

ABSTRACT
Females have a more severe clinical course than males in terms of several inflammatory lung conditions. Notably, females with cystic fibrosis (CF) suffer worse outcomes, particularly in the setting of Pseudomonas aeruginosa infection. Sex hormones have been implicated in experimental and clinical studies; however, immune mechanisms responsible for this sex-based disparity are unknown and the specific sex hormone target for therapeutic manipulation has not been identified. The objective of this study was to assess mechanisms behind the impact of female sex hormones on host immune responses to P. aeruginosa. We used wild-type and CF mice, which we hormone manipulated, inoculated with P. aeruginosa, and then examined for outcomes and inflammatory responses. Neutrophils isolated from mice and human subjects were tested for responses to P. aeruginosa. We found that female mice inoculated with P. aeruginosa died earlier and showed slower bacterial clearance than males (P < 0.0001). Ovariectomized females supplemented with 17β-estradiol succumbed to P. aeruginosa challenge earlier than progesterone- or vehicle-supplemented mice (P = 0.0003). 17β-Estradiol-treated ovariectomized female mice demonstrated increased lung levels of inflammatory cytokines, and when rendered neutropenic the mortality difference was abrogated. Neutrophils treated with 17β-estradiol demonstrated an enhanced oxidative burst but decreased P. aeruginosa killing and earlier cell necrosis. The estrogen receptor (ER) antagonist ICI 182,780 improved survival in female mice infected with P. aeruginosa and restored neutrophil function. We concluded that ER antagonism rescues estrogen-mediated neutrophil dysfunction and improves survival in response to P. aeruginosa. ER-mediated processes may explain the sex-based mortality gap in CF and other inflammatory lung illnesses, and the ER blockade represents a rational therapeutic strategy.
KEYWORDS: estrogen, neutrophil, Pseudomonas aeruginosa
INTRODUCTION
In numerous inflammatory lung diseases, there is an increased severity in females relative to males (1, 2). Asthmatics show a female predominance of the disease specifically after puberty, accompanied by more physician visits, hospitalizations, and a 30% increase in asthma-related deaths in females compared to males (3–5). In chronic obstructive pulmonary disease (COPD), the prevalence and overall mortality is greater in females than in males (6, 7). In bronchiectasis, which is marked clinically by chronic lung infections, females are hospitalized significantly more often than males and have an increased risk for infection with pathogens associated with impaired immunity, such as nontuberculous mycobacteria (2). In cystic fibrosis (CF), a genetic disease which also causes bronchiectasis, the prevalence is equal in males and females, but a clear sex-specific mortality difference exists (8, 9). In fact, the life expectancy of females with CF is nearly 3 years less than that of males (8, 9). CF mortality is strongly linked to chronic infection with pathogens such as Pseudomonas aeruginosa, which frequently targets the CF lung. Of note, females with CF also acquire P. aeruginosa at an earlier age and have worse outcomes than males once infected (9, 10). These differences are present despite investigators accounting for potential confounding factors such as morphometrics, nutrition, and comorbidities (9). Previous studies have suggested that sex hormones play a role in all of these diseases; here, we propose that this hormone effect occurs via modulation of innate immune responses that can be evaluated in P. aeruginosa lung infection models.
Levels of the major female sex hormones, estrogen and progesterone (P4), vary during ovulatory cycles, pregnancy, and menopause (11, 12). The predominant form of estrogen, 17β-estradiol (E2), varies in concentration from 20 to 400 pg/ml in serum, while tissue expression varies. There are two major estrogen receptor (ER) subtypes, ER-α and ER-β, each with a high affinity for E2 (13). Their classic actions are genomic; however, nongenomic membrane-associated ER effects are now also widely recognized (13–15). The other major female sex hormone, P4, ranges from 300 to 10,000 pg/ml in serum and acts via two main nuclear receptors, progesterone receptors A and B (PR-A and PR-B) (16). ER and PR are expressed in lung tissue (17, 18).
E2 behaves differently in various tissues and diseases and has been shown to induce a proinflammatory state in some settings and an anti-inflammatory state in others (19–22). Thus, the impact of sex hormones is highly contextual and the direct effects of ER and PR activation on innate immune responses in a P. aeruginosa model of lung infection have not been thoroughly evaluated. Neutrophils, key drivers of innate immunity, are one of the first responders in the infectious and inflammatory process and are known to be critical for the body's defense against P. aeruginosa (23). ER are expressed in neutrophils, though PR expression has not been well described (24–27). Very limited data exist regarding the effect of sex hormones on neutrophil functions. ER and PR activation has been shown to modulate degranulation and apoptosis of neutrophils (27–29), but no work in this field has been done related to the neutrophil killing effect and other responses to P. aeruginosa.
In this study, we sought to use a hormone-manipulated mouse model of P. aeruginosa infection to test the impact of female sex hormones on survival and bacterial clearance. We specifically focused on the impact of sex hormones on inflammation and neutrophil responses to P. aeruginosa, using neutrophils from mice and humans with and without CF. The ultimate goal of these studies was to improve the understanding of the mechanism of sex differences in inflammatory lung diseases associated with bacterial infection, such as CF, and determine the specific therapeutic target for sex hormone manipulation.
RESULTS
Female mice are more susceptible to P. aeruginosa infection than male mice.
To investigate the impact of sex on lung infection and inflammation, we inoculated male and female wild-type (WT) mice with P. aeruginosa, using an inoculum of 8.5 × 105 CFU. Weight-matched female mice died significantly earlier than male mice, with survival differences noted as early as 30 h postinfection (P < 0.0001) (Fig. 1A). Females had an almost 4-fold greater mortality rate than males and a markedly steeper decline in weight than males (P < 0.0001) (Fig. 1C). Additional experiments with age-matched mice of both sexes showed results that were consistent with the data for weight-matched animals (see Fig. S1 in the supplemental material). To determine if the rate of bacterial clearance in these mice was different between sexes, we infected male and female mice with a 0% lethal dose (LD0; 3.0 × 105 CFU) and measured the remaining CFU in lungs over time. By 6 h after infection, male mice displayed a significantly decreased bacterial burden and an overall higher rate of bacterial clearance from their lungs compared to females (P = 0.0100) (Fig. 1D). These experiments were repeated using a mucoid clinical isolate of P. aeruginosa, M57-15, and the results were consistent with those seen with strain PAO1, with female mice dying significantly earlier than male mice (Fig. 1B). Bacteremia was not detected in studies using nonlethal doses of PAO1 or lethal doses of M57-15 (Table S1).
FIG 1.

Female mice are more susceptible to P. aeruginosa infection than male mice. (A) Male and female C57BL/6 wild-type mice were infected with P. aeruginosa (8.5 × 105 CFU) and followed for up to 96 h (n = 18 per group). Displayed are Kaplan-Meier survival plots (P < 0.0001). (B) Male and female C57BL/6 wild-type mice were infected with a mucoid P. aeruginosa strain, M57-15 at 1.2 × 106 CFU and followed for up to 96 h (n = 6 per group). Displayed are Kaplan-Meier survival plots. *, P < 0.05. (C) Body weights for the animals whose survival data are shown in panel A. *, P < 0.0001. (D) Bacterial CFU remaining in the lungs of male versus female mice after infection with P. aeruginosa at 3 ×105 CFU (n = 6 to 8 mice per time point per group). *, P < 0.05. Data in panels B and C are mean results ± SEM.
Estrogen-supplemented male and female mice demonstrated decreased P. aeruginosa clearance, independent of progesterone.
To determine whether the principal sex hormones, E2 and P4, could account for the male versus female differences seen previously, female mice were ovariectomized and supplemented with vehicle, E2, or P4 at physiologic doses or sham ovariectomized, 10 to 14 days prior to challenge with P. aeruginosa. The E2 effect was confirmed by measuring uterine wet weights, which were significantly greater in the E2-supplemented mice than in ovariectomized mice (Fig. S2A). Serum measurements of E2 and P4 in hormone-treated mice confirmed significant differences from untreated ovariectomized mice (Fig. S2B and C). Following P. aeruginosa inoculation, E2-supplemented ovariectomized female mice died significantly earlier than vehicle-treated (P = 0.0023) or P4-supplemented mice (P = 0.0007) (Fig. 2A), with a median survival of 29.75 h for the E2 group, 25.00 h for the sham ovariectomy group, 53.87 h for the vehicle group, and >72 h for the P4 group. There was no detectable difference between vehicle-treated and P4-treated groups (P = 0.4405), or between sham-ovariectomized and E2-treated groups (P = 0.2393). We further evaluated clearance of P. aeruginosa 18 h after infection, as we had previously observed that this was the time point with the greatest difference noted between males and females in Fig. 1C. Ovariectomized mice supplemented with E2 demonstrated impaired bacterial clearance compared to vehicle-supplemented ovariectomized mice, a difference that was greater than 10-fold (P = 0.0027), but no difference relative to P4-treated ovariectomized mice (P = 0.0693) (Fig. 2B). Sham-ovariectomized mice showed clearance profiles similar to those of E2-treated ovariectomized mice (P = 0.9641). Finally, male mice were supplemented with E2 or vehicle, and they similarly demonstrated decreased survival in the E2-treated group (P = 0.0018) (Fig. 2C). These experiments were repeated using M57-15 (30), and results were consistent with those seen using PAO1, where ovariectomized female mice supplemented with estrogen died significantly earlier than ovariectomized female mice supplemented with vehicle (Fig. S3). Together, these findings suggest that E2 provides a mortality risk associated with an impaired ability to clear bacteria.
FIG 2.
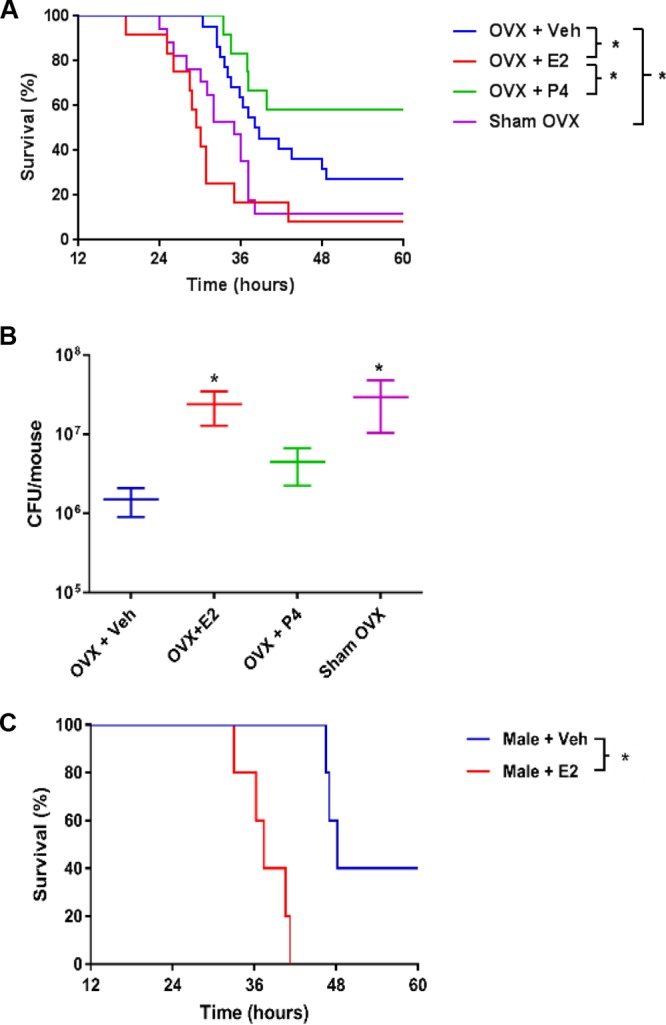
Estrogen-supplemented male and female mice demonstrated decreased P. aeruginosa clearance, independent of progesterone. Wild-type C57BL/6 female mice were ovariectomized (OVX) and supplemented with physiologic doses of hormones. Veh, vehicle; Sham OVX, sham ovariectomized. The E2 dose was 0.5 μg/day, and P4 was administered at 2 mg/day. (A) Mice were infected with P. aeruginosa at 8.5 × 105 CFU (n = 14 per group). Displayed are Kaplan-Meier survival plots. *, P < 0.05. (B) Bacterial CFU remaining in the lungs 18 h after infection with P. aeruginosa (3 × 105 CFU) in the groups described for panel A (n = 8 to 10 mice per group). Data are mean results ± SEM. *, P < 0.05 (relative to vehicle or relative to ovariectomized mice with vehicle; indicated by brackets to the right of the key on the right). (C) Kaplan-Meier survival plots for male mice supplemented with vehicle (Veh) or E2 (0.5 μg/day). *, P < 0.05.
Estrogen-dependent susceptibility to P. aeruginosa requires nuclear estrogen receptors.
To determine if the difference in ovariectomized mice supplemented with E2 versus vehicle was ER specific, mice were supplemented with an ER antagonist, ICI 182,780, which blocks both nuclear and nonnuclear ERs. P. aeruginosa-infected ovariectomized mice treated with both E2 and ICI 182,780 had a longer survival time than mice treated with E2 alone (P = 0.0399). As such, the poorer survival in E2-supplemented mice was attenuated in the presence of ICI 182,780 (Fig. 3A). To further evaluate the effect of nuclear versus nonnuclear ER, we used an estrogen dendrimer conjugate (EDC), which is an extranuclear ER agonist. Female mice were ovariectomized and supplemented with E2 or EDC and compared to control mice. EDC-treated ovariectomized mice had no detectable difference in survival relative to the dendrimer vehicle-treated group (P = 0.9627) (Fig. 3B), demonstrating that selective activation of nonnuclear ERs does not impact the host response to P. aeruginosa infection. We subsequently evaluated the role of the two well-established nuclear ERs, ER-α and ER-β, using propyl pyrazole triol (PPT; ER-α agonist) and diarylpropionitrile (DPN; ER-β agonist), and we observed that both ER agonist treatments decreased survival relative to the vehicle-treated group (P = 0.0340 for PPT, P = 0.0237 for DPN) (Fig. 3C), suggesting a role for both nuclear ERs in the impact of E2 on P. aeruginosa infection. Finally, female mice with intact endogenous sex hormones were treated with ICI 182,780 and notably displayed improved survival in response to P. aeruginosa infection (P = 0.0013) (Fig. 3D). Taken together, these findings support a specific role for nuclear E2 signaling in mediating sex-dependent differences in mortality following P. aeruginosa infection.
FIG 3.

Estrogen-dependent susceptibility to P. aeruginosa requires nuclear estrogen receptors. Survival of wild-type C57BL/6 female mice with the indicated hormone treatment were infected with P. aeruginosa at 8.5 × 105 CFU. Kaplan-Meier survival plots are shown. (A) Ovariectomized (OVX) female mice were treated with vehicle (Veh), E2 (0.5 μg/day), ER antagonist (ICI 182,780 [500 μg/day]), or a combination (n = 8 per group). (B) Ovariectomized female mice were treated with vehicle, E2, membrane estrogen receptor agonist (EDC, 0.5 μg/day), or dendrimer vehicle (Dend Veh) (n = 6 per group). (C) Ovariectomized female mice were treated with vehicle, E2, ER-α agonist (PPT, 250 μg/day), or ER-β agonist (DPN, 250 μg/day) (n = 12 per group). (D) Female mice with intact endogenous sex hormones were treated with vehicle or ICI 182,780 (500 μg/day). *, P < 0.05 (for data in all four panels).
Estrogen induces neutrophilic inflammation in the setting of P. aeruginosa infection.
We first evaluated the direct effect of E2 and P4 on P. aeruginosa, and we found no quantitative change in growth rate or qualitative changes to mucoid conversion (Fig. S4). Having identified no direct effect of E2 on P. aeruginosa infection, we sought to determine if E2 impacted the host inflammatory response. We examined gross sections and cytokine expression in E2-treated murine lungs harvested 9 h after P. aeruginosa infection, a time after which a difference in bacterial clearance was observed (Fig. 2). Examination of lung histopathology showed a notable increase in inflammation in the setting of E2 treatment (Fig. 4A). Using a lung inflammation scoring system previously described (31), we evaluated sections from vehicle- and E2-treated ovariectomized mice infected with P. aeruginosa. E2-treated mice had higher peribronchial and perivascular inflammatory scores (P < 0.0001 [total]) (Fig. 4B). Lung homogenates were also evaluated 9 h after P. aeruginosa infection for cytokine expression in ovariectomized mice treated with vehicle compared to E2. We observed significant increases in the inflammatory cytokines tumor necrosis factor alpha (TNF-α), KC, interleukin-10 (IL-10), and IL-6, as well as in chemokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein 1 (MCP-1), macrophage inhibitory protein 1α (MIP-1α), and MIP-1β (Fig. 4C). Cytokines that showed no difference in expression at this time point included gamma interferon (IFN-γ), IL-1β, RANTES, granulocyte colony-stimulating factor (G-CSF), eotaxin, IL-17, IL-13, IL-12(p70), IL-12(p40), IL-9, IL-5, IL-4, IL-3, IL-2, and IL-1α (data not shown). Furthermore, the neutrophil granular proteins myeloperoxidase (MPO) and neutrophil elastase (NE) were significantly more abundant in lung tissue from E2-treated mice (Fig. 4C). Lung homogenates were also evaluated 18 h after infection and showed similar results to those seen at 9 h (Fig. S5). The histologic changes and elevated inflammatory cytokine responses in the E2-treated mice were consistent with a neutrophil-predominant response.
FIG 4.

Estrogen induces neutrophilic inflammation in the setting of P. aeruginosa infection. Wild-type C57BL/6 mice were ovariectomized and treated with vehicle or E2 at 0.5 μg/day and then infected with P. aeruginosa at 3.0 ×105 CFU/ml for 9 h. (A) Lung inflammation in representative histological images stained with hematoxylin and eosin. Scale bars, 100 μm. (B) Peribronchial, perivascular, and total lung inflammation scores were measured from lung sections taken 9 h after infection (treatment groups as described for panel A) (n = 7 mice per group; examination of 12 fields per slide). (C) Cytokine expression in whole lung samples of female mice ovariectomized and hormone treated as for panel A. Cytokines were assyed 9 h after infection (n = 8 mice per group). Data are expressed as mean results ± SEM. *, P < 0.05.
The neutrophil response to P. aeruginosa infection is regulated by 17β-estradiol.
Given that neutrophils play a critical role in the host response to bacterial infection, we quantified the total number of neutrophils in the lungs of E2-treated versus vehicle-treated mice exposed to P. aeruginosa. We used ovariectomized mice treated with vehicle or E2 and infected them with P. aeruginosa at the LD0 of 3 × 105 CFU. Whole lung tissue and bronchoalveolar lavage (BAL) fluid samples demonstrated no differences in neutrophil numbers by flow cytometry between vehicle and E2 treatment at any time point (P > 0.05) (Fig. 5A and B). Although no quantifiable difference in neutrophil counts was seen, neutrophil granular proteins, including MPO and NE, were significantly higher in E2-treated mice (Fig. 4C), suggesting an important role for neutrophils in ER modulation of the host response to P. aeruginosa infection. To test the impact of neutrophils, we studied ovariectomized female mice supplemented with vehicle and E2 and rendered them neutropenic using a monoclonal LY6G clone 1A8 anti-granulocyte receptor antibody. After confirming neutropenia in peripheral blood (white blood cell counts of <250/μl), we inoculated mice with P. aeruginosa at a dose of 8 × 104 CFU, which preliminary experiments had shown was a lethal dose for neutropenic female mice. After neutropenia, the effect of E2 on survival following P. aeruginosa infection was abrogated (P = 0.655) (Fig. 5C), suggesting that neutrophils are a target of E2 activation in the hormonal modulation of the host response to P. aeruginosa infection. To more directly test the effect of E2 on neutrophil function, we isolated neutrophils from bone marrow cells of female mice and exposed the cells to vehicle and E2 before performing killing assays with P. aeruginosa. The ability of neutrophils to kill P. aeruginosa was hindered in the presence of escalating doses of E2, as demonstrated by increased CFU seen with E2 (P = 0.0310) (Fig. 5D).
FIG 5.

The neutrophil response to P. aeruginosa infection is regulated by 17β-estradiol. (A) Percentage and number of absolute neutrophils from whole lungs of C57BL/6 mice 9 h after P. aeruginosa infection with 3 ×105 CFU, quantified by flow cytometry (n = 6 mice per group per time point). (B) Percentage and absolute neutrophil counts from bronchoalveolar lavage fluid, quantified by flow cytometry (n = 6 per group per time point). (C) Kaplan-Meier survival plots of C57BL/6 ovariectomized and hormone-treated female mice rendered neutropenic by using the 1A8 monoclonal antibody (anti-Ly6G antibody). Ovariectomized mice were infected with 8 × 104 CFU of P. aeruginosa (n = 8 per group). (D) Neutrophils were isolated from mice and used ex vivo for P. aeruginosa killing experiments at an MOI of 1:4 under the indicated treatment conditions (n = 5 per treatment group). Data in panels A, B, D, and E are mean results ± SEM. *, P < 0.05 compared to vehicle.
Estrogen receptor antagonists improve survival in CFTR−/− female mice infected with P. aeruginosa.
To determine the impact of ER modulation in a clinically relevant model of P. aeruginosa lung infection, a CFTR-deficient mouse model (CFTRtm1UNC) was tested. Hormone-intact male and female mice were treated with P. aeruginosa at a dose of 3 × 105 CFU, which we determined is a lethal dose for this strain of mice. Median survival in female mice was 32.1 h, versus 48.0 h in male mice (P < 0.0001) (Fig. 6A). Ovariectomized female mice supplemented with vehicle or E2 showed worse survival in the E2-treated mice, consistent with the results in WT mice (P = 0.0008) (Fig. 6B). Treatment with ICI 182,780 improved survival in CFTR-null intact females infected with P. aeruginosa (P = 0.0013), similar to the effect observed in WT mice (Fig. 3D) and suggesting a potential role for ER antagonists in the treatment of P. aeruginosa infection in CF (Fig. 6C). Survival differences in male and female CFTR-null mice or between vehicle-treated and estrogen-supplemented, ovariectomized female mice were not notably greater than those seen in wild-type mice, suggesting that the E2 effect is not mediated by CFTR.
FIG 6.
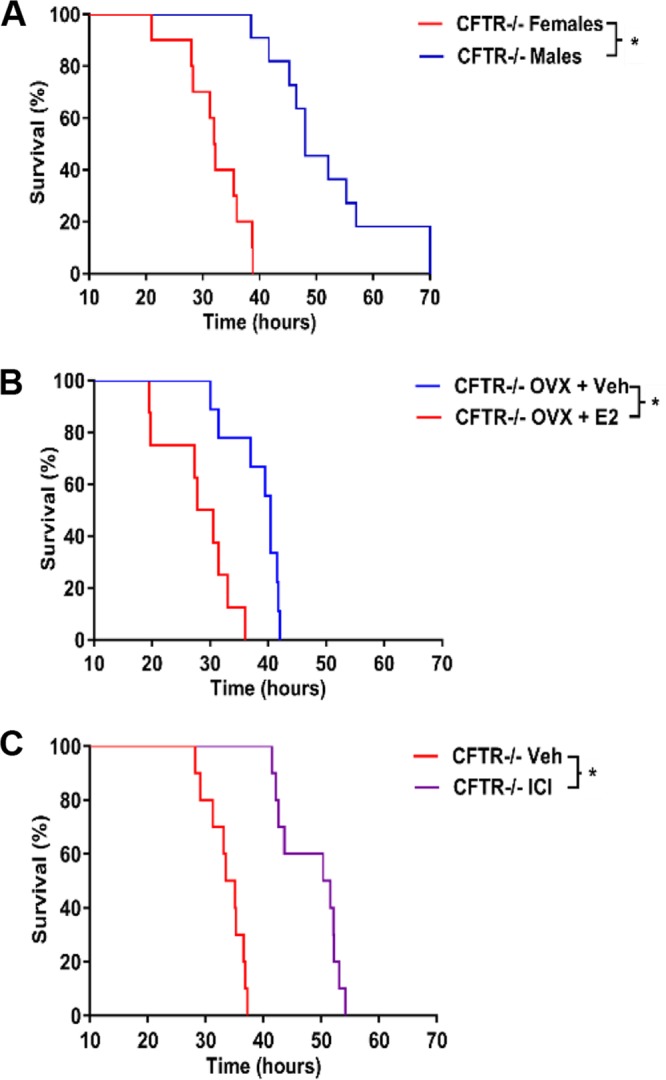
Estrogen receptor antagonists improve survival in CFTR−/− female mice infected with P. aeruginosa. Kaplan-Meier survival plots of CFTR−/− mice hormonally manipulated as indicated and then treated with 3 × 105 CFU of P. aeruginosa. (A) Hormonal intact male and female CFTR−/− mice (n = 10 per group). (B) Ovariectomized (OVX) CFTR−/− mice treated with Vehicle or E2 (n = 9 per group). (C) CFTR−/− intact females treated with vehicle or ICI 182,780 (ICI) (n = 10 per group). *, P < 0.05.
17β-Estradiol impacts responses to P. aeruginosa infection of neutrophils from subjects with CF.
Having demonstrated that E2 inhibits neutrophil function in mice (Fig. 5D), we sought to determine if this was present in humans and if the E2 effect on neutrophils was ER specific. We therefore used neutrophils from female subjects with CF to evaluate the effect of ER antagonists on the neutrophil killing capacity against P. aeruginosa. Stable CF subjects with a wide range of forced expiratory volume levels, genotypes, and bacterial profiles were recruited (Table S2). The effect of E2 on neutrophil killing was attenuated in a dose-dependent fashion with ER antagonism via ICI 182,780 (Fig. 7A). Consistent with our mouse data (Fig. 3D), we found that both nuclear ERs contributed to the ER effect, based on a significantly lower CFU at all concentrations of E2 (P < 0.0001). We also evaluated bacterial killing of neutrophils from female subjects without CF (healthy females), and the results were similar to those seen in females with CF (Fig. S6). We further evaluated neutrophils from males with CF and found P. aeruginosa killing responses similar to those seen in females. However, the neutrophils of males did not have a reduction in killing with E2 stimulation to the same extent as seen with female neutrophils (Fig. 7B). To further determine how E2 impacts the neutrophil response to infection, we evaluated the respiratory burst, which is a critical measure of the oxygen-dependent pathway of the neutrophil response to invading pathogens. We found E2 enhanced oxidative burst activity with both an earlier peak and a more robust response (P = 0.0235) (Fig. 7B). This was confirmed by demonstrating increased release of NE when the cells were stimulated with E2 (P = 0.0088) (Fig. 7C). Furthermore, E2 promoted earlier neutrophil cell death upon stimulation with P. aeruginosa (P = 0.0005) (Fig. 7D). Together, these findings implicate E2 as a cause for premature and exaggerated neutrophil activation and earlier death of neutrophils, which translate to a reduced ability of neutrophils to kill P. aeruginosa.
FIG 7.

17β-Estradiol impacts responses to P. aeruginosa infection of neutrophils from subjects with CF. (A) Neutrophils from female subjects with CF treated ex vivo with E2 and ICI 182,780 (ICI) at the indicated doses were subjected to P. aeruginosa killing assays at an MOI of 1:4 (n = 20 per group). (B) Neutrophils from male subjects with CF treated ex vivo with E2 and ICI 182,780 (ICI) at the indicated doses were analyzed in P. aeruginosa killing assays at an MOI of 1:4 (n = 5). Data shown are mean results ± SEM. *, P < 0.05 relative to vehicle. (C) Neutrophils similar to those desribed for panel A were treated with E2 and ICI 182,780 for oxidative burst assays induced by P. aeruginosa. Chemiluminescence (CLU) is expressed as the mean result ± SEM (n = 6 per group). *, P < 0.05 (AUC). (D) NE expression in CF neutrophils treated with vehicle or E2. Data shown are the mean results ± SEM (n = 6). *, P < 0.05. (E) Neutrophil death was assayed via LDH release at the indicated time points (n = 6 per group). *, P < 0.05 relative to vehicle with PAO1 and vehicle without PAO1. Shown in panels A, C, and D are the mean results ± SEM. *, P < 0.05 relative to vehicle.
DISCUSSION
This study demonstrated that ER activation interferes with the host response to P. aeruginosa infection, in part through a neutrophil-dependent mechanism, and is attenuated by an ER antagonist. The E2 effect is driven by both nuclear ER-α and ER-β activation, but not by nonnuclear ER activation. We showed that the effect was consistent in mice infected with P. aeruginosa, in both wild-type mice and in the clinically relevant model of CFTR-deficient mice. In each model, E2 stimulation resulted in increased inflammatory cytokine expression and decreased survival and ability to clear bacteria. The E2 effect was abrogated when mice were rendered neutropenic, suggesting a central role for neutrophils in the mechanism affected by ER activation.
Findings in mice were validated in human samples, again using a clinically relevant population that was infected with P. aeruginosa. E2-stimulated neutrophils isolated from female relative to male CF individuals showed early and overexuberant activation, demonstrated by a heightened oxidative burst and increased release of the intracellular granular proteins MPO and NE. Ultimately, under E2 treatment conditions, neutrophils underwent earlier death, which correlated with an impaired functional killing capacity for P. aeruginosa. Together, these data suggest that there is a dysregulated proinflammatory response and altered neutrophil function in the setting of ER activation during P. aeruginosa infection, and they provide a rational basis for the observed sex differences in both non-CF and CF subjects. Attenuation of this dysregulated response with the use of an ER antagonist may present a novel therapeutic approach to such infections.
Previous studies suggested that estrogens may impact P. aeruginosa infection in CF patients. An association was reported between pulmonary exacerbations and high E2 levels in women with CF and that study further showed that E2 promoted the in vitro conversion of P. aeruginosa from a nonmucoid to mucoid form after 4 weeks of daily E2 treatment (32). The relevance of these finding is unclear, as the other authors did not employ an ER antagonist to demonstrate ER specificity. We did not observe a morphological conversion of P. aeruginosa from a nonmucoid to mucoid type with E2 or P4 treatment, nor did we see a change in the rate of bacterial growth. The lack of mucoid conversion may be explained by the fact that our study periods were less than 96 h, whereas the previous study noted mucoid conversion after several weeks. We were also unable to find ER or PR expression in the PAO1 genome. Our observations indicate that the impact of E2 on P. aeruginosa infection is primarily due to an alteration in the host response rather than a direct effect on the bacterium.
E2 has been reported to have both pro- and anti-inflammatory effects in different organ systems (19, 33–35). The role of sex hormones has been investigated in experimental models of infection and inflammation with varied results. Inflammatory responses were worse in female than male mice injected with intrapleural carrageenan in a model that implicated the ER-α signaling pathway (21). A lung allergen-induced mouse model showed that progesterone influences the Th2 inflammatory response (22). In a model of pneumococcal pneumonia, macrophages from male and female mice treated with estrogen were more efficient in ingesting bacteria; however, the specific sex hormone signaling pathway was not identified (36). The specific inflammatory cell that leads to increased morbidity and mortality in infectious and inflammatory lung diseases has not been identified. More importantly, if sex hormone pathways were to be modulated pharmacologically in human trials, the specific estrogen receptor to be blocked also has not been defined. Consistent with our findings, ER activation has been shown to induce a proinflammatory response in CFTR−/− male mice infected with P. aeruginosa (37). However, we further revealed that the E2 response is driven by nuclear ER-α and ER-β receptor activation and does not involve nonnuclear ER involvement and also that the increase in inflammation results in diminished bacterial clearance and increased death in E2-treated mice. PPT and DPN were employed to assess ER-α and ER-β activities, rather than ER knockout mice, because ER-β knockout mice in particular are known to have decreased numbers of alveoli in adult female mice and altered surfactant homeostasis, which we felt would complicate interpretation of studies (38). We also showed that the death rate could be improved by using the ER-α and ER-β antagonist ICI 182,780. In contrast to prior studies, we did not find a quantitative difference in neutrophils in the lungs of E2-treated mice (37). We instead demonstrated that the neutrophils recruited to sites of inflammation are less capable of clearing P. aeruginosa and undergo overexuberant activation and enhanced cell death upon E2 treatment. Furthermore, we did not find a direct connection to CFTR and host susceptibility to infection under E2 stimulation. Therefore, our findings are also relevant to the sex-dependent differences in the outcomes of those with airway diseases where neutrophils also play a role in the pathophysiology of airway inflammation, including asthma and COPD (39, 40).
Historically, elevated levels of the neutrophil intragranular proteins MPO and NE in the CF lung were thought to be directly proportional to increased numbers of neutrophils rather than increased release from individual neutrophils. Importantly, TNF-α and IL-8 have been shown to promote increased release of NE from neutrophils (41). Our data support this finding in that we showed increased expression of several cytokines, including TNF-α and IL-8, in the presence of ER activation but no quantitative differences in neutrophil counts despite higher levels of MPO and NE in lung tissues. We further showed that E2 induces an earlier and more exaggerated oxidative burst and NE elastase release in neutrophils isolated from CF female subjects, supporting a role for estrogens in the complex mechanisms governing neutrophil activation and degranulation. Increased release of reactive oxygen species results in a protease-antiprotease imbalance that leads to long-term tissue destruction (42). Excessive NE in CF as well as COPD is associated with more severe disease and may explain in part the worse outcomes in females with these diseases (40, 43).
In conclusion, we have shown in a murine model of P. aeruginosa respiratory infection and in human neutrophils that ER activation has a pivotal role in enhancing inflammation and activating neutrophils but that it hinders important neutrophil killing functions, which can be attenuated with an ER antagonist. Together, these data suggest a dysregulated host immune response to P. aeruginosa infection in the presence of E2. Modulation of ER activation or downstream ER pathways may represent novel treatment targets to improve host immunity and thereby narrow the gap in outcomes between males and females with CF and other inflammatory lung diseases.
MATERIALS AND METHODS
Mice.
WT and CF (CFTRtm1UNC) male and female mice in a C57BL/6 background were used for all studies. Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern. Female mice were ovariectomized at 4 to 6 weeks of age and treated with hormones. Pellets were inserted subcutaneously in the nape of the neck that released vehicle only, E2 (0.5 μg/day), or P4 (2 mg/day) (Innovative Research of America, Sarasota, FL). To modulate ER, ovariectomized mice were treated with E2 (0.5 μg/day), ICI 182,780 (500 μg/day), PPT (250 μg/day), or DPN (250 μg/day) by subcutaneous infection (Tocris, Minneapolis, MN). To modulate the nongenomic ER, we used EDC and its dendrimer control (44). These ovariectomized mice were implanted with intraperitoneal osmotic minipumps (model 2006; Durect Corp., Cupertino, CA) that delivered EDC at an estrogen dose equivalent to 0.5 μg/day (45). Male mice were treated daily with subcutaneous injections of E2 (0.5 μg/day) or vehicle.
Pseudomonas aeruginosa infection.
P. aeruginosa strain PAO1 (ATCC, Manassas, VA) was used for all described bacterial studies (46). Bacteria were streaked from frozen glycerol stocks onto Trypticase soy agar (TSA) plates and incubated overnight at 37°C. Overnight cultures were washed in 150 nM sodium chloride, and final pellets were resuspended in 1 ml of Luria-Bertani (LB) broth. Culture concentrations were calculated based on the optical density at 600 nm (OD600) and the previously determined number of CFU for each strain, to give the CFU/OD600. Mice were inoculated intranasally with 50 ul of P. aeruginosa at low doses (3.5 × 105 CFU) for bacterial clearance assays and at high doses (8.5 × 105) for survival assays. Right and left lungs were harvested and homogenized for quantification of CFU per lung, histological assessment, determination of inflammatory cytokines in the lung, and cell counts via flow cytometry (47). Survival assays were also conducted in mice infected with the mucoid clinical P. aeruginosa isolate M57-15 at 1.2 × 106 CFU (described further in the supplemental material) (30, 48). Male and female mice were infected with PAO1 at 3.5 × 105 CFU and 8.5 × 105 CFU as well as M57-15 at 1.2 × 106 CFU. A subset of mice were euthanized 24 h postinfection and blood was drawn by cardiac puncture and sent to IDEXX Bioresearch (Columbia, MO) for microbiologic evaluation. Results are shown in Table S1.
Mucoid Pseudomonas aeruginosa infection.
A mucoid P. aeruginosa clinical isolate, M57-15, was provided by Carolyn Cannon (Texas A&M University, College Station, TX). Bacteria were streaked from frozen glycerol stocks onto Trypticase soy agar plates and incubated overnight at 37°C. Overnight cultures were washed in 150 mM sodium chloride, and final pellets were resuspended in 1 ml Tryptic soy broth. Concentrations were calculated using the CFU/OD600 with the previously determined number of CFU for each strain. Mice were inoculated with 1.2 × 106 CFU of M57-15 for survival experiments based on reports in the previous literature (30, 48). Male and female C57BL/6 wild-type mice were infected with M57-15 and followed for up to 96 h (n = 6 per group) (Fig. S3). A subset of mice were euthanized 24 h postinfection and blood was drawn by cardiac puncture and sent to IDEXX Bioresearch (Columbia, MO) for culture to detect bacteremia. Results are shown in Table S1.
Bronchoalveolar lavage fluid and tissue collection.
Two weeks after ovariectomy and hormone treatments, mice were infected with P. aeruginosa and then euthanized at different time points. BAL was performed by surgically exposing the trachea and inserting a plastic catheter (21 gauge). To lavage, 1 ml of cold phosphate-buffered saline (PBS) was introduced slowly into the lungs, and fluid was withdrawn three times and kept on ice. Lungs were then processed for flow cytometry and histology. Lungs were washed with PBS, homogenized, and kept on ice. In separate experiments, the lung vasculature was perfused, inflation fixed with 4% paraformaldehyde (PFA), and then paraffin embedded. Inflammatory cytokines in the lung were determined using a multianalyte assay (Bio-Rad, Hercules, CA). Select cytokines, MPO, and NE were measured based on colorimetric activity and enzyme-linked immunosorbent assay results, respectively.
Flow cytometry.
Cells were obtained from BAL fluid and minced lung homogenates by centrifugation, pelleted, and fixed for 1 min in 2 ml of intracellular fixative (eBioscience, San Diego, CA). The fixation reaction was stopped by the addition of 10 ml of ice-cold flow cytometry wash buffer (FWB) composed of PBS containing 2% fetal calf serum (FCS), 0.1% sodium azide, and 5 mM EDTA. The suspension was subsequently filtered through a 40-μm nylon membrane and washed again with 10 ml of ice-cold FWB. The single-cell suspension was centrifuged at 4°C at 1,282 × g. The cell pellet was resuspended in 1 ml of flow cytometry wash buffer and placed on ice. For in vitro antibody staining, single-cell suspensions were incubated with fluorochrome-conjugated antibodies for 30 min in a dark at room temperature (RT). Antibodies used to identify myeloid cells included CD45 (clone 30-F1; BD Pharmigen, San Jose, CA), CD24 (clone M1/69; BD Pharmigen), Ly6G (clone 1A8; BD Pharmigen), and F4/80 (clone BM8; Biolegend); propidium iodide (PI) staining solution was used to label dead cells (BD Biosciences, San Jose, CA). After incubation, 4 ml of FWB was added to the single-cell suspensions and centrifuged at 1,282 × g at 4°C for 5 min. Cells were resuspended in FWB and analyzed with a four-channel FACSCalibur flow cytometer (Beckman Coulter, Brea, CA). Data were analyzed with FlowJo software (Treestar, Ashland, OR).
Histology and immunohistochemistry.
Lung tissues from ovariectomized and E2-supplemented WT mice inoculated with vehicle or P. aeruginosa were fixed with 4% PFA and embedded in paraffin to produce 3-μm-thick sections. The sections were stained with hematoxylin and eosin (H&E). Histology was evaluated by two different parties, blinded to the treatment groups and using a scoring system modified from those previously described (31, 49, 50). The degree of peribronchial and perivascular inflammation was evaluated on a scale of 0 to 3. A value of 0 was assigned when no inflammation was detectable, a value of 1 was designated for occasional cuffing with inflammatory cells, a value of 2 was designated when the surrounding area of most bronchi and vessels showed loosely arranged inflammatory cells, and a value of 3 was given when the surrounding area of most bronchi and vessels contained a thick layer of densely packed inflammatory cells. Total lung inflammation was determined as the average of the peribronchial and perivascular inflammation scores.
Murine neutrophil isolation.
Murine bone marrow leukocyte isolation was performed using a modification of a previously described protocol, and cells were kept on ice prior to use (47). Briefly, mice were anesthetized using Avertin injection prior to cervical dislocation. The skin was cleansed with alcohol and the femur and tibia were removed bilaterally. The bones were cleaned of debris, and washed in Hanks' balanced salt solution (HBSS; HyClone, Pittsburgh, PA). Cells were isolated at 500 × g for 10 min at RT. Cells were then resuspended in 45% Percoll–Dulbecco's phosphate-buffered saline solution (Sigma, St. Louis, MO) and aspirated twice through a 22-gauge needle and syringe to break up dense bone marrow tissue. Cell-Percoll solutions were layered atop a Percoll density gradient (prepared by underlaying 2 ml of 52, 69, and 78% Percoll–Dulbecco's phosphate-buffered saline solutions) and centrifuged at 500 × g for 30 min at RT. Neutrophils were collected from the 78% Percoll solution interface, washed twice with 10 ml of HBSS supplemented with bovine serum albumin and glucose (1,400 × g for 5 min at RT), and then resuspended in buffer. Cells were counted using a hemocytometer, attached to glass slides by centrifugation, and stained for differential analysis.
Human neutrophil isolation and assays.
Human sample collection and data collection for these studies were approved by the Institutional Review Board at UT Southwestern and required signed consent from each participant. Blood was collected in acid citrate dextrose anticoagulant (Vacutainer ACD solution A; BD Biosciences, San Jose, CA) and centrifuged at 425 × g for 10 min to separate the plasma from the hemocytes. The hemocytes were diluted with calcium, magnesium-free HBSS (HBSS−) to the lesser of 25 or 50 ml, and mixed with an equal volume of 3% dextran 500 (MP Biochemicals, Santa Ana, CA) dissolved in HBSS−. While the erythrocytes were settling in dextran, the plasma from the first spin was spun again to enhance leukocyte recovery. The resulting plasma was then centrifuged at 1,847 × g for 10 min to remove platelets. After the erythrocytes formed a distinct phase (20 to 30 min), the supernatant was removed from the aggregated erythrocytes by centrifugation for 10 min at 425 × g. The resulting pellet was diluted in 10 ml of 33 mM NaCl followed 30 s later by 10 ml of 267 mM NaCl to restore isotonicity. The tube was filled with HBSS−, spun as before, and the lysis was repeated. The pellet was then resuspended in 2 ml of HBSS− and overlaid on a discontinuous gradient of Percoll, to 51% (bottom layer) and 42% (top layer). The gradient was centrifuged for 10 min at 425 × g, and the neutrophil-rich pellet was resuspended and washed once in HBSS−. Purity of neutrophils prepared by this method was typically 95 to 99% (51). Neutrophil killing assays, luminol-enhanced neutrophil chemiluminescence, and cytotoxicity assays were performed as previously described (51–55).
Killing assays.
Neutrophil killing assays were performed in nonpyrogenic polystyrene 96-well plates (Falcon tissue culture plates; BD Biosciences). Neutrophils were preincubated for 2 h with vehicle, E2, and ICI 182,780, an estrogen receptor antagonist, at different concentrations. P. aeruginosa was added at a multiplicity of infection (MOI) of 1:4 in a total volume of 300 μl that contained Roswell Park Memorial Institute (RPMI) 1640 medium with l-glutamine (Invitrogen, Carlsbad, CA) and 25 mmol/liter HEPES (pH 7.5; HyClone, Pittsburgh, PA) in the presence or absence of 10% autologous serum. The doubling time of PAO1 in our experiments was 20 to 25 min, consistent with previous reports in the literature, such that our final concentrations of P. aeruginosa reached ranges of 109 CFU/ml, which is within physiologic concentrations described in sputum of patients with CF (56, 57). Plates were incubated at 37°C with 5% carbon dioxide. After 4 h, the cells were placed on ice and treated with saponin (final concentration, 0.1%; Sigma-Aldrich, St. Louis, MO) for 10 min, followed by mechanical shearing with a 28-gauge needle and with a 0.5-ml syringe. Supernatants were diluted and spread on LB plates and then CFU were counted (52).
Luminol-enhanced neutrophil chemiluminescence.
Neutrophils were suspended at 1 × 106/ml in RPMI 1640 medium with 5% autologous serum, 27.5 mM HEPES (pH 7.4), and 55 μM luminol (Fisher Scientific, Pittsburg, PA), and 90 μl was aliquoted into a 96-well plate. Neutrophils were preincubated for 2 h with vehicle or E2 at doses of 1, 10, and 100 nM at 37°C in a 5% CO2 incubator, and 10 μl of washed bacteria (MOIs, 100:1, 10:1, 1:1) was suspended in saline and added to the plate. Saline alone was added as a control, and the luminometer (Anthos Zenyth 3100, Hilliston, MA) was set at 37°C, with readings of 1 s per well acquired every 2.4 min (52, 53, 58). Results are expressed kinetically or converted to the area under the concentration-time curve (AUC) using the Prism 5.0 software package (GraphPad, La Jolla, CA) (52).
Neutrophil cytotoxicity assay.
Neutrophils were suspended at 2 × 106/ml in RPMI 1640 medium with 1% human AB serum (Atlanta Biologicals, Flowery Branch, GA). Neutrophils were preincubated with vehicle and E2 at concentrations of 1 to 100 nM for 2 h. After preincubation, washed P. aeruginosa cells were added to each well at an MOI of 1:10 (52, 58). Two sets of replicates for each condition were used: one for the high control and one for the sample. After 3, 6, 9, and 12 h, the wells for maximal lysis control were treated with 5% Triton X-100 (Sigma, St. Louis, MO) and mixed thoroughly using a multichannel pipette to ensure the cells membranes were properly degraded. The plate was then centrifuged for 10 min at 51 × g. Supernatant (100 μl) from the top of all the wells was added to a new assay plate in a volume of 100 μl for determination of lactate dehydrogenase (LDH) levels according to the manufacturer's instructions (cytotoxicity detection kit; Roche, Branchburg, NJ) and using a standard plate reader with a reference wavelength of 490 nM (54, 55). The cytotoxicity percentage was determined as follows: (experimental value) − [(low control)/(high control − low control)] × 100.
Statistical analysis.
Data were analyzed using Prism 6.0 (GraphPad Software, San Diego, CA). Survival analysis was performed by the Kaplan-Meier method using log rank analysis. Experimental results were analyzed using Student's t test for two samples or a two-way analysis of variance (ANOVA) with Tukey's multiple-comparison test for evaluation of three or more groups. Results are expressed as means ± standard errors of the means (SEM) of separate replicates as indicated in the figure legends. P values were considered statistically significant if P was <0.05.
Further details concerning our methods are provided in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank the subjects at the University of Texas Southwestern Cystic Fibrosis Center who participated in this study. We also thank Kimberly Batty, Carolyn Cannon, Parth Shah, Justin Smollen, Deborah Clegg, Sima Zein, and Sirisha Kodeboyina for their support with these studies.
This work was supported in part by the NIH (K08HL105671 to R.J.), the Cystic Fibrosis Foundation (JAIN13I0 to R.J.), Gilead Sciences (Research Scholars Award to R.J.), and Children's Medical Center of Dallas (CCRAC to R.J.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00422-17.
REFERENCES
- 1.Townsend EA, Miller VM, Prakash YS. 2012. Sex differences and sex steroids in lung health and disease. Endocr Rev 33:1–47. doi: 10.1210/er.2010-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raghavan D, Jain R. 2016. Increasing awareness of sex differences in airway diseases. Respirology 21:449–459. doi: 10.1111/resp.12702. [DOI] [PubMed] [Google Scholar]
- 3.Vink NM, Postma DS, Schouten JP, Rosmalen JG, Boezen HM. 2010. Gender differences in asthma development and remission during transition through puberty: the TRacking Adolescents' Individual Lives Survey (TRAILS) study. J Allergy Clin Immunol 126:498–504.e1-6. doi: 10.1016/j.jaci.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, Liu X. 2012. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. NCHS Data Brief 94:1–8. [PubMed] [Google Scholar]
- 5.Hoyert DL. 2012. 75 years of mortality in the United States, 1935–2010. NCHS Data Brief 88:1–8. [PubMed] [Google Scholar]
- 6.Akinbami LJ, Liu X. 2011. Chronic obstructive pulmonary disease among adults aged 18 and over in the United States, 1998–2009. NCHS Data Brief 63:1–8. [PubMed] [Google Scholar]
- 7.Aryal S, Diaz-Guzman E, Mannino DM. 2013. COPD and gender differences: an update. Transl Res 162:208–218. doi: 10.1016/j.trsl.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B. 1997. Gender gap in cystic fibrosis mortality. Am J Epidemiol 145:794–803. doi: 10.1093/oxfordjournals.aje.a009172. [DOI] [PubMed] [Google Scholar]
- 9.Harness-Brumley CL, Elliott AC, Rosenbluth DB, Raghavan D, Jain R. 2014. Gender differences in outcomes of patients with cystic fibrosis. J Womens Health (Larchmt) 23:1012–1020. doi: 10.1089/jwh.2014.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demko CA, Byard PJ, Davis PB. 1995. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol 48:1041–1049. doi: 10.1016/0895-4356(94)00230-N. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham GF, Leveno JK, Bloom SL, Sponge CY, Dashe JS, Hoffman BL, Casey BM, Sheffield JS. 2014. Williams obstetrics, 24th ed. McGraw-Hill, New York, NY. [Google Scholar]
- 12.Speroff L, Glass RH, Kase NG. 1999. Clinical gynecologic endocrinology and infertility, 6th ed. Lippincott, Williams and Wilkins, Baltimore, MD. [Google Scholar]
- 13.Pietras RJ, Levin ER, Szego CM. 2005. Estrogen receptors and cell signaling. Science 310:51–53. doi: 10.1126/science.310.5745.51. [DOI] [PubMed] [Google Scholar]
- 14.Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER. 2004. Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol 18:2854–2865. doi: 10.1210/me.2004-0115. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee S, Chambliss KL, Mineo C, Shaul PW. 2014. Recent insights into non-nuclear actions of estrogen receptor alpha. Steroids 81:64–69. doi: 10.1016/j.steroids.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Conneely OM, Jericevic BM. 2002. Progesterone regulation of reproductive function through functionally distinct progesterone receptor isoforms. Rev Endocr Metab Disord 3:201–209. doi: 10.1023/A:1020020308980. [DOI] [PubMed] [Google Scholar]
- 17.Coakley RD, Sun H, Clunes LA, Rasmussen JE, Stackhouse JR, Okada SF, Fricks I, Young SL, Tarran R. 2008. 17β-Estradiol inhibits Ca2+-dependent homeostasis of airway surface liquid volume in human cystic fibrosis airway epithelia. J Clin Invest 118:4025–4035. doi: 10.1172/JCI33893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cagle PT, Mody DR, Schwartz MR. 1990. Estrogen and progesterone receptors in bronchogenic carcinoma. Cancer Res 50:6632–6635. [PubMed] [Google Scholar]
- 19.Straub RH. 2007. The complex role of estrogens in inflammation. Endocr Rev 28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 20.Speyer CL, Rancilio NJ, McClintock SD, Crawford JD, Gao H, Sarma JV, Ward PA. 2005. Regulatory effects of estrogen on acute lung inflammation in mice. Am J Physiol Cell Physiol 288:C881–C890. doi: 10.1152/ajpcell.00467.2004. [DOI] [PubMed] [Google Scholar]
- 21.Vegeto E, Cuzzocrea S, Crisafulli C, Mazzon E, Sala A, Krust A, Maggi A. 2010. Estrogen receptor-alpha as a drug target candidate for preventing lung inflammation. Endocrinology 151:174–184. doi: 10.1210/en.2009-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchell VL, Van Winkle LS, Gershwin LJ. 2012. Environmental tobacco smoke and progesterone alter lung inflammation and mucous metaplasia in a mouse model of allergic airway disease. Clin Rev Allergy Immunol 43:57–68. doi: 10.1007/s12016-011-8280-0. [DOI] [PubMed] [Google Scholar]
- 23.Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. 1999. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med 160:1532–1539. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 24.Blesson CS, Sahlin L. 2012. Expression pattern and signalling pathways in neutrophil like HL-60 cells after treatment with estrogen receptor selective ligands. Mol Cell Endocrinol 361:179–190. doi: 10.1016/j.mce.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Molero L, Garcia-Duran M, Diaz-Recasens J, Rico L, Casado S, Lopez-Farre A. 2002. Expression of estrogen receptor subtypes and neuronal nitric oxide synthase in neutrophils from women and men: regulation by estrogen. Cardiovasc Res 56:43–51. doi: 10.1016/S0008-6363(02)00505-9. [DOI] [PubMed] [Google Scholar]
- 26.Lamote I, Meyer E, De Ketelaere A, Duchateau L, Burvenich C. 2006. Expression of the estrogen receptor in blood neutrophils of dairy cows during the periparturient period. Theriogenology 65:1082–1098. doi: 10.1016/j.theriogenology.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 27.Buyon JP, Korchak HM, Rutherford LE, Ganguly M, Weissmann G. 1984. Female hormones reduce neutrophil responsiveness in vitro. Arthritis Rheum 27:623–630. doi: 10.1002/art.1780270604. [DOI] [PubMed] [Google Scholar]
- 28.Chiang K, Parthasarathy S, Santanam N. 2004. Estrogen, neutrophils and oxidation. Life Sci 75:2425–2438. doi: 10.1016/j.lfs.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 29.Molloy EJ, O'Neill AJ, Grantham JJ, Sheridan-Pereira M, Fitzpatrick JM, Webb DW, Watson RW. 2003. Sex-specific alterations in neutrophil apoptosis: the role of estradiol and progesterone. Blood 102:2653–2659. doi: 10.1182/blood-2003-02-0649. [DOI] [PubMed] [Google Scholar]
- 30.Shah PN, Lin LY, Smolen JA, Tagaev JA, Gunsten SP, Han DS, Heo GS, Li Y, Zhang F, Zhang S, Wright BD, Panzner MJ, Youngs WJ, Brody SL, Wooley KL, Cannon CL. 2013. Synthesis, characterization, and in vivo efficacy of shell cross-linked nanoparticle formulations carrying silver antimicrobials as aerosolized therapeutics. ACS Nano 7:4977–4987. doi: 10.1021/nn400322f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwak YG, Song CH, Yi HK, Hwang PH, Kim JS, Lee KS, Lee YC. 2003. Involvement of PTEN in airway hyperresponsiveness and inflammation in bronchial asthma. J Clin Invest 111:1083–1092. doi: 10.1172/JCI16440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chotirmall SH, Smith SG, Gunaratnam C, Cosgrove S, Dimitrov BD, O'Neill SJ, Harvey BJ, Greene CM, McElvaney NG. 2012. Effect of estrogen on Pseudomonas mucoidy and exacerbations in cystic fibrosis. N Engl J Med 366:1978–1986. doi: 10.1056/NEJMoa1106126. [DOI] [PubMed] [Google Scholar]
- 33.Josefsson E, Tarkowski A, Carlsten H. 1992. Anti-inflammatory properties of estrogen. I. In vivo suppression of leukocyte production in bone marrow and redistribution of peripheral blood neutrophils. Cell Immunol 142:67–78. [DOI] [PubMed] [Google Scholar]
- 34.Ghisletti S, Meda C, Maggi A, Vegeto E. 2005. 17β-Estradiol inhibits inflammatory gene expression by controlling NF-κB intracellular localization. Mol Cell Biol 25:2957–2968. doi: 10.1128/MCB.25.8.2957-2968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller AP, Feng W, Xing D, Weathington NM, Blalock JE, Chen YF, Oparil S. 2004. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation 110:1664–1669. doi: 10.1161/01.CIR.0000142050.19488.C7. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Huang YC, Koziel H, de Crom R, Ruetten H, Wohlfart P, Thomsen RW, Kahlert JA, Sorensen HT, Jozefowski S, Colby A, Kobzik L. 2014. Female resistance to pneumonia identifies lung macrophage nitric oxide synthase-3 as a therapeutic target. eLife 3:e03711. doi: 10.7554/eLife.03711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Cela E, Gagnon S, Sweezey NB. 2010. Estrogen aggravates inflammation in Pseudomonas aeruginosa pneumonia in cystic fibrosis mice. Respir Res 11:166. doi: 10.1186/1465-9921-11-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patrone C, Cassel TN, Pettersson K, Piao YS, Cheng G, Ciana P, Maggi A, Warner M, Gustafsson JA, Nord M. 2003. Regulation of postnatal lung development and homeostasis by estrogen receptor beta. Mol Cell Biol 23:8542–8552. doi: 10.1128/MCB.23.23.8542-8552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monteseirin J. 2009. Neutrophils and asthma. J Investig Allergol Clin Immunol 19:340–354. [PubMed] [Google Scholar]
- 40.Hoenderdos K, Condliffe A. 2013. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 48:531–539. doi: 10.1165/rcmb.2012-0492TR. [DOI] [PubMed] [Google Scholar]
- 41.Taggart C, Coakley RJ, Greally P, Canny G, O'Neill SJ, McElvaney NG. 2000. Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-alpha and interleukin-8. Am J Physiol Lung Cell Mol Physiol 278:L33–L41. [DOI] [PubMed] [Google Scholar]
- 42.Griese M, Kappler M, Gaggar A, Hartl D. 2008. Inhibition of airway proteases in cystic fibrosis lung disease. Eur Respir J 32:783–795. doi: 10.1183/09031936.00146807. [DOI] [PubMed] [Google Scholar]
- 43.Gifford AM, Chalmers JD. 2014. The role of neutrophils in cystic fibrosis. Curr Opin Hematol 21:16–22. doi: 10.1097/MOH.0000000000000009. [DOI] [PubMed] [Google Scholar]
- 44.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. 2006. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 45.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, Madak-Erdogan Z, Maggi A, Dineen SP, Roland CL, Hui DY, Brekken RA, Katzenellenbogen JA, Katzenellenbogen BS, Shaul PW. 2010. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest 120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klockgether J, Munder A, Neugebauer J, Davenport CF, Stanke F, Larbig KD, Heeb S, Schock U, Pohl TM, Wiehlmann L, Tummler B. 2010. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J Bacteriol 192:1113–1121. doi: 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klesney-Tait J, Keck K, Li X, Gilfillan S, Otero K, Baruah S, Meyerholz DK, Varga SM, Knudson CJ, Moninger TO, Moreland J, Zabner J, Colonna M. 2013. Transepithelial migration of neutrophils into the lung requires TREM-1. J Clin Invest 123:138–149. doi: 10.1172/JCI64181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Heeckeren AM, Tscheikuna J, Walenga RW, Konstan MW, Davis PB, Erokwu B, Haxhiu MA, Ferkol TW. 2000. Effect of Pseudomonas infection on weight loss, lung mechanics, and cytokines in mice. Am J Respir Crit Care Med 161:271–279. doi: 10.1164/ajrccm.161.1.9903019. [DOI] [PubMed] [Google Scholar]
- 49.Tournoy KG, Kips JC, Schou C, Pauwels RA. 2000. Airway eosinophilia is not a requirement for allergen-induced airway hyperresponsiveness. Clin Exp Allergy 30:79–85. doi: 10.1046/j.1365-2222.2000.00772.x. [DOI] [PubMed] [Google Scholar]
- 50.Braber S, Henricks PA, Nijkamp FP, Kraneveld AD, Folkerts G. 2010. Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir Res 11:99. doi: 10.1186/1465-9921-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. 2007. Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J Immunol 178:6367–6373. doi: 10.4049/jimmunol.178.10.6367. [DOI] [PubMed] [Google Scholar]
- 52.Zarember KA, Marshall-Batty KR, Cruz AR, Chu J, Fenster ME, Shoffner AR, Rogge LS, Whitney AR, Czapiga M, Song HH, Shaw PA, Nagashima K, Malech HL, DeLeo FR, Holland SM, Gallin JI, Greenberg DE. 2012. Innate immunity against Granulibacter bethesdensis, an emerging Gram-negative bacterial pathogen. Infect Immun 80:975–981. doi: 10.1128/IAI.05557-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dahlgren C, Karlsson A. 1999. Respiratory burst in human neutrophils. J Immunol Methods 232:3–14. doi: 10.1016/S0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 54.Matthias KA, Roche AM, Standish AJ, Shchepetov M, Weiser JN. 2008. Neutrophil-toxin interactions promote antigen delivery and mucosal clearance of Streptococcus pneumoniae. J Immunol 180:6246–6254. doi: 10.4049/jimmunol.180.9.6246. [DOI] [PubMed] [Google Scholar]
- 55.Smith SM, Wunder MB, Norris DA, Shellman YG. 2011. A simple protocol for using a LDH-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time. PLoS One 6:e26908. doi: 10.1371/journal.pone.0026908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, Haagensen JA, Jelsbak L, Johansen HK, Sternberg C, Hoiby N, Molin S. 2008. In situ growth rates and biofilm development of Pseudomonas aeruginosa populations in chronic lung infections. J Bacteriol 190:2767–2776. doi: 10.1128/JB.01581-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palmer KL, Mashburn LM, Singh PK, Whiteley M. 2005. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J Bacteriol 187:5267–5277. doi: 10.1128/JB.187.15.5267-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greenberg DE, Marshall-Batty KR, Brinster LR, Zarember KA, Shaw PA, Mellbye BL, Iversen PL, Holland SM, Geller BL. 2010. Antisense phosphorodiamidate morpholino oligomers targeted to an essential gene inhibit Burkholderia cepacia complex. J Infect Dis 201:1822–1830. doi: 10.1086/652807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.