

Abstract
The construction of quaternary centers is a common challenge in the synthesis of complex materials and natural products. Current cross-coupling strategies that can be generalized for setting these centers are sparse and, when known, are typically predicated on the use of reactive organometallic reagents. To address this shortcoming a new, photoredox-Ni dual catalytic strategy for the cross-coupling of tertiary organoboron reagents with aryl halides is reported. In addition to details on the cross-coupling scope and limitations, full screening efforts and mechanistic experiments are communicated.
Graphical abstract
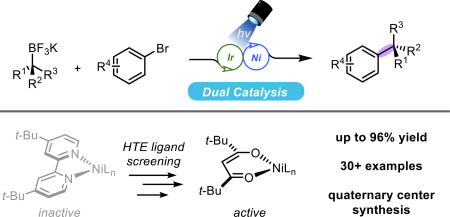
The installation of quaternary centers has been a long-standing challenge going back to the earliest days in the art of steroid synthesis. Woodward and coworkers succeeded in setting some of the first all-carbon stereocenters through cycloaddition chemistry over 60 years ago.1 Although methods have evolved dramatically, there are still relatively few ways to install these centers reliably across a broad array of functionalized partners.2–4 Among the most commonly employed methods, three predominate: 1) alkylation of 1,1-disubstituted allylic leaving groups,5a 2) conjugate addition to α,β-unsaturated carbonyls,5b and 3) alkylation of substituted enolate equivalents.5c In many cases, each of these subclasses can cleanly access the requisite quaternary center, but are constrained by the requirement and natural reactivity pattern for a particular structural element (e.g., α,β-unsaturated carbonyls, allyl, or carbonyl systems). Most notably, none of these methods adequately address the synthesis of arylated quaternary centers.
In addition to these common approaches, a variety of niche metal-catalyzed reactions have also been developed for installing such centers between various nucleophile and electrophile combinations. Key contributions to highlight include Tsuji-Trost allylations,6a Heck couplings,6b Pauson-Khand cyclizations,6c redox relay arylations,6d and carbene C-H insertion reactions.6e These examples are by no means comprehensive, but represent landmark catalytic approaches to the installation of challenging quaternary centers.
Although other metal-mediated catalytic reactions have enabled access to quaternary centers, there are, by contrast, vanishingly few examples of tertiary nucleophiles in classical cross-coupling chemistry. Seminal contributions from the Biscoe7a and Glorius7b groups have highlighted the success of Grignard reagents with N-heterocyclic carbene-ligated nickel complexes. Unfortunately, these strongly basic nucleophiles are poorly tolerant of electrophilic and protic functional groups and are often accompanied by isomerization, generating byproducts when the temperature of these reactions is not carefully controlled. Beyond these seminal reports, there are additional isolated examples using Kumada8 or Negishi9 reagents; however, these do not represent a unified strategy for tertiary cross-couplings. Particularly from a diversification perspective, approaches using these sensitive, pyrophoric reagents are wholly impractical when applied to a wide array of tertiary nucleophiles paired with an equally broad palette of aryl electrophiles. The clear absence of methods predicated on the use of bench-stable partners inspired the laboratory to explore tertiary organoboron nucleophiles more closely – especially given the recent surge in methods for their preparation.10
Studies were initiated using tert-butyltrifluoroborate 13 as the chosen nucleophile with 4’-bromoacetophenone as the aryl electrophile. Application of conditions11 (Figure 1A) successful in previous studies proved completely ineffective for achieving the desired cross-coupling. For all bipyridyl ligands, only starting material or protodehalogenation products were observed. Furthermore, the alkyltrifluoroborate was often minimally consumed after 24 h. Given the lack of conversion under our initially developed conditions, we sought to understand in a more comprehensive manner the divergent reactivity of these tertiary radicals.
Figure 1.

Plausible catalytic cycle and key mechanistic experiments. (A) Previously developed conditions for alkyltrifluoroborate coupling. (B) Oxidation experiment and cyclic voltammetry analysis of 13. (C) Stoichiometric experiments with Ni(II) oxidative addition complex 15.
First, oxidation of the reagent was considered (Figure 1B). Notably, in previous work static quenching of alkyltrifluoroborates in the presence of a cationic photocatalysts has been observed,12 indicating the formation of a salt pre-complex that precedes a single-electron transfer (SET) event. Because of the enhanced steric bulk of these tertiary precursors, it seemed prudent to confirm these reagents were indeed generating radical intermediates. Gratifyingly, both cyclic voltammetry analysis of the alkyltrifluoroborate (Ered1/2 = +1.26 V vs SCE) and Giese-type trapping experiments13 contained the viability of this oxidation, the latter affording the desired alkylated product 14 in 74% yield.
Next, attention was turned to the key bond-forming reductive elimination step in these couplings (Figure 1C). Here, the desired NiIII intermediate was deemed to be accessible through the use of a preformed NiII(dtbbpy) oxidative addition complex 15 in the presence of alkyltrifluoroborate 13 and an appropriate oxidant.14 Surprisingly, stoichiometric experiments exposing this complex to a variety of oxidant combinations in the presence of alkyltrifluoroborate 13 failed to afford any of the desired coupling product even under prolonged reaction times. The only products observed were THF-adduct 18 (known to be generated under the reaction conditions through C-H abstraction15 or halide abstraction pathways16) and protodehalogenation. Given these results, it appeared unlikely that the established bipyridyl ligand systems would be broadly applicable to their tertiary counterparts.
Although daunted by the failures here, we did achieve targeted success with adamantyltrifluoroborate and other constrained ring systems because of their unique structural properties. As a tied back (tetrahedral) radical possessing no sites for possible β-hydride elimination, slight modification of the previously developed conditions allowed access to an array of compounds (Figure 2). Electron-poor (19–23, 25), electron-rich (24) and heteroaryl halides (20) all proved viable. Unfortunately, this rather niche reactivity was not unprecedented17,9a and by no means represented a general solution to tertiary radical coupling.
Figure 2.

Constrained cyclic alkyltrifluoroborates in cross-coupling.
To identify a set of more robust general conditions, a screening effort was initiated to evaluate a wide variety of ligand classes that might be amenable to tertiary radical progenitors. Representative ligands from the Ni cross-coupling literature ranging from phosphines,18 pyridines,19 terpyridines,20 amino alcohols,21 N-heterocyclic carbenes,6 olefins,22 to 1,3-diketones23 were all introduced to both a Ni(0) and Ni(II) source in parallel. The reactions were then followed by UPLC to observe the desired coupling product (see Supporting Information for full details). Across all ligand classes, only diketonate-type species showed significant product formation. Interestingly, these ligands have seen use in mechanistically analogous reductive coupling chemistry reported by the Gong group in 2015.23
Further optimization of this ligand architecture identified a more bulky tetramethylheptanedione (TMHD) scaffold as being suitable for promoting the desired coupling. Additional evaluation of bases showed that the addition of inorganic bases served to reduce protodehalogenation byproducts and increase yield. A solvent screen showed that DMA was essential for coupling success. Use of other polar, aprotic solvents such as DMF, DMSO, or NMP resulted in poorer conversions. Use of ZnBr2 and other Lewis acids were tested as additives to facilitate the coupling. Although these additives were deemed non-essential, rate studies show that these salts reduce the initial induction period observed for the coupling, particularly at larger reaction scales. A summary of the effect of deviations from the optimized conditions can be found in Table 1. It should be noted that although reactions run in front of the low wattage LEDs were sluggish (48–72 h), increasing the light intensity through the use of blue aquarium lamps as in Figure 1, entries 27 and 35, substantially reduced the reaction times (24 h).
Table 1.
Optimized conditions for tert-butyltrifluoroborate cross-coupling and effects of deviation from standard conditions.
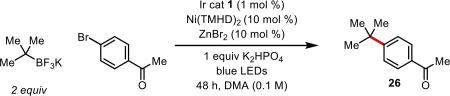
| ||||
|---|---|---|---|---|
|
| ||||
| entry | deviation | % Ar-Bra | % Ar-Ha | % product 26a |
| 1 | no change | <5 | <5 | >95 |
| 2 | no light | >95 | <5 | <5 |
| 3 | no PC | 51 | 12 | <5 |
| 4 | no [Ni] | 88 | 12 | <5 |
| 5 | no K2HPO4 | 15 | 36 | 36 |
| 6 | no ZnBr2 | <5 | 9 | 81 |
| 7 | Ni(COD)2 (10%) Zn(THMD)2 (20%) | 27 | 9 | 57 |
| 8 | NiBr2 (10%) Zn(THMD)2 (20%) | 79 | <5 | 8 |
| 9 | CzIPNb instead of Ir cat. 1 | <5 | 40 | 35 |
HPLC yield with 4,4'-di-tert-butylbiphenyl as internal standard.
CzIPN = 2,4,5,6-Tetra-9 H-carbazol-9-yl-1,3-benzenedicarbonitrile.
With suitable conditions in hand, an evaluation of this coupling across an array of aryl bromides was undertaken (Figure 3). The use of numerous electron-poor aryl bromides resulted in moderate to excellent yields (26–34). Of note, a number of electrophilic functional groups that are intolerant of more reactive Kumada conditions could be employed to afford nitrile 27, aldehyde 28, sulfonyl fluoride 30, Weinreb amide 34, and ketones 26 and 36. A bromo sulfonamide could also be engaged in the coupling to give 31, demonstrating tolerance of some protic functional groups. Product 30 demonstrates electronic tolerance of ortho-substitution; attempts to use a more sterically demanding ortho-methyl substituted bromide, however, resulted in trace conversion. A heteroaryl thiophene and benzofuran were acceptable partners to give products 35 and 36. Unfortunately, the more highly desirous N-heteroaryl bromides were ineffective under the coupling conditions (see 37). At this time, we surmise that the pyridine serves to ligate the metal center competitively, inhibiting the active catalyst. This is supported by doping experiments where addition of 10 mol % of pyridine to the active catalyst mixture arrests conversion to desired product (see Supporting Information).
Figure 3.

Aryl bromide scope in the cross-coupling of potassium tert-butyltrifluoroborate under the developed conditions. ‡Reactions irradiated with four H150 blue LED Kessil lamps.
To evaluate the scope further, an enone-derived alkyltrifluoroborate 38 was used to ease separation of reactions with incomplete conversion (Figure 4). Here, electron-poor systems worked well to give products 39–44. Meta substitution was tolerated, giving compounds 43 and 46 in moderate yields. Overall, the use of electron-neutral bromides resulted in poorer yields (45–46) and truly electron-rich systems such as 4-bromoanisole afforded trace product (47).
Figure 4.

Further aryl bromide scope evaluation with enone derived alkyltrifluoroborate 38
With this survey of electrophiles established, we next turned our attention to more complex tertiary alkyltrifluoroborate precursors, using electron-poor bromides as model electrophiles (Figure 5A). Cyclohexanone- and cyclopentanone- based ring systems could be coupled with an array of substituted aryl bromides (aldehydes, sulfones, ketones) to afford products 48–51. Notably, the products obtained from the coupling of these enone- and enoate-derived trifluoroborates are analogous to those accessed through 1,4 addition chemistry of aryl nucleophiles to α,β-unsaturated carbonyl systems.24 As such, the coupling here represents an umpolung approach to these methods, where the carbon β to the carbonyl has been rendered nucleophilic (Figure 5B). Considering the far greater number of commercially-available aryl electrophiles in comparison to the corresponding aryl nucleophiles, this alternative approach has clear advantages for diversity oriented synthesis. Other functionalized trifluoroborates could be employed to give quaternary centers in products 52–56. Here, ester-, ether-, and ketone-containing trifluoroborates all work well in the established protocol. The incorporation of an amide in a nearby position shut down the reaction, returning only β-hydride elimination and reduction products (see 57), suggesting that changes in the ligation around the nickel have drastic effects on the reaction profile for this tertiary cross-coupling.
Figure 5.

(A) Tertiary alkyltrifluoroborate scope with electron poor aryl bromides. (B) Comparison of enone- and enoate-derived trifluoroborate cross-coupling to metal-catalyzed 1,4-addition chemistry.
In summary, the first general method for the cross-coupling of tertiary organotrifluoroborates has been described. The development of this protocol hinged on the use of photoredox high-throughput screening technology to identify a new ligand architecture amenable to more bulky tertiary radical nucleophiles. Using the conditions identified therein, an array of functionalized, unactivated tertiary organoboron reagents have been employed in transition metal-catalyzed cross-coupling for the first time.25 These reactions generate notoriously challenging to access quaternary centers that are typically forged in cross-coupling through the use of highly reactive Kumada and Negishi partners. In contrast, these mild conditions are tolerant of a number of electrophilically-sensitive functional groups such as aldehydes, ketones, esters, and amides.
At present, a few key limitations of this protocol still remain, as the aryl bromide scope for this cross-coupling is currently limited to electron-poor and electron-neutral systems. From the standpoint of organoboron coupling, this limitation actually serves to complement existing metal-free tertiary coupling work by Aggarwal and others, wherein tertiary alkyl pinacol boronates can be best coupled with electron-rich arene systems.26 The current method also exhibits a notable absence of N-containing heteroaryl partners. In some ways, this failure of heteroaryl systems is mitigated by the successful implementation of tertiary alkyltrifluoroborates in Minisci processes,27 but extension to other electrophilic sites on heteroaryl compounds would add significant value. Further refinement to the ligand scaffold and conditions will likely address these two limitations, as well as the ortho-substitution challenge; efforts in the laboratory toward this end are an ongoing endeavor.
Given the growing suite of methods for the synthesis of tertiary organoboron compounds,10 this photoredox cross-coupling serves as a timely addition, enabling tertiary organoboron reagents as partners in the installation of arylated quaternary centers. Although limitations remain, the ligand screening data and mechanistic interrogation will allow practitioners seeking to employ other tertiary radical feedstocks (carboxylic acids, halides, aldehydes, silicates, etc.) to channel these lessons into developing new tertiary alkyl cross-couplings of broad interest. Taken together, these findings introduce tertiary alkyl radicals to the photoredox cross-coupling portfolio.
Supplementary Material
Acknowledgments
We thank NIGMS (R01 GM113878) for support of this research. We thank the NIH (S10 OD011980) for supporting the University of Pennsylvania (UPenn) Merck Center for High-Throughput Experimentation, which funded the equipment used in screening efforts.
Footnotes
ASSOCIATED CONTENT
Experimental details and spectra data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
References
- 1.Woodward RB, Sondheimer F, Taub D, Heusler K, McLamore WM. J. Am. Chem. Soc. 1952;74:4223. [Google Scholar]
- 2.Quasdorf KW, Overman LE. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das JP, Marek I, Lam JK, Wang D-H, Yu J-Q, Monde K, Harada N, Harada N. Chem. Commun. 2011;47:4593. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]
- 4.Christoffers J, Baro A. Adv. Synth. Catal. 2005;347:1473. [Google Scholar]
- 5.(a) Wu C, Yue G, Nielsen CD-T, Xu K, Hirao H, Zhou J. J. Am. Chem. Soc. 2016;138:742. doi: 10.1021/jacs.5b11441. [DOI] [PubMed] [Google Scholar]; (b) d'Augustin M, Palais L, Alexakis A. Angew. Chem. Int. Ed. 2005;44:1376. doi: 10.1002/anie.200462137. [DOI] [PubMed] [Google Scholar]; (c) Minko Y, Marek I. Chem. Commun. 2014;50:12597. doi: 10.1039/c4cc04391j. [DOI] [PubMed] [Google Scholar]
- 6.(a) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. Int. Ed. 2005;44:6924. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; (b) Atsuyuki A, Bachand B, Overman LE, Poon DJ. J. Am. Chem. Soc. 1998;120:6477. [Google Scholar]; (c) Jiang B, Xu M. Angew. Chem. Int. Ed. 2004;43:2543. doi: 10.1002/anie.200353583. [DOI] [PubMed] [Google Scholar]; (d) Mei T-S, Patel HH, Sigman MS. Nature. 2014;508:340. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kubialc RW, Mighion JD, Wilkerson-Hill SM, Alford JS, Yoshidomi T, Davies HML. Org. Lett. 2016;18:3118. doi: 10.1021/acs.orglett.6b01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Joshi-Pangu A, Wang C-Y, Biscoe MR. J. Am. Chem. Soc. 2011;133:8478. doi: 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lohre C, Dröge T, Wang C, Glorius F. Chem. -A Eur. J. 2011;17:6052. doi: 10.1002/chem.201100909. [DOI] [PubMed] [Google Scholar]
- 8.(a) Nugent WA. Org. Lett. 2002;4:2133. doi: 10.1021/ol0259488. [DOI] [PubMed] [Google Scholar]; (b) Hayashi T, Konishi M, Yokota K-I, Kumada M. Chem. Lett. 1980:767. [Google Scholar]; (c) Hintermann L, Xiao L, Labonne A. Angew. Chem. Int. Ed. 2008;47:8246. doi: 10.1002/anie.200803312. [DOI] [PubMed] [Google Scholar]
- 9.(a) Sämann C, Dhayalan V, Schreiner PR, Knochel P. Org. Lett. 2014;16:2418. doi: 10.1021/ol500781j. [DOI] [PubMed] [Google Scholar]; (b) Gurung SK, Thapa S, Kafle A, Dickie DA, Giri R. Org. Lett. 2014;16:1264. doi: 10.1021/ol500310u. [DOI] [PubMed] [Google Scholar]
- 10.(a) Silvi M, Sandford C, Aggarwal VK. J. Am. Chem. Soc. 2017;139:5736. doi: 10.1021/jacs.7b02569. [DOI] [PubMed] [Google Scholar]; (b) Li C, Wang J, Barton LM, Yu S, Tian M, Peters DS, Kumar M, Yu AW, Johnson KA, Chatterjee AK, Yan M, Baran PS. Science. 2017 doi: 10.1126/science.aam7355. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lo JC, Gui J, Yabe Y, Pan C-M, Baran PS. Nature. 2014;516:343. doi: 10.1038/nature14006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kerchner HA, Montgomery J. Org. Lett. 2016;18:5760. doi: 10.1021/acs.orglett.6b03090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Atack TC, Cook SP. J. Am. Chem. Soc. 2016;138:6139. doi: 10.1021/jacs.6b03157. [DOI] [PubMed] [Google Scholar]; (f) Kischkewitz M, Okamoto K, Mück-Lichtenfeld C, Studer A. Science. 2017;355:936. doi: 10.1126/science.aal3803. [DOI] [PubMed] [Google Scholar]
- 11.(a) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Primer DN, Karakaya I, Tellis JC, Molander GA. J. Am. Chem. Soc. 2015;137:2195. doi: 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsui JK, Molander GA. Org. Lett. 2017;19:950. doi: 10.1021/acs.orglett.7b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasu Y, Koike T, Akita M. Adv. Synth. Catal. 2002;354:3414. [Google Scholar]
- 14.Experiments using Cy-BF3K with oxidative addition complex (15) result in product despite its enhanced sterics. Note that only one mechanistic pathway is represented with this experiment, but as reduction products are observed, we strongly believe that oxidative addition is not rate limiting. However, it is also possible that the radical addition to NiII could be rate limiting instead, for supporting studies see Supporting Information.
- 15.(a) Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]; (b) Laar-hoven LJJ, Mulder P. J. Phys. Chem. B. 1997;101:73. [Google Scholar]
- 16.(a) Heitz DR, Tellis JC, Molander GA. J. Am. Chem. Soc. 2016;138:12715. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shields BJ, Doyle AG. J. Am. Chem. Soc. 2016;138:12719. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.A single example of tert-butyl radical coupling has been reported previously: Zhang P, Le C, MacMillan DWC. J. Am. Chem. Soc. 2016;138:8084. doi: 10.1021/jacs.6b04818.
- 18.Yu D-G, Wang X, Zhu R-Y, Luo S, Zhang X-B, Wang B-Q, Wang L, Shi Z-J. J. Am. Chem. Soc. 2012;134:14638. doi: 10.1021/ja307045r. [DOI] [PubMed] [Google Scholar]
- 19.Liao L-Y, Kong X-R, Duan X-F. J. Org. Chem. 2014;79:777. doi: 10.1021/jo402084m. [DOI] [PubMed] [Google Scholar]
- 20.Joshi-Pangu A, Ganesh M, Biscoe MR. Org. Lett. 2011;13:1218. doi: 10.1021/ol200098d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González-Bobes F, Fu GC. J. Am. Chem. Soc. 2006;128:5360. doi: 10.1021/ja0613761. [DOI] [PubMed] [Google Scholar]
- 22.Huang C-Y, Doyle AG. J. Am. Chem. Soc. 2012;134:9541. doi: 10.1021/ja3013825. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Wang S, Xue W, Gong H. J. Am. Chem. Soc. 2015;137:11562. doi: 10.1021/jacs.5b06255. [DOI] [PubMed] [Google Scholar]
- 24.Shockley SE, Holder JC, Stoltz BM. Org. Process Res. Dev. 2015;19:974. doi: 10.1021/acs.oprd.5b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The use of tertiary cyclopropyltrifluoroborates has been achieved previously: Harris MR, Li Q, Lian Y, Xiao J, Londregan AT. Org. Lett. 2017;19:2450. doi: 10.1021/acs.orglett.7b01097.
- 26.Sandford C, Aggarwal YK. Chem. Commun. 2017;53:5481. doi: 10.1039/c7cc01254c. [DOI] [PubMed] [Google Scholar]
- 27.Matsui JK, Primer DN, Molander GA. Chem. Sci. 2017;8:3512. doi: 10.1039/c7sc00283a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.