

Abstract
Fixed chromosomal inversions can reduce gene flow and promote speciation in two ways: by suppressing recombination and by carrying locally favored alleles at multiple loci. However, it is unknown whether favored mutations slowly accumulate on older inversions or if young inversions spread because they capture preexisting adaptive Quantitative Trait Loci (QTLs). By genetic mapping, chromosome painting and genome sequencing we have identified a major inversion controlling ecologically important traits in Boechera stricta. The inversion arose since the last glaciation and subsequently reached local high frequency in a hybrid speciation zone. Furthermore, the inversion shows signs of positive directional selection. To test whether the inversion could have captured existing, linked QTLs, we crossed standard, collinear haplotypes from the hybrid zone and found multiple linked phenology QTLs within the inversion region. These findings provide the first direct evidence that linked, locally adapted QTLs may be captured by young inversions during incipient speciation.
Chromosome inversions play an important role in local adaptation and speciation1,2, and selectively important inversions have been identified in many species3,4. Selection due to different environmental factors or stages in the life cycle1 may favor inversions carrying locally adapted alleles at several loci. In addition, established inversions are predicted to accumulate selectively important genetic differences, which may contribute to reproductive isolation during speciation1.
Although few studies have identified the actual loci that influence selection on inversions2,4,5, rearrangements may be favored due to gene alterations near breakpoints6, chromatin changes7, or combinations of advantageous, coadapted alleles8. Inversions suppress recombination, so locally advantageous alleles may segregate together, causing higher fitness than recombinant haplotypes9. Most evolutionary studies have focused on widespread, older inversions, so we have little knowledge of the evolutionary processes that guide their initial increase in frequency. For example, do inversions drift to higher frequency, and then acquire new advantageous mutations after they are common? Or are multiple linked, advantageous alleles captured in a new inversion, allowing them to spread together? Analysis of younger inversions may elucidate the evolutionary forces controlling the initial spread of chromosome inversions, which therefore influence their role in adaptation and speciation4,8.
Related species often differ for chromosome inversions that carry locally-favored alleles at multiple loci10,11. A key distinction among models for the evolution of inversions is whether early frequency increase is due to genetic drift or natural selection. Genetic drift might predominate initially, with subsequent accumulation of advantageous variants12. Alternatively, the Kirkpatrick-Barton (“KB”) model9 argues that linked, locally adapted alleles exist first, and subsequently are captured within a new, selectively-favored inversion13. In this “inversion-late” evolutionary sequence1,5, linked QTLs, similar to the ancestral haplotype that gave rise to the inversion, may still exist in non-inverted genotypes9. Here, we test these predictions of the KB model. First, we introduce ecologically-diverged subspecies of Boechera stricta. Next, we examine a young inversion to infer the selective forces controlling its early increase in frequency. Finally, we cross collinear, standard genotypes from the hybrid zone to ask whether old, linked QTLs can be found within the inversion region.
Young Inversion in a hybrid zone
Previously, we showed that Boechera stricta (a close relative of Arabidopsis14) has two ecologically differentiated subspecies (“EAST” and “WEST”)15,16, which form a contact zone across >200 km in the Northern Rocky Mountains. Throughout our study area, local water availability is the best predictor of habitat divergence between these subspecies15, with WEST genotypes growing in sites with more constant and abundant water supply. In and near the hybrid zone, the EAST and WEST subspecies show significant ecological differentiation across local environmental gradients, and the geographic distribution of the inversion falls within the typical range of hybrid zone habitats. The WEST subspecies has significantly faster growth rate, larger leaf area, less succulent leaves, delayed reproductive time, and longer flowering duration16, and WEST × EAST crosses found QTLs for flowering traits, leaf number, defensive chemistry, herbivore resistance, cold tolerance, overwinter survival, fecundity, and lifetime fitness17–20. In addition, QST-FST analysis showed that phenology and some morphological traits have experienced divergent selection between subspecies16.
In our initial cross17, genetic mapping in WEST × EAST recombinant inbred lines (RILs) identified a region of suppressed recombination (Supplementary Fig. 1) on linkage group 1 (LG1). Here, we combine short-read sequencing, end sequencing of Bacterial Artificial Chromosomes (BACs), linkage mapping, and physical mapping by Whole Genome Profiling to assemble a high quality genomic sequence (Supplementary Fig. 2), which enabled evolutionary analysis of the inversion. Chromosome painting (Fig. 1; Supplementary Fig. 3) cytogenetically verified the presence of an 8.4 Mb paracentric inversion in the WEST genotype (Bsi1; Fig. 2a; Supplementary Fig. 4), spanning ~31 cM (~4% of the genome, the “inversion region”, Supplementary Table 1). We developed primers to score the inversion using PCR (Fig. 2b), showing that the common, non-inverted allele (Bsi1-std) has the ancestral orientation found in closely-related Capsella and Arabidopsis lyrata21, and is found in >200 EAST and WEST populations across the species range (Fig. 3a; Dryad Data Archive). The derived, inverted allele (Bsi1-inv) was identified in a cluster of WEST and hybrid populations in the contact zone (Fig. 3b), where it has risen to high frequency. Genotypes carrying the inversion span a narrow geographic range (~14 km, the “inversion zone”), where pollen and geological analyses22,23 suggest that conditions suitable for B. stricta existed during the Last Glacial Maximum ~21 kya. SNPs in the inversion region show that the most similar Bsi1-inv and Bsi1-std haplotypes (Supplementary Fig. 5) are located only 1.7 km apart (Fig. 3b, left). Using measured mutation rates24 and assuming two years per generation, the inversion dates to ~2,100 – 8,800 years (Methods). Because these widely-distributed subspecies are ecologically diverged16, while the Bsi1 inversion originated very recently, this polymorphism is compatible with the inversion-late KB model.
Fig. 1. Comparative Chromosome Painting of chromosome 1 in Bst1-std and Bst1-inv haplotypes.
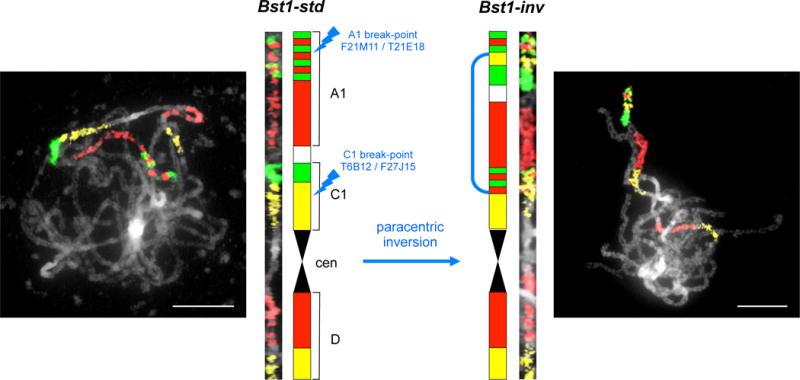
Shown are in situ chromosomal localization of painting probes on B. stricta pachytene chromosomes, their straightened images and diagrammatic representations of the Bst1-std and Bst1-inv haplotypes. Differential labeling of BAC contigs (F21M11, T21E18, T6B12, F27J15) identifies the breakpoints and extent of the paracentric inversion in the Bsi1-inv genotype. Scale bars, 10 μm. See Supplementary Fig. 3 for a detailed cytogenetic analysis.
Fig. 2. Inversion region and PCR genotyping.
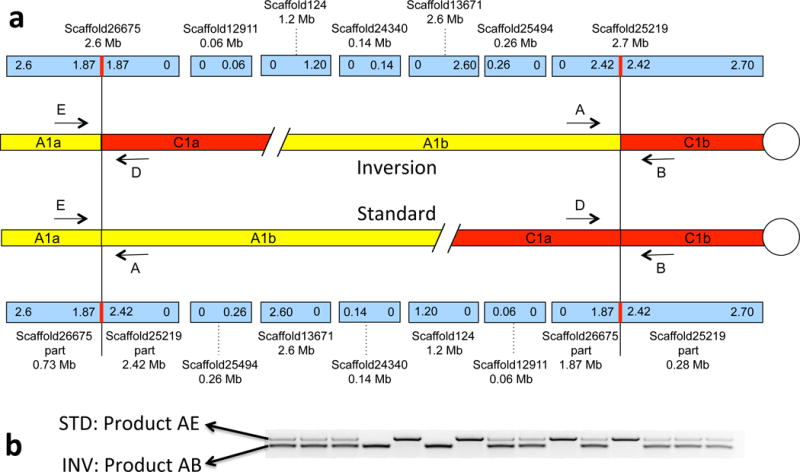
(a) Chromosome blocks (yellow and red) are shown for the derived (Bst1-inv, upper) and ancestral (Bst1-std, lower) haplotypes. Centromeres are indicated by white circles. Scaffolds (blue, above and below) are labeled with their name and size. Breakpoints are shown as vertical red lines within the scaffolds. Arrows indicate PCR primers. There are 1,591 annotated genes in the inversion region. (b) Gel image showing PCR products, allowing codominant identification of inversion genotypes.
Figure 3. Geographic locations of subspecies collections and inversion zone genotypes. Geographic locations of genotyped accessions used in this study.
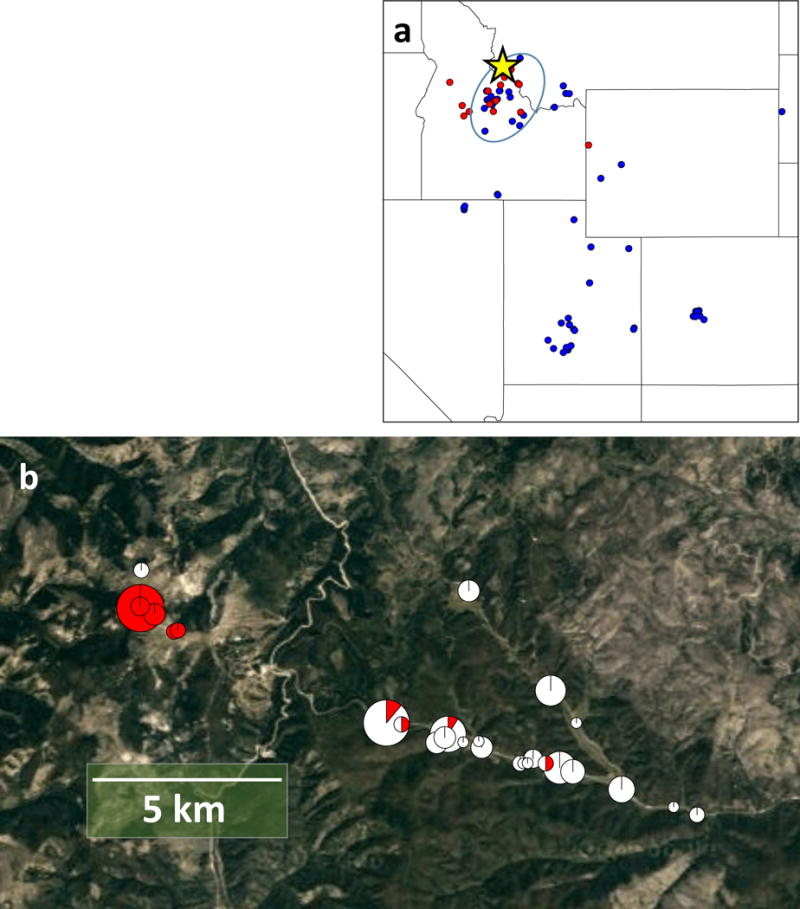
(a) Species-wide samples, with each dot indicating one genotype from a population. Subspecies assignments show EAST (blue) and WEST (red). Ellipse indicates the contact zone where subspecies overlap, and the star shows the inversion zone. N = 83. The collinear cross comes from within the hybrid zone (oval), outside the inversion zone (star). (b) Close-up of populations near the inversion zone (the star in Fig. 3a), with pie-diagrams indicating the inversion frequency (red) in each population, sized in proportion to sample size. Forested, unsuitable habitat likely limits gene flow. N = 122 genotypes. Individual populations range in size from 1 to 19.
Previous analysis of a WEST-inv × EAST-std mapping population grown in the inversion zone found many QTLs controlling components of fitness25 and strong selection on flowering time17, influenced by recent climate warming26. Native WEST Bsi1-inv alleles had higher fitness than EAST Bsi1-std alleles25. The inversion (spanning ~4% of the genome) explains 7.5% of heritable variation for lifetime fecundity, and inversion homozygotes differ by 0.56 genetic standard deviations for this trait (t = 3.64, df = 165, P = 0.0004). Here, we focused on effects of the inversion by examining flowering time in two F7 Near-Isogenic Line (NIL) families from this cross. We found no segregation distortion or deviation from Mendelian ratios (Supplementary Table 2) in either family, and the inversion had significant effects on flowering time in these NIL families in the greenhouse (F2, 282 = 21.81, P < 0.001), where the Bsi1-std haplotype flowered ~2.0 days faster than the Bsi1-inv haplotype (Supplementary Fig. 6). Thus, the inversion controls 40% of the average 5.0 day difference in flowering time between subspecies (Supplementary Table 2d). While this pedigree cannot resolve QTLs within the inversion, this cross shows that the inversion has significant effects on an ecologically important trait that experiences natural selection17. Gene(s) within the Bsi1 inversion control flowering in the field, and thus may contribute to reproductive isolation during speciation27.
To test for phenotypic effects of the inversion or transcription changes near the breakpoints, we compared closely-related inverted and standard haplotypes from the inversion zone, using a sympatric WEST-inv × WEST-std F2 cross (Fig. 3b, left). We found high seed germination (>99%), and no Bsi1 segregation distortion or deviation from Mendelian ratios (Supplementary Table 3). MANOVA showed a significant effect of Bsi1 on a suite of phenological and morphological traits (P = 0.005 by permutation test, R2 ≈ 4%; Supplementary Table 4), so we analyzed individual traits by ANOVA. In this cross, flowering time is delayed by 0.9 days (P = 0.030 by permutation test) in Bsi1-inv homozygotes (accounting for 18% of the difference between subspecies), which may increase reproductive isolation from the early-flowering EAST genotypes16. Finally, we tested for functional changes attributable to the inversion breakpoints; these breakpoints do not disrupt existing or create new open reading frames (Supplementary Fig. 7). Using replicated F3 homozygotes from this cross, we found no significant expression differences between inversion and standard haplotypes for any of the seven genes flanking the inversion breakpoints (Supplementary Table 5), although power to detect subtle, quantitative differences is limited. Thus, this comparison of WEST-inv vs. WEST-std Bsi1 haplotypes found genetic differences for ecologically-important traits, but little evidence for functional changes at the inversion breakpoints.
Evidence for Natural Selection
To test whether selection had favored this inversion, we analyzed patterns of population genetic variation (Fig. 4; Supplementary Fig. 8). Bsi1-inv genotypes show lower LG1 polymorphism, lower Tajima’s D, and more linkage disequilibrium (LD) than Bsi1-std individuals. These patterns are compatible both with neutral drift in this partially inbreeding species, or with a selective sweep on the inversion28. In contrast, young, derived mutations at high frequency suggest positive directional selection29, so we asked whether the frequency of Bsi1-inv is typical of frequencies of all private derived SNP alleles in the same population (the 54 similar genotypes in the left portion of Supplementary Fig. 9c and the left portion of Fig. 3b). The inversion is at relatively high frequency (0.63) in this group, but it is not fixed. We found 2,416 SNPs that, like the inversion, are confined to this population. Among these, only 63 SNPs (2.6%) have derived allele frequencies greater than the frequency of the Bsi1-inv inversion. Hence, the frequency of the Bsi1-inv inversion allele is higher than 97.4% of comparable derived alleles in this population – the inversion is a high frequency outlier, supporting the hypothesis of positive directional selection.
Figure 4. Population genetic variation for inversion and standard groups.
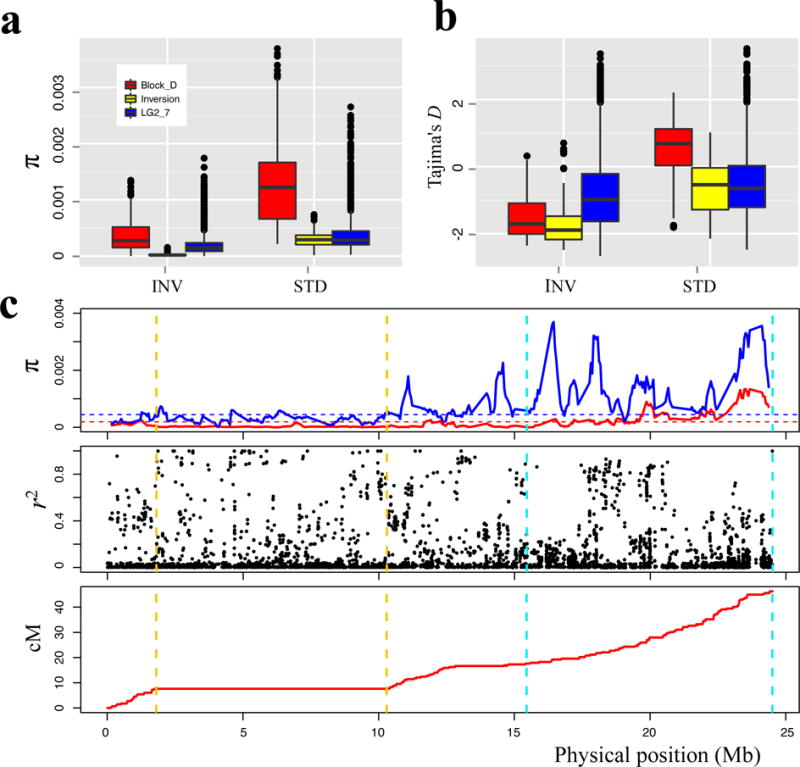
(a, b) Nucleotide diversity (π) and Tajima’s D of inverted genomic region (yellow), block D of chromosome 1 (red) and chromosome 2–7 (blue) in inversion (INV) and standard (STD) genotypes. Block D (south end of LG1) is treated separately because it has unusually high polymorphism in related species. In these box plots, the median is shown by a horizontal line, while the bottom and top of each box represents the first and third quartiles. The whiskers extend to 1.5 times the interquartile range. Outliers are represented by black dots. (c) Distribution of population genetic statistics along chromosome 1. Nucleotide diversity (π) in INV (red) and STD (blue) genotypes, with genome-wide averages as dashed horizontal lines. Linkage disequilibrium (R2) between the inversion and SNPs is shown in all INV and STD genotypes from the inversion zone, and the relationship between physical and linkage maps. The dashed vertical lines mark the inverted (golden) and block D (light blue) regions. N = 122, except four admixed individuals were excluded from LD analysis. LD for 10 comparator SNPs with derived allele frequencies similar to Bsi1-inv is shown in Supplementary Fig. 8b.
QTLs within the Inversion
During speciation, coadapted gene complexes within inversions might reduce gene flow if they preserve favorable combinations of alleles at multiple loci, reducing the frequency of disadvantageous allelic combinations. Although inversions can be engineered for proof of function in some organisms30, this is infeasible in Boechera. However, the KB model predicts that relatives of the ancestral haplotype that gave rise to the inversion might still exist as non-inverted genotypes nearby. To test for such linked QTLs, we crossed collinear WEST-std × EAST-std genotypes from the contact zone and tested for QTLs within the inversion region, using freely-recombining F3:F4 families. We found several multivariate QTLs altering ecologically-important phenology and development traits (Fig. 5a). To clarify differences among these QTLs, we examined differences in their pleiotropic effects (Fig. 5b), using Discriminant Function Analysis (DFA) to find the axis of greatest divergence between the multivariate trait means at each QTL peak. Each DFA axis quantifies the direction of pleiotropy that controls effects of a locus, and QTLs influencing these composite traits were mapped across the inversion region. We found several distinct QTL peaks, which show divergent patterns of pleiotropy among these linked loci. Finally, we estimated the time of divergence between these parental genotypes (Fig. 5c). These results show that the QTL haplotypes diverged at least 50 – 100 Kya, and are therefore much older than the inversion, which arose less than 10 Kya.
Figure 5. QTLs within the inversion.
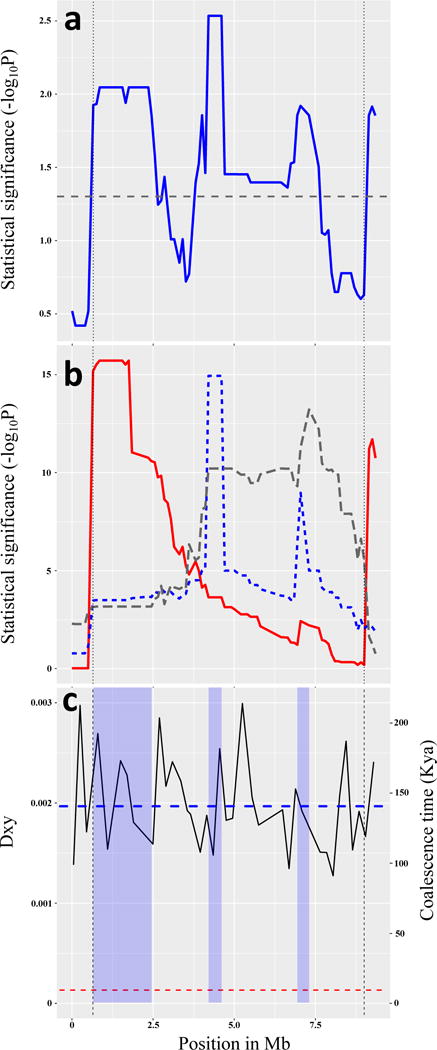
A collinear cross shows that several QTLs in the inversion region influence phenology and development traits. Inversion breakpoints are indicated by vertical lines. (a) Multivariate QTL mapping finds several QTL peaks (blue line) exceeding the significance threshold (horizontal dashed line); N = 1,714 F4 individuals in 153 families. (b) Testing the hypothesis that three linked QTLs have different pleiotropic effects. Plots for three QTLs (left, center, and right, shown in solid red, dotted blue, and dashed gray, respectively) quantify evidence that a locus with different patterns of pleiotropy occurs at each peak. Discriminant Function Analysis was used to identify new composite trait axes defined at each peak, and evidence for these composite traits was mapped across the inversion region. The left and center QTLs show little overlap, suggesting different patterns of pleiotropy. (c) Comparison of molecular divergence and time to coalescence of the East and West genotypes in the collinear QTL mapping cross. The horizontal red dashed line at 8.8 Kya is the upper confidence interval for age of the inversion. The vertical axes show nucleotide divergence (Dxy, left axis), and coalescence time (Kya, right axis). The blue dashed line indicates the mean genome-wide values of Dxy and coalescence time between these East and West alleles. The horizontal axis shows position across the inversion region in Mb, beginning at marker Scaf26675_2450000. Vertical blue shading indicates the QTL regions, +/− logP > 1.6 confidence intervals, with 408 annotated genes in these QTL regions. Divergence (Dxy) between these genotypes was calculated in 200kb non-overlapping windows, and the coalescence time was estimated using T = Dxy/2μ, where μ is 7E-9 per site per generation. Mean Dxy in the three QTLs (left to right) are 0.00206, 0.00169 and 0.00186, corresponding to coalescent times of 147 kya, 121 kya and 132 kya, respectively. The mean Dxy in the whole inversion region is 0.00191, and the average coalescence time is 136 kya.
In summary, population genetic evidence for a selective sweep corresponds with our ecological findings: the inversion affects multiple ecologically-important traits, including flowering differences that are expected to increase reproductive isolation between subspecies. The inversion occurs in a hybrid zone, and this genomic region contains multiple ecologically important QTLs, as predicted by the KB model. However, beyond the potential advantage conferred by recombination suppression9, closely related WEST-inv and WEST-std haplotypes show divergent phenotypic effects, which might contribute to selection in the hybrid zone. Our analysis of a young inversion is compatible with evolutionary predictions that linked, locally adapted QTLs may be captured by new inversions and contribute to local adaptation or incipient speciation9.
METHODS
Study Area
The inversion was originally detected21 at the Lost Trail Meadow site (~2,500m elevation) about four km NW from Lost Trail Pass on the Montana-Idaho border. Pollen and geological analyses22,23 show that climate and vegetation in this area was strongly influenced by Pleistocene climatic changes, although the site apparently was unglaciated during the Last Glacial Maximum (LGM) about 21,000 years ago. Inferred plant communities22,23 suggest conditions suitable for Boechera stricta may have existed at or near our study site during the LGM. We searched extensively in nearby B. stricta habitat; The Dryad Data Archive contains information on 122 genotypes from the inversion zone (Fig. 3), and 83 comparison genotypes from the across the species range.
Experimental Pedigrees
Details on experimental crosses and genotype locations are in the Dryad Data Archive.
DNA extraction for genomic sequencing
Seed sterilization
Seeds of Boechera stricta ecotypes (LTM and SAD12) were initially surface sterilized with ethanol (200 proof) for 2 min followed by treatment with 15% sodium hypochlorite solution (with a few drops of Tween-20) for 25 min on a rotatory shaker at room temperature. Surface sterilized seeds were thoroughly washed multiple times with sterile water and were suspended in 0.1% agar. Seeds were kept for stratification in a cold room at 4°C for 4 d under dark conditions.
Raising of aseptic seedling cultures
To raise aseptic seedlings of LTM and SAD12 ecotypes, surface sterilized and stratified seedlings were inoculated in 250 ml flasks containing 50 ml of sterile ½ MS liquid medium (pH 5.7) with 0.5 g/L MES and 2% sucrose. About 70–80 seeds were inoculated per flask. To obtain etiolated seedlings, the flasks were covered with aluminum foil and kept on a bench top rotatory shaker at 110 rpm for 20 d at room temperature.
Extraction of genomic DNA from etiolated seedlings
Freshly grown 20 d old etiolated seedlings of both ecotypes were used for extraction of nuclear DNA. Nuclei isolation was performed essentially according to Prasad et al18 with slight modifications, as it allowed isolation of clean high molecular size nuclear DNA with minimal contamination from organelle DNA. Briefly, about 10 flasks of etiolated seedling cultures were removed from the growing medium and thoroughly washed with ice cold water and placed in ice cold ethyl ether for 3 min. Subsequently the seedlings were washed five times with ice cold TE buffer (pH 7.0). The plant material was quickly blotted on sterile filter paper and homogenized in MEB buffer using a commercial blender. The homogenate was filtered through four layers of cheesecloth followed by two layers of mira cloth. Triton-X-100 was added to the filtrate at 0.5% concentration and centrifuged to collect the pellet. The pellet was suspended in MPDB solution and gently layered on 37.5% percoll made with MPDB. Nuclei were collected as a pellet after centrifugation, and the pellet was washed by resuspending it in 20 ml of MPBD followed by centrifugation. The nuclei pellet was again suspended in a MPDB solution and high molecular weight nuclear DNA was extracted using QIAGEN genomic tip 100/G protocol as per the manufacturer’s instructions, with some modifications. The pellet consisting of nuclei was resuspended in the lysis buffer and incubated at 40°C for 15 min. RNAse A and T1 were added to the suspension and further incubated at 37°C for additional 30 min. Subsequently, Proteinase K was added to the suspension at final concentration of 150 μg per ml and incubated at 3h at 45°C with gentle shaking. The suspension was cleared by centrifugation at 8,000 rpm for 15 min. The cleared suspension was added to the pre-equilibrated G-100 column and further purification was performed as per the protocol provided by the manufacturer. The eluate consisting of genomic DNA was further precipitated using isopropanol, washed with 70% ethanol and subsequently suspended in the TE (pH 8.0) buffer.
Total RNA isolation
To collect RNA from roots, the LTM-genotype seeds were surface sterilized with 12% bleach and grown under aseptic conditions on a Whatman filter paper bridges in glass tissue culture tubes containing 1/2 strength MS media with 1% sucrose. Each tube was inoculated with 2 to 3 seeds and allowed to grow. The roots obtained from the multiple seedlings were pooled and RNA was isolated with plant RNAeasy kit from Qiagen. For other tissues, we extracted RNA from soil-grown plants at several stages: 15 day old seedlings, rosette leaves from juvenile plants, cauline leaves from plants aged 2 – 3 months. Finally, from 7 month-old plants we extracted RNA from inflorescences, stems, flowers, and siliques.
For isolation of total RNA from various tissues at different developmental stages, the tissues were snap frozen in liquid nitrogen and stored at −80°C. The frozen tissue was homogenized with mortar and pestle using liquid nitrogen. About 150 mg of homogenized tissue was used for isolation. Total RNA was isolated using a Plant RNA-easy kit (Qiagen, USA). Each of the independent total RNA preparations was subjected to DNAse treatment to remove traces of contaminating genomic DNA using RNAse-free DNAse (Promega, USA). For each tissue type, three independent isolations were pooled together and used for further analysis.
Sequencing, Assembly, and Annotation
Eighteen genomic DNA libraries and one transcriptome cDNA library were prepared (Dryad Data Archive) for sequencing using Illumina and Roche 454. The genome assembly of B. stricta employed Meraculous build May 201331, as described on Phytozome: (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Bstricta),
Contigs were assembled with a k-mer size of 51 and a minimum depth of 8 from 2 × 150 Illumina HiSeq reads from an unamplified 250 bp whole genome shotgun fragment library. Several discrete rounds of scaffolding were performed with libraries containing inserts ranging in size from 250bp to 40kb sequenced with various Illumina technologies. At each round of scaffolding the minimum pairing threshold was chosen by exhaustive optimization of N50 scaffold length. Assembly gaps were closed with 2×150 Illumina HiSeq reads. The resulting assembly is comprised of 171.9 MB of contigs in 196.5 MB of scaffold.
The genome assembly was soft-masked to highlight consensus repeat family sequences predicted de novo by RepeatModeler. 37,384 RNAseq transcript assemblies were constructed from 118.7 million 2×150 paired-end Illumina RNAseq reads using PERTRAN (Shengqiang Shu, Joint Genome Institute in-house pipeline, http://jgi.doe.gov/wp-content/uploads/2013/11/CSHL-PERTRAN-Shengqiang-Shu-FINAL.pdf). Loci were determined by BLAT transcript assembly alignments, BLASTX alignments of proteins from arabi (Arabidopsis thaliana), rice, soybean, grape, maize, or Chlamydomonas reinhardtii genomes, and/or BLASTX alignments of UniProtKB/Swiss-Prot to the B. stricta genome. Gene models were predicated by homology-based predictors, mainly FGENESH+ and GenomeScan.
The best scored predictions for each locus were selected using multiple positive factors including EST and protein support, and one negative factor: overlap with repeats. The selected gene predictions were improved by PASA. Improvement includes adding UTRs, splicing correction, and adding alternative transcripts. PASA-improved gene model proteins were subject to protein homology analysis to above mentioned proteomes to obtain Cscore and protein coverage. Cscore is a protein BLASTP score ratio to MBH (mutual best hit) BLASTP score and protein coverage is highest percentage of protein aligned to the best of homologs. PASA-improved transcripts were selected based on Cscore, protein coverage, EST coverage, and its CDS overlapping with repeats. The transcripts were selected if its Cscore is larger than or equal to 0.5 and protein coverage larger than or equal to 0.5, or it has EST coverage, but its CDS overlapping with repeats is less than 20%. For gene models whose CDS overlaps with repeats for more that 20%, its Cscore must be at least 0.9 and homology coverage at least 70% to be selected. The selected gene models were subject to Pfam analysis and gene models whose protein is more than 30% in Pfam TE domains were removed.
There are 1,591 annotated genes in the inversion region (below), of which 408 are found in the QTL regions within the inversion.
Whole Genome Profiling (WGP)
A Hind III BAC library of B. stricta (Bs_LBa) was built from fresh leaf tissues from the LTM reference genotype18. The library contains 18,432 clones, arrayed into 48 – 384 well plates, with an average insert size of 150 Kb and an estimated genome coverage of 11X. The library or clones of interest from it are publicly available at the Arizona Genomics Institute (AGI) web page (http://www.genome.arizona.edu/orders/).
A Whole Genome Profiling (WGP) physical map of B. stricta was built following the protocol described by van Oeveren et al.32. Briefly, BAC library clone plates were arrayed into two and three dimensional pools, BAC DNA was extracted from pooled plates, BAC DNA was digested with EcoRI/MseI and ligated to their respective adapters (P5-EcoRI-tag barcode and P7-MseI), followed by PCR amplification and sequencing with an Illumina HiSeq 2500.
Resulting sequences were deconvoluted with KeyGene® proprietary scripts (licensed to AGI), to generate the tag file (Dryad Data Archive), which were assembled with FPC v9.433. Four test assemblies were performed with a fixed tolerance value = 0 and cutoff values 1e−25, 1e−20, 1e−15 and 1e−10, to choose the one producing best results, based on the number of contigs and number of clones included in those contigs.
Questionable clones were eliminated from the best project (1e−15) and the contigs were merged at a cutoff value of 1e−09. Singletons (clones that do not assemble with these parameters), were added at reduced stringency (1e−09). This new project was subject to manually editing, after linking the tags, BAC end and genome scaffolds (v 1.0) sequences to the physical map34. Using the SyMAP package35, the final edited map was aligned to pseudomolecules of B. stricta and Capsella rubella36. Synteny with A. lyrata was visualized with MUMmer37.
The integration of the WGP physical map with the genetic map and sequence scaffolds (via the WGP tags and BAC end sequencing) showed high concordance among these three genomic resources. WGP enabled regions with low recombination in the linkage map to be ordered and oriented based on their correspondence with the physical map contigs. In addition, we were able to integrate some scaffolds that were not linked in the genetic map, providing more robust pseudomolecules.
Variant discovery
We aligned each genotype read to the Boechera stricta LTM hardmasked reference (B. strict_278_ hardmasked) with BWA38. We used GATK39 for base quality recalibration, indel realignment, simultaneous SNP and INDEL discovery via HaplotypeCaller and joint genotyping using the default hard filtering parameters as prescribed by GATK Best Practices recommendations39.
Ordering and orienting scaffolds into linkage groups
We created a genetic map based on 159 sixth-generation recombinant inbred lines (RILs) bred from two parent individuals: LTM (the reference individual) and SAD12. We (re-)sequenced LTM to a genomic coverage of ~400X using paired-end Illumina sequencing. These reads were aligned to the reference sequence using bwa mem40 and samtools mpileup41 to visualize the alignment. We conservatively verified 105,074,494 positions as homozygous by requiring unambiguous alignment of a single base type with a depth ranging from 160–600. Next, we performed a similar analysis on a set of paired-end Illumina reads from SAD12 (depth ~170X) and compiled a catalog of 442,637 discriminatory positions throughout the genome. These markers were homozygous for different bases in LTM and SAD12, allowing identification of ancestry within the RILs. These results imply nucleotide divergence equals 0.000425 between these two accessions.
We initially aligned sequence from 199 barcoded RILs sequenced to modest (~a few X) coverage to the reference sequence and genotyped these at all sites possible within the discriminatory catalog. During this process we eliminated a number of contaminated RILs, RILs with too little sequence for reliable genotyping, and RILs that appeared to be heterozygous over most of the genome, suggesting that the source DNA had not originated from a single RIL. The scaffolds in the assembly with sizes ≥ 20 kb were binned into 20kb blocks and each block with at least 3 genotypeable sites were genotyped as either LTM, SAD12, or heterozygous, based on the consensus of genotypeable sites. A catalog of breakpoints was constructed, as a list of scaffolds and bins where one or more RILs changed genotype. From this data we identified 9 redundant RILs, after the removal of which we were ultimately left with 159 RILs used in the analyses. Also, we found two misjoins in the original assembly, which identified themselves as being apparently unlinked neighboring bins. These were Scaffold25219, between bin 134 and 137 (removed bins 135,136 and renamed bins 137–139 to Scaffold100000 bins 0–2) and Scaffold19424 between bins 344 and 345 (renamed Scaffold19424 bins 345–391 to Scaffold200000 bins 0–46). On the final data set, 2,664 crossovers were located (16.7 per RIL), and 2.4% of all blocks were genotyped as heterozygous (an average of 3.12% is expected for F6 RILs, with a large variance).
Next, we calculated a matrix of recombination fractions, r, between each pair of bins, defined as the fraction of RILs for which the bins differ in genotype. Bins within each pair with r < 0.15 were clustered by a single-linkage approach. (Briefly, all pairs of bins were compared, and pairs with r < 0.15 were assigned to the same cluster. Two clusters were merged if any pair of members had r < 0.15.) This algorithm resulted in seven distinct linkage groups. The bins in each linkage group were used as markers for input into MSTmap42. Finally, individual bins within assembled scaffolds were re-arranged into the order on which they occur on the scaffolds, and in the orientation, if known, consistent with the genetic map. This step is necessary as the size of the 20 kb bins are often smaller that the map resolution, resulting in the bins appearing in random order on the map. During this step, the cumulative physical length of the map (in bases) was also inserted into the map. In addition, occasional unmapped bins, the position of which could be inferred from the context of adjacent mapped bins, were inserted at the appropriate locations (Dryad Data Archive). Finally, from the matrix of markers and genotypes, we inferred the locations of recombination events near and within the inversion (Supplementary Fig. 1).
Identification of centromere positions in the Boechera stricta reference genome
To identify the centromere position for each B. stricta linkage group, we downloaded sequences of 16 A. thaliana centromeric BAC clones and looked for their homologues in the B. stricta reference genome using BLAST. These 16 clones were mapped on pericentromeric regions of B. stricta by chromosome painting43, thus positions of their homologues on B. stricta linkage group indicate margins of centromeres. The lengths of predicted centromere regions are 2 – 3 Mb for linkage groups 1, 2, 5, 6 and 7, and 6 – 7 Mb for linkage groups 3 and 4. As expected, all of these predicted centromere regions show low recombination rates, except parts of these regions on linkage groups 3 and 4 (Supplementary Fig. 4 and Supplementary Table 6).
Identification of a paracentric inversion by comparative chromosome painting
Painting probes
Previous F2 genetic mapping in the LTM × SAD12 F2 population found an extensive block of recombination suppression on LG1 (Bs1). Based on the published karyotype structure of B. stricta SAD1243 we designed BAC-based painting probes to verify the position and orientation of ancestral genomic blocks on the seven chromosomes of B. stricta LTM by comparative chromosome painting (CCP). A. thaliana BAC contigs used as painting probes are listed in Mandáková and Lysak44. Chromosome-specific BAC contigs were arranged and differentially labeled following the organization of genomic blocks within the reconstructed karyotype of B. stricta43.
Whereas chromosomes Bs2-Bs7 have the identical structure, chromosome Bs1 differs between the two B. stricta accessions. In LTM, the upper arm of Bs1 was restructured by a paracentric inversion of approximately 8.4 Mb (~4% of the genome). In order to more precisely map the two inversion breakpoints to approximately 100 kb (approximate size of Arabidopsis BAC clones) we carried out BAC FISH experiments on pachytene chromosomes of the accessions SAD12 and LTM. The following Arabidopsis BACs were used for mapping of A1 breakpoint: F21M11 (AC003027), F20D22 (AC002411), F19P19 (AC000104), T1G11 (AC002376), F13M7 (AC004809), T7A14 (AC005322), T25N20 (AC005106), F3F20 (AC007153), T20M3 (AC009999), and T21E18 (AC024174). The A1 breakpoint was mapped between BACs F19P19 (1,125,000 – 1,230,000 bp) and T1G11 (1,233,000 – 1,333,000 bp). The following BACs were used to narrow down the C1 breakpoint: F27J15 (AC016041), F27K7 (AC084414), T27P22 (GenBank Accession missing), F11I4 (AC073555), F9P7 (AC074308), T1N15 (AC020889), F11A17 (AC007932), F21D18 (AC023673), T2J15 (AC051631), and T6B12 (AC079679); the C1 breakpoint was mapped between BACs: F9P7 (17,975,000 – 17,987,000 bp) and F11I4 (17,982,000 – 18,062,000 bp). The A. thaliana BAC clone T15P10 (AF167571) bearing 35S rRNA genes was used for chromosomal localization of nucleolar organizer regions (NORs). Clone pCT 4.2 [a 500-bp 5S rRNA repeat (M65137)] was used to identify 5S rDNA loci.
Chromosome preparations
Chromosome spreads were prepared according to the protocol of Lysak and Mandáková45 with minor modifications. Entire inflorescences were fixed in ethanol: acetic acid fixative (3:1) overnight and stored in 70% ethanol at 20°C until further use. Prior to chromosome spreading, closed flower buds with white (young) anthers were rinsed in distilled water and in 1 × citrate buffer (10 × citrate buffer: 40 ml of 100 mM citric acid and 60 ml of 100 mM trisodium citrate, pH 4.8) and digested by a pectolytic enzyme mixture (0.3% cellulase, cytohelicase, and pectolyase; all Sigma Aldrich) in 1 × citrate buffer at 37°C for 3 to 6 h, and then kept in citrate buffer until used. An individual flower bud was put on a microscope slide and dissected by needles in a drop of 1 × citrate buffer to form a fine suspension. Then 15 to 30 μl of 60% acetic acid was pipetted to the cell suspension, which was spread over the slide placed on a hot plate at 50°C for ca. 30 sec to 2 min. The spread chromosomes and nuclei were then fixed by pipetting 100 μl of ethanol:acetic acid fixative (3:1) around the suspension drop. The slide was tilted to remove the fixative, dried using a hair dryer and postfixed with 4% formaldehyde in distilled water for 10 min and left to air dry.
Nick translation
DNA probes were labeled by nick translation with biotind-UTP, digoxigenin-dUTP, or Cy3-dUTP as follows: 1 μg of DNA diluted in distilled water to 29 μl, 5 μl of nucleotide mixture (2 mM dATP, dCTP, and dGTP, 400 μM dTTP; all Roche), 5 μl of 10 × NT buffer (0.5 M Tris–HCl, pH 7.5, 50 mM MgCl2, and 0.05% bovine serum albumin), 4 μl of 1 mM custom-made ×-dUTP (in which × is biotin, digoxigenin, or Cy3), 5 μl of 0.1 M b-mercaptoethanol, 1 μl of DNase I (2000 U mg−1 diluted to 4 μg ml−1; Roche), and 1 μl of DNA polymerase I (10 U μl−1, Fermentas). The nick translation mixture was incubated at 15°C for 90 min (or longer) to obtain fragments of ~200–500 bp. The reaction was stopped by 1 μl of 0.5 M EDTA (pH 8.0) and by incubation at 65°C for 10 min. The labeled probes were stored at −20°C until use.
FISH and CCP
Prior to pipetting the probe to selected slides, the slides were treated with pepsin (0.1 mg ml−1; Sigma Aldrich) in 0.01 M HCl for 3 – 6 min, postfixed in 4% formaldehyde in 2 × SSC (20 × saline sodium citrate: 3 M sodium chloride, 300 mM trisodium citrate, pH 7.0) for 10 min, dehydrated in an ethanol series (70, 80, and 96%; 3 min each), and air dried. The labeled BAC clones (and rDNA probes) were pipetted together and the DNA precipitated by adding 0.1 volume of 3 M sodium acetate (pH 5.2) and 2.5 volume of ice-cold 96% ethanol, kept at −20°C for at least 30 min, and centrifuged at 13,000 g at 4°C for 30 min. The pellet was dried using a desiccator and resuspended in 20 μl of hybridization buffer (50% formamide and 10% dextran sulfate in 2 × SSC) per slide. The probe and chromosomes were denatured together on a hot plate at 80°C for 2 min and hybridization was carried out by placing the slides into a moist chamber at 37°C for 40 to 63 h. Post-hybridization washing was performed in 20% formamide in 2 × SSC at 42°C. Signal detection and amplification were as follows: biotin-dUTP was detected by avidin–Texas Red (1:1000; Vector Laboratories) and amplified by goat anti-avidin–biotin (1:200, Vector Laboratories) and avidin–Texas Red; digoxigenin-dUTP was detected by mouse anti-digoxigenin (1:250; Jackson Immuno-Research Laboratories) and goat anti-mouse Alexa Fluor 488 (1:200; Molecular Probes); Cy3-dUTP was observed directly. DNA was counterstained with 4′,6-diamidino-2-phenylindole (2 μg ml−1) in Vectashield (Vector Laboratories). The hybridization signals were analyzed with an Olympus BX-61 epifluorescence microscope equipped with fluorochrome-specific excitation and emission filters (AHF Analysentechnik), and Zeiss Axio-Cam charged-coupled device (CCD) camera. The monochromatic images were pseudo-colored and processed using the Adobe Photoshop CS5 software (Adobe Systems).
Mapping SNPs to pseudomolecules
In the genetic map, 208 scaffolds were ordered and oriented into 7 linkage groups (LGs). Misjoins were found in original assembly of Scaffold25219 and 19424, which were split into four scaffolds, two with the original names (Scaffold25219 and 19424) and two with new names Scafffold100000 and 200000. In Scafffold25219, the first 135 bins (bin: 0 – 134; position: 12,700,000) were found to be unlinked with the last three bins (bin: 137 – 139; position: 2,740,0012,797,113 bp), while position of bins 135–136 (position: 2,700,0001 – 2,740,000) between them could not be determined. Thus, the first 135 bins were retained in Scaffold25219 (a shorter one, position: 1 – 2,720,000), the last three bins were moved to a new Scaffold100000 (position 2,740,001 – 2,797,113 of Scaffold25219 was changed to position 1 – 57,113 of Scaffold100000), and bins 135 – 136 were excluded and not used in map. In Scaffold19424, the first 345 bins (bin: 0344; position: 1 – 6,900,000) were found to be unlinked to the remaining 47 bins (bin: 345 – 391; position: 6,900,001 – 7,828,947), thus bins 0 – 134 were kept in Scaffold19424 (position: 16,900,000) and the rest were moved into a new scaffold200000 (position 6,900,001 – 7,828,947 of Scaffold19424 was changed to position 1 – 928,947 on Scaffold200000).
Accordingly, SNP coordinates were extracted from the vcf file (scaffold name and position on scaffold) and examined whether they fell in a bin of the linkage map based on its scaffold position. If so, its position on the pseudomolecules (g′) was calculated by g′ = A − |g − c| where A is the end position of the bin that the SNP fell in, g is the SNP’s position on the scaffold, and c is the end coordinate of the bin on scaffold. For SNPs that did not fall in any bins but were located on regions 2,740,001 – 2,797,113 of Scaffold25219 or 6,900,001 – 7,828,947 of Scaffold19424, their positions on Scaffold100000 and Scaffold200000 were calculated by g = g1 – 2,740,000 or g = g1 – 6,900,000, respectively (g1 is the position on original scaffold and g is the position on new scaffold). After that, their positions on pseudo-molecules (g′) were calculated as described above (g′ = A − |g − c|).
Delimiting and genotyping the inversion
After chromosome painting narrowed down possible locations of the inversion breakpoints in A. thaliana, we blasted these Arabidopsis genes onto the B. stricta LTM genome and identified putative scaffolds containing the two breakpoints. To identify the exact breakpoints, we mapped paired-end Illumina reads from SAD12 onto the LTM scaffolds with BWA38, followed by de novo assembly with Velvet46 for read-pairs having one or both ends near the putative breakpoint region in the LTM genome.
Primers Inv-A, Inv-B and Inv-E were designed to amplify the inversion region using Primer 347. Primer Inv-D was used for Sanger sequencing the breakpoints in the two karyotypes for the confirmation of breakpoints. Locations of the breakpoints and related primers are shown in the Dryad Data Archive.
Thermocycling consisted of 94° C for 3 minutes, then 33 cycles of 94° C for 20 seconds, 55° C for 1 minute, 72° C for 30 seconds, followed by 72° C for 6 minutes. Amplified fragments were separated by electrophoresis on a 1% agarose gel stained with SYBR Safe (ThermoFisher Scientific, Waltham, MA). Fragments were ~300bp in accessions containing the inversion, and ~600bp accessions without the inversion.
Differences between subspecies
From our earlier study of ecological divergence between EAST and WEST subspecies16, we used least-square means (LSMEANS) from 24 genotypes (Table S1) to quantify differences in flowering time. In the greenhouse, EAST genotypes flower an average of 5.0 days before WEST genotypes.
Here we report on three EAST × WEST crosses, which all gave healthy advanced generation progeny in F2, F4, or F6 generations. More generally, intrinsic reproductive isolation might be present in some instances, especially if complicated by occasional polyploidy or apomixis48.
Effect of the inversion on NIL flowering time
In the LTM × SAD12 (MTWEST-inv × CO East-std)cross17), we generated two F7 NIL families from the F6 population to investigate the inversion effect. Two F6 heterogeneous inbred families (HIFs: 116A and 120A)49 heterozygous for the inversion and mostly homozygous elsewhere in the genome were self-fertilized, and 144 F7 plants from each parent were grown in a completely randomized design in the greenhouse. After three months, plants were vernalized in 4°C for six weeks, and flowering time was recorded. We also genotyped the inversion status in all individuals using inversion primers Inv-A, Inv-B, and Inv-C. We used fixed effects ANOVA to test Flowering Time = Family + Inversion + Family*Inversion, and verified compatibility with parametric statistical assumptions.
For families from this MTWEST-inv × COEast-std cross, we also analyzed lifetime fecundity in the field, using Dryad-archived data (http://dx.doi.org/10.5061/dryad.rp3pc) from the Lost Trail field site in the inversion zone20. We analyzed lifetime fecundity in an EAST × WEST recombinant inbred F6 population grown in nature for three years.
Effects of the inversion in a sympatric cross
The inversion might be favored if this rearrangement alters genes around the break points and gives it higher fitness over its ancestral standard haplotypes from the same population. To test this, we generated crosses between the reference accession LTM (inversion) and accession SDM, which has the standard haplotype. These genotypes are the same subspecies (WEST) and were collected from similar habitats ~1.7 km apart. Two crosses were generated, one with LTM as mother (CL9.1) and the other with SDM as mother (CL10.1). The F1 hybrids were self-fertilized, and 1,000 F2 plants were grown in the greenhouse. The inversion was genotyped in all individuals, and we measured 11 life history traits (Survival after vernalization [Binary], Flowering [Binary], Width at 4 weeks, Width at 10 weeks, Flowering Time [days after vernalization], Flowering Width, Flowering Height, Flowering Rosette Number, Flowering Leaf Number, Fruit Number, Lifetime Fitness, and Inversion Genotype) on 994 individuals.
To test the phenotypic effects of the inversion, we permuted the inversion genotypes within crosses 1,000 times, and performed MANOVA on the vector of traits. Because R only computes Type I SS for MANOVA, we obtained Type III SS by performing several MANOVAs, and reported the effect of each factor when added to the model last. We modeled MULTIVARIATE-TRAITS = CROSS + INVERSION + CROSS*INVERSION, with cross type (either CL9.1 or CL10.1 as maternal parent), three inversion genotypes (inversion, standard, or heterozygote), and their interaction as fixed-effect predictor variables. P-values were obtained by comparing the true Wilk’s Lambda statistic to those from 1,000 permutations (Supplementary Table 4). Because MANOVA showed that multivariate means differed between Bsi1 genotypes, the univariate traits were analyzed by ANOVA with a P = 0.05 significance threshold50.
Gene prediction
We obtained the 10kb region on each side of each breakpoint in the LTM reference genome (inversion) and created pseudomolecules corresponding to the corresponding regions around each breakpoint in the standard haplotype. All four sequences were submitted to the Augustus gene prediction algorithm (http://bioinf.uni-greifswald.de/augustus/), and we found no open reading frames spanning the breakpoints in either the inversion or standard haplotypes. Thus, the chromosomal inversion does not disrupt existing or create new open reading frames (Supplementary Fig. 7). Genes predicted by Augustus largely correspond to the transcriptome-assisted annotation in B. Stricta genome v1.2.
Gene expression
To investigate whether the inversion event changed the expression of flanking genes, we compared two nearby populations using F2 progeny from the LTM × SDM cross (representing inversion and standard haplotypes, respectively). Based on the inversion genotypes, we chose 11 F2 plants homozygous for the inversion and 12 homozygous for standard haplotypes, providing sample sizes well above recommendations based on power analysis51. All 23 plants were from cross CL9.1 where LTM is the mother. These F2 plants were self-fertilized for one generation, and two-month-old rosette leaves from Bsi1 homozygote F3 plants were used to compare the expression of seven genes flanking the two breakpoints and syntenic to the region in A. thaliana. We used Sigma Spectrum Plant Total RNA Kit to extract RNA and Thermo Scientific DyNAmo cDNA Synthesis Kit to synthesize cDNA. We used Thermo Scientific DyNAmo SYBR Green qPCR Kits for quantitative PCR (qPCR; primers in Dryad Data Archive). As in previous gene expression studies in B. stricta18, we used ACTIN2 (ACT2) as reference and calculated the expression as ΔCt = Ct ACT2 – Ct gene, where Ct ACT2 is the Ct value of ACT2 and Ct gene is the Ct value of each gene. Fold gene expression relative to ACT2 is calculated as 2ΔCt. Since 2ΔCt has a skewed distribution, we analyzed results from both 2ΔCt and ΔCt. For each gene separately, ANOVA was performed with inversion status as fixed effect (Supplementary Table 5). In addition, we examined statistical power with JMP and with simulations in R.
Dissecting the inversion region in a collinear cross
The EAST × WEST “Parker × Ruby” cross used for QTL mapping was developed from two parents in the East-West contact zone: one in Parker Meadow (RP105, Parker, EAST subspecies, 44°37′ N, 114°31′ W) and one at Ruby Creek (RP109, Ruby, WEST subspecies, 45°33′ N, 113°46′ W). Based on nucleotide similarity (Supplementary Figure 8a, left portion), the WEST-std parent (RP109) used in this cross is very similar to the most recent common ancestor that gave rise to the inversion and its closest WEST-std relatives (IN086 and IN087). The F1 hybrid was self-fertilized to produce F2 plants, and subsequent generations were propagated by self-fertilization and single-seed descent to create 153 independent genetic lines (families). In each line, multiple F4 progeny from the same F3 plant were used in a randomized complete block design totaling 1,714 individuals. The phenotypic LSMEANS were calculated in JMP 8 (SAS, Cary, NC, USA) to represent the genotypic value for their F3 parent.
Genotyping by Sequencing
We used genotyping by sequencing (GBS)52 to identify SNPs in this cross. In each family, DNA from each accession was extracted from 0.1g of young leaf tissue following a dark treatment of about three days. Tissue was stored at −80 c, flash-frozen in liquid nitrogen, and homogenized prior to genomic DNA extraction using Qiagen DNeasy Plant Mini Kits (Qiagen, Valencia, CA). DNA concentration was measured using a Qubit Fluorometer (Turner Biosystems, Invitrogen, Carlsbad, CA). (Qiagen DNeasy Plant Mini Kit) from at least ten pooled F4 individuals to represent the genotype of their F3 parent. This protocol uses an adaptor design which is compatible with TruSeq adaptors and indexes while allowing paired-end sequencing. The combination of 48 unique barcodes with four different TruSeq indexes allowed multiplexing of 192 samples (153 F3:F4 families, 19 inbred individuals of the Parker parent, and 20 inbred individuals of the Ruby parent). Sequencing used Illumina HiSeq-2000 or HiSeq-2500 at the Duke Genome Sequencing & Analysis Core Resource. From this population we obtained ~249 million reads with unambiguous barcodes (SRA accession SRP075905). Read pairs were assigned to genotypes and parents by custom Perl code, and low-quality bases in the end of reads were trimmed. SNPs were called using GATK Best Practices (above). In addition, these protocols were used to assay GBS SNPs (SRA accession SRP075997) in 122 genotypes across the inversion area.
Because the LTM × SAD12 cross does not recombine in the inversion on chromosome 1, we used the Parker × Ruby cross to infer the linkage map for bins within the inversion. The mean sequencing depth of GBS SNPs in the Parker and Ruby parents was ~20x, with standard deviation ~40x. We focused on 52,827 bi-allelic SNPs where the two parents have sequencing depth ≥ 6x and ≤ 100x (the mean depth plus two standard deviations), and are homozygous for different alleles. We binned the genome into 100kb windows, and for each individual we counted the number of reads from the two parents, and calculated the proportion of PAR-derived reads. For an individual, if the cumulative depth of all SNPs in a window is less than 20x, it was classified as missing, after which we removed windows with ≥ 30% missing data. We used a hard-genotype cutoff based on the portion of PAR reads: PAR proportion ≤ 0.2 was defined as homozygous Ruby, PAR proportion ≥ 0.7 as homozygous PAR, and the remaining windows were classified as heterozygous. These cutoffs give the correct hard-genotype proportions for an F3 population (about 37.5% of either homozygote and 25% heterozygous genotypes). After determining the ‘hard genotype calls’, we calculated the proportion of the three genotypes in each window. To remove windows with excessive proportions of heterozygotes or either homozygote, we excluded windows where the proportion of the three genotypes are beyond the upper or lower 5% of their respective whole-genome distribution. Given this is a cross in the F3 generation, which has only experienced two generations of recombinations, we regarded it as extremely unlikely to have two recombination events within 1 Mb. Therefore, within each individual, if two recombination events were identified within 1 Mb, we changed the genotype calls of markers between two breakpoints to missing. The linkage map was construction with MSTmap42. With only three windows (markers) dropped out, MSTmap constructed seven linkage groups, with scaffold-chromosome assignment consistent with the LTM × SAD12 cross. The final linkage map was refined in multiple steps: we first removed markers that are > 5cM away from both flanking markers and re-constructed the map. In the new map, we imputed missing marker genotype if the up-stream and down-stream markers with data have the same genotype call and are not both > 10 cM away from the missing position. Another linkage map was constructed, and the imputation process was repeated since the new map had slightly changed the order of some markers, making some missing genotypes now imputable. The final linkage map was then constructed with 1010 markers. Thus, we used the Parker × Ruby cross to infer the linkage map for bins within the inversion and we inferred the position of polymorphisms within the inversion from their positions in each ordered and oriented contig.
This collinear EAST × WEST “Parker × Ruby” greenhouse experiment consists of 12 blocks, each with one F4 plant from each of the 153 lines and multiple Parker and Ruby individuals (N = 1,714). Seeds were stratified in 4°C for four weeks and planted in “Cone-tainers” (Ray Leach SC10, Stuewe & Sons Inc., Tangent, OR, USA), with soil composition and greenhouse conditions as previously described16. When rosettes were 11-weeks old, all leaves from three blocks of plants were harvested for rosette- and leaf-morphology measurements as described16. At 12 weeks of age, the remaining nine blocks were vernalized in 4°C for 6 weeks, then returned to the same greenhouse conditions for phenology measurements. All traits were measured as previous described16, except: 1) no physiological traits were measured; 2) leaf width/length ratio was used instead of leaf shape morphometrics because the leaf-shape landscape points were highly correlated16 (Supplementary Table 7).
For QTL mapping, all individual-level measurements were transformed to family-level LSMEANS in JMP (N = 153 families). Due to the skewed distribution of most phenotypes, all traits (except binomial traits or plant stages) were log-transformed at the individual level. For greenhouse measurements, BLOCK and GENOTYPE were considered as random effects. We used R53 libraries qvalue and MASS for MANOVA, False Discovery Rate, and Discriminant Function Analysis. For each marker across the inversion interval and adjacent contigs we fit MANOVA model: MULTIVARIATE-PHENOLOGY-TRAITS = BLOCK + SINGLE-MARKER-GENOTYPE. At each marker we computed Wilkes Lambda, permuted the vector of phenotypes with respect to marker genotypes 1,000 times, and finally corrected for false discovery rates across the inversion region54.
Discriminant Function Analysis (DFA) was performed in R for a marker at the peak of each multivariate QTL (bins Scaffold-25219_50000, Scaffold-13671_1150000, and Scaffold26675_450000), defining the direction of trait variation along the axis of greatest differentiation among the multivariate means of these marker genotypes. Subsequently, at these three markers we used the eigenvector for the first DFA axis to compute the phenotypic projection of each genotype on the trait axis identified by DFA. These new trait values were used for univariate QTL mapping across the inversion region, with correction for false discovery rates across the inversion region.
Finally, we annotated SNPs by using snpEff v.4.255, and found 17 genes that are orthologues of flowering-time genes of Arabidopsis thaliana56 within the inversion interval. See candidate genes in Supplementary Table 8 and annotated SNPs in Dryad Data Archive.
Population genetics
SNP filtering
SNPs called by GATK were filtered in each of three groups: sets consisting of 35 inversion genotypes (GINV) and 87 standard genotypes (GSTD) from the inversion zone, and a “Reference Population” set of 83 genotypes from across the species range, outside the inversion zone (GRP). Next, data from these single groups were pooled together into two datasets, the complete data set (GINV+GSTD+GRP) and the inversion zone genotypes (GINV+GSTD). In each dataset, genotypes supported by less than 2 reads were assigned as missing, and SNPs were discarded if they met the following criteria in any of the groups: 1) detected in fewer than 50% of individuals; 2) mean depth more than 20; 3) more than one variant allele was observed; 4) sites with a proportion of heterozygous genotypes more than 15% (B. stricta is predominantly inbred17, hence high heterozygosity may indicate paralogous loci); 5) reference or variant alleles are indels. After filtering, 75,737 and 43,722 SNPs were retained for the complete set of genotypes GINV+GSTD+GRP and for the inversion zone set of genotypes GINV+GSTD, respectively. The following analyses used these SNPs.
Ancestral allelic state
Several population genetic statistics require information on the ancestral state of segregating variants. Therefore, we sequenced a Boechera holboellii (= B. retrofracta; reference genotype “Panther”, location N45 18.198, W114 22.599) individual with mean depth of ca. 400× (JGI Project ID: 1051698). Short reads were mapped to the B. stricta reference genome using BWA, genotypes were called using GATK with default settings, and sites were filtered out if they had depth less than 20, higher than 800, or if they were heterozygous. A Python script called ancestral alleles as the shared allele between B. holboellii and B. stricta. In total, we obtained information for ancestral states for 63,182 (83.4%) of 75,737 and 35,100 (80.3%) of 43,722 SNPs for GINV+GSTD+GRP and GINV+GSTD, respectively.
Windows
We partitioned the genome into 300kb windows. The sequence coverage of each window was calculated by counting the number of available sites (both variant and non-variant sites obtained from GenotypeGVCF by using –allSite) per window passing the quality filters, i.e., detected in at least 50% individuals, mean depth no more than 20, and proportion of heterozygous genotypes no more than 15% in each of the three groups (GINV, GSTD, and GRP). Windows were distributed on a per-scaffold basis beginning at position 1 of a scaffold and were then oriented along pseudo-molecules according to the linkage map. After excluding windows with sequence coverage less than 5 kb, 2,801 windows were retained for downstream analyses.
Phylogenetic relationship and population structure analyses
Python scripts were used to generate alignments from genotypes in the vcf file, with missing and heterozygous loci coded as “N”. With genome-wide SNPs, we verified EAST-WEST population structure using principal component analysis (PCA) with EIGENSOFT v6.057.
To investigate the origin of the Bsi1-inv haplotype we used data from 901 SNPs in the inversion region of the genome to examine relationships among 35 Bsi1-inv and 83 Bsi1-std haplotypes. (Four admixed Bsi1-std genotypes were excluded.) First, we constructed neighbor-joining (NJ) trees using MEGA v6.06 with 1,000 bootstrap steps. Second, maximum-likelihood trees were constructed using RAxML version 8.0.058. One thousand ML trees were generated to find the best-scoring ML tree, and topological robustness was investigated using 1,000 nonparametric bootstrap replicates. NJ and ML trees were displayed by Figtree (http://tree.bio.ed.ac.uk/software/figtree/).
Site frequency spectrum and summary statistics
To control for error rates and variable coverage of short-read sequencing, we used a probabilistic method implemented in the software package ANGSD v0.91159 to estimate the site frequency spectrum (SFS) and related population genetic statistics. A number of filtering steps were performed: 1) removed reads with a minimal mapping quality of 30 and bases with a minimal quality score of 30 (-minMapQ and -minQ); 2) removed sites with information from less than 50% individuals (-minInd); 3) removed sites with P-value higher than 1e-6 (-snp_pval); 4) assigned genotypes as missing if the depth is less than two for an individual; 5) only used genotypes with a posterior probability higher than 0.95; 6) removed sites that did not pass previous filtering criteria (above). Because B. stricta is predominantly inbreeding17, we estimated inbreeding coefficients for each individual in ngsTools60 and incorporated them into the calculation of SFS in ANGSD. Using genotype likelihoods based on the GATK genotyping model61, we estimated folded and unfolded SFS and derived a set of population genetic summary statistics in 300kb windows. For the GINV and GSTD groups, we estimated Tajima’s D62 based on both folded and unfolded SFS, and calculated Fay & Wu’s H29, Fu and Li’s D and F63 based on unfolded SFS. The inferred ancestral allelic state based on short reads from B. retrofracta genotype “Panther”, aligned to the B. stricta reference genome was used to estimate the unfolded SFS. We compared population genetic statistics in the inversion region, on other chromosomes (LG2 – LG7), and in Block D64, a chromosome region with unusually high polymorphism in related species, perhaps due to clusters of NB-LRR genes65.
FST, dXY, dA and π
Genetic differentiation (FST)66 between GINV and GSTD groups was estimated using VCFtools v0.1.1267, and we used Python scripts to estimate nucleotide diversity (π)68 within each group, pairwise nucleotide divergence (dXY)69 and net pairwise nucleotide divergence (dA)69 between groups. All parameters were calculated on a per site basis, and then averaged to obtain window-based estimates (300 kb windows). The window-based FST was calculated by averaging per site FST across all variable loci, and the window-based π, dXY and dA were estimated by averaging all sites (both variable and monomorphic) passing the initial quality filters for each window (above).
Linkage disequilibrium
The level of linkage disequilibrium (LD) between the inversion and all SNPs on chromosome 1 (including those in the inverted region) was estimated as the mean squared correlation (r2) for 118 samples from inversion zone accessions (35 Bsi1-inv and 83 Bsi1-std) using plink v1.9070. (Four admixed Bsi1-std samples were excluded, see above.) In addition, to compare LD between the inversion and flanking SNPs, we identified ten randomly chosen comparator SNPs from LG2 – LG7, having similar derived allele frequencies (25 – 35%, close to the 29.7% frequency of Bsi1-inv in the inversion zone), and then calculated LD between them and flanking SNPs along the chromosomes. For LG1 (Fig 5), strong LD (r2 > 0.4) between the inversion and SNPs within and outside the inverted region extended to the end of the chromosome (more than 10 Mb). In contrast, LD with the ten comparator SNPs from LG2 – LG7 declined quickly to background levels (Supplementary Fig. 8b).
Age of the inversion
To estimate age of the inversion, we phased the 35 Bsi1-inv genotypes using Beagle v4.171 with default settings, and estimated pairwise genetic distances among haplotypes on the inverted genome region. After that, the divergence time between haplotypes was calculated by T = d/2μ, where d is pairwise genetic distance and μ is the mutation rate. We used a mutation rate of 7 ×10−9 per site per generation in Arabidopsis thaliana24) with a generation time of two years in B. stricta. To avoid biases due to missing data, we only considered 1,675 haplotype pairs with less than 50% missing sites in the inversion region. We approximated the lower bound for age of the inversion based on the mean pairwise distances72, and the upper bound from the maximum of all pairwise distances among INV genotypes.
Divergence time between QTL alleles in the collinear cross
Using the same mutation rate and generation time, we estimated DA = Dxy = 2μT, and solved for T between these two alleles in 200kb non-overlapping windows69. This window size was chosen to ensure sufficient polymorphic sites within each interval.
Frequency distribution of derived alleles
We tested whether Bsi1-inv has unusually high frequency among derived SNP alleles in the inversion zone population. For this analysis, we excluded one admixed, genetically divergent inversion individual (IN019, right half of Supplementary Fig. 9c), and analyzed Bsi1-inv and closely related Bsi1-std genotypes from the inversion zone (left half of Supplementary Fig. 9c, PC1 < 0.0). These genotypes comprise 54 samples (34 Bsi1-inv and 20 Bsi1-std), with 2,416 SNPs that, like the inversion, segregate among these genotypes and are monomorphic in the rest of our collection.
Supplementary Material
Acknowledgments
CRL was supported by NSF Doctoral Dissertation Improvement Grant 1110445, EMBO Long-Term Fellowship, and 105-2311-B-002-040-MY2 from Ministry of Science and Technology, Taiwan. BW was supported by the Swedish Research Council (VR). RAW was supported by the Bud Antle Endowed Chair of Excellence in Agriculture and Life Sciences, and the AXA Chair in Genome Biology and Evolutionary Genomics. MAL and TM were supported by grant P501/10/1014 from the Czech Science Foundation. TMO was supported by grant R01 GM086496 from the National Institutes of Health. Work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We thank Lauren Carley, Kathleen Donohue, Dan Hartl, Mohamed Noor, Sally Otto, Mark Rausher, Maggie Wagner, and John Willis for helpful discussion and comments.
Footnotes
AUTHOR CONTRIBUTIONS
CRL and TMO conceived these efforts. CRL, TMO, YY, DK, JZ, and JLG designed the study. TM and ML performed chromosome painting. CRL, KVSKP, NP, HNH, DK, YY, JT, WG, JZ, and JJ worked on molecular biology and sequencing. SF, UH, JWJ, JS, DSR, RAW, and KB; and JLG, DK, YY, JZ, JT, WG, and RAW planned and analyzed genomic and physical mapping experiments, respectively. CRL, BW, JM, UH, JLG, and TMO performed bioinformatic and evolutionary analyses. KG, CRL, and NP performed experiments with plants. CRL, BW, JM, MES, and TMO drafted the manuscript. All authors read, revised, and approved the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
DATA AVAILABILITY
The assembly and annotation of the Boechera stricta genome are available at https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Bstricta and have been deposited under GenBank accession number MLHT00000000. The BAC end sequences have been deposited under GenBank dbGSS accession numbers KS412618 – KS448200. The short reads of B. stricta (LTM, SAD12 and recombinant inbred lines) have been deposited under GenBank accession numbers SRP078672, SRP048481 and SRP079414, respectively. The short reads from the RNA-seq transcriptome have been deposited under GenBank accession numbers SRX1971488. The short reads of outgroup Boechera holboellii (= B. retrofracta) have been deposited under GenBank accession number SRP078889, and the short reads of genotyping-by-sequencing data of B. stricta have been deposited under GenBank accession numbers SRP075905 and SRP075997. All SNPs used in population genetic analyses have been deposited into dbSNP under accession numbers ss2136554982– ss2136641742. All other data are available in the Dryad Data Archive XXX.
CODE AVAILABILITY
All code is available in in the Dryad Data Archive.
Contributor Information
Cheng-Ruei Lee, Department of Biology, Box 90338, Duke University, Durham, NC, 27708, USA and Institute of Ecology and Evolutionary Biology & Institute of Plant Biology, National Taiwan University, Taipei 10617, Taiwan ROC.
Baosheng Wang, Department of Biology, Box 90338, Duke University, Durham, NC, 27708, USA, and Department of Plant Ecology and Genetics, Uppsala University, Norbyvägen 18D, SE-752 36 Uppsala, Sweden.
Julius Mojica, Department of Biology, Box 90338, Duke University, Durham, NC, 27708, USA.
Terezie Mandáková, Plant Cytogenomics Group, CEITEC – Central European Institute of Technology, Masaryk University, Kamenice 5, Brno CZ-62500, Czech Republic.
Kasavajhala V. S. K. Prasad, Department of Biology, Colorado State University, Fort Collins, Colorado 80523, USA
Jose Luis Goicoechea, Arizona Genomics Institute and BIO5 Institute, School of Plant Sciences, University of Arizona, Tucson, AZ, 85721, USA.
Nadeesha Perera, Department of Biology, Box 90338, Duke University, Durham, NC, 27708, USA.
Uffe Hellsten, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA.
Hope N. Hundley, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA
Jenifer Johnson, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA.
Jane Grimwood, HudsonAlpha Institute for Biotechnology, Huntsville, Alabama, USA.
Kerrie Barry, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA.
Stephen Fairclough, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA.
Jerry W. Jenkins, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA
Yeisoo Yu, Phyzen Genomics Institute, Phyzen Inc., Seoul, 151-836, South Korea.
Dave Kudrna, Arizona Genomics Institute and BIO5 Institute, School of Plant Sciences, University of Arizona, Tucson, AZ 85721, USA.
Jianwei Zhang, Arizona Genomics Institute and BIO5 Institute, School of Plant Sciences, University of Arizona, Tucson, AZ 85721, USA.
Jayson Talag, Arizona Genomics Institute and BIO5 Institute, School of Plant Sciences, University of Arizona, Tucson, AZ 85721, USA.
Wolfgang Golser, Arizona Genomics Institute and BIO5 Institute, School of Plant Sciences, University of Arizona, Tucson, AZ 85721, USA.
Katherine Ghattas, Department of Biology, Box 90338, Duke University, Durham, NC, 27708, USA.
M. Eric Schranz, Biosystematics Group, Wageningen University & Research Center, Droevendaalsesteeg 1, 6708PB, Wageningen, The Netherlands.
Rod Wing, Arizona Genomics Institute and BIO5 Institute, School of Plant Sciences, University of Arizona, Tucson, AZ, 85721, USA.
Martin A. Lysak, Plant Cytogenomics Group, CEITEC – Central European Institute of Technology, Masaryk University, Kamenice 5, Brno CZ-62500, Czech Republic
Jeremy Schmutz, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA.
Daniel S. Rokhsar, Department of Energy Joint Genome Institute, Walnut Creek, California, 94598, USA
Thomas Mitchell-Olds, Department of Biology, Box 90338, Duke University, Durham, NC, 27708, USA.
References
- 1.Kirkpatrick M. How and why chromosome inversions evolve. PLoS Biol. 2010;8:e1000501. doi: 10.1371/journal.pbio.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huber B, et al. Conservatism and novelty in the genetic architecture of adaptation in Heliconius butterflies. Heredity. 2015;114:515–524. doi: 10.1038/hdy.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowry DB, Willis JHA. Widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biology. 2010;8:e1000500. doi: 10.1371/journal.pbio.1000500. doi:1000510.1001371/journal.pbio.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corbett-Detig RB, Hartl DL. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet. 2012;8:e1003056. doi: 10.1371/journal.pgen.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirkpatrick M, Kern A. Where’s the money? Inversions, genes, and the hunt for genomic targets of selection. Genetics. 2012;190:1153–1155. doi: 10.1534/genetics.112.139899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guillen Y, Ruiz A. Gene alterations at Drosophila inversion breakpoints provide prima facie evidence for natural selection as an explanation for rapid chromosomal evolution. BMC Genomics. 2012;13 doi: 10.1186/1471-2164-13-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith AC, et al. Maternal gametic transmission of translocations or inversions of human chromosome 11p15.5 results in regional DNA hypermethylation and downregulation of CDKN1C expression. Genomics. 2012;99:25–35. doi: 10.1016/j.ygeno.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennington WJ, Partridge L, Hoffmann AA. Patterns of diversity and linkage disequilibrium within the cosmopolitan inversion In(3R)Payne in Drosophila melanogaster are indicative of coadaptation. Genetics. 2006;172:1655–1663. doi: 10.1534/genetics.105.053173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173:419–434. doi: 10.1534/genetics.105.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieseberg LH. Chromosomal rearrangements and speciation. Trends in Ecology & Evolution. 2001;16:351–358. doi: 10.1016/s0169-5347(01)02187-5. [DOI] [PubMed] [Google Scholar]
- 11.Noor MAF, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12084–12088. doi: 10.1073/pnas.221274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarro A, Barton NH. Accumulating postzygotic isolation genes in parapatry: A new twist on chromosomal speciation. Evolution. 2003;57:447–459. doi: 10.1111/j.0014-3820.2003.tb01537.x. [DOI] [PubMed] [Google Scholar]
- 13.Lohse K, Clarke M, Ritchie M, Etges W. Genome-wide tests for introgression between cactophilic Drosophila implicate a role of inversions during speciation. Evolution. 2015 doi: 10.1111/evo.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang CH, et al. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol Biol Evol. 2016;33:394–412. doi: 10.1093/molbev/msv226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CR, Mitchell-Olds T. Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Molec Ecol. 2011;20:4631–4642. doi: 10.1111/j.1365-294X.2011.05310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CR, Mitchell-Olds T. Complex trait divergence contributes to environmental niche differentiation in ecological speciation of Boechera stricta. Molec Ecol. 2013;22:2204–2217. doi: 10.1111/mec.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson J, Lee CR, Mitchell-Olds T. Life history QTLs and natural selection on flowering time in Boechera stricta, a perennial relative of Arabidopsis. Evolution. 2010;65:771–787. doi: 10.1111/j.1558-5646.2010.01175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prasad K, et al. A gain-of-function polymorphism controlling complex traits and fitness in nature. Science. 2012;337:1081–1084. doi: 10.1126/science.1221636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heo JY, et al. Identification of quantitative trait loci and a candidate locus for freezing tolerance in controlled and outdoor environments in the overwintering crucifer Boechera stricta. Plant, Cell & Environment. 2014;37:2459–2469. doi: 10.1111/pce.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson JT, Lee CR, Mitchell-Olds T. Strong selection genome-wide enhances fitness tradeoffs across environments and episodes of selection. Evolution. 2014;68:16–31. doi: 10.1111/evo.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schranz ME, Windsor AJ, Song B-H, Lawton-Rauh A, Mitchell-Olds T. Comparative genetic mapping in Boechera stricta, a close relative of Arabidopsis. Plant Physiol. 2007;144:286–298. doi: 10.1104/pp.107.096685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehringer PJ, Arno SF, Petersen KL. Postglacial history of Lost Trail Pass Bog, Bitterroot Mountains, Montana. Arctic and Alpine Research. 1977;9:345–368. [Google Scholar]
- 23.Mumma SA, Whitlock C, Pierce K. A 28,000 year history of vegetation and climate from Lower Red Rock Lake, Centennial Valley, Southwestern Montana, USA. Palaeogeography Palaeoclimatology Palaeoecology. 2012;326:30–41. doi: 10.1016/j.palaeo.2012.01.036. [DOI] [Google Scholar]
- 24.Ossowski S, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson J, Lee CR, Rushworth C, Colautti R, Mitchell-Olds T. Genetic tradeoffs and conditional neutrality contribute to local adaptation. Molec Ecol. 2013;22:699–708. doi: 10.1111/j.1365-294X.2012.05522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson JT, Inouye DW, McKinney AM, Colautti RI, Mitchell-Olds T. Phenotypic plasticity and adaptive evolution contribute to advancing flowering phenology in response to climate change. Proceedings Biological Sciences/The Royal Society. 2012;279:3843–3852. doi: 10.1098/rspb.2012.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smadja CM, Butlin RK. A framework for comparing processes of speciation in the presence of gene flow. Molecular Ecology. 2011;20:5123–5140. doi: 10.1111/j.1365-294X.2011.05350.x. [DOI] [PubMed] [Google Scholar]
- 28.Guerrero RF, Rousset F, Kirkpatrick M. Coalescent patterns for chromosomal inversions in divergent populations. Philosophical Transactions of the Royal Society B-Biological Sciences. 2012;367:430–438. doi: 10.1098/rstb.2011.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naseeb S, et al. Widespread impact of chromosomal inversions on gene expression uncovers robustness via phenotypic buffering. Molecular Biology and Evolution. 2016;33:1679–1696. doi: 10.1093/molbev/msw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapman JA, et al. Meraculous: De Novo genome assembly with short paired-end reads. PLoS ONE. 2011;6:e23501. doi: 10.1371/journal.pone.0023501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Oeveren J, et al. Sequence-based physical mapping of complex genomes by whole genome profiling. Genome Research. 2011;21:618–625. doi: 10.1101/gr.112094.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soderlund C, Humphray S, Dunham A, French L. Contigs Built with Fingerprints, Markers, and FPC V4.7. Genome Research. 2000;10:1772–1787. doi: 10.1101/gr.GR-1375R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson W, Soderlund C. Integrating sequence with FPC fingerprint maps. Nucleic Acids Research. 2009;37:e36. doi: 10.1093/nar/gkp034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soderlund C, Bomhoff M, Nelson WM. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Research. 2011;39:e68. doi: 10.1093/nar/gkr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slotte T, et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat Genet. 2013;45:831–835. doi: 10.1038/ng.2669. [DOI] [PubMed] [Google Scholar]
- 37.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biology. 2004;5 doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013:1303.3997. [Google Scholar]
- 41.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Y, Bhat PR, Close TJ, Lonardi S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. 2008;4:e1000212. doi: 10.1371/journal.pgen.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandáková T, Schranz ME, Sharbel TF, de Jong H, Lysak MA. Karyotype evolution in apomictic Boechera and the origin of the aberrant chromosomes. The Plant Journal. 2015;82:785–793. doi: 10.1111/tpj.12849. [DOI] [PubMed] [Google Scholar]
- 44.Mandáková T, Lysak MA. Chromosomal phylogeny and karyotype evolution in x=7 crucifer species (Brassicaceae) Plant Cell. 2008;20:2559–2570. doi: 10.1105/tpc.108.062166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lysak MA, Mandáková T. Analysis of plant meiotic chromosomes by chromosome painting. Humana Press; 2013. [DOI] [PubMed] [Google Scholar]
- 46.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 48.Rushworth CA, Song BH, Lee CR, Mitchell-Olds T. Boechera, a model system for ecological genomics. Molecular Ecology. 2011;20:4843–4857. doi: 10.1111/j.1365-294X.2011.05340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuinstra RM, Ejeta G, Goldsbrough BP. Heterogeneous inbred family (HIF) analysis: a method for developing near-isogenic lines that differ at quantitative trait loci. Theoretical and Applied Genetics. 1997;95:1005–1011. doi: 10.1007/s001220050654. [DOI] [Google Scholar]
- 50.Scheiner SM. In: Design and analysis of ecological experiments. Scheiner SM, Gurevitch J, editors. Chapman and Hall; 2001. pp. 99–115. [Google Scholar]
- 51.Schurch N, et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA (New York) 2016;22:839–851. doi: 10.1261/rna.053959.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cande J, Andolfatto P, Prud’homme B, Stern DL, Gompel N. Evolution of multiple additive loci caused divergence between Drosophila yakuba and D. santomea in wing rowing during male courtship. PLoS ONE. 2012;7:e43888. doi: 10.1371/journal.pone.0043888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.R_Core_Team. R Foundation for Statistical Computing. Vienna, Austria: 2013. [Google Scholar]
- 54.Benjamini Y, Yekutieli D. Quantitative trait loci analysis using the false discovery rate. Genetics. 2005;171:783–790. doi: 10.1534/genetics.104.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bouche F, Lobet G, Tocquin P, Perilleux C. FLOR-ID: an interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Research. 2016;44:D1167–D1171. doi: 10.1093/nar/gkv1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:1–13. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fumagalli M, Vieira FG, Linderoth T, Nielsen R. ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics. 2014;30:1486–1487. doi: 10.1093/bioinformatics/btu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenna A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee C, et al. Selection in a hybrid zone: evidence for linked QTLs in a young inversion. Nature Ecology and Evolution. 2016 doi: 10.1038/s41559-017-0119. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark RM, et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–342. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- 66.Weir B, Cockerham C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 67.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nei M. Molecular Evolutionary Genetics. Columbia University Press; 1987. [Google Scholar]
- 70.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics. 2013;194:459–471. doi: 10.1534/genetics.113.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Long Q, et al. Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nature Genetics. 2013;45:884–U218. doi: 10.1038/ng.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.