

Abstract
Background
Exon-targeted microarrays can detect small (<1000 bp) intragenic copy number variants (CNVs), including those that affect only a single exon. This genome-wide high-sensitivity approach increases the molecular diagnosis for conditions with known disease-associated genes, enables better genotype–phenotype correlations, and facilitates variant allele detection allowing novel disease gene discovery.
Methods
We retrospectively analyzed data from 63,127 patients referred for clinical chromosomal microarray analysis (CMA) at Baylor Genetics laboratories, including 46,755 individuals tested using exon-targeted arrays, from 2007 to 2017. Small CNVs harboring a single gene or two to five non-disease-associated genes were identified; the genes involved were evaluated for a potential disease association.
Results
In this clinical population, among rare CNVs involving any single gene reported in 7200 patients (11%), we identified 145 de novo autosomal CNVs (117 losses and 28 intragenic gains), 257 X-linked deletion CNVs in males, and 1049 inherited autosomal CNVs (878 losses and 171 intragenic gains); 111 known disease genes were potentially disrupted by de novo autosomal or X-linked (in males) single-gene CNVs. Ninety-one genes, either recently proposed as candidate disease genes or not yet associated with diseases, were disrupted by 147 single-gene CNVs, including 37 de novo deletions and ten de novo intragenic duplications on autosomes and 100 X-linked CNVs in males. Clinical features in individuals with de novo or X-linked CNVs encompassing at most five genes (224 bp to 1.6 Mb in size) were compared to those in individuals with larger-sized deletions (up to 5 Mb in size) in the internal CMA database or loss-of-function single nucleotide variants (SNVs) detected by clinical or research whole-exome sequencing (WES). This enabled the identification of recently published genes (BPTF, NONO, PSMD12, TANGO2, and TRIP12), novel candidate disease genes (ARGLU1 and STK3), and further confirmation of disease association for two recently proposed disease genes (MEIS2 and PTCHD1). Notably, exon-targeted CMA detected several pathogenic single-exon CNVs missed by clinical WES analyses.
Conclusions
Together, these data document the efficacy of exon-targeted CMA for detection of genic and exonic CNVs, complementing and extending WES in clinical diagnostics, and the potential for discovery of novel disease genes by genome-wide assay.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0472-7) contains supplementary material, which is available to authorized users.
Keywords: Exon targeted array CGH, Intragenic copy number variants, CNVs, de novo variants
Background
Clinical application of genome-wide assay by chromosomal microarray analysis (CMA) has significantly improved the detection rate for molecular diagnoses in clinical genomics diagnostics [1], enabling the elucidation of pathogenic copy number variants (CNVs) in individuals with various conditions, including congenital anomalies, intellectual disability/developmental delay (ID/DD), autism spectrum disorder (ASD), epilepsy, heart defects, and neuropsychiatric diseases [2–11]. CNVs smaller than 400 kb in size are challenging for clinical interpretation and, when not involving known genes [2, 12, 13], they are often not reported in routine clinical CMA. Nevertheless, such CNVs have been documented to contribute to cognitive phenotypes in population studies [14, 15].
Whereas the positive correlation between the size of a CNV and likelihood of pathogenicity guided the size cutoffs used for the clinical reporting of CNVs [5, 13], gene content is also an important factor in the determination of potential CNV pathogenicity. Disease-causing CNVs may be as small as a single exon [16–24], which still remain beyond the detection limits of whole-exome sequencing (WES) [25]. To improve the detection rate for such pathogenic CNVs, several groups developed exon-focused arrays with a sufficient number of interrogating oligo probes to target single exons of both known disease-associated genes and developmentally important genes that are not yet associated with human disease [12, 13, 26–33].
In 2010, we reported the CMA results in 3743 patients using our first version of a clinical exon-targeted array (OLIGO V8) [29]. We demonstrated that increasing array resolution to single exons not only allowed detection of small CNVs in the known disease genes, but also provided new opportunities for novel gene discoveries and the ability to detect somatic mosaicism for intragenic CNVs [29, 34].
Despite the advances in molecular diagnostics using genome-wide assays [1], WES and targeted next-generation sequencing (NGS) studies have limited ability to detect small intragenic CNVs [25] and therefore cannot currently reveal the totality of disease genes and pathogenic alleles. To further investigate this hypothesis and to assess the efficacy of exon-targeted CMA to identify putative novel disease genes, we queried our database of exon-targeted clinical CMA performed in 63,127 patients at Baylor Genetics (BG) Laboratories, with a particular focus on small de novo autosomal and X-linked (in males) CNVs involving genes that are currently not associated with human disease. These variants have increased likelihood to be pathogenic and may lead to the identification of the novel candidate disease genes [3, 23, 27, 35, 36]. We also cross-referenced these “gene level” data with single nucleotide variants (SNV) in the same gene detected in the clinical WES laboratory at BG and research exomes from the Baylor Hopkins Center for Mendelian Genomics (BHCMG).
Previous works demonstrating increased performance of exon-targeted CGH arrays focused mostly on the improvements in detection of CNVs involving known disease genes. Our results show that the systematic study of the large clinical cohort using exon-targeted CMA can be successfully used to discover novel disease genes, especially utilizing CNVs involving the candidate or potential new disease genes and with further integration of SNV data from WES.
Methods
Microarrays
In 2008, we designed and clinically implemented array comparative genomic hybridization (aCGH) with exonic coverage of over 1700 disease and candidate disease genes (OLIGO V8) and demonstrated its efficacy for detection of pathogenic exonic losses or gains as small as a few hundred base pairs in size [29]. This work suggested that as many as 10% (3/30 in known disease genes) of small intragenic CNVs might represent mosaic mutant alleles. Subsequently, we expanded our custom-designed oligo array to include > 4800 genes, including autosomal recessive disease genes (OLIGO V9 [37], V10, and V11).
We retrospectively analyzed CMA data from 63,127 patients (most diagnosed with neurodevelopmental defects) referred for CMA between April 15, 2007 and February 17, 2017 using six different versions of customized oligonucleotide arrays (OLIGO V6-V11; Agilent Technologies Inc., Santa Clara, CA, USA) developed at BG Laboratories. Among 63,127 patients, 46,755 were analyzed using microarrays targeting 1700 genes (OLIGO V8) or the subsequent microarray versions targeting > 4800 genes (OLIGO V9, V10, and V11) [12, 29]. In these microarrays, more than 90% of the exons in targeted genes were covered with at least three interrogating oligonucleotides, with an average of > 4.2 probes per exon, whereas intronic probes were uniformly distributed every 10 kb. In addition, for the purposes of normalization and statistical analyses of raw data, the design included unique sequence interrogating oligonucleotide probes for the entire genome covered at an average resolution of 30 kb (excluding segmental duplications). The procedures for DNA digestion, labeling, and hybridization for the oligo arrays were performed according to the manufacturers’ instructions, with minor modifications [38–40].
We used an in-house developed software to detect CNVs from aCGH data. The algorithm requires at least three consecutive probes with a log2 ratio < – 0.6 to detect deletion or at least three consecutive probes with a log2 ratio > 0.4 to detect duplication. CNVs < 500 kb with no RefSeq genes within the intervals or CNVs within segmental duplication (SDs) and CNVs in benign polymorphic regions, such as those listed in Database of Genomic Variants (DGV) are generally not reported. CNVs located in the regions known to be disease-associated and likely exhibiting incomplete penetrance, variable expressivity, or representing a potential recessive carrier state can be reported despite their overlap with DGV CNVs. The remaining CNVs are then classified as: (1) pathogenic = pathogenic CNVs includes aneuploidy, known microdeletion/microduplication, deletion, and intragenic duplication genomic intervals involving known dosage-sensitive autosomal-dominant (AD) disease genes, or deletion of any size involving a dosage sensitive gene associated with AD, haploinsufficiency, or other likely pathogenic consequence. Moreover large duplications (> 2 Mb) with genes or duplications > 1 Mb with genes known to be dosage-sensitive are also classified as pathogenic CNVs; (2) likely benign = < 1 Mb CNVs with no genes in the intervals or CNVs in the DGV database and have been reported to be inherited multiple times in our CMA database; (3) loss/gain in non-disease region = rare CNVs < 1 Mb with genes not implicated in disease phenotypes; (4) loss/gain of uncertain clinical significance = CNVs that have genes, but there is no supporting evidence of pathogenicity. CNVs of uncertain significance are usually investigated by parental studies. CNVs that have been shown to exhibit incomplete or uncertain penetrance were classified as such.
Whole-exome sequencing data
WES was performed in ~ 9000 individuals sequenced at BG and in the cohort of ~ 6000 samples sequenced in the Human Genome Sequencing Center (HGSC) at BCM through the BHCMG research initiative [41] as described previously [23, 42–44].
Computational parsing of clinical CMA database
The main aim of our retrospective analyses was to identify potential pathogenic or likely pathogenic CNVs affecting genes that thus far were not disease-associated. Subsequently, we queried the BG CMA database for other overlapping submicroscopic CNV deletions up to 5 Mb in size (de novo, inherited, or of unknown parental origin) that encompassed at least one gene that we consider as novel or recently published candidate disease gene.
Genes affected by single-gene CNVs and those included in de novo or hemizygous CNVs affecting 2–5 genes were classified according to their disease status using the lists of known AD, autosomal-recessive (AR), or X-linked (XL) disease genes, as defined in the Online Mendelian Inheritance in Man (OMIM) database (http://omim.org/) [29, 45, 46]. For each gene, we performed an extensive literature search for genotype–phenotype correlations. Moreover, all genes were cross-referenced with the Simons Foundation Autism Research Initiative (SFARI) database (https://sfari.org/) of ASD-related genes [47].
In addition, we searched the BG and BHCMG exome databases for predicted loss-of-function (LOF) (i.e. stop-gain, frameshift, or splicing) variants in the candidate genes to potentially provide further evidence of their disease-association. From the initial set, we excluded variants with a total coverage of < 20 reads or the ratio of variant to total reads below 0.2. We also removed variants with minor allele frequency (MAF) > 0.001 in ESP [48], 1000 Genomes [49], or our local exome databases or with MAF > 0.0001 in ExAC (Exome Aggregation Consortium, http://exac.broadinstitute.org) [50].
To assess the predicted probability of exhibiting haploinsufficiency for a given gene, we used haploinsufficiency scores calculated by Huang et al. [51]. These predictions were generated using classification models trained on known haploinsufficient genes and genes disrupted by unambiguous LOF variants in at least two apparently healthy individuals. Genes with high rank scores (0–10%) indicate that the gene is likely to exhibit haploinsufficiency. In our evaluation of a particular gene, we also consider the pLI score, i.e. the probability that the gene is intolerant to LOF variants. This score was generated based on the analysis of the ratio of the number of observed vs. expected LOF variants in the ExAC population [50, 52]. Genes with high pLI scores (pLI ≥ 0.9) are extremely LOF-intolerant, whereby genes with low pLI scores (pLI ≤ 0.1) are LOF-tolerant.
Fluorescent in situ hybridization (FISH)
FISH analyses were performed with bacterial artificial chromosome (BAC) or fosmid clones using standard procedures [53].
Breakpoint junction sequencing
We designed a customized high-density CGH array (HD-aCGH, AMADID 081888) for analyzing CNVs detected by CMA to further resolve breakpoint junctions and determine the nucleotide sequence of selected breakpoints. Long-range polymerase chain reaction (PCR) followed by Sanger sequencing of the PCR products was performed, as described, to obtain base-pair resolution of the breakpoint junctions [54]. Breakpoint junction sequencing was performed for small deletions in which the involved exons could not be determined due to a limited resolution of our clinical array.
Parental studies
The inheritance status of CNVs was determined by analyzing parental DNA using CMA or FISH. Note that in the case of de novo events, we did not formally confirm paternity and maternity by molecular testing.
Results
Out of 24,373 non-polymorphic CNVs detected in 18,708 patients, genome-wide clinical aCGH studies identified 7200 individuals with 8094 CNVs involving a single gene, including 145 de novo autosomal (117 losses and 28 intragenic gains) and 257 X-linked (in males) deletion CNVs, and 1049 inherited CNVs (878 losses and 171 intragenic gains) on autosomes (Fig. 1). Sizes of CNVs overlapping a single gene were in the range of ~ 100 bp to ~ 5.8 Mb with a median size of 94 kb. Importantly, 1857 (23%) of single gene CNVs affected only a single exon. We also found 6287 individuals with 6897 CNVs encompassing 2–5 genes (including disease-associated and non-disease-associated genes), including 127 de novo autosomal, 63 X-linked (in males), and 414 inherited autosomal CNV deletions (Fig. 1).
Fig. 1.
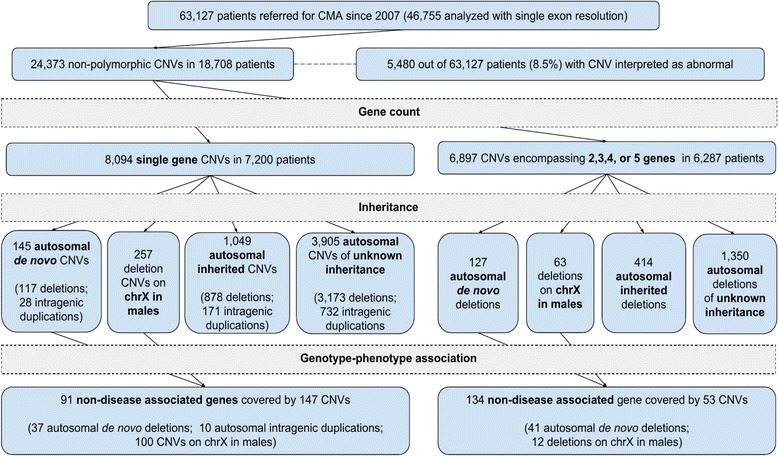
Overview of the CNV filtering strategy
Most common single-gene CNVs affect primarily known disease genes
In our clinical cohort, the most common single-gene CNVs include CHRNA7 (OMIM* 118511) (14 deletions/312 duplications), IMMP2L (OMIM* 605977) (151 deletions/1 duplication), TMLHE (OMIM* 300777) (125 deletions/21 duplications), RBFOX1 (OMIM* 605104) (60 deletions/62 duplications), DMD (OMIM* 300377) (74 deletions/32 duplications), NRXN1 (OMIM* 600565) (90 deletions/9 duplications), CNTN6 (OMIM* 607220) (22 deletions/63 duplications), and PARK2 (OMIM* 602544) (50 deletions/17 duplications). Of those, events in CHRNA7, DMD, and NRXN1 [34] were interpreted as directly causative for the patients’ phenotypes, whereas CNVs involving TMLHE [32], PARK2 [55], and RBFOX1 [56, 57] may confer susceptibility to disease or represent an allele for a recessive carrier state [58]. In total, we identified 111 known disease genes that were potentially disrupted by de novo autosomal or X-linked (in males) single-gene CNVs.
Recently proposed and potential novel disease genes
We identified 91 genes that were either recently proposed to be candidate disease-associated or non-disease-associated genes. These were disrupted by 37 de novo autosomal single-gene deletions, 100 X-linked CNVs (97 deletions and three intragenic duplications in males), and ten de novo intragenic duplications on autosomes (see Fig. 1 and Additional file 1).
To search for additional candidate disease genes, we extended our analyses to 6897 CNV deletions harboring 2–5 non-disease genes. We identified 134 distinct recently proposed or not yet disease-associated genes involved in 41 de novo autosomal and 12 X-linked (in males) deletions (see Fig. 1 and Additional file 2).
To further narrow the list of candidate disease-causing genes, we considered the following factors: (1) the number of de novo CNVs determined for each gene (Additional files 1 and 2); (2) additional CNVs < 5 Mb in size found in our cohort; (3) LOF variants found in ~ 15,000 WES cases (from BG and BHCMG “disease cohorts”); (4) phenotypic overlap among patients; (5) literature records supporting disease association; (6) predictions of haploinsufficiency [51] and intolerance to LOF [50] of the identified variants. Using these criteria, we found evidence supporting the contention of recently published disease genes, including BPTF (OMIM* 601819) [59], NONO (OMIM* 300084) [60], PSMD12 (OMIM* 604450) [61], TANGO2 (OMIM* 616830) [62, 63], TRIP12 (OMIM* 604506) [64, 65], and likely MAGED1 (OMIM* 300224) [66]. Furthermore, we found genes recently reported as disease-associated, including TBR1 (OMIM* 604616) [67] and CLTCL1 (OMIM* 601273) [68], as well as genes not yet associated with diseases.
We selected two novel non-disease-associated genes, STK3 (OMIM* 605030) mapping to 8q22.2 and ARGLU1 (OMIM* 614046) that maps in 13q33 for further molecular and clinical analyses, to determine whether these could be novel disease genes. In addition, we attempted to expand the genotype–phenotype correlations for two recently proposed candidate disease genes, MEIS2 (OMIM* 601740) on 15q14 [69–74], and PTCHD1 (OMIM* 3008280) on Xp22.11 [75–80]. In total, we found 17 small CNV deletions (3.2 kb to 4.9 Mb in size with three CNVs < 50 kb) overlapping these four candidate disease genes, including eight de novo events (Figs. 2, 3, 4 and 5). However, paternity was not tested formally (i.e. by molecular markers) and thus non-paternity could not be ruled out for most of the cases. The investigation of ~ 15,000 WES samples from BG and BHCMG revealed rare LOF variants, providing additional support for potential genotype-phenotype correlations (Tables 1, 2, 3 and 4, patients with variants in ARGLU1, STK3, MEIS2, and PTCHD1, respectively; see also Additional file 3 for discussion on two other candidate disease genes, AGBL4 (OMIM* 616476) and CSMD1 (OMIM* 608397); Additional files 4, 5 and 6 for information on patients with additional variants in ARGLU1/EFNB2, AGBL4, and CSMD1, respectively; and Additional files 7 and 8 for visualization of CNVs in AGBL4 and CSMD1, respectively).
Fig. 2.
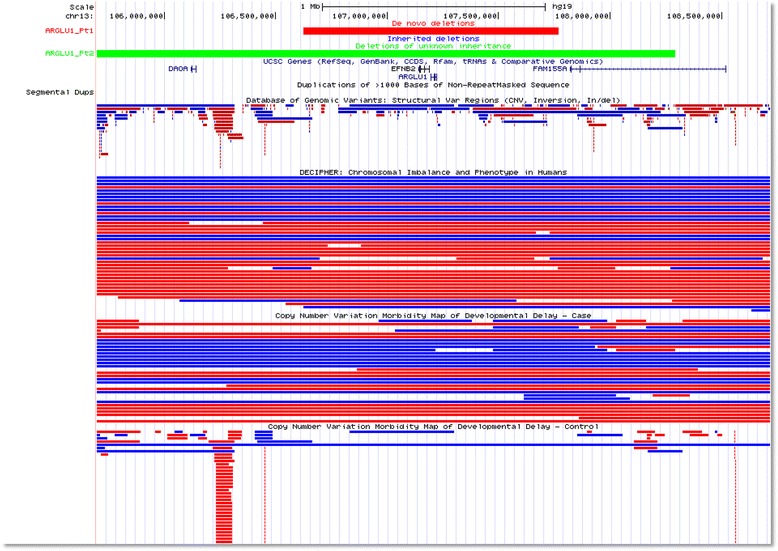
CNVs in ARGLU1/EFNB2, including de novo (red) and deletions of unknown inheritance (green)
Fig. 3.
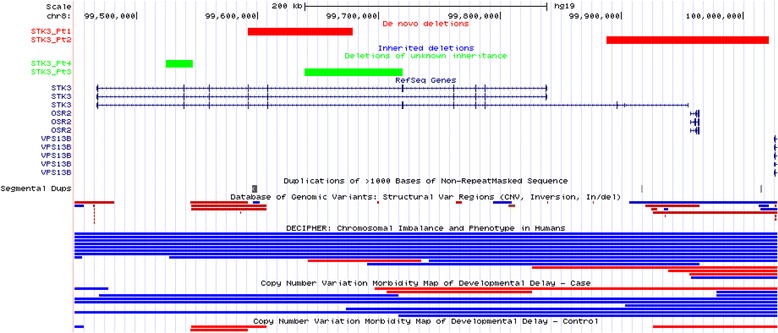
CNVs in STK3, including de novo (red) and deletions of unknown inheritance (green)
Fig. 4.
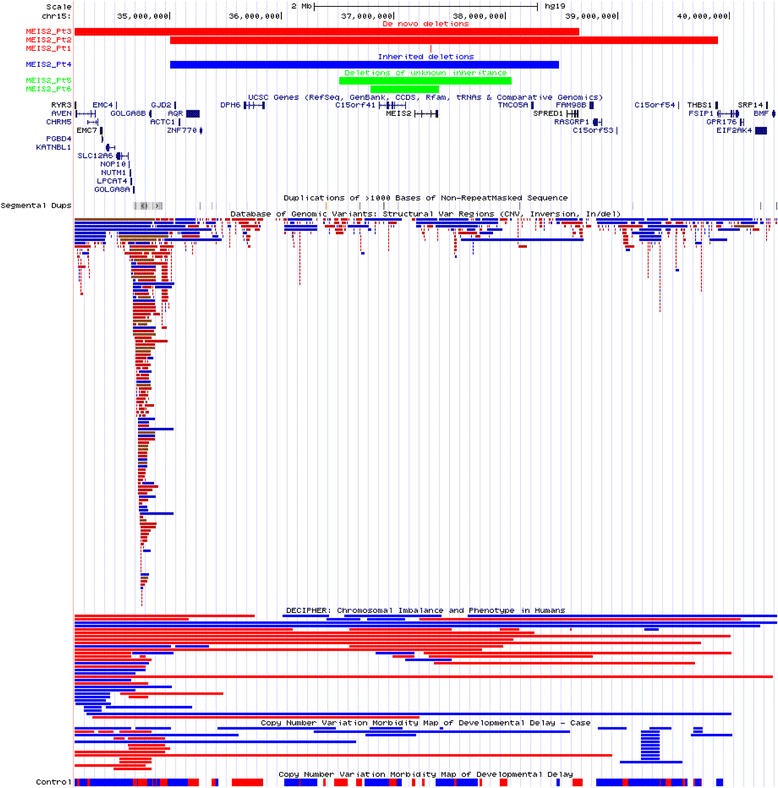
CNVs in MEIS2, including de novo (red), inherited (blue), and deletions of unknown inheritance (green)
Fig. 5.
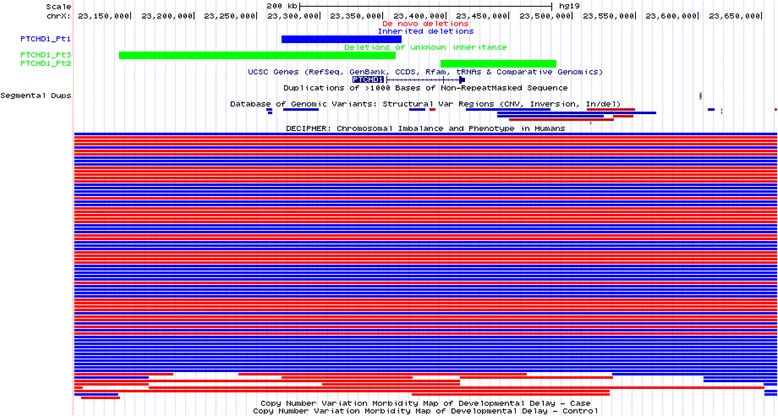
CNVs in PTCHD1, including inherited (blue) and deletions of unknown inheritance (green)
Table 1.
Clinical information on patients with ARGLU1 and EFNB2 variants
| Case number | Pt1 | Pt2 | Pt3 | Pt4 (DECIPHER 280488) |
|---|---|---|---|---|
| Sex | Male | Female | Male | Female |
| Variant | chr13:106,624,717-107,768,458 1.1 Mb del | chr13:104,114,620-108,292,078 4.1 Mb del | chr13:107211843delA; NM_018011:c.509delA; p.K170fs frameshift | chr13:106,884,343-110,711,191 3.8 Mb del |
| Confirmation method | FISH | N/A | PCR + Sanger | FISH |
| Affected genes | ARGLU1, EFNB2 | DAOA, FAM155A, ARGLU1, EFNB2 | ARGLU1 | ABHD13, ARGLU1, EFNB2, FAM155A, IRS2, LIG4, MYO6, TNFSF13B |
| Inheritance | De novo | Unknown | De novo | De novo |
| Parental studies | FISH | N/A | PCR + Sanger | FISH |
| Developmental delay/intellectual disability | + | + | + | + |
| Developmental regression | + | N/A | N/A | N/A |
| Autistic spectrum | + | N/A | N/A | N/A |
| Abnormal movement | N/A | N/A | + | N/A |
| Cerebellar hypoplasia | N/A | N/A | + | N/A |
| Oculomotor apraxia | N/A | N/A | + | N/A |
| Seizures/epilepsy | N/A | + | N/A | N/A |
| Other | N/A | Horse shoe kidney/ectopic kidney, dysmorphic features | N/A | N/A |
N/A not available
Candidate genes are indicated in bold
Table 2.
Clinical information on patients with STK3 variants
| Case number | Pt1 | Pt2 | Pt3 | Pt4 |
|---|---|---|---|---|
| Sex | Female | Male | Female | Male |
| Variant | chr8: 99,591,666-99,678,567 87 kb, exons 7-8 del | chr8: 99,883,084-100,026,306a 143 kb, exons 1-3 del | chr8: 99,638,463-99,719,599 81 kb, exons 5-6 del | chr8:99,524,409-99,546,574a 22 kb, exon 10 del |
| Confirmation method | FISH | FISH, PCR + Sanger | High-density CGH array | PCR + Sanger |
| Affected genes | STK3 | STK3, OSR2 | STK3 | STK3 |
| Inheritance | De novo | De novo | Unknown | Unknown |
| Parental studies | FISH | FISH | N/A | N/A |
| Developmental delay/intellectual disability | N/A | + | N/A | + |
| Multiple congenital anomalies | + | N/A | Skull, face, and neck anomalies | N/A |
| Other | Dysmorphic features | Radioulnar synostosis, hypospadias | N/A | Failure to thrive, hypotonia |
aCNV coordinates obtained using PCR and Sanger sequencing of breakpoint junction
N/A not available
Table 3.
Clinical information on patients with MEIS2 variants
| Reference | Previous cases | |||||||
|---|---|---|---|---|---|---|---|---|
| Case number | Erdogan et al.; Chen et al.; Crowley et al.; Johansson et al. (5 cases in 2 families); Louw et al. (4 cases); Fujita et al. | Pt1 | Pt2 | Pt3 | Pt4 | Pt5 | Pt6 | Pt7 (DECIPHER 286841) |
| Gender | 8 females; 6 males | Male | Male | Female | Male | Male | Male | Female |
| Variant | 6 CNV del (123 kb – 5.6 Mb); 1 CNV dup (58 kb); 1 stop-gain; 1 frameshift | chr15: 37,328,986-37,332,249a | chr15: 35,001,138-39,899,594 | chr15: 33,894,032-38,659,166 | chr15: 35,001,138-38,474,933 | chr15: 36,512,757-38,052,959a | chr15: 36,790,702-37,404,359a | chr15:36,606,006-37,515,525 |
| 3.2 kb del | 4.9 Mb del | 4.8 Mb del | 3.47 Mb del | 1.54 Mb del | 0.6 Mb del | 0.9 Mb del | ||
| Confirmation method | PCR + Sanger | FISH | FISH | FISH | PCR + Sanger | PCR + Sanger | Unknown | |
| Affected genes | MEIS2 | MEIS2 + 14 other genes | MEIS2 + 21 other genes | MEIS2 + 7 other genes | MEIS2 + 1 other gene | MEIS2 + 1 other gene | MEIS2 | |
| Inheritance | 9 de novo; 4 inherited; 1 mosaic | De novo | De novo | De novo | Mat | Unknown | Unknown | De novo |
| Parental studies | CMA | FISH | FISH | FISH | N/A | N/A | Unknown | |
| Cleft lip and cleft palate | 12 out of 14 | + | N/A | N/A | N/A | Bifid uvula | N/A | Bifid uvula |
| Cardiac malformation | Ventricular septal defect (7); atrial septal defect (2); LVOTO; CoA | NR | N/A | N/A | NR | NR | NR | Learning problems, aggressive behavior |
| Cognitive and behavioral phenotype | ID (7); delayed (6); ASD (2); LD (2) | NR | N/A | N/A | ASD | Global DD, ASD, ADHD | ASD? | Slower verbal development |
| Verbal developmental delay | 4 out of 4 | Possibly | N/A | N/A | NR | Delayed verbal milestones | Delayed language skills | NR |
| Motor developmental delay | 12 out of 12 | NR | N/A | N/A | NR | Delayed motor milestones | NR | NR |
| Walked at age | 14 months – 3 years | NR | N/A | N/A | NR | 2 years | NR | NR |
| Gastro-esophageal reflux | 2 out of 2 | NR | N/A | N/A | NR | NR | NR | NR |
| Other features | Hypotonia; prolapse of epiglottis; bilateral moderate hearing loss; agenesis of the right tympanic membrane; a gracile corpus callosum; congenital lobar emphysema, syndactyly; severe hypermetropia, severe constipation | NR | Hypertonia | MCA | NR | Asthma; sister with Ebstein’s cardiac anomaly; family history of ID; maternal prenatal cocaine use | NR | Calcaneovalgus, velopharyngeal insufficiency, asymmetric chest |
aCNV coordinates obtained using PCR and Sanger sequencing of breakpoint junction; note that Sanger sequencing of breakpoint junction PCR amplification product in Pt1 was affected by the homonucleotide tracts (poly-A/T) close by the breakpoint junction; therefore, the CNV coordinates in Pt1 were determined by the coordinates of the poly A/T tracts
N/A not available, NR not reported, ID intellectual disability, ASD Autism Spectrum Disorder, LD learning disability, LVOTO left ventricular outflow tract obstruction, CoA coarctation of the aorta, CHD coronary heart disease, MCA multiple congenital anomalies, Mat maternal
Table 4.
Clinical information on patients with PTCHD1 variants
| Clinical features | Pt1 | Pt2 | Pt3 |
|---|---|---|---|
| Gender | Male | Male | Male |
| Variant | chrX: 23,269,452-23,364,920 | chrX: 23,395,713-23,487,393a | chrX: 23,140,737-23,360,470a |
| 95 kb, exon 1 deletion | 92 kb, exons 2–3 deletion | 220 kb, exon 1 deletion | |
| Confirmation method | N/A | PCR + Sanger | PCR + Sanger |
| Affected genes | PTCHD1 | PTCHD1 | PTCHD1 |
| Inheritance | Mat | Unknown | Unknown |
| Parental studies | CMA | N/A | N/A |
| ASD | + | N/A | Autistic features |
| DD/ID | + | + | + |
| ADHD | + | N/A | + |
| Other | Hypotonia, speech impairment | N/A | N/A |
a - CNV coordinates obtained using PCR and Sanger sequencing of breakpoint junction
N/A not available, DD developmental delay, ID intellectual disability, Mat maternal
ARGLU1 and EFNB2 that map to 13q33 as potential new disease genes
In the BG CMA database, we found two CNVs involving ARGLU1 and EFNB2 (OMIM* 600527), including one ~ 1.1 Mb de novo deletion encompassing ARGLU1 and EFNB2 and one ~ 4.2 Mb deletion of unknown inheritance harboring ARGLU1, DAOA, FAM155A, and EFNB2 (Table 1). In addition, one de novo ~ 3.8 Mb deletion encompassing ARGLU1 and EFNB2 was found in one DECIPHER (https://decipher.sanger.ac.uk/) patient, 280488. Moreover, in the BG WES database, we identified one de novo frameshift variant (g.13:107211843delA; NM_018011:c.509delA; p.K170fs) in ARGLU1. Our further investigation of five novel or very rare missense variants: one in ARGLU1 (c.350G > A p.R117Q) and four in EFNB2 (c.498A > C, p.Q166H; c.503C > T, p.A168V; c.796A > G, p.T266A; c.803C > T, p.S268L) revealed that these five variants were all inherited (Additional file 4). Importantly, putative LOF variants in ARGLU1 and EFNB2 have been rarely seen in the BG and BHCMG databases (one variant mentioned above for ARGLU1 and zero for EFNB2), indicating their intolerance to haploinsufficiency. Prediction algorithms indicate that both ARGLU1 and EFNB2 are sensitive to LOF with haploinsufficiency scores of 5.27 and 1.35 and the probabilities of intolerance to LOF mutations (pLI scores) of 0.99 and 0.94, respectively.
STK3 may be associated with human disease phenotypes
We identified four different-sized CNV deletions involving STK3 (Table 2). Two of these deletions (87 kb and 143 kb in size) were confirmed to be de novo; inheritance of two other CNVs (81 kb and 22 kb in size) could not be determined. In the DECIPHER database, one patient (258095) had an intragenic deletion of exons 5 and 6 of unknown parental origin. Although computationally determined haploinsufficiency and pLI scores for STK3 (0.12 and 0, respectively) do not favor a haploinsufficiency pathomechanism for this gene, the identification of de novo variants suggest a potential disease association.
Novel MEIS2 and PTCHD1 variants
We identified new variants in MEIS2 and PTCHD1, which have been recently recognized as disease-associated genes. We found six different-sized deletions involving MEIS2 (Table 3). Three of them, including one small (~ 3.2 kb) and two large (~ 4.8 Mb and ~ 4.9 Mb) deletions were confirmed to be de novo; one deletion ~ 3.47 Mb was maternally inherited and the inheritance of two other (~ 1.54 Mb and ~ 0.6 Mb) CNVs remains unknown. Moreover, an ~ 909 kb de novo deletion encompassing MEIS2 and three other genes was found in the DECIPHER database (patient 286841). Prediction algorithms indicate MEIS2 as sensitive to LOF with a haploinsufficiency score of 0.68 and the pLI score of 0.99.
We found three male patients with hemizygous deletions varying in size between 92 kb and 220 kb and encompassing exon 1 (two cases) or exons 2 and 3 of the three-exon PTCHD1 gene on Xp22.11 (Table 4). In the DECIPHER database, there are at least two males with an inherited PTCHD1 deletion. pLI score of 0.95 strongly suggest that PTCHD1 is intolerant to LOF variants.
Discussion
Clinical WES studies showed that pathogenic variants occur de novo in ~ 87% of patients with an established molecular diagnosis for an autosomal dominant disease trait [42]. Moreover, de novo mutations, both SNV and CNV, have been demonstrated to represent an important cause of ID/DD [81, 82] and damaging de novo mutations are significantly enriched (P = 8.0 × 10-9; odds ratio [OR] = 1.84) in patients with ASD when compared to controls [83]. CNV alleles, whether intragenic or gene-encompassing, represent a key modality of disease causing variation as heterozygous alleles associated with dominant disease traits or contributing to carrier state for recessive traits [58]. Genomic CNV deletions and frameshifting intragenic duplication CNVs can lead to allele LOF. Genic CNV can be responsible for 14–60% of disease alleles in selected recent studies of novel disease genes (BPTF [59], NONO [60], PSMD12 [61], TANGO2 [62], TRIP12 [65]) and 7–26% of families of different disease cohorts (Bardet Biedl ciliopathies [84], primary immune deficiency disorders [85], brain malformations [36], an Arabic DD cohort [86], unsolved clinical exomes [87]). To advance the clinical investigation of CNVs as pathogenic alleles, we studied de novo CNVs and hemizygous deletion CNVs in males, involving both known and candidate disease genes using high-resolution exon-targeted clinical CMA data from over 62,000 patients. Our study indicated four genes ARGLU1, STK3, MEIS2, and PTCHD1 as having the strongest evidence for disease association.
ARGLU1 (arginine and glutamate rich protein 1) was reported to play a regulatory role in gene transcription through its interaction with MED1 Mediator complexes [88]. The highest expression level of this gene was found in the cerebellum (GTEx database). The neighboring EFNB2 (Ephrin B2) encodes a member of the Eph receptor family. In previous cytogenetic studies, two large 13q de novo deletions (9 Mb and 28 Mb in size) involving both ARGLU1 and EFNB2 were reported in patients with mild anorectal malformations and EFNB2 was proposed as a good candidate disease gene [89]. In support of this notion, studies in mice revealed that 28% of heterozygous Efnb2 knockout mice presented with mild anorectal malformations [90], closely resembling those observed in patients with EFNB2. However, none of our patients with CNV deletions involving EFNB2 had anorectal malformations. Most patients manifested neurological anomalies including DD confirmed in three out of four individuals. In addition, clinical evaluation of Pt.1 with a de novo 1 Mb deletion revealed developmental regression and ASD, whereas features of Pt.3 who carries a LOF point mutation in ARGLU1 included abnormal movement, cerebellar hypoplasia, and oculomotor apraxia. Given that we did not find additional evidence supporting EFNB2 as a disease-associated gene (i.e. all validated missense variants in EFNB2 were inherited from healthy parents), we propose that ARGLU1 rather than EFNB2 is a better candidate gene responsible for the neurological anomalies.
STK3 (also known as MST2) encodes serine/threonine-protein kinase 3, a component of the Hippo pathway that plays an important role in organ size regulation and tumor suppression by restricting proliferation and promoting apoptosis. Loss of Hpo (homologue of MST1 and MST2) in Drosophila causes tissue overgrowth [91]. Mouse studies showed that loss of Mst1 and Mst2 leads to severe growth retardation and other embryonic abnormalities, suggesting that both genes are crucial in early mouse development [92]. We identified four small-sized deletions encompassing 1–3 exons of the STK3 gene in individuals with different congenital anomalies (Table 2). Importantly, two of those deletions were de novo events. However, the STK3 haploinsufficiency score of 0.12 and pLI score of 0 do not support its pathogenicity. The contradictory findings between haploinsufficiency prediction scores and identification of two de novo exonic deletions, which suggested likely pathogenicity of LOF variants in STK3, could potentially be explained by incomplete penetrance or alternative disease contributing mechanisms other than haploinsufficiency.
MEIS2 is expressed during early fetal brain development in humans [93] and was proposed to contribute to the development of tissues originating from the neural crest, similarly to the mouse orthologue found to be expressed in neural crest cells. Homozygous Meis2 deficiency in mice results in perturbed development of the craniofacial skeleton and abnormalities in the heart and cranial nerves [74, 94]. Thus far, nine deletion CNVs of this gene were identified in patients with cleft palate (seven individuals), atrial or ventricular septal defect (four individuals), and mild to severe ID (eight individuals) [69, 70, 72]. Recently, a more severe phenotype of ID, cleft palate, and heart defects was associated with de novo frameshift deletion (p.Arg333del) and a de novo stop-gain SNV (p.Ser204*), suggesting that a truncated protein may cause a more severe clinical consequence than haploinsufficiency through a potential dominant-negative mechanism [71, 73, 74]. Our patients with a CNV deletion have a relatively milder phenotype, including DD, ASD (three patients), delayed verbal (three patients) and motor milestones (one patient), cleft palate (one patient), and bifid uvula (one patient). In contrast to previously reported cases, none of our patients with CNV deletions had cardiac defects. Similar phenotypic features (asymmetry of the thorax, bifid uvula, and a specific learning disability) were found in one patient with MEIS2 deletion reported in the DECIPHER database. MEIS2 maps to 15q14 distal to the Angelman/Prader-Willi syndromes genomic region on 15q11.2q12 and the CHRNA7 gene on 15q13.3 (located 5 Mb from MEIS2). Thus, better understanding of the MEIS2 alternations may help to elucidate the phenotypic spectrum of patients with larger-sized deletions encompassing proximal chromosome 15q.
PTCHD1 on Xp22.11 is highly expressed in the brain, especially in the cerebellum [80]. Recent functional studies reported that during early postnatal mouse development, Ptchd1 is selectively expressed in the thalamic reticular nucleus (TRN), a group of GABAergic neurons that regulate thalamo-cortical transmission, sleep rhythms, and attention [95]. It was suggested that Ptchd1 plays a role in the hedgehog signaling pathway [75]. Moreover, it was shown that a conditional TRN Ptchd1 deletion causes attention deficit and hyperactivity, whereas the constitutional deletion of this gene leads to a potentially more severe phenotype, including learning impairment, hyper-aggression, and motor defects [95]. Deletions involving PTCHD1, the upstream regulatory region encoding PTCHD1-AS and DDX53, or both were initially reported in individuals with ASD [75–77], ID [78], or both. Identification of additional male patients with CNV deletions or truncating SNVs, involving PTCHD1, further support LOF of this gene as disease-contributing for non-syndromic neurodevelopmental disorders, including ID, ASD, hypotonia, and behavioral abnormalities [79, 80]. Importantly, PTCHD1 is listed in the SFARI gene database (https://gene.sfari.org) as a strong candidate gene for ASD and DD/ID. Consistent with this, we identified three male patients with deletions of PTCHD1 who all manifested ASD, DD, or both (Table 4).
In summary, we applied computational scores that consider the vulnerability of genes to LOF and variant burden. We found that genes with pathogenic variants based on de novo occurrence also tend to have extreme values of haploinsufficiency scores and variant damage or pathogenicity burden. However, there were some genes that appear to harbor pathogenic variants but had prediction scores arguing against their pathogenicity due to LOF. Although the pattern between computational prediction and de novo variants are generally concordant, the lack of fit reinforces that computational predictions alone are perhaps insufficient for interpreting variation in the absence of transmission information and additional functional studies.
Conclusions
CNVs are a key class of disease causing variation. Our data further document the efficacy of exon-targeted CMA for the detection of genic and exonic CNVs, complementing WES in clinical diagnostics, and its potential for discovery of novel disease genes. Notably, exon-targeted CMA detected several pathogenic heterozygous and homozygous single-exon CNVs missed by clinical WES analyses. Technological advances and decreasing costs of whole-genome sequencing (WGS) may eventually make this approach a method of choice for detection of both SNVs and small CNVs, thus replacing CMA and WES; nevertheless, the clinical utility and implementation of WGS remains stymied by lack of objective studies documenting improved molecular diagnosis in comparison to WES plus CMA [87].
Additional files
Supplementary table reporting single gene de novo or hemizygous deletions or intragenic duplications in recently proposed candidate or not yet associated disease genes. (DOCX 39 kb)
Supplementary table reporting de novo or hemizygous deletions or intragenic duplications encompassing 2–5 genes in recently proposed candidate or not yet associated disease genes. (DOCX 29 kb)
Supplementary text discussing AGBL4 and CSMD1 as potential novel candidate disease genes. (DOCX 40 kb)
Supplementary table containing clinical information on patients with additional variants in ARGLU1/EFNB2. (DOCX 14 kb)
Supplementary table containing clinical information on patients with AGBL4 variants. (DOCX 19 kb)
Supplementary table containing clinical information on patients with CSMD1 variants. (DOCX 21 kb)
Supplementary figure presenting CNVs in AGBL4, including de novo (red), inherited (blue), and deletions of unknown inheritance (green). (PDF 104 kb)
Supplementary figure presenting CNVs in CSMD1, including de novo (red), inherited (blue), and deletions of unknown inheritance (green). (PPTX 95 kb)
Acknowledgments
This study makes use of data generated by the DECIPHER Consortium. A full list of centers who contributed to the generation of the data is available from https://decipher.sanger.ac.uk/ and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
Funding
Supported by the National Institutes of Health (National Human Genome Research Institute–National Heart, Lung, and Blood Institute grant U54 HG006542 to the Baylor Hopkins Center for Mendelian Genomics; National Institute of Neurological Disorders and Stroke grant R01 NS058529 to Dr. Lupski). Also funded in part from Polish budget funds for science in years 2016–2019 (Iuventus Plus grant IP2015 019874). RM is supported by the Osteogenesis Imperfecta Foundation Michael Geisman Fellowship. CPS is generously supported by the Joan and Stanford Alexander Family. HTC is supported by the AAN Neuroscience Research Scholarship from the American Academy of Neurology and the CNCDP-K12 Fellowship from the National Institute of Neurological Disorders and Stroke grant 1K12NS098482-01.
Availability of data and materials
The CNV calls presented in Tables 1, 2, 3 and 4 and Additional files 5 and 6 from BG CMA can be accessed through the NCBI dbVar database (http://www.ncbi.nlm.nih.gov/dbvar/) under accession number nstd149.
Abbreviations
- AD
Autosomal dominant
- AR
Autosomal recessive
- ASD
Autism Spectrum Disorders
- BAC
Bacterial artificial chromosome
- BCM
Baylor College of Medicine
- BG
Baylor Genetics
- BHCMG
Baylor-Hopkins Center for Mendelian Genomics
- CGH
Comparative genomic hybridization
- CMA
Chromosomal microarray analysis
- CNV
Copy number variant
- DD
Developmental delay
- ExAC
The Exome Aggregation Consortium
- HGSC
Human Genome Sequencing Center
- ID
Intellectual disability
- LOF
Loss of function
- MAF
Minor allele frequency
- OMIM
Online Mendelian Inheritance in Man
- PCR
Polymerase chain reaction
- SFARI
Simons Foundation Autism Research Initiative
- SNV
Single nucleotide variant
- TRN
Thalamic reticular nucleus
- WES
Whole-exome sequencing
- WGS
Whole-genome sequencing
- XL
X-linked
Authors’ contributions
TG, BY, CAS, ALB, SWC, JRL, AP, and PS conceived the project and designed the experiments. TG, BY, WB, MW, S-HLK, SRL, CAB, ALB, YY, AMB, JLS, SWC, JRL, AP, CAS, and PS performed the experiments and analyzed the data. ZCA, PL, JAR, and ANP advised on data analysis. TG, BY, JRL, and PS, wrote the manuscript. MBP, DTD, CB, LE, SG, LD, HGP, RMar, HTC, AC, HK, CH, NU, RMat, SB, ERR, KMN, PIB, GB, SCSN, MC, HN, MA, RWe, RWi, AEB, LI, LE, SV, RMo, EB, LF, ML, and CPS identified and collected patients. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Approval for this study, including written informed consents (H-22769) and waivers of informed consent (H-37568 and H-36612), were obtained from the Baylor College of Medicine Institutional Review Board. The Baylor College of Medicine IRB (IORG number 0000055) is recognized by the United States Office of Human Research Protections (OHRP) and Food and Drug Administration (FDA) under the federal-wide assurance program. The Baylor College of Medicine IRB is also fully accredited by the Association for the Accreditation of Human Research Protection Programs (AAHRPP).
Consent for publication
The Editor has waived consent to publish the clinical information in the manuscript due to the minimal risk of identification.
Competing interests
BCM and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories (BMGL), which performs chromosomal microarray analysis and clinical exome sequencing. PL, CAB, WB, JAR, SRL, MW, YY, AMB, JLS, SWC, AP, CAS, and PS are employees of BCM and derive support through a professional services agreement with the BG. JRL serves on the Scientific Advisory Board of the BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc., and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0472-7) contains supplementary material, which is available to authorized users.
References
- 1.Lupski JR. Clinical genomics: from a truly personal genome viewpoint. Hum Genet. 2016;135:591–601. doi: 10.1007/s00439-016-1682-6. [DOI] [PubMed] [Google Scholar]
- 2.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–46. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med Off J Am Coll Med Genet. 2011;13:777–84. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46:1063–71. doi: 10.1038/ng.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2013;17:589–99. doi: 10.1016/j.ejpn.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet. 2013;92:221–37. doi: 10.1016/j.ajhg.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mefford HC. CNVs in Epilepsy. Curr Genet Med Rep. 2014;2:162–7. doi: 10.1007/s40142-014-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–41. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefansson H, Rujescu D, Cichon S, Pietiläinen OPH, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green EK, Rees E, Walters JTR, Smith K-G, Forty L, Grozeva D, et al. Copy number variation in bipolar disorder. Mol Psychiatry. 2016;21:89–93. doi: 10.1038/mp.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiszniewska J, Bi W, Shaw C, Stankiewicz P, Kang S-HL, Pursley AN, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet. 2014;22:79–87. doi: 10.1038/ejhg.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallespín E, Palomares Bralo M, Mori MÁ, Martín R, García-Miñaúr S, Fernández L, et al. Customized high resolution CGH-array for clinical diagnosis reveals additional genomic imbalances in previous well-defined pathological samples. Am J Med Genet A. 2013;161A:1950–60. doi: 10.1002/ajmg.a.35960. [DOI] [PubMed] [Google Scholar]
- 14.Lupski JR. Cognitive Phenotypes and genomic copy number variations. JAMA. 2015;313:2029–30. doi: 10.1001/jama.2015.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Männik K, Mägi R, Macé A, Cole B, Guyatt AL, Shihab HA, et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA. 2015;313:2044–54. doi: 10.1001/jama.2015.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–53. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F, Seeman P, Liu P, Weterman MAJ, Gonzaga-Jauregui C, Towne CF, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thevenon J, Lopez E, Keren B, Heron D, Mignot C, Altuzarra C, et al. Intragenic CAMTA1 rearrangements cause non-progressive congenital ataxia with or without intellectual disability. J Med Genet. 2012;49:400–8. doi: 10.1136/jmedgenet-2012-100856. [DOI] [PubMed] [Google Scholar]
- 19.D’Souza L, Cukras C, Antolik C, Craig C, Lee J-Y, He H, et al. Characterization of novel RS1 exonic deletions in juvenile X-linked retinoschisis. Mol Vis. 2013;19:2209–16. [PMC free article] [PubMed] [Google Scholar]
- 20.Nagamani SCS, Erez A, Ben-Zeev B, Frydman M, Winter S, Zeller R, et al. Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur J Hum Genet EJHG. 2013;21:343–6. doi: 10.1038/ejhg.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vatta M, Niu Z, Lupski JR, Putnam P, Spoonamore KG, Fang P, et al. Evidence for replicative mechanism in a CHD7 rearrangement in a patient with CHARGE syndrome. Am J Med Genet A. 2013;0:3182–6. doi: 10.1002/ajmg.a.36178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boone PM, Yuan B, Campbell IM, Scull JC, Withers MA, Baggett BC, et al. The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am J Hum Genet. 2014;95:143–61. doi: 10.1016/j.ajhg.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–9. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayer DK, Martinez CA, Sorte HS, Forbes LR, Demmler-Harrison GJ, Hanson IC, et al. Vaccine-associated varicella and rubella infections in severe combined immunodeficiency with isolated CD4 lymphocytopenia and mutations in IL7R detected by tandem whole exome sequencing and chromosomal microarray. Clin Exp Immunol. 2014;178:459–69. doi: 10.1111/cei.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gambin T, Akdemir ZC, Yuan B, Gu S, Chiang T, Carvalho CMB, et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45:1633–48. doi: 10.1093/nar/gkw1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tayeh MK, Chin ELH, Miller VR, Bean LJH, Coffee B, Hegde M. Targeted comparative genomic hybridization array for the detection of single- and multiexon gene deletions and duplications. Genet Med Off J Am Coll Med Genet. 2009;11:232–40. doi: 10.1097/GIM.0b013e318195e191. [DOI] [PubMed] [Google Scholar]
- 27.Tucker T, Zahir FR, Griffith M, Delaney A, Chai D, Tsang E, et al. Single exon-resolution targeted chromosomal microarray analysis of known and candidate intellectual disability genes. Eur J Hum Genet EJHG. 2014;22:792–800. doi: 10.1038/ejhg.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiśniowiecka-Kowalnik B, Kastory-Bronowska M, Bartnik M, Derwińska K, Dymczak-Domini W, Szumbarska D, et al. Application of custom-designed oligonucleotide array CGH in 145 patients with autistic spectrum disorders. Eur J Hum Genet. 2013;21:620–5. doi: 10.1038/ejhg.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–42. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruno DL, Stark Z, Amor DJ, Burgess T, Butler K, Corrie S, et al. Extending the scope of diagnostic chromosome analysis: detection of single gene defects using high-resolution SNP microarrays. Hum Mutat. 2011;32:1500–6. doi: 10.1002/humu.21581. [DOI] [PubMed] [Google Scholar]
- 31.Aradhya S, Lewis R, Bonaga T, Nwokekeh N, Stafford A, Boggs B, et al. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet Med Off J Am Coll Med Genet. 2012;14:594–603. doi: 10.1038/gim.2011.65. [DOI] [PubMed] [Google Scholar]
- 32.Celestino-Soper PBS, Shaw CA, Sanders SJ, Li J, Murtha MT, Ercan-Sencicek AG, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360–70. doi: 10.1093/hmg/ddr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Retterer K, Scuffins J, Schmidt D, Lewis R, Pineda-Alvarez D, Stafford A, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med Off J Am Coll Med Genet. 2015;17:623–9. doi: 10.1038/gim.2014.160. [DOI] [PubMed] [Google Scholar]
- 34.Schaaf CP, Boone PM, Sampath S, Williams C, Bader PI, Mueller JM, et al. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur J Hum Genet EJHG. 2012;20:1240–7. doi: 10.1038/ejhg.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vulto-van Silfhout AT, Hehir-Kwa JY, van Bon BWM, Schuurs-Hoeijmakers JHM, Meader S, Hellebrekers CJM, et al. Clinical significance of de novo and inherited copy-number variation. Hum Mutat. 2013;34:1679–87. doi: 10.1002/humu.22442. [DOI] [PubMed] [Google Scholar]
- 36.Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron. 2015;88:499–513. doi: 10.1016/j.neuron.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dittwald P, Gambin T, Szafranski P, Li J, Amato S, Divon MY, et al. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013;23:1395–409. doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ou Z, Kang S-HL, Shaw CA, Carmack CE, White LD, Patel A, et al. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med Off J Am Coll Med Genet. 2008;10:278–89. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw CJ, Shaw CA, Yu W, Stankiewicz P, White LD, Beaudet AL, et al. Comparative genomic hybridisation using a proximal 17p BAC/PAC array detects rearrangements responsible for four genomic disorders. J Med Genet. 2004;41:113–9. doi: 10.1136/jmg.2003.012831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med Off J Am Coll Med Genet. 2005;7:422–32. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 41.Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto S, Jaiswal M, Charng W-L, Gambin T, Karaca E, Mirzaa G, et al. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159:200–14. doi: 10.1016/j.cell.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gambin T, Jhangiani SN, Below JE, Campbell IM, Wiszniewski W, Muzny DM, et al. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med. 2015;7:54. doi: 10.1186/s13073-015-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blekhman R, Man O, Herrmann L, Boyko AR, Indap A, Kosiol C, et al. Natural selection on genes that underlie human disease susceptibility. Curr Biol CB. 2008;18:883–9. doi: 10.1016/j.cub.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berg JS, Adams M, Nassar N, Bizon C, Lee K, Schmitt CP, et al. An informatics approach to analyzing the incidentalome. Genet Med Off J Am Coll Med Genet. 2013;15:36–44. doi: 10.1038/gim.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism. 2013;4:36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–9. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–50. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaffer LG, Kennedy GM, Spikes AS, Lupski JR. Diagnosis of CMT1A duplications and HNPP deletions by interphase FISH: implications for testing in the cytogenetics laboratory. Am J Med Genet. 1997;69:325–31. doi: 10.1002/(SICI)1096-8628(19970331)69:3<325::AID-AJMG20>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 54.Yuan B, Harel T, Gu S, Liu P, Burglen L, Chantot-Bastaraud S, et al. Nonrecurrent 17p11.2p12 rearrangement events that result in two concomitant genomic disorders: the PMP22-RAI1 contiguous gene duplication syndrome. Am J Hum Genet. 2015;97:691–707. doi: 10.1016/j.ajhg.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yin C-L, Chen H-I, Li L-H, Chien Y-L, Liao H-M, Chou MC, et al. Genome-wide analysis of copy number variations identifies PARK2 as a candidate gene for autism spectrum disorder. Mol Autism. 2016;7:23. doi: 10.1186/s13229-016-0087-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2007;144B:869–76. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- 57.Lal D, Pernhorst K, Klein KM, Reif P, Tozzi R, Toliat MR, et al. Extending the phenotypic spectrum of RBFOX1 deletions: Sporadic focal epilepsy. Epilepsia. 2015;56:e129–33. doi: 10.1111/epi.13076. [DOI] [PubMed] [Google Scholar]
- 58.Boone PM, Campbell IM, Baggett BC, Soens ZT, Rao MM, Hixson PM, et al. Deletions of recessive disease genes: CNV contribution to carrier states and disease-causing alleles. Genome Res. 2013;23:1383–94. doi: 10.1101/gr.156075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stankiewicz P, Khan TN, Szafranski P, Slattery L, Streff H, Vetrini F, et al. Haploinsufficiency of the chromatin remodeler BPTF causes syndromic developmental and speech delay, postnatal microcephaly, and dysmorphic features. Am J Hum Genet. 2017;101 in press. [DOI] [PMC free article] [PubMed]
- 60.Scott DA, Hernandez-Garcia A, Azamian MS, Jordan VK, Kim BJ, Starkovich M, et al. Congenital heart defects and left ventricular non-compaction in males with loss-of-function variants in NONO. J Med Genet. 2017;54:47–53. doi: 10.1136/jmedgenet-2016-104039. [DOI] [PubMed] [Google Scholar]
- 61.Küry S, Besnard T, Ebstein F, Khan TN, Gambin T, Douglas J, et al. De novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am J Hum Genet. 2017;100:352–63. doi: 10.1016/j.ajhg.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lalani SR, Liu P, Rosenfeld JA, Watkin LB, Chiang T, Leduc MS, et al. Recurrent muscle weakness with rhabdomyolysis, metabolic crises, and cardiac arrhythmia due to bi-allelic TANGO2 mutations. Am J Hum Genet. 2016;98:347–57. doi: 10.1016/j.ajhg.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kremer LS, Distelmaier F, Alhaddad B, Hempel M, Iuso A, Küpper C, et al. Bi-allelic truncating mutations in TANGO2 cause infancy-onset recurrent metabolic crises with encephalocardiomyopathy. Am J Hum Genet. 2016;98:358–62. doi: 10.1016/j.ajhg.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bramswig NC, Lüdecke H-J, Pettersson M, Albrecht B, Bernier RA, Cremer K, et al. Identification of new TRIP12 variants and detailed clinical evaluation of individuals with non-syndromic intellectual disability with or without autism. Hum Genet. 2017;136:179–92. doi: 10.1007/s00439-016-1743-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Gambin T, Yuan B, Szafranski P, Rosenfeld JA, Balwi MA, et al. Haploinsufficiency of the E3 ubiquitin-protein ligase gene TRIP12 causes intellectual disability with or without autism spectrum disorders, speech delay, and dysmorphic features. Hum Genet. 2017;136:377–86. doi: 10.1007/s00439-017-1763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, et al. Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability. PLoS One. 2017;12:e0175962. doi: 10.1371/journal.pone.0175962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burrage LC, Eble TN, Hixson PM, Roney EK, Cheung SW, Franco LM. A mosaic 2q24.2 deletion narrows the critical region to a 0.4 Mb interval that includes TBR1, TANK, and PSMD14. Am J Med Genet A. 2013;161A:841–4. doi: 10.1002/ajmg.a.35751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nahorski MS, Al-Gazali L, Hertecant J, Owen DJ, Borner GHH, Chen Y-C, et al. A novel disorder reveals clathrin heavy chain-22 is essential for human pain and touch development. Brain J Neurol. 2015;138:2147–60. doi: 10.1093/brain/awv149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crowley MA, Conlin LK, Zackai EH, Deardorff MA, Thiel BD, Spinner NB. Further evidence for the possible role of MEIS2 in the development of cleft palate and cardiac septum. Am J Med Genet A. 2010;152A:1326–7. doi: 10.1002/ajmg.a.33375. [DOI] [PubMed] [Google Scholar]
- 70.Johansson S, Berland S, Gradek GA, Bongers E, de Leeuw N, Pfundt R, et al. Haploinsufficiency of MEIS2 is associated with orofacial clefting and learning disability. Am J Med Genet A. 2014;164A:1622–6. doi: 10.1002/ajmg.a.36498. [DOI] [PubMed] [Google Scholar]
- 71.Louw JJ, Corveleyn A, Jia Y, Hens G, Gewillig M, Devriendt K. MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am J Med Genet A. 2015;167A:1142–6. doi: 10.1002/ajmg.a.36989. [DOI] [PubMed] [Google Scholar]
- 72.Conte F, Oti M, Dixon J, Carels CEL, Rubini M, Zhou H. Systematic analysis of copy number variants of a large cohort of orofacial cleft patients identifies candidate genes for orofacial clefts. Hum Genet. 2016;135:41–59. doi: 10.1007/s00439-015-1606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fujita A, Isidor B, Piloquet H, Corre P, Okamoto N, Nakashima M, et al. De novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J Hum Genet. 2016;61:835–8. doi: 10.1038/jhg.2016.54. [DOI] [PubMed] [Google Scholar]
- 74.Takai R, Ohta T. A commentary on de novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J Hum Genet. 2016;61:773–4. doi: 10.1038/jhg.2016.81. [DOI] [PubMed] [Google Scholar]
- 75.Noor A, Whibley A, Marshall CR, Gianakopoulos PJ, Piton A, Carson AR, et al. Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci Transl Med. 2010;2:49ra68. doi: 10.1126/scitranslmed.3001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK, et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87:173–88. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chaudhry A, Noor A, Degagne B, Baker K, Bok LA, Brady AF, et al. Phenotypic spectrum associated with PTCHD1 deletions and truncating mutations includes intellectual disability and autism spectrum disorder. Clin Genet. 2015;88:224–33. doi: 10.1111/cge.12482. [DOI] [PubMed] [Google Scholar]
- 80.Filges I, Röthlisberger B, Blattner A, Boesch N, Demougin P, Wenzel F, et al. Deletion in Xp22.11: PTCHD1 is a candidate gene for X-linked intellectual disability with or without autism. Clin Genet. 2011;79:79–85. doi: 10.1111/j.1399-0004.2010.01590.x. [DOI] [PubMed] [Google Scholar]
- 81.Lupski JR. New mutations and intellectual function. Nat Genet. 2010;42:1036–8. doi: 10.1038/ng1210-1036. [DOI] [PubMed] [Google Scholar]
- 82.de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 83.Yuen RKC, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genomic Med. 2016;1:160271–1602710. doi: 10.1038/npjgenmed.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lindstrand A, Frangakis S, Carvalho CMB, Richardson EB, McFadden KA, Willer JR, et al. Copy-number variation contributes to the mutational load of Bardet-Biedl syndrome. Am J Hum Genet. 2016;99:318–36. doi: 10.1016/j.ajhg.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139:232–45. doi: 10.1016/j.jaci.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Charng W-L, Karaca E, Coban Akdemir Z, Gambin T, Atik MM, Gu S, et al. Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med Genomics. 2016;9:42. doi: 10.1186/s12920-016-0208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eldomery MK, Coban-Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray-Pedersen A, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26. doi: 10.1186/s13073-017-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang D, Jiang P, Xu Q, Zhang X. Arginine and glutamate-rich 1 (ARGLU1) interacts with mediator subunit 1 (MED1) and is required for estrogen receptor-mediated gene transcription and breast cancer cell growth. J Biol Chem. 2011;286:17746–54. doi: 10.1074/jbc.M110.206029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dworschak GC, Draaken M, Marcelis C, de Blaauw I, Pfundt R, van Rooij IALM, et al. De novo 13q deletions in two patients with mild anorectal malformations as part of VATER/VACTERL and VATER/VACTERL-like association and analysis of EFNB2 in patients with anorectal malformations. Am J Med Genet A. 2013;161A:3035–41. doi: 10.1002/ajmg.a.36153. [DOI] [PubMed] [Google Scholar]
- 90.Dravis C, Yokoyama N, Chumley MJ, Cowan CA, Silvany RE, Shay J, et al. Bidirectional signaling mediated by ephrin-B2 and EphB2 controls urorectal development. Dev Biol. 2004;271:272–90. doi: 10.1016/j.ydbio.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 91.Thompson BJ, Sahai E. MST kinases in development and disease. J Cell Biol. 2015;210:871–82. doi: 10.1083/jcb.201507005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oh S, Lee D, Kim T, Kim T-S, Oh HJ, Hwang CY, et al. Crucial role for Mst1 and Mst2 kinases in early embryonic development of the mouse. Mol Cell Biol. 2009;29:6309–20. doi: 10.1128/MCB.00551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Larsen KB, Lutterodt MC, Laursen H, Graem N, Pakkenberg B, Møllgård K, et al. Spatiotemporal distribution of PAX6 and MEIS2 expression and total cell numbers in the ganglionic eminence in the early developing human forebrain. Dev Neurosci. 2010;32:149–62. doi: 10.1159/000297602. [DOI] [PubMed] [Google Scholar]
- 94.Machon O, Masek J, Machonova O, Krauss S, Kozmik Z. Meis2 is essential for cranial and cardiac neural crest development. BMC Dev Biol. 2015;15:40. doi: 10.1186/s12861-015-0093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wells MF, Wimmer RD, Schmitt LI, Feng G, Halassa MM. Thalamic reticular impairment underlies attention deficit in Ptchd1Y/− mice. Nature. 2016;532:58. doi: 10.1038/nature17427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table reporting single gene de novo or hemizygous deletions or intragenic duplications in recently proposed candidate or not yet associated disease genes. (DOCX 39 kb)
Supplementary table reporting de novo or hemizygous deletions or intragenic duplications encompassing 2–5 genes in recently proposed candidate or not yet associated disease genes. (DOCX 29 kb)
Supplementary text discussing AGBL4 and CSMD1 as potential novel candidate disease genes. (DOCX 40 kb)
Supplementary table containing clinical information on patients with additional variants in ARGLU1/EFNB2. (DOCX 14 kb)
Supplementary table containing clinical information on patients with AGBL4 variants. (DOCX 19 kb)
Supplementary table containing clinical information on patients with CSMD1 variants. (DOCX 21 kb)
Supplementary figure presenting CNVs in AGBL4, including de novo (red), inherited (blue), and deletions of unknown inheritance (green). (PDF 104 kb)
Supplementary figure presenting CNVs in CSMD1, including de novo (red), inherited (blue), and deletions of unknown inheritance (green). (PPTX 95 kb)
Data Availability Statement
The CNV calls presented in Tables 1, 2, 3 and 4 and Additional files 5 and 6 from BG CMA can be accessed through the NCBI dbVar database (http://www.ncbi.nlm.nih.gov/dbvar/) under accession number nstd149.