahydrofuran (MeTHF) was distilled over Na/benzophenone under Ar and deoxygenated with Ar before use. [CuI(AN)](BArF) and F8FeII (BArF = B(C6F5)4−; AN = bis(3-(dimethylamino)propyl)-amine; F8 = 5,10,15,20-tetrakis(2,6-difluorophenyl)porphyrinate) were synthesized as previously described.38,39 Deuterated (d-2,3,5,6-OD) 4-methoxyphenol was purchased from CDN Isotopes, and the radical trap, 5-diisopropoxy-phosphoryl-5-methyl-1-pyrroline-N-oxide (DIPPMPO), was purchased from Santa Cruz Biotechnology and stored at −80 °C.
UV–Vis Experiments and Kinetics
All UV–vis measurements were carried out using a Hewlett-Packard 8453 diode array spectrophotometer with a 1 cm path length quartz Schlenk cuvette cell. The spectrometer was equipped with HP Chemstation software and a Unisoku thermostated cell holder for low temperature experiments. For kinetic measurements, spectra were recorded every 3.0 s and fit with the HP Chemstation to obtain first order rate data. The complex 1 was generated at 0.1 mM concentration: A Schlenk cuvette was charged with 3 mL of an equimolar solution of F8FeII and [CuI(AN)]BArF in dry MeTHF in a glovebox under N2 atmosphere and sealed with a rubber septum to maintain an inert reaction atmosphere before removing to be cooled in the UV–vis cryostat chamber to −80 °C. Dioxygen was bubbled through the cold solution generating high-spin, F8FeIII-(O22−)-CuII(AN),32 and excess dioxygen was removed with vacuum/Ar cycles to purge the cuvette headspace while Ar was bubbled through the solution. Addition of 1 equiv of DCHIm via gastight syringe, and mixing via bubbling of Ar, afforded the low-spin complex, 1.26,31 For reactions with 4-OMe-phenol, 1 was generated at −80 °C, and then warmed anaerobically to the desired temperature (between −70 and −77 °C) before adding the desired amount of phenol (10–200 equiv to reach pseudo-first-order conditions), which was added using ≤100 μL (infinite dilution was therefore assumed). Following addition of phenol, spectral features attributed to 1 (λmax = 533, 790 nm) slowly decayed isosbestically to yield the F8FeIII–OH (λmax = 555 nm) product. Kinetic saturation (maximum rate) of the reaction was achieved with 50+ equiv of phenol, where the average rate across >5 trials was (4.18 ± 0.2) × 10−4 s−1 for PhOH and (2.41 ± 0.4) × 10−4 s−1 for PhOD. These allow for calculation of a deuterium KIE of 1.7 ± 0.3. Kinetic data were collected at least in triplicate across four temperatures and a wide range of concentrations, and fit to the Michaelis–Menten model. Tabulated results are given in the SI.
Radical Trapping Experiments with DIPPMPO
The following radical trap experiments are adapted from a previously published procedure.40 A 5 mm outer-diameter NMR tube was prepared in a glovebox under N2 atmosphere with 0.5 mL of a MeTHF solution containing 1:1, F8FeII and [CuI(AN)]BArF, and sealed with a rubber septum and parafilm to maintain an inert reaction atmosphere. The sample was removed from the glovebox and cooled in a −80 °C bath (acetone/LiqN2) and dioxygen was bubbled through the solution, followed by addition of a 25 μL solution containing 1 equiv of DCHIm via gastight syringe to form 1 at 5 mM concentration. Before addition of excess phenol (50 equiv in 50 μL) in the same manner, a 25 μL solution containing 5 equiv DIPPMPO was added and the solution was mixed by bubbling Ar. The sample was allowed to react at −80 °C for 1 h under Ar atmosphere (with intermittent mixing by bubbling Ar) when it was presumed to be complete. The reaction sample was then worked up in one of two ways to obtain either mass spectrometric or 31P NMR resonance analysis (see below and SI, Figure S19).
Electrospray Ionization–Mass Spectrometry
ESI-MS data were acquired using a Finnigan LCQ Duo ion-trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan, San Jose, CA), a heated capillary temperature of 250 °C, and a spray voltage of 5 kV. Samples were prepared by removing the solvent from the DIPPMPO samples described above by rotary evaporation and redissolving the obtained solid in dry methanol. This sample was introduced into the instrument at a rate of 10 uL/min using a syringe pump via a silica capillary line. The phenol dimer, 2,2′- dihydroxy-5,5′-dimethoxybiphenyl, 246.00 m/z (expected: 246.09), as well as phenol-DIPPMPO trapped adducts, 385.49, 374.09, 508.75 m/z were observed by ESI-MS.
Phosphorus NMR
31P NMR spectra were obtained at room temperature using a Bruker Avance 400 MHz FT-NMR spectrometer tuned to optimize for the 31P nucleus (161.978 MHz) and set in proton-decoupled mode, with 1600 scans. A sample of trimethylphosphate (TMP) in dry, degassed THF-d8 was added to the DIPPMPO samples described above prior to spectral acquisition as an internal standard (calibrated to 0 ppm) as well as for instrumental solvent locking purposes. Three new peaks are observed in the product mixture spectrum (6.0, 12.8, 20.6 ppm) indicative of three distinct phenol-trap adducts due to the radical localizing on C2, C4, or the hydroxy O-atom (δPDIPPMPO = 18.5 ppm relative to the internal standard, TMP).
3. Results
3.1. Reaction Coordinate of 1 with Exogenous Phenol
In order to investigate the observed reactivity of 1 toward exogenous phenolic substrates, a model containing both reactants was constructed (1·PhOH) using an unsubstituted phenol. The geometric and electronic structure of 1 has been defined previously (using the BP86 functional, shown to provide good agreement with EXAFS and resonance Raman data),33 and is employed as a starting point for this study. The broken symmetry MS = 0 state, achieved by AF coupling between the low-spin FeIII and CuII ions, is lowest in energy by 1.7 kcal/mol (ΔG) (consistent with experiment). The starting point for surveying this reaction was obtained from an unconstrained optimization of 1 with an approaching PhOH, which yielded an energetic local minimum denoted the docked reactant, D (Figure 5A), that is 7.0 kcal/mol more stable in ΔE than the separated species (3.3 kcal/mol less stable in ΔG, owing to the entropic cost of bringing the components together).41 In the docked reactant structure, the phenol is H-bonded to the peroxo O on the Cu (OCu) with an OCu⋯O(H)Ph separation of ∼2.8 Å.42 The H-bonding interaction induces a slight elongation of the peroxo O–O bond from 1.40 Å (in 1) to 1.43 Å. The low-spin Fe(III) has a doubly-occupied dyz orbital (in the Fe–O–O plane) and a singly occupied dxz orbital (perpendicular to the Fe–O–O plane), with the latter having π overlap with the peroxo π* orbital (by convention the Fe is designated as having α-spin). Note that the dxz and dyz orbitals on Fe have π overlap with the O22− π* and σ* orbitals, respectively (Figure S1).
Figure 5.
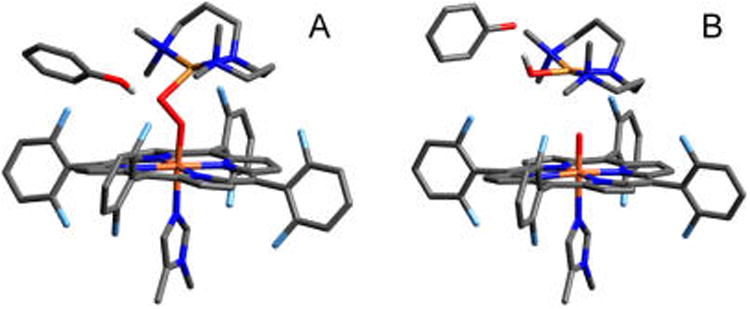
(A) Starting structure (D) for 1·PhOH, having O⋯O, OCu⋯ H(OPh), and OCu⋯OPh(H) distances of 1.43, 1.75, and 2.73 Å, respectively. (B) Product structure (P) for 1·PhOH, with distances of 3.54, 0.99, and 2.79 Å, respectively. H-atoms have been removed for clarity (except the phenolic H).
Given that the experimentally observed products include a phenoxyl radical (vide infra), transfer of a proton and an electron from the phenol to 1, followed by an unconstrained optimization yielded a product structure, P, consisting of three fragments: [(F8)(DCHIm)FeIV=O], [(AN)CuII(OH)]+, and the associated phenoxyl radical (Figure 5B). On the MS = 0 surface, these fragments have spins of MS = 1, MS = −1/2, and MS = −1/2, respectively. Thus, over the course of the reaction, a β electron has transferred from the Fe dyz orbital, and an α electron (and H+) has transferred from the PhOH. This phenoxy-associated product has an energy ΔG = −12.0 kcal/mol relative to the docked reactants (ΔE = −6.1 kcal/mol). Separating the phenoxyl radical yields the overall thermodynamics for the separated reactants to separated products of ΔG = −7.9 kcal/mol (ΔE = −5.3 kcal/mol), comparable to the values obtained for the associated species.
3.1.1. The Overall Reaction Landscape
The PES connecting the reactant and product minima described above was calculated as a function of three coordinates: the OFe—OCu, OCu⋯H(OPh), and OCu⋯OPh distances. Two representative surface slices with the OCu⋯OPh coordinate fixed at 2.4 and 2.6 Å (which are close to the OCu⋯OPh distance for two key saddle points identified on the 3D surface, vide infra) are shown in Figures 6A and 6B, respectively. Examination of the combined potential surface revealed two possible reaction pathways, approximated by the red and blue curves in Figure 6. As can be seen from the figure, one pathway (red curve) involves an initial, nearly complete decrease in the OCu⋯H distance, followed by OFe–OCu elongation, therefore indicating that the H+ transfer to OCu occurs early in O–O cleavage. The second pathway (blue curve) involves nearly complete OFe–OCu elongation followed by the decrease in OCu⋯H, indicating that the H+ transfer occurs late in O–O cleavage. With respect to the current literature on CcO,19,27,43–46 the mechanism depicted by the first pathway is closely representative of the process that has generally been considered in the enzyme. To evaluate which mechanism is kinetically favored in {1+PhOH} (i.e., which has the lowest barrier), a TS was found for each pathway.
Figure 6.
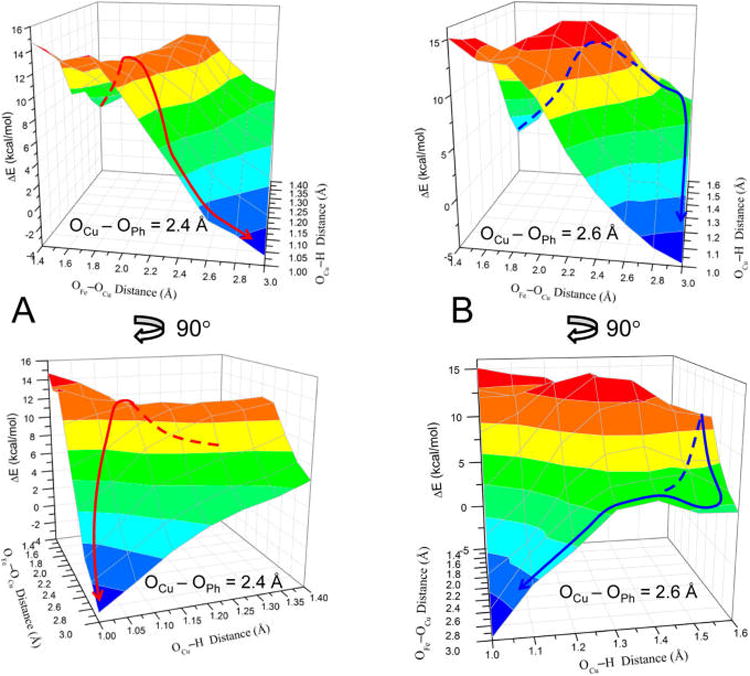
PESs for OFe⋯OCu vs OCu⋯H(OPh) distances (with fixed OCu⋯OPh), where the top/bottom plots are rotated ∼90° from each other to show the full surface. (A) OCu⋯OPh = 2.4 Å; the red line represents an approximate reaction pathway through the proton-initiated TS (TSPI). (B) OCu⋯OPh = 2.6 Å; the blue line represents an approximate reaction pathway through the H-bonded TS (TSHB). All energies are relative to the structure with OCu⋯OPh = 2.6 Å, OFe–OCu = 1.4 Å, and OCu⋯H(OPh) = 1.6 Å (which is set to 0 kcal/mol). Dashed lines are used to indicate that the path is behind the surface.
Using the highest energy point along the first reaction pathway (red curve) as an initial guess, an unconstrained TS was found (shown in Figure 7A) with an energy of ΔGǂ = 7.9 kcal/mol above the docked structure (ΔEǂ = +11.7 kcal/mol, which neglects zero-point corrections to the total energy that lower the barrier by ∼3.5 kcal/mol). This TS structure has the H+ almost completely transferred to the OCu (the OCu⋯H distance is 1.12 Å, relative to 0.99 Å in the Cu–OH product shown in Figure 5B), yet is relatively early in the O–O coordinate with a bond length of 1.88 Å, compared to 1.43 Å in D (Figure 5A). Note that the OCu⋯OPh separation is 2.45 Å, significantly shorter than in D (2.73 Å). The imaginary frequency in the TS is predominantly H motion between OCu and OPh, and to a lesser extent, O–O elongation and phenolate rotation. Importantly, the phenolic electron still resides on the phenolate ring at the TS (from a population analysis, vide infra), indicating only the proton is transferred, rather than an H-atom. Given the nature of the transition state, this mechanism for O–O cleavage is referred to as “proton-initiated” (PI), and the transition state is denoted TSPI.
Figure 7.
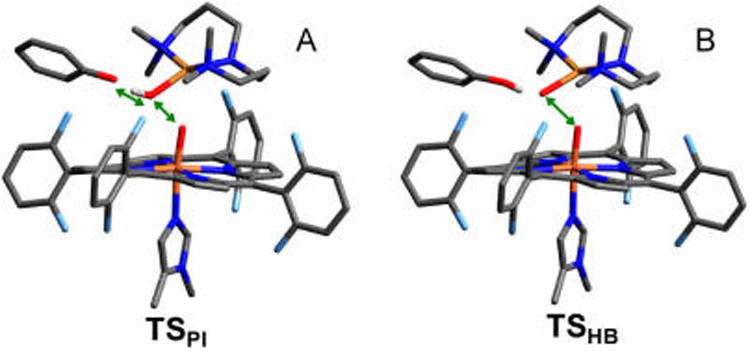
Geometric structures for (A) the transition state along the proton transfer pathway (TSPI), and (B) the transition state along the H-bond-assisted O–O homolysis pathway (TSHB). Green arrows illustrate the dominant motion in the imaginary frequency. Bond distances are given in the SI, Table S1. H-atoms have been removed for clarity (except the phenolic H).
In a similar manner, a TS for the second reaction pathway (blue curve in Figure 6B) was found having an energy of ΔGǂ = +10.0 kcal/mol above the docked reactant (ΔEǂ = +10.9 kcal/mol). The structure of this TS is depicted in Figure 7B, where the H is still predominantly on the phenol (the H–OPh bond length has increased to 1.04 Å, from 1.00 Å in the docked reactant), although the OCu⋯H distance has decreased from 1.75 to 1.54 Å due to the phenol moving closer to the peroxo core (the OCu⋯OPh distance has decreased from 2.73 Å in D to 2.57 Å in the TS). Thus, in this TS the proton has only minimally transferred and the phenol is effectively serving as an H-bond donor as the O–O bond is homolytically cleaved. The imaginary mode in this TS primarily involves O–O elongation, while the phenol moves in concert with OCu, such that the OCu–H distance exhibits very little change. Given the nature of the transition state, this reaction pathway is referred to as “H-bond assisted O–O homolysis” (or “HB”), and the transition state is denoted TSHB.
Comparing the reaction barriers calculated for these two pathways, the PI reaction coordinate is predicted to be kinetically favored, as it is lower in ΔGǂ by 2.1 kcal/mol. However, given that they are fairly similar in energy, and have sufficiently low barriers to be kinetically feasible, we examined each reaction coordinate to understand how and why they differ, and to confirm that both lead to the correct products.
3.1.2. Proton-Initiated O–O Cleavage (PI Mechanism)
The PES for the PI reaction pathway (Figure 8) was generated by following the intrinsic reaction coordinate (IRC) from TSPI in the reverse and forward directions, which yielded D and P, respectively. The reaction coordinate up to the barrier (from D to TSPI, Figure 8) involves three key geometric changes: the OFe–OCu bond lengthens, the Fe–O bond shortens, and the OCu⋯H distance shortens. These changes in bonding are coupled to electron transfer from Fe into the peroxo (predominantly β-spin, yielding an S = 1 ferryl heme) (Figure S3), which derives from the Fe dyz orbital via π–backbonding into the peroxo σ* orbital (Fe dπ(σ*) in Figure 9 and Figure S2). The O–O bond elongation from 1.43 to 1.88 Å parallels the increase in occupation of the σ* orbital (Figure 9B,C) and decrease in O–O Mayer bond order (MBO) (from 0.85 to 0.43, Figure 9A), suggesting that the bond is approximately halfway cleaved of TSHB. Meanwhile, the Fe–O bond shortens from 1.82 to 1.68 Å and the respective MBO increases from 0.68 to 1.11, indicating significant Fe–O double bond character at the TS. Finally, the OCu–H distance shortens from 1.75 to 1.12 Å and the MBO increases from 0.12 to 0.45. This is assisted by the electron density transferred from Fe, which localizes primarily on OCu (Figure S3A,B), thereby increasing the negative charge on OCu and strengthening its interaction with the approaching H(OPh).
Figure 8.
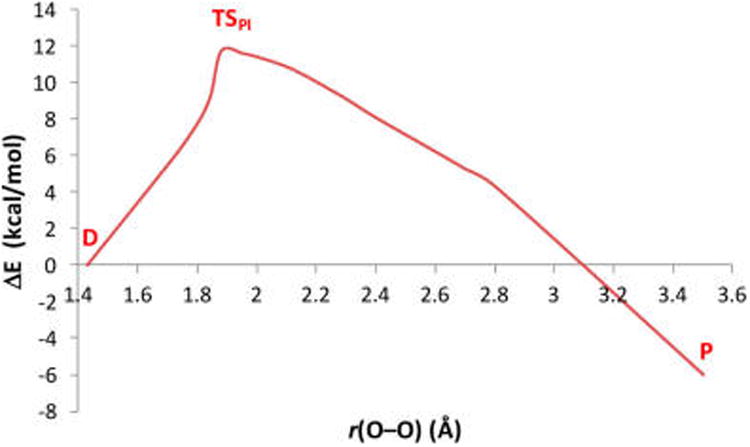
PES vs OFe–OCu distance for the PI pathway, generated by an intrinsic reaction coordinate from the TS (TSPI), with the OCu⋯H and OCu⋯OPh vectors unconstrained. The structure at TSPI is shown in Figure 7A and described in the text.
Figure 9.
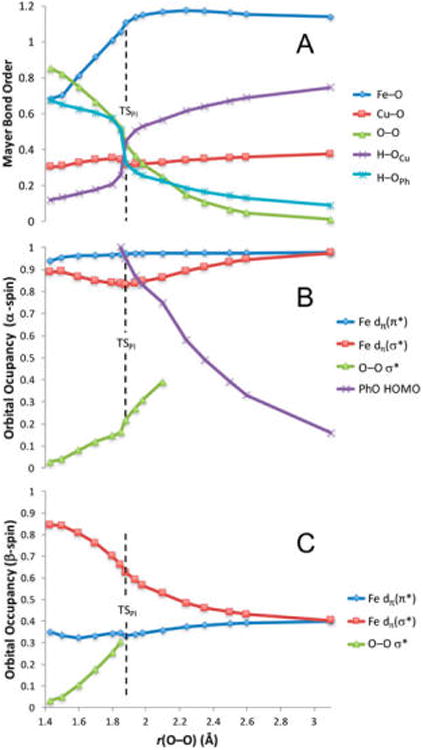
(A) Mayer bond orders for Fe–O, O–O, Cu–O, OCu–H, and OPh–H. Population densities for (B) α-spin and (C) β-spin Fe dπ orbitals, O–O σ*, and PhO− HOMO correlated to O–O distance over the IRC. The Fe dπ orbitals are differentiated by their overlap with the peroxo MOs. O–O σ* orbital occupancy is only reported up to ∼40% occupation. The vertical dashed lines mark the O–O distance in TSPI.
A key characteristic of the TSPI electronic structure is that both α and β HOMOs of phenolate (331α and 331β in Figure S2) are fully occupied, indicating that only the proton, and not the electron, has transferred from the phenol at the TS (Figure S2). Additionally, the charge on the porphyrin ring increases (becomes less negative) prior to the TS, resulting from polarization of the Fe–N σ bonds toward Fe (see Figure S5 and text) to compensate charge donated out of Fe. Taken together, these results establish that Fe is the primary source of e− transfer into the σ*O–O up to the TS. The overall electronic structure of TSPI is therefore best described as FeIV=(O—OH)2−—CuII/PhO−, where the O2 moiety is now three-electron reduced.
The PI reaction coordinate after the TS comprises two key processes: transfer of the second electron required for full reduction of O22−, and completion of the O–O bond cleavage. Since the electron transferred from Fe (which is mostly complete by the TS) has β-spin, the second electron has α-spin. From Figure 9B (also Figure S3B), the α e− derives from phenolate, which transfers into the σ*O–O over the remainder of the reaction coordinate. This involves donation out of the phenolate HOMO, which, following a rotation of the phenol ring (∼30°, see Figure S4), has direct overlap with the σ*O–O through the out-of-plane p(π)-orbital on the phenolate oxygen (depicted in the MO contours in Figure 10). Since this completes O2 reduction, the O–O bond cleaves and the FeIV= O, CuII—OH, and PhO• fragments move apart. An unconstrained optimization thus yields the products shown in Figure 5B, giving an overall ΔG = −12 kcal/mol.
Figure 10.
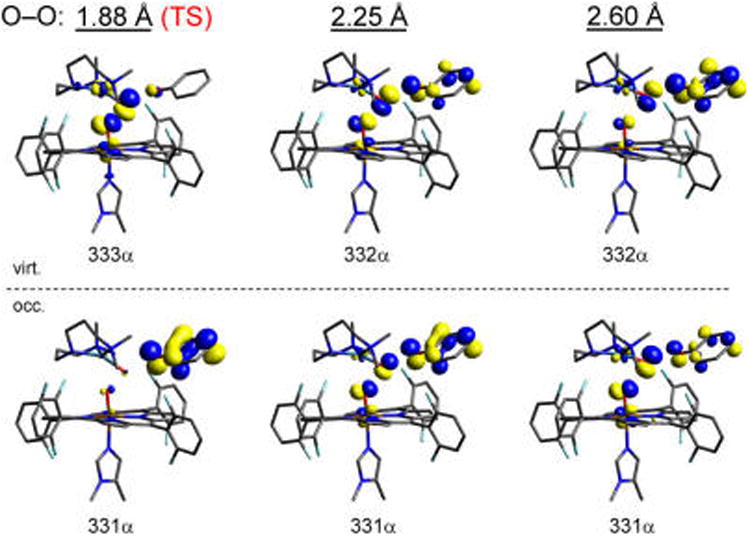
Occupied (bottom) and virtual (top) α-spin molecular orbitals comprised primarily of the Fe dyz, O–O σ*, and PhO− HOMO, shown at increasing O–O distances of 1.88, 2.25, and 2.6 Å. The change in population density illustrates the progressive transfer of an α electron from phenolate (occupied at the TS) into the O–O σ* (which increases in occupation).
3.1.3. H-Bond-Assisted O–O Homolysis (HB Mechanism)
The second pathway for the reaction of {1+PhOH} (blue in Figure 6B) involves O–O bond cleavage with minimal H+ transfer from phenol at the transition state (TSHB, Figure 7B). During the reaction coordinate from D to TSHB (generated from the reverse IRC from TSHB, shown as the blue curve in Figure 11), the O–O bond elongates while the Fe–O and Cu–O bonds shorten, and the phenol moves closer to OCu but remains an H-bond donor. These changes are coupled to the transfer of a β electron from Fe into the σ*O–O orbital, which polarizes toward OCu (similar to the PI reaction pathway, see Figure S7). The O–O bond has elongated from 1.42 Å in D to 2.30 Å in TSHB (longer than the 1.88 Å bond length in TSPI), although the energy rises minimally after reaching an r(O–O) of ∼2.0 Å. Based on an O–O MBO of 0.19 in TSHB (decreased from 0.85 in D, Figure 12), the O–O bond is nearly cleaved (although the fourth e− has not yet transferred). In concert, the Fe–O bond has shortened from 1.82 to 1.66 Å, and the Cu–O bond has shortened from 1.95 to 1.84 Å, with bond orders likewise reflecting much stronger bonds at the TS (Figure 12). In addition, the OCu⋯H distance of 1.54 Å in TSHB indicates that the phenol is H-bonded to the peroxo moiety, though it is worth noting that this interaction strengthens as the reaction proceeds to the TS, as evidenced by the increase in OCu–H MBO from 0.12 to 0.21. The overall electronic structure of TSHB is best described as an FeIV=O/CuII—O•/PhOH, where both metal fragments are triplet species (α for Fe, β for Cu). Due to the orthogonality of the singly occupied orbitals involved (Figure S8), a triplet Cu-oxyl is 3.8 kcal/mol more stable47 than the singlet species that would form on the overall triplet surface.
Figure 11.
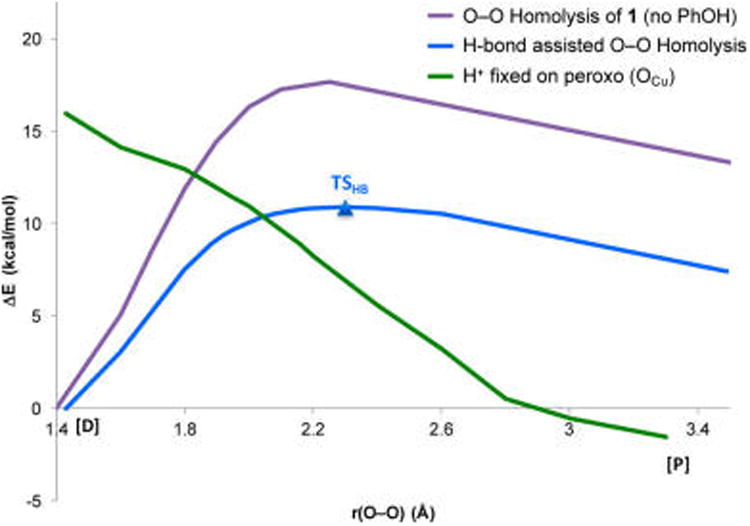
PESs for O–O cleavage without phenol (purple), with phenol H-bonded (blue), and with the phenol H+ transferred to the peroxo (green) vs OFe–OCu separation.
Figure 12.
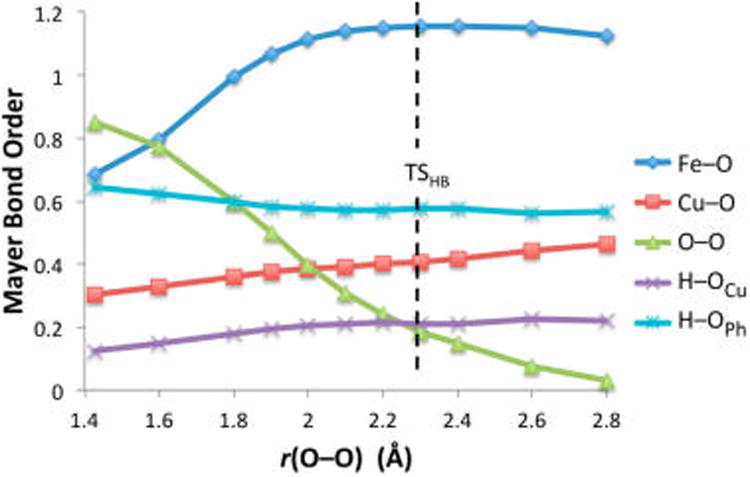
Mayer bond orders along the HB reaction coordinate, without H+/e− transfer from phenol.
Continuing in O–O elongation past TSHB, an IRC calculation in the forward direction yields a structure having a fully cleaved (3.5 Å, far right on blue curve in Figure 11) O–O bond, an FeIV-oxo porphine, a CuII-oxyl, and a phenol still H-bonded to the oxyl. To clarify the electronic structure of this product, we note that oxidizing the porphyrin (to form a porphyrin radical and CuII-oxo) would be 43 kcal/mol higher in energy, and oxidizing the phenol would be 66 kcal/mol higher. Thus, without H+ transfer from phenol, Cu-oxyl formation is driven by thermodynamics. Nevertheless, the energy of this structure is 4.1 kcal/mol (ΔG, 7.4 in ΔE) above D, which confirms that H+ transfer from phenol is necessary for O–O cleavage to become thermodynamically favorable.
Therefore, to investigate the process of transferring the H+ and e− to the Cu-oxyl to reach the favorable phenoxyl radical-containing products that are experimentally observed, an additional PES scan was generated. This involved fixing the proton initially from the phenol on the peroxide OCu in the reactant and elongating O–O bond (Figure 11, green curve). Comparing this to the IRC containing TSHB (Figure 11, blue curve), these surfaces become isoenergetic at r(O–O) ≈2 Å, after which it is thermodynamically favored for the H+ to transfer to OCu. However, transferring the H+ at the crossing point involves an additional barrier (∼1.5 kcal/mol) that is greater than the ∼0.5 kcal/mol required to reach TSHB (at r(O–O) = 2.3 Å), and therefore the lowest energy reaction pathway follows the H-bonding O–O homolysis surface (blue curve). As the O–O bond continues to elongate and it becomes increasingly more favorable for the H+ to reside on OCu, the H+ eventually transfers without contributing to the barrier (see SI for details and evaluation of this process). However, when the H+ can transfer, the energy (along the blue curve) has already reached that of TSHB (which is therefore ΔGǂ). Interestingly, when the H+ is halfway transferred, the phenolate still has <10% radical character (from a Mulliken analysis, Figure S17), indicating that the net H-atom transfer occurs as PT, followed by ET, similar to the “proton-initiated” pathway through TSPI described in section 3.1.2. Following H+ and e− transfer, a full optimization of the resultant structure yielded the final products P in Figure 5B.
Given that this reaction pathway essentially represents O–O homolysis with an H-bond donor interacting with OCu, it is instructive to compare this (blue curve in Figure 11) to O–O homolysis without the phenol present (purple curve in Figure 11). From Figure 11, the H-bond lowers the barrier from 17.5 to 10.0 kcal/mol in ΔGǂ (from 17.6 to 10.9 in ΔEǂ, purple and blue curves, respectively), which is due to the H-bond enhancing electron donation from Fe to promote O–O cleavage (see section 3.1.4 below).
Overall, this reaction pathway illustrates how an H-bond donor can lower the barrier to O–O homolysis by increasing donation from Fe and thereby raising the H+ affinity of the peroxo moiety, enabling fast PT-ET to form the thermodynamically favorable FeIV=O, CuII—OH, and PhO•, where the reaction barrier is defined by the H-bonded O–O homolysis and not the H+/e− transfer. While these calculations yield a slightly higher energy barrier relative to the “PI pathway” in which the proton transfers much earlier in O–O cleavage (10.0 vs 7.9 kcal/mol), both pathways demonstrate that the phenolic proton serves to lower the barrier to O–O cleavage, and that the PT precedes ET, which occurs after the barrier.
3.1.4. Effect of Metal–Ligand Covalency on the Reaction Surface
The above PESs, structures, and thermodynamics were obtained using the pure density functional BP86, which was shown to provide reasonable agreement with structural (EXAFS) and vibrational data at a lower computational cost than a hybrid functional (such as B3LYP). However, it is well documented that introducing the Hartree–Fock (HF)exchange in a hybrid functional will alter the bonding such that metal–ligand (M–L) interactions become less covalent.48–52 Indeed, when compared to BP86, a calculation of 1 in B3LYP yields lower Mayer bond orders for the Fe–O, Fe–N, O–O, and Cu–N interactions (Cu–O is unchanged), along with greater charges on these atoms, demonstrating the less covalent M–L bonding (see Table S2 for comparison of B3LYP and BP86). Applying B3LYP to 1·PhOH yields the same trends in the bonding of the docked reactant structure, D. We therefore examined how the inclusion of 20% HF exchange in a B3LYP calculation qualitatively and quantitatively impacts the {1+PhOH} reaction surface.
As summarized in Table 1, there is a generally good agreement in thermodynamics (ΔG°) calculated using the two functionals. We note that the difference in thermodynamics calculated for the fully optimized interacting products (“Proton-initiated O–O cleavage” in the table) is attributable to the difference in docked product structures predicted by the two functionals (see Figure S9 for details).
Table 1. Comparison of Barriers and Thermodynamics for O–O Cleavage Using BP86 and B3LYP.
| reactionb | ΔGǂ (ΔEǂ)a | G° (ΔE°)a | ||
|---|---|---|---|---|
|
|
|
|||
| BP86 | B3LYP | BP86 | B3LYP | |
| O–O homolysis of 1 (no PhOH, yielding CuII–O•) | 17.5 (17.6) | 20.2 (20.6) | +8.8 (+9.8) | +10.1 (+10.9) |
| H-bond-assisted O–O cleavage (without PT from PhOH, yielding CuII–O•) | 10.0 (10.9) | 16.4 (17.5) | +4.1 (+7.4) | +5.6 (+8.3) |
| Proton-initiated O–O cleavage (yielding P) | 7.9 (11.7) | 24.3 (25.7) | −12.0 (−6.1) | −5.8 (−2.0) |
| {1} + {PhOH} → {FeIV=O + CuII—OH} + {PhO•} | −7.9 (−5.3) | −9.9 (−7.4) | ||
All energies are in kcal/mol, at −80 °C.
Braces indicate separately calculated species.
To systematically evaluate how the barriers and PESs are affected, we first consider the homolytic O–O cleavage without phenol present (plotted as the purple curves in Figures 13A and 13B for B3LYP and BP86, respectively). A comparison between the two functionals reveals that there is only a small energetic difference in the barriers, where B3LYP yields a TS that is 2.7 kcal/mol higher in ΔGǂ (Table 1) and occurs at a shorter O– O distance (1.95 Å, vs 2.3 Å in BP86).
Figure 13.
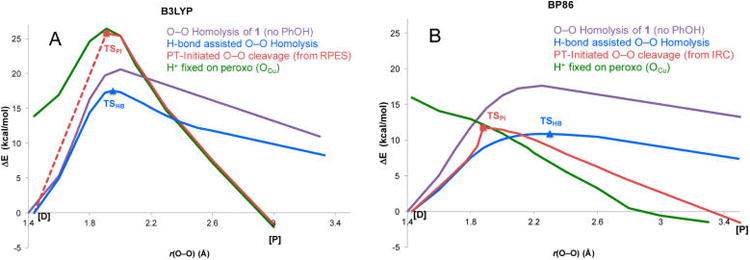
Comparison of B3LYP and BP86 PESs for the O−O homolysis without phenol (purple), PI pathway (red), H-bond assisted O−O homolysis (blue), and the phenolic H+ transferred and optimized on the peroxo (green) vs OFe−OCu separation. Note that the TSPI in B3LYP was obtained by increasing and fixing the OCu−OPh distance to 2.6 Å to prevent the H+ from returning to the phenolate, as an unconstrained transition state search converged to the much lower energy TSHB. Changing the OCu−OPh distance was found to have only a minor effect on the energy when the phenol was deprotonated. The dotted line denotes that the relaxed PES through TSPI requires an additional structural constraint and therefore is not generated from an IRC.
We next evaluated the reaction pathway in {1+PhOH} where the phenol is H-bonded to the cleaving peroxo for the two functionals (blue curves in Figure 13). The H-bond lowers the barrier (relative to O–O homolysis) by only 3.8 kcal/mol in B3LYP, compared to 7.5 kcal/mol in BP86. Since the homolysis of 1 was already slightly higher in B3LYP, the net result is that the barrier to H-bond assisted O–O homolysis (forming the FeIV=O/CuII—O•/PhOH species) is 6.4 kcal/mol higher in B3LYP than BP86, which is due to the difference in covalency (vide infra).
Finally, comparing the barrier to O–O cleavage when PT occurs prior to the homolysis barrier (red curves through TSPI in Figure 13), B3LYP predicts a much higher energy for TSPI (ΔGǂ = 24.3 kcal/mol) than TSHB (ΔGǂ = 16.5 kcal/mol), while in BP86 the TSPI barrier was lowest (2.3 kcal/mol lower than TSHB, vide supra). In fact, the TSPI in B3LYP (in which the H+ is almost completely transferred to OCu) is at sufficiently higher energy that it is unstable to the H+ returning to the phenolate (thus requiring a structural constraint, see Figure 13 caption). Therefore, the lower covalency not only quantitatively changes the barrier heights, but also qualitatively changes the relative barriers and therefore the predicted mechanism, by causing the PI pathway to be higher in energy in the less covalent B3LYP calculation.
Insight into how the interaction with the proton and the change in covalency each affect the barrier to O–O cleavage can be gained from an evaluation of the PESs generated by transferring the H+ to OCu in the reactant and elongating the O–O bond (green curves in Figure 13). Comparing these surfaces shows that in BP86 the barrier to cleave the peroxo O–O bond is effectively removed, while in B3LYP there is still a significant barrier. This is because the interaction with the proton (or H-bond) lowers the energy of the peroxide MOs, therefore allowing easier electron donation from Fe. Since cleaving the O–O bond requires ET from Fe into σ*O–O, this results in a lower barrier.
The more covalent M–L bonding (in BP86) also facilitates donation from Fe into σ*O–O, likewise resulting in lower barriers to O–O cleavage (compared to B3LYP). The difference in the PESs for cleaving the protonated peroxo (green curves in Figure 13), where only the B3LYP calculation shows a barrier, reflects the fact that there is initially little electron transfer from Fe into σ*O–O in B3LYP (from MBO and Mulliken analyses, see SI section 5), while in BP86 the H+ enhances electron flow and therefore accelerates O–O homolysis. Additionally, greater backbonding from Fe (due to higher covalency) increases negative charge on the peroxo, which raises the proton affinity of the OCu-atom relative to the phenolate (making H+ transfer favorable earlier in O–O cleavage).
Overall, the BP86 calculation yields two possible reaction pathways that have energetic barriers defined by TSPI and TSHB, which are similar in energy (with TSPI slightly lower) and lead to the same products. In contrast, upon altering the bonding description to be less covalent by using B3LYP, the latter barrier is significantly lower and therefore the H-bond assisted pathway is favored. The relative barrier heights depend on two competing factors: the degree to which protonation of the peroxo lowers the barrier to O–O cleavage (compared to H-bonding), and the energetic cost of deprotonating the phenol. That is to say, in BP86, protonating the peroxo lowers the barrier to O–O cleavage enough to overcome the proton affinity of the phenolate. Conversely, in B3LYP, protonation of the peroxo does not sufficiently lower the barrier, so the proton transfers later in O–O cleavage, after the O–O homolysis barrier.
3.2. Experimental Evaluation of the O–O Cleavage Mechanism
Given that two DFT functionals (BP86 and B3LYP) provided qualitatively and quantitatively different descriptions of {1+PhOH}, a computational evaluation alone cannot reliably elucidate how an exogenous phenol induces O–O bond cleavage in 1. Therefore, further experimental data were obtained to determine the mechanism by which this occurs and evaluate the reaction barrier.
To first consider which functional more accurately models the covalency of 1, we can turn to spectroscopic data. In our earlier study,33 we found that TD-DFT calculations performed using B3LYP more accurately predicted the O22− to Fe3+ charge-transfer transition and provided good agreement with the resonance Raman profile, indicating that B3LYP offers a better description of the covalency in 1. It was suggested that B3LYP provided better agreement with the data because BP86 predicted the porphyrin orbitals to be too high in energy, resulting in overly covalent Fe-porphyrin bonding.
To evaluate the possible reaction mechanisms presented in section 3.1, we investigated the reaction of 1 with an H-atom-donating phenol, 4-OMe-PhOH, which accomplishes this reaction at low temperature (−70 to −80 °C, where 1 is stable, Scheme 1).53 To verify that the observed reactivity is comparable to the reaction modeled by DFT, we have demonstrated that (1) the phenoxyl radical is formed, (2) the addition of 4-OMe-PhOH accelerates the disappearance of 1, and (3) decomposed 1 is unreactive toward phenol (see SI).
Scheme 1. Reaction of 1 with 4-OMe-PhOH Under Substrate-Saturated Conditionsa,b.
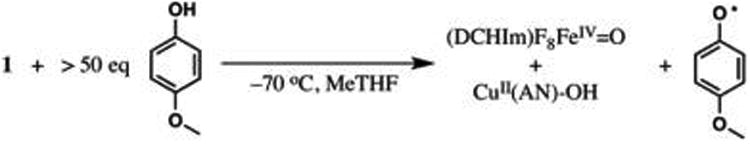
aUnder these conditions, kinetic measurements are independent of [4-OMe-PhOH]. bWe postulate that the (DCHIm)F8FeIV=O product is highly reactive and rapidly abstracts an H-atom from an additional phenol, yielding the FeIII—OH product observed experimentally.
Under substrate-saturated conditions where the rate is independent of phenol concentration (Figure 14), we obtain a reaction rate of kobs = 4.18 × 10−4 s−1 at −70 °C, corresponding to a barrier of ΔGǂ = 14.9 kcal/mol. This value is very similar to the ΔGǂ (14.8 kcal/mol) obtained from an Eyring analysis of the rates measured at −70, −72, −74, and −77 °C (Figure 14B, inset). Accordingly, the reaction barriers for the PI and HB pathways were recalculated (at −70 °C) with 4-OMe-PhOH, yielding values for BP86 (ΔGǂPI = 7.2 kcal/mol, ΔGǂHB = 8.3 kcal/mol) and B3LYP (ΔGǂPI = 20.9 kcal/mol, ΔGǂHB = 16.2 kcal/mol) that were comparable (in both energy and TS geometry) to the unsubstituted phenol used in section 3.1. Note that these values are calculated relative to an optimized “docked” structure, as the experimental rate is saturated in [4-OMe-PhOH] (analogous to Michaelis–Menten conditions in steady-state kinetics).54 Thus, the barrier predicted by BP86 (7.2 kcal/mol) underestimates the experimental value of 14.9 kcal/mol, while the barrier in B3LYP (16.2 kcal/mol) overestimates it.
Figure 14.
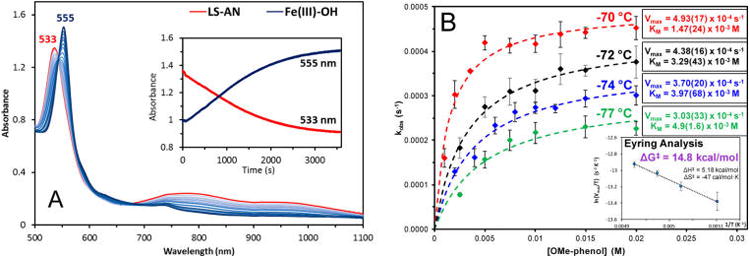
(A) Absorption spectra during the reaction of the LS-AN complex (1) (0.1 mM, −70 °C, MeTHF) (red) with >50 equiv 4-methoxyphenol to form the F8FeIII–OH final products (blue). (A, inset) Time evolution of absorption changes corresponding to 1 (533 nm) and FeIII–OH (555 nm). (B) Plots of initial reaction rate (monitored at 555 nm) dependence on phenol concentration at −70, −72, −74, and −77 °C, including Michaelis–Menten parameters for each temperature. (B, inset) Eyring analysis of the kinetic data, using the Vmax for each temperature.
However, given that the two mechanisms differ qualitatively by the extent of proton transfer from phenol involved in reaching the TS (where one involves predominantly proton transfer, while the other involves minimal proton transfer), these can be experimentally distinguished by KIE measurements. Employing a deuterated 4-OMe-PhOD slows the reaction rate to kobs = 2.41 × 10−4 s−1 at −70 °C, yielding a KIE of kH/kD = 1.7 (Table 2). As expected from the nature of TSPI and TSHB, a larger primary KIE (kH/D = 7.7 in BP86, 10.2 in B3LYP) is calculated for the former, while a smaller secondary KIE (kH/D = 1.6 in BP86, 1.2 in B3LYP) is calculated for the latter, based on the PhOH/PhOD effect on ΔGǂ. Thus, while the barrier calculated for each mechanism varies greatly with functional, a KIE >5 is predicted for the PI mechanism, compared to a KIE <2 for the HB mechanism, independent of functional (Table S7).55 Taken together, these results indicate that the reaction of 1 with 4-OMe-PhOH proceeds via the HB mechanism.
Table 2. Summary of Kinetics Data Using the Reaction Conditions Given in Scheme 1.
| {1 + p-OMe-PhOH} kinetics data (50 equiv, –70 °C)a | |
|---|---|
| kobs(p-OMe-PhOH) (s−1) | 4.18 (0.2) × 10−4 |
| kobs(p-OMe-PhOD) (s−1) | 2.41 (0.4) × 10−4 |
| KIE (kH/kD) | 1.7 (0.3) |
| ΔGǂ (kcal/mol) | 14.9 (0.1) |
Error margins are noted in parentheses. Data for individual trials are given in Table S4.
In the interest of determining a method that accurately reproduces the experimental results, we calculated the barriers to both mechanisms for {1 + 4-OMe-PhOH} using several functionals that are commonly employed for first-row transition metal complexes in the literature and vary in the amount of HF exchange (M06-L, TPSSh, ωB97X-D, and PBE0). These results (Tables S6 and S7) further illustrate the trends observed between BP86 and B3LYP, where an increase in HF exchange yields a higher barrier and more strongly favors the HB mechanism. Based on these calculations, TPSSh and B3LYP appear to most accurately reproduce the experimental barrier, while BP86 provides the closest estimate for the KIE (for the HB mechanism).
3.3. Correlation to CcO
The above calculations and experimental data establish that the transfer of a H+/e− pair from phenol enables favorable O–O bond cleavage in 1. It is therefore valuable to consider how these results relate to cytochrome c oxidase, in which a cross-linked Tyr residue (that is ∼6 Å away from CuB) is widely proposed to participate in O–O cleavage. As a benchmark comparison to {1+PhOH}, we first evaluate the reaction of an exogenous phenol with a bridging peroxo species in CcO, using a model of the active site that includes the cross-linked Tyr (Figure 15). Similar to {1+PhOH}, if the exogenous phenol is allowed to provide both the H+ and e−, the reaction is exergonic by 3.1 kcal/mol (calculated in a dielectric of 4.0 at room temperature), compared to –7.9 kcal/mol for 1. The key implication of this for the cross-linked Tyr residue (i.e., a separated phenol) is that it could induce thermodynamically favorable O–O cleavage in CcO by supplying both the H+ and e−, analogous to the reaction of {1+PhOH}.
Figure 15.
Computational model of the cytochrome c oxidase active site, based on the crystal structure from bovine heart (PDB code 1V54). The α carbons of all included residues are frozen to mimic constraints imposed by the protein backbone.
However, given that the calculations in section 3.1 indicate that the net H-atom transfer from phenol in {1+PhOH} occurs in a stepwise manner as PT followed by ET, independent of the timing of H+ transfer (before or after the barrier), it is important to consider that in CcO, multiple proton and electron donors are available. This is in contrast to {1+PhOH}, where the phenol is the only possible source of the proton. Following PT from phenol to 1, the electron can derive from either the resultant phenolate or the heme fragment. Oxidation of the heme would generate an FeIV=O/porphyrin radical species (called “Compound I”) that is known to form in many porphyrin systems in biology such as cytochrome P450 enzymes and peroxidases, and has even been invoked in some computational studies on CcO.19 A straightforward explanation for why a porphyrin radical species is not observed in {1+PhOH} lies in the thermodynamics of phenolate oxidation versus porphyrin oxidation. Calculation of the ionization potentials for the individual PhO− and [(DCHIm)-(F8)FeIV=O] fragments reveals that the heme is 36 kcal/mol harder to oxidize than the phenolate. Since the PhO− is oriented such that its electron can easily transfer to the peroxy moiety (Figure 10), the thermodynamics govern the reaction products and a Compound I intermediate would therefore not form in {1+PhOH}.
In CcO, while the cross-linked Tyr residue is generally implicated in the O–O cleavage step of the catalytic cycle, the nature of the proton donor remains unknown. However, given that in the reaction of reduced CcO with O2, no proton uptake or translocation to the active site occurs before forming PM (the first intermediate after the O–O bond is cleaved),56,57 the H+ must derive from an amino acid or other donor within the enzyme. Furthermore, since a peroxo-level intermediate is not observed in CcO during the generation of PM, the H+ donor must be near the active site for the protonation and reduction step to occur rapidly. The possible proton sources within a ∼10 Å radius of the active site include Tyr244, Thr309, Thr316, Asp364, His368, Arg438, and propionic acid pendants of heme a3.
Since the source of the fourth electron required for O2 reduction has likewise been disputed in the literature,58,59 we next considered possible electron donors around the active site. Based on thermodynamics (summarized in Table 3 and ref 60), none of the available proton donors are of sufficiently low energy to drive O–O cleavage via oxidation of heme a3 (forming a porphyrin radical),61 indicating that the electron must also derive from a protein residue.62
Table 3. Overall Thermodynamics of O–O Bond Rupture Forming an FeIV=O/Por•/CuII—OH Species in CcO, Employing Several Common Amino Acids and Some Small-Molecule Donors for Reference.
| {[(heme a3)(His)Fe–O2–Cu(His)3]+} + {HA} → {[(heme a3)FeO(His)]+ + [(His)3Cu(OH)]+} + {A−} | |
|---|---|
|
| |
| H+ donor ({HA}) | ΔG° (kcal/mol)a |
| Arg+ | +10.3b |
| Asp | +29.9 |
| His+ | 0b |
| Thr | +55.2 |
| Tyr | +41.6 |
| CH3COOH | +40.6 |
| PhOH | +44.4 |
Calculated in ε = 4.0 at 298 K. Independently calculated components are denoted by braces.
See ref 60.
Examining the crystal structure, the possible electron donors include Trp126, Trp236, and the cross-linked Tyr244, all three of which have been proposed to participate in the redox chemistry of CcO.58,59 Evaluating the thermodynamics for O–O cleavage driven by protonation (from one of the possible H+ donors) and reduction by each of these two amino acids, it was found that the only e− source that enables a favorable reaction is a deprotonated Tyr−. As shown in Table 4, even employing the lowest energy H+ donor that is realistically possible in CcO (Arg+, from Table 3), oxidation of Trp to cleave the O–O bond would be unfavorable by 12.7 kcal/mol (oxidation of a protonated Tyr is an additional 4.5 kcal/mol uphill).66 These results indicate that the deprotonated Tyr residue likely serves as the active reductant during the O–O cleavage step. Importantly, this in turn necessitates that the cross-linked Tyr is deprotonated at the time that the peroxo is cleaved. Furthermore, if Tyr serves as both the H+ and e− donor, O–O cleavage and formation of the tyrosyl radical is favorable by 4.6 kcal/mol, which is in agreement with the ∼4 kcal/mol estimate for the driving force of this step in CcO based on studies calibrated to kinetic data on the enzyme.67 Overall, the thermodynamics indicate that the cross-linked Tyr is required to effectively act as the proton donor in order to generate a tyrosinate, the only accessible electron source that leads to favorable O–O cleavage.
Table 4. Overall Thermodynamics of O–O Bond Rupture in CcO for Different Combinations of Possible e− Source with the Lowest Energy H+ Donor Available (Arg+) and Tyra.
| {[(heme a) (His)Fe–O2-Cu(His)3]+} + {HA} + {E−} → {[(heme a) FeO(His)]+ + [(His)3Cu(OH)]+} + {A−} + {E} | ||
|---|---|---|
|
| ||
| H+ donor ({HA}) | e− donor ({E−}) | ΔG° (kcal/mol) |
| Arg+ | Trp | +12.7 |
| TyrH | +17.2 | |
| Tyr− | −35.9 | |
| Tyr | Trp | +44.0 |
| Tyr− | −4.6 | |
Calculated in ε = 4.0 at 298 K. Note that a carboxylic acid H+ donor yields a ΔG° in between the values obtained for Arg+ and Tyr. Independently calculated components are denoted by braces.
4. Discussion
4.1. Two Possible Mechanisms for Phenol-Induced O–O Cleavage
While a peroxy-level intermediate has not been observed in HCOs, it has often been proposed that such a species serves as the active proton acceptor during the O–O cleavage step of the catalytic cycle (between Intermediates A and PM, see Figure 2).19,45,68 This has led to great interest in model studies of heme–copper complexes, in particular 1, the focus of this study, where a stable peroxo species can be synthesized and spectroscopically characterized. In the presence of a phenolic substrate, 1 (a low-spin, μ-1,2-peroxo complex) undergoes O–O cleavage at low temperatures to yield a phenoxyl radical product.32 This important finding highlights the reactive nature of a low-spin heme–peroxo–copper species with an H-atom-donating phenol (or more generally, a readily accessible H+/e− pair), giving direct relevance to HCOs in which a Tyr residue lies 6 Å away from the O2-binding heme/Cu center and is widely implicated as having a role in the catalytic cycle.
The present investigation of the overall PES for the reaction of 1 with phenol identified two pathways by which an exogenous phenol can induce O–O cleavage (summarized in Figure 16). The first pathway involves PT from phenol early in O–O cleavage (PI pathway), where the proton is almost fully transferred by the TS. The second pathway involves O–O homolysis where the energy barrier is lowered by the phenol H-bonding to the distal O of the peroxo (HB pathway), and the proton transfers after reaching the TS. In both cases, the electron transfer from phenol occurs after the proton has transferred (and after the barrier). This highlights a key element in the O–O cleavage of 1: the interaction with the proton is the primary mode through which phenol impacts the barrier. The relative barriers calculated for these two mechanisms depend on the functional (which tunes the covalency), such that an increase in covalency lowers the PI barrier relative to the HB barrier by enhancing electron transfer from Fe into the peroxo σ* orbital early in O–O cleavage. In fact, upon changing from BP86 to the less-covalent B3LYP, the predicted mechanism shifts from the PI to the HB pathway.
Figure 16.
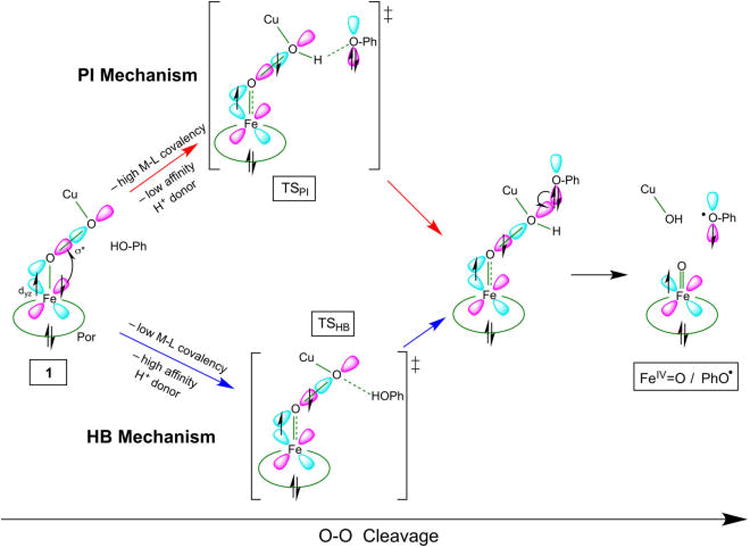
Schematic representation of the two mechanisms for O–O cleavage of 1, where the favored mechanism is dictated by M–L covalency and H+ affinity of the donor. Both mechanisms involve β ET from Fe before the barrier, and α ET from phenolate after the barrier (see text).
Since the calculated barriers vary greatly between functionals, comparison with only an experimental barrier cannot reliably distinguish the correct mechanism (although it can serve to calibrate the DFT-derived energies). However, the two mechanisms differ fundamentally by the degree of proton transfer in the TS, and therefore exhibit different KIEs from PhOH/PhOD substitution. Employing an H-atom-donating phenol (4-OMePhOH) that was shown to accomplish the reaction modeled by our calculations, an experimental KIE of 1.7 was obtained, along with a barrier of ΔGǂexpt = 14.9 kcal/mol (at −70 °C). Compared to the KIEs predicted from DFT (<2 for the HB mechanism, and >5 for the PI mechanism), these data strongly support that the reaction of 1 with 4-OMePhOH proceeds via the H-bond assisted O–O homolysis mechanism.
These results also offer valuable insight for effectively modeling the reactivity in heme-peroxo-Cu systems, as the predicted barriers and KIEs vary between functionals (an effect that has been observed in many studies in the literature).69–72 The B3LYP functional predicts the correct mechanism and provides reasonable agreement with the experimental barrier (ΔGǂcalc = 16.2 kcal/mol, which lowers to ΔGǂcalc = 15.7 kcal/mol with Grimme empirical dispersion (D3) corrections; ΔGǂexp = 14.9 kcal/mol), although it underestimates the observed KIE (1.16 in B3LYP). Note that while a value of 1.7 is large for a secondary KIE, this reaction is measured at low temperatures, which can increase KIEs.73 The BP86 and TPSSh functionals offer the closest agreement with the observed KIE (whether calculated using an H/D free energy difference, or using Streitwieser's approximation for secondary KIEs, see Table S7), which suggests that they most accurately model the nature of the TS (in particular, the strength of the H-bonding interaction). However, unlike BP86, the TPSSh calculations correctly predicted the HB mechanism and provided reasonable agreement with the barrier (ΔGǂcalc = 12.7 kcal/mol).
An interesting consequence of the mechanistic dependence on covalency is that it presents possibilities for tuning the reactivity in heme–peroxo–copper model systems. An increase in the Fe backbonding into σ* would favor the PI mechanism, which can be enhanced by increasing the donation from the trans axial ligand and, to a lesser extent, the porphyrin ring of the heme. Conversely, using a less acidic proton source (HA) would favor the HB mechanism by making the H–A bond cleavage too costly to allow H+ transfer before the O–O homolysis barrier. Furthermore, since only the H+ (but not the e−) has a significant impact in lowering the barrier,74 the H+ and e− could derive from different sources without raising the barrier. A key implication of this is that faster reaction rates may be achieved by employing an exceptionally low energy proton source, without requiring it to also be redox active. Finally, an alternative approach that could alter the reactivity is modification the Cu ligation in such a way that it influences the stability of a Cu-oxyl species. Efforts are currently underway to examine how altering both the ligand scaffold and the proton donor affects the reactivity.
4.2. Correlation to CcO
In CcO, the first intermediate observed following O–O cleavage (Pm) is widely believed to be comprised of an FeIV=O and CuII—OH, implying that a proton and an electron have transferred from an external source after binding O2. Furthermore, kinetic studies have indicated that if a peroxy intermediate forms in CcO, it must have an O–O cleavage barrier of <12.4 kcal/mol at room temperature in order to not be observed.11,75 Considering our results on {1+PhOH} presented herein, this value suggests that the barrier is somehow lowered relative to homolytic cleavage (>20 kcal/mol using the model in Figure 15). However, in contrast to {1+PhOH}, where the phenol is the only proton source available and can approach the bound O2 molecule directly, the O–O cleavage reaction in CcO is more complex because a variety of proton sources are possible, each of which is held >5 Å away from the heme-Cu center by the protein tertiary structure. In CcO, water molecules present around the active site (crystallographically identified in the resting enzyme, and generated during catalytic turnover)76–78 could interact directly with the bound O2 molecule, providing a possible means for PT or hydrogen bonding to facilitate O–O cleavage. Since both mechanisms involve PT before ET, and only the proton impacts the barrier, this suggests that a water molecule interacting with the peroxide could assume the role played by the phenol in {1+PhOH} in lowering the barrier. While the literature has generally focused on a reaction mechanism where the peroxo is already protonated before the barrier, we consider below the process of O–O bond cleavage in CcO within the context of both the PI and HB reaction pathways described for {1+PhOH}.79
In a manner analogous to the PI pathway, water could induce O–O cleavage by transferring a proton to the peroxide before the homolysis barrier. However, while water would act as the initial proton donor, lower energy sources are available near the CcO active site that can serve as the effective proton donor. The timing of proton transfer would then depend on the relative proton affinities of the cleaving peroxo, the OH− (from H2O), and the effective donor. Alternatively, a water molecule could lower the barrier by H-bonding to the peroxo moiety, and then assisting with transferring the proton after the barrier. Thus, both mechanisms suggest that water can lower the barrier to O–O cleavage without contributing to the overall thermodynamics.
While these mechanisms suggest ways in which an active-site water could make O–O cleavage kinetically accessible, a sufficiently low-energy proton/electron pair is required for the reaction to be thermodynamically favorable. Over the range of proton sources available around the active site (Table 3), none is of sufficiently low energy to allow oxidation of the porphyrin ring, necessitating that the fourth electron required to fully reduce O2 derive from an external donor (i.e., an amino acid). Additionally, while other electron donors are possible in CcO (Table 4), only a tyrosinate can yield an energetically favorable net reaction (given the accessible proton donors). In {1+PhOH}, phenolate oxidation is more straightforward after transferring the proton (phenol is the only H+ source), as the resultant phenolate is easier to oxidize than the porphyrin (the only other electron source), and the phenolate is oriented to readily transfer an electron into σ*O–O. This raises two key unresolved questions in CcO: (1) how does the cross-linked tyrosinate electron reach the BNC, and (2) what is the biological function of the covalent linkage that fixes the Tyr >5 Å away? Nevertheless, these results suggest that the similarity between {1+PhOH} and CcO, with respect to phenolate being the electron source, is dictated by thermodynamics.
Considering that the tyrosinate is required to serve as the electron donor in CcO, this residue must be deprotonated prior to fully cleaving the O–O bond. Furthermore, since a proton is delivered to the active site during each of the reduction steps from intermediate F to R, and the cross-linked Tyr residue is widely considered protonated by state R,86 it will therefore be protonated when O2 binds to the active site. Thus, in order for the required electron donor (Tyr−) to be available, the Tyr must effectively serve as the proton donor as well (which has been proposed from FTIR data).30 The net result is that the overall reaction in CcO should closely resemble that of {1+PhOH} in proton plus electron transfer from phenol. It is interesting to consider, however, whether the role of the tyrosyl proton lies primarily in the thermodynamics, or if it also participates in lowering the barrier (i.e., whether the mechanism in CcO is HB or PI).
5. Conclusion
In this study, we have applied DFT calculations to understand the overall potential energy surface of a low-spin heme-peroxo-Cu complex, 1, reacting with phenol, in which a net H-atom transfer from phenol results in cleavage of the O–O bond and formation of FeIV=O, CuII—OH, and PhO• products. These calculations show that the phenol can facilitate O–O cleavage via two possible mechanisms, which differ by the amount of proton transfer prior to the TS. One mechanism involves nearly complete proton transfer from the phenol to the peroxo before the barrier. The second mechanism involves O–O homolysis facilitated by the phenol H-bond to peroxo, with the proton transfer occurring after the barrier. For both mechanisms: (1) the interaction of the proton with the peroxo (via protonation or H-bonding) lowers the barrier to O–O cleavage by increasing donation from Fe into the σ* orbital of the peroxide, (2) electron transfer from phenol occurs after the proton transfer (and after the barrier), and (3) only the proton is involved in lowering the barrier to O–O cleavage. The relative barriers for these mechanisms greatly depend on the M–L bond covalency, and as a result the lowest energy mechanism predicted for {1+PhOH} varies between different density functionals.
Through KIE data for an H-atom-donating phenol inducing O–O cleavage of 1, it is determined that this reaction proceeds via the H-bond-assisted O–O homolysis mechanism (based on the finding of kH/kD = 1.7). The barrier for this process is ∼15 kcal/mol, indicating that the B3LYP and TPSSh functionals provide the best estimate of the reaction barrier.
To correlate the reaction of {1+PhOH} to O–O cleavage in cytochrome c oxidase, it is found that the only accessible electron source capable of yielding favorable O–O cleavage in CcO is the deprotonated nearby tyrosinate of the cross-link. However, given that the tyrosine is protonated when O2 binds to the active site, this residue likely also serves as the proton donor for O–O cleavage in CcO. Considering that water molecules present in the active site could function to impact the barrier in a manner analogous to the phenol in {1+PhOH}, this study offers an excellent opportunity to apply experimentally calibrated DFT to understand how a spectroscopically unobserved peroxo intermediate in CcO can undergo rapid O–O bond cleavage.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) (DK031450 to E.I.S.; GM60353 to K.D.K.). M.K.E. was supported by an NIH postdoctoral fellowship (GM085914). Computational resources were provided in part by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation under grant number ACI-1053575 (TG-CHE130047 to M.K.E. and TG-CHE160096 to E.I.S.).
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b03292.
Additional analysis of DFT results for {1+PhOH} reaction coordinate; characterization of phenoxyl radical product; kinetic data for reaction of 1 with 4-OMephenol; Figures S1–S21 and Tables S1–S8 (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Yoshikawa S, Shimada A. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 3.Cracknell JA, Vincent KA, Armstrong F. A Chem Rev. 2008;108:2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- 4.Kaila VRI, Verkhovsky MI, Wikstrom MA. R Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 5.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 6.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 7.Yano N, Muramoto K, Shimada A, Takemura S, Baba J, Fujisawa H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Tsukihara T, Yoshikawa S. J Biol Chem. 2016;291:23882–23894. doi: 10.1074/jbc.M115.711770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adam SM, Garcia-Bosch I, Schaefer AW, Sharma SK, Siegler MA, Solomon EI, Karlin KD. J Am Chem Soc. 2017;139:472–481. doi: 10.1021/jacs.6b11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han SW, Ching YC, Rousseau DL. Proc Natl Acad Sci U S A. 1990;87:2491–2495. doi: 10.1073/pnas.87.7.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varotsis C, Woodruff WH, Babcock GT, Woodruffi H, Babcock T. J Biol Chem. 1990;265:11131–11136. [PubMed] [Google Scholar]
- 12.Gennis RB. Biochim Biophys Acta, Bioenerg. 1998;1365:241–248. [Google Scholar]
- 13.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 14.White GF, Field S, Marritt S, Oganesyan VS, Gennis RB, Yap LL, Katsonouri A, Thomson AJ. Biochemistry. 2007;46:2355–2363. doi: 10.1021/bi062265h. [DOI] [PubMed] [Google Scholar]
- 15.Iwaki M, Puustinen A, Wikstrom MAR, Rich P. RBiochemistry. 2003;42:8809–8817. doi: 10.1021/bi034522d. [DOI] [PubMed] [Google Scholar]
- 16.Koutsoupakis C, Kolaj-Robin O, Soulimane T, Varotsis CJ. Biol Chem. 2011;286:30600–30605. doi: 10.1074/jbc.M111.252213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR. J Phys Chem A. 2008;112:13172–13179. doi: 10.1021/jp8017875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blomberg M. J Inorg Biochem. 2000;80:261–269. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 19.Blomberg MRA, Siegbahn PEM, Wikstrom MAR. Inorg Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 20.Liu JG, Naruta Y, Tani F. Angew Chem, Int Ed. 2005;44:1836–1840. doi: 10.1002/anie.200462582. [DOI] [PubMed] [Google Scholar]
- 21.Naruta Y, Chishiro T, Shimazaki Y, Tani F, Liu JG, Tachi Y. J Inorg Biochem. 2003;96:36. [Google Scholar]
- 22.Del Río D, Sarangi R, Chufan EE, Karlin KD, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2005;127:11969–11978. doi: 10.1021/ja043374r. [DOI] [PubMed] [Google Scholar]
- 23.Chufan EE, Puiu SC, Karlin KD. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 24.Ghiladi RA, Ju TD, Lee DH, et al. J Am Chem Soc. 1999;121:9885–9886. [Google Scholar]
- 25.Chishiro T, Shimazaki Y, Tani F, Tachi Y, Naruta Y, Karasawa S, Hayami S, Maeda Y. Angew Chem, Int Ed. 2003;42:2788–2791. doi: 10.1002/anie.200351415. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Bosch I, Adam SM, Schaefer AW, Sharma SK, Peterson RL, Solomon EI, Karlin KD. J Am Chem Soc. 2015;137:1032–1035. doi: 10.1021/ja5115198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noodleman L, Han Du WG, Fee JA, Gotz AW, Walker RC. Inorg Chem. 2014;53:6458–6472. doi: 10.1021/ic500363h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshioka Y, Satoh H, Mitani M. J Inorg Biochem. 2007;101:1410–1427. doi: 10.1016/j.jinorgbio.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Proshlyakov DA, Pressler MA, Babcock GT. Proc Natl Acad Sci U S A. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorbikova EA, Belevich I, Wikstrom M, Verkhovsky MI. Proc Natl Acad Sci U S A. 2008;105:10733–10737. doi: 10.1073/pnas.0802512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kieber-Emmons MT, Qayyum MF, Li Y, Halime Z, Hodgson KO, Hedman B, Karlin KD, Solomon EI. Angew Chem, Int Ed. 2012;51:168–172. doi: 10.1002/anie.201104080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halime Z, Kieber-Emmons MT, Qayyum MF, Mondal B, Gandhi T, Puiu SC, Chuf a n EE, Sarjeant AAN, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Inorg Chem. 2010;49:3629–3645. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kieber-Emmons MT, Li Y, Halime Z, Karlin KD, Solomon EI. Inorg Chem. 2011;50:11777–11786. doi: 10.1021/ic2018727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli CJ, Ochterski W, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. revision D.01. [Google Scholar]
- 35.Kieber-Emmons MT. Lumo, Version 1.3. Salt Lake City, UT: 2013. [Google Scholar]
- 36.Tenderholt AL. QMForge, Version 2.4. 2015 [Google Scholar]
- 37.Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO, Version 3.1 [Google Scholar]
- 38.Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbuhler AD, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2002;124:4170–4171. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
- 39.Ghiladi RA, Kretzer RM, Guzei I, Rheingold AL, Neuhold YM, Hatwell KR, Zuberbuhler AD, Karlin KD. Inorg Chem. 2001;40:5754–5767. doi: 10.1021/ic0105866. [DOI] [PubMed] [Google Scholar]
- 40.Zoia L, Argyropoulos DS. J Phys Org Chem. 2009;22:1070–1077. [Google Scholar]
- 41.Note that for comparison to experimental data, the ender-gonicity of “docking” the reactants was removed by measuring the kinetics under conditions where the rate is saturated in phenol concentration, indicating that equilibrium in the reactants is fully shifted to the bound complex.
- 42.Geometry optimizations starting from a structure where the phenol was oriented to H-bond to the peroxo OFe resulted in the phenol either reorienting and binding the OCu or optimizing to a local minimum that was >3 kcal/mol higher in energy than the OCu-bound “docked” reactant. Over the reaction coordinate, the OCu-atom maintained a greater affinity for the phenol.
- 43.Yoshikawa S, Muramoto K, Shinzawa-Itoh K, Aoyama H, Tsukihara T, Ogura T, Shimokata K, Katayama Y, Shimada H. Biochim Biophys Acta, Bioenerg. 2006;1757:395–400. doi: 10.1016/j.bbabio.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 44.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 45.Han Du WG, Götz AW, Yang L, Walker RC, Noodleman L. Phys Chem Chem Phys. 2016;18:21162–21171. doi: 10.1039/c6cp00349d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshioka Y, Mitani M. Bioinorg Chem Appl. 2010;2010:1–18. doi: 10.1155/2010/182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decker A, Solomon EI. Curr Opin Chem Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 48.Atanasov M, Comba P, Martin B, Muller V, Rajaraman G, Rohwer H, Wunderlich S. J Comput Chem. 2006;27:1263–1277. doi: 10.1002/jcc.20412. [DOI] [PubMed] [Google Scholar]
- 49.Niu S, Huang DL, Dau PD, Liu HT, Wang LS, Ichiye T. J Chem Theory Comput. 2014;10:1283–1291. doi: 10.1021/ct400842p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomson NC, Williams KD, Dai X, Sproules S, DeBeer S, Warren TH, Wieghardt K. Chemical Science. 2015;6:2474–2487. doi: 10.1039/c4sc03294b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guell M, Luis JM, Rodríguez-Santiago L, Sodupe M, Sola M. J Phys Chem A. 2009;113:1308–1317. doi: 10.1021/jp8031379. [DOI] [PubMed] [Google Scholar]
- 52.Qayyum MF, Sarangi R, Fujisawa K, Stack TDP, Karlin KD, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2013;135:17417–17431. doi: 10.1021/ja4078717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.4-OMe-PhOH possesses chemical properties comparable to those of PhOH (e.g., pKa and O–H bond dissociation energy) and was found to react with 1 slightly faster than unsubstituted phenol (data not shown) at −70 °C. 4-OMe-PhOH was therefore used for the kinetic studies to minimize competition with autodecomposition of 1 (<5% of observed decay), improve kinetic data quality at lower temperatures, and simplify radical product characterization.
- 54.While phenol binding to a heme–O2–Cu complex has been shown to induce changes in the peroxo core vibrational modes, samples to which >50 equiv of p-OMe-PhOH was added below −80 °C (to minimize product formation) tha were mixed for several minutes and then frozen in liquid N2 showed no perturbation of 1 by rR However, a Van't Hoff analysis of the binding affinity indicates this is due to a low Keq(<10−4) when the samples froze (f.p.(MeTHF) = −136 °C) (see SI).
- 55.These calculations were done on the singlet surface; calculation on the triplet surface yields KIEs similar to those for the singlet, but the barriers for the PI and HB mechanisms are ∼0.5 and ∼2 kcal/mol higher in energy, respectively (using both BP86 and B3LYP).
- 56.Belevich I, Verkhovsky MI, Wikstrom MAR. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 57.Jasaitis A, Verkhovsky MI, Morgan JE, Verkhovskaya ML, Wikstrom M. Biochemistry. 1999;38:2697–2706. doi: 10.1021/bi982275l. [DOI] [PubMed] [Google Scholar]
- 58.Wiertz FGM, Rchter OMH, Ludwig B, de Vries S. J Biol Chem. 2007;282:31580–31591. doi: 10.1074/jbc.M705520200. [DOI] [PubMed] [Google Scholar]
- 59.Yu MA, Egawa T, Shinzawa-Itoh K, Yoshikawa S, Yeh SR, Rousseau DL, Gerfen G. J Biochim Biophys Acta, Bioenerg. 2011;1807:1295–1304. doi: 10.1016/j.bbabio.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.(a) While a protonated His residue is calculated to yield a thermoneutral reaction to form a Compound I-like species, histidine has a pKa ≈ 6–7, yet CcO exhibits fast conversion of A to PM up to pH 9.63,64 Additionally, the only nearby His residue (excluding those bound to a metal) is located ∼10 Å from the BNC. (b) The positively charged donors in Table 3 have a lower energy cost due to the low dielectric of 4 used to model the protein environment. (c) Using the energy of a water-solvated H+ (a recent consensus value is ΔG =-265.9 kcal/mol) to represent uptake from the bulk solution affords thermodynamically favorable Compound I formation (FeIV=O/Por•/CuII—OH in CcO) with a net ΔG =−1.6 kcal/mol; however, noproton uptake is observed in CcO between A and PM.
- 61.While some peroxidase enzymes (which likewise possess a His-ligated heme) do access a FeIV=O/Por• intermediate for O–O cleavage, the proton in this case derives from H2O2, which upon binding to the ferric heme has an extremely low pKa. It is interesting to note, however, that the Compound I in other peroxidases involves a Trp• rather than a Por•. In contrast, P450 enzymes are axially ligated by an anionic thiolate donor, which can stabilize the highly oxidized FeIV=O/Por•.
- 62.O–O bond cleavage still occurs rapidly when O2 binds to the partially reduced “mixed-valent” state, in which heme a (the low-spin heme responsible for ET to the BNC) remains oxidized and therefore cannot serve as the source of the fourth electron.
- 63.Hallén S, Nilsson T. Biochemistry. 1992;31:11853–11859. doi: 10.1021/bi00162a025. [DOI] [PubMed] [Google Scholar]
- 64.Ädelroth P, Ek M, Brzezinski P. Biochim Biophys Acta, Bioenerg. 1998;1367:107–117. doi: 10.1016/s0005-2728(98)00142-x. [DOI] [PubMed] [Google Scholar]
- 65.Kelly CP, Cramer CJ, Truhlar DG. J Phys Chem B. 2006;110:16066–16081. doi: 10.1021/jp063552y. [DOI] [PubMed] [Google Scholar]
- 66.Furthermore, given the substantial number of charged residuesnearby, and the fact that water molecules must be able to move freelyinto and out of the O2 reduction site, a dielectric of 4.0 may representa lower estimate for the local environment. However, increasing to a dielectric of 8.0, the tyrosinate is still the only electron source capable of driving favorable O–O cleavage (Table S8).
- 67.Krab K, Kempe H, Wikstrom M. Biochim Biophys Acta, Bioenerg. 2011;1807:348–358. doi: 10.1016/j.bbabio.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 68.Kaukonen M. J Phys Chem B. 2007;111:12543–12550. doi: 10.1021/jp070578w. [DOI] [PubMed] [Google Scholar]
- 69.Liu F, Proynov E, Yu JG, Furlani TR, Kong J. J Chem Phys. 2012;137:114104. doi: 10.1063/1.4752396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pabis A, Paluch P, Szala J, Paneth P. J Chem Theory Comput. 2009;5:33–36. doi: 10.1021/ct800412h. [DOI] [PubMed] [Google Scholar]
- 71.Guner V, Khuong KS, Leach AG, Lee PS, Bartberger MD, Houk KN. J Phys Chem A. 2003;107:11445–11459. [Google Scholar]
- 72.Goldstein E, Beno B, Houk KN. J Am Chem Soc. 1996;118:6036–6043. [Google Scholar]
- 73.The KIE derives from the zero-point energy contribution to ΔGǂ, which has a greater effect on the rate (larger kH/kD) at lower T.
- 74.Using a redox-inactive proton source (HOAc) in place of phenol for the “H-bonded O–O homolysis” and “H+ fixed on OCu” curves (shown in Figure 13) reproduces the potential surfaces obtained with phenol through the barrier in the HB pathway (i.e., TSHB), with the “H+ fixed on OCu” shifted down in energy due to a slightly lower H+ affinity (see SI, Figure S21).
- 75.Takahashi T, Kuroiwa S, Ogura T, Yoshikawa S. J Am Chem Soc. 2005;127:9970–9971. doi: 10.1021/ja051761a. [DOI] [PubMed] [Google Scholar]
- 76.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Proc Natl Acad Sci U S A. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koepke J, Olkhova E, Angerer H, Muller H, Peng G, Michel H. Biochim Biophys Acta, Bioenerg. 2009;1787:635–645. doi: 10.1016/j.bbabio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 78.Aoyama H, Muramoto K, Shinzawa-Itoh K, Hirata K, Yamashita E, Tsukihara T, Ogura T, Yoshikawa S. Proc Natl Acad Sci U S A. 2009;106:2165–2169. doi: 10.1073/pnas.0806391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.It is worth noting that the porphyrin a1u and a2u HOMOs are known to be highly sensitive to porphyrin ring substitution.80 While this effect has generally been studied with regard to porphyrin radical formation, it is possible that differences in electronic structure between the meso-substituted porphyrin in 1 and biological heme could contribute to differences in reactivity.
- 80.Fujii H. J Am Chem Soc. 1993;115:4641–4648. [Google Scholar]
- 81.While possible functions of the cross-link have been proposed in the literature, which include modulating the binding affinity and geometry of CuB,82 as well as the redox potential and pKa of both Tyr83,84 and CuB,85 its role is not yet fully understood. Therefore, as both questions given in the text represent critical components of HCO chemistry, studies aimed to resolve these issues are currently underway.
- 82.Uchida T, Mogi T, Nakamura H, Kitagawa T. J Biol Chem. 2004;279:53613–53620. doi: 10.1074/jbc.M409719200. [DOI] [PubMed] [Google Scholar]
- 83.Cappuccio JA, Ayala I, Elliott GI, Szundi I, Lewis J, Konopelski JP, Barry BA, Einarsdóttir O. J Am Chem Soc. 2002;124:1750–1760. doi: 10.1021/ja011852h. [DOI] [PubMed] [Google Scholar]
- 84.MacMillan F, Kannt A, Behr J, Prisner T, Michel H. Biochemistry. 1999;38:9179–9184. doi: 10.1021/bi9911987. [DOI] [PubMed] [Google Scholar]
- 85.Kaila VRI, Johansson MP, Sundholm D, Laakkonen L, Wiström MAR. Biochim Biophys Acta, Bioenerg. 2009;1787:221–233. doi: 10.1016/j.bbabio.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 86.Gorbikova EA, Wikstrom M, Verkhovsky MI. J Biol Chem. 2008;283:34907–34912. doi: 10.1074/jbc.M803511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.