

Abstract
Nitrogen-containing bisphosphonates have anti-tumor activity in certain breast cancer and myeloma patients. However, these drugs have limited oral absorption, tumor cell entry and activity, and cause bone side effects. The potencies of phosphorylated antiviral drugs have been increased by administering them as prodrugs in which the negative charges on the phosphate moieties are masked to make them lipophilic. We synthesized heterocyclic bisphosphonate (BP) prodrugs in which the phosphonate moieties are derivatized with pivaloyloxymethyl (pivoxil) groups and that lack the hydroxyl “bone hook” on the geminal carbon. When the lipophilic BP prodrugs enter tumor cells, they are converted into their active forms by intracellular esterases. The most active prodrug, tetrakispivaloyloxymethyl 2-(thiazole-2-ylamino)ethylidene-1,1- bisphosphonate (7), was found to potently inhibit the in vitro growth of a variety of tumor cell lines, especially hematopoietic cell lines, at nanomolar concentrations. Consistent with this fact, compound 7 inhibited the prenylation of the RAP1A small GTPase signaling protein at concentrations as low as 1-10 nM. In preclinical studies, 7 slowed the growth of human bladder cancer cells in an immunodeficient mouse model. Thus, 7 is significantly more active than zoledronic acid, the most active FDA-approved BP, and a potential anti-cancer therapeutic.
Keywords: bisphosphonate, lipophilic, prodrug, anti-cancer, farnesyl diphosphate synthase
Graphical abstract
A novel bisphosphonate prodrug that has its P-C-P group esterified with pivaloyloxymethyl shows potent in vitro and in vivo activity against tumor cells, especially lymphoma and myeloid leukemia hematopoietic cells and certain solid tumor cells.
Introduction
Geminal bisphosphonates (BPs) are synthetic compounds containing P-C-P groups that are metabolically stable analogs of diphosphate.[1] BPs were first synthesized more than 100 years ago and have a number of industrial uses.[2] Because BPs bind to bone hydroxyapatite[3] and inhibit bone resorption,[4] a variety of BPs have been developed as therapeutics for the treatment of bone-related diseases, such as Paget's disease, osteoporosis, and hypercalcemia of malignancy.[5]
First generation BPs are simple non-nitrogen-containing compounds, such as etidronate and clodronate, that can be metabolically incorporated into a β-γ-methylene analog of ATP. The non-hydrolyzable ATP analog inhibits ATP-dependent intracellular enzymes like mitochondrial ADP/ATP translocase, leading to cell death.[6] Second generation BPs, such as pamidronate and alendronate, contain alkylamine moieties and function to inhibit farnesyl diphosphate synthase (FDPS) in the mevalonate pathway, blocking the synthesis of downstream farnesyl and geranylgeranyl diphosphate metabolites. Transfer of the aliphatic chains of the diphosphates is required for the activation of small guanosine triphosphate (GTP)-binding proteins, such as Ras, Rho, Rac, Rab, and Rap, because the aliphatic chains function to anchor the proteins to the cell membrane for signaling.These GTP-binding signaling proteins are essential for the proliferation, survival, and migration of cells. Release of BPs from bone and uptake into osteoclasts causes osteoclast apoptosis and decreases bone resorption.[7-9] Third generation BPs, including risedronate and zoledronic acid (Zol) that have nitrogen-containing heterocyclic moieties, exhibit more potent antiresorptive activity than second generation BPs through stronger inhibition of FDPS.[10]
The addition of Zol to standard treatments for breast cancer and multiple myeloma improved disease-free survival and overall survival, respectively.[11-15] These improvements appeared to be independent of preventing bone-related events, suggesting that BPs have anti-tumor activity besides preventing bone metastases. Our goal was to develop BPs specifically designed as cancer therapeutics for improved activity. However, heterocyclic BPs, such as Zol and minodronate, fit well into the active site of FDPS[16] making it difficult to design BPs with higher activity.
An alternative approach to increase BP activity is to improve BP cell entry. Cellular uptake of BPs through fluid-phase endocytosis is enhanced by Ca2+ suggesting that the negatively-charged, hydrophilic P-C-P structure limits BP entry into cellular vesicles.[17] Oldfield and co-workers have reported several approaches to improving cellular uptake through the addition of a 9-12 carbon acyl chain to Zol and other BPs[18, 19] or by masking the negatively-charged P-C-P structure with pivoxil esters.[20] For Zol, addition of a 9-12 carbon acyl chain decreases FDPS inhibition by 2- to 3-fold while increasing activity in cellular assays by up to 22-fold.[19] Similarly, the pivoxil derivatives of mono- and dialky-BPs that inhibit FDPS or geranylgeranyl diphosphate synthase (GGDPS) were consistently more potent than their acid forms at inhibiting the prenylation of RAP1A in K562 cells and K562 proliferation.[21] In this study, we have increased the cellular uptake and activity of nitrogen-containing BPs by masking the phosphonate charges with pivoxil groups resulting in novel BP prodrugs. These prodrugs have greatly increased bioactivity against hematopoietic tumor cells.
Results and Discussion
We synthesized BP prodrugs where the phosphonate charges are masked by pivoxil groups, and evaluated the effect of pivoxil addition on their biological activity in vitro and in vivo. We first synthesized seven BP prodrugs and their corresponding active acid forms (Figure 1). Five of the pairs (2-6/9-13) were based on NE11809 (compound 8)[22, 23] by varying the position of the methyl group on the heterocyclic ring, by the addition of a halogen group, or by addition of a nitrogen atom to the heterocyclic ring. A final compound substituted a thiazole for the pyridine group. The synthesis of BP pivoxil derivatives is detailed in Supporting Information, Figure S1. All of the BP prodrugs inhibited the growth of U937, a human histiocytic lymphoma cell line, more efficiently than their acid forms (Table 1 and Supporting Information, Figure S2). For example, compound 7 was found to be 100-fold more potent than its acid form, 14, (IC50 against U937 was 260 nM for 7 versus 26,000 nM for 14). 1 inhibited U937 cells 185-fold more efficiently than its acid form, 8. However, the effect of pivoxil-derivatization of BPs was variable ranging from 5- to 1,618-fold (mean increase in potency was 291- ± 588-fold). Similar increases in activity of BP pivoxil derivatives compared with their acid forms were noted with EJ-1 bladder carcinoma cells (Table 2 and Supporting Information, Figure S3). Again, the effect was variable depending on the BP ranging from 1.2- to 422-fold (mean increase was 123- ± 163-fold). For EJ-1, 7 inhibited 142-fold more than 14. Given the high activity of 7, growth inhibition by 7 and 14 were determined for cell lines derived from solid tumors. Again, 7 was 28- to 455-fold more potent than its acid form, 14 (Table 3 and Supporting Information, Figure S4). Similarly, growth inhibition of hematopoietic cell lines was enhanced by pivoxil protection with 7 being 100- to 3,714-fold more active than 14 (Table 4 and Supporting Information, Figure S5).
Figure 1.

Structure of pivoxil esters and their acid forms. Details of synthesis are summarized in the Experimental Section.
Table 1.
Comparison of pivoxil esters to their acid forms in growth inhibition of the U937 histiocytic lymphoma cell line.a
| Pivoxil : H # | Pivoxil IC50 : H IC50 (μM) | Ratio IC50 Pivoxil : IC50 H |
|---|---|---|
| 1 : 8 | 5.3 ± 0.4 : 980 ± 80 | 1 : 185 |
| 2 : 9 | 0.68 ± 0.05 : 1,100 ± 200 | 1: 1,618 |
| 3 : 10 | 5.6 ± 0.7 : 29 ± 1 | 1 : 5 |
| 4 : 11 | 3.6 ± 0.3 : 200 ± 3 | 1 : 56 |
| 5 : 12 | 5.3 ± 0.4 : 68 ± 1 | 1 : 13 |
| 6 : 13 | 2.4 ± 0.0 : 150 ± 6 | 1 : 63 |
| 7 : 14 | 0.26 ± 0.01 : 26 ± 1 | 1 : 100 |
Experimental details are described in Supporting Information, Figure S2.
Table 2.
Comparison of pivoxil esters to their acid forms in growth inhibition of the EJ-1 bladder carcinoma cell line.a
| Pivoxil : H # | Pivoxil IC50 : H IC50 (μM) | Ratio IC50 Pivoxil : IC50 H |
|---|---|---|
| 1 : 8 | 11 ± 0 : 13 ± 2 | 1 : 1.2 |
| 2 : 9 | 2.3 ± 0.1 : 970 ± 30 | 1 : 422 |
| 3 : 10 | 5.4 ± 0.6 : 7.9 ± 0.6 | 1 : 1.5 |
| 4 : 11 | 7.8 ± 0.2 : 290 ± 10 | 1 : 37 |
| 5 : 12 | 5.7 ± 1.3 : 22 ± 2 | 1 : 3.9 |
| 6 : 13 | 0.09 ± 0.01 : 23 ± 2 | 1 : 256 |
| 7 : 14 | 0.026 ± 0.014 : 3.7 ± 0.2 | 1 : 142 |
Experimental details are described in Supporting Information, Figure S3.
Table 3.
Comparison of 7 (pivoxil ester) to 14 (acid form) in growth inhibition of solid tumor cell lines.a
| Tumor cell line | Origin | Pivoxil IC50 : H IC50 (μM) | Ratio IC50 Pivoxil : IC50 H |
|---|---|---|---|
| 786-0 | Renal cell carcinoma | 0.88 ± 0.15 : 25 ± 1 | 1 : 28 |
| MKN1 | Gastric carcinoma | 1.2 ± 0.8 : 100 ± 60 | 1 : 83 |
| OST | Osteosarcoma | 0.81± 0.20 : 66 ± 5 | 1 : 81 |
| PC-3 | Prostate carcinoma | 0.22 ± 0.06 : 100 ± 20 | 1 : 455 |
| PK1 | Pancreatic cancer | 0.34 ± 0.06 : 110 ± 20 | 1 : 324 |
| LK-2 | Squamous non-small cell lung carcinoma | 0.80 ± 0.33 : 35 ± 1 | 1 : 44 |
| G-361 | Melanoma | 0.12 ± 0.02 : 20 ± 4 | 1 : 167 |
| TFK-1 | Cholangiocarcinoma | 0.10 ± 0.05 : 30 ± 1 | 1 : 300 |
| MRK-nu-1 | Mammary carcinoma | 0.74 ± 0.03 : 210 ± 10 | 1 : 284 |
| EJ-1 | Bladder carcinoma | 0.026 ± 0.014 : 3.7 ± 0.2 | 1 : 142 |
Experimental details are described in Supporting Information, Figure S4.
Table 4.
Comparison of 7 (pivoxil ester) to 14 (acid form) in growth inhibition of lymphoma and leukemia hematopoietic cell lines.a
| Tumor cell line | Origin | Pivoxil IC50 : H IC50 (μM) | Ratio IC50 Pivoxil : IC50 H |
|---|---|---|---|
| MOLT-3 | T cell acute lymphoblastic leukemia | 0.13 ± 0.09 : 180 ± 20 | 1 : 1,385 |
| MOLT-4 | T cell acute lymphoblastic leukemia | 0.16 ± 0.12 : 120 ± 10 | 1 : 750 |
| PEER | T cell acute lymphocytic leukemia | 0.023 ± 0.009 : 82 ± 6 | 1 : 3,565 |
| C1R | B cell lymphoma | 0.078 ± 0.018 : 79 ± 6 | 1 : 1,013 |
| SCC-3 | Non-Hodgkin's lymphoma | 0.18 ± 0.08 : 66 ± 3 | 1 : 367 |
| Daudi | Burkitt's lymphoma | 0.43 ± 0.52 : 76 ± 5 | 1 : 177 |
| Raji | Burkitt's lymphoma | 0.27 ± 0.01 : 120 ± 10 | 1 : 444 |
| Ramos-RA1 | Burkitt's lymphoma | 0.11 ± 0.07 : 75 ± 5 | 1 : 682 |
| THP-1 | Acute monocytic leukemia | 0.035 ± 0.061 : 130 ± 10 | 1 : 3,714 |
| U937 | Histiocytic lymphoma | 0.26 ± 0.01 : 26 ± 1 | 1 : 100 |
| K562 | Erythroleukemia | 0.029 ± 0.006 : 16 ± 2 | 1 : 552 |
Experimental details are described in Supporting Information, Figure S5.
Based on these findings, we synthesized 28 additional BP pivoxil and other esters (compounds 15-42, Figure 2), and assessed their ability to inhibit the growth of the U937 and EJ-1 cell lines (Table 5). However, again, 7 was the most potent inhibitor of U937 and EJ-1 cell growth. Therefore, we focused further studies on 7. To determine its potential clinical usefulness, 7 was tested against a variety of tumor cell lines in comparison with Zol, the most potent FDA-approved BP (Figure 3 and Supporting Information, Figure S6 and Table S1). Compound 7 was found to be, on average, 27-fold more active than Zol for 52 solid tumor cell lines. Several tumor cells lines, such as the bladder cancer cell line, EJ-1, and the gastric tumor cell lines, GCIY and MKN45, were highly sensitive to 7. Only two solid tumor cell lines were more sensitive to Zol than Compound 7. Compound 7 was found to be even more active on average against hematopoietic cell lines. Hematopoietic cell lines averaged 796-fold (± 1,442-fold) more sensitive to growth inhibition with 7 than Zol. For example, HL60 (promyelocytic leukemia) was 5,679-fold and PEER (T cell lymphoma) was 2,391-fold more sensitive to 7 than Zol. The IC50 for 7 averaged 515 nM for B cell malignancies, 116 nM for T cell malignancies, and 107 nM for myeloid malignancies versus 85,100 nM, 55,000 nM, and 85,557 nM, respectively, for Zol (Figure 3 and Supporting Information, Figure S6 and Table S1). Thus, while solid tumors were 27-fold more susceptible to 7 than Zol, lymphoma and myeloid hematopoietic cell lines were markedly more susceptible to 7 than to Zol.
Figure 2.

Structure of pivoxil esters. Details of synthesis are summarized in the Experimental Section and Supporting Information (Chemistry and MNR spectral information).
Table 5.
Inhibition of U937 and EJ-1 cell proliferation by additional pivoxil esters. a
| Pivoxil Compound | U937 IC50% (μM) | EJ-1 IC50% (μM) | Pivoxil Compound | U937 IC50% (μM) | EJ-1 IC50% (μM) |
|---|---|---|---|---|---|
| 7 | 0.26 ± 0.01 | 0.026 ± 0.014 | 30 | 6.6 ± 0.3 | 10 ± 1 |
| 39 | 0.65 ± 0.11 | 0.059 ± 0.006 | 19 | 6.7 ± 0.3 | 9.5 ± 0.5 |
| 35 | 1.1 ± 0.3 | 3.5 ± 0.5 | 16 | 6.8 ± 0.4 | 15 ± 1 |
| 31 | 2.2 ± 0.1 | 1.7 ± 0.2 | 20 | 6.9 ± 0.6 | 16 ± 1 |
| 34 | 2.5 ± 0.2 | 1.1 ± 0.4 | 26 | 7.0 ± 0.3 | 8.7 ± 0.4 |
| 17 | 2.6 ± 0.1 | 4.3 ± 0.5 | 15 | 7.1 ± 0.5 | 16 ± 0 |
| 24 | 3.0 ± 0.2 | 6.3 ± 0.5 | 28 | 7.1 ± 0.3 | 6.0 ± 0.4 |
| 18 | 3.3 ± 0.2 | 4.8 ± 0.5 | 36 | 7.9 ± 0.2 | 15 ± 1 |
| 22 | 3.4 ± 0.1 | 3.7 ± 0.4 | 37 | 7.9 ± 0.6 | 16 ± 2 |
| 23 | 3.5 ± 0.6 | 4.5 ± 0.4 | 27 | 8.9 ± 1.0 | 13 ± 1 |
| 33 | 5.0 ± 0.5 | 4.2 ± 0.4 | 38 | 11 ± 2 | 8.5 ± 0.3 |
| 21 | 6.1 ± 0.5 | 9.0 ± 0.6 | 42 | 17 ± 1 | 16 ± 1 |
| 25 | 6.2 ± 0.5 | 8.8 ± 0.7 | 41 | 50 ± 3 | 56 ± 2 |
| 32 | 6.3 ± 0.2 | 12 ± 2 | 40 | 70 ± 1 | 79 ± 7 |
| 29 | 6.6 ± 0.1 | 12 ± 3 |
Experimental details are described in Supporting Information.
Figure 3.

Tumor cell growth inhibition by compound 7 in comparison to Zol. (A-F) Dose-dependent inhibition of tumor cell growth by compound 7. Tumor cells were grown, harvested and resuspended at 1 × 104 cells/mL in complete RPMI 1640 medium. A total of 0.05 mL of the cell suspension was added to flat-bottomed 96-well plates, followed by 0.05 mL of three-fold serial dilutions of compound 7. After incubation at 37°C with 5% CO2 for 4 days, 0.1 mL of CellTiter-Glo reagent (Promega, Madison, WI, USA) was added and the luminescence resulting from ATP was measured using an ARVO luminometer (PerkinElmer, Foster City, CA, USA). All experiments were performed in triplicate: (A) lymphomas, ● MOLT-3, ▲ J.RT3-T3.5, ■ Raji, ◆ Ramos (RA 1), (B) myeloid leukemias, ● HL60, ▲ SCC-3, ■ P31/FUJ, ◆ NOMO-1, (C) mammary carcinomas, ● YMB-1-E, ▲ HMC-1-8, ■ MCF-7, ◆ MDA-MB-231, (D) renal cell carcinomas, ● VMRC-RCW, ▲ UOK121, ■ Caki-1, ◆ A-704, (E) pancreatic carcinomas, ● KP4-1, ▲ KP4-2, ■ KP4-3, ◆ MiaPaCa2, and (F) other tumor cells, ● 24TKB, ▲ MKN1, ■ Colo320, ◆ TAKAO. (G) Comparison of compound 7 (left panel) to Zol (right panel) concentrations required for half-maximal growth inhibition of various tumor cell lines. Ly = lymphomas, My = myeloid leukemia, Ma = mammary carcinomas, and Others = other tumor cells. Cut-off values were arbitrarily set at 300 nM for 7 and 20,000 nM for Zol. Details of Zol growth inhibition are given in Supporting Information, Figure S6.
To determine its mechanism of action, the effect of 7 on RAP1A prenylation in tumor cells was assessed. Compound 7 strongly inhibited RAP1A geranylgeranylation in six tumor cell lines with inhibition detected at concentrations as low as 1-10 nM (Figure 4). As expected based on its inhibition of FDPS, Zol also inhibited prenylation but required concentrations of at least 10,000-100,000 nM. For example, whereas unprenylated RAP1A was detected in EJ-1 with 1 nM of 7, Zol required ∼10,000 nM. The differences between 7 and Zol are similar to those noted with growth inhibition, albeit somewhat greater in magnitude. Thus, the activity of 7 is consistent with inhibition of isoprenoid biosynthesis. Given that most strong inhibitors of GGDPS contain mono- or diacyl chains[24-26], the activity of 7 is likely due to inhibition of FDPS, although dual FDPS and GGDPS inhibition remains possible,[27] as does direct or indirect inhibition of signaling kinases in addition to the inhibition of FDPS[28, 29].
Figure 4.

Inhibition of geranylgeranylation of RAP1A in tumor cells by compound 7. Tumor cells were cultured with 7 or Zol for 16 h, lysed, and the proteins were separated by SDS-PAGE, transferred onto PVDF membranes and probed with anti-unprenylated RAP1A Ab. Data for inhibition of geranylgeranylation of RAP1A by Zol are from Idress et al.[30] and are included for comparison. Note that the maximum concentration used for 7 was 100-fold less than that used for Zol.
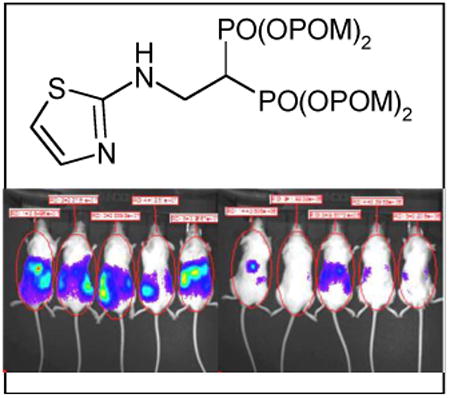
To assess the activity of 7 against tumors in vivo, the effect of 7 on the growth of EJ-1 bladder carcinoma cells was assessed in immunodeficient NOG mice. Luciferase-expressing EJ-1 bladder carcinoma cells were intraperitoneally inoculated into NOG mice and the mice were then treated with compound 7 twice a week. Compound 7 significantly inhibited the growth of EJ-1 from weeks 4 through 7 such that at 7 weeks, EJ-1 tumor growth was decreased by 78% in mice treated with compound 7 relative to control mice (Figure 5). Thus, compound 7 has direct therapeutic benefits in the treatment of human cancer in this preclinical mouse model.
Figure 5.

Compound 7 inhibits EJ-1 bladder carcinoma growth in immunodeficient NOG mice. (A) Whole-body bioluminescence imaging of tumor cells. Immunodeficient NOG mice were intraperitoneally (i.p.) inoculated with 1 × 106 EJ-1 cells transfected with pGL4.10[luc2] on day 0 and treated with 2 μg/0.1 mL compound 7 (97.1 μg/kg) or 0.1 mL of PBS i.p. on days 3 and 6. On day 7, 0.1 mL of 15 mg/mL VivoGlo™ luciferin (Xenogen, Alameda, CA, USA) was injected i.p. and the mice were placed in the specimen chamber and photon emissions transmitted from the mice were then measured. This treatment regimen was repeated 7 times and the images from day 28 are shown. Luciferase units are photons/second/mouse. (B) Inhibition of tumor growth by compound 7 in vivo. The growth of EJ-1 was monitored for weeks 2 through 7 as described in (A) and the average photon flux/sec for four to five mice is plotted ± SEM. *p < 0.05 by Mann-Whitney U test. ● PBS and ○ compound 7.
The development of phosphate and phosphonate prodrugs has been an important advance in anticancer and antiviral therapy.[31-33] Esterification of phosphate- and phosphonate-containing drugs can increase their gastrointestinal absorption, survival in the systemic circulation, and cell penetration. A number of different FDA approved drugs contain the pivoxil group used here or the related disoproxil group with acceptable levels of toxicity. Adefovir dipivoxil and tenofovir disoproxil fumarate have been used to treat hepatitis B virus and HIV while cefditoren pivoxil is a broad-spectrum cephalosporin antibiotic. Besides pivoxil acyloxyalkyl esters, it may be possible to further improve targeting of hematopoietic tumors using aryloxy phosphoramidate esters.[33-35] The tenofovir alafenamide aryloxy phosphoramidate prodrug has increased levels in plasma and lymphatic tissue and is 1000-fold more active against HIV compared with tenofovir[36] and is more active in patients with fewer side effects than its disoproxil derivative.[37, 38] Unlike nucleoside prodrugs, nitrogen-containing BP prodrugs would be given intermittently and their predicted reduced deposition into bone would minimize jaw osteonecrosis and atypical fractures.
The mechanism for the high activity of 7 for hematopoietic tumors may reflect increased uptake by 7 coupled with increased dependence of hematopoietic tumors on prenylated Ras or other GTPases for their growth. Whereas hematopoietic cancer cells were more resistant to Zol than non-hematopoietic cancer cells (mean EC50% of 77,800 nM versus 9,600 nM), they were more sensitive to 7 (mean EC50% of 240 nM versus 770 nM). This suggests that BP uptake by hematopoietic tumors is lower than with other tumors; therefore, the increased uptake of the 7 prodrug results in higher anti-proliferative activity. To further improve the biological activity of nitrogen-containing BPs, it is necessary to examine and compare their cell penetration, lipophilicity, and metabolic stability in detail.
Conclusions
In conclusion, masking the negatively-charged P-C-P structure of BPs with pivoxil esters greatly increases their capacity to inhibit tumor cell growth. The most active BP pivoxil ester, compound 7, was found to be particularly effective at inhibiting the growth of hematopoietic cells with IC50 values generally between 20 to 200 nM whereas the IC50 values for Zol were up to 5,679-fold higher being generally greater than 20,000 nM. Besides the direct effect of 7 on tumor growth, 7 also expands cytotoxic Vγ2Vδ2 T cells in vitro and can be used in combination with adoptively transferred Vγ2Vδ2 T cells in vivo to enhance tumor control in the NOG mouse model (Tanaka et al., manuscript in preparation). Moreover, we speculate that 7 may exhibit less bone deposition due to its lack of free phosphonate moieties as well as the absence of the hydroxyl group (bone hook) on the germinal carbon of the P-C-P structure. Although further research is required, BP prodrugs could increase the effectiveness of BP treatment for both hematopoietic and non-hematopoietic solid tumors.
Experimental Section
General Chemistry
Thin-layer chromatography (TLC) was performed on precoated plates (0.25 mm, silica gel plate 60F245, Merck Millipore, MA). Column chromatography was conducted using silica gel (Kanto Chemical Co., Inc., Chuo-ku, Tokyo, Japan). All reactions were conducted under an air atmosphere unless otherwise noted. Unless otherwise stated, reagents were purchased from chemical sources and used without further purification. 1H NMR and 13C NMR spectra were obtained in CDCl3 solution on a JNM-AL-400 spectrometer (1H NMR at 400 MHz, 13C NMR at 100 MHz) and a JNM-ECA-500 spectrometer (1H NMR at 500 MHz, 13C NMR at 125 MHz) (JEOL Ltd., Akishima, Tokyo, Japan). 1H NMR chemical shifts were referenced to tetramethylsilane (TMS) (0.00 ppm) and 13C NMR chemical shifts to CDCl3 (77.0 ppm). The chemical sifts were expressed in parts per million (ppm). The following abbreviations were used for peak multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; sept, septet; m, multiplet; and br, broad. IR spectra were recorded on FT/IR-4100 (JASCO Corp., Hachioji, Tokyo, Japan). Mass spectra and high-resolution mass spectra were recorded on JMS-HX/HX 110A (JEOL Ltd.). NMR spectral information and physical data are shown in Supporting Information (Chemistry and NMR spectral information).
General procedure for the synthesis of 1,1-bisphosphonic acid pivoxil esters
Tetramethyl vinylidene-1,1-bisphosphonate (S1)
Folowing the protocol of Degenhart,[39] diethylamine (6.3 mL, 60 mmol) was added to a solution of paraformaldehyde (9.0 g, 300 mmol) in methanol (230 mL) at room temperature and stirred at 65°C for 30 min. A solution of tetramethyl methylenediphosphonate (14 g, 60 mmol) in methanol (10 mL) was added and the reaction mixture was refluxed for 1.5 hr. The resulting mixture was concentrated in vacuo to give a crude product, which was used for the next reaction without further purification. The crude product was dissolved in toluene (200 mL) and treated with p-toluenesulfonic acid monohydrate (114 mg, 0.6 mmol). The reaction mixture was refluxed using a Dean-Stark trap for 16 hr and then diluted with CHCl3. The resulting mixture was washed with brine, dried over MgSO4, filtered and concentrated in vacuo to give a crude product that was purified by silica gel column chromatography (MeOH/ CHCl3 = 10%) to yield 8.9 g (60%) of the title compound as a colorless oil.
Tetrakispivaloyloxymethyl vinylidene-1,1-bisphosphonate (S2)
NaI (10.2 g, 68 mmol) and chloromethyl pivalate (POMCl, 12.4 mL, 85 mmol) were added to a solution of tetramethyl vinylidene-1,1-bisphosphonate (4.2 g, 17 mmol) in MeCN (85 mL) and then refluxed for 14 hr. After addition of H2O, the resulting mixture was extracted with EtOAc and the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to give a crude product that was purified by silica gel column chromatography (AcOEt/ hexane = 50%) to yield 3.8 g (35%) of the title compound as a pale yellow oil.
Synthesis of 1,1-bisphosphonic acid tetrakispivoxil esters
Tetrakispivaloyloxymethyl 2-(thiazole-2-ylamino)ethylidene-1,1-bisphosphonate (7)
2-Aminothiazole (20 mg, 0.2 mmol) was added to a solution of tetrakispivaloyloxymethyl vinylidene-1,1-bisphosphonate (64 mg, 0.1 mmol) in CHCl3 (0.4 mL) and stirred at room temperature for 1 hr. The resulting mixture was concentrated in vacuo to give a crude product, which was purified using silica gel column chromatography (AcOEt/CHCl3 = 50%) to yield 69 mg (92%) of the title compound as colorless solid. The following compounds were prepared using the same synthetic procedure; 1-6 and 15-26.
Tetrakispivaloyloxymethyl 2-(3-hydroxypyridine-2-ylamino) ethylidene-1,1-bisphosphonate (27)
To a solution of tetrakis-pivaloyloxymethyl 2-(3-benzyloxypyridine-2-ylamino)ethylidene-1,1- bisphosphonate (15, 253 mg, 0.30 mmol) in AcOEt (15 ml) was added 10% Pd/C (62 mg, 0.058 mmol) and stirred under H2 (1 atm) at room temperature for 1.5 hour. The resulting mixture was filtered and concentrated in vacuo to give a crude product, which was purified by silica gel column chromatography (AcOEt) to afford 196 mg (87%) of the title compound. The following compounds were prepared using the same synthetic procedure; 28-39.
Tetramethyl 2-(pyridine-2-ylamino)ethylidene-1,1-bisphosphonate (40)
2-Aminopyridine (47 mg, 0.50 mmol) was added to a solution of tetramethyl vinylidene-1,1-bisphosphonate (122 mg, 0.50 mmol) in THF (1 mL) and stirred at room temperature for 34 hr. The resulting mixture was concentrated in vacuo to give a crude product that was purified by silica gel column chromatography (MeOH/ AcOEt = 5% to 100%) to yield 171 mg (99%) of the title compound.
Tetraethyl 2-(pyridine-2-ylamino) ethylidene-1,1-bisphosphonate (41)
2-Aminopyridine (376 mg, 4.0 mmol) was added to a solution of tetraethyl vinylidene-1,1-bisphosphonate (600 mg, 2.0 mmol) in CHCl3 (8 mL) and stirred at room temperature for 3 hr. The resulting mixture was concentrated in vacuo to give a crude product, which was purified using silica gel column chromatography (MeOH/ CHCl3 = 10%) to yield 772 mg (98%) of the title compound. Compound 42 was prepared using the same synthetic procedure.
General procedure for the synthesis of 1,1-bisphosphonic acids
2-(5-Methylpyridine-2-ylamino) ethylidene 1,1-bisphosphonic acid (10)
2-Amino-5-methylpyridine (108 mg, 1.0 mmol) was added to a solution of tetraethyl vinylidene-1,1-bisphosphonate (150 mg, 0.50 mmol) in CHCl3 (2 mL) and stirred at room temperature for 1 hr. The resulting mixture was concentrated in vacuo to give a crude product that was purified by silica gel column chromatography (MeOH/ CHCl3 = 10%) to yield 212 mg (99%) of tetraethyl 2-aminoethylidene-1,1-bisphosphonate. Tetraethyl 2-aminoethylidene-1,1-bisphosphonate (102 mg, 0.25 mmol) was dissolved in MeCN (2 mL) and treated with bromotrimethylsilane (TMSBr, 0.20 ml, 1.5 mmol). The resulting mixture was stirred at room temperature for 4 hr. The mixture was concentrated in vacuo to give a crude product that was purified by recrystallization from acetone-H2O to yield 55 mg (74%) of the title compound. The following compounds were prepared using the same synthetic procedure; 8-9 and 11-14.
Biological Assays
In vitro tumor cell growth inhibition assay
Tumor cells were grown, harvested, and resuspended at 1 × 104 cells/ml in complete RPMI 1640 medium. A total of 0.05 ml of the cell suspension was added to flat bottomed 96-well plates, followed by 0.05 ml of three-fold serial dilutions of POM esters. After incubation at 37°C with 5% CO2 for 4 days, 0.1 ml of CellTiter-Glo reagent (Promega, Madison, WI, USA) was added and the luminescence resulting from ATP was measured using an ARVO luminometer (PerkinElmer, Foster City, CA, USA). All experiments were performed in triplicate. The concentrations required for 50% tumor cell growth inhibition (IC50 values) are shown. The sources for the cell lines are detailed in Idrees et al.[30]
Assessing inhibition of geranylgeranylation of RAP1A in tumor cells
Tumor cells were resuspended in 90 mL of complete RPMI 1640 medium supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO), 10-5 M 2-mercaptoethanol (Invitrogen corp., Carlsbad, CA), 100 IU/mL of penicillin (Meiji Seika Kaisha, Ltd., Chuo-Ku, Tokyo, Japan), and 100 μg/ml of streptomycin (Meiji Seika Kaisha) and grown overnight at 37°C with 5% CO2 in 225 cm2 flasks. Compound 7 was then added to the flasks to the concentrations indicated. After incubation for 16 h, the cells were harvested and resuspended in 100 μL of lysis solution containing 1% NP-40 (Wako Pure Chemical Industries Ltd., Chuo-ku, Osaka, Japan), 0.1% sodium dodecyl sulfate (Tokyo Chemistry Industry Co., Ltd., Chuo-Ku, Tokyo, Japan), and 0.5% sodium deoxycholate (Wako) in microcentrifuge tubes. After centrifugation at 15,000 rpm for 10 min, the supernatants were transferred to new tubes and SDS-urea buffer containing 6.7 M urea (Wako), 5% sodium dodecyl sulfate (Tokyo Chemistry Industry), 100 mM Tris–HCl buffer, pH 7.4 (Wako), 0.25% bromophenol blue (Wako), and 50 mM dithiothreitol (Wako) were added to give a protein concentration of 5 mg/mL. The samples were loaded on 15% polyacrylamide slab gels (Daiichi Pure Chemicals Co., Ltd., Chuo-ku, Tokyo, Japan) at 50 μg/lane, and electrophoresed at 120 mA/h. The proteins were then transferred onto Polyscreen (R) PVDF Transfer Membranes (PerkinElmer Inc., Waltham, MA) treated with goat anti-unprenylated RAP1A antisera (×500, Santa Cruz Biotechnology Inc., Santa Cruz, CA), and horse radish peroxydase-conjugated anti-goat IgG antisera (×5,000, KPL Inc., Gaithersburg, MD), followed by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL). Although not shown, controls using goat anti-RAP1A and anti-GAPDH antisera (Santa Cruz Biotechnology) were included in this study. Chemiluminescence was detected on Amersham HyperfilmTM MP (GE Healthcare Ltd., Little Chalfont, Buckinghamshire, UK) using a Fuji Medical Film Processor FPM100 (Fuji Film Co., Ltd., Ashigara, Kanagawa, Japan).
Inhibition of EJ-1 bladder tumor cell growth by 7 in NOG immunodeficient mice
EJ-1 tumor cells (1 × 106) stably transfected with pGL4.10[luc2] (Promega, Madison, WI, USA) were intraperitoneally (i.p.) inoculated into immunodeficient NOG mice (obtained from the Central Institute for Experimental Animals, Kawasaki, Kanagawa, Japan). Mice were treated i.p. with 2 μg of compound 7 in 0.1 mL of PBS (97.1 μg/kg body weight for mice weighing 20.6 g), on day 3 and 6. On day 7, 0.1 mL of 15 mg/ml VivoGlo™ luciferin (Xenogen, Alameda, CA, U.S.A.) was i.p. injected and the mice placed in a specimen chamber mounted with a CCD camera cooled to -120°C (In Vivo Imaging System, Waltham, MA, U.S.A). Then, the photon emission transmitted from the mice was measured. The grey scale photographic images and bioluminescence color images were superimposed. This treatment regimen was repeated for seven weeks and the images for week 4 are shown. Arbitrary luciferase units were expressed as average flux (photons/second/mouse) and the photon intensity values per mouse were plotted with time. The growth of EJ-1 was monitored for weeks 2 through 7 and the average photon flux/sec for four to five mice is plotted ± SEM. Statistical significance was assess by the non-parametric Mann-Whitney U test. The animal studies were conducted in accordance with the relevant laws and institutional guidelines, and were approved by the local IACUC committee.
Supplementary Material
Scheme 1.

Structure of representative BPs.
Scheme 2.

General procedure for the synthesis of 1,1-bisphosphonic acid pivoxil esters.
Scheme 3.

General procedure for the synthesis of 1,1-bisphosphonic acids.
Acknowledgments
The authors thank Ms. Chiyomi Inoue for her technical assistance. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Culture, Sports, and Technology of Japan (MEXT), by “Special Coordination Funds for Promoting Science and Technology” from MEXT and Astellas Pharma Inc. through the “Formation of Center for Innovation by Fusion of Advanced Technologies” program, by “Platform for Drug Discovery, Informatics, and Structural Life Science” from MEXT, by Grant-in Aid for Translational Research from Kyoto University and Japan Agency for Medical Research and Development (AMED) (to Y.T.), and by the Department of Veterans Affairs (Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development) Grant 1 I01 BX000972-01A1, National Cancer Institute Grants CA097274 (University of Iowa/Mayo Clinic Lymphoma Specialized Program of Research Excellence) and P30 CA086862 (Core Support), and the University of Iowa Carver College of Medicine Carver Medical Research Initiative Grant (to C.T.M.). C.T.M. is the Kelting Family Scholar in Rheumatology.
Grant support: Weiner, George PI NIH CA097274
Footnotes
Supporting information for this article can be found under http://dx.doi.org/.
References
- 1.Fleisch H. Endocr Rev. 1998;19:80–100. doi: 10.1210/edrv.19.1.0325. [DOI] [PubMed] [Google Scholar]
- 2.Fleisch H. Breast Cancer Res. 2002;4:30–34. doi: 10.1186/bcr414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleisch H, Russell RG, Francis MD. Science. 1969;165:1262–1264. doi: 10.1126/science.165.3899.1262. [DOI] [PubMed] [Google Scholar]
- 4.Jung A, Bisaz S, Fleisch H. Calcif Tissue Res. 1973;11:269–280. doi: 10.1007/BF02547227. [DOI] [PubMed] [Google Scholar]
- 5.Russell RG. Pediatrics. 2007;119(2):S150–162. doi: 10.1542/peds.2006-2023H. [DOI] [PubMed] [Google Scholar]
- 6.Lehenkari PP, Kellinsalmi M, Näpänkangas JP, Ylitalo KV, Mönkkönen J, Rogers MJ, Azhayev A, Väänänen HK, Hassinen IE. Mol Pharmacol. 2002;61:1255–1262. doi: 10.1124/mol.61.5.1255. [DOI] [PubMed] [Google Scholar]
- 7.van Beek E, Pieterman E, Cohen L, Lowik C, Papapoulos S. Biochem Biophys Res Commun. 1999;264:108–111. doi: 10.1006/bbrc.1999.1499. [DOI] [PubMed] [Google Scholar]
- 8.Bergstrom JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G. Arch Biochem Biophys. 2000;373:231–241. doi: 10.1006/abbi.1999.1502. [DOI] [PubMed] [Google Scholar]
- 9.Rogers MJ, Gordon S, Benford HL, Coxon FP, Luckman SP, Monkkonen J, Frith JC. Cancer. 2000;88(Suppl 12):2961–2978. doi: 10.1002/1097-0142(20000615)88:12+<2961::aid-cncr12>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 10.Ebetino FH, Hogan AML, Sun S, Tsoumpra MK, Duan X, Triffitt JT, Kwaasi AA, Dunford JE, Barnett BL, Oppermann U, Lundy MW, Boyde A, Kashemirov BA, McKenna CE, Russell RGG. Bone. 2011;49:20–33. doi: 10.1016/j.bone.2011.03.774. [DOI] [PubMed] [Google Scholar]
- 11.Gnant M, Mlineritsch B, Schippinger W, Luschin-Ebengreuth G, Pöstlberger S, Menzel C, Jakesz R, Seifert M, Hubalek M, Bjelic-Radisic V, Samonigg H, Tausch C, Eidtmann H, Steger G, Kwasny W, Dubsky P, Fridrik M, Fitzal F, Stierer M, Rücklinger E, Greil R, Marth C. N Engl J Med. 2009;360:679–691. doi: 10.1056/NEJMoa0806285. [DOI] [PubMed] [Google Scholar]
- 12.Gnant M, Mlineritsch B, Stoeger H, Luschin-Ebengreuth G, Heck D, Menzel C, Jakesz R, Seifert M, Hubalek M, Pristauz G, Bauernhofer T, Eidtmann H, Eiermann W, Steger G, Kwasny W, Dubsky P, Hochreiner G, Forsthuber EP, Fesl C, Greil R. Lancet Oncol. 2011;12:631–641. doi: 10.1016/S1470-2045(11)70122-X. [DOI] [PubMed] [Google Scholar]
- 13.Coleman RE, Marshall H, Cameron D, Dodwell D, Burkinshaw R, Keane M, Gil M, Houston SJ, Grieve RJ, Barrett-Lee PJ, Ritchie D, Pugh J, Gaunt C, Rea U, Peterson J, Davies C, Hiley V, Gregory W, Bell R. N Engl J Med. 2011;365:1396–1405. [Google Scholar]
- 14.Coleman R, de Boer R, Eidtmann H, Llombart A, Davidson N, Neven P, von Minckwitz G, Sleeboom HP, Forbes J, Barrios C, Frassoldati A, Campbell I, Paija O, Martin N, Modi A, Bundred N. Ann Oncol. 2013;24:398–405. doi: 10.1093/annonc/mds277. [DOI] [PubMed] [Google Scholar]
- 15.Morgan GJ, Davies FE, Gregory WM, Cocks K, Bell SE, Szubert AJ, Navarro-Coy N, Drayson MT, Owen RG, Feyler S, Ashcroft AJ, Ross F, Byrne J, Roddie H, Rudin C, Cook G, Jackson GH, Child JA. Lancet. 2010;376:1989–1999. doi: 10.1016/S0140-6736(10)62051-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jahnke W, Rondeau JM, Cotesta S, Marzinzik A, Pellé X, Geiser M, Strauss A, Götte M, Bitsch F, Hemmig R, Henry C, Lehmann S, Glickman JF, Roddy TP, Stout SJ, Green JR. Nat Chem Biol. 2010;6:660–666. doi: 10.1038/nchembio.421. [DOI] [PubMed] [Google Scholar]
- 17.Thompson K, Rogers MJ, Coxon FP, Crockett JC. Mol Pharmacol. 2006;69:1624–1632. doi: 10.1124/mol.105.020776. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Cao R, Yin F, Lin FY, Wang H, Krysiak K, No JH, Mukkamala D, Houlihan K, Li J, Morita CT, Oldfield E. Angew Chem Int Ed. 2010;49:1136–1138. doi: 10.1002/anie.200905933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhu W, Liu YL, Wang H, Wang K, Li K, No JH, Ayong L, Gulati A, Pang R, Freitas-Junior L, Morita CT, Oldfield E. ACS Med Chem Lett. 2013;4:423–427. doi: 10.1021/ml4000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Leon A, Song Y, Studer D, Haase C, Koscielski LA, Oldfield E. J Med Chem. 2006;49:5804–5814. doi: 10.1021/jm060280e. [DOI] [PubMed] [Google Scholar]
- 21.Wiemer AJ, Yu JS, Shull LW, Barney RJ, Wasko BM, Lamb KM, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2008;16:3652–3660. doi: 10.1016/j.bmc.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 22.Rogers MJ, Xiong X, Brown RJ, Watts DJ, Russell RG, Bayless AV, Ebetino FH. Mol Pharmacol. 1995;47:398–402. [PubMed] [Google Scholar]
- 23.Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, Ebetino FH, Rogers MJ. J Pharmacol Exp Ther. 2001;296:235–242. [PubMed] [Google Scholar]
- 24.Szabo CM, Matsumura Y, Fukura S, Martin MB, Sanders JM, Sengupta S, Cieslak JA, Loftus TC, Lea CR, Lee HJ, Koohang A, Coates RM, Sagami H, Oldfield E. J Med Chem. 2002;45:2185–2196. doi: 10.1021/jm010412y. [DOI] [PubMed] [Google Scholar]
- 25.Wiemer AJ, Yu JS, Lamb KM, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2008;16:390–399. doi: 10.1016/j.bmc.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 26.Chen CKM, Hudock MP, Zhang Y, Guo RT, Cao R, No JH, Liang PH, Ko TP, Chang TH, Chang SC, Song Y, Axelson J, Kumar A, Wang AHJ, Oldfield E. J Med Chem. 2008;51:5594–5607. doi: 10.1021/jm800325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Mukherjee S, Gao YG, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang TK, Lin FY, Van Beek E, Papapoulos S, Wang AH, Kubo T, Ochi M, Mukkamala D, Oldfield E. J Am Chem Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuen T, Stachnik A, Iqbal J, Sgobba M, Gupta Y, Lu P, Colaianni G, Ji Y, Zhu LL, Kim SM, Li J, Liu P, Izadmehr S, Sangodkar J, Bailey J, Latif Y, Mujtaba S, Epstein S, Davies TF, Bian Z, Zallone A, Aggarwal AK, Haider S, New MI, Sun L, Narla G, Zaidi M. Proc Natl Acad Sci USA. 2014;111:17989–17994. doi: 10.1073/pnas.1421410111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stachnik A, Yuen T, Iqbal J, Sgobba M, Gupta Y, Lu P, Colaianni G, Ji Y, Zhu LL, Kim SM, Li J, Liu P, Izadmehr S, Sangodkar J, Scherer T, Mujtaba S, Galsky M, Gomez J, Epstein S, Buettner C, Bian Z, Zallone A, Aggarwal AK, Haider S, New MI, Sun L, Narla G, Zaidi M. Proc Natl Acad Sci USA. 2014;111:17995–18000. doi: 10.1073/pnas.1421422111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Idrees AS, Sugie T, Inoue C, Murata-Hirai K, Okamura H, Morita CT, Minato N, Toi M, Tanaka Y. Cancer Sci. 2013;104:536–542. doi: 10.1111/cas.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hecker SJ, Erion MD. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 32.Wiemer AJ, Wiemer DF. Top Curr Chem. 2014 doi: 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehellou Y. ChemMedChem. 2016;11:1114–1116. doi: 10.1002/cmdc.201600156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehellou Y, Balzarini J, McGuigan C. ChemMedChem. 2009;4:1779–1791. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 35.Gao LJ, De Jonghe S, Daelemans D, Herdewijn P. Bioorg Med Chem Lett. 2016;26:2142–2146. doi: 10.1016/j.bmcl.2016.03.076. [DOI] [PubMed] [Google Scholar]
- 36.Lee WA, He GX, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, Cundy KC. Antimicrob Agents Chemother. 2005;49:1898–1906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markowitz M, Zolopa A, Squires K, Ruane P, Coakley D, Kearney B, Zhong L, Wulfsohn M, Miller MD, Lee WA. J Antimicrob Chemother. 2014;69:1362–1369. doi: 10.1093/jac/dkt532. [DOI] [PubMed] [Google Scholar]
- 38.Sax PE, Wohl D, Yin MT, Post F, DeJesus E, Saag M, Pozniak A, Thompson M, Podzamczer D, Molina JM, Oka S, Koenig E, Trottier B, Andrade-Villanueva J, Crofoot G, Custodio JM, Plummer A, Zhong L, Cao H, Martin H, Callebaut C, Cheng AK, Fordyce MW, McCallister S. Lancet. 2015;385:2606–2615. doi: 10.1016/S0140-6736(15)60616-X. [DOI] [PubMed] [Google Scholar]
- 39.Degenhardt CR, Burdsall DC. J Org Chem. 1986;51:3488–3490. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.