

Abstract
Intramolecular chirality transfer [2 + 2] cyclo-addition of enantiomerically enriched allenoates and alkenes is presented. The use of a chiral catalyst was found to be critical to achieve high levels of diastereoselectivity compared to use of an achiral catalyst. The method developed leads to highly substituted cyclobutanes that would be difficult to prepare by alternative methods.
Keywords: Traumatic brain injury, social communication, theory of mind, executive function, speech language pathology
Graphical Abstract
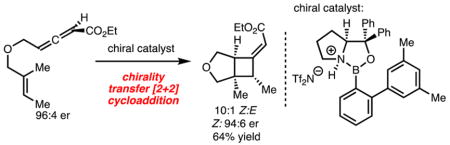
Cyclobutane synthesis by [2 + 2] cycloaddition represents an important class of reaction for chemical synthesis.1 Despite widespread application of these reactions, enantioselective variants have been slower to develop.2 Accordingly, our laboratory has taken an interest in the development of methods for the enantioselective synthesis of cyclobutanes by [2 + 2] cycloaddition (Scheme 1).
Scheme 1.

[2 + 2] Cycloadditions
Recently, our laboratory disclosed a method for catalytic enantioselective [2 + 2] cycloaddition of allenoates and alkenes.3 In our efforts to extend the utility of this process, we became interested in developing variants that utilized γ-substituted allenoates. This substitution pattern renders the allene chiral and therefore presents the possibility for a chirality-transfer [2 + 2] cycloaddition.4 This would represent an attractive approach for the synthesis of cyclobutanes, provided the chiral allene can be readily prepared in high enantioselectivity. Several examples of chirality transfer [2 + 2] cycloaddition have been reported.5 While high chirality transfer has been observed in related chemistry, the use of enantiomerically enriched allenoates has not been reported. This would be significant as enantioselective synthesis of allenoates is readily achieved by enantioselective isomerization of β,γ-unsaturated alkynyl esters.6,7 Therefore, development of chirality transfer [2 + 2] cycloaddition of allenoates and alkenes would be complementary to existing approaches. Herein, we disclose an intramolecular chirality transfer [2 + 2] cycloaddition of allenoates and alkenes.8
To initiate our studies, the chirality transfer [2 + 2] cycloaddition of allenoate 3 was investigated (Scheme 2A). The synthesis of the γ-substituted allenoate 3 was accomplished by application of an enantioselective isomerization of non-conjugated alkynyl ketone 2 developed by Tan and coworkers.6 However, we elected to employ chiral thiourea catalyst 29,10 because it is readily available and delivers allenoate 3 in good enantioselectivity (96:4 er). Treatment of chiral allenoate 3 with EtAlCl2 delivered the product in good yield and transfer of chirality, albeit as a 1.4:1 mixture of alkene isomers (Scheme 2A).
Scheme 2.

Initial Findings
Upon determination of the absolute configuration of each alkene isomer, it was revealed that the products were epimeric at the bridgehead positions.11 The nonselective nature of this reaction can be rationalized by analysis of the putative transition states shown in Scheme 2B. The key difference between the two models is the approach of the tethered alkene on the same face, or opposite face, of the ester unit. Upon coordination of the Lewis acid to the ester group, the steric effects from the ester group are similar in both cases (weak interaction with H or Me), which led to the low diastereoselectivity of the cycloaddition. Computational evaluation (B3LYP/6-31G(d))11 of the two transition-state structures reveals that they are nearly equal in energy, which is consistent with the experimentally observed low diastereoselectivity.3,12
Two strategies were pursued to improve the diastereoselectivity of the cycloaddition. The first was to utilize a t-Bu ester (vs Et ester) to increase the steric interaction between the Me and ester unit in TS-B, thus favoring TS-A. However, use of the analogous t-Bu ester of allenoate 3 resulted in nearly identical diastereoselectivity (1.4:1 vs 1.2:1 Z/E). It is likely that the steric interaction between the t-Bu ester and the Me group was still negligible.
The second strategy investigated was to use a Lewis acid catalyst that projects substituents toward the incoming alkene to perturb either TS-A or TS-B. Along these lines, N-activated oxazaborolidines were investigated due to their success in our prior efforts3 and for their ability to project substituents toward the allene.
Catalyst 5 was evaluated in the chirality transfer [2 + 2] cycloaddition and afforded 4a,b in excellent yield and chirality transfer. More importantly, improved and reversed diastereoselectivity was observed (as compared to reactions using EtAlCl2 illustrated in Scheme 2A). Encouraged by this initial result, a series of N-activated oxazaborolidines with different steric and electronic properties were evaluated (Scheme 3, catalysts 6–8); however, only minimal perturbation in selectivity was observed. Further steric tuning of the catalyst revealed that reaction with catalyst 9, which incorporates an o-PhC6H3 unit, led to improved diastereoselectivity. Additional modification of the B–Ar o-aryl group revealed that reaction with catalyst 10 delivered the product in 5.2:1 E/Z. Increasing the steric demand on the B–Ar o-aryl group (catalyst 11) led to no improvement in diastereoselectivity but a reduction in yield. Finally, to probe a match/mismatched effect, ent-3 was evaluated with catalyst 10. In this case, lower diastereoselectivity (E/Z) was observed, indicating that proper pairing of the enantiomers of substrate and catalyst is important.
Scheme 3. Evaluation of Chiral Catalystsa.

aSee the Supporting Information for experimental details. Isolated yields and crude E/Z selectivity are reported. Enantiomeric ratios determined by HPLC analysis with a chiral column.
Based on the result shown in Scheme 3 with catalyst 10, the substrate scope of this method was explored and is illustrated in Table 1. Several points are noteworthy: (1) Reaction with allene 12 bearing an NTs substituent generated 13a,b with 8:1 E/Z selectivity (Table 1, entry 1). (2) With increased substitution of the alkene of starting material, the Z-alkene isomer of product was preferentially generated with good chirality transfer (compare Table 1, entry 1 with entries 2 and 3).13 It is likely that the Z-alkene isomer of product was generated because of an unfavorable steric interaction between the β-methyl group of alkene and the ester group in a model analogous to TS-A (Scheme 2B).14 (3) Reaction with Z-alkene isomer 18 (Table 1, entry 4) provided 19a,b in 26% yield. Comparison of the results from reactions of 14 and 18 also served to confirm the stereospecificity of the process and suggests the reaction proceeded via a concerted asynchronous transition state or involves a short-lived intermediate.3,12 To rationalize the poor reactivity of substrate 18, inspection of the models 4a and 4b in Scheme 2B reveals that if a Z-β-methyl group is present an unfavorable steric interaction with the tether likely occurs, raising the energy of the TS.15 (4) Reactions with substrates bearing cyclic alkenes (Table 1, entries 5 and 6) required the use of catalyst 6 to achieve high yield. It appears that use of catalyst 10 is too sterically demanding with substrates bearing increased substitution. (5) Using the terminal dimethyl allenic ester (Table 1, entry 7), a cyclization via an ene reaction pathway was observed, albeit without chirality transfer. (6) With respect to the known limitations of the method, use of either a substrate that lacks the α-methyl group or a substrate that would lead to a six-membered ring resulted in poor conversion (not shown).11 The lack of reactivity of these substrates is likely due to reduced nucleophilicity of the alkene and poor cyclization kinetics, respectively.
Table 1.
Evaluation of Substratesa

|
See the Supporting Information for experimental details. Yields reported are the average of two experiments (0.2 mmol scale). Enantiomeric ratios determined by HPLC analysis with a chiral column. The E/Z selectivity was determined by analysis of the unpurified reaction mixture by 1H NMR.
Catalyst 6 was used for the reaction.
50 mol % of EtAlCl2 was used for the cycloaddition.
Reaction with substrate 26 was also attempted as it would lead to synthesis of novel benzofuran structures (Scheme 4). In this case, [2 + 2] cycloaddition occurred under the isomerization reaction conditions to generate 27 and 28 with high enantioselectivity. At this point, it is not clear if the thiourea catalyst also promotes the [2 + 2] cycloaddition.
Scheme 4.

Cycloaddition To Generate Benzofuran-Derived Cycloadducts
Finally, [2 + 2] cycloadditions of an allenic phosphonate have also been briefly investigated (Scheme 5). This process is significant due to the ease with which chiral allene 30 can be prepared from secondary alcohol 29 by [2,3]-rearrangement.16 Chirality transfer could be achieved using EtAlCl2 at 45 °C and afforded 31 in 13:1 Z/E selectivity and 90:10 er. It should be noted that the use of the chiral catalyst was ineffective in these reactions.
Scheme 5.

Chirality Transfer with Allenic Phosphonate
In summary, intramolecular chirality-transfer reaction of allenoates and alkenes using Lewis acids is disclosed. The use of chiral catalyst led to improved diastereoselectivity of the cycloadditions compared to the use of achiral catalysts. Future investigations aim to extend this method to intermolecular variants.
Supplementary Material
Acknowledgments
Indiana University and the NIH (R01GM110131) are acknowledged for generous financial support. M.K.B. is a 2017 Novartis Early Career Awardee. We thank Dr. Maren Pink and Dr. Chun-Hsing Chen (IU) for X-ray crystallography. The NSF XSEDE program is acknowledged for computational support.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.or-glett.7b01420.
Crystallographic data for S17 (PDB)
Crystallographic data for S18 (PDB)
Experimental procedures for all compounds and crystallographic data (PDF)
Analytical data for all compounds (PDF)
References
- 1.(a) Lee-Ruff E, Mladenova G. Chem Rev. 2003;103:1449. doi: 10.1021/cr010013a. [DOI] [PubMed] [Google Scholar]; (b) Namyslo JC, Kaufmann DE. Chem Rev. 2003;103:1485. doi: 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]; (c) Hoffmann N. Chem Rev. 2008;108:1052. doi: 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]; (d) Bach T, Hehn JP. Angew Chem, Int Ed. 2011;50:1000. doi: 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]; (e) Yoon TP. ACS Catal. 2013;3:895. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Poplata S, Tröster A, Zou YQ, Bach T. Chem Rev. 2016;116:9748–9815. doi: 10.1021/acs.chemrev.5b00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For reviews, see: Xu Y, Conner ML, Brown MK. Angew Chem, Int Ed. 2015;54:11918. doi: 10.1002/anie.201502815.Fructos MR, Prieto A. Tetrahedron. 2016;72:355.
- 3.Conner ML, Xu Y, Brown MK. J Am Chem Soc. 2015;137:3482. doi: 10.1021/jacs.5b00563. [DOI] [PubMed] [Google Scholar]
- 4.For reviews regarding chirality transfer reactions with allenes, see: Campolo D, Gastaldi S, Roussel C, Bertrand MP, Nechab M. Chem Soc Rev. 2013;42:8434. doi: 10.1039/c3cs60182j.Neff RK, Frantz DE. Tetrahedron. 2015;71:7.
- 5.(a) Carreira EM, Hastings CA, Shepard MS, Yerkey LA, Millward DB. J Am Chem Soc. 1994;116:6622. [Google Scholar]; (b) Shepard MS, Carreira EM. Tetrahedron. 1997;53:16253. [Google Scholar]; (c) Shepard MS, Carreira EM. J Am Chem Soc. 1997;119:2597. [Google Scholar]; (d) Gulías M, Collado A, Trillo B, López F, Oñate E, Esteruelas MA, Mascareñas JL. J Am Chem Soc. 2011;133:7660. doi: 10.1021/ja200784n. [DOI] [PubMed] [Google Scholar]; (e) Brummond KM, Osbourn JM. Beilstein J Org Chem. 2011;7:601. doi: 10.3762/bjoc.7.70. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Noucti NN, Alexanian EJ. Angew Chem, Int Ed. 2015;54:5447. doi: 10.1002/anie.201411737. [DOI] [PubMed] [Google Scholar]
- 6.(a) Liu H, Leow D, Huang KW, Tan CH. J Am Chem Soc. 2009;131:7212. doi: 10.1021/ja901528b. [DOI] [PubMed] [Google Scholar]; (b) Inokuma T, Furukawa M, Uno T, Suzuki Y, Yoshida K, Yano Y, Matsuzaki K, Takemoto Y. Chem - Eur J. 2011;17:10470. doi: 10.1002/chem.201101338. [DOI] [PubMed] [Google Scholar]
- 7.For selected examples of alternative syntheses of chiral allenoates, see: Li CY, Wang XB, Sun XL, Tang Y, Zheng JC, Xu ZH, Zhou YG, Dai LX. J Am Chem Soc. 2007;129:1494. doi: 10.1021/ja068642v.Crouch IT, Neff RK, Frantz DE. J Am Chem Soc. 2013;135:4970. doi: 10.1021/ja401606e.Wang Y, Zhang W, Ma S. J Am Chem Soc. 2013;135:11517. doi: 10.1021/ja406135t.Wang Y, Zhang W, Ma S. Org Chem Front. 2014;1:807.
- 8.Intramolecular [2 + 2] cycloadditions of racemic electron-deficient allenes and alkenes are known. For reactions of allenoates, see: Snider BB, Ron E. J Org Chem. 1986;51:3643.For reactions of allenyl sulfones, see: Padwa A, Lipka H, Watterson SH, Murphree SS. J Org Chem. 2003;68:6238. doi: 10.1021/jo0345796.
- 9.Vakulya B, Varga S, Csámpai A, Soós T. Org Lett. 2005;7:1967. doi: 10.1021/ol050431s. [DOI] [PubMed] [Google Scholar]
- 10.For selected reviews regarding enantioselective reactions using thiourea catalysts, see: Schreiner PR. Chem Soc Rev. 2003;32:289. doi: 10.1039/b107298f.Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713. doi: 10.1021/cr068373r.
- 11.See the Supporting Information for details.
- 12.[π2s + π2a], [(π2s + π2s) + π2s], or a quasipericyclic process with carbenoid character have all been proposed for related ketene–alkene [2 + 2] cycloadditions. For a lead reference, see: Wang X, Houk KN. J Am Chem Soc. 1990;112:1754.
- 13.The reaction to prepare 17a/b on a 1.0 mmol scale proceeded with similar yield and selectivity to that shown in Table 1, entry 3, on a 0.2 mmol scale. See the Supporting Information for details.
- 14.Reaction of (±)-14 with EtAlCl2 led to formation of (±)-15a/b in 1:6 E/Z.
- 15.Using EtAlCl2 to promote the cycloaddition of (±)-18 provided (±)-19a/b in 76% yield but only 1:1.2 E/Z selectivity.
- 16.For representative examples, see: Muller M, Mann A, Taddei M. Tetrahedron Lett. 1993;34:3289.Kalek M, Stawinski J. Adv Synth Catal. 2011;353:1741.Xin N, Ma S. Eur J Org Chem. 2012;2012:3806.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.