

Abstract
There has been significant progress in our understanding of the molecular mechanisms by which calcium (Ca2+) ions mediate various types of cardiac arrhythmias. A growing list of inherited gene defects can cause potentially lethal cardiac arrhythmia syndromes, including catecholaminergic polymorphic ventricular tachycardia, congenital long QT syndrome, and hypertrophic cardiomyopathy. In addition, acquired deficits of multiple Ca2+-handling proteins can contribute to the pathogenesis of arrhythmias in patients with various types of heart disease. In this review article, we will first review the key role of Ca2+ in normal cardiac function - in particular, excitation-contraction coupling and normal electrical rhythms. The functional involvement of Ca2+ in distinct arrhythmia mechanisms will be discussed, followed by various inherited arrhythmia syndromes caused by mutations in Ca2+-handling proteins. Finally, we will discuss how changes in the expression of regulation of Ca2+ channels and transporters can cause acquired arrhythmias, and how these mechanisms might be targeted for therapeutic purposes.
Subject codes: Arrhythmias, Calcium Cycling/Excitation-Contraction Coupling, Genetics, Cardiomyopathy
Keywords: Atrial fibrillation, arrhythmias, calcium channels, RyR2, ventricular tachycardia
The bivalent cation calcium (Ca2+) represents one of the most ubiquitous signal transduction molecules known.1 It mediates a diverse array of biological functions including the muscle contraction, cellular exocytosis, neuronal activity, and triggering of programmed cellular death. Since the first observation by Ringer in 1883 that Ca2+ was required for cardiac contraction, the role of Ca2+ as a signaling ion in the heart has become increasingly appreciated.2 In addition, it has become clear that abnormalities of Ca2+ homeostasis can play a key role in the pathogenesis of common cardiovascular disorders, including cardiac arrhythmias. Human genetic studies of patients with inherited arrhythmia syndromes have uncovered inherited mutations in various Ca2+ channels and Ca2+ transporters, directly implicating dysfunction of these proteins in the disease mechanisms. Moreover, acquired modifications of various Ca2+-handling proteins have been associated with cardiac arrhythmias, including atrial fibrillation (AF) and ventricular arrhythmias in failing hearts. In this review, we provide a comprehensive overview of the potential contributions of Ca2+ in arrhythmia mechanisms, and highlight various gaps in knowledge and controversies in the field.
OVERVIEW OF EXCITATION-CONTRACTION COUPLING IN THE HEART
Regular contraction of the heart requires the conversion of electrical activation (excitation) into mechanical force (contraction). This process, known as excitation-contraction (EC) coupling, requires coordinated movement of Ca2+ ions at the cardiomyocyte level (Figure 1A). Each action potential (AP), triggered by influx of sodium (Na+) through the voltage-gated sodium channel (Nav1.5), thereby generating the INa current, induces Ca2+ influx through voltage-activated L-type Ca2+ channels (LTCCs, Cav1.2), creating the ICa,L current. This Ca2+ triggers a much larger Ca2+ release from the sarcoplasmic reticulum (SR), the principal intracellular Ca2+ storage organelle.3 SR Ca2+ release is mediated by specialized Ca2+-release channels known as ryanodine receptor type-2 (RyR2).4 This process of Ca2+-sensitive RyR2-mediated SR release is known as Ca2+-induced Ca2+-release (CICR). The cytosolic Ca2+ binds to and activates cardiac troponin C (TnC), the Ca2+-sensing protein of the contractile apparatus, and initiates myofilament contraction. During diastole, cardiac muscle relaxation occurs when Ca2+ is removed from the cytosol either by sequestration into the SR by the SR Ca2+-ATPase type-2a (SERCA2a) or out into the extracellular space by the Na+/Ca2+-exchanger type-1 (NCX1). In addition, there is a minor contribution by the plasmalemmal Ca2+-ATPase (PMCA). NCX1 is electrogenic, as it imports three Na+ ions into the cell for each extruded Ca2+ ion, thereby creating a depolarizing transient inward current (INCX). The rapid release of Ca2+ from the SR into the cytosol, followed by rapid reuptake into the SR or extrusion from the cell, creates a Ca2+ wave that runs through the cardiomyocyte and is known as the Ca2+ transient. The amount of Ca2+ released from the SR via RyR2 largely determines the Ca2+-transient amplitude, which correlates with the strength of systolic contraction.
Figure 1. Role of calcium-handling in excitation-contraction (EC) coupling.
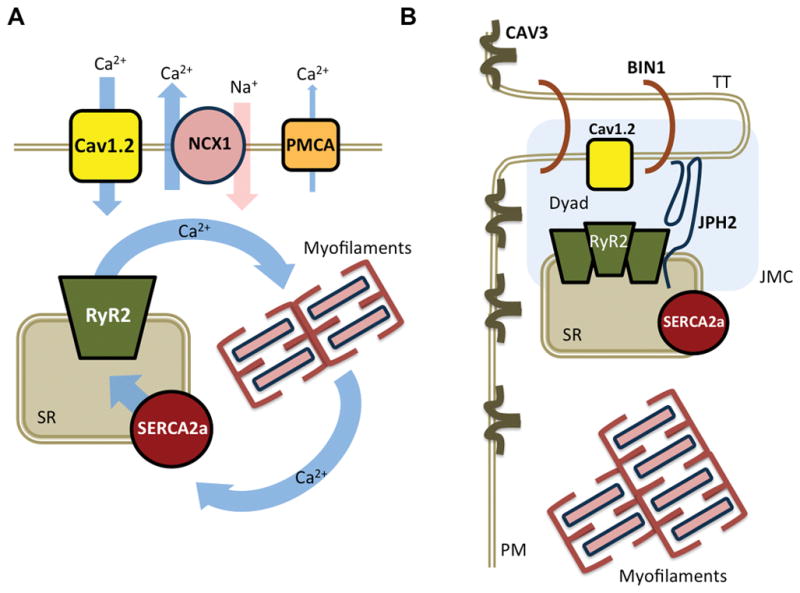
A. Schematic overview of key Ca2+-handling proteins involved in EC coupling. B. Schematic diagram of Ca2+ the release unit and major components of the JCM. The transverse tubule (TT) and SR membranes approximate to form the dyad. BIN1, bridging integrator 1; Cav1.2, L-type Ca2+ channel; CAV3, caveolin-3; JMC, junctional membrane complex; JPH2, juncophilin-2; NCX1, Na+/Ca2+ exchanger; PM, plasma membrane; PMCA, plasmalemmal Ca2+-ATPase; RyR2, ryanodine receptor type-2; SERCA2a, sarco/endoplasmic reticulum ATPase type-2a; SR, sarcoplasmic reticulum.
EC coupling occurs within specialized subcellular structures called junctional membrane complexes (JMCs), where LTCCs on transverse T-tubules – plasmalemmal invaginations that reach deep into myocytes – are positioned in close proximity of the RyR2 channels on the SR membranes (Figure 1B).5 The movement of Ca2+ within these dyadic cleft domains is, in part, regulated by junctophilin-2 (JPH2), a protein that provides a structural bridge between the plasmalemma and SR ensuring appropriate proximity between the LTCC and RyR2 channels6, 7. JPH2 is also necessary for bridging integrator 1 (BIN1) recruitment to develop the T-tubule forming the dyad. There are important differences in the organization of the JMC between atrial and ventricular cardiomyocytes.8 In ventricular myocytes, almost all Ca2+ release events (i.e., sparks and transients/waves) are activated directly by LTCC on T-tubules which leads to synchronized SR Ca2+ release and a rapid upstroke of the Ca2+ transient. In atrial cardiomyocytes, in which TTs are relatively underdeveloped, the Ca2+ transient begins with LTCC-triggered local SR Ca2+-release events at the cell periphery that propagate slowly as Ca2+ waves towards the cell center.9, 10 In addition, atrial cardiomyocytes possess larger and more heterogeneous axial tubules and much more Ca2+-buffering mitochondria than ventricular cardiomyocytes.11, 12 Finally, another class of Ca2+ release channels known as inositol 1,4,5-trisphosphate type 2 receptors (IP3R2) may also contribute to CICR.13
REGULATION OF INTRACELLULAR CALCIUM-HANDLING
The activity of Ca2+ channels and exchangers involved in EC coupling is regulated by several mechanisms and signaling pathways in response to changing demands for cardiac output. For example, the ‘fight-or-flight’ response activates the sympathetic portion of the autonomous nervous system with downstream effects on Ca2+ signaling (recently reviewed).14 Activation of the β-adrenoceptor (βAR) causes a rise in the intracellular concentration of the second messenger cyclic adenosine monophosphate (cAMP). Downstream effectors of cAMP include cAMP-dependent protein kinase A (PKA), which in turn can phosphorylate Ca2+ transporters including LTCC, RyR2, and SERCA2a regulatory proteins like phospholamban (PLN) and sarcolipin (SLN). In addition, the Ca2+/calmodulin-dependent protein kinase II (CaMKII) can modulate Ca2+ homeostasis in response to changes in heart rate, cellular oxidation levels, and persistent βAR stimulation.4, 15
Each of the Ca2+ channels and transporters consist of pore-forming proteins and various accessory subunits that modulate the amount of Ca2+ that is moved through the pore. These channels and exchangers have been extensively reviewed elsewhere.16, 17 Perhaps one of the most well studied multiprotein complexes is the RyR2 macromolecular complex. A diverse array of RyR2-interacting proteins directly regulate RyR2 channel activity by binding to the pore subunit (e.g., FK506-binding protein-12.6 (FKBP12.6), calmodulin (CaM), calsequestrin-2 (CASQ2), junctin (JCTN), triadin (TRDN), βII-spectrin (Figure 2).18, 19 CASQ2 binds to RyR2 via both JCTN and TRDN. RyR2 is strongly regulated by luminal (within the SR) Ca2+ levels, either by direct Ca2+ binding to RyR2, or by luminal Ca2+ interacting with CASQ2, JCTN, and TRDN.20 Other proteins in the RyR2 macromolecular complex regulate the level of RyR2 posttranslational modification. Examples include the protein kinases PKA, CaMKII, and newly discovered striated preferentially expressed protein kinase (SPEG), and protein phosphatases type-1 and type-2A (PP1, PP2A), that regulate the actual level of RyR2 phosphorylation. 21–24 The RyR2 channel is also regulated by S-nitrosylation and oxidation.25
Figure 2. RyR2 macromolecular complex.
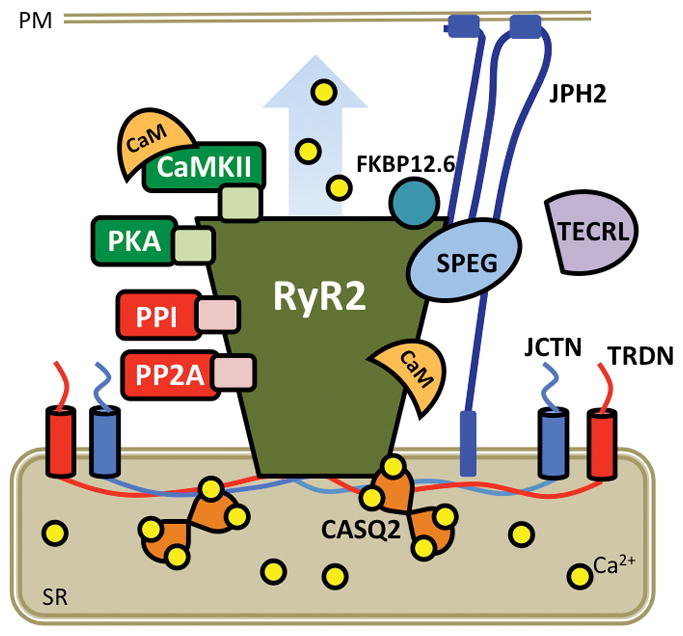
Cartoon representing RyR2 pore-forming subunits with accessory proteins that bind to and/or modulate channel function. CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CASQ2, calsequestrin-2; FKBP12.6, FK506-binding protein 12.6; JCTN, junctin; JPH2, juncophilin-2; PKA, protein kinase A; PM, plasma membrane; PP, protein phosphatase; SR, sarcoplasmic reticulum; TECRL, trans-2,3-enoyl-CoA reductase-like protein; TRDN; triadin.
The LTCC, responsible for voltage-dependent Ca2+ entry into the cells, consists of a macromolecular protein complex comprised of pore-forming Cav1.2 (β subunit) and various auxillary subunits (β2, β, δ, and γ) that modulate channel function (Figure 3). Similar to RyR2, the LTCC is regulated by protein kinases such as CaMKII and PKA as well as protein phosphatases PP1, PP2A and calcineurin (also known as protein phosphatase 2B, PP2B), which can modulate channel gating. In addition, regulatory subunits, like CaM, are embedded within the channel complex.26 LTCC are localized to rafts of other sarcolemmal ion channels and membrane-limited proteins.27, 28 A critical mediator of this membrane clustering is caveolin 3 (CAV3) which binds and interacts with the N-terminal part of JPH2.29
Figure 3. LTCC macromolecular complex.
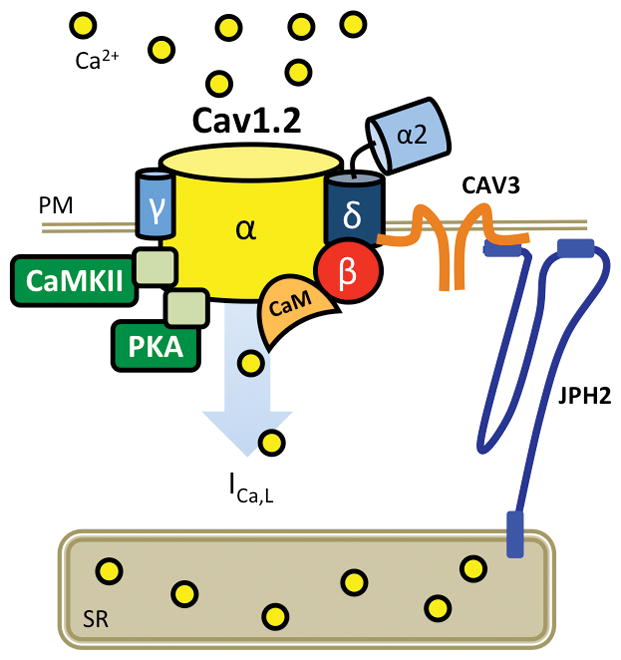
Cartoon representing Cav1.2 pore-forming β subunit with accessory β2, β, γ, δ subunits. CaMKII, Ca2+/calmodulin-dependent protein kinase II; CAV3, caveolin-3; ICa,L, L-type Ca2+ current; JPH2, juncophilin-2; PKA, protein kinase A; PM, plasma membrane; PP, protein phosphatase; SR, sarcoplasmic reticulum.
Finally, SERCA2a is a macromolecular complex required for Ca2+-reuptake into the SR (Figure 4). It is allosterically regulated by phosphorylation-mediated conformational shifts of its regulatory subunit PLN, that can be phosphorylated by PKA and CaMKII.30, 31 These post-translational modifications can relieve the PLN-mediated inhibition of SERCA2a, allowing for rapid Ca2+ reuptake. Histidine-rich Ca2+-binding protein (HRC) has been shown to bind the SR luminal side of SERCA2a and to interact with TRDN, potentially coordinating Ca2+-reuptake with SR Ca2+-release along with S100A1.32, 33 Moreover, calreticulin (CALR) may play a role in the inactivation and degradation of SERCA2a under oxidative stress.34
Figure 4. SERCA2a macromolecular complex.
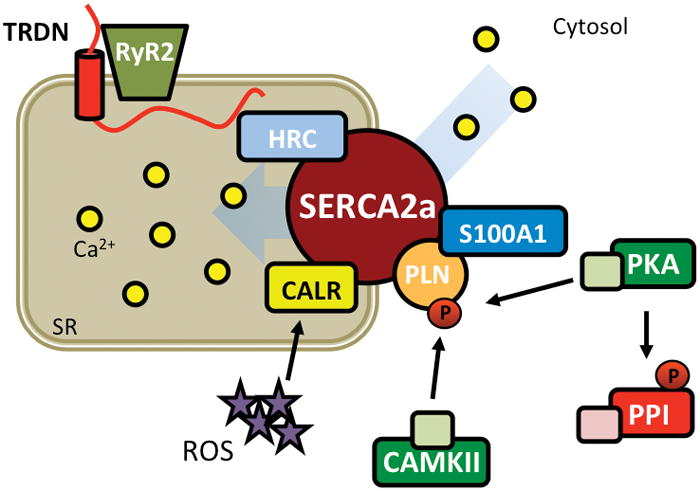
Cartoon representing SERCA2a complex required for reuptake of Ca2+ from the cytosol to the SR. CALR, calreticulin; CaMKII, Ca2+/calmodulin-dependent protein kinase II; HRC, histidine-rich Ca2+ binding protein; PKA, protein kinase A; PP, protein phosphatase; ROS, reactive oxygen species; RyR2, ryanodine receptor type-2; SERCA2a, sarco/endoplasmic reticulum ATPase type-2a; SR, sarcoplasmic reticulum; TRDN, triadin
FUNDAMENTAL ARRHYTHMIA MECHANISMS
The mechanisms responsible for cardiac arrhythmias are generally divided into two major categories – enhanced or abnormal impulse generation (i.e., focal activity), and conduction disturbances (i.e., reentry).35, 36 Focal activity includes enhanced automaticity and triggered activity. Automaticity causes spontaneous generation of APs that do not require induction by previous beats. Healthy myocardium is not normally automatic, but disease conditions (e.g. heart failure, HF) can lead to resting membrane potential depolarization to more positive values causing abnormal automaticity. The most common causes of focal arrhythmias are early afterdepolarizations (EADs) that precede full repolarization (typically corresponding to phase-2 and phase-3 repolarization of the human AP) and delayed afterdepolarizations (DADs) that occur after full repolarization.
EADs cause focal firing by depolarizing surrounding tissue to excitation threshold EADs and are most characteristic of Purkinje-fiber tissue and ventricular tachyarrhythmias associated with HF and long-QT syndrome (Figure 5A). EADs are usually, but not exclusively, associated with excessive AP prolongation (e.g. by increased inward ICa,L 37 and late Na+-current INa,L or INCX,38, 39 or by reduced K+-currents (IK), allowing ICa,L to recover from inactivation and depolarize the cardiomyocyte by allowing Ca2+ to enter.40 CaMKII-dependent ICa,L phosphorylation slows inactivation and accelerates recovery from inactivation, further enhancing the likelihood of EADs.37 At membrane potentials negative to the threshold of ICa,L activation, spontaneous SR Ca2+ release-activated NCX favors the non-equilibrium reactivation of INa, driving phase-3 EADs induction.41, 42 Finally, EADs have also been associated with APD shortening, occurring late in phase-3 of the AP.43 If the intracellular Ca2+ concentration is still high (e.g., due to a large Ca2+ transient) when the membrane potential is negative to the equilibrium potential for NCX, INCX can be activated leading to membrane depolarization. This type of EADs typically occurs after termination of ventricular tachycardia (VT), ventricular fibrillation (VF), and AF. Overall, in regions where EADs reach the threshold to propagate, they generate triggers that initiate reentry.
Figure 5. Key electrophysiological mechanisms leading to cardiac arrhythmias.
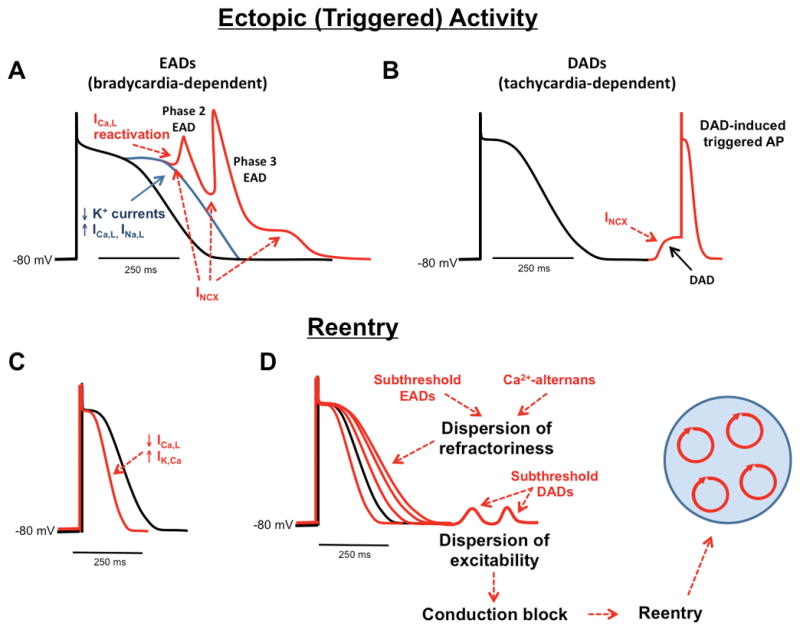
Ectopic (triggered) activity is primarily caused by A) early after-depolarizations (EADs) that occur mainly during bradycardia or following a pause, and B) delayed after-depolarizations (DADs) that occur using during tachycardia. Reentry requires a vulnerable substrate, which can be caused by C) action potential shortening or D) dispersion of refractoriness. ICa,L, L-type Ca2+ current; IK,Ca, Ca2+ dependent K+ current; INa,L, late Na+ current; INCX, Na+/Ca2+ exchanger current;
DADs typically occur during diastole and conditions of elevated cellular Ca2+-loading (Figure 5B). They are caused by spontaneous rises in cytoplasmic Ca2+-concentration, which activate NCX, generating forward mode INCX, although other Ca2+-sensitive currents (nonselective cationic currents and chloride currents) might also contribute to DAD formation.44 The amplitude of the DAD depends on the size of the resting K+ conductance, mainly determined by the inward-rectifier K+-current IK1, relative to INCX amplitude. When IK1 is low, the same INCX will produce a larger DAD and vice versa.45 When DADs reach excitation-threshold, INa is activated and spontaneous APs can arise. DAD-mediated triggered activity contributes the arrhythmogenesis associated with catecholaminergic polymorphic ventricular tachycardia (CPVT), HF and AF.
Reentry can occur around a fixed anatomical obstacle or in a substrate in which functional properties permit initiation and maintenance of reentrant circuits.46 The likelihood of reentry formation is determined by the tissue properties of conduction and refractoriness, with abnormal conduction (slowing and/or local block) and refractoriness (abbreviated or prolonged) making reentry more likely (see Figure 5C–D). Refractory period depends on AP duration (APD), whereas conduction velocity largely depends on INa, expression and localization of gap-junction proteins, and composition of extracellular matrix (e.g. fibrosis). When the refractory period decreases (like in AF), the circuits are smaller and more numerous, simultaneous termination of all circuits is unlikely and the arrhythmia is sustained. When the refractory period is prolonged (like in HF), the heterogeneity (dispersion) of refractoriness is increased and the occurrence of reentry promoting conduction block is more likely. The reentry-promoting substrate can be caused by disease-related cardiac remodeling or predisposing genetic factors, but can also be produced by altered restitution dynamics and subcellular Ca2+-alternans (SR Ca2+ load and release alternans).47 Altered Ca2+ signaling can contribute to the formation of a reentry substrate by two mechanisms: promoting dispersion of excitability, and promoting dispersion of refractoriness.35 For example, DADs that do not reach the threshold to trigger an AP can cause resting membrane potential depolarization, increasing Na+-channel inactivation and promoting dispersion of excitability. The latter might lead to regional conduction block of impulses arising from regions with supra-threshold DADs, thereby promoting reentry initiation. EADs that remain below the threshold to propagate may increase dispersion of refractoriness, also creating a reentry substrate. The rapid rates during DAD-induced triggered activity can promote Ca2+ transient alternans, which can cause spatially discordant APD alternans, thereby enhancing the dispersion of refractoriness and the likelihood of reentry.48 Thus depending on the cellular and tissue context, EADs, DADs and Ca2+ alternans can provide the trigger and may contribute to the formation of the reentry-promoting substrate. A deep understanding of the detailed molecular mechanisms by which abnormal Ca2+-signaling increases the susceptibility to cardiac arrhythmias is key for the development of novel therapeutic options for prevention and treatment of cardiac arrhythmias.
ARRHYTHMIAS CAUSED BY HERITABLE DEFECTS IN CALCIUM-HANDLING GENES
The discovery of the first inherited mutations in genes encoding Ca2+-regulatory proteins has provided the best evidence to date that defects in intracellular Ca2+-handling can directly cause different cardiac arrhythmias (Figure 6). In the following section, we will review several inherited arrhythmia syndromes that are often caused by mutations in genes encoding Ca2+ channels, transporters, or related proteins.49
Figure 6. Diagram showing which genes have been linked to genetic arrhythmia disorders.
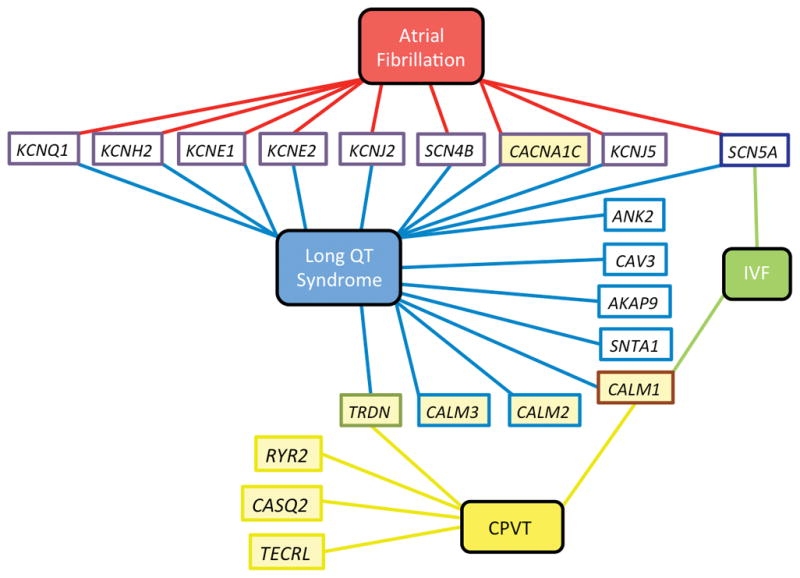
Yellow fill indicates gene that encodes a Ca2+-sensitive or Ca2+-handling protein. CPVT, catecholaminergic polymorphic ventricular tachycardia; IVF, idiopathic ventricular fibrillation.
Catecholaminergic polymorphic ventricular tachycardia (CPVT)
The inherited arrhythmia disorder CPVT is one of the most-deadly arrhythmias known, and it classically manifests with βAR-induced syncope or sudden cardiac death (SCD).50, 51 CPVT was first described in 1978 as a distinct syndrome associated with syncope and arrhythmia in the setting of a structurally normal heart. This condition can present with premature ventricular contractions (PVCs) at rest, or with exercise, and this ventricular ectopy can degenerate into bidirectional VT or VF. The majority of CPVT cases present in childhood and have normal cardiac repolarization on ECG measurement of the QT interval.52 The estimated prevalence of CPVT is around 1:5,000 to 1:10,000 depending on the population studied.53
RYR2-encoded ryanodine receptor type-2 (CPVT-1)
In the first major case series of children described to have CPVT, the authors noted the presence of bidirectional VT as an arrhythmia previously often associated with digitalis toxicity.52 This led to the hypothesis that the etiology of CPVT may be due to DADs induced by increased SR Ca2+ load exacerbated by catecholamines.54 A genetic locus associated with CPVT was first mapped to 1q42-43 of the genome in a large study of two unrelated families with a heritable, autosomal-dominant syndrome manifesting as stress-induced polymorphic VT, syncope, and SCD with structurally normal hearts.55 Two years later, inherited mutations in the RYR2 gene were identified as the most common genetic subtype of CPVT (CPVT-1; Table 1).56, 57
Table 1.
Summary of CPVT-associated genes
| Type | MIM* | Gene | Protein | Genetic Locus | Frequency | Inheritance |
|---|---|---|---|---|---|---|
| CPVT 1 | 604772 | RYR2 | Ryanodine receptor 2 | 1q42.1-q43 | 50–60% | AD |
| CPVT 2 | 611938 | CASQ2 | Calsequestrin 2 | 1p13.1 | Rare | AR |
| CPVT 3 | 614021 | TECRL | Trans-2,3-enoyl-CoA reductase-like | 7p22-p14 | Rare | AR |
| CPVT 4 | 614916 | CALM1 | Calmodulin 1 | 14q31-q32 | Rare | AD |
| CPVT 5 | 603283 | TRDN | Triadin | 6q22.31 | Rare | AR |
Phenotype MIM number; AD, autosomal dominant; AR, autosomal recessive
Subsequent studies rapidly expanded the spectrum of RYR2 mutations in CPVT which account for approximately 50 to 60% of all cases.56 Pathogenic mutations most often alter a single amino acid (missense mutations) and are inherited in autosomal-dominant pattern. CPVT-associated mutations in RYR2 almost always result in increased SR Ca2+ leak which is amplified in the setting of increased sympathetic drive.58 This increased propensity to SR Ca2+ leak can be detected as an increase in the frequency of elementary Ca2+-release events (i.e., Ca2+ sparks).59 It is believed that diastolic SR Ca2+ leak can lead to increased intracellular Ca2+ which activates NCX during diastole, leading to DADs and triggering of ventricular arrhythmias.60 Several aspects of the pathophysiology of CPVT caused by RyR2 mutations remain controversial, including the potential role of reduced binding of FKBP12.6 to RyR2, channel gating deficits in the absence of βAR stimulation, and the potential involvement of SR Ca2+ overload as an additional mechanism. For example, the role of FKBP12.6 in regulating RyR2 Ca2+-release and the role of PKA-mediated phosphorylation on RyR2 in cardiac arrhythmia and HF are subjects of on-going debate.61
Early studies demonstrated that FKBP12.6 was expressed in the heart, associated with RyR2, and modulated CICR.62 Further, studies found that FKBP12.6 directly bound RyR2 and stabilized the closed conformational state of the protein such that removal caused SR Ca2+ leak63, 64. This stabilizing property of FKBP12.6 was not universally observed.65. As this line of exploration was developing, a separate body of evidence was emerging that RyR2 phosphorylation at serine 2808 (S2808) by PKA could increase channel opening probability as part of the “fight or flight” mechanism.66, 67 These studies converged with the observation that PKA-mediated increased channel sensitivity to Ca2+ was based on partial dissociation of FKBP12.6 binding following S2808 phosphorylation, and identified lethal exercise-induced arrhythmias in FKBP12.6 knockout mice (Fkbp12.6−/−).58 This observation was expanded to other forms of cardiac disease, including HF, whereby elevated βAR signaling through PKA resulted in hyperphosphorylated S2808 and dissociation of FKBP12.6.68, 69 These findings have not been universally observed by other investigators have catalyzed a number of follow-up studies which have introduced debate in the field.70, 71 Some have argued that reduced Ca2+ reuptake into the SR led is the predominant mechanism underlying HF72 or that PLN activity and increased SR Ca2+ load is involved.73 There is also evidence that CaMKII phosphorylation of RyR2 may contribute to the development of HF and arrhythmogenesis through increased Ca2+ leak.74 For in-depth review of this topic, please refer to prior articles.75–77 Overall, these studies highlight the complexity of Ca2+ release regulation in the cardiac myocyte.
Studies of several knock-in mouse models of human RYR2 mutations have provided additional insights into the pathogenesis of CVPT.59, 78–80 Based on some of these studies, it has been proposed that Purkinje cells in a mouse model of CPVT exhibited a higher frequency and amplitude of spontaneous SR Ca2+-release events, suggesting that focal arrhythmias might originate from the specialized conduction system.81 More sophisticated genetic studies are needed to confirm whether Purkinje cells are truly the source of triggered arrhythmias in CPVT mutant mice as well as in patients with this condition. Finally, recent studies in human induced pluripotent stem cells (iPSC) have confirmed prior studies on recombinantly expressed channels and studies in mouse models, while providing additional mechanistic insights. For example, it has been shown that iPSC-derived cardiomyocytes (iPSC-CM) from CPVT patients exhibit an increased susceptibility to DADs due to abnormal SR Ca2+-release events82. Overall, these studies demonstrate that exacerbation of DADs following sympathetic stimulation is the key mechanism and that β-blockers, dantrolene, CaMKII inhibitors like KN-93s, and RyR2-inhibiting compounds such a S107 all represent potential therapeutic options for CPVT.82–84 Subsequent clinical studies in CPVT patients confirmed the anti-arrhythmic potential of dantrolene.85 Thus, iPSC-CM from CPVT patients may represent a valuable system for preclinical drug screening.
CASQ2-encoded calsequestrin type-2 (CPVT-2)
A second rare genetic subtype of CPVT (CPVT-2) is caused by autosomal-recessive variants in CASQ2-encoded CASQ2, the most abundant Ca2+-buffering protein in the SR. These mutations are relatively rare among CPVT cases, accounting for only about 3–5% of all patients with CPVT.86 This genetic subtype, initially identified in 7 families of a Bedouin tribe in northern Israel, is characterized by resting bradycardia and VT by treadmill or βAR activation with isoprenaline infusion.87 Recently, a family with a unique autosomal-dominant form of CPVT was found to be caused by a CASQ2 mutation.88. CASQ2 is the cardiac-specific isoform of a family of proteins which directly and indirectly regulate SR Ca2+ storage and release.89 CASQ2 has a high-binding capacity (40–50 mol of Ca2+/mol) but a moderate affinity (Kd of 1 mM) for free Ca2+ and serves as molecular sink for Ca2+ that has been sequestered into SR following cardiac contraction.90 With an increased prevalence of acidic amino acid residues, it is believed that the negatively charged CASQ2 directly binds free Ca2+.91
All CASQ2 mutations identified so far are missense, deletion, or nonsense mutations which lead to a severe reduction, or complete loss of, the CASQ2 protein.92 RyR2 channels that lack CASQ2 open spontaneously without being triggered by ICa,L-mediated Ca2+ influx 93 Studies in isolated rat cardiomyocytes transfected with mutant CASQ2 protein revealed a reduced SR store Ca2+ capacity with spontaneous Ca2+ transient generation and evidence of DADs.94 This effect was abrogated by addition of citrate, a low-affinity Ca2+ buffer, suggesting that mutant CASQ2 destabilizes SR-store Ca2+ capacity which alters the Ca2+-sensitivity of RyR2 resulting in pro-arrhythmic DADs.94. Other studies utilizing knock-in mice carrying missense or radical loss-of-function human mutations demonstrated reduced CASQ2 expression with elevated resting cytosolic Ca2+ levels and reduced SR-store Ca2+ which was further exacerbated by βAR stress.95 CASQ2 is part of the RyR2 macromolecular complex which also involves the SR-proteins TRDN and JCTN.96, 97. The levels of these proteins are often dramatically altered when CASQ2 is genetically ablated or mutated.96, 98 Moreover, increased levels of CALR and RyR2, which increases SR Ca2+ leak have been reported in mice with mutant CASQ2.95 Therefore, it cannot be excluded that some of the RyR2 functional changes in CASQ2 mutant mice are, at least in part, mediated by changes in TRDN, JCTN, or CALR levels. Finally, the mechanisms of increased arrhythmogenesis have been confirmed in human iPSC-CM. For instance, the βAR agonist isoprenaline caused DADs, oscillatory arrhythmic prepotentials, and after-contractions in cardiomyocytes derived from CPVT patients with CASQ2 variants but not from individuals with normal CASQ2.99, 100
TECRL-encoded trans-2,3-enoyl-CoA reductase-like protein (CPVT-3)
A third genetic subtype of CPVT is the gene encoding trans-2,3-enoyl-CoA reductase-like protein (TECRL). Initially identified by linkage analysis in a consanguineous Sudanese family with multiple SCDs among children while playing, subsequent whole exome sequencing (WES) identified mutations in a handful of families and probands in TECRL.101,102 Each patient demonstrated VT and VF, particularly with exertion, and had SCD. Interestingly, while the subjects had normal QT intervals at baseline, adrenergic stimulation caused QT interval prolongation. As such, mutations in the TECRL gene appear to cause an overlap syndrome with features clearly associated with CPVT but also congenital long QT syndrome (LQTS, see below).
Creation of a mouse model of TECRL mutations is necessary to examine arrhythmia mechanisms in the experimental setting. Studies of iPSC-CM generated from the Sudanese proband demonstrated reduced systolic Ca2+-transient amplitudes and reduced caffeine-stimulated Ca2+ transient amplitudes (an index of SR Ca2+ content) along with elevated resting cytosolic Ca2+ levels, consistent with presence of SR Ca2+ leak as seen in CPVT-1 and CPVT-2.82, 99 In addition, mutant iPSC-CMs demonstrated slower Ca2+-transient upstroke velocity and impaired SR Ca2+-reuptake when compared to both heterozygous and wild-type (WT) controls. Interestingly, stimulation with norepinephrine resulted in an increased propensity for DADs, which was suppressed by flecainide. AP recordings revealed prolonged APD also suggesting a clinical overlap between TECRL mutation-positive individuals with features of both CPVT and LQTS.102 At present, the mechanisms by which loss of TECRL function alters SR Ca2+-handling or ionic currents resulting in prolonged APD remain unknown.
CALM1-encoded calmodulin type-1 (CPVT-4)
A fourth subtype of CPVT (CPVT-4) is caused by inherited mutations in CALM1-encoded calmodulin (CaM). The locus for this variant, 14q31-q31, was initially found by linkage analysis in a large, multigenerational Swedish family.103. Family members demonstrated multiple episodes of syncope and sudden death, particularly with exercise and exertion. On clinical evaluation, affected individuals demonstrated ventricular ectopy and evidence of VT/VF that was suppressed by β-blockers. The genetic haplotype was inherited in an autosomal dominant fashion and was completely penetrant. Subsequent genetic analysis of the approximately 70 known genes within the locus demonstrated a heterozygous CaM-N53I mutation that segregated with incidence of disease within the family. A second mutation, CaM-N97S, was identified in an unrelated proband from Iraq who was diagnosed with CPVT and was negative for mutations in RYR2.
Calmodulin is a ubiquitously expressed Ca2+-sensitive signaling molecule which is found in all eukaryotic cells.104. There are three CAM genes in humans, CALM1, CALM2, and CALM3 which all encode a single protein – CaM. CaM is a relatively small, 148 amino acid alpha-helical protein with four classical Ca2+ binding EF hands that each bind to a single Ca2+ cation. This direct Ca2+-binding property allows conformational shifts in the N- and C-terminal domains of the protein which mediate a variety of interactions with a large number of intracellular binding targets.105. A dumbbell-shaped molecule, CaM can sense both local and global Ca2+ levels, which allows for exquisite sensitivity to a variety of Ca2+-signaling events with downstream regulation of a number of Ca2+-handling proteins.106 Within the heart, CaM plays a key role in EC coupling and is critical for the SR Ca2+ release and subsequent Ca2+ re-uptake into the SR. The LTCC and RyR2 are both important binding partners of CaM.107 Ca2+ entering the cardiomyocyte via LTCCs binds to CaM which, in turn, binds to the C-terminal IQ domain of the Cav1.2 channel α pore subunit (α1C) of LTCC. This process allows Cav1.2 channels to cluster and interact with each other, allowing for sufficient Ca2+ entry to initiate EC coupling.108 CaM also binds to RyR2, and binding of CaM reduces the open probability of RyR2. Conversely, impaired binding of CaM to RyR2 due to a mutated binding domain on RyR2 leads to a variety of cardiac pathologies.109.
In vitro experiments have revealed that mutations in the gene encoding CaM compromise Ca2+-binding and result in an aberrant interaction with the CaM-binding domain of RyR2.103 Subsequent studies revealed that the CaM-N97S mutation in the C-domain reduced Ca2+-binding affinity of the C-domain and impaired binding to RyR2 at low Ca2+ concentrations, which was predicted to lead to an increased RyR2 open state. This impaired inhibitory gating regulation was confirmed by subsequent studies of RyR2 single channel recordings in the presence of mutant CaM and functionally resulted in an increased susceptibility for RyR2-mediated store overload-induced Ca2+ release (SOICR).110, 111 In contrast, the CaM-N53I variant, which localized to the opposing N-domain, demonstrated a small yet significant increase in the Ca2+-saturation of the C-domain with an alteration to RyR2 binding affinity. These findings demonstrated that mutations in CALM1 are associated with CPVT through two distinct mechanisms of RyR2 dysregulation and support a model whereby the Ca2+-saturated C-lobe is constitutively bound to RyR2 while the N-lobe senses fluctuations in cellular Ca2+.111
TRDN-encoded triadin (CPVT-5)
Finally, a fifth subtype of CPVT (CPVT-5) is caused by mutations in TRDN-encoded TRDN. Mutations in TRDN were first identified by candidate gene approach, and a small number of probands were identified with either homozygous loss-of-function (LOF) mutations or compound heterozygous mutations. For example, a homozygous frame-shift mutation, TRDN-D18Afs*13, was noted in a proband with cardiac arrest at age of 2 years who was found to have polymorphic VT.112 A second independent proband hosted two mutations, TRDN-Q205X and –T59R, and demonstrated proximal muscle weakness, syncope with exertion and bidirectional ventricular ectopy.112 Thus, TRDN mutations can cause CPVT in an autosomal-dominant manner.
As discussed above, TRDN is a transmembrane protein on the SR that forms a macromolecular complex with RyR2, CASQ2, and JCTN.113 TRDN is a multiprotein family arising from alternative splicing of a single TRDN gene. Two isoforms are exclusively expressed in skeletal muscle, whereas a third isoform (also known as Trisk 32 or CT1) is expressed mainly in cardiac muscle.114 Interestingly, all three TRDN mutations localized to a region of the protein that is common to all isoforms, including skeletal muscle isoforms.112 The link between TRDN mutations and skeletal myopathy remains unknown. In vitro functional analysis of the TRDN-T59R mutation in non-muscle COS-7 cells demonstrated intracellular retention and degradation of the mutation protein. Further, viral transduction of TRDN-T59R mutant protein into Trdn−/− mice demonstrated no expression of the protein by immunofluorescence of isolated cardiomyocytes.112 Thus, functionally CPVT-associated mutations lead to a severe TRDN function in cardiomyocytes. Electron microscopy studies of cardiomyocytes from Trdn−/− mice revealed fragmentation and overall reduction in contacts between the junctional SR and T-tubules.115 The function of CRU channels was impaired with reduced negative feedback of SR Ca2+ release on ICa,L. This uninhibited sarcolemmal Ca2+ influx via ICa,L likely caused SR Ca2+ overload leading to spontaneous SR Ca2+-release events upon βAR stimulation.
Congenital long QT syndrome (LQTS)
Congenital long QT syndrome (LQTS) refers to a distinct group of cardiac channelopathies characterized by delayed cardiac repolarization, which places affected individuals at risk for syncope, seizures, and SCD. A relatively common arrhythmia syndrome, affecting as many as 1 in 2,500 persons, this delay in cardiac repolarization occurs in the absence of an underlying syndrome or structural heart disease.116, 117 Approximately 75% of LQTS cases are due to mutations in three genes: KCNQ1-encoded IKs potassium channel (Kv7.1, LQTS-1), KCNH2-encoded IKr potassium channel (Kv11.1, LQTS-2), and SCN5A-encoded INa sodium channel (NaV1.5, LQTS-3).118 These ion channels play key roles in the cardiac AP and genetic defects in these channels delay repolarization. Several channel interacting proteins, such as ANK2-encoded ankyrin B (LQTS-4), KCNE1-encoded min-K (LQTS-5), and KCNE2-encoded min-K related protein 1 (LQTS-6), among others, interact with these major channels and have been implicated as rare causes of LQTS.119, 120 To date, hundreds of mutations have been identified in 17 LQTS-susceptibility genes (Table 2). In addition, large population-based GWAS analysis exploring common genetic variants associated with QT prolongation have identified a number of loci which encode Ca2+-signaling proteins that were associated with longer QT durations.121 While the majority of the accepted LQTS genes encode proteins which govern the flux of Na+ and K+ about the sarcolemma, there is mounting evidence that Ca2+ fluxes and intracellular Ca2+ signaling are associated with prolonged cardiac repolarization and LQTS.
Table 2.
Summary of LQTS-associated genes
| Type | MIM* | Gene | Protein | Genetic Locus | Frequency | Inheritance |
|---|---|---|---|---|---|---|
| LQTS 1 | 192500 | KCNQ1 | Kv7.1 | 11p15.5-p15.4 | 30–35 | AD |
| LQTS 2 | 613688 | KCNH2 | KV11.1 | 7p36.1 | 25–30 | AD |
| LQTS 3 | 603830 | SCN5A | NaV1.5 | 3p22.2 | 5–10 | AD |
| LQTS 4 | 600919 | ANK2 | Ankyrin B | 4q25-q26 | Rare | AD |
| LQTS 5 | 613695 | KCNE1 | MinK | 21q22.12 | Rare | AD |
| LQTS 6 | 613693 | KCNE2 | MinK related protein 1 | 21q22.12 | Rare | AD |
| LQTS 7 | 170390 | KCNJ2 | Kir2.1 | 17q24.3 | Rare | AD |
| LQTS 8 | 601005 | CACNA1C | CaV1.2 | 12p13.33 | Rare | AD |
| LQTS 9 | 611818 | CAV3 | Caveolin 3 | 3p25.3 | Rare | AD |
| LQTS 10 | 611819 | SCN4B | Sodium channel β4 | 11p23 | Rare | AD |
| LQTS 11 | 611820 | AKAP9 | Yotiao | 7p21.2 | Rare | AD |
| LQTS 12 | 612955 | SNTA1 | Syntrophin α1 | 20q11.21 | Rare | AD |
| LQTS 13 | 613485 | KCNJ5 | Kir3.4 | 11q24.3 | Rare | AD |
| LQTS 14 | 616247 | CALM1 | Calmodulin 1 | 14q32.11 | Rare | AD |
| LQTS 15 | 616249 | CALM2 | Calmodulin 2 | 2p21 | Rare | AD |
| LQTS 16 | 114183^ | CALM3 | Calmodulin 3 | 19q13.32 | Rare | AD |
| LQTS 17 | 603283^ | TRDN | Triadin | 6q22.31 | Rare | AR |
Phenotype MIM number;
Gene MIM number; AD, autosomal dominant; AR, autosomal recessive; lines in bold are Ca2+-sensitive proteins or involved in Ca2+-signaling
CACNA1C-encoded L-type calcium channel (LQTS-8)
The CACNA1C gene encodes the Cav1.2 (β1C) channel subunit of the LTCC, a macromolecular channel complex responsible for ICa,L and EC coupling.3 The Cav1.2 protein is comprised of 4 homologous domains (DI through DIV) that are connected by intracellular linker regions (I-II, II-III, and III-IV loops) and 6 transmembrane segments (S1 through S6).122 Mutations in CACNA1C have been associated with a number of human diseases that have cardiac manifestations. Classically, mutations in CACNA1C have been associated with Timothy syndrome (TS) – a disease characterized by extreme QT interval prolongation, syndactyly, neurodevelopmental delay, and SCD predisposition.123–126 Expansion of clinical genetic testing has identified a number of CACNA1C mutations in individuals demonstrating only cardiac abnormalities (QT prolongation, structural heart disease, and cardiomyopathy), without extracardiac abnormalities, so-called cardiac-only Timothy syndrome (COTS).127 Individuals with only QT prolongation, and a diagnosis of LQTS, have been identified in a large number of independent cohorts.
Many CACNA1C mutations have been characterized in vitro through heterologous expression in cell lines such as HEK293 and TSA201 cells, and demonstrate either increased peak ICa,L, decreased current density with increased window current, or negative activation/positive inactivation shifts.128 Experimental and modeling studies have demonstrated that mutant CACNA1C can lead to enhanced ICa,L, and DAD-mediated triggered activity.129 In addition, they can steepen the APD restitution curve, disrupt rate-dependent cardiac excitation dynamics, and promote the development of alternans.130 Finally, CACNA1C mutations can amplify dispersion of repolarization across the tissue, which produces T-wave alternans and T-wave inversion on the ECG.130, 131
While the overall functional impact of these mutations is the prolongation of phase-2 of the AP causing delayed repolarization, there does not appear to be a clear mechanistic difference between the CACNA1C mutations that lead to TS, COTS, or LQTS. Indeed, this is reflected in the recent identification of a CACNA1C-I1166T in a proband with TS, and independently identified CACNA1C-I1166V mutation, localizing to the identical residue, in a patient with LQTS.132, 133 Given the lack of robust mechanistic studies, it remains unclear how a near-identical genetic substrate can lead to variable expressivity and severity of a disease phenotype. It is likely that genetic modifiers contribute to the differential phenotype manifestations. Additional mechanistic studies, perhaps utilizing iPSC-CM derived from TS, COTS, and LQTS patients with CACNA1C mutations may yield insight into genomic, epigenomic, molecular, and biophysical changes that are specific to each disease presentation.
CALM1, 2, and 3-encoded calmodulin 1, 2, and 3 (LQTS-14-16)
In 2013, the first mutations in the CALM1 and CALM2 genes were associated with LQTS.134 Two unrelated infant probands were described with a severe phenotype of recurrent cardiac arrests with markedly elevated QTc intervals. They were each found to host a heterozygous mutation – CaM-D130G and CaM-D96V mutations, respectively.134 Subsequent validation genotyping in a cohort of LQTS patients yielded an unrelated proband with CaM-D130G and a second subject with CaM-F142L. These mutations were all found to localize either within, or immediately adjacent to, the third and fourth EF hand domains of the C-terminal lobe, resulting in impaired Ca2+-binding of the domain.134 Interestingly, subsequent biochemical investigations have elucidated two distinct mechanisms of CaM and RyR2 dysregulation. CaM-D130G and -D96V both impaired CaM-dependent inhibition of RyR2, resulting in an increased open state when single channels were recorded and an increased propensity for SOICR.110 In contrast, while CaM-F142L demonstrated reduced Ca2+ binding, it was unexpectedly found to enhance CaM-dependent gating inhibition of RyR2 and related RyR2-mediated SOICR. Specialized thermodynamic and NMR spectral analysis of the interaction between CaM-F142L and the reciprocal binding domain of RyR2 demonstrated unique alterations in the protein-protein interface suggesting that the mutation does not disrupt the negative regulatory role of CaM despite an impaired ability to bind free Ca2+.110 In addition to mutations identified in CALM1 and 2, the first reports of LQTS-associated mutations in CALM3 have been recently reported. Specifically, a CaM-D130G mutation was identified in a neonate with a profoundly elevated QTc interval.135 To date, mutations in CALM3 have not been widely identified and there have been no robust mechanistic studies to evaluate the role of CaM in LQTS. Taken together, these studies identify divergent mechanisms of disease pathogenesis that can, nonetheless, result in altered RyR2 inhibition by CaM.
As previously described, the loss of RyR2 gating inhibition is classically associated with the development of CPVT, and the link between these mutations and LQTS remains unexplored. One explanation for this dichotomy is that there are additional molecular effects to impaired CaM activity, such as increased ICa,L, which can prolong the APD. This possibility is supported by early studies in guinea pig cardiomyocytes which demonstrate reduced CaM-dependent inactivation of ICa,L with expression of LQTS-associated CAM mutations. In addition, LQTS-associated CAM mutations result in electrical alternans in a high dispersed manner across and within cells consistent with the electrical remodeling observed in canonical LQTS-associated mutations.136 Given the clear role of these mutations on RyR2 gating, it is likely that there is significant molecular overlap between the LQTS- and CPVT-associated mutations. However, the effect of CPVT-associated mutations in other sarcolemmal ionic currents that shape APD and cardiac repolarization are largely unexplored, although the Nav1.5 and delayed rectifying IK currents are strong candidates.
Recently, the first attempts to derive patient-specific therapies to mitigate the abnormally prolonged repolarization have been reported. In 2017, human iPSC-CMs were derived from a subject who was diagnosed with LQTS shortly after birth following a cardiac arrest with a markedly elevated QTc of 740 who hosted a CaM-D130G mutation.137 Human iPSC-CMs derived from dermal fibroblasts demonstrated prolonged APD and larger Ca2+ transients with slower rise and decay kinetics when compared to WT iPSCs from an unrelated ostensibly healthy donor.138 Further, CaM-D130G imparted a significant decrease to CaM-dependent inactivation of the LTCC. The authors utilized CRISPR-mediate interference of the transcription of CALM2, which specifically reduced expression of the mutant protein without altering expression of either CALM1 or CALM3. This selective expression inhibition rescued the prolonged APD in iPSC-derived cardiomyocytes.138 While this study represents a major step forward in gene therapy-approaches to altering monogenic disease expression, translating this technique to an in vivo model of arrhythmia remains an active and challenging area of exploration.
TRDN-encoded triadin (LQTS-17)
The most recent gene associated with LQTS is TRDN-encoded TRDN, which has been previously also linked to CPVT-5. Identified following WES of probands negative for the known LQTS-associated genes, a handful of TRDN null variants were identified. As with CPVT-5, each mutation-positive proband demonstrated either homozygous inheritance of LOF allele or a compound heterozygous mutation with a LOF allele. 112, 139 Both entities clinically manifest as SCD with either QT prolongation (LQTS-17) or signs of ventricular ectopy in the absence of QT prolongation (CPVT-5) diagnosed at an early age. This combination of clinical findings, in addition to the skeletal muscle weakness occasionally noted with CPVT-5, and the TRDN genetic substrate has been labeled the so-called triadin knockout syndrome. To date, there have been no mechanistic studies involving LQTS-associated TRDN mutations, and while Trdn−/− mice have a known propensity for arrhythmogenesis with βAR stimulation, QT prolongation has not been detected.115, 140 Given the previously described possibility that Trdn−/− mice likely demonstrate reduced negative feedback of RyR2-mediated SR Ca2+-inhibition of ICa,L, an interesting possibility is that the increased Cav1.2 current might lead to APD prolongation. This would be an indirect mechanism of QT prolongation that is analogous to the CACNA1C mutations described in LQTS-8 which produce an increased ICa,L current. Further extensive studies are needed to delineate these hypotheses.
Idiopathic Ventricular Fibrillation (IVF)
IVF is a genetic disease characterized by a documented VF event that is otherwise unexplained. Comprising approximately 1% of out-of-hospital cardiac arrest survivors presenting with a shockable rhythm, IVF can often be challenging to diagnosis.141, 142 Further, in the setting of a normal ECG, the affected status of an individual can only be known following an arrhythmic event, which makes genetic studies challenging. Traditionally associated with mutations in the SCN5A-encoded Nav1.5, the first IVF-associated mutations were often described in sporadic cases presenting with VF and had significant clinical overlap with a group of Nav1.5-mediated channelopathies known as Brugada syndrome (BrS).143, 144 New genetic testing platforms have allowed for the identification of other IVF genes implicated in families with the arrhythmia (Figure 5) and recent advances in WES have identified the first genes encoding Ca2+-handling proteins in children with IVF.
In 2014, a family with a history of VF and SCD with normal ECG and echocardiograms was subjected to WES after kindred were found to be genotype-negative for the major LQTS, CPVT, and arrhythmogenic right ventricular cardiomyopathy (ARVC)-associated genes. This identified a CALM1-F90L mutation in a proband who experienced out-of-hospital arrest due to VF at age 16 with no clinical evidence of LQTS, CPVT, cardiomyopathy, or other SCD-predisposing etiology.145 Subsequent functional evaluation of the CALM1-F90L mutation demonstrated impaired CaM stability and impaired Ca2+ binding cooperativity.109 It was concluded that the F90L mutation likely perturbs the position of two Ca2+ EF hands within the C-lobe relative to each. As a result, the ability of the first occupied site to induce a favorable conformational shift in the second, which is needed to facilitate Ca2+-binding, is impaired. The authors concluded that this creates a relatively insensitive CaM protein which is not responsive over small changes in Ca2+ concentration.109
While the impact of the F90L on the function of CaM is known, the ultimate effect of this perturbation on RyR2 gating or other Ca2+-handling proteins are still unknown. While there has been some incremental progress in identifying the genetic and molecular etiology of IVF, mutations remain rare and IVF remains enigmatic as a disease entity. As with the development of CPVT and LQTS, one possibility is that altered CaM function associated with IVF selectively impairs some ion channels while leaving other channels unaltered. A tempting target is Nav1.5, which contains a number of IVF-associated mutations. SCN5A-associated IVF and BrS mutations demonstrate a diverse array of biophysical effects in heterologous cell line over-expression models. For example, some SCN5A IVF/BrS mutations create depolarizing shifts in channel inactivation while others create hyperpolarization shifts in both activation and inactivation, all with the ultimate effect of loss-of-function effect on Nav1.5 and VF predisposition.143, 146 It is possible that IVF-associated CAM mutations results in loss of Nav1.5 depolarizing current and dispersion of excitability – a known molecular substrate for reentry-mediated VF. This possibility is supported by structural evidence that CaM directly binds Nav1.5 and is a critical player in channel inactivation and permitting channel activation. However, a direct link between CaM mutations and VF has not been clearly demonstrated.105 Ultimately, subsequent studies are needed to link CAM mutations to INa current and a reentry substrate in IVF.
Hypertrophic Cardiomyopathy (HCM)
HCM is an inherited cardiac disorder characterized by asymmetrical hypertrophy of the heart, with a prevalence of 1 in 500.147 This disease represents the most common cause of arrhythmogenic SCD in the young, particularly in young athletes.148 HCM is not only associated with lethal ventricular arrhythmias, but also with AF.149, 150 Since the association of the first mutation gene with HCM, MYH7-encoded β-myosin heavy chain, multiple studies have determined that the majority of HCM cases are due to mutations in genes encoding components of the cardiac sarcomere.151–153 While the cardiac myofilaments are the major molecular cause of HCM, Ca2+ dysregulation plays a significant role in the pathologic remodeling and hypertrophy. Further, abnormal Ca2+-signaling and the myofilament sensitivity to Ca2+, are both known triggers for ventricular arrhythmias. Sarcomeric HCM genes are divided into sub-groups based location of the encoded protein in the cardiac sarcomere consisting of the thick, intermediate, and thin myofilaments. Mutations in genes encoding the thick myofilament (MYH7-encoded beta myosin heavy chain, MYL2-encoded regulatory myosin light chain, and MYL3-encoded essential myosin light chain), the intermediate myofilament (MYPBC3-encoded cardiac myosin binding protein C), and the thin filament proteins (ACTC-encoded actin, TPM1-encoded alpha-tropomyosin, TNNT2-encoded cardiac troponin T (TnT), TNNI3-encoded cardiac troponin I (TnI), and TNNC1-encoded cardiac troponin C (TnC)) have been linked with development of HCM.154–160 Mechanisms of sarocomeric HCM pathogenesis have been extensively reviewed.161, 162
Arrhythmia predisposition in sarcomeric HCM
Early in the exploration of the sarcomeric gene-association with HCM, it was proposed that the arrhythmia burden, manifest in SCD, might be higher with certain mutations. For example, early studies identified individuals in large families of HCM hosting either the MYH7-R403Q or -R453C missense mutations with increased sudden deaths compared to those hosting a -V606M mutation.163 Further, early genotype-phenotype studies of TNNT2 suggested an association with decreased life expectancy and a high incidence of SCD despite minimal cardiac hypertrophy.164 These studies proposed that individual mutations, or mutations in specific genetic loci, may predispose to lethal arrhythmic events in HCM. As the field has matured, these associations were not universally observed, and there is significant heterogeneity in the expression and penetrance of sarcomeric HCM disease.165–168 This controversy has been previously reviewed.169, 170 Overall, these genotype-phenotype correlations did not have mechanistic support for the arrhythmia burden observed in some cases; however, a growing body of evidence suggests myofilament Ca2+-sensitivity as a major arrhythmic mechanism which is independent of gene mutation. As a molecular unit, the troponin complex and thin filament proteins are responsible for sensing intracellular Ca2+ fluctuations and triggering sarcomeric contraction.171 While many myofilament proteins have been linked to HCM arrhythmogenesis, alterations in Ca2+-sensitivity of the components of the thin filament have been most clearly linked with potentially fatal ventricular arrhythmias.172 This is detailed below.
While HCM carries an increased risk of lethal ventricular arrhythmias,173 atrial fibrillation (AF) is found commonly with a frequency of 20–25% of all patients with HCM.174 The hemodynamic mechanism of this may be related to atrial dilation secondary to elevated left ventricular filling pressures resulting in left atrial dilation; however, the cellular mechanism of this is unexplored among sarcomeric HCM. Further, while there have been some suggestions the sarcomeric mutations may predispose individuals for early and more severe AF,175, 176 there have not been conclusive studies linking specific genotype to AF predisposition.150
TNNT2-encoded cardiac troponin T and TNNC1-encoded cardiac troponin C
Troponins are the Ca2+-sensing molecule of the myofilament. Following CICR, free Ca2+ binds TnC which increases its binding affinity for TnI, pulling the TnI inhibitory domain away from its binding site on actin through its interaction with the molecular linker TnT.177 This permits the troponin-tropomyosin complex to move further into the actin groove fully exposing the myosin binding sites on actin. Active actin-myosin cross-bridging then occurs and contraction begins.177
Traditionally, mutations in TNNT2 were believed to be more arrhythmogenic compared to other genetic subtypes of HCM.178 While this belief has been called into question recently,169 a significant body of evidence has linked HCM-associated TNNT2 mutations with the development of fatal arrhythmias in the absence of other known predictors of arrhythmia predisposition such as significant hypertrophy or fibrosis. A number of TnT mutations have been described that nearly universally increase Ca2+ sensitivity, and thus Ca2+-binding of TnT and the sarcomeric thin filament. It is believed that TnT serves as a molecular sink for dynamic Ca2+-buffering, and that increased Ca2+-sensitivity may lead to altered Ca2+-transient dynamics. Overall, the degree of arrhythmia susceptibility appears to be directly correlated to the degree of increased Ca2+ sensitivity.179
A mouse model of HCM (transgenic over-expression of I79N mutant TnT) exhibits increased cardiac contractility with reduced diastolic relaxation in the absence of significant fibrosis, as well as increased myofilament Ca2+ sensitivity.180 This increased Ca2+-sensitivity was associated with increased diastolic Ca2+ levels and intracellular Ca2+ overload in isolated cardiomyocyte studies.181 Further, TnT-I79N was associated with decreased Ca2+-transient amplitudes in the face of elevated resting Ca2+ levels which caused ventricular ectopy and VT.182 While the precise mechanism has not been clarified, the increased TnT Ca2+ sensitivity may lead to DAD-mediated VT resulting from reduced myofilament Ca2+ buffering or could cause reentrant arrhythmia through a still undefined mechanism. The first option is supported in other models of increased thin filament Ca2+ sensitivity. The transgenic expression of fetal slow skeletal troponin I (ssTnI) in place of TnI increased Ca2+ sensitivity in a manner analogous to the electrical remodeling found in pathologic hypertrophy.183 In this model, constitutive increase in Ca2+ sensitivity is associated with increased expression of NCX which might result in increased INCX current to maintain Ca2+ homeostasis during diastole when SERCA2a is also reduced.184 Interestingly, this observation was noted in younger but not in older mice, reflecting the early age of onset of arrhythmias in TNNT2-positive subjects. The reentry hypothesis is supported by evidence that TNNI3 mutations can increase spatial dispersion of activation times across the myocardium, thereby promoting reentry. For example, TNNT2 mutations, including TnT-179N, have been shown to associate with a short effective refractory period along with beat-to-beat variability in APD with increased spatial dispersion of conduction velocity.179 Additional studies are required to directly prove two suggested hypotheses and delineate the underlying arrhythmogenic mechanisms associated with TNNT2 mutations.
Mutations in TNNC1, a rare cause of HCM, have been also linked with a predisposition to fatal arrhythmias. A TnC-A31S mutation was identified in 3-year-old boy who had HCM and an out-of-hospital VF event. Despite being on β-blocker therapy, he had multiple breakthrough VF events with appropriate ICD discharged. This mutation is located within the inactive Ca2+-binding domain of TnC. When reconstituted in skinned porcine cardiac fibers, this resulted in increased Ca2+-sensitivity of both TnC and the thin filament compared to WT.185 Should future studies confirm the presence of increased cellular Ca2+ levels, this also raises the possibility of either a DAD-mediated trigger or formation of an arrhythmogenic substrate for reentry. In addition, while rare, identification and characterization of human mutations affecting other thin filament components will add additional mechanistic understanding to this process.
JPH2-encoded junctophilin type 2
A small subset of patients without mutations in sarcomeric genes host a genetic mutation in genes encoding Ca2+-handling proteins, and some have been linked with a predisposition to arrhythmia.186 JPH2-encoded JPH2, is a member of the junctophilin family of proteins which plays a critical role in maintaining the JMC in excitable cells, including striated muscle.6, 7 JPH2 is the major family member found in the heart and spans the JMC, tethering the SR to the sarcolemma creating a fixed cardiac dyad distance as well as serving a key role in negatively regulating RyR2 opening (Figure 1B).7, 187 Reflective of the critical role that this protein plays in maintain CICR as well as Ca2+ homeostasis by RyR2 gating regulation, JPH2 plays a prominent role in cardiomyopathy development, HF progression, and development of EC coupling in the immature myocyte.188–191 These diverse roles have been previously reviewed.192, 193 An emerging role of JPH2 is the development of Ca2+-mediated arrhythmias, in particular congenital AF. While the vast majority of AF is acquired, reviewed in detail below, a specific mutation (E169K) in JPH2 was linked with AF development in a small family with HCM.194 Expression of JPH2-E169K in mice demonstrated a higher incidence of pacing-induced AF with increased SR Ca2+ leak and propensity of ectopic Ca2+ transients following rapid pacing.194 This was associated with increased RyR2-mediated SR Ca2+ leak due to loss of direct binding between RyR2 and JPH2.189, 194 This contributed to increased diastolic Ca2+, increased NCX activity, and a predisposition to DADs. Additional studies are needed to more thoroughly dissect the molecular underpinnings of JPH2-mediated atrial electrical remodeling.
CASQ2-encoded calsequestrin 2 and CALR3-encoded calreticulin type 3
Rare mutations in other members of the JMC and RyR2 macromolecular complex have been linked to HCM. Genetic interrogation of an Australian cohort of 252 unrelated individuals with HCM revealed a single mutation, D63E in CASQ2 as well as 2 mutations in the CALR3 gene (CALR3-R73Q and -K82R) that were not identified in the ostensibly healthy control population.195 To our knowledge, these are the only mutations described in these genes among individuals with cardiomyopathy. The CASQ2-D633 was found in compound heterozygosity with two MYBPC3 mutations, which decrease the likelihood of a truly causative biomarker. Conversely, the 2 CALR3 mutations were found in genotype-negative individuals. CALR is a Ca2+-binding chaperone in the sarcoplasmic/endoplasmic reticulum, where it buffers Ca2+ and plays an important role in the quality control of intracellular secretory pathway processes.196 CALR has two isoforms, and little is known about the expression levels of CALR3 in myocardial tissue. The functional implications of the CALR3 variants are presently unknown. In embryonic stem cell knockout model, CALR3 deficiency compromised the nuclear pore complex and disrupted the nuclear import of the cardiac transcription factor MEF2C in a Ca2+-dependent manner.186, 197
Arrhythmogenic Cardiomyopathy (ARVC)
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/C), also referred to as arrhythmogenic cardiomyopathy, is a relatively rare type primary myocardial disease characterized by fibro-fatty replacement of myocardial tissue, cardiac arrhythmias, and an increased risk of SCD. This disease process has been recently reviewed.198 Traditionally, ARVC is considered a disease of the cardiac desmosome, whereby mutations in components of this cell-cell adhesion structure are commonly identified in individuals with disease.199, 200 There is some evidence that RYR2 mutations may be a rare cause of ARVC or are present with a prevalence that is significantly higher than rare RYR2 variants in a control population.201, 202 These clinical observations suggests that RYR2 variants play a role in the genetic basis of traditional ARVC as either disease-causing mutations or as a modifier susceptibility allele. In a mouse model of an ARVC-linked RYR2 variant, a reduced right ventricular end-diastolic volume was observed, but pathognomic fibrofatty infiltration or structural abnormalities seen in ARVC patients were absent.59 Despite the possible link between RyR2 and ARVC, there currently is insufficient evidence to implicate primary defects in Ca2+-signaling in the pathogenesis of this disorder.203
Dilated cardiomyopathy (DCM)
Familial dilated cardiomyopathy (DCM) is a genetic heart muscle disease characterized by progressive dilation and dysfunction of the left or both ventricles. Mutations in over 30 genes can cause congenital DCM, and most of these genes encode proteins that are part of the sarcomere or are structural proteins needed to conduct mechanical force in the cardiomyocyte.204 The remaining genes encode proteins that play various roles within cardiomyocytes to ensure proper contractile function. Various studies suggest that mutations in sarcomeric genes205–207 as well as non-sarcomeric genes208, 209 can alter Ca2+ homeostasis, although the affected proteins are not directly involved in Ca2+-handling. On the other hand, there is a clear role of defective Ca2+-handling in DCM pathogenesis in patients with inherited mutations in phospholamban (PLN) and histidine-rich Ca2+-binding protein (HRC).
PLN-encoded phospholamban
Rare mutations and polymorphisms localizing to PLN have been linked to patients with inherited cardiomyopathy. In a large family with multiple generations of cardiomyopathy, kindred homozygous for a PLN-L39X nonsense (early stop) mutation developed DCM and HF requiring cardiac transplantation as adolescents.210 Interestingly, individuals heterozygous for this mutation tended to demonstrate HCM. This raises the possibility of a dose-dependent effect with loss of PLN expression. In addition, multiple small genotyping studies identified a handful of heterozygous PLN mutations in individuals that were missense.211, 212 Moreover, PLN mutations have been identified in individuals with HCM exclusively. These include a handful rare PLN promoter have been identified in multiple independent cohorts.213–215 Overall, it appears that mutations in PLN are a rare cause of both DCM and HCM accounting for less than 1% of all individuals with disease.213
As previously discussed, myocyte relaxation during diastole is an active process mediated by ATP-expending pumping of cytosolic Ca2+ into the SR lumen via SERCA2a, which is negatively regulated by PLN. Moreover, PLN inhibitory action can be reduced by PKA and CaMKII-mediate phosphorylation.216, 217 Since the discovery of cardiomyopathy-associated mutations, several mouse models have been made expressing putatively pathogenic mutations. For example, transgenic overexpression of the PLN-R14del mutation, initially identified in a large family of DCM, lead to cardiac dilation, myocyte disarray, fibrosis and early death in a mouse model.211 In vitro studies of PLN-R14del expressed in HEK293 cells demonstrated super-inhibition of Ca2+ affinity for SERCA2a that was not relieved by PKA phosphorylation. While the precise mechanism of PLN-mediated DCM remains unclear, it is possible that chronic suppression of SERCA2a activity leads to increased cytosolic Ca2+ and a substrate for DAD arrhythmogenesis and, perhaps concurrently, pathologic myocardial remodeling resulting in HF. This possibility is supported by recent work exploring a PLN-R25C mutation. Originally identified by WES of a DCM family with significant ventricular arrhythmias requiring ICD placement, this mutation caused super-inhibition of SERCA2a when virally over-expressed in adult cardiomyocytes.218 This resulted in decreased SR Ca2+ content and reduced Ca2+ transient amplitude which is consistent with the loss of systolic function observed in DCM. Further, increased Ca2+ spark frequency and spontaneous Ca2+ waves were seen, suggesting DAD-type arrhythmia susceptibility.218 While the mechanism of increased SR Ca2+ leak is unknown, it is possible that CaMKII activity is increased in the setting of elevated myocyte Ca2+ levels. Additional studies specifically exploring the interaction between SERCA2a function and RyR2 gating are needed to clarify this relationship.
HRC-encoded histidine-rich calcium-binding protein
Candidate gene-based genetic interrogation of a cohort of DCM patients for HRC-encoded HRC identified a S96A polymorphism that was statistically associated with the development of ventricular arrhythmias. Presence of the minor allele variant conferred a hazard ratio of 4.2 for VT or VF among individuals with DCM.219 HRC is part of the SERCA2a macromolecular complex and serves as a regulator of SR Ca2+ reuptake (Figure 4). It has been shown to bind the SR luminal side of SERCA2a and interact with TRDN.32, 33 Subsequent studies utilizing adenoviral overexpression in rat ventricular myocytes demonstrated reduced SR store Ca2+ reuptake and increased Ca2+ sparks with HRC-S96A expression compared to WT.220 Interestingly, this phenotype was exacerbated following myocardial ischemia and resulted in spontaneous Ca2+ waves.220 This suggested an arrhythmia susceptibility allele that may alter RyR2 gating function in the setting of ischemic stress. While in vivo studies are needed to validate these observations, the findings together suggest a relatively common variant that may be clinically silent until an acquired myocardial stress or injury. Further, these findings suggest a molecular mechanism of cross-talk between SR reuptake via SERCA2a and SR release via RyR2. Whether this association is direct through common binding partners, indirect through signal transduction molecules, or a combination of both, remains unknown.
CALCIUM-DEPENDENT ACQUIRED ARRHYTHMIAS
While a number of arrhythmias can be caused by heritable mutations in cardiac ion channels and channel interacting proteins, Ca2+-mediated arrhythmias can also develop in the setting of acquired diseases of the myocardium. These common types of arrhythmias include atrial fibrillation (AF) and ventricular tachyarrhythmias encountered in patients with structural heart disease.75, 221 In this review, we will not discuss arrhythmias that occur in conjunction with structural heart disease.
Heart Failure (HF)
Heart failure is a clinical diagnosis, which is defined as any abnormality in cardiac structure or function which results in failure of the heart to meet the metabolic demands of the body. Affecting millions of people worldwide, an estimated 1 in 5 people will develop heart failure during their lifetime, making it one of the most deadly, morbid, and expensive diseases known.222 While gains have been made in reducing mortality, there is recent evidence that these gains have plateaued and that global burden of HF remains high.223, 224 Given this, there have been rapid advances in the pharmacologic management of HF, as well as guidelines shifts for recommended therapies, which target a growing number of molecular mechanisms.225 Pathologic alterations in cardiomyocyte Ca2+ cycling have emerged as a prominent component of the molecular dysfunction that occurs in HF. Understanding these mechanisms has been central to the development of recent novel therapies. These topics have been heavily reviewed.226–228 Further, a substantial body of evidence exists linking these pathologic alterations in Ca2+ cycling to arrhythmic predisposition during HF remodeling. These arrhythmias are the cause of a significant proportion of SCD which occurs during HF.229, 230. Just as HF is a complex and varied disease, the arrhythmic substrate from aberrant Ca2+ signaling is a broad subject and has been the topic of multiple comprehensive reviews.15, 231, 232
Atrial fibrillation (AF)
AF represents the most common type of cardiac arrhythmia observed in the general population.233 This disease often progresses from a more intermittent form (paroxysmal AF; pAF) to persistent (chronic) AF (cAF) which lasts for more than 7 days at a time.36, 221 Numerous factors can promote the occurrence of AF, including genetic determinants (Figure 6), extra-cardiac factors (e.g., sleep apnea, obesity, hypertension, autonomic imbalance), as well as remodeling of the cardiac tissue.36, 234 In this section, we will focus on the potential involvement of Ca2+ in the development of AF. The primary arrhythmia mechanisms contributing to AF are focal ectopic firing and reentrant activity (Figure 7).
Figure 7. Calcium-dependent arrhythmia mechanisms in atrial fibrillation (AF).
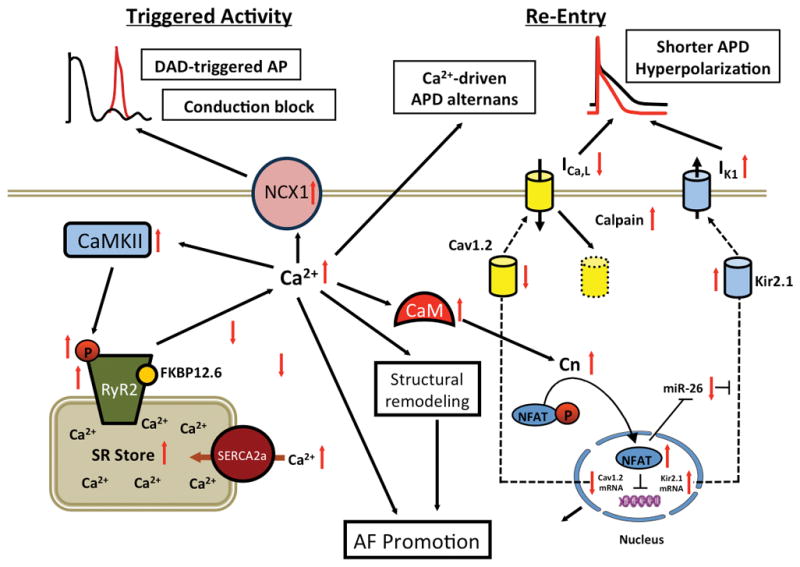
Schematic diagram delineating which changes in intracellular Ca2+-handling promote arrhythmia mechanisms leading to AF. Enhanced RyR2-mediated Ca2+ release leads to activation of NCX, which in turn can cause a DAD-mediated triggered action potential (AP). Shortening of the AP duration (APD) due to reduction of ICa,L (L-type Ca2+ current) and membrane hyperpolarization due to upregulation of IK,1 (inward rectifier K+ current) promote reentry. CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent protein kinase II; Cav1.2, L-type Ca2+ channels; Cn, calcineurin; FKBP12.6, FK506-binding protein 12.6; miR-26, micro-RNA-26; mRNA, messenger RNA; NFAT, nuclear factor of activated T-cells; RyR2, ryanodine receptor type-2; SERCA2a, sarco/endoplasmic reticulum ATPase type-2a.
Ca2+-dependent triggered activity in AF
Experimental studies in animal AF models and atrial samples from AF patients revealed that abnormal atrial Ca2+-signaling likely plays a role in AF pathophysiology by contributing to afterdepolarization-mediated triggered activity, conduction block, and Ca2+-driven subcellular alternans.36, 235 Cellular DAD-mediated triggered activity was demonstrated in atrial myocytes from patients with pAF (Figure 7A).236 These patients were in sinus rhythm at the time of tissue collection for weeks, thus excluding confounding effects of high atrial rate-induced atrial remodeling. Several factors contribute to the increased incidence of spontaneous SR Ca2+ release events, including increased SR Ca2+ load and enhanced RyR2-mediated SR Ca2+ release. The SR Ca2+ stores are overloaded due to increased SERCA2a activity secondary to PLN phosphorylation resulting in inactivation of the inhibitory protein.236 Increased SR Ca2+ leak was caused by increased RyR2 protein levels and RyR2 activity levels, whereas RyR2 phosphorylation levels were unaltered.194, 237 Enhanced RyR2 protein expression during pAF appears to be caused in part by a reduced expression of the microRNA cluster miR-106b-25, which enhances post-transcriptional regulation of RyR2.238 Consistent with these data is the finding that miR-106b-25 deficient mice are more susceptible to pacing-induced AF, atrial ectopy, and increased SR Ca2+ release events.238 Recent transcriptomic analyses suggest that there may be additional alterations in miRNA and mRNA which have not been fully explored in patients with pAF.239 Taken all of these studies together, it is clear that additional investigation is needed to assess the potential effects of intracellular Ca2+ modulation on the pathogenesis and progression of AF.
In patients with persistent AF, an increased prevalence of spontaneous SR Ca2+ release events and DADs have also been reported.235, 240, 241 The activity of single RyR2 channels was found to be increased in patients with cAF.242, 243 Increased levels of PKA and CaMKII-mediated phosphorylation of RyR2 have been reported in patients and large animal models of cAF.242, 244, 245 Functionally, however, it appears that mainly CaMKII phosphorylation of RyR2 promotes excessive channel activation and SR Ca2+ leak.243 In addition, reduced interactions of RyR2 with channel-stabilizing subunits such as FKBP12.6 and JPH2 may contribute to increased diastolic SR Ca2+ leak and triggered activity.194, 246 The enhanced SR Ca2+ leak is more likely to lead to triggered activity due to upregulation of NCX in patients with cAF.243
Finally, AF has been reported in patients with CPVT, which is not surprising since mutant RyR2 channels cause SR Ca2+ leak in both the atria and ventricles.247, 248 Studies in mouse models of CPVT caused by an RyR2 mutant confirmed enhanced SR Ca2+ leak in atrial myocytes, consistent with DADs and triggered activity.249, 250 In addition, atrial conduction slowing has been reported in an RyR2 knock-in mouse model of CVPT, which may be caused by acute Ca2+-dependent inhibition of Na+-channels and a chronic downregulation of Nav1.5 expression.251, 252 This study suggests a possible mechanistic link between abnormal SR Ca2+ release and reduced conduction velocity and a slower action potential upstroke, which might contribute to reentry. Overall, abnormal Ca2+ signaling and enhanced diastolic SR Ca2+ leak along with cellular DAD-mediated triggered activity may support AF induction by producing DADs and could promote AF persistence by increasing heterogeneity (dispersion) of excitability, thereby causing conduction block that increases the susceptibility to AF-maintaining reentry.
In addition to DADs, late phase-3 EADs have been observed in dogs after rapid atrial pacing (which causes Ca2+ loading of the cells) (Figure 7B).239 This is somewhat surprising since EADs typically occur in the setting of APD prolongation, whereas the atrial APD is usually abbreviated in most models of AF. Several changes favor the development of EADs in cAF, including SR Ca2+ leak via RyR2 can promote ICa,L reactivation, the upregulation of INa,L, and enhancement of INCX.243, 253–255 Nevertheless, the potential role of late phase-3 EADs in the development of AF requires further investigation. Other mechanisms may contribute to the formation of triggered activity, including cytosolic Ca2+ alternans (see below), which play a critical role in the initiation of AF in humans.256
Ca2+-dependent reentry in AF
Reentry requires a suitable vulnerable substrate, as well as a trigger that acts on the substrate to initiate reentry (discussed above). Atrial remodeling is induced by atrial arrhythmias, and has the potential to increase the likelihood of ectopic activity as well as reentry through multiple mechanisms. The persistence of abnormal Ca2+ signaling and enhanced diastolic SR Ca2+ leak can activate ion channels and trigger Ca2+-dependent signaling pathways, thereby promoting the evolution of atrial remodeling and the progression of AF to more persistent forms.257 For example, small-conductance Ca2+-dependent K+-channels (SK channels) govern the risk of human AF likely by decreasing APD and promoting reentry,258 and the association between SK channels and RyR2 as the potential internal source of SK channel activation, position SK channels as an important Ca2+-dependent link between triggered activity with reentry.259, 260 Furthermore, reduced ICa,L in AF causes APD shortening and promotes reentry.235 Its reduction is complex and involves downregulation of the Cav1.2 subunit expression by the calcineurin-NFAT system and Cav1.2 breakdown by Ca2+-dependent calpain proteases.235 Reduced Ca1.3 might also contribute.261 Reentry-promoting increased IK1 may result from a Ca2+-dependent enhancement in expression of Kir2.1-subunits due to a calcineurin-NFAT-mediated decrease in micro-RNA-26.235
In atria, the primary mechanism leading to alternans results from abnormalities in Ca2+ signaling (Ca2+-driven alternans), with APD alternans being secondary to Ca2+-alternans.262 Despite some controversies about whether Ca2+-alternans results from fluctuations in SR Ca2+ content or from changes in RyR2 refractoriness, Ca2+-alternans can be observed in both animal models of AF and in humans with AF. For instance, SR Ca2+ leak increases the susceptibility to Ca2+-alternans and atrial arrhythmias in mice with CPVT mutations in RyR2,263 and in rabbits with chronic myocardial infarction or hypertension induced atrial remodeling Ca2+-alternans is a prominent feature of the arrhythmogenic substrate.264, 265 Atrial cardiomyocytes from patients with cAF are also more prone to Ca2+-alternans, an effect which appears to involve an increased activation of adenosine A2A receptors with subsequent enhancement of RyR2-mediated SR Ca2+ leak.266 Overall, computer modeling clearly suggests that elevated Ca2+-driven APD alternans leads to increased arrhythmia vulnerability, complexity, and persistence due to increased heterogeneity of repolarization in atria.267
There is evidence that abnormal intracellular Ca2+ handling promotes atrial remodeling. Mice with cardiac-restricted overexpression of a repressor form of the cAMP-response element modulator (CREM-Tg mice) develop atrial dilatation, abnormal cardiomyocyte growth, atrial fibrosis along with conduction disturbance leading to spontaneous AF.268 By crossing the CREM-Tg mice with RyR2-S2814A mice, in which RyR2 phosphorylation by CaMKII is inhibited, the development of a substrate for spontaneous AF was prevented.269 These studies suggest that RyR2-mediated SR Ca2+ leak is involved in atrial remodeling, potentially by activation of calcineurin-NFAT-mediated changes in gene transcription.269
In addition, there is emerging evidence that intracellular Ca2+ signaling regulates the proliferation and the transition of fibroblasts to collagen-secreting myofibroblasts, thereby promoting reentry.235 Transient-receptor potential canonical-3 channels (TRPC3) are key mediators of the fibroblast-to myofibroblast transition and their increase in AF involves the NFAT-microRNA-26 pathway.270 Overall these findings indicate that abnormal RyR2-mediated SR Ca2+ leak and the related Ca2+-dependent signaling may drive AF progression via these and possibly other unrecognized remodeling pathways, leading to the evolution of AF-maintaining substrate for reentry (Figure 7).
In summary, despite some controversies about the precise role of RyR2 in AF there is good evidence for contribution of abnormal Ca2+ signaling to the formation of the trigger and the substrate for reentry in both animal models and humans with AF.221 However, it is unknown whether intracellular Ca2+ oscillations are required and sufficient to sustain high-frequency foci once the arrhythmia persists. During high-frequency pacing of normal Langendorff-perfused whole rabbit hearts, RyR2 refractoriness initiates SR Ca2+-release alternans in the ventricle without concomitant changes in diastolic SR Ca2+ alternans, which points to a potential role of RyR2 dysfunction in Ca2+ alternans during pacing.271 However, in this model RyR2-related Ca2+ alternans did not play a major role for the transition of spatially concordant to spatially discordant alternans and the transition of alternans to VF, which rather depended on APD and CV restitution.271 These findings can be interpreted to suggest that while RyR2-related Ca2+ alternans is involved in the initiation of arrhythmias, the maintenance of VF might be less dependent on intracellular SR Ca2+-release oscillations. Of note, studies employing optical mapping of voltage and Ca2+ were not yet performed in the diseased ventricle or atrium, thus the consequences of dysfunctional RyR2 (and other ECC components) for arrhythmia maintenance, particularly in the diseased atrium, remain unknown and require thorough addressing in subsequent studies. Simultaneous high resolution optical mapping of voltage and Ca2+ in perfused intact human atria of sinus rhythm and AF patients should be performed to obtain first hints about the putative role of intracellular Ca2+ oscillations for the fibrillatory process during pacing-induced AF.272
THERAPEUTIC APPROACHES TO CORRECTING CALCIUM MISHANDLING
Ca2+-handling within cardiomyocytes has been recognized as a potential target for the treatment of cardiac disease for a long time. One class of drugs known as ‘Ca2+ channel antagonists’ target the voltage-gated sarcolemmal Ca2+ channels, and are currently being used clinically to treat hypertension, angina pectoris, cardiomyopathy, and cardiac arrhythmias. Fleckenstein described the first Ca2+ channel blockers as new drugs for the treatment of coronary disease about 50 years ago.273 During decades of subsequent studies, the role of Ca2+ channels in cardiac muscle contraction was elucidated [for review, see274]. Moreover, the biophysical and genetic identities of various voltage-gated Ca2+ channels were subsequently described.275, 276 Several classes of antagonists have been described (i.e., benzothiazepines, phenylalkylamines, and dihydropyridines), and are now part of the formulary for the treatment of cardiac diseases including arrhythmias. Ca2+ channel blockers are able to decrease the automaticity of ectopic foci in the heart and have emerging uses in a number of arrhythmia. For example, T-type Ca2+ channel blockers and LTCC blockers have efficacy in reducing AF arrhythmia burden and can prevent electrical remodeling.277–279 Moreover, while mainstay for treatment of CPVT is beta-blockade, there has been early evidence that also blocking ICa,L with the LTCC blocker verapamil prevented ventricular arrhythmias.279. Overall, it is believed that reduced ICa,L results in less Ca2+ overload of the myocyte, reducing predisposition to ectopy which can trigger arrhythmias.
During the past 15 years or so, several groups including our own have tried to develop pharmaceutical agents that target the intracellular Ca2+ release channel. To our knowledge, the first example of an experimental small molecule is K201 (also referred to as JTV519), a 1,4-benzothiazepine shown to normalize RyR2 gating in a canine model with tachycardia-induced HF.280 Subsequently, K201 was shown to prevent lethal ventricular tachyarrhythmias in a mouse model of CPVT by stabilizing RyR2 channels.281 Studies using recombinantly expressed RyR2 channels with CPVT-linked missense mutations showed that K201 can normalize mutant channel activity.282 In addition, K201 was shown to exert anti-arrhythmic effects against AF in an experimental guinea pig model.283 Although K201 normalizes defective RyR2 channels, this compound also inhibits various other targets including annexin V and K+ channels, raising concerns about potential off-target and pro-arrhythmic side-effects.284, 285 The proposed mechanism of RyR2 stabilization, through normalization of the binding stoichiometry of RyR2 and FKBP12.6, remains controversial. For example, the role of FKBP12.6 in reducing RyR2-mediated Ca2+ leak has been debated and this topic has been robustly reviewed.77, 286 Further, dissociating FKBP12.6 from RyR2 by FK506 did not affect suppression of spontaneous Ca2+ release events in rat ventricular myocytes questioning the role of FKBP12.6 binding in the mechanism of FK506.287 While debate exists in the field, there is a preponderance of datam, which suggests that RyR2 stabilization can be achieved by small molecules. Since discovery of this first molecule, newer generations of RyR2 stabilizing molecules have been developed. For example, a 1,4-benzothiazepine named S107 - a more specific RyR2-blocker - was shown to prevent ventricular arrhythmias in a CPVT mouse model heterozygous for mutation R2474S.80 Moreover, S107 has been shown to inhibit the RyR2-mediated diastolic SR Ca2+ leak in atrial myocytes in a number of RYR2 mutation knock-in models and decreased the incidence of burst pacing induced AF.250 While believed to have less off-target effects on a host of other receptors than K201, there have yet to be comprehensive studies on the major ion channels responsible for EC-coupling and Ca2+ homeostasis.80
Although flecainide is a class IC anti-arrhythmic drug with Na+ channel blocking properties, this drug has also been shown to inhibit RyR2 and exert therapeutic effects in mouse models of and patients with CPVT.288 This is most salient in patients for whom beta blockade is less effective. For example, flecainide has been shown to inhibit aberrant RyR2 activity and reduce spontaneous Ca2+ waves in both patients with CPVT refractory to beta blocker therapy and in Casq2−/− mouse models of CPVT in numerous independent studies.289–291. While the suppression of aberrant SR Ca2+ release seems a consistent effect of flecainide, other investigators have questioned whether RyR2 is the primary molecular target. For example, increasing the threshold for triggered activity by action on the cardiac Na+ channels with minimal effect on intracellular Ca2+ flux has been proposed.291 In addition, reducing elevated intracellular Ca2+ levels through reduction of INa leading to increased net Ca2+ influx via NCX has also been proposed.292 In an attempt to reconcile these various mechanisms, a so-called “triple mode of action” of flecainide has been proposed whereby all these various mechanisms are incorporated with the predominant effect being reduction of spontaneous Ca2+ release from RyR2.293, 294
In addition to these molecules, other classes of RyR2 inhibitors with anti-arrhythmic effects have been described, including dantrolene,295 carvedilol analogues,296 and tetracaine derivatives.297 In addition to LTCC and RyR2, other Ca2+ channels, Ca2+ transporters, and Ca2+-dependent signaling molecules (such as CaM, CaMKII) are potential therapeutic targets.
CONCLUDING REMARKS
The last 3 decades have seen a remarkable expansion in identifying the genetic and molecular etiologies of both congenital and acquired cardiac arrhythmias. While the specific molecular mechanisms of arrhythmic remodeling of the heart are as diverse as the many ways in which arrhythmia can present, Ca2+ is a critical and central player in many. Progress into identifying the role of Ca2+ in arrhythmias has led to novel understanding of the physiologic and pathologic regulation of the cardiomyocyte. When coupled to rapid advancement in genetic sequencing platforms, and recent breakthroughs in the development of both in vitro and in vivo models of disease, these advances offer the possibility of revolutionizing the diagnosis and treatment of these common and potentially life-threatening conditions.
Acknowledgments
SOURCES OF FUNDING
A.P.L. is supported by the National Institutes of Health (NIH) L40-HL129273, Baylor College of Medicine Department of Pediatrics Pilot Grant Award, and the Pediatric and Congenital Electrophysiology Society Paul C. Gillette Award. D.D. is supported by DZHK (German Center for Cardiovascular Research), the German Research Foundation DFG (Do 769/4-1), and a National Institutes of Health grant (NIH R01-HL131517). X.H.T.W. is supported by NIH grants R01-HL089598, R01-HL091947, R01-HL117641, and R41-HL129570, and American Heart Association grant 13EIA14560061.
Footnotes
DISCLOSURES
X.H.T.W. is a founding partner of Elex Biotech, a start-up company that developed drug molecules that target ryanodine receptors for the treatment of cardiac arrhythmia disorders.
References
- 1.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 2.Ringer S. A further Contribution regarding the influence of the different Constituents of the Blood on the Contraction of the Heart. J Physiol. 1883;4:29–42. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 4.Heijman J, Voigt N, Wehrens XH, Dobrev D. Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front Pharmacol. 2014;5:30. doi: 10.3389/fphar.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Boston: Kluwer Academic Publisher; 2001. [Google Scholar]
- 6.Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273:920–927. doi: 10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- 7.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 8.Dobrev D. Unique cardiomyocyte ultrastructure in atria: Role of T tubules in subcellular Ca2+ signaling and atrial arrhythmogenesis. Heart Rhythm. 2017;14:282–283. doi: 10.1016/j.hrthm.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circ Physiol. 2011;301:H1996–2005. doi: 10.1152/ajpheart.00284.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greiser M, Kerfant BG, Williams GS, et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest. 2014;124:4759–4772. doi: 10.1172/JCI70102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandenburg S, Kohl T, Williams GS, et al. Axial tubule junctions control rapid calcium signaling in atria. J Clin Invest. 2016;126:3999–4015. doi: 10.1172/JCI88241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaami T, Ishida H, Seguchi H, Hirota Y, Kadono T, Genka C, Nakazawa H, Barry WH. Difference in propagation of Ca2+ release in atrial and ventricular myocytes. Jpn J Physiol. 2005;55:81–91. doi: 10.2170/jjphysiol.R2077. [DOI] [PubMed] [Google Scholar]
- 13.Zima AV, Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias. J Physiol. 2004;555:607–615. doi: 10.1113/jphysiol.2003.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catterall WA. Regulation of Cardiac Calcium Channels in the Fight-or-Flight Response. Curr Mol Pharmacol. 2015;8:12–21. doi: 10.2174/1874467208666150507103417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohler PJ, Wehrens XH. Mechanisms of human arrhythmia syndromes: abnormal cardiac macromolecular interactions. Physiology (Bethesda) 2007;22:342–350. doi: 10.1152/physiol.00018.2007. [DOI] [PubMed] [Google Scholar]
- 17.Abriel H, Rougier JS, Jalife J. Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ Res. 2015;116:1971–1988. doi: 10.1161/CIRCRESAHA.116.305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCauley MD, Wehrens XH. Ryanodine receptor phosphorylation, calcium/calmodulin-dependent protein kinase II, and life-threatening ventricular arrhythmias. Trends Cardiovasc Med. 2011;21:48–51. doi: 10.1016/j.tcm.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Valle G, Furlan S, Nani A, Gyorke S, Fill M, Volpe P. Mechanism of calsequestrin regulation of single cardiac ryanodine receptor in normal and pathological conditions. J Gen Physiol. 2013;142:127–136. doi: 10.1085/jgp.201311022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heijman J, Ghezelbash S, Wehrens XH, Dobrev D. Serine/Threonine Phosphatases in Atrial Fibrillation. J Mol Cell Cardiol. 2017;103:110–120. doi: 10.1016/j.yjmcc.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang DY, Lebesgue N, Beavers DL, Alsina KM, Damen JM, Voigt N, Dobrev D, Wehrens XH, Scholten A. Alterations in the interactome of serine/threonine protein phosphatase type-1 in atrial fibrillation patients. J Am Coll Cardiol. 2015;65:163–173. doi: 10.1016/j.jacc.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, Showell J, McCauley MD, Scholten A, Wehrens XH. SPEG (Striated Muscle Preferentially Expressed Protein Kinase) Is Essential for Cardiac Function by Regulating Junctional Membrane Complex Activity. Circ Res. 2017;120:110–119. doi: 10.1161/CIRCRESAHA.116.309977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heijman J, Dewenter M, El-Armouche A, Dobrev D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol. 2013;64:90–98. doi: 10.1016/j.yjmcc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Shaw RM, Colecraft HM. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res. 2013;98:177–186. doi: 10.1093/cvr/cvt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galbiati F, Razani B, Lisanti MP. Caveolae and caveolin-3 in muscular dystrophy. Trends Mol Med. 2001;7:435–441. doi: 10.1016/s1471-4914(01)02105-0. [DOI] [PubMed] [Google Scholar]
- 28.Balijepalli RC, Kamp TJ. Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol. 2008;98:149–160. doi: 10.1016/j.pbiomolbio.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. doi: 10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 30.Gustavsson M, Verardi R, Mullen DG, Mote KR, Traaseth NJ, Gopinath T, Veglia G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc Natl Acad Sci U S A. 2013;110:17338–17343. doi: 10.1073/pnas.1303006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am J Physiol Heart Circ Physiol. 2007;293:H1581–1589. doi: 10.1152/ajpheart.00278.2007. [DOI] [PubMed] [Google Scholar]
- 33.Kiewitz R, Acklin C, Schafer BW, Maco B, Uhrik B, Wuytack F, Erne P, Heizmann CW. Ca2+-dependent interaction of S100A1 with the sarcoplasmic reticulum Ca2+-ATPase2a and phospholamban in the human heart. Biochem Biophys Res Commun. 2003;306:550–557. doi: 10.1016/s0006-291x(03)00987-2. [DOI] [PubMed] [Google Scholar]
- 34.Ihara Y, Kageyama K, Kondo T. Overexpression of calreticulin sensitizes SERCA2a to oxidative stress. Biochem Biophys Res Commun. 2005;329:1343–1349. doi: 10.1016/j.bbrc.2005.02.112. [DOI] [PubMed] [Google Scholar]
- 35.Weiss JN, Garfinkel A, Karagueuzian HS, Nguyen TP, Olcese R, Chen PS, Qu Z. Perspective: a dynamics-based classification of ventricular arrhythmias. J Mol Cell Cardiol. 2015;82:136–152. doi: 10.1016/j.yjmcc.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol. 2016;13:575–590. doi: 10.1038/nrcardio.2016.118. [DOI] [PubMed] [Google Scholar]
- 37.Benitah JP, Alvarez JL, Gomez AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 38.Karagueuzian HS, Pezhouman A, Angelini M, Olcese R. Enhanced Late Na and Ca Currents as Effective Antiarrhythmic Drug Targets. Front Pharmacol. 2017;8:36. doi: 10.3389/fphar.2017.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sipido KR, Bito V, Antoons G, Volders PG, Vos MA. Na/Ca exchange and cardiac ventricular arrhythmias. Ann N Y Acad Sci. 2007;1099:339–348. doi: 10.1196/annals.1387.066. [DOI] [PubMed] [Google Scholar]
- 40.Chiamvimonvat N, Chen-Izu Y, Clancy CE, et al. Potassium currents in the heart: functional roles in repolarization, arrhythmia and therapeutics. J Physiol. 2017;595:2229–2252. doi: 10.1113/JP272883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szabo B, Sweidan R, Rajagopalan CV, Lazzara R. Role of Na+:Ca2+ exchange current in Cs(+)-induced early afterdepolarizations in Purkinje fibers. J Cardiovasc Electrophysiol. 1994;5:933–944. doi: 10.1111/j.1540-8167.1994.tb01133.x. [DOI] [PubMed] [Google Scholar]
- 42.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Heller Brown J, Bers DM, McCulloch AD. Nonequilibrium reactivation of Na+ current drives early afterdepolarizations in mouse ventricle. Circ Arrhythm Electrophysiol. 2014;7:1205–1213. doi: 10.1161/CIRCEP.113.001666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, Scherlag BJ, Po SS. Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol. 2006;47:1196–1206. doi: 10.1016/j.jacc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 44.Asakura K, Cha CY, Yamaoka H, Horikawa Y, Memida H, Powell T, Amano A, Noma A. EAD and DAD mechanisms analyzed by developing a new human ventricular cell model. Prog Biophys Mol Biol. 2014;116:11–24. doi: 10.1016/j.pbiomolbio.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Sung RJ, Wu SN, Wu JS, Chang HD, Luo CH. Electrophysiological mechanisms of ventricular arrhythmias in relation to Andersen-Tawil syndrome under conditions of reduced IK1: a simulation study. Am J Physiol Heart Circ Physiol. 2006;291:H2597–2605. doi: 10.1152/ajpheart.00393.2006. [DOI] [PubMed] [Google Scholar]
- 46.Zipes DP. Mechanisms of clinical arrhythmias. J Cardiovasc Electrophysiol. 2003;14:902–912. doi: 10.1046/j.1540-8167.2003.03228.x. [DOI] [PubMed] [Google Scholar]
- 47.Shkryl VM, Maxwell JT, Domeier TL, Blatter LA. Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol Heart Circ Physiol. 2012;302:H2310–2320. doi: 10.1152/ajpheart.00079.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato D, Shiferaw Y, Garfinkel A, Weiss JN, Qu Z, Karma A. Spatially discordant alternans in cardiac tissue: role of calcium cycling. Circ Res. 2006;99:520–527. doi: 10.1161/01.RES.0000240542.03986.e7. [DOI] [PubMed] [Google Scholar]
- 49.Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. doi: 10.1038/nrcardio.2012.93. [DOI] [PubMed] [Google Scholar]
- 50.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 51.Tester DJ, Kopplin LJ, Will ML, Ackerman MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2005;2:1099–1105. doi: 10.1016/j.hrthm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 52.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 53.Paludan-Muller C, Ahlberg G, Ghouse J, Herfelt C, Svendsen JH, Haunso S, Kanters JK, Olesen MS. Integration of 60,000 exomes and ACMG guidelines question the role of Catecholaminergic Polymorphic Ventricular Tachycardia-associated variants. Clin Genet. 2017;91:63–72. doi: 10.1111/cge.12847. [DOI] [PubMed] [Google Scholar]
- 54.Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol. 1990;258:H1796–1805. doi: 10.1152/ajpheart.1990.258.6.H1796. [DOI] [PubMed] [Google Scholar]
- 55.Swan H, Piippo K, Viitasalo M, Heikkila P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035–2042. doi: 10.1016/s0735-1097(99)00461-1. [DOI] [PubMed] [Google Scholar]
- 56.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 57.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 58.Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 59.Kannankeril PJ, Mitchell BM, Goonasekera SA, et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kujala K, Paavola J, Lahti A, Larsson K, Pekkanen-Mattila M, Viitasalo M, Lahtinen AM, Toivonen L, Kontula K, Swan H, Laine M, Silvennoinen O, Aalto-Setala K. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS One. 2012;7:e44660. doi: 10.1371/journal.pone.0044660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houser SR. Does protein kinase a-mediated phosphorylation of the cardiac ryanodine receptor play any role in adrenergic regulation of calcium handling in health and disease? Circ Res. 2010;106:1672–1674. doi: 10.1161/CIRCRESAHA.110.221853. [DOI] [PubMed] [Google Scholar]
- 62.Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334–338. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- 63.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered Stoichiometry of FKBP12.6 Versus Ryanodine Receptor as a Cause of Abnormal Ca2+ Leak Through Ryanodine Receptor in Heart Failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 64.Xiao RP, Valdivia HH, Bogdanov K, Valdivia C, Lakatta EG, Cheng H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. J Physiol. 1997;500( Pt 2):343–354. doi: 10.1113/jphysiol.1997.sp022025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Timerman AP, Onoue H, Xin HB, Barg S, Copello J, Wiederrecht G, Fleischer S. Selective binding of FKBP12.6 by the cardiac ryanodine receptor. J Biol Chem. 1996;271:20385–20391. doi: 10.1074/jbc.271.34.20385. [DOI] [PubMed] [Google Scholar]
- 66.Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153:699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 69.Walweel K, Molenaar P, Imtiaz MS, Denniss A, Dos Remedios C, van Helden DF, Dulhunty AF, Laver DR, Beard NA. Ryanodine receptor modification and regulation by intracellular Ca2+ and Mg2+ in healthy and failing human hearts. J Mol Cell Cardiol. 2017;104:53–62. doi: 10.1016/j.yjmcc.2017.01.016. [DOI] [PubMed] [Google Scholar]
- 70.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, Berretta RM, Koch WJ, Chen X, Gao E, Valdivia HH, Houser SR. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–840. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houser SR. Role of RyR2 phosphorylation in heart failure and arrhythmias: protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ Res. 2014;114:1320–1327. doi: 10.1161/CIRCRESAHA.114.300569. discussion 1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 74.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 75.Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114:1311–1319. doi: 10.1161/CIRCRESAHA.114.300568. discussion 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCauley MD, Wehrens XH. Targeting ryanodine receptors for anti-arrhythmic therapy. Acta Pharmacol Sin. 2011;32:749–757. doi: 10.1038/aps.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 79.Goddard CA, Ghais NS, Zhang Y, Williams AJ, Colledge WH, Grace AA, Huang CL. Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol (Oxf) 2008;194:123–140. doi: 10.1111/j.1748-1716.2008.01865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herron TJ, Milstein ML, Anumonwo J, Priori SG, Jalife J. Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2010;7:1122–1128. doi: 10.1016/j.hrthm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jung CB, Moretti A, Mederos y Schnitzler M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–191. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang XH, Haviland S, Wei H, Saric T, Fatima A, Hescheler J, Cleemann L, Morad M. Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium. 2013;54:57–70. doi: 10.1016/j.ceca.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Di Pasquale E, Lodola F, Miragoli M, Denegri M, Avelino-Cruz JE, Buonocore M, Nakahama H, Portararo P, Bloise R, Napolitano C, Condorelli G, Priori SG. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4:e843. doi: 10.1038/cddis.2013.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Penttinen K, Swan H, Vanninen S, Paavola J, Lahtinen AM, Kontula K, Aalto-Setala K. Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS One. 2015;10:e0125366. doi: 10.1371/journal.pone.0125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B, Pras E. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation. 2001;103:2822–2827. doi: 10.1161/01.cir.103.23.2822. [DOI] [PubMed] [Google Scholar]
- 88.Gray B, Bagnall RD, Lam L, Ingles J, Turner C, Haan E, Davis A, Yang PC, Clancy CE, Sy RW, Semsarian C. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2016;13:1652–1660. doi: 10.1016/j.hrthm.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. J Biol Chem. 1983;258:1197–1204. [PubMed] [Google Scholar]
- 90.Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem. 1994;135:61–70. doi: 10.1007/BF00925961. [DOI] [PubMed] [Google Scholar]
- 91.Slupsky JR, Ohnishi M, Carpenter MR, Reithmeier RA. Characterization of cardiac calsequestrin. Biochemistry. 1987;26:6539–6544. doi: 10.1021/bi00394a038. [DOI] [PubMed] [Google Scholar]
- 92.Faggioni M, Kryshtal DO, Knollmann BC. Calsequestrin mutations and catecholaminergic polymorphic ventricular tachycardia. Pediatr Cardiol. 2012;33:959–967. doi: 10.1007/s00246-012-0256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Knollmann BC, Chopra N, Hlaing T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, Napolitano C, Nori A, Williams SC, Gyorke S. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471–477. doi: 10.1161/01.RES.0000115944.10681.EB. [DOI] [PubMed] [Google Scholar]
- 95.Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner DA, Berul CI, Eldar M, Seidman CE, Seidman JG. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2007;117:1814–1823. doi: 10.1172/JCI31080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dulhunty AF, Wium E, Li L, Hanna AD, Mirza S, Talukder S, Ghazali NA, Beard NA. Proteins within the intracellular calcium store determine cardiac RyR channel activity and cardiac output. Clin Exp Pharmacol Physiol. 2012;39:477–484. doi: 10.1111/j.1440-1681.2012.05704.x. [DOI] [PubMed] [Google Scholar]
- 98.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A, Itskovitz-Eldor J, Binah O. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J Cell Mol Med. 2012;16:468–482. doi: 10.1111/j.1582-4934.2011.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Novak A, Barad L, Lorber A, Gherghiceanu M, Reiter I, Eisen B, Eldor L, Itskovitz-Eldor J, Eldar M, Arad M, Binah O. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J Cell Mol Med. 2015;19:2006–2018. doi: 10.1111/jcmm.12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhuiyan ZA, Hamdan MA, Shamsi ET, Postma AV, Mannens MM, Wilde AA, Al-Gazali L. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J Cardiovasc Electrophysiol. 2007;18:1060–1066. doi: 10.1111/j.1540-8167.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 102.Devalla HD, Gelinas R, Aburawi EH, et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016;8:1390–1408. doi: 10.15252/emmm.201505719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nyegaard M, Overgaard MT, Sondergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, Fosdal I, Christiansen M, Borglum AD. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stevens FC. Calmodulin: an introduction. Can J Biochem Cell Biol. 1983;61:906–910. doi: 10.1139/o83-115. [DOI] [PubMed] [Google Scholar]
- 105.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 106.Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 108.Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, Navedo MF, Santana LF. Graded Ca(2)(+)/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife. 2015:4. doi: 10.7554/eLife.05608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamaguchi N, Xu L, Pasek DA, Evans KE, Meissner G. Molecular basis of calmodulin binding to cardiac muscle Ca(2+) release channel (ryanodine receptor) J Biol Chem. 2003;278:23480–23486. doi: 10.1074/jbc.M301125200. [DOI] [PubMed] [Google Scholar]
- 110.Sondergaard MT, Liu Y, Larsen KT, Nani A, Tian X, Holt C, Wang R, Wimmer R, Van Petegem F, Fill M, Chen SR, Overgaard MT. The Arrhythmogenic Calmodulin p.Phe142Leu Mutation Impairs C-domain Ca2+ Binding but Not Calmodulin-dependent Inhibition of the Cardiac Ryanodine Receptor. J Biol Chem. 2017;292:1385–1395. doi: 10.1074/jbc.M116.766253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sondergaard MT, Tian X, Liu Y, Wang R, Chazin WJ, Chen SR, Overgaard MT. Arrhythmogenic Calmodulin Mutations Affect the Activation and Termination of Cardiac Ryanodine Receptor-mediated Ca2+ Release. J Biol Chem. 2015;290:26151–26162. doi: 10.1074/jbc.M115.676627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Knudson CM, Stang KK, Jorgensen AO, Campbell KP. Biochemical characterization of ultrastructural localization of a major junctional sarcoplasmic reticulum glycoprotein (triadin) J Biol Chem. 1993;268:12637–12645. [PubMed] [Google Scholar]
- 114.Marty I, Faure J, Fourest-Lieuvin A, Vassilopoulos S, Oddoux S, Brocard J. Triadin: what possible function 20 years later? J Physiol. 2009;587:3117–3121. doi: 10.1113/jphysiol.2009.171892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chopra N, Yang T, Asghari P, et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:7636–7641. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Landstrom AP, Tester DJ, Ackerman MJ. Role of genetic testing for sudden death predisposing heart conditions in athletes. 2011 [Google Scholar]
- 117.Wehrens XH, Vos MA, Doevendans PA, Wellens HJ. Novel insights in the congenital long QT syndrome. Ann Intern Med. 2002;137:981–992. doi: 10.7326/0003-4819-137-12-200212170-00012. [DOI] [PubMed] [Google Scholar]
- 118.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 119.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 120.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 121.Arking DE, Pulit SL, Crotti L, et al. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. doi: 10.1038/ng.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ Res. 2011;108:607–618. doi: 10.1161/CIRCRESAHA.110.224279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reichenbach H, Meister EM, Theile H. The heart-hand syndrome. A new variant of disorders of heart conduction and syndactylia including osseous changes in hands and feet. Kinderarztl Prax. 1992;60:54–56. [PubMed] [Google Scholar]
- 124.Marks ML, Trippel DL, Keating MT. Long QT syndrome associated with syndactyly identified in females. Am J Cardiol. 1995;76:744–745. doi: 10.1016/s0002-9149(99)80216-1. [DOI] [PubMed] [Google Scholar]
- 125.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 127.Boczek NJ, Ye D, Jin F, Tester DJ, Huseby A, Bos JM, Johnson AJ, Kanter R, Ackerman MJ. Identification and Functional Characterization of a Novel CACNA1C-Mediated Cardiac Disorder Characterized by Prolonged QT Intervals With Hypertrophic Cardiomyopathy, Congenital Heart Defects, and Sudden Cardiac Death. Circ Arrhythm Electrophysiol. 2015;8:1122–1132. doi: 10.1161/CIRCEP.115.002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Landstrom AP, Boczek NJ, Ye D, Miyake CY, De la Uz CM, Allen HD, Ackerman MJ, Kim JJ. Novel long QT syndrome-associated missense mutation, L762F, in CACNA1C-encoded L-type calcium channel imparts a slower inactivation tau and increased sustained and window current. Int J Cardiol. 2016;220:290–298. doi: 10.1016/j.ijcard.2016.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thiel WH, Chen B, Hund TJ, Koval OM, Purohit A, Song LS, Mohler PJ, Anderson ME. Proarrhythmic defects in Timothy syndrome require calmodulin kinase II. Circulation. 2008;118:2225–2234. doi: 10.1161/CIRCULATIONAHA.108.788067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu ZI, Clancy CE. L-type Ca2+ channel mutations and T-wave alternans: a model study. Am J Physiol Heart Circ Physiol. 2007;293:H3480–3489. doi: 10.1152/ajpheart.00476.2007. [DOI] [PubMed] [Google Scholar]
- 131.Bai J, Wang K, Li Q, Yuan Y, Zhang H. Pro-arrhythmogenic effects of CACNA1C G1911R mutation in human ventricular tachycardia: insights from cardiac multi-scale models. Sci Rep. 2016;6:31262. doi: 10.1038/srep31262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boczek NJ, Miller EM, Ye D, Nesterenko VV, Tester DJ, Antzelevitch C, Czosek RJ, Ackerman MJ, Ware SM. Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm. 2015;12:211–219. doi: 10.1016/j.hrthm.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wemhoner K, Friedrich C, Stallmeyer B, Coffey AJ, Grace A, Zumhagen S, Seebohm G, Ortiz-Bonnin B, Rinne S, Sachse FB, Schulze-Bahr E, Decher N. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J Mol Cell Cardiol. 2015;80:186–195. doi: 10.1016/j.yjmcc.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 134.Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Reed GJ, Boczek NJ, Etheridge SP, Ackerman MJ. CALM3 mutation associated with long QT syndrome. Heart Rhythm. 2015;12:419–422. doi: 10.1016/j.hrthm.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George AL, Jr, Yue DT. Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol. 2014;74:115–124. doi: 10.1016/j.yjmcc.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Boczek NJ, Gomez-Hurtado N, Ye D, et al. Spectrum and Prevalence of CALM1-, CALM2-, and CALM3-Encoded Calmodulin Variants in Long QT Syndrome and Functional Characterization of a Novel Long QT Syndrome-Associated Calmodulin Missense Variant, E141G. Circ Cardiovasc Genet. 2016;9:136–146. doi: 10.1161/CIRCGENETICS.115.001323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Limpitikul WB, Dick IE, Tester DJ, Boczek NJ, Limphong P, Yang W, Choi MH, Babich J, DiSilvestre D, Kanter RJ, Tomaselli GF, Ackerman MJ, Yue DT. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ Res. 2017;120:39–48. doi: 10.1161/CIRCRESAHA.116.309283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Altmann HM, Tester DJ, Will ML, Middha S, Evans JM, Eckloff BW, Ackerman MJ. Homozygous/Compound Heterozygous Triadin Mutations Associated With Autosomal-Recessive Long-QT Syndrome and Pediatric Sudden Cardiac Arrest: Elucidation of the Triadin Knockout Syndrome. Circulation. 2015;131:2051–2060. doi: 10.1161/CIRCULATIONAHA.115.015397. [DOI] [PubMed] [Google Scholar]
- 140.Marty I. Triadin regulation of the ryanodine receptor complex. J Physiol. 2014 doi: 10.1113/jphysiol.2014.281147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Conte G, Caputo ML, Regoli F, Marcon S, Klersy C, Adjibodou B, Del Bufalo A, Moccetti T, Auricchio A. True idiopathic ventricular fibrillation in out-of-hospital cardiac arrest survivors in the Swiss Canton Ticino: prevalence, clinical features, and long-term follow-up. Europace. 2017;19:259–266. doi: 10.1093/europace/euv447. [DOI] [PubMed] [Google Scholar]
- 142.Meyer L, Stubbs B, Fahrenbruch C, Maeda C, Harmon K, Eisenberg M, Drezner J. Incidence, causes, and survival trends from cardiovascular-related sudden cardiac arrest in children and young adults 0 to 35 years of age: a 30-year review. Circulation. 2012;126:1363–1372. doi: 10.1161/CIRCULATIONAHA.111.076810. [DOI] [PubMed] [Google Scholar]
- 143.Akai J, Makita N, Sakurada H, Shirai N, Ueda K, Kitabatake A, Nakazawa K, Kimura A, Hiraoka M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. doi: 10.1016/s0014-5793(00)01875-5. [DOI] [PubMed] [Google Scholar]
- 144.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 145.Marsman RF, Barc J, Beekman L, Alders M, Dooijes D, van den Wijngaard A, Ratbi I, Sefiani A, Bhuiyan ZA, Wilde AA, Bezzina CR. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2014;63:259–266. doi: 10.1016/j.jacc.2013.07.091. [DOI] [PubMed] [Google Scholar]
- 146.Rook MB, Bezzina Alshinawi C, Groenewegen WA, van Gelder IC, van Ginneken AC, Jongsma HJ, Mannens MM, Wilde AA. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44:507–517. doi: 10.1016/s0008-6363(99)00350-8. [DOI] [PubMed] [Google Scholar]
- 147.Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 149.Bos JM, Ommen SR, Ackerman MJ. Genetics of hypertrophic cardiomyopathy: one, two, or more diseases? Curr Opin Cardiol. 2007;22:193–199. doi: 10.1097/HCO.0b013e3280e1cc7f. [DOI] [PubMed] [Google Scholar]
- 150.Bongini C, Ferrantini C, Girolami F, Coppini R, Arretini A, Targetti M, Bardi S, Castelli G, Torricelli F, Cecchi F, Ackerman MJ, Padeletti L, Poggesi C, Olivotto I. Impact of Genotype on the Occurrence of Atrial Fibrillation in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2016;117:1151–1159. doi: 10.1016/j.amjcard.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 151.Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis-Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378. doi: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 152.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 153.Kapplinger JD, Landstrom AP, Bos JM, Salisbury BA, Callis TE, Ackerman MJ. Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J Cardiovasc Transl Res. 2014;7:347–361. doi: 10.1007/s12265-014-9542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 155.Olson TM, Karst ML, Whitby FG, Driscoll DJ. Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation. 2002;105:2337–2340. doi: 10.1161/01.cir.0000018444.47798.94. [DOI] [PubMed] [Google Scholar]
- 156.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nature Genetics. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 157.Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, Gregersen N, Hansen PS, Daandrup U, Borglum AD. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–R43. doi: 10.1172/JCI6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 159.Kimura A, Harada H, Park JE, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 160.Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, Bos JM, Tester DJ, Ommen SR, Potter JD, Ackerman MJ. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J Mol Cell Cardiol. 2008;45:281–288. doi: 10.1016/j.yjmcc.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 2011;9:91–100. doi: 10.1038/nrcardio.2011.159. [DOI] [PubMed] [Google Scholar]
- 162.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 163.Watkins H, Rosenzweig A, Hwang D, Levi T, McKenna W, Seidman C, Seidman J. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. New England Journal of Medicine. 1992;326:1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 164.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE. Mutations in the Genes for Cardiac Troponin T and alpha-Tropomyosin in Hypertrophic Cardiomyopathy. New England Journal of Medicine. 1995;332:1058–1065. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 165.Fananapazir L, Epstein ND. Genotype-phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation. 1994;89:22–32. doi: 10.1161/01.cir.89.1.22. [DOI] [PubMed] [Google Scholar]
- 166.Menon S, Michels V, Pellikka P, Ballew J, Karst M, Herron K, Nelson S, Rodeheffer R, Olson T. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet. 2008;74:445–454. doi: 10.1111/j.1399-0004.2008.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kokado H, Shimizu M, Yoshio H, Ino H, Okeie K, Emoto Y, Matsuyama T, Yamaguchi M, Yasuda T, Fujino N, Ito H, Mabuchi H. Clinical Features of Hypertrophic Cardiomyopathy Caused by a Lys183 Deletion Mutation in the Cardiac Troponin I Gene. Circulation. 2000;102:663–669. doi: 10.1161/01.cir.102.6.663. [DOI] [PubMed] [Google Scholar]
- 168.Brito D, Richard P, Isnard R, Pipa J, Komajda M, Madeira H. Familial hypertrophic cardiomyopathy: the same mutation, different prognosis. Comparison of two families with a long follow-up. Revista Portuguesa de Cardiologia. 2003;22:1445–1461. [PubMed] [Google Scholar]
- 169.Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation. 2010;122:2441–2449. doi: 10.1161/CIRCULATIONAHA.110.954446. discussion 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. doi: 10.1161/CIRCULATIONAHA.110.978924. discussion 2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 172.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006. Circulation. 2009;119:1085–1092. doi: 10.1161/CIRCULATIONAHA.108.804617. [DOI] [PubMed] [Google Scholar]
- 174.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 175.Eiras S, Narolska NA, van Loon RB, Boontje NM, Zaremba R, Jimenez CR, Visser FC, Stooker W, van der Velden J, Stienen GJ. Alterations in contractile protein composition and function in human atrial dilatation and atrial fibrillation. J Mol Cell Cardiol. 2006;41:467–477. doi: 10.1016/j.yjmcc.2006.06.072. [DOI] [PubMed] [Google Scholar]
- 176.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, Armentano C, Poggesi C, Torricelli F, Cecchi F. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83:630–638. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- 177.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 178.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 179.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Knollmann BC, Blatt SA, Horton K, de Freitas F, Miller T, Bell M, Housmans PR, Weissman NJ, Morad M, Potter JD. Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J Biol Chem. 2001;276:10039–10048. doi: 10.1074/jbc.M006745200. [DOI] [PubMed] [Google Scholar]
- 181.Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, Culbreath L, McCue J, Wang Y, Xu Y, Kerrick WG, Potter JD. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem. 2001;276:3743–3755. doi: 10.1074/jbc.M006746200. [DOI] [PubMed] [Google Scholar]
- 182.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res. 2003;92:428–436. doi: 10.1161/01.RES.0000059562.91384.1A. [DOI] [PubMed] [Google Scholar]
- 183.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol. 1999;517( Pt 1):143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Puglisi JL, Goldspink PH, Gomes AV, Utter MS, Bers DM, Solaro RJ. Influence of a constitutive increase in myofilament Ca(2+)-sensitivity on Ca(2+)-fluxes and contraction of mouse heart ventricular myocytes. Arch Biochem Biophys. 2014;552–553:50–59. doi: 10.1016/j.abb.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Parvatiyar MS, Landstrom AP, Figueiredo-Freitas C, Potter JD, Ackerman MJ, Pinto JR. A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J Biol Chem. 2012;287:31845–31855. doi: 10.1074/jbc.M112.377713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Landstrom AP, Ackerman MJ. Beyond the cardiac myofilament: hypertrophic cardiomyopathy-associated mutations in genes that encode calcium-handling proteins. Curr Mol Med. 2012;12:507–518. doi: 10.2174/156652412800620020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XH. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, Ma J, Wehrens XH, Ackerman MJ. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail. 2011;4:214–223. doi: 10.1161/CIRCHEARTFAILURE.110.958694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Quick AP, Landstrom AP, Wehrens XH. Junctophilin-2 at the intersection of arrhythmia and pathologic cardiac remodeling. Heart Rhythm. 2016;13:753–754. doi: 10.1016/j.hrthm.2015.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, Wehrens XH, Claycomb WC, Ko JK, Hwang M, Pan Z, Ma J, Ackerman MJ. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. doi: 10.1016/j.yjmcc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Beavers DL, Landstrom AP, Chiang DY, Wehrens XH. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res. 2014;103:198–205. doi: 10.1093/cvr/cvu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Landstrom AP, Beavers DL, Wehrens XH. The junctophilin family of proteins: from bench to bedside. Trends Mol Med. 2014;20:353–362. doi: 10.1016/j.molmed.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ, Wehrens XH. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 2013;62:2010–2019. doi: 10.1016/j.jacc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chiu C, Tebo M, Ingles J, Yeates L, Arthur JW, Lind JM, Semsarian C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2007;43:337–343. doi: 10.1016/j.yjmcc.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 196.Michalak M, Guo L, Robertson M, Lozak M, Opas M. Calreticulin in the heart. Mol Cell Biochem. 2004;263:137–142. doi: 10.1023/B:MCBI.0000041855.10149.5f. [DOI] [PubMed] [Google Scholar]
- 197.Faustino RS, Behfar A, Groenendyk J, Wyles SP, Niederlander N, Reyes S, Puceat M, Michalak M, Terzic A, Perez-Terzic C. Calreticulin secures calcium-dependent nuclear pore competency required for cardiogenesis. J Mol Cell Cardiol. 2016;92:63–74. doi: 10.1016/j.yjmcc.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 198.Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med. 2017;376:61–72. doi: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- 199.Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J Am Coll Cardiol. 2013;61:1945–1948. doi: 10.1016/j.jacc.2013.01.073. [DOI] [PubMed] [Google Scholar]
- 200.Kapplinger JD, Landstrom AP, Salisbury BA, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–2327. doi: 10.1016/j.jacc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 202.Roux-Buisson N, Gandjbakhch E, Donal E, et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: results of a systematic screening. Heart Rhythm. 2014;11:1999–2009. doi: 10.1016/j.hrthm.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 203.McCauley MD, Wehrens XH. Animal models of arrhythmogenic cardiomyopathy. Dis Model Mech. 2009;2:563–570. doi: 10.1242/dmm.002840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tayal U, Prasad S, Cook SA. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017;9:20. doi: 10.1186/s13073-017-0410-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vikhorev PG, Song W, Wilkinson R, Copeland O, Messer AE, Ferenczi MA, Marston SB. The dilated cardiomyopathy-causing mutation ACTC E361G in cardiac muscle myofibrils specifically abolishes modulation of Ca(2+) regulation by phosphorylation of troponin I. Biophys J. 2014;107:2369–2380. doi: 10.1016/j.bpj.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Klos M, Mundada L, Banerjee I, Morgenstern S, Myers S, Leone M, Kleid M, Herron T, Devaney E. Altered myocyte contractility and calcium homeostasis in alpha-myosin heavy chain point mutations linked to familial dilated cardiomyopathy. Arch Biochem Biophys. 2017;615:53–60. doi: 10.1016/j.abb.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 207.Ramratnam M, Salama G, Sharma RK, Wang DW, Smith SH, Banerjee SK, Huang XN, Gifford LM, Pruce ML, Gabris BE, Saba S, Shroff SG, Ahmad F. Gene-Targeted Mice with the Human Troponin T R141W Mutation Develop Dilated Cardiomyopathy with Calcium Desensitization. PLoS One. 2016;11:e0167681. doi: 10.1371/journal.pone.0167681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rowe GC, Asimaki A, Graham EL, Martin KD, Margulies KB, Das S, Saffitz J, Arany Z. Development of dilated cardiomyopathy and impaired calcium homeostasis with cardiac-specific deletion of ESRRbeta. Am J Physiol Heart Circ Physiol. 2017;312:H662–H671. doi: 10.1152/ajpheart.00446.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wyles SP, Hrstka SC, Reyes S, Terzic A, Olson TM, Nelson TJ. Pharmacological Modulation of Calcium Homeostasis in Familial Dilated Cardiomyopathy: An In Vitro Analysis From an RBM20 Patient-Derived iPSC Model. Clin Transl Sci. 2016;9:158–167. doi: 10.1111/cts.12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fish M, Shaboodien G, Kraus S, Sliwa K, Seidman CE, Burke MA, Crotti L, Schwartz PJ, Mayosi BM. Mutation analysis of the phospholamban gene in 315 South Africans with dilated, hypertrophic, peripartum and arrhythmogenic right ventricular cardiomyopathies. Sci Rep. 2016;6:22235. doi: 10.1038/srep22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am Heart J. 2011;161:165–171. doi: 10.1016/j.ahj.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Minamisawa S, Sato Y, Tatsuguchi Y, Fujino T, Imamura S, Uetsuka Y, Nakazawa M, Matsuoka R. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2003;304:1–4. doi: 10.1016/s0006-291x(03)00526-6. [DOI] [PubMed] [Google Scholar]
- 215.Medin M, Hermida-Prieto M, Monserrat L, Laredo R, Rodriguez-Rey JC, Fernandez X, Castro-Beiras A. Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN-42 C>G mutation. Eur J Heart Fail. 2007;9:37–43. doi: 10.1016/j.ejheart.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 216.Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 1986;261:13333–13341. [PubMed] [Google Scholar]
- 217.Inui M, Chamberlain BK, Saito A, Fleischer S. The nature of the modulation of Ca2+ transport as studied by reconstitution of cardiac sarcoplasmic reticulum. J Biol Chem. 1986;261:1794–1800. [PubMed] [Google Scholar]
- 218.Liu GS, Morales A, Vafiadaki E, Lam CK, Cai WF, Haghighi K, Adly G, Hershberger RE, Kranias EG. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res. 2015;107:164–174. doi: 10.1093/cvr/cvv127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Arvanitis DA, Sanoudou D, Kolokathis F, Vafiadaki E, Papalouka V, Kontrogianni-Konstantopoulos A, Theodorakis GN, Paraskevaidis IA, Adamopoulos S, Dorn GW, 2nd, Kremastinos DT, Kranias EG. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur Heart J. 2008;29:2514–2525. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Han P, Cai W, Wang Y, Lam CK, Arvanitis DA, Singh VP, Chen S, Zhang H, Zhang R, Cheng H, Kranias EG. Catecholaminergic-induced arrhythmias in failing cardiomyocytes associated with human HRCS96A variant overexpression. Am J Physiol Heart Circ Physiol. 2011;301:H1588–1595. doi: 10.1152/ajpheart.01153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dobrev D, Wehrens XH. Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol. 2017 doi: 10.1113/JP273048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Writing Committee M. Yancy CW, Jessup M, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:e240–327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 223.Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, Murabito JM, Vasan RS. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 224.Christiansen MN, Kober L, Weeke P, Vasan RS, Jeppesen JL, Smith JG, Gislason GH, Torp-Pedersen C, Andersson C. Age-Specific Trends in Incidence, Mortality, and Comorbidities of Heart Failure in Denmark, 1995 to 2012. Circulation. 2017;135:1214–1223. doi: 10.1161/CIRCULATIONAHA.116.025941. [DOI] [PubMed] [Google Scholar]
- 225.Writing Committee M. Yancy CW, Jessup M, et al. 2016 ACC/AHA/HFSA Focused Update on New Pharmacological Therapy for Heart Failure: An Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134:e282–293. doi: 10.1161/CIR.0000000000000435. [DOI] [PubMed] [Google Scholar]
- 226.Kaye DM, Hoshijima M, Chien KR. Reversing advanced heart failure by targeting Ca2+ cycling. Annu Rev Med. 2008;59:13–28. doi: 10.1146/annurev.med.59.052407.103237. [DOI] [PubMed] [Google Scholar]
- 227.Ikeda Y, Hoshijima M, Chien KR. Toward biologically targeted therapy of calcium cycling defects in heart failure. Physiology (Bethesda) 2008;23:6–16. doi: 10.1152/physiol.00033.2007. [DOI] [PubMed] [Google Scholar]
- 228.Loffredo FS, Nikolova AP, Pancoast JR, Lee RT. Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ Res. 2014;115:97–107. doi: 10.1161/CIRCRESAHA.115.302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Dudas K, Lappas G, Stewart S, Rosengren A. Trends in out-of-hospital deaths due to coronary heart disease in Sweden (1991 to 2006) Circulation. 2011;123:46–52. doi: 10.1161/CIRCULATIONAHA.110.964999. [DOI] [PubMed] [Google Scholar]
- 230.Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, Kottke TE, Giles WH, Capewell S. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med. 2007;356:2388–2398. doi: 10.1056/NEJMsa053935. [DOI] [PubMed] [Google Scholar]
- 231.Wagner S, Maier LS, Bers DM. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res. 2015;116:1956–1970. doi: 10.1161/CIRCRESAHA.116.304678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Nass RD, Aiba T, Tomaselli GF, Akar FG. Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Clin Pract Cardiovasc Med. 2008;5:196–207. doi: 10.1038/ncpcardio1130. [DOI] [PubMed] [Google Scholar]
- 233.Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. 2014;114:1453–1468. doi: 10.1161/CIRCRESAHA.114.303211. [DOI] [PubMed] [Google Scholar]
- 234.Goette A, Kalman JM, Aguinaga L, et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: definition, characterization, and clinical implication. Europace. 2016;18:1455–1490. doi: 10.1093/europace/euw161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114:1483–1499. doi: 10.1161/CIRCRESAHA.114.302226. [DOI] [PubMed] [Google Scholar]
- 236.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XH, Nattel S, Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156. doi: 10.1161/CIRCULATIONAHA.113.006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Waddell LB, Lemckert FA, Zheng XF, et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J Neuropathol Exp Neurol. 2011;70:302–313. doi: 10.1097/NEN.0b013e31821350b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N. Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol. 2014;7:1214–1222. doi: 10.1161/CIRCEP.114.001973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chiang DY, Zhang M, Voigt N, Alsina KM, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N. Identification of microRNA-mRNA dysregulations in paroxysmal atrial fibrillation. Int J Cardiol. 2015;184:190–197. doi: 10.1016/j.ijcard.2015.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 241.Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144. doi: 10.1161/CIRCRESAHA.109.203836. [DOI] [PubMed] [Google Scholar]
- 242.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- 243.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Greiser M, Neuberger HR, Harks E, El-Armouche A, Boknik P, de Haan S, Verheyen F, Verheule S, Schmitz W, Ravens U, Nattel S, Allessie MA, Dobrev D, Schotten U. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol. 2009;46:385–394. doi: 10.1016/j.yjmcc.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 245.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- 246.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047–1054. doi: 10.1016/j.hrthm.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sumitomo N, Sakurada H, Taniguchi K, Matsumura M, Abe O, Miyashita M, Kanamaru H, Karasawa K, Ayusawa M, Fukamizu S, Nagaoka I, Horie M, Harada K, Hiraoka M. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ J. 2007;71:1606–1609. doi: 10.1253/circj.71.1606. [DOI] [PubMed] [Google Scholar]
- 248.Pizzale S, Gollob MH, Gow R, Birnie DH. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol. 2008;19:1319–1321. doi: 10.1111/j.1540-8167.2008.01211.x. [DOI] [PubMed] [Google Scholar]
- 249.Chelu MG, Sarma S, Sood S, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, Marks AR. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012;111:708–717. doi: 10.1161/CIRCRESAHA.112.273342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.King JH, Zhang Y, Lei M, Grace AA, Huang CL, Fraser JA. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol (Oxf) 2013;207:308–323. doi: 10.1111/apha.12006. [DOI] [PubMed] [Google Scholar]
- 252.Heijman J, Wehrens XH, Dobrev D. Atrial arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia--is there a mechanistic link between sarcoplasmic reticulum Ca(2+) leak and re-entry? Acta Physiol (Oxf) 2013;207:208–211. doi: 10.1111/apha.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schondube FA, Hasenfuss G, Belardinelli L, Maier LS. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–2342. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 254.Fischer TH, Herting J, Mason FE, et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res. 2015;107:184–196. doi: 10.1093/cvr/cvv153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bogeholz N, Pauls P, Kaese S, Schulte JS, Lemoine MD, Dechering DG, Frommeyer G, Goldhaber JI, Seidl MD, Kirchhefer U, Eckardt L, Muller FU, Pott C. Triggered activity in atrial myocytes is influenced by Na+/Ca2+ exchanger activity in genetically altered mice. J Mol Cell Cardiol. 2016;101:106–115. doi: 10.1016/j.yjmcc.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Franz MR, Jamal SM, Narayan SM. The role of action potential alternans in the initiation of atrial fibrillation in humans: a review and future directions. Europace. 2012;14(Suppl 5):v58–v64. doi: 10.1093/europace/eus273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S. The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res. 2016;109:467–479. doi: 10.1093/cvr/cvv275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Heijman J, Voigt N, Ghezelbash S, Schirmer I, Dobrev D. Calcium Handling Abnormalities as a Target for Atrial Fibrillation Therapeutics: How Close to Clinical Implementation? J Cardiovasc Pharmacol. 2015;66:515–522. doi: 10.1097/FJC.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 259.Terentyev D, Rochira JA, Terentyeva R, Roder K, Koren G, Li W. Sarcoplasmic reticulum Ca(2)(+) release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2014;306:H738–746. doi: 10.1152/ajpheart.00621.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Qi XY, Diness JG, Brundel BJ, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, Grunnet M, Nattel S. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430–440. doi: 10.1161/CIRCULATIONAHA.113.003019. [DOI] [PubMed] [Google Scholar]
- 261.Cunha SR, Hund TJ, Hashemi S, et al. Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation. 2011;124:1212–1222. doi: 10.1161/CIRCULATIONAHA.111.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kanaporis G, Blatter LA. The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ Res. 2015;116:846–856. doi: 10.1161/CIRCRESAHA.116.305404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Xie W, Santulli G, Guo X, Gao M, Chen BX, Marks AR. Imaging atrial arrhythmic intracellular calcium in intact heart. J Mol Cell Cardiol. 2013;64:120–123. doi: 10.1016/j.yjmcc.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kettlewell S, Burton FL, Smith GL, Workman AJ. Chronic myocardial infarction promotes atrial action potential alternans, afterdepolarizations, and fibrillation. Cardiovasc Res. 2013;99:215–224. doi: 10.1093/cvr/cvt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Pluteanu F, Hess J, Plackic J, Nikonova Y, Preisenberger J, Bukowska A, Schotten U, Rinne A, Kienitz MC, Schafer MK, Weihe E, Goette A, Kockskamper J. Early subcellular Ca2+ remodelling and increased propensity for Ca2+ alternans in left atrial myocytes from hypertensive rats. Cardiovasc Res. 2015;106:87–97. doi: 10.1093/cvr/cvv045. [DOI] [PubMed] [Google Scholar]
- 266.Molina CE, Llach A, Herraiz-Martinez A, Tarifa C, Barriga M, Wiegerinck RF, Fernandes J, Cabello N, Vallmitjana A, Benitez R, Montiel J, Cinca J, Hove-Madsen L. Prevention of adenosine A2A receptor activation diminishes beat-to-beat alternation in human atrial myocytes. Basic Res Cardiol. 2016;111:5. doi: 10.1007/s00395-015-0525-2. [DOI] [PubMed] [Google Scholar]
- 267.Chang KC, Trayanova NA. Mechanisms of arrhythmogenesis related to calcium-driven alternans in a model of human atrial fibrillation. Sci Rep. 2016;6:36395. doi: 10.1038/srep36395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Muller FU, Lewin G, Baba HA, Boknik P, Fabritz L, Kirchhefer U, Kirchhof P, Loser K, Matus M, Neumann J, Riemann B, Schmitz W. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem. 2005;280:6906–6914. doi: 10.1074/jbc.M407864200. [DOI] [PubMed] [Google Scholar]
- 269.Li N, Chiang DY, Wang S, et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. 2014;129:1276–1285. doi: 10.1161/CIRCULATIONAHA.113.006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Harada M, Luo X, Qi XY, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM, Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res. 2014;114:1410–1421. doi: 10.1161/CIRCRESAHA.114.302505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Li N, Csepe TA, Hansen BJ, et al. Adenosine-Induced Atrial Fibrillation: Localized Reentrant Drivers in Lateral Right Atria due to Heterogeneous Expression of Adenosine A1 Receptors and GIRK4 Subunits in the Human Heart. Circulation. 2016;134:486–498. doi: 10.1161/CIRCULATIONAHA.115.021165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Fleckenstein A. History of calcium antagonists. Circ Res. 1983;52:I3–16. [PubMed] [Google Scholar]
- 274.Fozzard HA. Cardiac sodium and calcium channels: a history of excitatory currents. Cardiovasc Res. 2002;55:1–8. doi: 10.1016/s0008-6363(02)00407-8. [DOI] [PubMed] [Google Scholar]
- 275.Nilius B, Hess P, Lansman JB, Tsien RW. A novel type of cardiac calcium channel in ventricular cells. Nature. 1985;316:443–446. doi: 10.1038/316443a0. [DOI] [PubMed] [Google Scholar]
- 276.Curtis BM, Catterall WA. Purification of the calcium antagonist receptor of the voltage-sensitive calcium channel from skeletal muscle transverse tubules. Biochemistry. 1984;23:2113–2118. doi: 10.1021/bi00305a001. [DOI] [PubMed] [Google Scholar]
- 277.Ohashi N, Mitamura H, Ogawa S. Development of newer calcium channel antagonists: therapeutic potential of efonidipine in preventing electrical remodelling during atrial fibrillation. Drugs. 2009;69:21–30. doi: 10.2165/00003495-200969010-00002. [DOI] [PubMed] [Google Scholar]
- 278.Fareh S, Benardeau A, Nattel S. Differential efficacy of L- and T-type calcium channel blockers in preventing tachycardia-induced atrial remodeling in dogs. Cardiovasc Res. 2001;49:762–770. doi: 10.1016/s0008-6363(00)00288-1. [DOI] [PubMed] [Google Scholar]
- 279.Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, Belhassen B, Viskin S. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149–1154. doi: 10.1016/j.hrthm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 280.Kohno M, Yano M, Kobayashi S, Doi M, Oda T, Tokuhisa T, Okuda S, Ohkusa T, Kohno M, Matsuzaki M. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol Heart Circ Physiol. 2003;284:H1035–1042. doi: 10.1152/ajpheart.00722.2002. [DOI] [PubMed] [Google Scholar]
- 281.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 282.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 283.Nakaya H, Furusawa Y, Ogura T, Tamagawa M, Uemura H. Inhibitory effects of JTV-519, a novel cardioprotective drug, on potassium currents and experimental atrial fibrillation in guinea-pig hearts. Br J Pharmacol. 2000;131:1363–1372. doi: 10.1038/sj.bjp.0703713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kaneko N, Matsuda R, Toda M, Shimamoto K. Inhibition of annexin V-dependent Ca2+ movement in large unilamellar vesicles by K201, a new 1,4-benzothiazepine derivative. Biochim Biophys Acta. 1997;1330:1–7. doi: 10.1016/s0005-2736(97)00132-6. [DOI] [PubMed] [Google Scholar]
- 285.Hasumi H, Matsuda R, Shimamoto K, Hata Y, Kaneko N. K201, a multi-channel blocker, inhibits clofilium-induced torsades de pointes and attenuates an increase in repolarization. Eur J Pharmacol. 2007;555:54–60. doi: 10.1016/j.ejphar.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 286.Thomas NL, Maxwell C, Mukherjee S, Williams AJ. Ryanodine receptor mutations in arrhythmia: The continuing mystery of channel dysfunction. FEBS Lett. 2010;584:2153–2160. doi: 10.1016/j.febslet.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 287.Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, Shimoni Y, Chen SR. K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J. 2007;404:431–438. doi: 10.1042/BJ20070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Yang PC, Moreno JD, Miyake CY, Vaughn-Behrens SB, Jeng MT, Grandi E, Wehrens XH, Noskov SY, Clancy CE. In silico prediction of drug therapy in catecholaminergic polymorphic ventricular tachycardia. J Physiol. 2016;594:567–593. doi: 10.1113/JP271282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.van der Werf C, Kannankeril PJ, Sacher F, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011;57:2244–2254. doi: 10.1016/j.jacc.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–383. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48:293–301. doi: 10.1016/j.yjmcc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Sikkel MB, Collins TP, Rowlands C, Shah M, O’Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Flecainide reduces Ca(2+) spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res. 2013;98:286–296. doi: 10.1093/cvr/cvt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Steele DS, Hwang HS, Knollmann BC. Triple mode of action of flecainide in catecholaminergic polymorphic ventricular tachycardia. Cardiovasc Res. 2013;98:326–327. doi: 10.1093/cvr/cvt059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sikkel MB, Collins TP, Rowlands C, Shah M, O’Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Triple mode of action of flecainide in catecholaminergic polymorphic ventricular tachycardia: reply. Cardiovasc Res. 2013;98:327–328. doi: 10.1093/cvr/cvt068. [DOI] [PubMed] [Google Scholar]
- 295.Hartmann N, Pabel S, Herting J, Schatter F, Renner A, Gummert J, Schotola H, Danner BC, Maier LS, Frey N, Hasenfuss G, Fischer TH, Sossalla S. Antiarrhythmic effects of dantrolene in human diseased cardiomyocytes. Heart Rhythm. 2017;14:412–419. doi: 10.1016/j.hrthm.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 296.Zhou Q, Xiao J, Jiang D, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17:1003–1009. doi: 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Li N, Wang Q, Sibrian-Vazquez M, Klipp RC, Reynolds JO, Word TA, Scott L, Jr, Salama G, Strongin RM, Abramson JJ, Wehrens XH. Treatment of catecholaminergic polymorphic ventricular tachycardia in mice using novel RyR2-modifying drugs. Int J Cardiol. 2017;227:668–673. doi: 10.1016/j.ijcard.2016.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]