

Abstract
The renin-angiotensin system (RAS) is undisputedly one of the most prominent endocrine (tissue-to-tissue), paracrine (cell-to-cell) and intracrine (intracellular/nuclear) vasoactive systems in the physiological regulation of neural, cardiovascular, blood pressure, and kidney function. The importance of the RAS in the development and pathogenesis of cardiovascular, hypertensive and kidney diseases has now been firmly established in clinical trials and practice using renin inhibitors, angiotensin-converting enzyme (ACE) inhibitors, type 1 (AT1) angiotensin II (ANG II) receptor blockers (ARBs), or aldosterone receptor antagonists as major therapeutic drugs. The major mechanisms of actions for these RAS inhibitors or receptor blockers are mediated primarily by blocking the detrimental effects of the classic angiotensinogen/renin/ACE/ANG II/AT1/aldosterone axis. However, the RAS has expanded from this classic axis to include several other complex biochemical and physiological axes, which are derived from the metabolism of this classic axis. Currently, at least five axes of the RAS have been described, with each having its key substrate, enzyme, effector peptide, receptor, and/or downstream signaling pathways. These include the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor, the ANG II/APA/ANG III/AT2/NO/cGMP, the ANG I/ANG II/ACE2/ANG (1–7)/Mas receptor, the prorenin/renin/prorenin receptor (PRR or Atp6ap2)/MAP kinases ERK1/2/V-ATPase, and the ANG III/APN/ANG IV/IRAP/AT4 receptor axes. Since the roles and therapeutic implications of the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor axis have been extensively reviewed, this article will focus primarily on reviewing the roles and therapeutic implications of the vasoprotective axes of the RAS in cardiovascular, hypertensive and kidney diseases.
Keywords: Angiotensin 1-converting enzyme, ACE2, alamandine, angioprotectin, angiotensin (1–7), angiotensin II, cardiovascular disease, dipeptidyl peptidase III, hypertension
Graphical abstract
1. Introduction
The renin-angiotensin system (RAS) is undisputedly one of the most important endocrine (tissue-to-tissue), paracrine (cell-to-cell) and intracrine (intracellular/nuclear) vasoactive systems in the physiological regulation of cardiovascular, blood pressure, and kidney function, and the development of cardiovascular, hypertensive, and renal diseases [1–5]. The discovery of each of key members of the RAS, uncovering their cardiovascular, blood pressure, and renal actions, and developing pharmacological drugs to target the key enzyme(s) and receptor(s) of the RAS to treat cardiovascular, hypertensive and renal diseases have become a successful story in the cardiovascular, hypertensive, and renal research [1–5]. The earliest and most significant member of the RAS, the rate-limiting enzyme renin, was first discovered by Robert Tigerstedt and P. G. Bergman at Karolinska Institute in 1898 [6]. The foremost contribution of Tigerstedt and Bergman to the RAS research was their discovery of renin as a pressor substance released from the kidney, which increased blood pressure systemically [6]. However, it took almost 40 years until 1934, when Harry Goldblatt demonstrated in a landmark study that a vasopressor substance from the kidney induced hypertension in a dog model of two-kidney, one-clip (2K1C) renovascular hypertension [7]. Subsequent studies suggested that renal ischemia not only caused the release of renin from the kidney, but also a heat-stable, short-lived plasma pressor substance, which was initially named angiotonin or hypertensin, but was later widely recognized as angiotensinogen [8–10]. The liver was then considered to be the primary source of plasma angiotensinogen, with the first twelve amino acids, Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile-..., being the most important for its biological activity. It was in early 1950s that Skeggs and colleagues purified this peptide and found that there were actually two peptides, termed angiotensin I (ANG I) with ten amino acids, Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu, and ANG II with eight amino acids, Asp-Arg-Val-Tyr-Ile-His-Pro-Phe, and that ANG I had to be cleaved to form ANG II to be active by angiotensin-converting enzyme (ACE) [11,12]. Interestingly, ACE is also known as Kininase II [13,14], which metabolizes the vasodepressor peptide bradykinin; thus ACE is not only responsible for ANG II formation, but also for degradation of bradykinin [14]. Therefore the RAS and the kinin–kallikrein-bradykinin system interact to regulate cardiovascular, blood pressure, and renal function in health and disease [15,16].
In the tissues which express an enzyme named aminopeptidase A (APA), especially in the kidney, ANG II is metabolized to form des-aspartyl1-ANG II, also termed ANG III with seven amino acids, Arg-Val-Tyr-Ile-His-Pro-Phe [17–19]. ANG III is further metabolized by the action of Aminopeptidase N (APN) to form ANG IV with six amino acids, Val-Tyr-Ile-His-Pro-Phe, ANG (3–8) [17,19,20]. Unlike ANG II, both ANG III and ANG IV were previously considered partial agonists for ANG II receptors with lesser vasopressor activity [21–23]. More recently, ANG III has been recognized as a major ligand for the AT2 receptor/NO/cGMP signaling cascade [18,24,25], whereas ANG IV is thought to activate the AT4 receptor [23,26–28]. In late 1980s and early 1990s, through the study of ANG II receptor pharmacology it was discovered that ANG II acted on at least two different classes of G protein-coupled receptors, which led to the development of two classes of nonpeptide ANG II receptor antagonists, losartan as a representative blocker for type 1 (AT1) and PD123319 as a representative type 2 (AT2) receptor blockers [1,29–31]. The AT1 receptor was successfully cloned in 1991 by Murphy et al. [32] and Sasaki et al [33], respectively, which shares the seven-transmembrane-region motif with the G protein-coupled receptor superfamily. The AT1 receptor mediates the well-recognized effects of ANG II on cardiovascular, blood pressure, and renal systems, such as vasopressor, cardiac hypertrophic, hypertensive, renal vasoactive and salt-retaining actions, as well as aldosterone biosynthesis and release [1,5,29,31]. The AT2 receptor was successfully cloned by Mukoyama M et al. [34], Nakajima M [35], and Kambayashi Y [36], respectively. The cloned AT2 receptor has 34% identical sequence to the cloned AT1 receptor, shares a seven-transmembrane domain topology [34], and appears to mediate ANG II-induced inhibition of protein tyrosine phosphatase in COS-7 cells [36]. The 3rd ANG receptor termed “AT3” was reportedly cloned to encode a Mr 40,959 protein with 95% amino acid identity to the rat smooth muscle AT1 receptor, which also mediates ANG II-induced Ca2+ mobilization [37]. However, the so-called “AT3” receptor has not been recognized in the field. The 4th ANG receptor was identified in 2001 as insulin-regulated aminopeptidase (IRAP) by Albiston et al. using protein purification and peptide sequencing [26]. ANG (3–8) was found to bind and activate this receptor in the brain and plays an important role in learning and memory [26,38]. Most recently, the Mas oncogene was identified by Santos et al. as the specific receptor for ANG (1–7) [39], playing a key role in mediating ANG (1–7)-induced cardiovascular, vasodepressor, and renal responses [39–42].
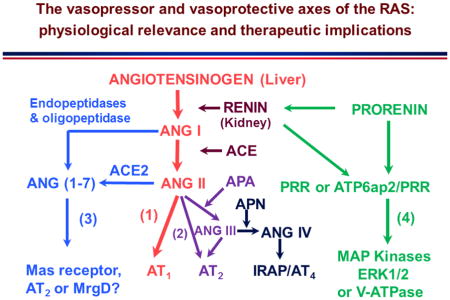
Collectively, it is now well understood that the classic RAS axis includes the substrate angiotensinogen primarily from the liver, which is cleaved by the rate-limiting enzyme renin released primarily from the juxtaglomerulus apparatus (JGA) of the kidney to form the biologically inactive ANG I. ACE, which is primarily localized in the vascular endothelium of major target organs including lung, heart, kidney, adrenal glands, brain etc., converts ANG I to the most potent vasopressor of the RAS, ANG II, by removing two amino acids, His-Leu, from ANG I. ANG II acts on two classes of G protein-coupled receptors (GPCR), AT1 and AT2 and possibly the Mas-related G-protein-coupled receptor (MrgD) as well, to play important “YIN” and “YANG” counter-regulatory roles in maintaining normal cardiovascular, blood pressure and renal function, as well as in the development of cardiovascular, hypertensive and renal diseases [1,5,29,43]. In addition to the classic axis, the RAS has evolved well beyond its classic paradigms primarily as a powerful vasopressor, growth/fibrosis promoter, a potent aldosterone stimulator, or a sodium-retaining hormone [25,39,41,42]. New members of the RAS are continuously discovered with different roles during last few decades, so that ANG II is no longer the only active peptide of the RAS. Figure 1 summarizes the current understanding on the entire RAS superfamily, including the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor axis, the prorenin/renin/prorenin receptor (PRR or Atp6ap2)/MAP kinases ERK1/2/V-ATPase axis, the ANG II/APA/ANG III/AT2/NO/cGMP axis, the ANG I/ANG II/ACE2/ANG (1–7)/Mas receptor axis, and the ANG III/APN/ANG IV/IRAP/AT4 receptor axis. The first two axes represent the powerful vasopressor systems, which are physiologically required to maintain normal cardiovascular, blood pressure, and renal homeostasis [1,5,29,44]. However, overactivation of these two axes of the RAS plays a critical role in the development of cardiovascular and kidney diseases and hypertension. By contrast, the other two axes of the RAS, including the ANG II/APA/ANG III/AT2/NO/cGMP axis [24,25,45] and the ANG I/ANG II/ACE2/ANG (1–7)/Mas receptor axis [39–42] may serve as the vasodepressor and cardiorenal protective arms of the RAS acting to counteract the detrimental effects of the renin/ACE/ANG II/AT1 receptor axis and the prorenin/renin/PRR/MAP kinases ERK1/2/V-ATPase axis. The ANG III/APN/ANG IV/IRAP/AT4 receptor axis is unlikely classified as an independent member of the vasopressor or vasodepressor system, since it appears to play a primary role in learning and memory [26,31,38]. Recently, a number of new enzyme(s) and peptides have been reported to possess the vasodepressor properties, which may have novel therapeutic implications in cardiovascular, hypertensive, and renal diseases. Among these newly described enzyme(s) or peptides, dipeptidyl peptidase III, or DPP III [46], alamandine [47–49], and angioprotectin [50,51], have been described as “new members” of the RAS or associated with the vasoprotective arms of the RAS. These enzyme(s) or peptides act to counter or opposing the actions of ANG II and may have a potential therapeutic role in treating hypertension associated with the activation of RAS [46–48].
Figure 1.

Classical and new paradigms of the evolving renin-angiotensin system. (1): receptor axis. (2): the ANG the classical angiotensinogen/renin/ACE/ANG II/AT1 receptor/NO/cGMP axis. (3): the ANG I/ANG II/ACE2/ANG (1–7)/Mas II/APA/ANG III/AT2 receptor axis. (4): the prorenin/renin/prorenin receptor (PRR or ATP6ap2)/MAP kinases receptor/IRAP axis. Modified ERK1/2/V-ATPase axis. (5): the ANG III/APN/ANG IV/AT4 from reference [5] with permission.
The roles and the therapeutic implications of the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor axis in the cardiovascular, blood pressure, and renal physiology and diseases have been extensively reviewed elsewhere, and therefore are not the subject of this review article. Instead, we focus our reviews and discussions primarily on recent advances in the research of the roles, insights, and potential therapeutic implications of the vasodepressor axes of the RAS.
2. New insights into the roles and therapeutic implications of the ANG II/APA/ANG III/AT2/NO/cGMP axis in cardiovascular, hypertensive and renal diseases
2.1. Role of aminopeptidase A
To maintain physiological cardiovascular, blood pressure and renal homeostasis by the RAS, a balance between the formation and degradation of the effector peptide ANG II is critical. ANG II is increased in the plasma and tissues either by increased ACE expression and actions or by decreasing the expression and activity of key enzymes that metabolize ANG II. Increased ANG II formation over its degradation leads to elevation of circulating and tissue ANG II levels, which play a key role in animal models of hypertension with 2-kidney, 1-clip [52–54] or ANG II infusion [55–57]. ACE is the primary enzyme responsible for ANG II formation, and its biochemistry, localization and its implications in cardiovascular, hypertensive and kidney diseases have been studied and reviewed extensively during last several decades in the context of the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor axis [58–60]. Thus the role of ACE in the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor axis is not the focus of discussion in this article. By comparison, the roles of ANG II degradation in cardiovascular, blood pressure and renal regulation have not been well studied. ANG II is metabolized to form des-aspartyl1-ANG II, ANG III, primarily by aminopeptidase A (APA) [17–19,61]. Aminopeptidase A, also named glutamyl aminopeptidase (EC 3.4.11.7), aspartate aminopeptidase, or angiotensinase A, is a zinc-dependent membrane-bound enzyme [17–19,61]. Its primary action is to cleave glutamatic and aspartatic amino acid residues from the N-terminus of polypeptides, such as ANG II. Aminopeptidase A circulates in the plasma and hydrolyzes ANG II reportedly at the rate of 4.1 ± 0.5 nmol/min/ml and a Km of 90.3 ± 14.3 μM in the rat plasma [62]. Aminopeptidase A is expressed widely in tissues including the kidney, vessels, and brain [17,63,64]. Within the kidney, the glomeruli and the proximal tubules in Spontaneously hypertensive rats (SHR) and ANG II-infused rats expresses high levels of APA and high APA activity [17,20,61]. In the extrarenal tissues, the pituitary and the circumventricular organs express high levels of APA or APA activity, followed by the median eminence, the area postrema, and paraventricular nuclei, which are associated with the central regulation of cardiovascular function and blood pressure [64,65]. Indeed, it is interesting that APA activity was higher in SHR in these brain nuclei, where AT1 receptors have been localized [65].
Since APA is the key enzyme for the degradation of ANG II, its implications in cardiovascular diseases have been studied in APA-KO mice [66,67]. Lin et al. first generated mice with deficiency of APA by gene targeting in embryonal stem cells to study its role in normal B and T cell development [66]. The absence of APA in these mice was confirmed in the lack of APA proteins and activity in bone marrow-derived stromal cells, thymic cortical epithelial cells, and renal proximal tubular cells [66]. This study showed that APA is not essential for normal B and T cell development, but it did not study any cardiovascular, blood pressure and kidney phenotypes [66]. Mitsui et al. showed that basal systolic blood pressure was significantly elevated in APA-KO mice, compared with heterozygous mutant and wild-type littermate mice [67]. Furthermore, infusion of ANG II further increased blood pressure in APA-KO mice, an enhanced response due to the lack of APA to metabolize ANG II [67]. Interestingly, deletion of APA did not increase the heart or kidney wt. to body wt. ratio, nor altered urinary sodium and water excretion and osmolality, in the presence of significant elevation of blood pressure [67]. But a later study reported that water consumption in APA-KO mice was significantly elevated, suggesting that ANG II rather than ANG III regulates water drinking behavior or thirst [68]. Kubota et al. demonstrated that ischemia-induced angiogenesis is impaired in APA-KO mice via down-regulation of HIF-1α, suggesting a potential role of ANG III in ischemia-induced angiogenesis [69]. While these findings from APA-KO mice provide the proof of the concept for a physiological role of APA in the blood pressure regulation, thirst response, or ischemia-induced angiogenesis, the potential therapeutic implications of APA in cardiovascular, hypertensive and kidney diseases remain to be further determined. However, the clinical benefits of APA as a therapeutic target may most likely be limited, since it acts to decrease circulating and tissue ANG II levels primarily by degrading ANG II, rather than by inhibiting ANG II formation. Although overexpression of APA in tissues may increase ANG II degradation to attenuate the effects of ANG II, such as in cultured mouse mesangial cells [70], its feasibility in humans remains uncertain. Indeed, if the goal is to decrease circulating and tissue ANG II levels either by inhibition of its formation or by increasing its degradation, the renin inhibitors and ACE inhibitors, especially the latter, have been widely used to block the RAS in treating cardiovascular, hypertensive and kidney diseases with proven clinical beneficial outcomes [71–74]. A potential pharmacological strategy to upregulate APA expression or enhance its activity to further decrease circulating and tissue ANG II levels may be warranted, if ACE inhibitors and/or renin inhibitors are inadequate to treat hypertension or cardiovascular and kidney diseases associated with the activation of the RAS.
2.2. Role of angiotensin III
Angiotensin III (ANG III) is the key metabolite of the effector peptide ANG II derived from the enzymatic action of APA to remove its 1st amino acid to des-aspartyl1-ANG II [20,64,75]. The metabolism of ANG II and generation of ANG III and their structure and activity relationship have been extensively investigated from 1970s to present [18,20,21,76,77]). Although ANG III is thought to circulate in the blood as well as is generated in tissues, few studies have specifically measured and compared the levels of ANG III in the plasma and tissues relative to ANG II. The reported levels of ANG III in the plasma and kidney varied widely from fmol/ml or fmol/g to nmol/ml or nmol/g due to different assays or approaches being used [78–80]. Using Liquid chromatography–tandem mass spectrometry (LC-MS/MS), Wysocki et al. reported that the level of plasma ANG III was remarkably higher than that of ANG II in wild-type mice on the C57BL6 genetic background (1,047±178 vs. 7.8±2.0 pg/ml), whereas the kidney ANG III level was much lower than that of plasma (172±12 pg/g) [81]. Also using LC-MS/MS, Olkowicz et al. reported the plasma ANG III level nearly 35-fold lower at 30.3 ± 3.8 pg/ml in wild-type mice [82]. More reliable ANG II levels in the plasma and kidney may be obtained by the gold-standard high-performance liquid chromatography (HPLC)-based RIA or ELISA approach [78–80]. Campbell and colleagues simultaneously measured and compared angiotensin peptides and their carboxy-truncated metabolites in human and rat plasma using N-terminal-directed antisera and HPLC [78,79]. In the plasma of normal male human subjects, the level of ANG II is reportedly five-fold higher than that of ANG III (13.9 ± 2.0 vs. 2.9 ± 1.0 fmol/ml) [78]. In normal rat plasma, the ANG II level is about 50% of that for ANG I, whereas the ANG III is about 50% of ANG II (47 ± 17 vs. 29 ± 8 fmol/ml), respectively [79]. In the normal rat kidney, ANG II is the major ANG peptide and the ANG II level is much higher than in the plasma, whereas the level of ANG III is much lower than ANG II (338 ±33 vs. <15 fmol/g) [79]. Kemp et al. appeared to confirm the finding of lower kidney ANG III, but the levels of renal cortical interstitial ANG II and ANG III were comparable (76±12.0 and 77.4±12.1 fmol/ml) [18]. In the rat proximal tubule fluid collected from free-flow micropuncture, ANG II and III concentrations were estimated at 6–8 nM and 14–25 nM, respectively [83]. Thus based on the available data, ANG III levels are most likely lower in the plasma and kidney than ANG II.
The physiological roles of ANG III as a vasopressor or a vasodepressor remain incompletely understood. It also remains unknown whether there is a specific receptor for ANG III [1,4,31]. One exception is that an “AT3” receptor was reportedly cloned with 95% amino acid identity to the AT1 receptor, but it remains to be recognized [1,37]. It is likely that ANG III may bind and activate AT1 and AT2 receptors with lower affinity than ANG II [1,84], and its vasopressor or vasodepressor effects may be dose-dependent on whether the AT1 or AT2 receptor is activated [4,18,76,85,86]. Previous studies in 1970s and 1980s have shown that although not as potent as ANG II, up to 40% to 50% of the pressor activity of ANG III was retained with removal of the aspartic acid from ANG II [21,87]. In a human study, Carey et al. carefully compared the effect of [des-Asp]-ANG II (ANG III) and ANG II on blood pressure and aldosterone production, and demonstrated that ANG III is less efficacious than ANG II in increasing blood pressure and stimulating aldosterone production [87]. In rats, Gardiner et al. showed that although less potent than ANG II, ANG III induced dose-dependent pressor and renal and mesenteric vasoconstrictor effects [88]. In the rat kidney, we previously found that unlike ANG II, equimolar ANG III failed to completely displace 125I-[Sar1,Ile8]-ANG II receptor binding in the glomeruli, proximal tubules, and the inner stripe of the outer medulla [84] or raise blood pressure induced by ACE inhibition with enalaprilat, suggesting that ANG III acts as a weaker agonist of ANG II [76].
More recently, Carey’s group has been instrumental to uncovering a novel role of ANG III in the kidney in inducing natriuresis via activation of AT2 receptors [18,89,90]. To demonstrate the roles of ANG III and AT2 receptors, Padia et al. first treated rats with candesartan to block AT1 receptors before ANG III was infused directly into the renal cortical interstitium to stimulate AT2 receptors [89]. Interestingly, ANG III significantly increased urinary sodium excretion in the presence of candesartan, suggesting a possible role of AT2 receptors in mediating ANG III-induced natriuretic response unmasked by AT1 receptor blockade [89]. Subsequent studies by Carey’s group further confirmed these ANG III-induced natriuretic responses in Wistar-Kyoto, but not in spontaneously hypertensive rats (SHR), implying an important role for a defect in ANG III/AT2 receptor signaling in the development of hypertension in SHR [90]. A further support for the interaction between AT2 receptor and ANG III came from a study that administration of a highly selective nonpeptide AT2 receptor agonist, C21, was able to recapitulate the ANG III-induced natriuretic effect [24]. These studies lead to the hypothesis that in the kidney, ANG III is the predominant ligand for proximal tubule AT2 receptors, and activation of which induces natriuresis responses and contributes to blood pressure regulation [18]. However, how ANG III preferably binds and stimulates AT2 receptors in the kidney remains to be elucidated.
2.3. Role of the AT2 receptor
The AT2 receptor plays a key role in mediating the vasodepressor responses, and cardiorenal protective effects to the activation of the APA/ANG III/AT2/cGMP axis [1,91–94]. While the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis acts as a “YANG” factor to induce vasoconstriction, increase blood pressure, and promote growth and fibrotic and proinflammatory responses, the APA/ANG III/AT2/cGMP axis acts as a “YIN” factor to counteract the vasopressor effect of ANG II/AT1 activation to induce vasodilation and decrease blood pressure to maintain normal blood pressure homeostasis [25,95–97]. Before the AT2 receptor was molecularly cloned, this class of ANG II receptors was primarily discriminated by using nonpeptide antagonists PD123177 and PD123319 or a peptide agonist CGP42112A in ANG II receptor binding assays [1,29,84,98]. Using radioreceptor binding assays and autoradiography with 125I-[Sar1,Ile8]-ANG II as the radioligand and losartan, PD123317 or PD123319 as ANG II receptor blockers, we have localized the distribution of AT2 receptors in the kidneys, heart, blood vessels, adrenal glands, and brain (Fig. 2) [98–102]. Notably, AT2 receptor binding is widely localized in fetal and neonatal tissues days after birth, but it was primarily localized in cell- or tissue-specific manners in certain tissues of adult humans and rodents. Inagami’s group first cloned the rat AT2 receptor cDNA from a rat pheochromocytoma cell line [36] and isolated the human AT2 receptor from a genomic DNA library of human placenta [103]. Both rat and human AT2 cDNAs encode a 363-amino acid protein with 7-transmembrane domains and a 32% identity of amino acid sequence of the rat and mouse AT1 receptor [36,103]. In COS-7 cells stably expressing the rat AT2 receptor, the AT2 receptor blocker PD123319, but not the AT1 blocker losartan blocked its binding to ANG II [36]. Although the signaling transduction mechanisms, upon which ANG II binds and activates AT2 receptors, have been extensively studied in cultured cells in vitro and in tissues in vivo, they remain incompletely understood. Unlike the well-established signaling pathways for the AT1 receptor, the most commonly described signaling pathways for the AT2 receptor include ANG II-induced inhibition of the protein phosphotyrosine phosphatase (PTP) activity, phospholipase A(2), nitric oxide, and cyclic guanosine monophosphate (cGMP) [1,36,95,96].
Figure 2.

In vitro autoradiographic localization of AT1 and AT2 receptors in the human kidney and renal artery using [125I]-[Sar1,Ile8]-ANG II as the radioligand. A & E: total ANG II (AT1 and AT2) receptors. C & F: AT1 receptor binding in the presence of 10 μM of the AT2 receptor blocker PD123319 (10 μM). D & G: AT2 receptor binding in the presence of 10 μM of the AT1 receptor blocker losartan. Modified from reference [100] with permission.
The physiological or vasodepressor role of the ANG II/ANG III/AT2/cGMP axis has been extensively investigated during last two decades. Although it was widely believed that the primary action of the AT2 receptor activation is to oppose the vasopressor action of the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis on the cardiovascular, blood pressure, and kidney functions, the results of early animal studies were not very conclusive [1,29,30]. Indeed, it is very difficult to elicit the vasodepressor effect of the ANG II/ANG III/AT2/cGMP axis due to the dominant role of the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis in cardiovascular, blood pressure and kidney regulation [92,104]. For example, activation of the AT2 receptor with the AT2 receptor-selective agonist CGP42112A or pharmacological blockade of the AT2 receptor with the AT2 receptor-selective antagonists, PD123177 or PD123319, alone doesn’t alter blood pressure in animal studies [1,29,92]. Barber et al. demonstrated that AT2 receptor–mediated depressor responses to CGP42112A were demonstrated only in the presence of AT1 receptor blockade in both SHR and WKY rats [104]. Li and Widdop further showed that in the presence of AT1 receptor blockade with candesartan, CGP42112 caused a marked depressor effect, which was abolished by the coadministration of the AT2 receptor antagonist, PD123319 (Fig. 3) [92]. The unmasking of the vasodepressor role of the AT2 receptor during AT1 receptor blockade has important clinical therapeutic implications, because AT1 receptor blockers (ARBs) are currently widely prescribed to treat cardiovascular, hypertensive and kidney diseases [92,94,104,105]. ARBs selectively blocks the actions of the angiotensinogen/ renin/ACE/ANG II/AT1 receptor axis by binding and occupying AT1 receptors in target tissues, which leads to several-fold increases in the circulating ANG II levels [106,107]. ANG II then may bind and activate the unopposed AT2 receptors in cardiovascular and kidney tissues to induce vasodepressor effects.
Figure 3.

In vitro autoradiographic localization of AT1 and AT2 receptors in bovine (A–D), monkey (E–H) and human adrenal glands (I–L) using [125I]-[Sar1,Ile8]-ANG II as the radioligand. A, E & I: total ANG II (AT1 and AT2) receptors. B, F & J: AT1 receptor binding in the presence of 10 μM of the AT2 receptor blocker PD123319 (10 μM). C, G & K: AT2 receptor binding in the presence of 10 μM of the AT1 receptor blocker losartan. D, H & L: nonspecific binding in the presence of 10 μM of unlabled ANG II. Modified from [99] with permission.
The evidence supporting the vasodepressor role of AT2 receptors is best appreciated in genetically modified mice lacking [91,108] or overexpressing AT2 receptors [109]. Although AT2 receptors are abundantly expressed in most of fetal tissues [1,36,99], one would expect that genetic deletion of AT2 receptors may lead to the impairment of the development and growth of organs or tissues. However, AT2 receptor-null mice develop normally and show no structural defects in the brain, heart, vessels, and kidneys [91,108]. The lack of developmental defects in AT2 receptor-null mice is surprising, given that the structural or developmental abnormalities in the kidney have been reported in mice lacking angiotensinogen [110,111], ACE [58,112], and both AT1a/AT1b receptor double-null mice [113,114]. One study shows that basal blood pressure remains unaltered [108], while the other shows elevated basal blood pressure in AT2 receptor-null mice [91]. Although these results on basal blood pressure seem conflicting, both studies demonstrated increased vasopressor response to ANG II stimulation in AT2 receptor-null mice [91,108]. Siragy et al. subsequently confirmed that basal systolic blood pressure in AT2-null mice was slightly elevated, compared with wild-type mice, but showed sustained hypertensive and antinatriuretic responses to ANG II [45]. In transgenic mice overexpressing AT2 receptors in vascular smooth muscle cells, the AT1-mediated pressor effect of ANG II was completely abolished in AT2 transgenic mice via the bradykinin/NO/cGMP signaling [109]. While the latter study may seem too perfect, all available data appear to be consistent with the concept that AT2 receptors may be physiologically important and beneficial in counteracting the vasopressor effect of ANG II [45,91,108].
Whether the ANG II/APA/ANG III/AT2/cGMP axis may be targeted in preventing and treating cardiovascular, hypertensive, and kidney diseases has been a subject of continuous debates for two decades [93,96,105]. The fundamental premise of therapeutically targeting the ANG II/APA/ANG III/AT2/cGMP axis is primarily based on the “YIN” and “YANG” balance hypothesis, in that as a “YANG” factor, the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis plays a key role in the pathogenesis of cardiovascular, hypertensive, diabetic and kidney diseases, whereas as an “YIN” factor, the ANG II/APA/ANG III/AT2/cGMP axis acts to counteract the detrimental effects of ANG II/AT1 activation [25,95–97]. Indeed, one of the most successful stories in the drug development for cardiovascular, hypertensive, diabetic and kidney diseases during last three decades is the successful development of ACE inhibitors, ARBs, renin inhibitors, and aldosterone receptor antagonists to treat hypertension, ischemic heart disease or heart failure, and diabetic nephropathy in humans [72,74,115–117]. However, it has been questioned whether AT2 receptor activation may have deleterious effects in cardiovascular diseases [105]. AT2 receptors have been implicated in programed cell death [118], cardiomyocyte enlargement during pressure overload [119], left ventricular hypertrophy and cardiac fibrosis during ANG II-induced hypertension [120], and cellular proliferation in the kidney [121]. Nevertheless, most of these reported “deleterious effects” of AT2 receptors were mainly deduced from animal studies using the AT1 and AT2 receptor-selective blockers or AT2 receptor-null mice as the experimental model.
2.4. Therapeutic role of CGP42112A as a peptide AT2 receptor agonist
Perhaps a more direct and clinically relevant approach will be to investigate whether either peptide or non-peptide AT2 receptor agonists are antihypertensive, cardiovascular and renal protective in animal models and human trials. Two different AT2 receptor-selective agonists, one peptide and the other nonpeptide form, have been developed for targeting AT2 receptors. CGP42112A, an AT2 receptor-selective peptide agonist, has been widely used along with the nonpeptide AT2 receptor antagonist PD123319 to localize and characterize AT2 receptors in cells and tissues [84,122,123]. In vivo administration of CGP42112A has been shown to induce renal natriuretic responses via AT2 receptor-mediated NO/cGMP signaling and inhibition of Na+/K+-ATPase activity and oxidative stress in obese Zucker rats [124–126], lower blood pressure in SHR [92;104], and inhibit vascular endothelial growth factor-induced migration in human endothelial cells [127]. However, while it is useful as an agonist for AT2 receptor binding assay and for studying the roles of AT2 receptors in vitro and in vivo animal models, CGP42112A, as a peptide, may not be practical in human studies to activate the ANG II/APA/ANG III/AT2/cGMP axis.
2.5. Therapeutic role of Compound 21 as a novel nonpeptide AT2 receptor agonist
Recently, Vicore Pharma in Sweden has designed and patented a unique nonpeptide AT2 receptor agonist, named Compound 21, to target AT2 receptors for therapeutic application to treat pulmonary fibrosis or hypertension, renal failure, spinal cord injury and acute ischemic stroke [128]. Wan et al. first reported that Compound 21 is an ANG II receptor agonist with a Ki value of 0.4 nM for the AT2 receptor and a Ki >10 μM for the AT1 receptor [128,129]. They found that Compound 21 has a bioavailability of 20–30% after oral administration and a half-life of 4 h in rat, induces neurite outgrowth, and lowers blood pressure in anesthetized SHR, some well-described effects of AT2 receptor activation [128]. Further studies in animal models and humans reveal some very encouraging cardiovascular, blood pressure and renal effects of Compound 21, showing some potential therapeutic implications. Compound 21 dose-dependently inhibits tumor necrosis factor-alpha (TNFα)-induced interleukin 6 production and nuclear factor κB in primary human and murine dermal fibroblasts [130], decreases early renal inflammatory responses in renovascular hypertension via production of NO and cGMP [131,132], inhibits endothelial inflammation and leukocyte adhesion in human umbilical vein endothelial cells (HUVECs) and in ApoE−/− mice [133], and prevents vascular dementia induced by bilateral common carotid artery stenosis in mice [134]. Additionally, Compound 21 is protective against high salt diet-induced kidney injury in obesity Zucker rats [135]. Whether Compound 21 is antihypertensive in animal models and humans in vivo remains uncertain despite the well-recognized vasodepressor and natriuretic effects of AT2 receptor activation [94,136]. Compound 21 has no significant effect on blood pressure in stroke-prone spontaneously hypertensive rats (SHR) [137] or on renal vasodilator responses in SHR [138]. High blood pressure in 2K1C rats is not altered by Compound 21 either [131]. By contrast, Kemp et al. demonstrated that C21 lowered blood pressure in ANG II-dependent hypertensive rats and mice through AT2 receptor-mediated natriuresis [24,94]. Taken together, the available evidence suggests that it is unlikely that Compound 21 may be useful as an antihypertensive agent, but it may be used as a potentially therapeutic agent to help prevent and treat hypertension-induced cardiovascular and kidney organ damages through its anti-fibrotic and anti-inflammatory actions [131,133,135,136], or to counteract AT1 receptor-mediated ANG II-dependent hypertension [24,94].
3. New insights into the roles and therapeutic implications of the ACE2/ANG (1–7)/Mas receptor axis in cardiovascular and renal diseases
3.1. ACE2
In addition to the well-recognized angiotensin-converting enzyme 1 (ACE), which converts the biologically inactive ANG I into the potent vasopressor ANG II, a new ACE isoform, named angiotensin-converting enzyme 2 (ACE2) was discovered in 2000 by two different groups of scientists [139,140]. The discovery of ACE2, along with the Mas receptor [39], as a key enzyme in generating the vasodepressor peptide ANG (1–7) was viewed as a scientific breakthrough in the RAS research [60,139,141]. Indeed, ACE2 is the key enzyme playing a crucial physiological role of the ACE2/ANG (1–7)/Mas receptor axis, the vasodepressor or cardiovascular, blood pressure and renal protective arm of the RAS [39,40,42]. Donoghue et al. were the first to clone ACE2, a human homologue of ACE, from 5' sequencing of a human heart failure ventricle cDNA library [139], found that ACE2 contains a signal peptide, a single metalloprotease active site, and a transmembrane domain [139]. Although ACE2 and ACE share only 42% of amino acid identity, they both act as carboxypeptidases to cleave amino acids from the peptides’ carboxyl terminal [139]. However, ACE and ACE2 differ in their enzymatic selectivity in that ACE cleaves two amino acids from ANG I to form ANG II at a time, whereas ACE2 cleaves only one amino acid from ANG I to form ANG (1–9), which is then converted to ANG (1–7) by ACE [60,139]. A further important enzymatic action of ACE2 is to metabolize ANG II to form the vasodepressor peptide ANG (1–7) to decrease the vasopressor peptide ANG II levels in the circulation and tissues [139,142]. Additionally, the activity and action of ACE2 are not affected by ACE inhibitors, further distinguishing ACE2 from the classic ACE [139,140].
3.2. Localization of ACE2 in cardiovascular and kidney tissues
Donoghue et al. first reported that ACE2 is expressed mainly in the heart, kidney, and testis of 23 human tissues in their landmark study [139]. In the heart, Northern blot analysis revealed that ACE2 mRNA is expressed as a 3.4-kb transcript, and ACE2 immunohistochemical analysis showed its primary localization in the ventricular myocardium, the endothelium of capillaries, and the coronary arteries [139]. Lower levels of ACE2 expression was found in vascular smooth muscle cells and in the adventitia of larger blood vessels. One interesting finding is that the distribution or level of ACE2 protein expression is not different in failing and nonfailing human hearts [139]. In the kidney, ACE2 is primarily localized in the endothelium of intrarenal blood vessels and the proximal tubule epithelial cells [139,143,144], with very low levels of ACE2 proteins in the glomeruli [139]. Similar findings were observed in biopsied samples obtained from normal and diseased human kidneys [145], though no differences in ACE2 expression was found in human kidneys of normal and various renal diseases [145]. In the mouse kidney, ACE2 was localized in glomerular podocytes, whereas ACE was localized in glomerular endothelial cells [143], consistent with the finding that ANG I or ANG II was metabolized to form ANG (1–7) in the glomerular podocytes [146]. In the rat kidney, ACE2 and ACE proteins were colocalized predominantly in the proximal tubule, in which ACE2 expression was downregulated in diabetes [144]. These animal studies were supported by findings that glomerular and tubular ACE2 expression was decreased in patients with type 2 diabetes and kidney disease [147,148]. In the mouse brain, ACE2 was localized in the cytoplasm of neuronal cells, especially in the subfornical organ (SFO), a neural structure outside the blood-brain barrier, the nucleus of tractus solitarius (NTS), dorsal motor nucleus of the vagus, and the ventrolateral medulla [149]. The localization of ACE2 in these brain structures is physiologically relevant, since these brain structures are closely involved in the neural control of cardiovascular function by the RAS.
3.3. Therapeutic implications of ACE2 in cardiovascular, hypertensive and kidney diseases
3.3.1. New insights from ACE2-KO mice
In contrast to the classic ACE, which converts inactive ANG I to the active ANG II, ACE2 cleaves ANG I to ANG (1–9) and ANG II to ANG (1–7) [139,140]. Thus, whilst ANG II is the effector peptide for the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis, ANG (1–7) is the effector peptide for the ANG I/ACE2/ANG (1–7)/Mas receptor axis [39,40,42]. The roles and therapeutic implications of ACE2 in cardiovascular, hypertensive and renal diseases are best appreciated from studying the phenotypes of ACE2 knockout mouse models [150–152]. ACE2 knockout mice were first generated by Crackower et al. in 2002 to determine its roles in the regulation of cardiac function [150]. ACE2 knockout in mice showed severe cardiac contractility defect with significantly increased ANG II levels in the heart and kidney [150]. The left ventricular wall of ACE2-null mice was slightly thinner, whereas the chamber dimension was increased, but no cardiac hypertrophy or dilated cardiomyopathy was observed [150]. Blood pressure was normal in 3 month-old male, but significantly decreased in 6 month-old male ACE2-null mice, though the reason is unknown. Since genetic ablation of ACE on an ACE2 mutant background completely rescues the cardiac phenotype of ACE2-null mice, increased cardiac ANG II levels may contribute to the cardiac phenotypes of ACE2-null mice [150]. However two subsequently studies showed completely different cardiac phenotypes in their independent ACE2-null mice [151,152]. Yamamoto et al. reported that their ACE2-null mice showed completely normal cardiac histology and function, including heart wt. and cardiac morphology, systolic left ventricular pressure, though the hypertrophic response to pressure overload induced by transverse aortic constriction was accelerated in these mice [152]. Gurley et al. generated ACE2-null mice on two different genetic background, one on C57BL/6 and the other on 129/SvEv [151]. No abnormal cardiac morphological and functional phenotypes were observed. Blood pressure was moderately increased in ACE2-null mice on the C57BL/6 genetic background, but not in ACE2-null mice on the 129/SvEv genetic background, although hypertension induced by ANG II infusion was more severe in ACE2-null than in wild-type mice [151]. The reasons underlying these significant differences in cardiac morphological and functional phenotypes between these studies remain unresolved, but likely due to the differences in target-deletion approaches, genetic backgrounds, age of animals, genders used, and experimental designs [150–152].
Although ACE2 is widely localized in the renal glomeruli, intrarenal blood vessels, and tubular epithelial cells, there are few studies have determined the roles of renal ACE2 on renal blood flow, glomerular filtration rate, or tubular transporter functions using ACE2-null mice [139,143,144]. The renal histological and functional phenotypes have not been studied in those three different lines of ACE2-null mice [150–152]. This is very surprising, given the importance of ACE2 in the metabolism of ANG I and ANG II to produce the vasodepressor and natriuretic ANG (1–7) in the kidney [39,42,153].
3.3.2. ACE2 as a therapeutic target
The therapeutic potentials of targeting ACE2 in treating cardiovascular, hypertensive and kidney diseases remain to be determined. It is attractive to speculate that any pharmacological approaches to upregulate ACE2 expression via overexpression or to stimulate ACE2 activity with chemical compounds to markedly increase the vasodepressor peptide ANG (1–7) would be clinically beneficial. Some proof of concept studies recently published have provided some support of the concept. In the mouse brain, Feng et al. used the adenovirus-mediated approach to overexpress the human ACE2 cDNA upstream of an enhanced green fluorescent protein (eGFP) reporter gene, Ad-hACE2-eGFP, in the subfornical organ, and reported attenuated pressor and drinking responses to ANG II [154]. In a further study, Feng et al. used a synapsin promoter to drive neuron-specific overexpression of hACE2 throughout the brain in mice, which also attenuated ANG II-induced hypertension [155]. In the kidney, Wysocki et al. tested whether a soluble human recombinant ACE2 (rACE2) may be used to decrease ANG II and increase ANG (1–7) levels in plasma and tissues, and whether rACE2 may be used to prevent ANG II-induced hypertension in mice [156]. Interestingly, this study found that rACE2 infusion induced a dose-dependent increase in serum ACE2 activity, but had no effect on kidney or cardiac ACE2 activity [156]. rACE2 infusion alone had no effect on blood pressure, but it normalized the hypertensive response to ANG II infusion [156]. Nadarajah et al. used a nephrin promoter to generate a transgenic mouse model with overexpression of human ACE2 specifically in podocytes of the glomerulus, and treated these transgenic mice with STZ to induce type 1 insulin-dependent diabetes [157]. Although urinary albumin excretion was significantly attenuated in hACE2 transgenic diabetic mice, compared with wild-type nondiabetic mice, at 4 weeks, this anti-albumin effect was not long-lasting [157]. Indeed, the differences in proteinuria were no longer observed between hACE2 transgenic and nontransgenic diabetic mice by 16 weeks [157]. The clinical relevance or practicality of using hACE2 to treat cardiovascular, hypertensive and kidney diseases has been evaluated in a clinical trial [158]. Haschke et al. designed a randomized, double-blind, placebo-controlled, single-dose, dose-escalation study in five cohorts of four subjects each, who were intravenously treated with single doses of rhACE2 at a starting dose of 100 μg/kg body weight in the first cohort over 30 min, whereas cohorts 2–4 were treated with 200, 400, and 800 μg /kg body weight, respectively [158]. Surprisingly, rhACE2 had no effect on Blood pressure and heart rate [158].
3.3.4. Therapeutic potentials of ACE2 activators
Recently, efforts have been made to identify so-called ACE2 activators for therapeutic application [159–161]. Hernandez et al. first used a novel conformation-based drug discovery strategy to identify compounds that increase ACE2 activity and to determine whether they may reverse hypertension-induced pathophysiologies [159]. Two compounds, xanthenone and resorcinolnaphthalein, were identified and found to increase ACE2 activity in a dose-dependent manner. Xanthenone showed a remarkable antihypertensive effect, as it acutely decreased blood pressure in SHR by a massive 71 mmHg, but its long-term antihypertensive effect was only moderate [159]. Xanthenone was also found to improve cardiac function and reverse myocardial, perivascular, and renal fibrosis in SHR [159]. However, despite of its impressive antihypertensive and cardiovascular protective effects, there are very few follow-up studies to confirm these findings by this and other groups. Additionally, a chemical compound, diminazene aceturate (DIZE), has been promoted as a potential ACE2 activator to treat ischemia-induced cardiac pathophysiology, pulmonary hypertension, and ischemic stroke [160–163]. DIZE is a widely used antiprotozoal agent to treat trypanosomiasis in domestic livestock, but rarely used in humans [164]. Mecca et al. first reported that intracerebroventricular infusion of either ANG (1–7) or DIZE significantly attenuated the cerebral infarct size and neurological deficits, which were reversed by co-intracerebroventricular administration of the Mas receptor inhibitor, A-779 [162]. This was interpreted as that DIZE may have cerebroprotective properties against endothelin 1-induced ischemic stroke [162]. Subsequent studies by the same group of investigators have demonstrated that DIZE significantly protected rats from monocrotaline (MCT)-induced pulmonary hypertension (PH) [163,165], and decreased myocardial infarction (MI)-induced infarct area, LV remodeling post-MI, and restored normal balance of the cardiac renin-angiotensin system [160]. Recently, the cardiovascular, hypertensive, and renal effects of targeting ACE2 with DIZE in various animal models have been comprehensively reviewed elsewhere often with mixed or conflicting results [161]. In twenty in vivo studies reviewed, only six studies reported a significant decrease in blood pressure in response to DIZE administration, whilst conflicting beneficial effects of DIZE on other cardiovascular, renal, or inflammatory responses were demonstrated [161]. Overall, the potential therapeutic use of DIZE as an ACE2 activator to treat cardiovascular, hypertensive, and diabetic, and kidney diseases remains to be determined for a number of reasons. One of the significant weaknesses of the DIZE story is that the off-target effects of DIZE on ACE2 activity have not been excluded in cultured cell models with absence of ACE2 expression in vitro or in ACE2-null mouse model, nor has it been compared with well-recognized renin and ACE inhibitors or ARBs. Another significant weakness is that most of these studies simply focused on the effect of DIZE on ACE2 activity, but paid no attention to the potential effects of DIZE on the expression of production of ANG I, Mas receptor, ACE, ANG II, AT1 or AT2 receptors. Increases in ANG (1–7) or Mas receptor expression, or downregulation of the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis would help explain the in vivo effects of DIZE. An easy argument against the use of DIZE as a therapeutic agent is that if the therapeutic goal of using DIZE is to activate ACE2 to increase the vasodepressor peptide ANG (1–7) level and lower the vasopressor peptide ANG II level, ACE inhibitors have already been widely used clinically, not mentioning that DIZE may not be safe for use in humans.
3.4. Localization, roles, and therapeutic implications of the Mas receptor in cardiovascular, hypertensive and kidney diseases
3.4.1. New insights from Mas-KO mice
Mas, or the Mas receptor, is the first discovered member of a subfamily of Mas-related G protein-coupled receptors (Mrgprs or Mas-related genes) [40,43], and a protooncogene encoding a protein with seven transmembrane domains [166]. In late 1980s, the Mas receptor was thought to mediate the effects of ANG II and ANG III on intracellular accumulation of inositol phosphates (IP3) and calcium (Ca2+) after in Mas-transfected cells, suggesting that the Mas oncogene may encode a different ANG receptor [167]. However, two subsequent studies in the early 1990s found no evidence of Mas-induced ANG II receptor and/or Ca2+ responses in Xenopus oocytes or in most of transfected cells [168,169]. Thus the Mas receptor and ANG II receptor are not identical in their molecular structure, function and signaling [168,169]. To determine whether the Mas receptor plays a central role in anxiety, spatial learning, and memory responses, Walther et al. performed targeted deletion of the Mas receptor in mice and demonstrated sustained long term potentiation and anxiety in mice lacking the Mas protooncogene [170]. No direct interactions or associations in the localization and function were found between the Mas receptor and the ANG II/AT1 receptor axis in the brain [170]. It was Santos et al. who first reported that ANG (1–7) is an endogenous ligand for the Mas receptor, thus uncovering for the first time that Mas is the specific receptor for the vasodepressor peptide ANG (1–7) [39]. However, there is evidence that some neuropeptides (beta-alanine, alamandine, GABA, cortistatin-14, metabolites of proenkephalin, pro-opiomelanocortin, prodynorphin), may also be potential ligands for the Mrgprs family including the Mas receptor [40,43]. These ligands activate the Mrgprs to initiate the signaling cascades coupled to Gq/11 and pertussis toxin (PTX)-sensitive Gi/o proteins, activation of which mobilizes PTX-insensitsitive intracellular Ca2+ and PTX-sensitive inhibition of forskolin-induced activation of adenylyl cyclase [43]. There is also evidence that additional receptors, such as AT2 receptors and MrgD receptors, may be involved in mediating the effects of ANG (1–7) [47,48,171–173].
Previous studies have shown that most of the Mrgprs family are expressed in primary sensory neurons in the brain [40,43,174]. Mas is also expressed in mast cells in other tissues, such as the testis, heart and kidney [39,40,43]. In the kidney, Santos et al. demonstrated that the Mas receptor was localized in all regions of the mouse kidney, though some of 125I-labelled ANG (1–7) receptor binding persisted in the kidney of Mas receptor-deficient mice [39]. Using the immunofluorescent approach with a Mas receptor antibody, Gwathmey et al. localized Mas antibody binding in the sheep kidney primarily in the proximal tubules, thick limb of Henle, and distal tubules [175], consistent with a natriuretic action of ANG (1–7) especially in the proximal tubules [176]. By contrast, we used [125I]-ANG (1–7) as a radioligand to map the distribution of ANG (1–7) receptor binding in the rat kidney, and demonstrated its predominant localization in the proximal tubules in the inner cortex (Fig. 4) [5]. Thus the results strongly suggest that ANG (1–7) may only be one of endogenous ligands for the Mas receptor, activation of which may lead to some off-target responses [39,40,43].
Figure 4.

In vitro autoradiographic localization of ANG (1–7) (A) and AT4 receptors (B) in the rat kidney using [125I]-ANG (1–7) and [125I]-ANG (3–8) as the radioligands, respectively. Modified from [5] and [23] with permission.
3.4.2. The Mas receptor as a therapeutic target
The roles and therapeutic implications of the Mas receptor in the cardiovascular, blood pressure and renal regulation may be best appreciated from our understanding the physiological roles of the ANG (1–7) (see discussion below) and the cardiovascular, blood pressure, and renal phenotypes demonstrated in Mas receptor-deficient mice [39.41,42,153]. The Mas receptor was first knocked out by Walther et al., but unfortunately cardiovascular, blood pressure and renal phenotypes were not studied [170]. In a subsequent study, Walther et al. reported that neither heart rate nor blood pressure were significantly different between adult Mas receptor-deficient mice and wild-type controls [177]. However, further analysis by these authors found that female Mas receptor-deficient mice showed reduced heart rate variability, and increased sympathetic tone in male and female Mas receptor-deficient mice [177]. Heringer-Walther et al. also failed to uncover any basal blood pressure phenotype in Mas receptor-deficient mice [178]. In the kidney, Santos reported that ANG (1–7) induced significant antidiuretic effect due to increased tubular water reabsorption, whereas the Mas receptor knockout in mice significantly attenuated this antidiuretic effect of ANG (1–7) [39]. The Mas receptor knockout abolished ANG (1–7)-induced, but not acetylcholine-induced endothelium-dependent relaxation in Mas receptor-deficient mice [39,179]. The lack of a blood pressure phenotype in Mas receptor-deficient mice is unexpected, because if the vasodepressor peptide ANG (1–7) is the endogenous ligand for the Mas receptor, targeted deletion of the Mas receptor is expected to elevate basal blood pressure. Likewise, the reported abolishment of ANG (1–7)-induced endothelium-dependent relaxation and ANG (1–7)-induced antinatriuretic effect is also expected to lead to increases in basal arterial pressure in Mas receptor-deficient mice [39]. The mechanisms underlying the absence of a clear blood pressure phenotype in Mas receptor-deficient mice remain to be determined. One reason may be due to the activation of other compensatory mechanisms to offset the effects of globally knocking out the Mas receptor. The other reason may be due to the different genetic backgrounds, mixed 129/C57Bl/6 or FVB/N, on which Mas receptor-deficient mice were generated [180]. To further uncover the cardiovascular and blood pressure phenotypes in Mas receptor-deficient mice, Xu et al. generated a new strain of Mas-deficient mice by backcrossing Mas-deficient mice for 7 generations onto the FVB/N background [180]. With the change in the genetic background, these authors were able to demonstrate that Mas- deficient mice had elevated basal blood pressure, endothelial dysfunction, and impaired NO production [180]. Furthermore, intracellular Ca2+ responses, eNOS expression and NO production in cardiomyocytes were significantly impaired in Mas-deficient mice on the FVB/N background [181], whereas PPARκ expression was decreased in adipocytes of Mas-deficient mice [182]. Taken together, these results are largely consistent with the well-recognized roles of ANG (1–7) in the cardiovascular, blood pressure and renal regulation, but need to be confirmed by others.
3.5. Roles and therapeutic implications of ANG (1–7) in cardiovascular, hypertensive and kidney diseases
3.5.1. ANG (1–7) as the effector peptide of the ACE2/ANG (-17)/Mas receptor axis
In the ACE2/ANG (1–7)/Mas receptor axis, ANG (1–7) is considered to be the most important effector peptide. The importance of ANG (1–7) in the neural, cardiovascular, blood pressure, and renal regulation and its therapeutic implications has been extensively investigated since 1970s [41,42,85,183]. Indeed, a careful search of PubMed easily yielded over 3300 published studies directly and indirectly involving ANG (1–7). ANG (1–7) was previously considered to be an inactive component of the RAS in the 1970s, because structure and activity studies showed that removal of phenylalanine (position 8) or the dipeptide, Pro-Phe (positions 7 and 8) from the potent vasopressor ANG II completely abolished the vasoconstrictor, pressor or thirst-promoting effects of ANG II [184). However, Ferrario’s group at Cleveland Clinic and then at Wake Forest University has been instrumental in uncovering the vasodepressor roles and mechanisms of ANG (1–7) in the neural, cardiovascular, blood pressure, and renal regulation due to their persistent effort from 1980s to present [41,42,85,183]. Recent identification of the Mas receptor for ANG (1–7) [39], and ACE2 as the key enzyme for the formation of ANG (1–7) [139] adds further enthusiasm to the ongoing research on the roles and therapeutic implications of the ACE2/ANG (1–7)/Mas receptor axis. Now it appears to be well accepted that the ACE2/ANG (1–7)/Mas receptor axis may act as the protective arm of the RAS, counteracting the detrimental effects of the activation of the angiotensinogen/renin/ACE/ANG II/AT1 receptor axis [41,185]. Whilst ANG II induces potent vasoconstriction and cardiac hypertrophy [186–189], renal vasoconstriction and antinatriuresis [3,190,191], promotes central thirst and sympathetic nerve activity [192,193], and increases aldosterone biosynthesis and release [194,195], ANG (1–7) appears to oppose and attenuate these harmful effects of ANG II [41,185,196]. Over the last two decades, the biochemistry, physiological roles and signaling mechanisms underlying the ANG (1–7) have been comprehensively reviewed elsewhere, and therefore not a focus of discussion in this article [41,42,183].
3.5.2. ANG (1–7) as a therapeutic agent
The potential implications in using exogenous ANG (1–7) as a therapeutic agent or increasing endogenous ANG (1–7) formation alone in treating hypertension, cardiovascular and kidney diseases remain a challenging issue. In theory, in vivo administration of ANG (1–7) would be beneficial to oppose or attenuate the harmful effects of ANG II, but in practice it may be difficult to administer a peptide in a clinical setting. The questions remain with respect to how much ANG (1–7) should be given and via what route the vasodepressor peptide ANG (1–7) should be administered to induce beneficial cardiovascular or renal protective effects. Campbell et al. have previously shown that under basal conditions, ANG (1–7) levels were about one-tenth of ANG II in both the rat plasma and the kidney, as measured by HLPC-based radioimmunoassays [78,79]. Chappell showed that ANG (1–7) and ANG II levels were largely similar in the plasma and kidney, but ANG II levels are higher than ANG (1–7) in the heart, adrenal glands, lung and liver [80]. However, plasma and tissue ANG (1–7) levels are significantly increased during ACE inhibition, which leads to the notion that the beneficial effects of ACE inhibitors may be mediated in part by ANG (1–7) [80,197].
Although global or cell-specific ACE2 overexpression may be beneficial in SHR [198,199] or diabetic nephropathy [200], not all animal studies in vivo support the therapeutic premise of the ACE2/ANG (1–7)/Mas receptor axis in protecting cardiovascular, pulmonary, and renal systems, or in treating hypertension. Indeed, inconsistent findings have been reported in mutant animals overexpressing ANG (1–7) [201–203] or ACE2 [157,200,204] with markedly increased circulating and tissue ANG (1–7) levels. Indeed, overexpression of ANG (1–7) had no effect on blood pressure in TGR(A1–7)3292 rats, despite of significant increased stroke volume and cardiac index and decrease in total peripheral resistance [201,202]. Rentzsch et al. overexpressed the human ACE2 gene selectively in rat vascular smooth muscle cells using of the SM22 promoter on a SHRSP genetic background [204]. Circulating ANG (1–7) was significantly elevated; whereas basal blood pressure was only slightly lower [204]. Liu et al. overexpressed ACE2 globally via the adenoviral gene delivery, and found no differences in blood pressure between control and ACE2-overexpressing rats [200]. Breitling et al. found insignificant therapeutic usefulness of ANG (1–7) in an animal model of experimental pulmonary hypertension [205], whereas Zhang et al. reported that activation of Mas during myocardial infarction contributed to, rather than attenuated, ischemia-reperfusion injury in rats [206]. In the latter study, inhibition of Mas G-protein signaling surprisingly improved coronary blood flow, reduced myocardial infarct size, and provided long-term cardioprotection [206].
3.5.3. (ANG (1–7) mimetics as therapeutic drugs
A novel nonpeptide compound, AVE0991, has been developed as a mimetic of ANG (1–7) for therapeutic application [207]. AVE0991 was found to be 10-fold more potent than ANG (1–7) in competing for [125I]-ANG (1–7) binding to, and 5-fold more potent than ANG (1–7) to induce NO release from bovine aortic endothelial cells [207], suggesting that AVE0991 may act as a nonpeptide agonist for the Mas receptor [208,209]. Subsequent in vivo studies have reported that AVE0991 significantly protected postischemic heart failure in rats [210], prevented diabetes-induced cardiovascular dysfunction [211], inhibited atherogenesis in apoE-knockout mice [212,213], ameliorated inflammation in experimental arthritis [214], and attenuated cardiac hypertrophy [215]. Despite these reported protective effects in different animal models or diseases, no clinical trial for AVE0991 is currently registered since it was developed more than 10 years ago [207]. Although these results from animal studies are very promising, they are basically very similar to those well-described responses to ACE inhibitors or ARBs. Whether these findings will be confirmed in humans in clinical trials remains unknown.
4. New insights into the roles and therapeutic implications of dipeptidyl peptidase III in cardiovascular, hypertensive and renal diseases
Recently, Dipeptidyl peptidase III (DPP III, EC 3.4.14.4) has been reported to have anti-hypertensive, anti-cardiac hypertrophic and anti-fibrotic effects in mice [46]. DPP III is zine-dependent aminopeptidase and acts to preferentially cleave two amino acids (dipeptide residues) from the N-terminus of the oligopeptides in the serum, placenta, liver and brain [46,216–218]. The oligopeptides subjected to metabolism by DPP III include ANG II and its metabolites ANG III and ANG IV, and enkephalins and endorphins [46]. Degradation of the vasopressor peptide ANG II is expected to lower circulating and tissue ANG II levels, leading to the same cardiovascular, blood pressure and renal protective effects induced by ACE inhibitors and ARBs. Although DPP III was discovered and characterized several decades ago [216–218], there is only one study investigating its roles and therapeutic implications in hypertension [46]. Pang et al. first studied the enzymatic properties of DPP III for degrading the vasopressor peptide ANG II, and found that DPP III potently metabolized ANG II with a Km of 3.7×10−6 mol/L and more effectively ANG IV with a Km of 1.7×10−6 mol/L in vitro [46]. In ANG II-induced hypertensive mice, intravenous bolus injection of DPP III at 8 μg/g body wt (or ~200 μg for a 25 g mouse) markedly decreased systolic blood pressure by almost 60 mmHg one hr after injection, and the response returned to pre-injection level one day later [46]. More interestingly, repeated DPP III injection every other day for 4 weeks significantly attenuated ANG II-induced cardiac fibrosis and hypertrophy, albuminuria and kidney injury were significantly attenuated [46]. These results strongly suggest that DPP III may be a therapeutic target for ANG II-dependent hypertension and target organ damage [46,219].
However, there are a number of caveats with the potential roles or therapeutic implications of DPP III that need to be further discussed. First, only high pressor doses of ANG II or high doses of DPP III were used in the above-mentioned study [46]. Second, DPP III not only metabolizes the vasopressor arm of the key RAS peptide ANG II, but likely also degrade the vasodepressor and protective arm of ANG fragments, ANG (1–7), ANG III, which offsets its potential therapeutic effects [220]. Third, as an enzyme DPP III has to be given intravenously and daily, it may not be practical in clinical settings, and perhaps a nonpeptide mimetic must be developed for long-term oral use. Fourth, the cardiovascular, antihypertensive and renal protective effects of DPP III were reportedly similar to those induced by the AT1 receptor blocker candesartan [46]. If DPP III mediates these effects by increasing ANG II degradation and lowering systemic and tissue ANG II levels, then ACE inhibitors and ARBs are readily available for use and should be more superior to DPP III as antihypertensive drugs. Nevertheless, more studies are required to determine whether DPP III may be suitable for treating other cardiovascular and renal diseases not associated with ANG II.
5. New insights into the roles and therapeutic implications of a novel ANG peptide alamandine in cardiovascular, hypertensive and renal diseases
Alamandine is another recently discovered ANG peptide reportedly having vasodepressor properties [47,48]. Alamandine is a heptapeptide derived from the catalytic hydrolysis of the octapeptide Ala1-ANG II (ANG A) by human ACE2 and structurally similar to ANG (1–7), except that aspartic acid in position 1 of ANG (1–7) is substituted with alanine in position 1 of alamandine [48,49,221]. Lautner et al. demonstrated that alamandine can be generated from ANG (1–7) in the rat heart and is circulating in human blood with increased levels in nephropathic patients [48]. Unlike ANG (1–7), which binds and activates the Mas receptor, alamandine reportedly binds and activates the Mas-related G-protein–coupled receptor, member D (MrgD), which may be blocked by D-Pro7-ANG (1–7), β-alanine, and PD123319 [48]. Since the Mas antagonist A-779 had no effect in blocking alamandine binding, this suggests that alamandine and ANG (1–7) act on two different receptors, yet they induce similar vasodepressor and cardiovascular protective effects [47,48]. In the in vivo setting, oral administration of alamandine/β-hydroxypropyl cyclodextrin decreased blood pressure by <20 mmHg in SHR, and showed antifibrotic effects in isoproterenol-treated rats, but not in human tumoral cell lines [48]. How this structural substitution of aspartic acid in position 1 of ANG (1–7) with alanine leads to the vasodepressor effect in alamandine remains poorly understood. Another newly identified ANG peptide, ANG A (Ala-Arg-Val-Tyr-Ile-His-Pro-Phe), with the substitution of alanine for aspartate acid in position 1 of ANG II, shows similar vasoconstrictive response to ANG II, rather than a vasodepressor effect [222]. This is also inconsistent with previous studies in 1970s, which consistently showed that substitution of the amino acid in position 1 had no effect on the vasopressor activity of ANG II [21]. An interesting study even suggests that like ANG II, alamandine acts on both AT1 and AT2 receptors to cause both vasopressor and vasodepressor effects in 2K1C hypertensive rats [223]. Taken together, the findings that the AT2 receptor antagonist PD123319 completely displaced alamandine receptor binding and blocked alamandine-induced aortic vasorelaxation, and that the Mas antagonist A-779 had no effect in blocking alamandine binding, raise more questions than answers on whether alamandine is a truly endogenous or physiological agonist for the alamandine/MrgD axis or the ANG II/ANG III/AT2 receptor/cGMP axis [48,49,224]. More studies are required to confirm the structure, biochemistry, the physiological role and therapeutic implications of alamandine in cardiovascular, hypertensive and kidney diseases.
6. New insights into the roles and therapeutic implications of angioprotectin in cardiovascular and renal diseases
In 2011, a novel endogenous ANG II-like octapeptide, angioprotectin (MW 1001.5 Da), was identified by Jankowski et al. in blood of healthy humans and patients with end-stage renal failure using chromatographic purification and structural analysis by matrix-assisted laser desorption/ionization time-of-flight/time-of-flight (MALDI-TOF/TOF) [50]. Compared with the sequence of ANG II, angioprotectin has the sequence of Pro-Glu-Val-Tyr-Ile-His-Pro-Phe, with its Pro1-Glu2 substituting Asp1 and Arg2 in ANG II, raising the possibility that angioprotectin may be derived from ANG II [50]. In fluorescent receptor binding assays in mouse endothelial cells, unlabeled ANG II did not displace Cy3-angioprotectin binding, but unlabeled angioprotectin blocked FAM-ANG (1–7) binding to the Mas receptor [50]. These results implicate that like ANG (1–7), angioprotectin may represent an additional agonist for the Mas receptor. Indeed, this study showed that angioprotectin induced a significant vasodilatory effect in wild-type mice at μmol/L concentrations, but it had no similar vasodilatory effect in Mas receptor-deficient mice [50]. This study also showed dose-dependent antihypertensive effects in SHR with a peak vasodepressor response to angioprotectin at 30–300 pmol/kg/min, i.v. [50]. In healthy humans, the plasma angioprotectin and ANG II levels were reportedly similar at 3–7 pmol/L, whereas they were about 10–12 pmol/L in patients with end-stage renal diseases [50]. These levels of angioprotectin and ANG II levels, as determined by MALDI-TOF/TOF, are largely similar to those measured using HPLC-based RIAs [78,80,225]. Finally, a cDNA encoding angioprotectin or RNA-splicing variants for angioprotectin has not been identified in the human genome, raising a doubt whether it is an endogenous vasodepressor peptide [50]. Since its discovery in 2011, there have been very few studies reporting its true levels in rodent and human plasma and tissues. Accordingly, the molecular, biochemical and physiological or pathological roles and therapeutic implications of angioprotectin remain to be further studied and independently confirmed by others [51].
7. Conclusions
In summary, the extensive research during last three decades has firmly established at least five axes of the RAS, with each having its primary substrate, key enzyme, the major effector peptide, specific receptor, and downstream signaling pathways. These RAS axes may be divided into two different classes of vasoactive systems, one “YIN” and one “YANG”. The “YANG” vasopressor system includes the classic angiotensinogen/renin/ACE/ANG II/AT1 receptor axis and the newly discovered prorenin/renin/prorenin receptor (PRR)/MAPK/V-ATPase axis, and possibly includes the newly described ANG fragment, ANG A, if the latter’s structure and role are confirmed by others. The ANG IV/AT4 receptor axis may also belong to the vasopressor system, since at the pharmacological concentrations, ANG IV activates not only the AT4 receptor, but also the AT1 receptor, in blood vessels and the kidney to increase blood pressure and induce renal vasoconstriction (Fig. 4 & Fig. 5) [21–23]. The “YIN” vasodepressor system includes the ANG II/APA/ANG III/AT2 receptor/NO/cGMP axis and the ANG I/ANG II/ACE2/ANG (1–7)/Mas receptor axis. The newly described DPP III, alamandine and angioprotectin, together may be “assigned” to the “YIN” vasodepressor system. It is now beyond any doubt that the “YANG” vasoactive system plays the most critical role, contributing to the overall physiological regulation of central, cardiovascular and kidney function, and blood pressure homeostasis. However, the upregulation or overactivation of the “YANG” vasoactive system contributes to the pathogenesis and underlying mechanisms of most well-recognized cardiovascular, hypertensive and kidney diseases. The development of orally active therapeutic drugs to target the “YANG” vasoactive system by inhibiting the rate-limiting enzyme, renin, and the key ANG II-generating enzyme, ACE, and blocking the receptors for ANG II and aldosterone, has been one of the most successful stories in pharmacological and pharmaceutical research during last three decades. By contrast, although tremendous efforts and progress have been made in identifying key members of the “YIN” vasoactive system, and uncovering its respective roles and signaling mechanisms of actions, the therapeutic implications of targeting the ANG II/APA/ANG III/AT2 receptor/NO/cGMP axis, the ANG I/ANG II/ACE2/ANG (1–7)/Mas receptor axis, DPP III, alamandine and angioprotectin in treating cardiovascular, hypertensive, and kidney diseases still remain to be determined. Nevertheless, recent research on the nonpeptide AT2 receptor agonists, ACE2 activators, ANG (1–7) mimetic, dipeptidyl peptidase III, alamandine and angioprotectin has provided some proof of concept support of using the “YIN” vasoactive system to counteract the detrimental effects of the vasopressor peptide ANG II and activation of AT1 receptors. However, to be as successful as the well-established renin and ACE inhibitors, ARBs and aldosterone receptor antagonists in treating cardiovascular, hypertensive and kidney diseases, the therapeutic usefulness and effectiveness of the nonpeptide AT2 receptor agonists, ACE2 activators, ANG (1–7) mimetic, DPP III, alamandine or angioprotectin will have to be confirmed in multi-center, randomized and double-blinded clinical trials in humans.
Figure 5.

Dose-dependent pressor and renal cortical and medullary vasoconstrictor responses to intravenous infusion of increasing concentrations of ANG II, ANG III, and ANG IV. ANG II, ANG III, and ANG IV increased mean arterial pressure (MAP) and decreased renal cortical blood flow (CBF) in a dose-dependent manner, exhibiting potency in the order: ANG II > ANG III > ANG IV. ANG II and ANG III also decreased renal medullary blood flow (MBF) at higher doses, whereas ANG IV had no effect. P < 0.05 vs. baseline ANG IV response (*), vs. baseline ANG III response (+), and vs. baseline ANG II response (#). Reproduced from [23] with permission.
Acknowledgments
This work was supported in part by grants from National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (2RO1DK067299-06A2), and NIDDK/National Institute of General Medical Sciences (1R01DK102429-01 and 2R01DK102429-03A1), and National Heart, Lung, And Blood Institute (1R56HL130988-01) to Dr. Jia L. Zhuo. Dr. Jianfeng Zhang was a Visiting Professor at the University of Mississippi Medical Center and supported by a grant from National Natural Science Foundation of China (#81360290). We sincerely apologize to many other outstanding investigators whose work were not cited in this article due to its primary focus on the vasoprotective arm of the renin-angiotensin-aldosterone system.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52(3):415–472. [PubMed] [Google Scholar]
- 2.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17(11):1402–1409. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 3.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003;24:261–271. doi: 10.1210/er.2003-0001. [DOI] [PubMed] [Google Scholar]
- 5.Zhuo JL, Ferrao FM, Zheng Y, Li XC. New frontiers in the intrarenal Renin-Angiotensin system: a critical review of classical and new paradigms. Front Endocrinol (Lausanne) 2013;4:166. doi: 10.3389/fendo.2013.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tigerstedt R, Bergman PG. Niere und Kreislauf. Scand Arch Physiol. 1898;8:223–271. [Google Scholar]
- 7.Goldblatt H, Lynch J, Hanzal RF, Summerville WW. Studies on experimental hypertension. 1. The production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med. 1934;59:347–379. doi: 10.1084/jem.59.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.SKEGGS LT, Jr, KAHN JR, LENTZ K, SHUMWAY NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med. 1957;106(3):439–453. doi: 10.1084/jem.106.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tewksbury DA, Frome WL, Dumas ML. Characterization of human angiotensinogen. J Biol Chem. 1978;253(11):3817–3820. [PubMed] [Google Scholar]
- 10.Ohkubo H, Kageyama R, Ujihara M, Hirose T, Inayama S, Nakanishi S. Cloning and sequence analysis of cDNA for rat angiotensinogen. Proc Natl Acad Sci U S A. 1983;80(8):2196–2200. doi: 10.1073/pnas.80.8.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skeggs LT, Dorer FE, KAHN JR, Lentz KE, Levine M. The biological production of angiotensin. In: Eichler O, Farah A, Herken H, Welch AD, editors. Handbook of Experimental Pharmacology XXXVII: Angiotensin. Berlin, Heidelberg, New York: Springer-Verlag; 1974. pp. 1–16. [Google Scholar]
- 12.SKEGGS LT, Jr, KAHN JR, SHUMWAY NP. The preparation and function of the hypertensin-converting enzyme. J Exp Med. 1956;103(3):295–299. doi: 10.1084/jem.103.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang HY, Erdos EG, Levin Y. Characterization of a dipeptide hydrolase (kininase II: angiotensin I converting enzyme) J Pharmacol Exp Ther. 1971;177(1):291–300. [PubMed] [Google Scholar]
- 14.Carretero OA, Oza NB, Piwonska A, Ocholik T, Scicli AG. Measurement of urinary kallikrein activity by kinin radioimmunoassay. Biochem Pharmacol. 1976;25(20):2265–2270. doi: 10.1016/0006-2952(76)90008-3. [DOI] [PubMed] [Google Scholar]
- 15.Linz W, Wiemer G, Gohlke P, Unger T, Scholkens BA. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol Rev. 1995;47(1):25–49. [PubMed] [Google Scholar]
- 16.Rhaleb NE, Yang XP, Carretero OA. The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr Physiol. 2011;1(2):971–993. doi: 10.1002/cphy.c100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ardaillou R. Active fragments of angiotensin II: enzymatic pathways of synthesis and biological effects. Curr Opin Nephrol Hypertens. 1997;6(1):28–34. doi: 10.1097/00041552-199701000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Kemp BA, Bell JF, Rottkamp DM, Howell NL, Shao W, Navar LG, Padia SH, Carey RM. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension. 2012;60:387–395. doi: 10.1161/HYPERTENSIONAHA.112.191403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padia SH, Kemp BA, Howell NL, Siragy HM, Fournie-Zaluski MC, Roques BP, Carey RM. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension. 2007;49(3):625–630. doi: 10.1161/01.HYP.0000254833.85106.4d. [DOI] [PubMed] [Google Scholar]
- 20.Healy DP, Song L. Kidney aminopeptidase A and hypertension, part I: spontaneously hypertensive rats. Hypertension. 1999;33(2):740–745. doi: 10.1161/01.hyp.33.2.740. [DOI] [PubMed] [Google Scholar]
- 21.Khosla MC, Smeby RR, Bumpus FM. Structure-activity relationship in angiotensin analogs. In: Page IH, Bumpus FM, editors. Handbook of Experimental Pharmacology XXXVII: Angiotensin. Berlin.Heidelberg.New York: Springer-Verlag; 1974. pp. 126–161. [Google Scholar]
- 22.Fitzgerald SM, Evans RG, Bergstrom G, Anderson WP. Renal hemodynamic responses to intrarenal infusion of ligands for the putative angiotensin IV receptor in anesthetized rats. J Cardiovasc Pharmacol. 1999;34(2):206–211. doi: 10.1097/00005344-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Li XC, Campbell DJ, Ohishi M, Yuan S, Zhuo JL. AT1 receptor-activated signaling mediates angiotensin IV-induced renal cortical vasoconstriction in rats. Am J Physiol Renal Physiol. 2006;290(5):F1024–F1033. doi: 10.1152/ajprenal.00221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ Res. 2014;115(3):388–399. doi: 10.1161/CIRCRESAHA.115.304110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carey RM. Update on angiotensin AT2 receptors. Curr Opin Nephrol Hypertens. 2017;26(2):91–96. doi: 10.1097/MNH.0000000000000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, Lee J, Mendelsohn FA, Simpson RJ, Connolly LM, Chai SY. Evidence that the angiotensin IV (AT4) receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem. 2001;276(52):48623–48626. doi: 10.1074/jbc.C100512200. [DOI] [PubMed] [Google Scholar]
- 27.Zhuo JL, Moeller I, Jenkins T, Chai SY, Allen AM, Ohishi M, Mendelsohn FA. Mapping tissue angiotensin-converting enzyme and angiotensin AT1, AT2 and AT4 receptors. J Hypertens. 1998;16(12 Pt 2):2027–2037. doi: 10.1097/00004872-199816121-00026. [DOI] [PubMed] [Google Scholar]
- 28.Lew RA, Mustafa T, Ye S, McDowall SG, Chai SY, Albiston AL. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP) J Neurochem. 2003;86(2):344–350. doi: 10.1046/j.1471-4159.2003.01852.x. [DOI] [PubMed] [Google Scholar]
- 29.Timmermans PB, Smith RD. Angiotensin II receptors and functional correlates. Am J Hypertens. 1992;5(12 Pt 2):221S–235S. doi: 10.1093/ajh/5.12.221s. [DOI] [PubMed] [Google Scholar]
- 30.Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JA, Smith RD. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45(2):205–251. [PubMed] [Google Scholar]
- 31.Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, Thomas WG International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin receptors: interpreters of pathophysiological angiotensinergic stimuli [corrected] Pharmacol Rev. 2015;67(4):754–819. doi: 10.1124/pr.114.010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature. 1991;16(351):233–236. doi: 10.1038/351233a0. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki K, Yamano Y, Bardhan S, Iwai N, Murray JJ, Hasegawa M, Matsuda Y, Inagami T. Cloning and expression of a complementary DNA encoding a bovine adrenal angiotensin II type-1 receptor. Nature. 1991;351(6323):230–233. doi: 10.1038/351230a0. [DOI] [PubMed] [Google Scholar]
- 34.Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, Dzau VJ. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem. 1993;268(33):24539–24542. [PubMed] [Google Scholar]
- 35.Nakajima M, Mukoyama M, Pratt RE, Horiuchi M, Dzau VJ. Cloning of cDNA and analysis of the gene for mouse angiotensin II type 2 receptor. Biochem Biophys Res Commun. 1993;197(2):393–399. doi: 10.1006/bbrc.1993.2492. [DOI] [PubMed] [Google Scholar]
- 36.Kambayashi Y, Bardhan S, Takahashi K, Tsuzuki S, Inui H, Hamakubo T, Inagami T. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J Biol Chem. 1993;268(33):24543–24546. [PubMed] [Google Scholar]
- 37.Sandberg K, Ji H, Clark AJ, Shapira H, Catt KJ. Cloning and expression of a novel angiotensin II receptor subtype. J Biol Chem. 1992;267(14):9455–9458. [PubMed] [Google Scholar]
- 38.Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FA, Jenkins TA, Albiston AL. The angiotensin IV/AT4 receptor. Cell Mol Life Sci. 2004;61(21):2728–2737. doi: 10.1007/s00018-004-4246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de BI, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100(14):8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bader M, Alenina N, Andrade-Navarro MA, Santos RA. MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol Rev. 2014;66(4):1080–1105. doi: 10.1124/pr.113.008136. [DOI] [PubMed] [Google Scholar]
- 41.Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1–7)-MAS receptor axis: more than regulation of blood pressure? Hypertension. 2007;50(4):596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- 42.Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1–7): an evolving story in cardiovascular regulation. Hypertension. 2006;47(3):515–521. doi: 10.1161/01.HYP.0000196268.08909.fb. [DOI] [PubMed] [Google Scholar]
- 43.Solinski HJ, Gudermann T, Breit A. Pharmacology and signaling of MAS-related G protein-coupled receptors. Pharmacol Rev. 2014;66(3):570–597. doi: 10.1124/pr.113.008425. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen G. The (pro)renin receptor: pathophysiological roles in cardiovascular and renal pathology. Curr Opin Nephrol Hypertens. 2007;16(2):129–133. doi: 10.1097/MNH.0b013e328040bfab. [DOI] [PubMed] [Google Scholar]
- 45.Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci U S A. 1999;96(11):6506–6510. doi: 10.1073/pnas.96.11.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pang X, Shimizu A, Kurita S, Zankov DP, Takeuchi K, Yasuda-Yamahara M, Kume S, Ishida T, Ogita H. Novel Therapeutic Role for Dipeptidyl Peptidase III in the Treatment of Hypertension. Hypertension. 2016;68(3):630–641. doi: 10.1161/HYPERTENSIONAHA.116.07357. [DOI] [PubMed] [Google Scholar]
- 47.Villela DC, Passos-Silva DG, Santos RA. Alamandine: a new member of the angiotensin family. Curr Opin Nephrol Hypertens. 2014;23(2):130–134. doi: 10.1097/01.mnh.0000441052.44406.92. [DOI] [PubMed] [Google Scholar]
- 48.Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V, Sousa F, Alzamora A, Soares E, Barbosa C, Kjeldsen F, Oliveira A, Braga J, Savergnini S, Maia G, Peluso AB, Passos-Silva D, Ferreira A, Alves F, Martins A, Raizada M, Paula R, Motta-Santos D, Klempin F, Pimenta A, Alenina N, Sinisterra R, Bader M, Campagnole-Santos MJ, Santos RA. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013;112(8):1104–1111. doi: 10.1161/CIRCRESAHA.113.301077. [DOI] [PubMed] [Google Scholar]
- 49.Qaradakhi T, Apostolopoulos V, Zulli A. Angiotensin (1–7) and Alamandine: Similarities and differences. Pharmacol Res. 2016;111:820–6. doi: 10.1016/j.phrs.2016.07.025. [DOI] [PubMed] [Google Scholar]
- 50.Jankowski V, Tolle M, Santos RA, Gunthner T, Krause E, Beyermann M, Welker P, Bader M, Pinheiro SV, Sampaio WO, Lautner R, Kretschmer A, van der Giet M, Zidek W, Jankowski J. Angioprotectin: an angiotensin II-like peptide causing vasodilatory effects. FASEB J. 2011;25(9):2987–2995. doi: 10.1096/fj.11-185470. [DOI] [PubMed] [Google Scholar]
- 51.Balakumar P, Jagadeesh G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell Signal. 2014;26(10):2147–2160. doi: 10.1016/j.cellsig.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 52.Guan S, Fox J, Mitchell KD, Navar LG. Angiotensin and angiotensin converting enzyme tissue levels in two-kidney, one-clip hypertensive rats. Hypertension. 1992;20:763–767. doi: 10.1161/01.hyp.20.6.763. [DOI] [PubMed] [Google Scholar]
- 53.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57(3):355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Navar LG, Zou L, Von Thun A, Tarng WC, Imig JD, Mitchell KD. Unraveling the mystery of Goldblatt hypertension. News Physiol Sci. 1998;13:170–176. doi: 10.1152/physiologyonline.1998.13.4.170. [DOI] [PubMed] [Google Scholar]
- 55.von Thun AM, Vari RC, El Dahr SS, Navar LG. Augmentation of intrarenal angiotensin II levels by chronic angiotensin II infusion. Am J Physiol. 1994;266(1 Pt 2):F120–F128. doi: 10.1152/ajprenal.1994.266.1.F120. [DOI] [PubMed] [Google Scholar]
- 56.Zou LX, Hymel A, Imig JD, Navar LG. Renal accumulation of circulating angiotensin II in angiotensin II- infused rats. Hypertension. 1996;27(3 Pt 2):658–662. doi: 10.1161/01.hyp.27.3.658. [DOI] [PubMed] [Google Scholar]
- 57.Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT1 receptor. Hypertension. 2002;39(1):116–121. doi: 10.1161/hy0102.100780. [DOI] [PubMed] [Google Scholar]
- 58.Esther CR, Marino EM, Howard TE, Machaud A, Corvol P, Capecchi MR, Bernstein KE. The critical role of tissue angiotensin-converting enzyme as revealed by gene targeting in mice. J Clin Invest. 1997;99:2375–2385. doi: 10.1172/JCI119419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Villalobos RA, Billet S, Kim C, Satou R, Fuchs S, Bernstein KE, Navar LG. Intrarenal angiotensin-converting enzyme induces hypertension in response to angiotensin I infusion. J Am Soc Nephrol. 2011;22(3):449–459. doi: 10.1681/ASN.2010060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bernstein KE. Two ACEs and a heart. Nature. 2002;417(6891):799–802. doi: 10.1038/417799a. [DOI] [PubMed] [Google Scholar]
- 61.Song L, Healy DP. Kidney aminopeptidase A and hypertension, part II: effects of angiotensin II. Hypertension. 1999;33(2):746–752. doi: 10.1161/01.hyp.33.2.746. [DOI] [PubMed] [Google Scholar]
- 62.Ahmad S, Ward PE. Role of aminopeptidase activity in the regulation of the pressor activity of circulating angiotensins. J Pharmacol Exp Ther. 1990;252(2):643–650. [PubMed] [Google Scholar]
- 63.Mentzel S, Dijkman HB, Van Son JP, Koene RA, Assmann KJ. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. J Histochem Cytochem. 1996;44(5):445–461. doi: 10.1177/44.5.8627002. [DOI] [PubMed] [Google Scholar]
- 64.Healy DP, Wilk S. Localization of immunoreactive glutamyl aminopeptidase in rat brain. II. Distribution and correlation with angiotensin II. Brain Res. 1993;606(2):295–303. doi: 10.1016/0006-8993(93)90997-2. [DOI] [PubMed] [Google Scholar]
- 65.Zini S, Masdehors P, Lenkei Z, Fournie-Zaluski MC, Roques BP, Corvol P, Llorens-Cortes C. Aminopeptidase A: distribution in rat brain nuclei and increased activity in spontaneously hypertensive rats. Neuroscience. 1997;78(4):1187–1193. doi: 10.1016/s0306-4522(96)00660-4. [DOI] [PubMed] [Google Scholar]
- 66.Lin Q, Taniuchi I, Kitamura D, Wang J, Kearney JF, Watanabe T, Cooper MD. T and B cell development in BP-1/6C3/aminopeptidase A-deficient mice. J Immunol. 1998;160(10):4681–4687. [PubMed] [Google Scholar]
- 67.Mitsui T, Nomura S, Okada M, Ohno Y, Kobayashi H, Nakashima Y, Murata Y, Takeuchi M, Kuno N, Nagasaka T, Wang J, Cooper MD, Mizutani S. Hypertension and angiotensin II hypersensitivity in aminopeptidase A-deficient mice. Mol Med. 2003;9(1–2):57–62. [PMC free article] [PubMed] [Google Scholar]
- 68.Faber F, Gembardt F, Sun X, Mizutani S, Siems WE, Walther T. Lack of angiotensin II conversion to angiotensin III increases water but not alcohol consumption in aminopeptidase A-deficient mice. Regul Pept. 2006;136(1–3):130–137. doi: 10.1016/j.regpep.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Kubota R, Numaguchi Y, Ishii M, Niwa M, Okumura K, Naruse K, Murohara T. Ischemia-induced angiogenesis is impaired in aminopeptidase A deficient mice via down-regulation of HIF-1alpha. Biochem Biophys Res Commun. 2010;402(2):396–401. doi: 10.1016/j.bbrc.2010.10.043. [DOI] [PubMed] [Google Scholar]
- 70.Wolf G, Assmann KJ, Stahl RA. Overexpression of aminopeptidase A abolishes the growth promoting effects of angiotensin II in cultured mouse mesangial cells. Kidney Int. 1997;52(5):1250–1260. doi: 10.1038/ki.1997.450. [DOI] [PubMed] [Google Scholar]
- 71.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, Jones DW, Materson BJ, Oparil S, Wright JT, Jr, Roccella EJ. Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206–1252. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 72.Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, Sharma D, Thiyagarajan B. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial--the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355(9215):1582–1587. doi: 10.1016/s0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- 73.Zhuo JL, Froomes P, Casley D, Liu JJ, Murone C, Chai SY, Buxton B, Mendelsohn FA. Perindopril chronically inhibits angiotensin-converting enzyme in both the endothelium and adventitia of the internal mammary artery in patients with ischemic heart disease. Circulation. 1997;96(1):174–182. [PubMed] [Google Scholar]
- 74.O'Brien E, Barton J, Nussberger J, Mulcahy D, Jensen C, Dicker P, Stanton A. Aliskiren reduces blood pressure and suppresses plasma renin activity in combination with a thiazide diuretic, an angiotensin-converting enzyme inhibitor, or an angiotensin receptor blocker. Hypertension. 2007;49(2):276–284. doi: 10.1161/01.HYP.0000253780.36691.4f. [DOI] [PubMed] [Google Scholar]
- 75.Ledingham JG, Leary WP. Handbook of Experimental Pharmacology XXXVII. Berlin-Heidelberg-New York: Springer-Verlag; 1974. Catabolism of angiotensin II; pp. 111–125. [Google Scholar]
- 76.Harris PJ, Zhuo JL, Skinner SL. Effects of angiotensins II and III on glomerulotubular balance in rats. Clin Exp Pharmacol Physiol. 1987;14(6):489–502. doi: 10.1111/j.1440-1681.1987.tb01505.x. [DOI] [PubMed] [Google Scholar]
- 77.Hus-Citharel A, Gasc JM, Zini S, Marchetti J, Roques B, Corvol P, Llorens-Cortes C. Aminopeptidase A activity and angiotensin III effects on [Ca2+]i along the rat nephron. Kidney Int. 1999;56(3):850–859. doi: 10.1046/j.1523-1755.1999.00634.x. [DOI] [PubMed] [Google Scholar]
- 78.Campbell DJ, Kladis A. Simultaneous radioimmunoassay of six angiotensin peptides in arterial and venous plasma of man. J Hypertens. 1990;8:165–172. doi: 10.1097/00004872-199002000-00011. [DOI] [PubMed] [Google Scholar]
- 79.Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijn AJ. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension. 1991;18(6):763–773. doi: 10.1161/01.hyp.18.6.763. [DOI] [PubMed] [Google Scholar]
- 80.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310(2):H137–H152. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wysocki J, Ye M, Batlle D. Plasma and kidney angiotensin peptides: importance of the aminopeptidase A/angiotensin III axis. Am J Hypertens. 2015;28(12):1418–1426. doi: 10.1093/ajh/hpv054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olkowicz M, Radulska A, Suraj J, Kij A, Walczak M, Chlopicki S, Smolenski RT. Development of a sensitive, accurate and robust liquid chromatography/mass spectrometric method for profiling of angiotensin peptides in plasma and its application for atherosclerotic mice. J Chromatogr A. 2015;1393:37–46. doi: 10.1016/j.chroma.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 83.Seikaly MG, Arant BS, Jr, Seney FD., Jr Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest. 1990;86(4):1352–1357. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhuo JL, Song K, Harris PJ, Mendelsohn FA. In vitro autoradiography reveals predominantly AT1 angiotensin II receptors in rat kidney. Ren Physiol Biochem. 1992;15(5):231–239. doi: 10.1159/000173458. [DOI] [PubMed] [Google Scholar]
- 85.Chappell MC. Nonclassical renin-angiotensin system and renal function. Compr Physiol. 2012;2(4):2733–2752. doi: 10.1002/cphy.c120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yugandhar VG, Clark MA. Angiotensin III: A physiological relevant peptide of the renin angiotensin system. Peptides. 2013;46:26–32. doi: 10.1016/j.peptides.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 87.Carey RM, Vaughan ED, Jr, Peach MJ, Ayers CR. Activity of (des-Aspartyl1)-angiotensin II and angiotensin II in man. Differences in blood pressure and adrenocortical response during normal and low sodium intake. J Clin Invest. 1978;61(1):20–31. doi: 10.1172/JCI108919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gardiner SM, Kemp PA, March JE, Bennett T. Regional haemodynamic effects of angiotensin II (3–8) in conscious rats. Br J Pharmacol. 1993;110(1):159–162. doi: 10.1111/j.1476-5381.1993.tb13786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Padia SH, Howell NL, Siragy HM, Carey RM. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin II type 1 receptor-blocked rat. Hypertension. 2006;47(3):537–544. doi: 10.1161/01.HYP.0000196950.48596.21. [DOI] [PubMed] [Google Scholar]
- 90.Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension. 2009;53(2):338–343. doi: 10.1161/HYPERTENSIONAHA.108.124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ichiki T, Labosky PA, Shiota C, Okuyama S, Imagawa Y, Fogo A, Niimura F, Ichikawa I, Hogan BL, Inagami T. Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature. 1995;377(6551):748–750. doi: 10.1038/377748a0. [DOI] [PubMed] [Google Scholar]
- 92.Li XC, Widdop RE. AT2 receptor-mediated vasodilatation is unmasked by AT1 receptor blockade in conscious SHR. BrJ Pharmacol. 2004;142:821–830. doi: 10.1038/sj.bjp.0705838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Steckelings UM, Paulis L, Namsolleck P, Unger T. AT2 receptor agonists: hypertension and beyond. Curr Opin Nephrol Hypertens. 2012;21(2):142–146. doi: 10.1097/MNH.0b013e328350261b. [DOI] [PubMed] [Google Scholar]
- 94.Carey RM. AT2 receptors: potential therapeutic targets for yypertension. Am J Hypertens. 2017;30(4):339–347. doi: 10.1093/ajh/hpw121. [DOI] [PubMed] [Google Scholar]
- 95.Siragy HM, Carey RM. The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3', 5'-monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J Clin Invest. 1996;97(8):1978–1982. doi: 10.1172/JCI118630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carey RM, Jin XH, Siragy HM. Role of the angiotensin AT2 receptor in blood pressure regulation and therapeutic implications. Am J Hypertens. 2001;14(6 Pt 2):98S–102S. doi: 10.1016/s0895-7061(01)02076-3. [DOI] [PubMed] [Google Scholar]
- 97.Siragy HM, Carey RM. Angiotensin type 2 receptors: potential importance in the regulation of blood pressure. Curr Opin Nephrol Hypertens. 2001;10(1):99–103. doi: 10.1097/00041552-200101000-00015. [DOI] [PubMed] [Google Scholar]
- 98.Zhuo JL, Alcorn D, Harris PJ, Mendelsohn FA. Localization and properties of angiotensin II receptors in rat kidney. Kidney Int Suppl. 1993;42:S40–S46. [PubMed] [Google Scholar]
- 99.Zhuo JL, Allen AM, Alcorn D, MacGregor D, Aldred GP, Mendelsohn FA. The distribution of angiotensin II receptors. In: Laragh JH, Brenner BM, editors. Hypertension: Pathology, Diagnosis & Management. 2. New York: Raven Press; 1995. pp. 1739–1762. [Google Scholar]
- 100.Zhuo JL, Dean R, MacGregor D, Alcorn D, Mendelsohn FA. Presence of angiotensin II AT2 receptor binding sites in the adventitia of human kidney vasculature. Clin Exp Pharmacol Physiol Suppl. 1996;3:S147–S154. [PubMed] [Google Scholar]
- 101.Zhuo JL, Yamada H, Allen AM, Sun Y, Mendelsohn FA. Localization and properties of angiotensin converting enzyme and angiotensin II receptors in the heart. In: Lindpaintner K, Ganten D, editors. The Cardiac Renin-Angiotensin System. New York: Futura Scientific Publishing; 1994. pp. 63–88. [Google Scholar]
- 102.Allen AM, Zhuo J, Mendelsohn FA. Localization of angiotensin AT1 and AT2 receptors. J Am Soc Nephrol. 1999;10(Suppl 11):S23–S29. [PubMed] [Google Scholar]
- 103.Tsuzuki S, Ichiki T, Nakakubo H, Kitami Y, Guo DF, Shirai H, Inagami T. Molecular cloning and expression of the gene encoding human angiotensin II type 2 receptor. Biochem Biophys Res Commun. 1994;200(3):1449–1454. doi: 10.1006/bbrc.1994.1613. [DOI] [PubMed] [Google Scholar]
- 104.Barber MN, Sampey DB, Widdop RE. AT2 receptor stimulation enhances antihypertensive effect of AT1 receptor antagonist in hypertensive rats. Hypertension. 1999;34(5):1112–1116. doi: 10.1161/01.hyp.34.5.1112. [DOI] [PubMed] [Google Scholar]
- 105.Levy BI. Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation. 2004;109(1):8–13. doi: 10.1161/01.CIR.0000096609.73772.C5. [DOI] [PubMed] [Google Scholar]
- 106.Zhuo JL, Song K, Abdelrahman A, Mendelsohn FA. Blockade by intravenous losartan of AT1 angiotensin II receptors in rat brain, kidney and adrenals demonstrated by in vitro autoradiography. Clin Exp Pharmacol Physiol. 1994;21(7):557–567. doi: 10.1111/j.1440-1681.1994.tb02555.x. [DOI] [PubMed] [Google Scholar]
- 107.Zhuo JL, Alcorn D, McCausland J, Casley D, Mendelsohn FA. In vivo occupancy of angiotensin II subtype 1 receptors in rat renal medullary interstitial cells. Hypertension. 1994;23(6 Pt 2):838–843. doi: 10.1161/01.hyp.23.6.838. [DOI] [PubMed] [Google Scholar]
- 108.Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK. Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature. 1995;377(6551):744–747. doi: 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- 109.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104(7):925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Niimura F, Labosky PA, Kakuchi J, Okubo S, Yoshida H, Oikawa T, Ichiki T, Naftilan AJ, Fogo A, Inagami T. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest. 1995;96(6):2947–2954. doi: 10.1172/JCI118366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, Jennette JC, Coffman TM, Maeda N, Smithies O. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci U S A. 1995;92(7):2735–2739. doi: 10.1073/pnas.92.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Krege JH, John SW, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O'Brien DA, Smithies O. Male-female differences in fertility and blood pressure in ACE- deficient mice. Nature. 1995;375(6527):146–148. doi: 10.1038/375146a0. [DOI] [PubMed] [Google Scholar]
- 113.Tsuchida S, Matsusaka T, Chen X, Okubo S, Niimura F, Nishimura H, Fogo A, Utsunomiya H, Inagami T, Ichikawa I. Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J Clin Invest. 1998;101(4):755–760. doi: 10.1172/JCI1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci U S A. 1998;95(26):15496–15501. doi: 10.1073/pnas.95.26.15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC, Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT, Jr, Narva AS, Ortiz E. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8) JAMA. 2014;311(5):507–520. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 116.Casas JP, Chua W, Loukogeorgakis S, Vallance P, Smeeth L, Hingorani AD, MacAllister RJ. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet. 2005;366(9502):2026–2033. doi: 10.1016/S0140-6736(05)67814-2. [DOI] [PubMed] [Google Scholar]
- 117.Wing LM, Reid CM, Ryan P, Beilin LJ, Brown MA, Jennings GL, Johnston CI, McNeil JJ, Macdonald GJ, Marley JE, Morgan TO, West MJ. A comparison of outcomes with angiotensin-converting--enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med. 2003;348(7):583–592. doi: 10.1056/NEJMoa021716. [DOI] [PubMed] [Google Scholar]
- 118.Yamada T, Horiuchi M, Dzau VJ. Angiotensin II type 2 receptor mediates programmed cell death. Proc Natl Acad Sci U S A. 1996;93(1):156–160. doi: 10.1073/pnas.93.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Senbonmatsu T, Ichihara S, Price E, Jr, Gaffney FA, Inagami T. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest. 2000;106(3):R25–R29. doi: 10.1172/JCI10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ichihara S, Senbonmatsu T, Price E, Jr, Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation. 2001;104(3):346–351. doi: 10.1161/01.cir.104.3.346. [DOI] [PubMed] [Google Scholar]
- 121.Cao Z, Kelly DJ, Cox A, Casley D, Forbes JM, Martinello P, Dean R, Gilbert RE, Cooper ME. Angiotensin type 2 receptor is expressed in the adult rat kidney and promotes cellular proliferation and apoptosis. Kidney Int. 2000;58(6):2437–2451. doi: 10.1046/j.1523-1755.2000.00427.x. [DOI] [PubMed] [Google Scholar]
- 122.Balla T, Baukal AJ, Eng S, Catt KJ. Angiotensin II receptor subtypes and biological responses in the adrenal cortex and medulla. Mol Pharmacol. 1991;40(3):401–406. [PubMed] [Google Scholar]
- 123.Goldfarb DA, Diz DI, Tubbs RR, Ferrario CM, Novick AC. Angiotensin II receptor subtypes in the human renal cortex and renal cell carcinoma. J Urol. 1994;151(1):208–213. doi: 10.1016/s0022-5347(17)34918-2. [DOI] [PubMed] [Google Scholar]
- 124.Ali Q, Wu Y, Hussain T. Chronic AT2 receptor activation increases renal ACE2 activity, attenuates AT1 receptor function and blood pressure in obese Zucker rats. Kidney Int. 2013;84(5):931–939. doi: 10.1038/ki.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hakam AC, Hussain T. Angiotensin II type 2 receptor agonist directly inhibits proximal tubule sodium pump activity in obese but not in lean Zucker rats. Hypertension. 2006;47(6):1117–1124. doi: 10.1161/01.HYP.0000220112.91724.fc. [DOI] [PubMed] [Google Scholar]
- 126.Sabuhi R, Ali Q, Asghar M, Al-Zamily NR, Hussain T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: opposing effects in lean and obese Zucker rats. Am J Physiol Renal Physiol. 2011;300(3):F700–F706. doi: 10.1152/ajprenal.00616.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Benndorf R, Boger RH, Ergun S, Steenpass A, Wieland T. Angiotensin II type 2 receptor inhibits vascular endothelial growth factor-induced migration and in vitro tube formation of human endothelial cells. Circ Res. 2003;93(5):438–447. doi: 10.1161/01.RES.0000088358.99466.04. [DOI] [PubMed] [Google Scholar]
- 128.Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, Johansson B, Holm M, Botoros M, Karlen A, Pettersson A, Nyberg F, Fandriks L, Gallo-Payet N, Hallberg A, Alterman M. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47(24):5995–6008. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- 129.Unger T, Dahlof B. Compound 21, the first orally active, selective agonist of the angiotensin type 2 receptor (AT2): implications for AT2 receptor research and therapeutic potential. J Renin Angiotensin Aldosterone Syst. 2010;11(1):75–77. doi: 10.1177/1470320309347792. [DOI] [PubMed] [Google Scholar]
- 130.Rompe F, Artuc M, Hallberg A, Alterman M, Stroder K, Thone-Reineke C, Reichenbach A, Schacherl J, Dahlof B, Bader M, Alenina N, Schwaninger M, Zuberbier T, Funke-Kaiser H, Schmidt C, Schunck WH, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension. 2010;55(4):924–931. doi: 10.1161/HYPERTENSIONAHA.109.147843. [DOI] [PubMed] [Google Scholar]
- 131.Matavelli LC, Huang J, Siragy HM. Angiotensin AT2 receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension. 2011;57(2):308–313. doi: 10.1161/HYPERTENSIONAHA.110.164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Matavelli LC, Zatz R, Siragy HM. A nonpeptide angiotensin II type 2 receptor agonist prevents renal inflammation in early diabetes. J Cardiovasc Pharmacol. 2015;65(4):371–376. doi: 10.1097/FJC.0000000000000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sampson AK, Irvine JC, Shihata WA, Dragoljevic D, Lumsden N, Huet O, Barnes T, Unger T, Steckelings UM, Jennings GL, Widdop RE, Chin-Dusting JP. Compound 21, a selective agonist of angiotensin AT2 receptors, prevents endothelial inflammation and leukocyte adhesion in vitro and in vivo. Br J Pharmacol. 2016;173(4):729–740. doi: 10.1111/bph.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Iwanami J, Mogi M, Tsukuda K, Wang XL, Nakaoka H, Kan-no H, Chisaka T, Bai HY, Shan BS, Kukida M, Horiuchi M. Direct angiotensin II type 2 receptor stimulation by compound 21 prevents vascular dementia. J Am Soc Hypertens. 2015;9(4):250–256. doi: 10.1016/j.jash.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 135.Patel SN, Ali Q, Hussain T. Angiotensin II type 2-receptor agonist C21 reduces proteinuria and oxidative stress in kidney of high-salt-fed obese Zucker rats. Hypertension. 2016;67(5):906–915. doi: 10.1161/HYPERTENSIONAHA.115.06881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Foulquier S, Steckelings UM, Unger T. Impact of the AT2 receptor agonist C21 on blood pressure and beyond. Curr Hypertens Rep. 2012;14(5):403–409. doi: 10.1007/s11906-012-0291-6. [DOI] [PubMed] [Google Scholar]
- 137.Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension. 2012;59(2):291–299. doi: 10.1161/HYPERTENSIONAHA.111.180158. [DOI] [PubMed] [Google Scholar]
- 138.Brouwers S, Smolders I, Massie A, Dupont AG. Angiotensin II type 2 receptor-mediated and nitric oxide-dependent renal vasodilator response to compound 21 unmasked by angiotensin-converting enzyme inhibition in spontaneously hypertensive rats in vivo. Hypertension. 2013;62(5):920–926. doi: 10.1161/HYPERTENSIONAHA.112.00762. [DOI] [PubMed] [Google Scholar]
- 139.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 140.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 141.Marian AJ. The discovery of the ACE2 gene. Circ Res. 2013;112(10):1307–1309. doi: 10.1161/CIRCRESAHA.113.301271. [DOI] [PubMed] [Google Scholar]
- 142.Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res. 2006;98(4):463–471. doi: 10.1161/01.RES.0000205761.22353.5f. [DOI] [PubMed] [Google Scholar]
- 143.Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17(11):3067–3075. doi: 10.1681/ASN.2006050423. [DOI] [PubMed] [Google Scholar]
- 144.Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, Cooper ME. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41(3):392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- 145.Lely AT, Hamming I, van GH, Navis GJ. Renal ACE2 expression in human kidney disease. J Pathol. 2004;204(5):587–593. doi: 10.1002/path.1670. [DOI] [PubMed] [Google Scholar]
- 146.Velez JC, Bland AM, Arthur JM, Raymond JR, Janech MG. Characterization of renin-angiotensin system enzyme activities in cultured mouse podocytes. Am J Physiol Renal Physiol. 2007;293(1):F398–F407. doi: 10.1152/ajprenal.00050.2007. [DOI] [PubMed] [Google Scholar]
- 147.Reich HN, Oudit GY, Penninger JM, Scholey JW, Herzenberg AM. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int. 2008;74(12):1610–1616. doi: 10.1038/ki.2008.497. [DOI] [PubMed] [Google Scholar]
- 148.Mizuiri S, Hemmi H, Arita M, Ohashi Y, Tanaka Y, Miyagi M, Sakai K, Ishikawa Y, Shibuya K, Hase H, Aikawa A. Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am J Kidney Dis. 2008;51(4):613–623. doi: 10.1053/j.ajkd.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 149.Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R373–R381. doi: 10.1152/ajpregu.00292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da CJ, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 151.Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest. 2006;116(8):2218–2225. doi: 10.1172/JCI16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M, Tatara Y, Shiota A, Sugano S, Takeda S, Rakugi H, Ogihara T. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension. 2006;47(4):718–726. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- 153.Ferrario CM, Varagic J. The ANG-(1–7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Renal Physiol. 2010;298(6):F1297–F1305. doi: 10.1152/ajprenal.00110.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Feng Y, Yue X, Xia H, Bindom SM, Hickman PJ, Filipeanu CM, Wu G, Lazartigues E. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res. 2008;102(6):729–736. doi: 10.1161/CIRCRESAHA.107.169110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RA, Speth RC, Sigmund CD, Lazartigues E. Brain-selective overexpression of human Angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2010;106(2):373–382. doi: 10.1161/CIRCRESAHA.109.208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55(1):90–98. doi: 10.1161/HYPERTENSIONAHA.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nadarajah R, Milagres R, Dilauro M, Gutsol A, Xiao F, Zimpelmann J, Kennedy C, Wysocki J, Batlle D, Burns KD. Podocyte-specific overexpression of human angiotensin-converting enzyme 2 attenuates diabetic nephropathy in mice. Kidney Int. 2012;82(3):292–303. doi: 10.1038/ki.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Haschke M, Schuster M, Poglitsch M, Loibner H, Salzberg M, Bruggisser M, Penninger J, Krahenbuhl S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52(9):783–792. doi: 10.1007/s40262-013-0072-7. [DOI] [PubMed] [Google Scholar]
- 159.Hernandez Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RA, Castellano RK, Lampkins AJ, Gubala V, Ostrov DA, Raizada MK. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51(5):1312–1317. doi: 10.1161/HYPERTENSIONAHA.107.108944. [DOI] [PubMed] [Google Scholar]
- 160.Qi Y, Zhang J, Cole-Jeffrey CT, Shenoy V, Espejo A, Hanna M, Song C, Pepine CJ, Katovich MJ, Raizada MK. Diminazene aceturate enhances angiotensin-converting enzyme 2 activity and attenuates ischemia-induced cardiac pathophysiology. Hypertension. 2013;62(4):746–752. doi: 10.1161/HYPERTENSIONAHA.113.01337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Velkoska E, Patel SK, Burrell LM. Angiotensin converting enzyme 2 and diminazene: role in cardiovascular and blood pressure regulation. Curr Opin Nephrol Hypertens. 2016;25(5):384–395. doi: 10.1097/MNH.0000000000000254. [DOI] [PubMed] [Google Scholar]
- 162.Mecca AP, Regenhardt RW, O'Connor TE, Joseph JP, Raizada MK, Katovich MJ, Sumners C. Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011;96(10):1084–1096. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rigatto K, Casali KR, Shenoy V, Katovich MJ, Raizada MK. Diminazene aceturate improves autonomic modulation in pulmonary hypertension. Eur J Pharmacol. 2013;713(1–3):89–93. doi: 10.1016/j.ejphar.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Peregrine AS, Mamman M. Pharmacology of diminazene: a review. Acta Trop. 1993;54(3–4):185–203. doi: 10.1016/0001-706x(93)90092-p. [DOI] [PubMed] [Google Scholar]
- 165.Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, Ferreira AJ, Fraga-Silva RA, Kearns P, Douglas JY, Agarwal D, Mubarak KK, Bradford C, Kennedy WR, Jun JY, Rathinasabapathy A, Bruce E, Gupta D, Cardounel AJ, Mocco J, Patel JM, Francis J, Grant MB, Katovich MJ, Raizada MK. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187(6):648–657. doi: 10.1164/rccm.201205-0880OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Young D, Waitches G, Birchmeier C, Fasano O, Wigler M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell. 1986;45(5):711–719. doi: 10.1016/0092-8674(86)90785-3. [DOI] [PubMed] [Google Scholar]
- 167.Jackson TR, Blair LA, Marshall J, Goedert M, Hanley MR. The mas oncogene encodes an angiotensin receptor. Nature. 1988;335(6189):437–440. doi: 10.1038/335437a0. [DOI] [PubMed] [Google Scholar]
- 168.Ambroz C, Clark AJ, Catt KJ. The mas oncogene enhances angiotensin-induced [Ca2+]i responses in cells with pre-existing angiotensin II receptors. Biochim Biophys Acta. 1991;1133(1):107–111. doi: 10.1016/0167-4889(91)90248-v. [DOI] [PubMed] [Google Scholar]
- 169.Monnot C, Weber V, Stinnakre J, Bihoreau C, Teutsch B, Corvol P, Clauser E. Cloning and functional characterization of a novel mas-related gene, modulating intracellular angiotensin II actions. Mol Endocrinol. 1991;5(10):1477–1487. doi: 10.1210/mend-5-10-1477. [DOI] [PubMed] [Google Scholar]
- 170.Walther T, Balschun D, Voigt JP, Fink H, Zuschratter W, Birchmeier C, Ganten D, Bader M. Sustained long term potentiation and anxiety in mice lacking the Mas protooncogene. J Biol Chem. 1998;273(19):11867–11873. doi: 10.1074/jbc.273.19.11867. [DOI] [PubMed] [Google Scholar]
- 171.De Souza AM, Lopes AG, Pizzino CP, Fossari RN, Miguel NC, Cardozo FP, Abi-Abib R, Fernandes MS, Santos DP, Caruso-Neves C. Angiotensin II and angiotensin-(1–7) inhibit the inner cortex Na+-ATPase activity through AT2 receptor. Regul Pept. 2004;120(1–3):167–175. doi: 10.1016/j.regpep.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 172.Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension. 2005;45(5):960–966. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- 173.Tetzner A, Gebolys K, Meinert C, Klein S, Uhlich A, Trebicka J, Villacanas O, Walther T. G-protein-coupled receptor MrgD is a receptor for angiotensin-(1–7) involving adenylyl cyclase, cAMP, and phosphokinase A. Hypertension. 2016;68(1):185–194. doi: 10.1161/HYPERTENSIONAHA.116.07572. [DOI] [PubMed] [Google Scholar]
- 174.Young D, O'Neill K, Jessell T, Wigler M. Characterization of the rat mas oncogene and its high-level expression in the hippocampus and cerebral cortex of rat brain. Proc Natl Acad Sci U S A. 1988;85(14):5339–5342. doi: 10.1073/pnas.85.14.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, Chappell MC. Nuclear angiotensin-(1–7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol. 2010;299(5):F983–F990. doi: 10.1152/ajprenal.00371.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Su Y, Bi J, Pulgar VM, Chappell MC, Rose JC. Antenatal betamethasone attenuates the angiotensin-(1–7)/Mas receptor/nitric oxide axis in isolated proximal tubule cells. Am J Physiol Renal Physiol. 2017 Feb 22; doi: 10.1152/ajprenal.00593.2016. ajprenal.00593.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Walther T, Wessel N, Kang N, Sander A, Tschope C, Malberg H, Bader M, Voss A. Altered heart rate and blood pressure variability in mice lacking the Mas protooncogene. Braz J Med Biol Res. 2000;33(1):1–9. doi: 10.1590/s0100-879x2000000100001. [DOI] [PubMed] [Google Scholar]
- 178.Heringer-Walther S, Gembardt F, Perschel FH, Katz N, Schultheiss HP, Walther T. The genetic deletion of Mas abolishes salt induced hypertension in mice. Eur J Pharmacol. 2012;689(1–3):147–153. doi: 10.1016/j.ejphar.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 179.Lemos VS, Silva DM, Walther T, Alenina N, Bader M, Santos RA. The endothelium-dependent vasodilator effect of the nonpeptide Ang(1–7) mimic AVE 0991 is abolished in the aorta of mas-knockout mice. J Cardiovasc Pharmacol. 2005;46(3):274–279. doi: 10.1097/01.fjc.0000175237.41573.63. [DOI] [PubMed] [Google Scholar]
- 180.Xu P, Costa-Goncalves AC, Todiras M, Rabelo LA, Sampaio WO, Moura MM, Santos SS, Luft FC, Bader M, Gross V, Alenina N, Santos RA. Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension. 2008;51(2):574–580. doi: 10.1161/HYPERTENSIONAHA.107.102764. [DOI] [PubMed] [Google Scholar]
- 181.Dias-Peixoto MF, Santos RA, Gomes ER, Alves MN, Almeida PW, Greco L, Rosa M, Fauler B, Bader M, Alenina N, Guatimosim S. Molecular mechanisms involved in the angiotensin-(1–7)/Mas signaling pathway in cardiomyocytes. Hypertension. 2008;52(3):542–548. doi: 10.1161/HYPERTENSIONAHA.108.114280. [DOI] [PubMed] [Google Scholar]
- 182.Mario EG, Santos SH, Ferreira AV, Bader M, Santos RA, Botion LM. Angiotensin-(1–7) Mas-receptor deficiency decreases peroxisome proliferator-activated receptor gamma expression in adipocytes. Peptides. 2012;33(1):174–177. doi: 10.1016/j.peptides.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 183.Ferrario CM, Brosnihan KB, Diz DI, Jaiswal N, Khosla MC, Milsted A, Tallant EA. Angiotensin-(1–7): a new hormone of the angiotensin system. Hypertension. 1991;18(5 Suppl):III126–III133. doi: 10.1161/01.hyp.18.5_suppl.iii126. [DOI] [PubMed] [Google Scholar]
- 184.Khosla MC, Smeby RR, Bumps FM. Structural-activity relationship in angiotensin II analogs. Angiotensin. Handbook of Experimental Pharmacology. 1974;XXXVII:126–161. [Google Scholar]
- 185.Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216(2):R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 186.De Mello WC, Danser AH. Angiotensin II and the heart : on the intracrine renin-angiotensin system. Hypertension. 2000;35(6):1183–1188. doi: 10.1161/01.hyp.35.6.1183. [DOI] [PubMed] [Google Scholar]
- 187.Xu J, Carretero OA, Lin CX, Cavasin MA, Shesely EG, Yang JJ, Reudelhuber TL, Yang XP. Role of cardiac overexpression of ANG II in the regulation of cardiac function and remodeling postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293(3):H1900–H1907. doi: 10.1152/ajpheart.00379.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103(47):17985–90. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Reudelhuber TL, Bernstein KE, Delafontaine P. Is angiotensin II a direct mediator of left ventricular hypertrophy? Time for another look Hypertension. 2007;49(6):1196–1201. doi: 10.1161/HYPERTENSIONAHA.106.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Navar LG, Inscho EW, Majid SA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76(2):425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- 191.Zhuo JL, Li XC. Proximal nephron. Comp Physiol. 2013;3(3):1079–1123. doi: 10.1002/cphy.c110061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jancovski N, Bassi JK, Carter DA, Choong YT, Connelly A, Nguyen TP, Chen D, Lukoshkova EV, Menuet C, Head GA, Allen AM. Stimulation of angiotensin type 1A receptors on catecholaminergic cells contributes to angiotensin-dependent hypertension. Hypertension. 2013;62(5):866–871. doi: 10.1161/HYPERTENSIONAHA.113.01474. [DOI] [PubMed] [Google Scholar]
- 193.Young CN, Davisson RL. Angiotensin-II, the brain, and hypertension: an update. Hypertension. 2015;66(5):920–926. doi: 10.1161/HYPERTENSIONAHA.115.03624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Romero DG, Gomez-Sanchez EP, Gomez-Sanchez CE. Angiotensin II-regulated transcription regulatory genes in adrenal steroidogenesis. Physiol Genomics. 2010;42A(4):259–266. doi: 10.1152/physiolgenomics.00098.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bassett MH, White PC, Rainey WE. The regulation of aldosterone synthase expression. Mol Cell Endocrinol. 2004;217(1–2):67–74. doi: 10.1016/j.mce.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 196.Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin-(1–7) Hypertension. 1997;30(3 Pt 2):535–541. doi: 10.1161/01.hyp.30.3.535. [DOI] [PubMed] [Google Scholar]
- 197.Campbell DJ, Kladis A, Duncan AM. Nephrectomy, converting enzyme inhibition, and angiotensin peptides. Hypertension. 1993;22(4):513–522. doi: 10.1161/01.hyp.22.4.513. [DOI] [PubMed] [Google Scholar]
- 198.Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension. 2007;49(4):926–931. doi: 10.1161/01.HYP.0000259942.38108.20. [DOI] [PubMed] [Google Scholar]
- 199.Diez-Freire C, Vazquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, Torres R, Raizada MK. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27(1):12–19. doi: 10.1152/physiolgenomics.00312.2005. [DOI] [PubMed] [Google Scholar]
- 200.Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB, Wang R, Wang XP, Dong B, Gao F, Zhang MX, Zhang Y. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med. 2011;17(1–2):59–69. doi: 10.2119/molmed.2010.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Santos RA, Ferreira AJ, Nadu AP, Braga AN, de Almeida AP, Campagnole-Santos MJ, Baltatu O, Iliescu R, Reudelhuber TL, Bader M. Expression of an angiotensin-(1–7)-producing fusion protein produces cardioprotective effects in rats. Physiol Genomics. 2004;17(3):292–299. doi: 10.1152/physiolgenomics.00227.2003. [DOI] [PubMed] [Google Scholar]
- 202.Botelho-Santos GA, Sampaio WO, Reudelhuber TL, Bader M, Campagnole-Santos MJ, Souza dos Santos RA. Expression of an angiotensin-(1–7)-producing fusion protein in rats induced marked changes in regional vascular resistance. Am J Physiol Heart Circ Physiol. 2007;292(5):H2485–H2490. doi: 10.1152/ajpheart.01245.2006. [DOI] [PubMed] [Google Scholar]
- 203.Ferreira AJ, Pinheiro SV, Castro CH, Silva GA, Silva AC, Almeida AP, Bader M, Rentzsch B, Reudelhuber TL, Santos RA. Renal function in transgenic rats expressing an angiotensin-(1–7)-producing fusion protein. Regul Pept. 2006;137(3):128–133. doi: 10.1016/j.regpep.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 204.Rentzsch B, Todiras M, Iliescu R, Popova E, Campos LA, Oliveira ML, Baltatu OC, Santos RA, Bader M. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52(5):967–973. doi: 10.1161/HYPERTENSIONAHA.108.114322. [DOI] [PubMed] [Google Scholar]
- 205.Breitling S, Krauszman A, Parihar R, Walther T, Friedberg MK, Kuebler WM. Dose-dependent, therapeutic potential of angiotensin-(1–7) for the treatment of pulmonary arterial hypertension. Pulm Circ. 2015;5(4):649–657. doi: 10.1086/683696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhang T, Li Z, Dang H, Chen R, Liaw C, Tran TA, Boatman PD, Connolly DT, Adams JW. Inhibition of Mas G-protein signaling improves coronary blood flow, reduces myocardial infarct size, and provides long-term cardioprotection. Am J Physiol Heart Circ Physiol. 2012;302(1):H299–H311. doi: 10.1152/ajpheart.00723.2011. [DOI] [PubMed] [Google Scholar]
- 207.Wiemer G, Dobrucki LW, Louka FR, Malinski T, Heitsch H. AVE 0991, a nonpeptide mimic of the effects of angiotensin-(1–7) on the endothelium. Hypertension. 2002;40(6):847–852. doi: 10.1161/01.hyp.0000037979.53963.8f. [DOI] [PubMed] [Google Scholar]
- 208.Pinheiro SV, Simoes e Silva AC, Sampaio WO, de Paula RD, Mendes EP, Bontempo ED, Pesquero JB, Walther T, Alenina N, Bader M, Bleich M, Santos RA. Nonpeptide AVE 0991 is an angiotensin-(1–7) receptor Mas agonist in the mouse kidney. Hypertension. 2004;44(4):490–496. doi: 10.1161/01.HYP.0000141438.64887.42. [DOI] [PubMed] [Google Scholar]
- 209.Faria-Silva R, Duarte FV, Santos RA. Short-term angiotensin(1–7) receptor MAS stimulation improves endothelial function in normotensive rats. Hypertension. 2005;46(4):948–952. doi: 10.1161/01.HYP.0000174594.17052.33. [DOI] [PubMed] [Google Scholar]
- 210.Ferreira AJ, Jacoby BA, Araujo CA, Macedo FA, Silva GA, Almeida AP, Caliari MV, Santos RA. The nonpeptide angiotensin-(1–7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;292(2):H1113–H1119. doi: 10.1152/ajpheart.00828.2006. [DOI] [PubMed] [Google Scholar]
- 211.Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1–7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292(1):H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- 212.Toton-Zuranska J, Gajda M, Pyka-Fosciak G, Kus K, Pawlowska M, Niepsuj A, Wolkow P, Olszanecki R, Jawien J, Korbut R. AVE 0991-angiotensin-(1–7) receptor agonist, inhibits atherogenesis in apoE-knockout mice. J Physiol Pharmacol. 2010;61(2):181–183. [PubMed] [Google Scholar]
- 213.Skiba DS, Nosalski R, Mikolajczyk TP, Siedlinski M, Rios FJ, Montezano AC, Jawien J, Olszanecki R, Korbut R, Czesnikiewicz-Guzik M, Touyz RM, Guzik TJ. Antiatherosclerotic effect of Ang-(1–7) non-peptide mimetic (AVE 0991) is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br J Pharmacol. 2016 doi: 10.1111/bph.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.da Silveira KD, Coelho FM, Vieira AT, Sachs D, Barroso LC, Costa VV, Bretas TL, Bader M, de Sousa LP, da Silva TA, dos Santos RA, Simoes e Silva AC, Teixeira MM. Anti-inflammatory effects of the activation of the angiotensin-(1–7) receptor, MAS, in experimental models of arthritis. J Immunol. 2010;185(9):5569–5576. doi: 10.4049/jimmunol.1000314. [DOI] [PubMed] [Google Scholar]
- 215.Ma Y, Huang H, Jiang J, Wu L, Lin C, Tang A, Dai G, He J, Chen Y. AVE 0991 attenuates cardiac hypertrophy through reducing oxidative stress. Biochem Biophys Res Commun. 2016;474(4):621–625. doi: 10.1016/j.bbrc.2015.09.050. [DOI] [PubMed] [Google Scholar]
- 216.Fukasawa K, Fukasawa KM, Kanai M, Fujii S, Hirose J, Harada M. Dipeptidyl peptidase III is a zinc metallo-exopeptidase. Molecular cloning and expression Biochem J. 1998;329(Pt 2):275–282. doi: 10.1042/bj3290275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Shimamori Y, Watanabe Y, Fujimoto Y. Purification and characterization of dipeptidyl aminopeptidase III from human placenta. Chem Pharm Bull (Tokyo) 1986;34(8):3333–3340. doi: 10.1248/cpb.34.3333. [DOI] [PubMed] [Google Scholar]
- 218.Ohkubo I, Li YH, Maeda T, Yamamoto Y, Yamane T, Du PG, Nishi K. Dipeptidyl peptidase III from rat liver cytosol: purification, molecular cloning and immunohistochemical localization. Biol Chem. 1999;380(12):1421–1430. doi: 10.1515/BC.1999.182. [DOI] [PubMed] [Google Scholar]
- 219.Ocaranza MP, Jalil JE. On Endogenous Angiotensin II Antagonism in Hypertension: The role of dipeptidyl peptidase III. Hypertension. 2016;68(3):552–554. doi: 10.1161/HYPERTENSIONAHA.116.07471. [DOI] [PubMed] [Google Scholar]
- 220.Cruz-Diaz N, Wilson BA, Pirro NT, Brosnihan KB, Marshall AC, Chappell MC. Identification of dipeptidyl peptidase 3 as the angiotensin-(1–7) degrading peptidase in human HK-2 renal epithelial cells. Peptides. 2016;83:29–37. doi: 10.1016/j.peptides.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dell'Italia LJ, Ferrario CM. The never-ending story of angiotensin peptides: beyond angiotensin I and II. Circ Res. 2013;112(8):1086–1087. doi: 10.1161/CIRCRESAHA.113.301246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jankowski V, Vanholder R, van der Giet M, Tolle M, Karadogan S, Gobom J, Furkert J, Oksche A, Krause E, Tran TN, Tepel M, Schuchardt M, Schluter H, Wiedon A, Beyermann M, Bader M, Todiras M, Zidek W, Jankowski J. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol. 2007;27(2):297–302. doi: 10.1161/01.ATV.0000253889.09765.5f. [DOI] [PubMed] [Google Scholar]
- 223.Soltani HA, Javanmardi K, Kouhpayeh A, Baharamali E, Farjam M. Differences in cardiovascular responses to alamandine in two-Kidney, one clip hypertensive and normotensive rats. Circ J. 2017:10–0958. doi: 10.1253/circj.CJ-16-0958. [DOI] [PubMed] [Google Scholar]
- 224.Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, Unger T, Bader M, Santos RA, Sumners C, Steckelings UM. Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin Sci (Lond) 2015;128(4):227–234. doi: 10.1042/CS20130515. [DOI] [PubMed] [Google Scholar]
- 225.Campbell DJ. Clinical relevance of local renin angiotensin systems. Front Endocrinol (Lausanne) 2014;5:113. doi: 10.3389/fendo.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]