

Abstract
Background
Combined retinal degeneration and sensorineural hearing impairment is mostly due to autosomal recessive Usher syndrome (USH1: congenital deafness, early retinitis pigmentosa (RP); USH2: progressive hearing impairment, RP).
Methods
Sanger sequencing and NGS of 112 genes (Usher syndrome, nonsyndromic deafness, overlapping conditions), MLPA, and array‐CGH were conducted in 138 patients clinically diagnosed with Usher syndrome.
Results
A molecular diagnosis was achieved in 97% of both USH1 and USH2 patients, with biallelic mutations in 97% (USH1) and 90% (USH2), respectively. Quantitative readout reliably detected CNVs (confirmed by MLPA or array‐CGH), qualifying targeted NGS as one tool for detecting point mutations and CNVs. CNVs accounted for 10% of identified USH2A alleles, often in trans to seemingly monoallelic point mutations. We demonstrate PTC124‐induced read‐through of the common p.Trp3955* nonsense mutation (13% of detected USH2A alleles), a potential therapy target. Usher gene mutations were found in most patients with atypical Usher syndrome, but the diagnosis was adjusted in case of double homozygosity for mutations in OTOA and NR2E3, genes implicated in isolated deafness and RP. Two patients with additional enamel dysplasia had biallelic PEX26 mutations, for the first time linking this gene to Heimler syndrome.
Conclusion
Targeted NGS not restricted to Usher genes proved beneficial in uncovering conditions mimicking Usher syndrome.
Keywords: Copy number variation, Heimler syndrome, next‐generation sequencing, phenocopies, translational read‐through, Usher syndrome
Introduction
The co‐occurrence of bilateral hearing impairment (here comprehensively termed “deafness”) and visual impairment, if due to retinal degeneration, is of genetic origin in most cases in industrial countries. Usher syndrome mutations account for approximately 11% of deaf and hard of hearing children, and the population prevalence was estimated to be 1/6000 (Kimberling et al. 2010). The by far most prevalent causes are mutations in the 11 genes (MYO7A, OMIM *276903; USH1C, OMIM *605242; CDH23, OMIM *605516; PCDH15, OMIM *605514; USH1G, OMIM *607696; CIB2, OMIM *605564; USH2A, OMIM *608400; ADGRV1, OMIM *602851; DFNB31/WHRN, OMIM *607928; CLRN1, OMIM *606397; PDZD7, OMIM *612971) associated with Usher syndrome (Besnard et al. 2014), an autosomal recessive trait characterized by congenital deafness and RP in the first decade (in type 1, USH1; about 35% of cases (Petit 2001)) or by progressive hearing loss and RP of later onset in USH2 (about two thirds of patients). Symptoms apart from deaf‐blindness, however, may indicate other (genetic) diagnoses (e.g., disease related to mutations in PEX1, OMIM *602136, or PEX6, OMIM *601498). Especially in consanguineous families, simultaneous presence of two non‐syndromic sensory deficits must be taken into account. In our comprehensive analysis of a large cohort of deaf‐blindness patients clinically diagnosed as Usher syndrome, we therefore conducted both conventional Sanger and next‐generation sequencing (NGS) of a large gene panel not only comprising the Usher genes but also the known genes for non‐syndromic deafness and for syndromes that may comprise both sensory deficits. We efficiently established identification of CNVs from NGS data, highlighting targeted NGS as a tool for diagnosing both point mutations and copy number alterations. Simultaneous homozygosity for mutations in genes associated with isolated retinal degeneration and hearing loss (OTOA, OMIM *607038 and NR2E3, OMIM *604485), and mutations in PEX26 (OMIM *608666) in patients with additional enamel dysplasia demonstrate how rare, genetically distinct entities may mimic Usher syndrome.
Materials and Methods
Ethical compliance
Samples were obtained with written informed consent. All investigations were conducted according to the Declaration of Helsinki, and the study was approved by the institutional review board of the Ethics Committee of the University Hospital of Cologne.
Patients
The patients had been referred to our diagnostic laboratory with the diagnosis of retinal degeneration and sensorineural hearing loss, and therefore in most cases with suspected Usher syndrome (see below for exceptions concerning deafness patients). About two third of the patients were of German descent, and the remaining one third were from Saudi Arabia (KSA) and other Middle East/North African (MENA) countries (Fig. 1D). Patients whose phenotype was compatible with USH1 or USH2 were grouped accordingly. Patients whose symptoms comprised retinal degeneration and hearing impairment but did neither correspond to USH1 nor to USH2 (either because of clinical course or “plus symptoms” that were unusual for Usher syndrome) were categorized as “atypical Usher syndrome”. In nine pediatric or adolescent patients with apparently non‐syndromic deafness who had been referred for genetic testing of hearing loss genes (including the most important syndrome genes like those for Usher syndrome), the diagnosis was reversed (to a syndrome with RP to develop in the future) due to the genetic findings. For clarity, and although these patients had not been referred as Usher syndrome patients, they were grouped retrospectively under the clinical subtype that is usually associated with the respective gene (Table 1).
Figure 1.
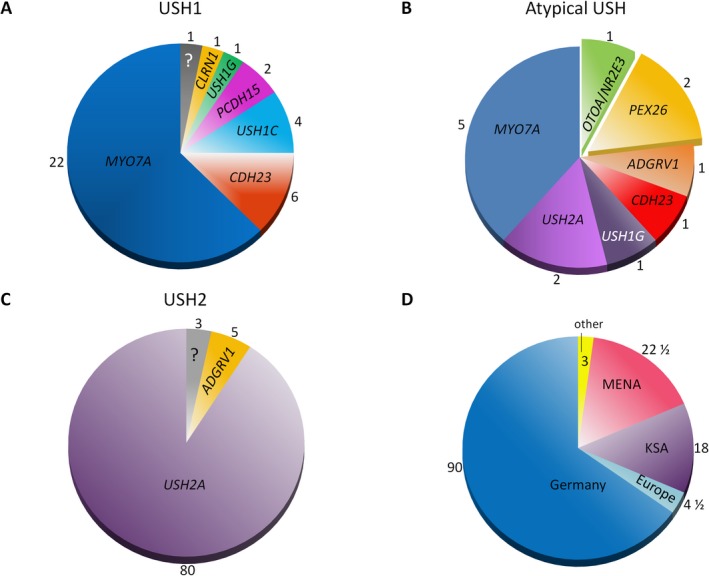
Diagnostic yield and mutational spectrum in patients clinically diagnosed with different types of Usher syndrome. Numbers correspond to patient numbers. ?, unsolved patients. (A) USH1. (B) Atypical Usher syndrome (including patients with additional, non‐sensory symptoms). (C) USH2. (D) Ethnic origin of patients. Patients were counted as 1/2 + 1/2 if parents had different ethnical backgrounds.
Table 1.
Mutations identified in our study
| Pat | Gene | Allele 1 | dbSNP | Ref | Met | Allele 2 | dbSNP | S | Ref | Met | Age (years) | Cons | Origin | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| USH1 | |||||||||||||||
| 101 | MYO7A | c.397C>T | p.His133Tyr | rs111033403 | Le Guedard‐Mereuze et al. (2010) | NGS | c.640G>A | p.Gly214Arg | rs111033283 | Adato et al. (1997); Aparisi et al. (2013) | NGS | 36 | No | KSA | |
| DEM289 | MYO7A | c.397dupC | p.His133Profs*7 | ‐‐‐ | nov | NGS | c.397dupC | p.His133Profs*7 | ‐‐‐ | nov | NGS | 29 | Yes | Pak | |
| DEM318 | MYO7A | c.397dupC | p.His133Profs*7s | ‐‐‐ | nov | NGS | c.397dupC | p.His133Profs*7 | ‐‐‐ | nov | NGS | n.d. | Yes | Pak | |
| 94 | MYO7A | c.470+1G>A | Splice | ‐‐‐ | Adato et al. (1997) | NGS | c.470+1G>A | Splice | ‐‐‐ | Adato et al. (1997) | NGS | n.d. | Yes | KSA | |
| 102 | MYO7A | c.470+1G>A | Splice | ‐‐‐ | Adato et al. (1997) | NGS | c.4818delG | p.Lys1606Asnfs*39 | ‐‐‐ | ‐‐‐ | NGS | 18 | Yes | KSA | |
| 110 | MYO7A | c.470+1G>A | Splice | ‐‐‐ | Adato et al. (1997) | NGS | c.470+1G>A | Splice | ‐‐‐ | x | Adato et al. (1997) | NGS | 5 | Yes | KSA |
| 87 | MYO7A | c.496delG | p.Glu166Argfs*5 | rs111033448 | Riazuddin et al. (2008) | SaS | c.5617C>T | p.Arg1873Trp | ‐‐‐ | x | Riazuddin et al. (2008) | SaS | 35 | No | Italy |
| DEM279 | MYO7A | c.745G>T | p.Glu249* | ‐‐‐ | nov | NGS | c.745G>T | p.Glu249* | ‐‐‐ | nov | NGS | 16 | Yes | Pak | |
| 1 | MYO7A | c.848T>C | p.Met283Thr | ‐‐‐ | nov | SaS | c.1093G>A | p.Asp365Asn | ‐‐‐ | nov | SaS | 30 | No | Turkey | |
| 63 | MYO7A | c.1189G>A | p.Ala397Thr | ‐‐‐ | Kimberling et al. (2010) | NGS | Del ex21‐27 | Truncation | ‐‐‐ | x | nov | NGS | 29 | No | Ger |
| 88 | MYO7A | c.1903T>C | p.Cys635Arg | ‐‐‐ | nov | NGS | c.1903T>C | p.Cys635Arg | ‐‐‐ | nov | NGS | 11 | Yes | KSA | |
| 53* | MYO7A | c.2181dupT | p.Leu728Serfs*6 | ‐‐‐ | nov | NGS | c.5749G>T | p.Glu1917* | ‐‐‐ | x | Jacobson et al. (2009); Aparisi et al. (2014) | NGS | 2 | No | Ger |
| 109 | MYO7A | c.2904G>T | p.Glu968Asp | rs111033233 | Bharadwaj et al. (2000) | SaS | c.6409G>A | p.Gly2137Arg | ‐‐‐ | x | nov | SaS | 29 | No | Ger/Per |
| 130 | MYO7A | c.2904G>T | p.Glu968Asp | rs111033233 | Bharadwaj et al. (2000) | NGS | c.2904G>T | p.Glu968Asp | rs111033233 | Bharadwaj et al. (2000) | NGS | 28 | Yes | Syria | |
| 104 | MYO7A | c.3547C>A | p.Pro1183Thr | ‐‐‐ | Cremers et al. (2007) | NGS |
c.5879_5880delAC (c.5637‐3C>G) |
Truncation (Splice?) |
‐‐‐ ‐‐‐ |
x | nov Cremers et al. (2007) | NGS | 58 | No | Ger |
| 5 | MYO7A | c.3719G>A | p.Arg1240Gln | rs111033178 | Janecke et a(l. 1999); Jacobson et al. (2011) | NGS | c.5320T>G | p.Phe1774Va | rs62625014 | x | Yoshimura et al. (2013) | NGS | 9 | No | Ger |
| 50* | MYO7A | c.3719G>A | p.Arg1240Gln | rs111033178 | Janecke et al. (1999); Jacobson et al. (2011) | NGS | c.3719G>A | p.Arg1240Gln | rs111033178 | x | Janecke et al. (1999); Jacobson et al. (2011) | NGS | 4 | No | Ger |
| 2 | MYO7A | c.3747delG | p.Leu1249Leufs*14 | ‐‐‐ | nov | SaS | c.3851_3878dup28 | p.Leu1293Leufs*24 | ‐‐‐ | nov | SaS | 47 | No | Turkey | |
| 99 | MYO7A | c.3764delA | p.Lys1255Argfs*8 | ‐‐‐ | Jaijo et al. (2007) | NGS | c.3764delA | p.Lys1255Argfs*8 | ‐‐‐ | x | Jaijo et al. (2007) | NGS | 34 | No | Ger |
| 90 | MYO7A | c.5886_5888del | p.Phe1963del | rs111033232 | Roux et al. (2006) | NGS | c.5886_5888del | p.Phe1963del | rs111033232 | Roux et al. (2006) | NGS | 37 | Yes | KSA | |
| 9237* | MYO7A | c.5958dupA | p.Leu1987Thrfs*89 | nov | NGS | c.5958dupA | p.Leu1987Thrfs*89 | nov | NGS | 9 | Yes | Egypt | |||
| 117 | MYO7A | c.6231dupG | p.Lys2078Glufs*50 | ‐‐‐ | nov | NGS | c.6231dupG | p.Lys2078Glufs*50 | ‐‐‐ | nov | NGS | 13 | Yes | KSA | |
| 6 | CDH23 | c.1411_1412delins | p.Glu471Ser | ‐‐‐ | nov | NGS | c.3862C>T | p.Gln1288* | ‐‐‐ | x | nov | NGS | 29 | No | Sri L. |
| 51* | CDH23 | c.1528A>T | p.Lys510* | ‐‐‐ | nov | NGS | c.1528A>T | p.Lys510* | ‐‐‐ | x | nov | NGS | 3 m | n.d. | KSA |
| DEM127 | CDH23 | c.1701_1702del | p.Gly568Cysfs*20 | ‐‐‐ | nov | MS, SaS | c.1701_1702del | p.Gly568Cysfs*20 | ‐‐‐ | nov | MS, SaS | n.d. | Yes | Pak | |
| 122 | CDH23 | c.4393dupG | p.Ala1465Glyfs*3 | ‐‐‐ | nov | NGS | c.4393dupG | p.Ala1465Glyfs*3 | ‐‐‐ | x | nov | NGS | 15 | n.d. | Syria |
| 12 | CDH23 | c.6047‐9G>A | Splice | ‐‐‐ | von Brederlow et al. (2002) | NGS | c.6047‐9G>A | Splice | ‐‐‐ | x | von Brederlow et al. (2002) | NGS | 13 | No | Italy |
| DEM296 | CDH23 | c.6047‐9G>A | Splice | ‐‐‐ | von Brederlow et al. (2002) | NGS | c.6047‐9G>A | Splice | ‐‐‐ | von Brederlow et al. (2002) | NGS | n.d. | Yes | Pak | |
| 95 | CLRN1 | c.301_305del | p.Val101Serfs*27 | ‐‐‐ | Akoury et al. (2011) | NGS | c.301_305del | p.Val101Serfs*27 | ‐‐‐ | Akoury et al. (2011) | NGS | 13 | Yes | Leban | |
| 7 | PCDH15 | c.401G>A | p.Arg134Gln | rs137853003 | Ahmed et al. (2003) | MS, SaS | Del ex1‐3 | Truncation | ‐‐‐ | x | Aller et al. (2010a,b) | MS, MLPA | 30 | Yes | Syria/Turkey |
| 8 | PCDH15 | Del ex1‐3 | Truncation | ‐‐‐ | Aller et al. (2010a,b) | MS, MLPA | Del ex1‐3 | Truncation | ‐‐‐ | Aller et al. (2010a,b) | MS, MLPA | n.d. | Yes | Syria | |
| 123 | USH1C | c.521+1G>A | Splice | ‐‐‐ | nov | NGS | c.521+1G>A | Splice | ‐‐‐ | nov | NGS | 18 | Yes | Syria | |
| 96 | USH1C | Del ex3‐27 | Truncation | ‐‐‐ | Bitner‐Glindzicz et al. (2000) | NGS | Del ex3‐27 | Truncation | ‐‐‐ | Bitner‐Glindzicz et al. (2000) | NGS | 9 | n.d. | KSA | |
| 97 | USH1C | Del ex3‐27 | Truncation | ‐‐‐ | Bitner‐Glindzicz et al. (2000) | CGH | Del ex3‐27 | Truncation | ‐‐‐ | Bitner‐Glindzicz et al. (2000) | CGH | 2 | n.d. | KSA | |
| 9140* | USH1C | c.1210+1G>C | Splice | ‐‐‐ | nov | NGS | c.1210+1G>C | Splice | ‐‐‐ | nov | NGS | 10 | Yes | Egypt | |
| 134* | USH1G | c.1311delG | p.Lys438Argfs*6 | ‐‐‐ | nov | NGS | c.1311delG | p.Lys438Argfs*6 | ‐‐‐ | nov | NGS | 9 | Yes | KSA | |
| USH1 | Unsolved | ||||||||||||||
| 56 | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS, NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS, NGS | 34 | No | Greece | |
| Atyp | |||||||||||||||
| 103 | CDH23 | Dup ex19‐27 | Truncation? | ‐‐‐ | nov | NGS | Dup ex19‐27 | Truncation? | ‐‐‐ | x | nov | NGS | 30 | Yes | KSA |
| DEM74 | USH1G | c.1373 A>T | p.Asp458Val | rs397517925 | Kalay et al. (2005 ) | GLA, SaS | c.1373 A>T | p.Asp458Val | rs397517925 | Kalay et al. (2005 ) | MS, SaS | Yes | Pak | ||
| 30 | ADGRV1 | c.6981delT | p.Gly2328Valfs*7 | ‐‐‐ | nov | NGS | c.14044‐1G>A | Splice | ‐‐‐ | x | nov | NGS | Ger | ||
| 9 | MYO7A | c.849+5G>A | Splice | ‐‐‐ | nov | NGS | c.3907_3910dup | p.Ala1304Aspfs*5 | ‐‐‐ | nov | NGS | 27 | No | Ger | |
| 11 | MYO7A | c.3262C>T | p.Gln1088* | ‐‐‐ | nov | NGS | c.6439‐2A>G | Splice | ‐‐‐ | Mutai et al. (2013); Glockle et al. (2014) | NGS | 26 | No | Ger | |
| 13 | MYO7A | c.3503G>A | p.Arg1168Gln | ‐‐‐ | nov | NGS | c.6025delG | Truncation | ‐‐‐ | Bharadwaj et al. (2000) | NGS | 48 | No | Ger | |
| 14 | MYO7A | c.3503G>A | p.Arg1168Gln | ‐‐‐ | Aparisi et al. (2014) | NGS | c.5573T>C | p.Leu1858Pro | ‐‐‐ | Bharadwaj et al. (2000) | NGS | 44 | No | Ger | |
| 10 | MYO7A | c.3718C>T | p.Arg1240Trp | ‐‐‐ | Janecke et al. (1999); Cremers et al. (2007) | SaS | c.4814C>A | p.Ser1605Tyr | ‐‐‐ | x | nov | SaS | 25 | No | Ger |
| 75 | USH2A | c.1036A>C | p.Asn346His | ‐‐‐ | Weston et al. (2000) | NGS | c.7967delA | p.Asn2656Ilefs*18 | ‐‐‐ | x | nov | NGS | 22 | No | Ger |
| 136* | PEX26 | c.3G>A | p.Met1? | ‐‐‐ | nov | NGS | c.292C>T | p.Arg98Trp | rs62641228 | Matsumoto et al. (2003); Furuki et al. (2006); Berendse et al. (2016) | NGS | 3 | No | Ger | |
| 135 | PEX26 | c.127G>C | p.Asp43His | ‐‐‐ | nov | NGS | c.292C>T | p.Arg98Trp | rs62641228 | Matsumoto et al. (2003); Furuki et al. (2006); Berendse et al. (2016) | NGS | 13 | No | Ger | |
| 93 | OTOA | gene deletion | Gene loss | ‐‐‐ | Shahin et al. (2010); Sloan‐Heggen et al. (2016) | NGS | Gene deletion | Gene loss | ‐‐‐ | Shahin et al. (2010); Sloan‐Heggen et al. (2016) | NGS | 6 | Yes | KSA | |
| NR2E3 | c.932G>A | p.Arg311Gln | ‐‐‐ | Haider et al. (2000) | SaS | c.932G>A | p.Arg311Gln | rs28937873 | Haider et al. (2000) | SaS | |||||
| Atyp | Monoallelic | ||||||||||||||
| 57 | USH2A | c.13316C>T | p.Thr4439Ile | ‐‐‐ | Dreyer et al. (2008) | NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | NGS | 86 | No | Ger | |
| USH2 | |||||||||||||||
| 89 | USH2A | c.486‐1G>C | Splice | ‐‐‐ | Cremers et al. (2007) | NGS | c.486‐1G>C | Splice | ‐‐‐ | Cremers et al. (2007) | NGS | 17 | Yes | KSA | |
| 118 | USH2A | c.486‐1G>C | Splice | ‐‐‐ | Cremers et al. (2007) | NGS | c.486‐1G>C | Splice | ‐‐‐ | Cremers et al. (2007) | NGS | 24 | Yes | KSA | |
| 15 | USH2A | c.486‐14G>A | Splice | ‐‐‐ | Le Guedard‐Mereuze et al. (2010); Neveling et al. (2012) | SaS | c.6805+1G>A | Splice | ‐‐‐ | nov | SaS | 44 | No | Ger | |
| 34 | USH2A | c.653T>A | p.Val218Glu | ‐‐‐ | Leroy et al. (2001) | SaS | c.949C>A | Silent/Splice | ‐‐‐ | Pennings et al. (2004) | SaS | 30 | No | Ger | |
| 52 | USH2A | c.653T>A | p.Val218Glu | ‐‐‐ | Leroy et al. (2001) | NGS | c.2276G>T | p.Cys759Phe | rs80338902 | x | Rivolta et al. (2000); Garcia‐Garcia et al. (2011) | NGS | 48 | No | Ger |
| 33 | USH2A | c.653T>A | p.Val218Glu | ‐‐‐ | Leroy et al. (2001) | SaS | c.8681+1G>T | Splice | ‐‐‐ | nov | SaS | 58 | No | Ger | |
| 37 | USH2A | c.775_776delAG | p.Ser259Phefs*63 | ‐‐‐ | Seyedahmadi et al. (2004) | SaS | c.9424G>T | p.Gly3142* | ‐‐‐ | x | Baux et al. (2007) | SaS | 40 | No | Ger |
| 125 | USH2A | c.802G>A | p.Gly268Arg | ‐‐‐ | Dreyer et al. (2008); Huang et al. (2015) | SaS | c.9346C>A | p.Pro3116Thr | ‐‐‐ | x | nov | SaS | 44 | No | Ger |
| 41 | USH2A | c.920_923dup | p.His308Glnfs*16 | ‐‐‐ | Weston et al. (2000) | SaS | Del ex38‐41 | Truncation | ‐‐‐ | nov | MLPA | 26 | No | Ger | |
| 132 | USH2A | c.920_923dup | p.His308Glnfs*16 | ‐‐‐ | Weston et al. (2000) | NGS | c.6084T>A | p.Tyr2028* | ‐‐‐ | Krawitz et al. (2014) | NGS | 44 | No | Ger | |
| 43 | USH2A | c.949C>A | Silent/Splice | ‐‐‐ | Pennings et al. (2004) | SaS | c.14131C>T | p.Gln4711* | ‐‐‐ | McGee et al. (2010) | SaS | 48 | No | Ger | |
| 58 | USH2A | c.1000C>T | p.Arg334Trp | ‐‐‐ | Adato et al. (2000) | NGS | c.6805+2T>C | Splice | ‐‐‐ | x | Krawitz et al. (2014) | NGS | 60 | No | Ger |
| 121 | USH2A | c.1036A>C | p.Asn346His | rs369522997 | Weston et al. (2000); Sadeghi et al. (2013); Wang et al. (2014); Lenassi et al. (2015a,b) | SaS | c.8723_8724del | p.Val2908Glyfs*29 | ‐‐‐ | x | van Wijk et al. (2004) | SaS | 32 | No | Ger |
| 73 | USH2A | c.1036A>C | p.Asn346His | rs369522997 | Weston et al. (2000); Sadeghi et al. (2013); Wang et al. (2014); Lenassi et al. (2015a,b) | SaS | c.10561T>C | p.Trp3521Arg | rs111033264 | x | Dreyer et al. (2008); McGee et al. (2010) | SaS | 60 | No | Ger |
| 42 | USH2A | c.1039G>C | p.Asp347His | ‐‐‐ | Pierrache et al. (2016) | SaS | c.14131C>T | p.Gln4711* | ‐‐‐ | McGee et al. (2010) | SaS | 38 | No | Ger | |
| 76 | USH2A | c.1606T>C | p.Cys536Arg | rs111033273 | Dreyer et al. (2000; Bhattacharya et al. (2004) | NGS | c.1606T>C | p.Cys536Arg | rs111033273 | Dreyer et al. (2000); Bhattacharya et al. (2004) | NGS | 19 | No | Ger | |
| 68 | USH2A | c.1752C>A | p.Cys584* | ‐‐‐ | nov | SaS | c.5122G>A | p.Gly1708Arg | ‐‐‐ | nov | SaS | 45 | No | Ger | |
| 131 | USH2A | c.1876C>T | p.Arg626* | rs534534437 | Weston et al. (2000) | NGS | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | NGS | 36 | No | Ger | |
| 8 | USH2A | c.2073C>A | p.Cys691* | ‐‐‐ | Seyedahmadi et al. (2004) | NGS | c.2209C>T | p.Arg737* | rs111033334 | Kaiserman et al. (2007) | NGS | 76 | No | Ger | |
| 100 | USH2A | c.2209C>T | p.Arg737* | rs111033334 | Kaiserman et al. (2007) | NGS | c.6657+3_6657+6del | Splice | ‐‐‐ | nov | NGS | 23 | No | Ger | |
| 28 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.8682‐9A>G | Splice | ‐‐‐ | x | Dreyer et al. (2008); Glockle et al. (2014) | SaS | 59 | No | Ger |
| 32 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.920_923dup | p.His308Glnfs*16 | ‐‐‐ | Weston et al. (2000) | SaS | 69 | No | Ger | |
| 46 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.949C>A | Silent/Splice | ‐‐‐ | Pennings et al. (2004) | SaS | 47 | No | Ger | |
| 31 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. 1998) | SaS | c.2299delG | p.Glu767Serfs*21 | rs80338903 | x | Eudy et al. (1998) | SaS | 35 | No | Ger |
| 59 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | 25 | No | Ger | |
| 47 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.2610C>A | p.Cys870* | ‐‐‐ | x | Le Quesne Stabej et al. (2012) | SaS | 25 | No | Ger/Bos |
| 127 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS |
c.4714C>T (c.5516T>A) |
p.Leu1572Phe (p.Val1839Glu) |
rs111033333 ‐‐‐ |
Song et al. (2011); Zhao et al. (2015); Sloan‐Heggen et al. (2016) nov | SaS | 33 | No | Ger | |
| 108 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.8522G>A | p.Trp2841* | ‐‐‐ | Sloan‐Heggen et al. (2016) | SaS | 30 | No | Ger | |
| 72 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | c.11831C>A | p.Ala3944Asp | ‐‐‐ | Krawitz et al. (2014) | SaS | 26 | No | Ger | |
| 79* | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | NGS | c.11864G>A | p.Trp3955* | rs111033364 | x | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | NGS | 15 | No | Ger |
| 44 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | Del ex22‐24 | Truncation | ‐‐‐ | Krawitz et al. (2014); Dad et al. (2015) | MLPA | 43 | No | Ger | |
| 78* | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | NGS | Del ex15‐21 | Truncation | ‐‐‐ | X | Baux et al. (2007) | NGS | 1 | No | Ger |
| 71 | USH2A | c.3648C>A | p.Tyr1216* | ‐‐‐ | nov | SaS | Del ex22‐24 | Truncation | ‐‐‐ | x | Krawitz et al. (2014); Dad et al. (2015) | MLPA | 19 | No | Ger |
| 40 | USH2A | c.4314delG | p.Ile1439Tyrfs*15 | ‐‐‐ | nov | SaS | c.4314delG | p.Ile1439Tyrfs*15 | ‐‐‐ | x | nov | SaS | 17 | Yes | Asia |
| 35 | USH2A | c.4773delA | p.Glu1591Glufs*2 | ‐‐‐ | nov | SaS | Del ex5‐11 | Truncation | ‐‐‐ | nov | MLPA | 30 | No | Ger | |
| 112 | USH2A | c.4933G>T | p.Gly1645* | ‐‐‐ | Sloan‐Heggen et al. (2016) | SaS | Del ex22‐24 | Truncation | ‐‐‐ | Krawitz et al. (2014); Dad et al. (2015) | MLPA | 33 | No | Ger | |
| 84 | USH2A | c.5776+1G>A | Splice | ‐‐‐ | Dreyer et al. (2008); Wang et al. (2014; Patel et al. (2016) | NGS | c.5776+1G>A | Splice | ‐‐‐ | x | Dreyer et al. (2008); Wang et al. (2014); Patel et al. (2016) | NGS | 13 | Yes | KSA |
| 85 | USH2A | c.7076T>G | p.Leu2359* | ‐‐‐ | Yang et al. (2013) | SaS | c.7595‐2144A>G | Splice | ‐‐‐ | x | Vache et al. (2012) | SaS | 26 | No | Ger |
| 82 | USH2A | c.7198delG | p.Asp2400Metfs*13 | ‐‐‐ | nov | SaS | c.7198delG | p.Asp2400Metfs*13 | ‐‐‐ | x | nov | SaS | 42 | No | Ger |
| 129 | USH2A | c.7595‐2144A>G | Splice | ‐‐‐ | Vache et al. (2012) | NGS | Del ex10‐11 | Truncation | ‐‐‐ | Baux et al. (2014) | NGS | 26 | No | Ger | |
| 60 | USH2A | c.8240delC | p.Pro2747Hisfs*22 | ‐‐‐ | nov | SaS | c.13898delT | p.Leu4633* | ‐‐‐ | nov | SaS | 71 | No | Ger | |
| 126 | USH2A | c.8834G>A | p.Trp2945* | McGee et al. (2010) | SaS | c.10561T>C | p.Trp3521Arg | rs111033264 | Dreyer et al. (2008); McGee et al. (2010) | SaS | 34 | No | Ger | ||
| 107* | USH2A | c.9258+1G>A | Splice | ‐‐‐ | nov | SaS | c.7595‐2144A>G | Splice | ‐‐‐ | x | Vache et al. (2012) | SaS | 1 | No | Ger |
| 98 | USH2A | c.8682‐9A>G | Splice | ‐‐‐ | Dreyer et al. (2008); Glockle et al. (2014) | SaS | c.12525G>A | p.Trp4175* | ‐‐‐ | x | Yan et al. (2009) | SaS | 34 | No | Ger |
| 115 | USH2A | c.8915delC | p.Ser2972Phefs*2 | ‐‐‐ | nov | SaS | c.12234_12235delGA | p.Asn4079Trpfs*19 | rs398124618 | Baux et al. (2007) | SaS | 25 | No | Ger | |
| 38 | USH2A | c.9424G>T | p.Gly3142* | ‐‐‐ | Baux et al. (2007) | SaS | c.9424G>T | p.Gly3142* | ‐‐‐ | Baux et al. (2007) | SaS | 31 | No | Ger | |
| 64 | USH2A | c.9676C>T | p.Arg3226* | ‐‐‐ | Katagiri et al. (2014) | NGS | c.7595‐2144A>G | Splice | ‐‐‐ | x | Vache et al. (2012) | SaS | 21 | No | Ger |
| 86 | USH2A | c.9815C>T | p.Pro3272Leu | ‐‐‐ | Herrera et al. (2008) | SaS | c.5607_5615del | p.Arg1870_Ala1872del | ‐‐‐ | Krawitz et al. (2014) | NGS | 44 | No | Ger | |
| 114 | USH2A | c.6928A>C | p.Thr2310Pro | ‐‐‐ | Le Quesne Stabej et al. (2012) | SaS | c.11864G>A | p.Trp3955* | rs111033364 | x | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | NGS | 16 | No | Ger |
| 62 | USH2A | c.10388‐1G>A | Splice | ‐‐‐ | nov | SaS | c.11054G>A | p.Trp3685* | ‐‐‐ | nov | SaS | 31 | No | Ger | |
| 69 | USH2A | c.10388‐1G>A | Splice | ‐‐‐ | nov | SaS | c.11054G>A | p.Trp3685* | ‐‐‐ | nov | SaS | 19 | No | Ger | |
| 66 | USH2A | c.10759C>T | p.Gln3587* | rs111033418 | Garcia‐Garcia et al. (2011) | NGS | c.11549‐1G>A | Splice | ‐‐‐ | Lenassi et al. (2015a,b) | NGS | 12 | No | Ger | |
| 111 | USH2A | c.11065C>T | p.Arg3689* | rs41314534 | Le Quesne Stabej et al. (2012); Aparisi et al. (2014) | SaS | c.12234_12235delGA | p.Asn4079Trpfs*19 | rs398124618 | x | Baux et al. (2007) | SaS | 36 | No | Ger |
| 22 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.775_776delAG | p.Ser259Phefs*63 | ‐ | x | Seyedahmadi et al. (2004) | SaS | 68 | No | Ger |
| 17 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.1036A>C | p.Asn346His | rs369522997 | Weston et al. (2000); Sadeghi et al. (2013); Wang et al. (2014); Lenassi et al. (2015a,b) | SaS | 38 | No | Ger | |
| 81 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.1271delT | p.Met424Argfs*34 | ‐‐‐ | nov | SaS | 14 | No | Ger | |
| 106 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.1807G>A | p.Gly603Arg | ‐‐‐ | x | nov | SaS | 30 | No | Ger |
| 24 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | 45 | No | Ger | |
| 25 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. 2012) | SaS | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS | 17 | No | Ger | |
| 83 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.4102C>T | p.Pro1368Ser | ‐‐‐ | x | nov | SaS | 19 | No | Ger |
| 23 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.6642dupT | p.Leu2215Serfs*16 | ‐‐‐ | nov | SaS | 43 | No | Ger | |
| 26 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.9270C>A | p.Cys3090* | ‐‐‐ | McGee et al. (2010) | SaS | 21 | No | Ger | |
| 48 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.11048‐2A>G | Splice | ‐‐‐ | nov | SaS | 77 | No | Ger | |
| 19 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.11549‐1G>A | Splice | ‐‐‐ | Lenassi et al. (2015a,b) | SaS | 26 | No | Ger | |
| 18 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | 41 | n.d. | Turkey | |
| 49 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | NGS | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. )2004); Le Quesne Stabej et al. (2012) | SaS | 41 | n.d. | Rus | |
| 20 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | Dup ex4 | Truncation? | ‐‐‐ | nov | MLPA | 49 | No | Ger | |
| 16 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | Del ex22‐24 | Truncation | ‐‐‐ | Krawitz et al. (2014); Dad et al. (2015) | MLPA | 47 | No | Ger | |
| 113 | USH2A | c.11864G>A | p.Trp3955* | rs111033364 | van Wijk et al. (2004); Le Quesne Stabej et al. (2012) | SaS | Del ex22‐24 | Truncation | ‐‐‐ | x | Krawitz et al. (2014); Dad et al. 2015) | MLPA | 34 | No | Ger |
| 80 | USH2A | c.12067‐2A>G | Splice | ‐‐‐ | Auslender et al. (2008); Aparisi et al. (2014) | NGS | c.12067‐2A>G | Splice | ‐‐‐ | Auslender et al. (2008); Aparisi et al. (2014) | NGS | 47 | No |
Jewish M‐Asia |
|
| 105 | USH2A | c.13010C>T | p.Thr4337Met | ‐‐‐ | Aller et al. (2006); Besnard et al. (2014); Ge et al. (2015) | SaS | c.14439_14454del | p.Cys4813* | ‐‐‐ | Koparir et al. (2015) | NGS | 21 | No | Ger | |
| 77 | USH2A | c.14439_14454del | p.Cys4813* | ‐‐‐ | Koparir et al. (2015) | NGS | c.14439_14454del | p.Cys4813* | ‐‐‐ | Koparir et al. (2015) | NGS | 44 | n.d. | Turkey | |
| 91 | USH2A | c.15017C>T | p.Thr5006Met | ‐‐‐ | Huang et al. (2015) | NGS | c.15017C>T | p.Thr5006Met | ‐‐‐ | Huang et al. (2015) | NGS | 37 | Yes | KSA | |
| 61 | USH2A | Del ex14 | Truncation | ‐‐‐‐ | Aparisi et al. (2014); Garcia‐Garcia et al. (2014); Glockle et al. (2014) | MLPA | Del ex14 | Truncation | ‐‐‐‐ | x | Aparisi et al. (2014); Garcia‐Garcia et al. (2014); Glockle et al. (2014) | MLPA | 21 | n.d. | Syria |
| 120 | USH2A | Del ex45‐47 | Truncation | ‐‐‐ | Baux et al. (2014) | NGS | Del ex45‐47 | Truncation | ‐‐‐ | Baux et al. (2014) | NGS | 25 | Yes | Syria | |
| 39 | USH2A | Del ex48 | Truncation | ‐‐‐ | Neveling et al. (2013) | SaS, MLPA | Del ex48 | Truncation | ‐‐‐ | x | Neveling et al. (2013) | SaS, MLPA | 21 | n.d. | Turkey |
| 133 | ADGRV1 | c.7606G>T | p.Glu2536* | ‐‐‐ | nov | NGS | c.7606G>T | p.Glu2536* | ‐‐‐ | nov | NGS | 28 | n.d. | Ger | |
| 124 | ADGRV1 | c.8749G>T | p.Glu2917* | ‐‐‐ | nov | NGS | Del ex85 | Truncation | ‐‐‐ | nov | NGS | 45 | No | Ger | |
| 45 | ADGRV1 | c.15716delA | p.Asn5239Thrfs*19 | ‐‐‐ | nov | NGS | c.17204+5G>C | Splice | ‐‐‐ | nov | NGS | 17 | No | Ger | |
| USH2 | Monoallelic | ||||||||||||||
| 27 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS, NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS | 50 | No | Ger | |
| 119 | USH2A | c.2299delG | p.Glu767Serfs*21 | rs80338903 | Eudy et al. (1998) | SaS, NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | x | ‐‐‐ | SaS, NGS | 55 | No | Ger |
| 29 | USH2A | c.2522C>A | p.Ser841Tyr | rs111033282 | Jaijo et al. (2010); Garcia‐Garcia et al. (2011) | SaS, NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS, NGS | 59 | No | Ger | |
| 128 | USH2A | c.8682‐9A>G | Splice | ‐‐‐ | Dreyer et al. (2008); Glockle et al. (2014) | SaS, NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS, NGS | 35 | No | Ger | |
| 65 | ADGRV1 | c.12895C>T | p.Arg4299* | ‐‐‐ | nov | NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | NGS | 27 | No | Tunisia | |
| 92 | ADGRV1 | c.11410C>T | p.Arg3804* | ‐‐‐ | nov | NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | NGS | 25 | n.d. | KSA | |
| USH2 | Unsolved | ||||||||||||||
| 55 | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS/NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS/NGS | 55 | No | Ger | |
| 70 | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS, NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | SaS, NGS | 50 | No | Ger | |
| 74 | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | NGS | ‐‐‐ | ‐‐‐ | ‐‐‐ | ‐‐‐ | NGS | 50 | No | Ger |
Pat, patient number; Ref, reference from the literature; Met, applied method(s); add. allele, heterozygous mutation in a secondary locus (in most cases reflecting carriership for a recessive mutation); nov, novel mutation (not previously reported); m, months; *, no retinal dystrophy diagnosed at the time the genetic diagnosis was made; SaS, Sanger sequencing; PEX1/PEX6, these genes were sequenced by the Sanger method before targeted panel‐NGS was applied; MS, locus‐specific polymorphic microsatellite markers for the known USH1 genes were genotyped. GLA, SNP‐array‐based genome‐wide linkage analysis. S, Segregation analysis performed. Ger, German; KSA, Kingdom of Saudi Arabia; Pak, Pakistan; Per, Persia; Sri L., Sri Lanka. In case of more than two potentially pathogenic alleles, the least likely causative one is in brackets.
Workflow of genetic analysis and determination of diagnostic yield
The analytic workflow depended on the assumed diagnosis and the request of the physician in charge of the patient. If the clinical diagnosis was USH2, Sanger sequencing (and possibly MLPA) of the USH2A exons was the initial step of genetic testing in most cases because of the high probability to identify the causative mutation with this approach, followed by NGS for patients without USH2A mutations. For most patients who were categorized as USH1 or atypical Usher syndrome, NGS was carried out without other precedent tests. In P135, whose symptoms indicated a peroxisome biogenesis disorder (PBD), Sanger sequencing of PEX1 and PEX6 was carried out, followed by NGS. MLPA or array‐CGH analysis was conducted to verify CNVs that were indicated by quantitative analysis of NGS data (see below). In a few cases, genotyping of Usher locus‐specific polymorphic microsatellite markers or genome‐wide linkage analysis (as reported previously (Zaki et al. 2016)) preceded gene analysis.
When calculating the diagnostic yield, we considered patients with monoallelic mutations in a gene compatible with the respective clinical subtype as “resolved”, assuming that the secondary mutations had escaped detection due to atypical extra‐exonic localizations (deep‐intronic, non‐coding regulatory regions).
Next‐generation sequencing (NGS)
Targeted next‐generation sequencing (NGS) was conducted for 112 genes (1914 coding exons) that have been associated with non‐syndromic (NSHL) and selected forms of syndromic hearing loss (SHL), including 11 genes associated with Usher syndrome (MYO7A/USH1B; USH1C; CDH23/USH1D; PCDH15/USH1F; USH1G; CIB2/USH1J; USH2A; ADGRV1/USH2C; WHRN/USH2D; CLRN1/USH3A; PDZD7/USH2A modifier, digenic contributor) and 14 linked to peroxisome biogenesis disorders (Table S1; including GenBank Accession Numbers of the wild‐type gene sequences), on a MiSeq or a HiSeq1500 system (Illumina), as previously described (Eisenberger et al. 2014). In brief, sheared DNA was ligated to barcoded adaptors for multiplexing. Exons were targeted by an in‐solution customized sequence capture library (NimbleGen). Amplified enriched DNA was subjected to NGS. Reads were mapped against the hg19 human reference genome using BWA (Li and Durbin 2009) and processed with SAMtools (Li et al. 2009), Picard (http://picard.sourceforge.net), and GATK (McKenna et al. 2010). Variants were filtered against dbNSFP v2.0 (Liu et al. 2011), dbSNP v137, the Human Gene Mutation Database (HGMD® Professional 2013.2) (Stenson et al. 2014), and our in‐house database. The cutoff for the maximum minor allele frequency (MAF) was set to 1% (Bamshad et al. 2011). Nonsense, frameshift, and canonical splice site variants were regarded likely pathogenic. SNVs were assessed using SIFT (Ng and Henikoff 2003), MutationTaster (Schwarz et al. 2010), PolyPhen‐2 (Adzhubei et al. 2013), AlignGVGD (Mathe et al. 2006; Tavtigian et al. 2006), Pmut (Ferrer‐Costa et al. 2005), NNSPLICE v0.9 (Reese et al. 1997), and NetGene2 (Brunak et al. 1991; Hebsgaard et al. 1996). SeqPilot SeqNext module (v4.0.1, JSI medical systems) was used for visualization and final assessment of SNVs. Verification of all point mutations identified by NGS was carried out by Sanger sequencing. If samples from other family members were available, segregation analyses were carried out to confirm biallelic constellations – in particular in case of compound‐heterozygous mutations, but also in case of apparent homozygosity to rule out large deletions in trans to point mutations. Because the identified mutations were clearly pathogenic in almost all cases, biallelic situations are very likely true also in cases where segregation analyses were not possible.
Copy number variation analysis from NGS data
We performed copy number variation (CNV) analysis on highly covered samples sequenced on the Illumina Hiseq1500TM system. Potential copy number alterations (CNA) were initially identified with the tools copy number and copyCaller from VarScan v2.3.6 (Koboldt et al. 2012) on mapped reads with a maximum segment size of 300. All other parameters were used with standard settings. Thereby, coverage of every target region of the sample of interest was internally normalized and compared versus normalized control data of other samples of the same run. CNVs were annotated using RefSeq gene file from UCSC (ftp://hgdownload.cse.ucsc.edu/golden-Path/hg19/database/refGene.txt.gz). CNVs were initially taken into account if indicated by VarScan against at least 85% of the control patients and if the log2 threshold was ≥0.6 (in case of an amplification) or ≤−0.6 (in case of a deletion).
MLPA and array‐CGH
Results from CNV analysis were verified by MLPA (multiplex ligation‐dependent probe amplification) analysis or, if corresponding MLPA kits were not available, by array‐CGH. The following SALSA MLPA probe mixes (MRC‐Holland, Amsterdam, The Netherlands) were applied: P361‐A1 and P362‐A1 for USH2A, and P292‐A2 for PCDH15 (USH1F). In every MLPA analysis, six samples without CNVs in the investigated locus were used as negative controls.
Molecular karyotyping (array‐CGH) was performed using Agilent Human Genome CGH 244A (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions. Genomic positions were defined using NCBI37/hg19. CNVs were considered if at least five contiguous oligonucleotides presented with an abnormal log2 ratio.
Translational read‐through approach for p.Trp3955*USH2A
HEK293T cells (cultured at 37°C and 5% CO2 in Dulbecco's Modified Eagle Medium with GlutaMax™, with 10% fetal bovine serum; Invitrogen, Karlsruhe, Germany) were transiently transfected (Lipofectamine™ with PLUS™ reagent; Invitrogen, Karlsruhe, Germany) with cDNAs coding for the FN3 domains 24 and 35, the transmembrane domain, and the cytoplasmic tail (residues p.3955‐4175 fused to residues p.4926‐5202) of wild‐type and mutant USH2A (USH2A_p.Trp3955*), respectively. A cDNA fragment from c.12250‐15996 of USH2A isoform b, encoding protein residues p.3955‐5202, was amplified and inserted into the pDest SP S/F‐C‐Tag vector with a C‐terminal Flag tag. The region of c.12910–14988 was deleted using the restriction enzymes BlpI and PmII (NEB, Frankfurt am Main, Germany). The reading frame was recovered by insertion of the bases G and C at position c.12907 of the wild‐type sequence using the QuickChange Lightning Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The p.Trp3955* mutation was generated using the QuickChange Lightning Site‐Directed Mutagenesis Kit.
After 6 h, PTC124 (Selleckchem, Houston, USA; dissolved in DMSO; Sigma‐Aldrich, Deisenhofen, Germany) was applied to the culture media for 48 hours. Read‐through of the nonsense mutation was validated by indirect immunofluorescence using antibodies against Flag (Sigma‐Aldrich) on methanol‐fixed HEK293T cells as previously described (Goldmann et al. 2012). The amount of restored USH2A protein expression was calculated as the ratio of Flag‐positive cells in PTC124‐treated p.Trp3955*‐transfected cells, normalized to the total amount of analyzed cells.
Cell cultures were grown on sterile cover slips and fixed using cold methanol. PBS‐washed cover slips were blocked with blocking solution (0.5% cold water fish gelatin, 0.1% ovalbumin in PBS) for 30 min, followed by incubation with primary antibodies overnight at 4°C. Cover slips were incubated with secondary antibodies conjugated to Alexa 488 (Molecular Probes, Leiden, Netherlands) and DAPI (4′,6‐diamidino‐2‐phenylindole, Sigma–Aldrich) for staining of the nuclear DNA for 1 h at room temperature. PBS‐washed cover slips were mounted in Mowiol 4.88 (Hoechst, Frankfurt, Germany).
Results
High diagnostic yield with predominance of Usher syndrome mutations
Biallelic mutations (in trans constellation was either proven by segregation analysis or very likely, see Methods) were identified in the vast majority of patients (97% of USH1, 90% of USH2, and 92% of atypical Usher syndrome). When considering patients with monoallelic mutations (USH1: none; USH2: six; atypical Usher: one) as resolved, the diagnostic yield was 97% for both USH1 and USH2, and 92% for atypical Usher syndrome. In one USH1 patient and in three USH2 patients, no mutation was identified despite NGS of the aforementioned extended gene panel. The genetic diagnosis was made before onset of RP in 10 young patients with apparently isolated hearing impairment: nine with Usher syndrome due to mutations in MYO7A and USH2A, and one with a peroxisome biogenesis disorder (PBD) due to compound heterozygous PEX26 mutations. Overall, 83 alleles carried a novel mutation, several of which were observed more than once. This was often the case in patients from the KSA and other MENA countries, then often in homozygous state. The “rarest” Usher syndrome genes with mutations were as follows: USH1G (1 patient), CLRN1 (1 patient), PCDH15 (2 patients), and USH1C (4x).
CNVs account for a significant proportion of Usher syndrome mutations
We have established quantitative analysis of NGS data to detect CNVs such as deletions or duplications of one or several exons. We have previously shown that this bioinformatic tool effectively uncovers such structural mutations which escape detection in conventional approaches (PCR and Sanger sequencing of exons) if present in heterozygous state (Eisenberger et al. 2013). In our cohort, CNVs in USH genes significantly contribute to the mutational load. Compatible with its prevalence, but probably also due to its large size, USH2A is most often affected (Fig. 2). In patients with biallelic USH2A mutations, CNVs account for 10% (16/157 alleles; Table 1, Fig. 4A). Some CNVs were observed more than once and likely represent regional founder alleles. For example, a deletion of PCDH15 exons 1–3 was found in two families from Syria.
Figure 2.
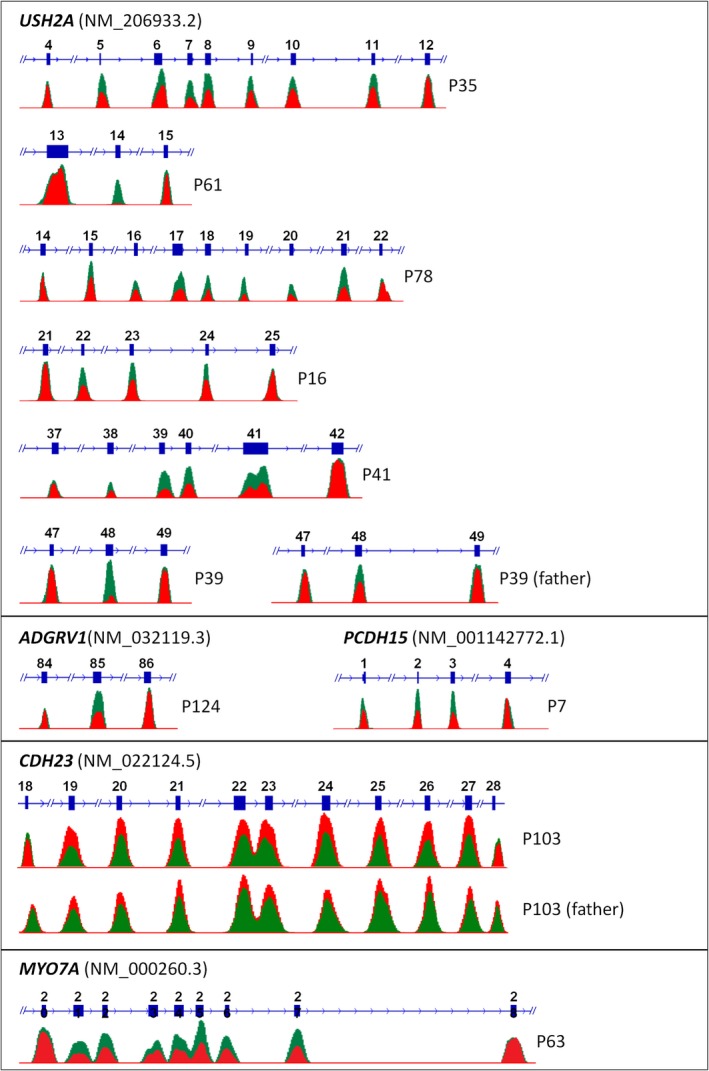
CNVs in USH genes detected by quantitative analysis of NGS data. The coverage plots illustrate the statistical readout, with the absolute coverage deduced from unique read count and as calculated by the CNV analysis mode in SeqNext (JSI Medical Systems). The coverage of affected and neighboring exons of patients (red) and controls (green) from the same NGS runs is shown in overlay schemes for comparison. While most patients harbor heterozygous deletions, reflected by approximately 50% reduction in coverage, patients P61 and P39 (the heterozygous father is shown for comparison) have homozygous deletions, reflected by virtually no coverage in the respective plot. Patient P103 had a homozygous duplication of nine CDH23 exons (19‐27; also see Fig. S1) the heterozygous father is depicted for comparison.
In two (not knowingly related) Saudi patients with Usher syndrome and hyperinsulinism (P96 and P97), we identified a homozygous deletion of the largest part of the USH1C gene (exons 3–27). One patient, P97, had a family history with likewise affected members and a deletion involving USH1C and ABCC8. Accordingly, high‐resolution array‐CGH revealed a homozygous microdeletion of approximately 123 kb on chromosome an 11p15.1 between genomic positions 17,439,772 and 17,546,526 bp (Fig. 3), defined by 14 contiguous oligomers (eight in ABCC8, MIM #600509; three in USH1C; three between ABCC8 and USH1C). The deletion breakpoints are located in intron 22 of ABCC8 and in intron 2 of USH1C, corresponding to the previously reported 11p15‐p14 deletion syndrome (MIM #606528 (Bitner‐Glindzicz et al. 2000)). In addition to Usher syndrome, patients with this condition present with congenital hyperinsulinism, severe enteropathy, and renal tubulopathy, and they may develop non‐autoimmune diabetes in adolescence (Hussain et al. 2004; Al Mutair et al. 2013). In P96, the USH1C/ABCC8 deletion was primarily detected by NGS and confirmed by array‐CGH.
Figure 3.
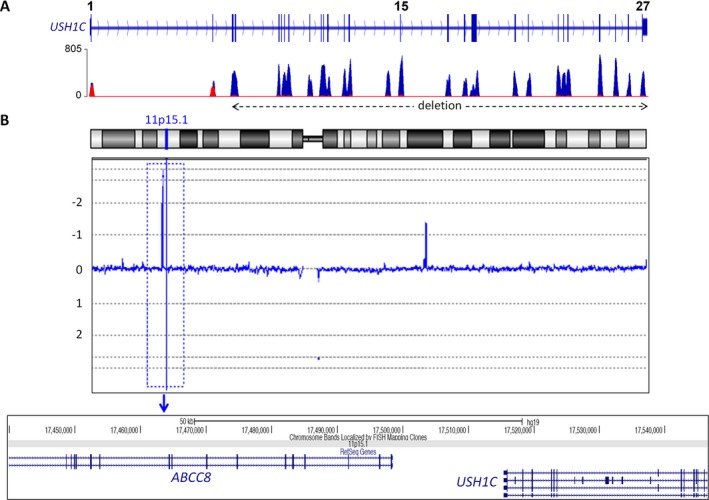
Contiguous gene syndrome due to a deletion of USH1C and ABCC8. (A) NGS indicated a homozygous deletion of USH1C exons 3–27 in two not knowingly related USH1 patients from Saudi Arabia, P96 and P97. (B) Array‐CGH revealed that the deletion also comprises the neighboring ABCC8 gene. Thus, the alteration corresponds to a contiguous gene syndrome previously described in the USH1C gene identification study (Bitner‐Glindzicz et al. 2000). The replication of this mutation in our study indicates that this is a founder mutation from the Arabian Peninsula.
p.Thr3977*USH2A: highly prevalent in USH2 and rectifiable by read‐through drugs
The USH2A mutation c.2299delG (p.Glu767Serfs*21) is the most prevalent USH mutation in several populations (Liu et al. 1999; Leroy et al. 2001; Pennings et al. 2004; Aller et al. 2006, 2010a,b; Dreyer et al. 2008). Unexpectedly, we found that the USH2A nonsense mutation, c.11864G>A (p.Trp3955*), previously reported in several studies (van Wijk et al. 2004; Le Quesne Stabej et al. 2012; Lenarduzzi et al. 2015), was even more common in our cohort, accounting for 13% of determined USH2A alleles (compared to 11% for c.2299delG; Fig. 4A). Both, c.2299delG and p.Trp3955*, have been annotated in dbSNP (rs80338903 and rs111033364, respectively), and c.2299delG has a higher minor allele frequency (MAF) than p.Trp3955* (0.07915 compared to 0.01071%; ExAC database), with no homozygotes annotated in the healthy population. Although our cohort consists of patients from diverse geographic regions and ethnic backgrounds, the largest group consists of patients of German descent (65%). The high prevalence of c.2299delG and p.Trp3955* is in accordance with the results of a recent large‐scale study on Usher syndrome (Bonnet et al. 2016). In contrast to that study, however, we found predominance of the p.Trp3955* mutation in German patients where it exceeds the prevalence of c.2299delG.
Figure 4.
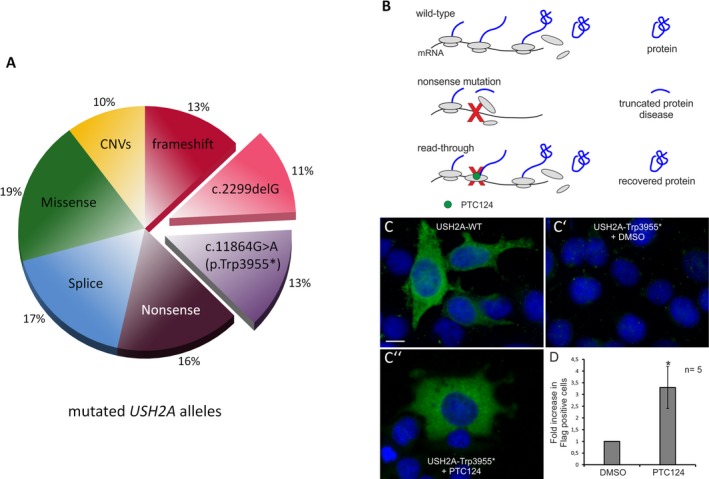
High prevalence of USH2A nonsense mutation p.Trp3955* and drug‐mediated read‐through. (A) USH2A alleles: Proportion of missense and small in‐frame alterations, truncating point mutations (nonsense, small deletions, and duplications), and large CNVs affecting one or more exons. Two mutations, p.Trp3955* and c.2299delG, are predominant. (B) Scheme of PTC124‐induced translational read‐through of a nonsense mutation. In the wild‐type situation, translation of mRNA results in functional full‐length protein. Nonsense mutations introduce a premature termination codon (red X) on the mRNA level, resulting in a truncated non‐functional protein. Read‐through‐inducing drugs like Ataluren (PTC124) bind to the ribosomes and promote the incorporation of an amino acid at the position of a PTC, resulting in the expression of full‐length protein. (C) Indirect immunofluorescence analyses of PTC124‐induced translational read‐through in cells transfected with wild‐type (WT) and mutant (Trp3955*) constructs (indirect immunofluorescence, anti‐Flag antibodies). Flag‐tagged USH2A (green) was detected in USH2A‐WT cells but not in (C′) DMSO‐treated USH2A‐p.Trp3955* cells. (C``) Application of PTC124 recovered USH2A expression in p.Trp3955*‐transfected cells. Nuclei were stained with DAPI (blue). (D) Increase in USH2A‐Flag‐positive cells after application of PTC124 (quantification of five independent experiments. Error bars represent SD; *<0.05; magnification bar: 10 μm).
Read‐through of p.Trp3955*USH2A
The most prevalent mutation in our study, p.Trp3955* mutation in USH2A, alters the TGG codon at position 11864 of the cDNA sequence into a premature UAG stop codon. Targeting of such nonsense mutations by small molecules such as PTC124 (Welch et al. 2007), known as translational read‐through‐inducing drugs (TRIDs), has become an important therapy approach (Fig. 4B; reviewed in Nagel‐Wolfrum et al. (2016)). To test this approach for the USH2A p.Trp3955* nonsense mutation, we transfected HEK293T cells with Flag‐tagged wild‐type (USH2A‐WT) and mutant USH2A plasmids (USH2A‐Trp3955*), and determined USH2A expression by indirect immunofluorescence using anti‐Flag antibodies. In contrast to USH2A‐WT cells (Fig. 4C), a low number of Flag‐positive cells was detected in DMSO‐treated USH2A‐Trp3955* cells (Fig. 4C′), most probably resulting from spontaneous read‐through of the p.Trp3955* mutation. Application of PTC124 to the Trp3955* cells resulted in a 3.3‐fold increase of USH2A expression (Fig. 4C′′) compared to DMSO‐treated Trp3955* cells.
Simultaneous homozygosity of mutations in non‐syndromic genes and a novel Heimler syndrome gene
In three patients with deaf‐blindness, disease was found to be due to “non‐Usher” gene mutations: Quantification of NGS reads in a Saudi patient from a consanguineous family, apparently affected by USH1, revealed a homozygous deletion of OTOA, a gene known to be associated with autosomal recessively inherited deafness, DFNB22. Because the patient's retinal phenotype (deep pigment deposits along the vascular arcades, subretinal fibrosis; delayed, depressed, and simplified scotopic flash response in the ERG) appeared compatible with a recessive NR2E3‐related dystrophy (Khan et al. 2007, 2010), this gene was sequenced. Indeed, a homozygous NR2E3 missense mutation, p.Arg311Gln, previously reported as a pathogenic mutation (Haider et al. 2000; Kanda and Swaroop 2009; von Alpen et al. 2015), was identified (Fig. 5).
Figure 5.
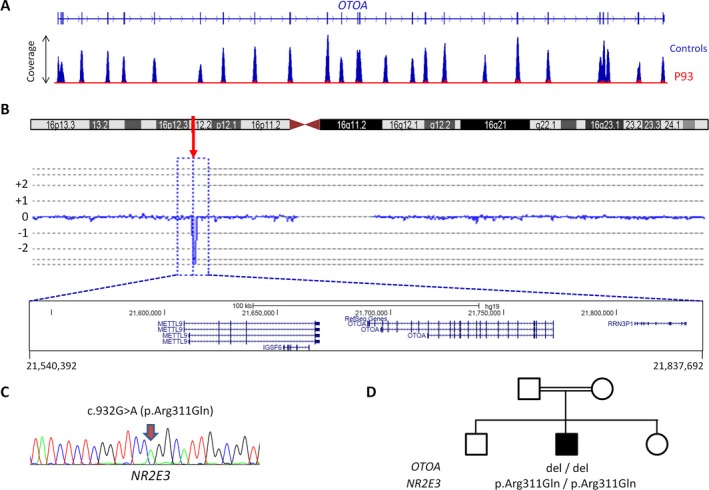
Double homozygosity for mutations in two genes associated with non‐syndromic disease simulates Usher syndrome in patient 93. (A) NGS indicated a homozygous deletion of the entire OTOA gene, the gene associated with recessive deafness DFNB22, in the patient. (B) This was confirmed by array‐CGH analysis. (C) Targeted analysis revealed a homozygous missense mutation of NR2E3. (D) Pedigree of the patient's consanguineous family summarizing the genetic findings.
Very recently, Heimler syndrome, characterized by the association of an “Usher‐like” presentation (retinal degeneration and hearing loss) with enamel dysplasia and nail abnormalities (Heimler et al. 1991), has been found to be caused by recessive mutations in two genes, PEX1 and PEX6, known to be associated with peroxisome biogenesis disorders, PBD (Ratbi et al. 2015; Smith et al. 2016; Zaki et al. 2016). PBD‐associated genes have therefore been considered in our analysis. Here, we identified compound‐heterozygous mutations in another PBD‐related gene, PEX26, in two patients from two families (Fig. 6A): A 14‐year‐old boy (P135), diagnosed with “Usher syndrome with additional abnormalities”, carries two missense mutations affecting evolutionarily highly conserved residues, p.Asp43His and p.Arg98Trp (Fig. 6B,C). After birth, lack of reaction to noise was noted. At 22 months, profound hearing loss (80 dB), hepatosplenomegaly, and elevation of liver enzymes (which persisted) were diagnosed. Retinitis punctata albescens with macular involvement and significant visual loss was diagnosed at 5 1/2 years. Opacities of deciduous teeth indicated thin enamel, and permanent teeth showed severe enamel dysplasia in terms of amelogenesis imperfecta combined with gingival hyperplasia and progressive preeruptive crown resorption (Fig. 6D,E). The other patient (P136), a 4‐year‐old girl with apparently non‐syndromic hearing loss, was found to carry a translation initiation codon mutation (p.Met1?) and, as P135, p.Arg98Trp. Subsequent detailed inspection of the deciduous teeth revealed enamel defects. Although development of retinal degeneration in this patient seems very likely, stressful in‐depth ophthalmological investigations were not carried out now.
Figure 6.
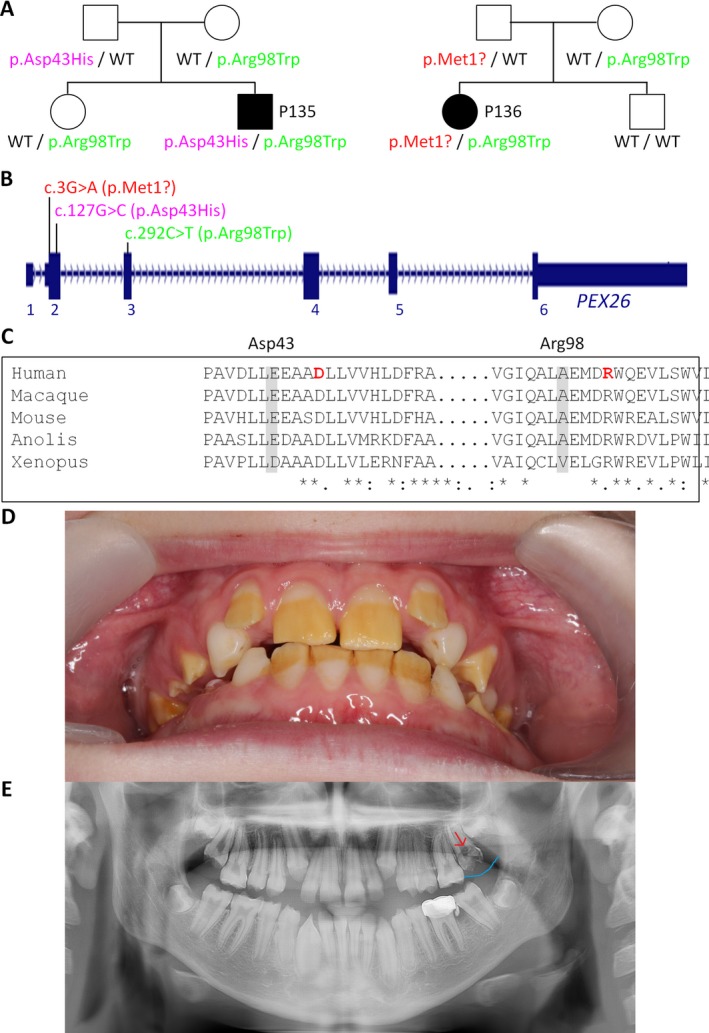
Biallelic PEX26 mutations cause deaf‐blindness with enamel dysplasia (Heimler syndrome). (A) Pedigrees of the two patients with PEX26 mutations. (B) Scheme of the PEX26 gene and localization of mutations. (C) Partial alignment of PEX26/Pex26 peptide sequences from various species, indicating high evolutionary conservation of the mutated residues. (D) Severe enamel dysplasia of permanent teeth of patient 135 at 117/12 years of age. (E) X‐ray of patient 135 showing preeruptive crown resorption in the upper left first molar (red arrow) and a local enlargement of the gingival tissue (blue line) at 1310/12 years of age.
Discussion
Deaf‐blindness is mostly of genetic origin in developed countries, and biallelic mutations in the genes for Usher syndrome, an autosomal recessive disorder, are the predominant cause. With 11 known, mostly very large genes whose mutations explain the majority of cases, Usher syndrome is a prime condition for targeted NGS. Several reports have accordingly shown that the method has matured into a powerful diagnostic tool (Aparisi et al. 2014; Besnard et al. 2014; Bujakowska et al. 2014; Krawitz et al. 2014; Jiang et al. 2015; Bonnet et al. 2016).
The diagnostic yield in our study is very high and similar to a recent large‐scale study on Usher syndrome patients (Bonnet et al. 2016). Many mutations were novel, confirming that the genetic basis is often “private” and not detectable by screens that focus on previously reported mutations (Cremers et al. 2007). Repeatedly observed mutations, however, were common, not confined to but particularly in patients from MENA countries where the rate of parental consanguinity is high (Table 2). This indicates regional founder mutations that may guide the genetic diagnostic approach. Among the recurrent mutations are several large CNVs, including the 11p15‐p14 deletion syndrome that involves USH1C and ABCC8 in two KSA families. Hyperinsulinism is therefore a symptom to be considered in deaf(‐blind) patients from the Arabian Peninsula.
Table 2.
Repeatedly observed mutations
| Patients (n) | Origin | |||
|---|---|---|---|---|
| MYO7A | ||||
| c.397dupC | p.His133Profs*7 | 2 | Pakistan | |
| c.470+1G>A | Splice | 2 | KSA | |
| c.2904G>T | p.Glu968Asp | rs111033233 | 2 | Germany, Persia, Syria |
| c.3719G>A | p.Arg1240Gln | rs111033178 | 2 | Germany |
| c.3503G>A | p.Arg1168Gln | 2 | Germany | |
| CDH23 | ||||
| c.6047‐9G>A | p.Leu728Serfs*6 | 2 | Italy, Pakistan | |
| PCDH15 | ||||
| Del ex1‐3 | p.Glu968Asp | 2 | Syria | |
| USH1C | ||||
| Del ex3‐27 | CNV | 2 | KSA | |
| USH2A | ||||
| c.11864G>A | p.Trp3955* | rs111033364 | 19 | Germany |
| c.2299delG | p.Glu767Serfs*21 | rs80338903 | 16 | Germany |
| Del ex22‐24 | CNV | 5 | Germany | |
| c.7595‐2144A>G | Splice | 4 | Germany | |
| c.653T>A | p.Val218Glu | 3 | Germany | |
| c.920_923dup | p.His308Glnfs*16 | 3 | Germany | |
| c.949C>A | p.Arg317Arg | 3 | Germany | |
| c.1036A>C | p.Asn346His | rs369522997 | 3 | Germany |
| c.8682‐9A>G | Splice | 3 | Germany | |
| c.486‐1G>C | Splice | 2 | KSA | |
| c.2209C>T | p.Arg737* | rs111033334 | 2 | Germany |
| c.9424G>T | p.Gly3142* | 2 | Germany | |
| c.10388‐1G>A | Splice | 2 | Germany | |
| c.14439_14454del | p.Cys4813* | 2 | Germany, Turkey | |
| c.14131C>T | p.Gln4711* | 2 | Germany | |
| c.10561T>C | p.Trp3521Arg | rs111033264 | 2 | Germany |
| c.12234_12235delGA | p.Asn4079Trpfs*19 | rs398124618 | 2 | Germany |
| c.11054G>A | p.Trp3685* | 2 | Germany | |
| c.11549‐1G>A | Splice | 2 | Germany | |
| PEX26 | ||||
| c.292C>T | p.Arg98Trp | rs62641228 | 2 | Germany |
Here, we aimed at a one‐method approach, based on targeted NGS. To achieve this, we a) established quantitative readout of NGS data to detect CNVs, with conventional methods like MLPA and array‐CGH only being used for verification of NGS‐based CNV detection; b) included genes mutated in clinically overlapping conditions like Heimler syndrome; and c) simultaneously enriched genes implicated in isolated deafness to recognize cases with co‐occuring non‐syndromic defects mimicking a single syndrome.
Because all CNVs detected by quantitative readout of NGS data could be confirmed by MLPA or array‐CGH and the majority of patients were found to carry two (either proven or very likely) biallelic mutations, we assume that probably no CNV escaped detection in our study. This eliminates a major dead corner in diagnostic testing of deaf‐blindness genes and enables highly efficient testing by a single technique, targeted NGS comprising all exons of genes for Usher syndrome, clinically overlapping conditions, and the repeatedly reported deep‐intronic c.7595‐2144A>G mutation in USH2A. Deep‐intronic mutations apart from c.7595‐2144A>GUSH2A (which accounted for only four alleles in our study) have been reported (Liquori et al. 2016) and would still escape detection in our non‐genomic approach but – given the small proportion of patients with monoallelic or no mutations – do not seem to play a significant role. Very recently, however, by whole‐genome sequencing, we found evidence that a deep intronic founder mutation in CLRN1 may significantly contribute to USH1 on the Arabian Peninsula (Khan et al. 2017). Such recurrent “hidden” splice mutations should be considered at least in patients with the respective ethnic background.
The clinical presentation of most patients with Usher syndrome corresponded to either USH1 or USH2. All genetically resolved patients from these two categories had mutations in Usher syndrome genes. The same applied to the nine patients diagnosed with “atypical Usher syndrome” in whom course and/or age of onset of sensorineural hearing impairment and RP did not allow for clear‐cut assignment to USH1 or USH2 and who had no additional abnormalities: They were found to have atypical expressions of USH1B (MYO7A), USH1D (CDH23), USH2A, and USH2C (ADGRV1). The diagnosis had to be reversed in the three remaining patients with apparent “atypical Usher syndrome”: They were found to have clinically similar conditions, a peroxisome biogenesis disorder (PBD; PEX26 mutations), or simultaneous presence of two non‐syndromic conditions. Retrospectively, these patients had additional abnormalities (in case of PEX26‐associated PBD) or a distinct, NR2E3‐typical form of retinopathy. Detailed clinical characterization before genetic testing could have largely ruled out Usher syndrome beforehand. However, patients undergoing genetic testing for deaf‐blindness usually represent a heterogeneous cohort and range from poor to excellent clinically characterized. Although most will have mutations in Usher syndrome genes, it is the geneticists’ role to anticipate this problem and to equally take rare differential diagnoses into account.
In patient 93, the specific retinopathy due to NR2E3 mutation homozygosity could have indicated a diagnosis distinct from Usher syndrome, but congenital deafness resulting from the homozygous deletion of OTOA is indistinguishable from the hearing impairment in USH1. If both components, deafness and RP, present as in Usher syndrome, as we have previously reported for patients with double homozygosity for mutations in DFNB59 (deafness) and MERTK (RP) (Ebermann et al. 2007), and if the co‐occurence of the two non‐syndromic conditions in the index patient is not uncovered by their division in siblings, the genetic diagnosis is essential. Although the disentanglement of the genetic basis of disease in patient 93 does not change his medical follow‐up, it makes a major difference in genetic counseling for the parents whose family planning was ongoing: Instead of a 25% recurrence risk for Usher syndrome in further children, the actual risk is 25% for non‐syndromic deafness, 25% for non‐syndromic retinopathy, and 6.25% for deaf‐blindness. With the increasing diagnostic application of large‐scale panel NGS, whole‐exome and whole‐genome sequencing, it has become apparent that the co‐occurence of two (or more) monogenic conditions in one patient is not so uncommon (Boycott and Innes 2017) – and hard to recognize if it resembles a syndrome. Given the relatively high prevalence of carriers for (mostly recessively inherited) monogenic retinal dystrophies (with about 20% of the general population assumed to be carriers (Nishiguchi and Rivolta 2012)) and hearing impairment (assuming a monogenic cause in about 80% of the affected newborns (1 in 500) (Shearer and Smith 2012)), mimicking of Usher syndrome by both conditions should be a recurrent scenario. Although this is more likely in offspring from consanguineous parents (as in case of patient 93), migration from regions with high rates of consanguinity, like the Middle East and North Africa (MENA regions), this phenomenon will increase, for example, in Central Europe. In summary, our results demonstrate the potential of extended NGS panels that include non‐Usher genes to resolve difficult genetic constellations.
Inherited retinal dystrophies are a major cause of blindness worldwide. There has been remarkable progress in different therapeutic approaches (Scholl et al. 2016), such as gene therapy and optogenetics. Gene addition with adeno‐associated viral (AAV) vectors has shown to be effective in case of RPE65 in patients with Leber's congenital amaurosis (LCA) type 2. In Usher syndrome, the retinopathy component would be the primary target of gene therapy, especially in patients who still have early‐stage RP or who are still non‐syndromic (“only” deaf). The identification of the causative gene will therefore be of utmost importance if gene therapy should become routinely available. However, the enormous size of many genes, including USH2A, represents a major hurdle for the packaging capacity of AAV vectors. Therefore, alternative gene‐based strategies for therapy or slowdown of retinal degeneration are necessary. Translational read‐through represents a promising alternative for patients with nonsense mutations (Nagel‐Wolfrum et al. 2014a,b). Here, we show that PTC124 effectively induces translational read‐through of the predominant p.Trp3955* USH2A nonsense mutation which accounted for 13% of determined mutant USH2A alleles in our cohort. Because 35% of Usher syndrome patients with determined mutations in our study carry nonsense mutations on at least one allele (48/138), translational read‐though could be a promising therapeutic strategy for Usher syndrome patients of all genetic subtypes.
Pinpointing the molecular diagnosis can be crucial for specific prophylaxis – as in case of patients P135 and P136 who both carry biallelic PEX26 mutations. While P135 expresses the full picture of Heimler syndrome (except specific nail abnormalities which do not seem to be obligate part of the syndrome (Ratbi et al. 2016; Witters et al. 2016)), the only “Heimler sign” so far in patient P136 was enamel dysplasia, probably because of her young age (3 years). This patient can benefit, in terms of early prophylaxis, from the molecular diagnosis by being monitored for signs of hepatopathy, elevation of fatty acids and retinopathy. To protect hepatic function and lipid metabolism, the patient should avoid certain medications and noxious substances (e.g., alcohol). Furthermore, and in contrast to Usher syndrome, therapeutic options could exist for patients with mild PBD who may benefit from a phytanic acid‐restricted diet and extracorporeal lipid apheresis (Baldwin et al. 2010; Ruether et al. 2010; Kohlschütter et al. 2012). In the ideal case, an effective diet could also counteract progression or even manifestation of retinal degeneration. Nine patients (in whom biallelic Usher or, in one case, Heimler syndrome gene mutations were found) had been diagnosed with non‐syndromic deafness. If AAV‐based gene addition or read‐through approaches should become available as regular therapies, such early diagnosed patients could benefit immensely from their early genetic diagnosis.
While exome sequencing has become a reasonable “one‐test‐solution” for genetically heterogeneous conditions with a significant proportion of patients lacking mutations in the known disease genes, we propose NGS (large) panel analysis for Usher(‐like) syndrome – with the advantage of technically feasible CNV discovery, a very high diagnostic yield, and uncovering conditions mimicking Usher syndrome.
Conflict of Interest
CN, TE, CD, SN, CBl, and HJB are employees of Bioscientia which is part of a publicly traded diagnostic company. The other authors have no competing interests.
Supporting information
Figure S1. Confirmation of a heterozygous intragenic deletion of CDH23 exons 19–27 in patient P103 by array‐CGH (244k Agilent Technologies microarray).
Table S1. Deafness genes analyzed by targeted NGS.
Acknowledgments
We thank the patients for participating in our study and Katja Busch, Anika Greiche, and Annika Heinemann‐Dott for excellent technical assistance. HJB was supported by Forschung contra Blindheit, Initiative Usher‐Syndrom e.V., and the GEERS‐Stiftung.
Molecular Genetics & Genomic Medicine 2017; 5(5): 531–552
References
- Adato, A. , Weil D., Kalinski H., Pel‐Or Y., Ayadi H., Petit C., et al. 1997. Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am. J. Hum. Genet. 61:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adato, A. , Weston M. D., Berry A., Kimberling W. J., and Bonne‐Tamir A.. 2000. Three novel mutations and twelve polymorphisms identified in the USH2A gene in Israeli USH2 families. Hum. Mutat. 15:388. [DOI] [PubMed] [Google Scholar]
- Adzhubei, I. , Jordan D. M., and Sunyaev S. R.. 2013. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr. Protoc. Hum. Genet. Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, Z. M. , Riazuddin S., Ahmad J., Bernstein S. 7., Guo Y., Sabar M. F., et al. 2003. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 12:3215–3223. [DOI] [PubMed] [Google Scholar]
- Akoury, E. , El Zir E., Mansour A., Megarbane A., Majewski J., and Slim R.. 2011. A novel 5‐bp deletion in Clarin 1 in a family with Usher syndrome. Ophthalmic Genet. 32:245–249. [DOI] [PubMed] [Google Scholar]
- Al Mutair, A. N. , Brusgaard K., Bin‐Abbas B., Hussain K., Felimban N., Al Shaikh A., et al. 2013. Heterogeneity in phenotype of usher‐congenital hyperinsulinism syndrome: hearing loss, retinitis pigmentosa, and hyperinsulinemic hypoglycemia ranging from severe to mild with conversion to diabetes. Diabetes Care 36:557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aller, E. , Jaijo T., Beneyto M., Najera C., Oltra S., Ayuso C., et al. 2006. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J. Med. Genet. 43:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aller, E. , Larrieu L., Jaijo T., Baux D., Espinos C., Gonzalez‐Candelas F., et al. 2010a. The USH2A c.2299delG mutation: dating its common origin in a Southern European population. Eur. J. Hum. Genet. 18:788–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aller, E. , Jaijo T., Garcia‐Garcia G., Aparisi M. J., Blesa D., Diaz‐Llopis M., et al. 2010b. Identification of large rearrangements of the PCDH15 gene by combined MLPA and a CGH: large duplications are responsible for Usher syndrome. Invest. Ophthalmol. Vis. Sci. 51:5480–5485. [DOI] [PubMed] [Google Scholar]
- von Alpen, D. , Tran H. V., Guex N., Venturini G., Munier F. L., Schorderet D. F., et al. 2015. Differential dimerization of variants linked to enhanced S‐cone sensitivity syndrome (ESCS) located in the NR2E3 ligand‐binding domain. Hum. Mutat. 36:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparisi, M. J. , Garcia‐Garcia G., Aller E., Sequedo M. D., Martinez‐Fernandez de la Camara C., Rodrigo R., et al. 2013. Study of USH1 splicing variants through minigenes and transcript analysis from nasal epithelial cells. PLoS ONE 8:e57506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparisi, M. J. , Aller E., Fuster‐Garcia C., Garcia‐Garcia G., Rodrigo R., Vazquez‐Manrique R. P., et al. 2014. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet. J. Rare. Dis. 9:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auslender, N. , Bandah D., Rizel L., Behar D. M., Shohat M., Banin E., et al. 2008. Four USH2A founder mutations underlie the majority of Usher syndrome type 2 cases among non‐Ashkenazi Jews. Genet. Test. 12:289–294. [DOI] [PubMed] [Google Scholar]
- Baldwin, E. J. , Gibberd F. B., Harley C., Sidey M. C., Feher M. D., and Wierzbicki A. S.. 2010. The effectiveness of long‐term dietary therapy in the treatment of adult Refsum disease. J. Neurol. Neurosurg. Psychiatry 81:954–957. [DOI] [PubMed] [Google Scholar]
- Bamshad, M. J. , Ng S. B., Bigham A. W., Tabor H. K., Emond M. J., Nickerson D. A., et al. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12:745–755. [DOI] [PubMed] [Google Scholar]
- Baux, D. , Larrieu L., Blanchet C., Hamel C., Ben Salah S., Vielle A., et al. 2007. Molecular and in silico analyses of the full‐length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum. Mutat. 28:781–789. [DOI] [PubMed] [Google Scholar]
- Baux, D. , Blanchet C., Hamel C., Meunier I., Larrieu L., Faugere V., et al. 2014. Enrichment of LOVD‐USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum. Mutat. 35:1179–1186. [DOI] [PubMed] [Google Scholar]
- Berendse, K. , Engelen M., Ferdinandusse S., Majoie C. B., Waterham H. R., Vaz F. M., et al. 2016. Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J. Inherit. Metab. Dis. 39:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard, T. , Garcia‐Garcia G., Baux D., Vache C., Faugere V., Larrieu L., et al. 2014. Experience of targeted Usher exome sequencing as a clinical test. Mol. Genet. Genomic Med. 2:30–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj, A. K. , Kasztejna J. P., Huq S., Berson E. L., and Dryja T. P.. 2000. Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp. Eye Res. 71:173–181. [DOI] [PubMed] [Google Scholar]
- Bhattacharya, G. , Kalluri R., Orten D. J., Kimberling W. J., and Cosgrove D.. 2004. A domain‐specific usherin/collagen IV interaction may be required for stable integration into the basement membrane superstructure. J. Cell Sci. 117:233–242. [DOI] [PubMed] [Google Scholar]
- Bitner‐Glindzicz, M. , Lindley K. J., Rutland P., Blaydon D., Smith V. V., Milla P. J., et al. 2000. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet. 26:56–60. [DOI] [PubMed] [Google Scholar]
- Bonnet, C. , Riahi Z., Chantot‐Bastaraud S., Smagghe L., Letexier M., Marcaillou C., et al. 2016. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 24:1730–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , and Innes A. M.. 2017. When one diagnosis is not enough. N. Engl. J. Med. 376:83–85. [DOI] [PubMed] [Google Scholar]
- von Brederlow, B. , Bolz H., Janecke A., La O. C. A., Rudolph G., Lorenz B., et al. 2002. Identification and in vitro expression of novel CDH23 mutations of patients with Usher syndrome type 1D. Hum. Mutat. 19:268–273. [DOI] [PubMed] [Google Scholar]
- Brunak, S. , Engelbrecht J., and Knudsen S.. 1991. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 220:49–65. [DOI] [PubMed] [Google Scholar]
- Bujakowska, K. M. , Consugar M., Place E., Harper S., Lena J., Taub D. G., et al. 2014. Targeted exon sequencing in Usher syndrome type I. Invest. Ophthalmol. Vis. Sci. 55:8488–8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers, F. P. , Kimberling W. J., Kulm M., de Brouwer A. P., van Wijk E., te Brinke H., et al. 2007. Development of a genotyping microarray for Usher syndrome. J. Med. Genet. 44:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dad, S. , Rendtorff N. D., Kann E., Albrechtsen A., Mehrjouy M. M., Bak M., et al. 2015. Partial USH2A deletions contribute to Usher syndrome in Denmark. Eur. J. Hum. Genet. 23:1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer, B. , Tranebjaerg L., Rosenberg T., Weston M. D., Kimberling W. J., and Nilssen O.. 2000. Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur. J. Hum. Genet. 8:500–506. [DOI] [PubMed] [Google Scholar]
- Dreyer, B. , Brox V., Tranebjaerg L., Rosenberg T., Sadeghi A. M., Moller C., et al. 2008. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum. Mutat. 29:451. [DOI] [PubMed] [Google Scholar]
- Ebermann, I. , Walger M., Scholl H. P., Charbel Issa P., Lüke C., Nürnberg G., et al. 2007. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction. Hum. Mutat. 28:571–577. [DOI] [PubMed] [Google Scholar]
- Eisenberger, T. , Neuhaus C., Khan A. O., Decker C., Preising M. N., Friedburg C., et al. 2013. Increasing the yield in targeted next‐generation sequencing by implicating CNV analysis, non‐coding exons and the overall variant load: the example of retinal dystrophies. PLoS ONE 8:e78496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger, T. , Di Donato N., Baig S. M., Neuhaus C., Beyer A., Decker E., et al. 2014. Targeted and genomewide NGS data disqualify mutations in MYO1A, the “DFNA48 gene”, as a cause of deafness. Hum. Mutat. 35:565–570. [DOI] [PubMed] [Google Scholar]
- Eudy, J. D. , Weston M. D., Yao S., Hoover D. M., Rehm H. L., Ma‐Edmonds M., et al. 1998. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 280:1753–1757. [DOI] [PubMed] [Google Scholar]
- Ferrer‐Costa, C. , Gelpi J. L., Zamakola L., Parraga I., de la Cruz X., and Orozco M.. 2005. PMUT: a web‐based tool for the annotation of pathological mutations on proteins. Bioinformatics 21:3176–3178. [DOI] [PubMed] [Google Scholar]
- Furuki, S. , Tamura S., Matsumoto N., Miyata N., Moser A., Moser H. W., et al. 2006. Mutations in the peroxin Pex26p responsible for peroxisome biogenesis disorders of complementation group 8 impair its stability, peroxisomal localization, and interaction with the Pex1p x Pex6p complex. J. Biol. Chem. 281:1317–1323. [DOI] [PubMed] [Google Scholar]
- Garcia‐Garcia, G. , Aparisi M. J., Jaijo T., Rodrigo R., Leon A. M., Avila‐Fernandez A., et al. 2011. Mutational screening of the USH2A gene in Spanish USH patients reveals 23 novel pathogenic mutations. Orphanet. J. Rare. Dis. 6:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Garcia, G. , Aller E., Jaijo T., Aparisi M. J., Larrieu L., Faugere V., et al. 2014. Novel deletions involving the USH2A gene in patients with Usher syndrome and retinitis pigmentosa. Mol. Vis. 20:1398–1410. [PMC free article] [PubMed] [Google Scholar]
- Ge, Z. , Bowles K., Goetz K., Scholl H. P., Wang F., Wang X., et al. 2015. NGS‐based Molecular diagnosis of 105 eyeGENE((R)) probands with Retinitis Pigmentosa. Sci. Rep. 15:18287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glockle, N. , Kohl S., Mohr J., Scheurenbrand T., Sprecher A., Weisschuh N., et al. 2014. Panel‐based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 22:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann, T. , Overlack N., Moller F., Belakhov V., van Wyk M., Baasov T., et al. 2012. A comparative evaluation of NB30, NB54 and PTC124 in translational read‐through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 4:1186–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider, N. B. , Jacobson S. G., Cideciyan A. V., Swiderski R., Streb L. M., Searby C., et al. 2000. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 24:127–131. [DOI] [PubMed] [Google Scholar]
- Hebsgaard, S. M. , Korning P. G., Tolstrup N., Engelbrecht J., Rouze P., and Brunak S.. 1996. Splice site prediction in Arabidopsis thaliana pre‐mRNA by combining local and global sequence information. Nucleic Acids Res. 24:3439–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimler, A. , Fox J. E., Hershey J. E., and Crespi P.. 1991. Sensorineural hearing loss, enamel hypoplasia, and nail abnormalities in sibs. Am. J. Med. Genet. 39:192–195. [DOI] [PubMed] [Google Scholar]
- Herrera, W. , Aleman T. S., Cideciyan A. V., Roman A. J., Banin E., Ben‐Yosef T., et al. 2008. Retinal disease in Usher syndrome III caused by mutations in the clarin‐1 gene. Invest. Ophthalmol. Vis. Sci. 49:2651–2660. [DOI] [PubMed] [Google Scholar]
- Huang, X. F. , Huang F., Wu K. C., Wu J., Chen J., Pang C. P., et al. 2015. Genotype‐phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next‐generation sequencing. Genet. Med. 17:271–278. [DOI] [PubMed] [Google Scholar]
- Hussain, K. , Bitner‐Glindzicz M., Blaydon D., Lindley K. J., Thompson D. A., Kriss T., et al. 2004. Infantile hyperinsulinism associated with enteropathy, deafness and renal tubulopathy: clinical manifestations of a syndrome caused by a contiguous gene deletion located on chromosome 11p. J. Pediatr. Endocrinol. Metab. 17:1613–1621. [DOI] [PubMed] [Google Scholar]
- Jacobson, S. G. , Aleman T. S., Sumaroka A., Cideciyan A. V., Roman A. J., Windsor E. A., et al. 2009. Disease boundaries in the retina of patients with Usher syndrome caused by MYO7A gene mutations. Invest. Ophthalmol. Vis. Sci. 50:1886–1894. [DOI] [PubMed] [Google Scholar]
- Jacobson, S. G. , Cideciyan A. V., Gibbs D., Sumaroka A., Roman A. J., Aleman T. S., et al. 2011. Retinal disease course in Usher syndrome 1B due to MYO7A mutations. Invest. Ophthalmol. Vis. Sci. 52:7924–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaijo, T. , Aller E., Beneyto M., Najera C., Graziano C., Turchetti D., et al. 2007. MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J. Med. Genet. 44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaijo, T. , Aller E., Garcia‐Garcia G., Aparisi M. J., Bernal S., Avila‐Fernandez A., et al. 2010. Microarray‐based mutation analysis of 183 Spanish families with Usher syndrome. Invest. Ophthalmol. Vis. Sci. 51:1311–1317. [DOI] [PubMed] [Google Scholar]
- Janecke, A. R. , Meins M., Sadeghi M., Grundmann K., Apfelstedt‐Sylla E., Zrenner E., et al. 1999. Twelve novel myosin VIIA mutations in 34 patients with Usher syndrome type I: confirmation of genetic heterogeneity. Hum. Mutat. 13:133–140. [DOI] [PubMed] [Google Scholar]
- Jiang, L. , Liang X., Li Y., Wang J., Zaneveld J. E., Wang H., et al. 2015. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet. J. Rare. Dis. 10:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiserman, N. , Obolensky A., Banin E., and Sharon D.. 2007. Novel USH2A mutations in Israeli patients with retinitis pigmentosa and Usher syndrome type 2. Arch. Ophthalmol. 125:219–224. [DOI] [PubMed] [Google Scholar]
- Kalay, E. , de Brouwer A. P., Caylan R., Nabuurs S. B., Wollnik B., Karaguzel A., et al. 2005. A novel D458V mutation in the SANS PDZ binding motif causes atypical Usher syndrome. J. Mol. Med. (Berl) 83:1025–1032. [DOI] [PubMed] [Google Scholar]
- Kanda, A. , and Swaroop A.. 2009. A comprehensive analysis of sequence variants and putative disease‐causing mutations in photoreceptor‐specific nuclear receptor NR2E3. Mol. Vis. 15:2174–2184. [PMC free article] [PubMed] [Google Scholar]
- Katagiri, S. , Akahori M., Sergeev Y., Yoshitake K., Ikeo K., Furuno M., et al. 2014. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE 9:e108721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. O. , Aldahmesh M., and Meyer B.. 2007. The enhanced S‐cone syndrome in children. Br. J. Ophthalmol. 91:394–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. O. , Aldahmesh M. A., Al‐Harthi E., and Alkuraya F. S.. 2010. Helicoid subretinal fibrosis associated with a novel recessive NR2E3 mutation p. S44X. Arch. Ophthalmol. 128:344–348. [DOI] [PubMed] [Google Scholar]
- Khan, A. O. , Becirovic E., Betz C., Neuhaus C., Altmüller J., Maria Riedmayr L., et al. 2017. A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci. Rep. 7:1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberling, W. J. , Hildebrand M. S., Shearer A. E., Jensen M. L., Halder J. A., Trzupek K., et al. 2010. Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet. Med. 12:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt, D. C. , Zhang Q., Larson D. E., Shen D., McLellan M. D., Lin L., et al. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlschütter, A. , Santer R., Lukacs Z., Altenburg C., Kemper M. J., and Rüther K.. 2012. A child with night blindness: preventing serious symptoms of Refsum disease. J. Child Neurol. 27:654–656. [DOI] [PubMed] [Google Scholar]
- Koparir, A. , Karatas O. F., Atayoglu A. T., Yuksel B., Sagiroglu M. S., Seven M., et al. 2015. Whole‐exome sequencing revealed two novel mutations in Usher syndrome. Gene 563:215–218. [DOI] [PubMed] [Google Scholar]
- Krawitz, P. M. , Schiska D., Kruger U., Appelt S., Heinrich V., Parkhomchuk D., et al. 2014. Screening for single nucleotide variants, small indels and exon deletions with a next‐generation sequencing based gene panel approach for Usher syndrome. Mol. Genet. Genomic Med. 2:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guedard‐Mereuze, S. , Vache C., Baux D., Faugere V., Larrieu L., Abadie C., et al. 2010. Ex vivo splicing assays of mutations at noncanonical positions of splice sites in USHER genes. Hum. Mutat. 31:347–355. [DOI] [PubMed] [Google Scholar]
- Le Quesne Stabej, P. , Saihan Z., Rangesh N., Steele‐Stallard H. B., Ambrose J., Coffey A., et al. 2012. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 49:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenarduzzi, S. , Vozzi D., Morgan A., Rubinato E., D'Eustacchio A., Osland T. M., et al. 2015. Usher syndrome: an effective sequencing approach to establish a genetic and clinical diagnosis. Hear. Res. 320:18–23. [DOI] [PubMed] [Google Scholar]
- Lenassi, E. , Robson A. G., Luxon L. M., Bitner‐Glindzicz M., and Webster A. R.. 2015a. Clinical heterogeneity in a family with mutations in USH2A. JAMA Ophthalmol. 133:352–355. [DOI] [PubMed] [Google Scholar]
- Lenassi, E. , Vincent A., Li Z., Saihan Z., Coffey A. J., Steele‐Stallard H. B., et al. 2015b. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease‐causing variants. Eur. J. Hum. Genet. 23:1318–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy, B. P. , Aragon‐Martin J. A., Weston M. D., Bessant D. A., Willis C., Webster A. R., et al. 2001. Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp. Eye Res. 72:503–509. [DOI] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori, A. , Vache C., Baux D., Blanchet C., Hamel C., Malcolm S., et al. 2016. Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum. Mutat. 37:184–193. [DOI] [PubMed] [Google Scholar]
- Liu, X. Z. , Hope C., Liang C. Y., Zou J. M., Xu L. R., Cole T., et al. 1999. A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am. J. Hum. Genet. 64:1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Jian X., and Boerwinkle E.. 2011. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe, E. , Olivier M., Kato S., Ishioka C., Hainaut P., and Tavtigian S. V.. 2006. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 34:1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, N. , Tamura S., Furuki S., Miyata N., Moser A., Shimozawa N., et al. 2003. Mutations in novel peroxin gene PEX26 that cause peroxisome‐biogenesis disorders of complementation group 8 provide a genotype‐phenotype correlation. Am. J. Hum. Genet. 73:233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee, T. L. , Seyedahmadi B. J., Sweeney M. O., Dryja T. P., and Berson E. L.. 2010. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non‐syndromic retinitis pigmentosa. J. Med. Genet. 47:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutai, H. , Suzuki N., Shimizu A., Torii C., Namba K., Morimoto N., et al. 2013. Diverse spectrum of rare deafness genes underlies early‐childhood hearing loss in Japanese patients: a cross‐sectional, multi‐center next‐generation sequencing study. Orphanet. J. Rare. Dis. 28:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel‐Wolfrum, K. , Baasov T., and Wolfrum U.. 2014a. Therapy strategies for Usher syndrome Type 1C in the retina. Adv. Exp. Med. Biol. 801:741–747. [DOI] [PubMed] [Google Scholar]
- Nagel‐Wolfrum, K. , Moller F., Penner I., and Wolfrum U.. 2014b. Translational read‐through as an alternative approach for ocular gene therapy of retinal dystrophies caused by in‐frame nonsense mutations. Vis. Neurosci. 31:309–316. [DOI] [PubMed] [Google Scholar]
- Nagel‐Wolfrum, K. , Moller F., Penner I., Baasov T., and Wolfrum U.. 2016. Targeting nonsense mutations in diseases with translational read‐through‐inducing drugs (TRIDs). BioDrugs 30:49–74. [DOI] [PubMed] [Google Scholar]
- Neveling, K. , Collin R. W., Gilissen C., van Huet R. A., Visser L., Kwint M. P., et al. 2012. Next‐generation genetic testing for retinitis pigmentosa. Hum. Mutat. 33:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveling, K. , Feenstra I., Gilissen C., Hoefsloot L. H., Kamsteeg E. J., Mensenkamp A. R., et al. 2013. A post‐hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum. Mutat. 34:1721–1726. [DOI] [PubMed] [Google Scholar]
- Ng, P. C. , and Henikoff S.. 2003. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi, K. M. , and Rivolta C.. 2012. Genes associated with retinitis pigmentosa and allied diseases are frequently mutated in the general population. PLoS ONE 7:e41902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, N. , Aldahmesh M. A., Alkuraya H., Anazi S., Alsharif H., Khan A. O., et al. 2016. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 18:554–562. [DOI] [PubMed] [Google Scholar]
- Pennings, R. J. , Te Brinke H., Weston M. D., Claassen A., Orten D. J., Weekamp H., et al. 2004. USH2A mutation analysis in 70 Dutch families with Usher syndrome type II. Hum. Mutat. 24:185. [DOI] [PubMed] [Google Scholar]
- Petit, C. 2001. Usher syndrome: from genetics to pathogenesis. Annu. Rev. Genomics Hum. Genet. 2:271–297. [DOI] [PubMed] [Google Scholar]
- Pierrache, L. H. , Hartel B. P., van Wijk E., Meester‐Smoor M. A., Cremers F. P., de Baere E., et al. 2016. Visual Prognosis in USH2A‐Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 123:1151–1160. [DOI] [PubMed] [Google Scholar]
- Ratbi, I. , Falkenberg K. D., Sommen M., Al‐Sheqaih N., Guaoua S., Vandeweyer G., et al. 2015. Heimler syndrome is caused by hypomorphic mutations in the peroxisome‐biogenesis genes PEX1 and PEX6. Am. J. Hum. Genet. 97:535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratbi, I. , Jaouad I. C., Elorch H., Al‐Sheqaih N., Elalloussi M., Lyahyai J., et al. 2016. Severe early onset retinitis pigmentosa in a Moroccan patient with Heimler syndrome due to novel homozygous mutation of PEX1 gene. Eur. J. Med. Genet. 59:507–511. [DOI] [PubMed] [Google Scholar]
- Reese, M. G. , Eeckman F. H., Kulp D., and Haussler D.. 1997. Improved splice site detection in Genie. J. Comput. Biol. 4:311–323. [DOI] [PubMed] [Google Scholar]
- Riazuddin, S. , Nazli S., Ahmed Z. M., Yang Y., Zulfiqar F., Shaikh R. S., et al. 2008. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum. Mutat. 29:502–511. [DOI] [PubMed] [Google Scholar]
- Rivolta, C. , Sweklo E. A., Berson E. L., and Dryja T. P.. 2000. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 66:1975–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, A. F. , Faugere V., Le Guedard S., Pallares‐Ruiz N., Vielle A., Chambert S., et al. 2006. Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J. Med. Genet. 43:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruether, K. , Baldwin E., Casteels M., Feher M. D., Horn M., Kuranoff S., et al. 2010. Adult Refsum disease: a form of tapetoretinal dystrophy accessible to therapy. Surv. Ophthalmol. 55:531–538. [DOI] [PubMed] [Google Scholar]
- Sadeghi, A. M. , Cohn E. S., Kimberling W. J., Halvarsson G., and Moller C.. 2013. Expressivity of hearing loss in cases with Usher syndrome type IIA. Int. J. Audiol. 52:832–837. [DOI] [PubMed] [Google Scholar]
- Scholl, H. P. , Strauss R. W., Singh M. S., Dalkara D., Roska B., Picaud S., et al. 2016. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 8:368rv6. [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Rodelsperger C., Schuelke M., and Seelow D.. 2010. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat. Methods 7:575–576. [DOI] [PubMed] [Google Scholar]
- Seyedahmadi, B. J. , Rivolta C., Keene J. A., Berson E. L., and Dryja T. P.. 2004. Comprehensive screening of the USH2A gene in Usher syndrome type II and non‐syndromic recessive retinitis pigmentosa. Exp. Eye Res. 79:167–173. [DOI] [PubMed] [Google Scholar]
- Shahin, H. , Walsh T., Rayyan A. A., Lee M. K., Higgins J., Dickel D., et al. 2010. Five novel loci for inherited hearing loss mapped by SNP‐based homozygosity profiles in Palestinian families. Eur. J. Hum. Genet. 18:407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer, A. E. , and Smith R. J.. 2012. Genetics: advances in genetic testing for deafness. Curr. Opin. Pediatr. 24:679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan‐Heggen, C. M. , Bierer A. O., Shearer A. E., Kolbe D. L., Nishimura C. J., Frees K. L., et al. 2016. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C. E. , Poulter J. A., Levin A. V., Capasso J. E., Price S., Ben‐Yosef T., et al. 2016. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 24:1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Smaoui N., Ayyagari R., Stiles D., Benhamed S., MacDonald I. M., et al. 2011. High‐throughput retina‐array for screening 93 genes involved in inherited retinal dystrophy. Invest. Ophthalmol. Vis. Sci. 52:9053–9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort M., Ball E. V., Shaw K., Phillips A. D., and Cooper D. N.. 2014. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S. V. , Deffenbaugh A. M., Yin L., Judkins T., Scholl T., Samollow P. B., et al. 2006. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 43:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vache, C. , Besnard T., le Berre P., Garcia‐Garcia G., Baux D., Larrieu L., et al. 2012. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum. Mutat. 33:104–108. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang V. W., Feng Y., Tian X., Li F. Y., Truong C., et al. 2014. Dependable and efficient clinical utility of target capture‐based deep sequencing in molecular diagnosis of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 55:6213–6223. [DOI] [PubMed] [Google Scholar]
- Welch, E. M. , Barton E. R., Zhuo J., Tomizawa Y., Friesen W. J., Trifillis P., et al. 2007. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447:87–91. [DOI] [PubMed] [Google Scholar]
- Weston, M. D. , Eudy J. D., Fujita S., Yao S., Usami S., Cremers C., et al. 2000. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am. J. Hum. Genet. 66:1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk, E. , Pennings R. J., te Brinke H., Claassen A., Yntema H. G., Hoefsloot L. H., et al. 2004. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 74:738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witters, P. , Regal L., Waterham H. R., De Meirleir L., Wanders R. J., and Morava E.. 2016. Normal cognitive outcome in a PEX6 deficient girl despite neonatal multisystem presentation. Am. J. Med. Genet. A 170:1642–1646. [DOI] [PubMed] [Google Scholar]
- Yan, D. , Ouyang X., Patterson D. M., Du L. L., Jacobson S. G., and Liu X. Z.. 2009. Mutation analysis in the long isoform of USH2A in American patients with Usher Syndrome type II. J. Hum. Genet. 54:732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. , Wei X., Chai Y., Li L., and Wu H.. 2013. Genetic etiology study of the non‐syndromic deafness in Chinese Hans by targeted next‐generation sequencing. Orphanet. J. Rare. Dis. 14:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, H. , Iwasaki S., Kanda Y., Nakanishi H., Murata T., Iwasa Y., et al. 2013. An Usher syndrome type 1 patient diagnosed before the appearance of visual symptoms by MYO7A mutation analysis. Int. J. Pediatr. Otorhinolaryngol. 77:298–302. [DOI] [PubMed] [Google Scholar]
- Zaki, M. S. , Heller R., Thoenes M., Nürnberg G., Stern‐Schneider G., Nürnberg P., et al. 2016. PEX6 is expressed in photoreceptor cilia and mutated in deafblindness with enamel dysplasia and microcephaly. Hum. Mutat. 37:170–174. [DOI] [PubMed] [Google Scholar]
- Zhao, L. , Wang F., Wang H., Li Y., Alexander S., Wang K., et al. 2015. Next‐generation sequencing‐based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum. Genet. 134:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Confirmation of a heterozygous intragenic deletion of CDH23 exons 19–27 in patient P103 by array‐CGH (244k Agilent Technologies microarray).
Table S1. Deafness genes analyzed by targeted NGS.