

SUMMARY
BET proteins commonly activate cellular gene expression, yet inhibiting their recruitment paradoxically reactivates latent HIV-1 transcription. Here we identify the short isoform of BET family member BRD4 (BRD4S) as a corepressor of HIV-1 transcription. We found that BRD4S was enriched in chromatin fractions of latently infected T cells and was more rapidly displaced from chromatin upon BET inhibition than the long isoform. BET inhibition induced marked nucleosome remodeling at the latent HIV-1 promoter, which was dependent on the activity of BAF, a SWI/SNF chromatin-remodeling complex with known repressive functions in HIV-1 transcription. BRD4S directly bound BRG1, a catalytic subunit of BAF, via its bromo- and ET domains and was necessary for BRG1 recruitment to latent HIV-1 chromatin. Using ChIP-seq combined with ATAC-seq data, we found that the latent HIV-1 promoter phenotypically resembles endogenous LTR sequences, pointing to a select role of BRD4S:BRG1 complexes in genomic silencing of invasive retroelements.
Keywords: Bromodomain, BET protein, BRD4, JQ1, HIV, LTR, latency, chromatin, SWI/SNF, BRG1, BAF250a
Graphical abstract
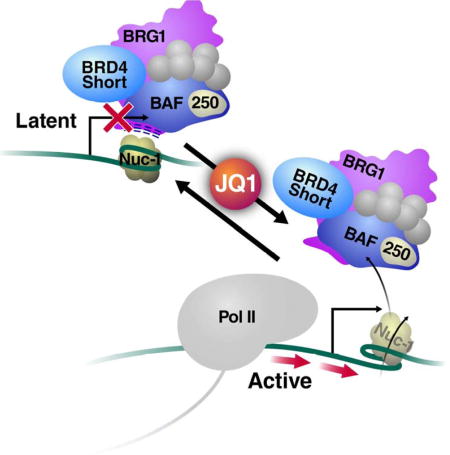
INTRODUCTION
BRD4 is a member of the bromodomain and extraterminal domain (BET) family of proteins. BET proteins are characterized by tandem N-terminal bromodomains that bind acetyl-lysine residues (Dhalluin et al., 1999; Filippakopoulos et al., 2012) and an extraterminal domain involved in protein:protein interactions (Rahman et al., 2011). BRD4 interacts with multiple transcriptional regulators, including Mediator (Jiang et al., 1998), P-TEFb (Bisgrove et al., 2007; Jang et al., 2005; Yang et al., 2005), and NF-kB (Huang et al., 2009), but also possesses intrinsic histone chaperone (Kanno et al., 2014) as well as kinase (Devaiah et al., 2012) and acetyltransferase (Devaiah et al., 2016) activities. BRD4 encodes at least two isoforms via alternative exon usage. A long isoform (BRD4L, isoform A, amino acids (aa) 1–1362) contains an extended C-terminus with the P-TEFb-interacting domain (PID) (Bisgrove et al., 2007). A second, short isoform (BRD4S, isoform C, aa 1–722) resembles other BET family members in that it lacks the extended C-terminus and PID. A third isoform (isoform B, aa 1–794) is slightly longer than BRD4S but so far has only been detected in osteosarcoma cells to restrain the propagation of DNA damage signaling along chromatin (Floyd et al., 2013). Here, we focus on the respective functional contributions of BRD4L and BRD4S proteins to the enforcement HIV-1 latency.
Latent infection by HIV-1 yields a transcriptionally inactive but replication-competent provirus integrated into host chromatin (Siliciano and Greene, 2011). It is well recognized that the host epigenetic machinery participates in transcriptional repression during HIV-1 latency by imposing reversible restrictions to the chromatinized provirus, which can be overcome or enforced by epigenetic drugs (Gallo, 2016). Such restrictions include the positioning of a nucleosome, termed nuc-1, immediately downstream of the HIV-1 transcription start site (TSS) that is specifically disrupted during transcriptional reactivation in response to proinflammatory cytokines (i.e., TNFα) or latency-reversing agents (LRAs) (i.e., histone deacetylase inhibitors) (Rafati et al., 2011; Van Lint et al., 1996; Verdin, 1991; Verdin et al., 1993). Positioning of nuc-1 during latent infection occurs over thermodynamically unfavorable cis-elements of the provirus and is actively mediated by the SWI/SNF remodeling complex BAF, characterized by the presence of the ATPases BRM or BRG1 and the ARID-domain containing BAF250a subunit (Rafati et al., 2011; Van Lint et al., 1996; Verdin, 1991; Verdin et al., 1993).
While BRD4L binds and activates P-TEFb complexes to positively support HIV-1 transcript elongation (Bisgrove et al., 2007; Jang et al., 2005; Yang et al., 2005), a function for BRD4S in HIV transcription is currently unknown. Reports from cellular genes point to considerable functional differences between BRD4 isoforms. For example, the two isoforms differentially regulate transcription of the neuronal genes Arc and GluA1 (Korb et al., 2015) and have opposing roles in the progression of metastatic breast cancer via divergent regulation of the TPX2 network (Hu et al., 2012). The two isoforms reportedly localize to distinct nuclear compartments and have unique histone-binding profiles (Alsarraj et al., 2013). Notably, overexpression and artificial tethering of N-terminal regions of BRD4 result in significant changes in chromatin structure (Floyd et al., 2013; Wang et al., 2012; Zhao et al., 2011), allusive of a potential yet undetermined function of BRD4S in chromatin biology.
Although BRD4L activates basal HIV-1 transcription in the absence of the viral transactivator Tat (Jang et al., 2005; Yang et al., 2005), it also competes with Tat for limiting reservoirs of P-TEFb, and as such negatively impacts HIV-1 transcription via P-TEFb sequestration (Bisgrove et al., 2007). Tat is a virally encoded transcription factor that recruits active P-TEFb to initiated HIV mRNAs to dramatically enhance transcript elongation and thereby support a Tat-fueled positive feed-forward loop (Ott et al., 2011). HIV-1 latency is characterized by the relative absence of Tat; as the virus exits latency, initial HIV-1 mRNAs must be produced in the absence of Tat to ignite its feed-forward loop (Ruelas and Greene, 2013). Relief of competition between BRD4L and Tat partially explains how BET inhibitors, such as the thienodiazepine JQ1 (Filippakopoulos et al., 2010), reactivate HIV-1 from latency across a variety of experimental models and in synergy with protein kinase C (PKC) agonists (Banerjee et al., 2012; Bartholomeeusen et al., 2012; Boehm et al., 2013a; Halper-Stromberg et al., 2014; Laird et al., 2015; Li et al., 2013; Zhu et al., 2012). However, BET inhibitors reverse latency also in lentiviral vectors lacking Tat, indicating a second yet unknown repressive function of BRD4 at the latent HIV-1 promoter that is distinct from interfering with the Tat:P-TEFb axis (Boehm et al., 2013b).
Here we characterized the Tat-independent repressive function of BRD4 at the latent HIV-1 promoter and identified its short isoform as a corepressor of HIV-1 transcription. BRD4S is greatly responsive to BET inhibition, highly enriched in T cell chromatin fractions, and binds the BRG1 catalytic subunit of the BAF chromatin-remodeling complex to maintain a repressive nucleosome structure at the latent HIV-1 promoter. Genome-wide ChIP- and ATAC-seq data indicate that the cooperative function of BRD4S and BRG1-containing BAF complexes may represent a select silencing mechanism to restrict expression of parasitic retroviral promoter elements such as HIV-1.
RESULTS
Short BRD4 Enforces HIV-1 Latency
Our initial results demonstrated that knockdown of BRD4 has no substantial effect on reactivation of HIV-1 from latency (Boehm et al., 2013a). However, the shRNAs used at the time exclusively targeted the 3′-UTR unique to BRD4L mRNAs, not affecting expression of the short isoform (Figure 1A, shRNA#1). We now tested shRNAs directed against the 3′-UTR unique to BRD4S (shRNA #2) and against coding sequences present in mRNAs of both isoforms (shRNA #3), resulting in efficient knockdown of one or both BRD4 isoforms (Figure 1A). Knockdown of BRD4S induced HIV-1 RNA production by ~20-fold, but knockdown of BRD4L only caused a fourfold induction (Figure 1A), similar to as previously reported (Boehm et al., 2013a). The same result was observed when GFP levels were measured by flow cytometry as a transcriptional reporter of HIV-1 latency reactivation (Figure S1A). These data indicate that BRD4S possesses an as-yet unknown function as a transcriptional repressor during HIV-1 latency. Interestingly, concomitant knockdown of both BRD4 isoforms using shRNA #3 reactivated latent HIV-1 RNA production by ~400-fold (Figure 1A). This synergistic effect was phenocopied by dual infection with shRNAs #1 and 2 targeting BRD4L and BRD4S individually as measured by GFP levels (Figure S1B). These results point to distinct, yet complementary molecular mechanisms associated with both BRD4 isoforms in latent HIV-1 infection. T cells depleted of short, but not long, BRD4 showed less reactivation in response to treatment with the BET inhibitor JQ1, and cells deprived of both isoforms failed to respond to JQ1 entirely (Figure 1B, Figure S1C), supporting a model wherein BRD4S, in conjunction with BRD4L, serves as relevant target of BET inhibitor treatment in HIV-1 latency.
Figure 1. Short BRD4 Promotes HIV-1 Latency.

(A) J-Lat A2 cells, Jurkat T cells harboring a latent HIV GFP reporter virus under the control of Tat (Jordan et al., 2003), were transduced with lentiviral shRNAs targeting BRD4 sequences as indicated or scramble non-targeting shRNAs as controls. RT-qPCR with primers specific for the GFP reporter indicating HIV transcriptional activity (relative to RPL13A, mean of three independent experiments analyzed in triplicate ± SEM) and representative western blotting of BRD4 to assess knockdown are shown. *ns denotes a non-specific band that was resistant to shRNA treatment.
(B) RT-qPCR of RNA isolated from J-Lat A2 cells transduced with shRNAs described in (A) and treated with JQ1 (625nM for 18h). Data are expressed relative to DMSO control cells for each respective shRNA transduction, and the mean of three independent experiments analyzed in triplicate ± SEM is shown.
(C) Western blotting of nuclear and chromatin fractions isolated from J-Lat A72 cells treated with JQ1 (625nM). Representative experiment of three replicates is shown.
(D) Quantification of chromatin fraction band intensities expressed relative to untreated controls from J-Lat A72 cells treated with increasing concentrations of JQ1. Average of two biological replicates is shown.
We also performed high-throughput sequencing of RNA (RNA-seq) isolated from T cells depleted of one or both BRD4 isoforms. Overall, we found ~3000 genes differentially expressed in cells depleted of any BRD4 isoform, as compared to cells treated with control shRNAs, demonstrating that the two BRD4 isoforms together regulate ~10–15% of genes (Figure S2A). Ontological analysis revealed significant enrichment of cellular pathways previously associated with BRD4, such as oncogenesis and cell cycle control (Figure S2B) (Zuber et al., 2011), and identified upstream regulators such as E2F, Myc, and p53, also formerly connected with BRD4 function (Figure S2C) (Belkina and Denis, 2012; Wu et al., 2013). Notably, a substantial number of genes were differentially affected by BRD4S and BRD4L knockdown (Figure S2A, clusters 2 and 4, n=1359), evidence that BRD4 executes isoform-specific transcriptional functions at certain loci. The smallest cluster (cluster 1, n=532) represented genes activated by depletion of either or both BRD4 isoforms (Figure S2A), indicating that only a minority of BRD4-target genes phenotypically resembles the latent HIV-1 promoter.
Next, we processed T cell nuclei into nucleoplasmic and chromatin fractions to examine the subnuclear localization of BRD4 isoforms. We found BRD4S characteristically enriched in the chromatin fraction of latently infected T cells, while BRD4L was present in both fractions (Figure 1C). Treatment with JQ1 induced a rapid decrease in BRD4S levels in chromatin fractions (<1h), with BRD4L levels decreasing more slowly and less markedly over time (Figure 1C). The same difference was observed when increasing concentrations of JQ1 were tested; chromatin-bound BRD4S responded to 10-fold lower JQ1 concentrations than BRD4L (0.18µM vs. 1.8µM, Figure 1D). Nucleoplasmic and chromatin-associated levels of Cyclin T1, a core component of P-TEFb, were not altered by JQ1 treatment over time (Figure 1C). Notably, cytoplasmic levels of both BRD4 isoforms increased in the presence of JQ1, indicating release of BRD4 into the cytoplasm under BET inhibitor treatment (Figure S1D). These results establish that BRD4S is abundant in chromatin fractions of T cells and is highly responsive to JQ1 treatment.
JQ1 Disrupts HIV-1 Chromatin Architecture Independently of Transcription
As nuc-1 is a key restraint to processive HIV-1 transcription during viral latency (Van Lint et al., 1996; Verdin, 1991; Verdin et al., 1993) and BRD4S is enriched in chromatin fractions (Figure 1C), we next examined whether JQ1 treatment is linked to nuc-1 remodeling. We performed enzymatic digestion of chromatin isolated from JQ1-treated J-Lat cells with the restriction enzyme AflII, whose cognate recognition site only becomes available when nuc-1 is remodeled (Van Lint et al., 1996). Increased cutting was captured as a decrease in successful PCR product amplification spanning this site (Figure 2A). Digestion of naked DNA was used as a control, and data were expressed as a fold increase in AflII cutting relative to untreated cells. We found that treatment with JQ1 doubled AflII accessibility in J-Lat A2 cells, indicating significant nuc-1 remodeling, which occurred to a similar extent in cells treated with TNFα, a well-known latency-reversing cytokine (Figure 2B, left panel). Importantly, remodeling was comparable when HIV-1 mRNA induction was blocked with the transcriptional CDK inhibitor flavopiridol (Figure 2B, C, right panels). This excludes the possibility that nuc-1 remodeling by JQ1 occurred as a mere consequence of transcriptional activation and polymerase passage rather than a direct influence on nuc-1 positioning.
Figure 2. Chromatin Disruption at the HIV-1 Promoter upon JQ1 Treatment.

(A) Schematic of AflII assay to measure nuc-1 accessibility. Nuclei were isolated from cells, digested in vivo with AflII, DNA was purified and subjected to qPCR using primers flanking the AflII site. Data are normalized to both uncut DNA and naked DNA cut in vitro.
(B) AflII accessibility of J-Lat A2 cells treated with TNFα (10ng/mL) or JQ1 (625nM) with or without flavopiridol (5µM) for 18h. Average of three independent experiments performed in triplicate (± SEM) is shown.
(C) RT-qPCR of HIV RNA levels relative to RPL13A from experiments (B). Average of three independent experiments performed in triplicate (± SEM) is shown.
(D) Schematic of DNase I digestion coupled to LM-PCR to measure nuc-1 positioning. Nuclei were digested with DNase I (20U/mL), DNA was purified, linkers were ligated to first-strand synthesized DNA, and PCR was performed using a linker primer and a primer specific for the 5′-LTR. Naked gDNA cut in vitro with 1U/mL DNase I was used to control for DNase I specificity and aberrant PCR amplification. PCR products were labeled with a 6-FAM-modified primer and product size, corresponding to DNase I cutting, was detected using capillary electrophoresis.
(E, F) Raw amplicon intensity as measured by fragment analysis of LM-PCR signals from J-Lat A72 cells treated with DMSO (E) or 625nM JQ1 (F) for 18h before DNase I digestion. Purple signal represents naked gDNA. One representative experiment is shown.
(G) LOWESS regression curves of LM-PCR signal for DMSO and JQ1 treatments as in (E, F). gDNA signal was subtracted from DNA cut in vivo. Data are representative of two biological replicates.
(H) ATAC-seq of viral chromatin in response to JQ1 treatment. Chromatin from J-Lat A72 cells treated with either DMSO or 625nM JQ1 for 18h was transposed and resultant libraries were paired-end sequenced. ATAC density (top) and inferred nucleosome density (bottom) for the integrated provirus are shown.
These data were confirmed by DNase I digestion coupled to ligation-mediated PCR (LM-PCR). We altered the original protocol, which relies on radiolabeled PCR probes (Carey et al., 2009b), and introduced a 6-carboxyfluorescein (6-FAM)-labeled probe, enabling sensitive detection of PCR amplicon sizes by fragment analysis with capillary electrophoresis (Figure 2D). Using a nested 6-FAM labeled primer anchored at nt +281 relative to the HIV TSS (Verdin et al., 1993), we captured PCR fragments corresponding to the 3′-edge of nuc-1 (Figure 2E, F). Upon smoothing of LM-PCR amplicon curves via LOWESS non-parametric regression and normalization to naked DNA, we observed in untreated cells a region of protection immediately downstream of the TSS corresponding to nuc-1 (Figure 2G, DMSO curve). This protected area was lost and DNase I accessibility was increased upon JQ1 treatment (Figure 2F, G), supporting the model that JQ1 increases chromatin accessibility downstream of the HIV TSS by altering nuc-1 positioning.
In addition, we performed the assay for transposase-accessible chromatin coupled to high throughput sequencing (ATAC-seq), a recent restriction enzyme-independent method relying on enhanced transposition into open regions of chromatin (Buenrostro et al., 2013). We chose paired-end sequencing of transposed J-Lat chromatin to enable proper assignment of homologous 5′- and 3′-LTR regions. Using this method, we observed a high degree of transposase accessibility centered just downstream of the HIV-1 TSS that sharply decreased over sequences occupied by nuc-1 (Figure 2H, top panel). This accessible area broadened and transposition density decreased under JQ1 treatment, indicating nuc-1 remodeling (Figure 2H, top panel). This was confirmed upon inference of nucleosome densities, which under basal conditions positioned nuc-1 at a very similar position downstream of the TSS as Verdin and colleagues reported (Verdin et al., 1993). Upon JQ1 treatment, nuc-1 shifted further downstream from the TSS with overall less density, indicative of repositioning and remodeling during transcriptional activation (Figure 2H, bottom panel). In addition, we observed considerable depletion of the upstream nucleosome, nuc-0, under JQ1 treatment. Interestingly, nuc-1 remodeling was not mirrored in the 3′-LTR, where the corresponding nucleosome shifted in the same direction as nuc-1, possibly disfavoring antisense transcription from the 3′-LTR. Collectively, these data detail previously unknown connections between chromatin remodeling processes at the latent HIV-1 provirus and BET inhibitor treatment.
JQ1-Mediated Latency Reversal and nuc-1 Remodeling Depend on the BAF Complex
The SWI/SNF family of chromatin remodeling machines has formerly been linked to the regulation of HIV transcription and latency via nucleosome positioning, both with activating and repressive effects (Easley et al., 2010b; Mahmoudi et al., 2006; Rafati et al., 2011; Treand et al., 2006; Van Duyne et al., 2011). To test the influence of these chromatin remodelers on JQ1 action, we first performed knockdown studies of the two catalytic subunits of SWI/SNF complexes, BRM and BRG1 (Kadoch and Crabtree, 2015). While BRM was dispensable for JQ1 to induce HIV-1 transcription, BRG1 was required to efficiently reverse latency in response to JQ1 treatment (Figure 3A). Notably, BRG1 was the most robust hit among eight different chromatin remodeling ATPases spanning all families (SWI/SNF, CHD, ISWI, and INO80) tested in knockdown studies with JQ1 (Figure S3).
Figure 3. The BAF Chromatin Remodeling Complex is Required for JQ1-Mediated Latency Reversal and nuc-1 Remodeling.

(A) Flow cytometry and western blotting of J-Lat A72 cells transduced with two lentiviral shRNAs targeting BRG1 or BRM to measure transcriptional reactivation. Selected cells were treated with 625nM JQ1 for 18h. Average of three independent experiments (± SEM), normalized to DMSO control per shRNA, is shown for flow cytometry.
(B, C) Flow cytometry and western blotting of J-Lat A72 cells transduced with two lentiviral shRNAs targeting BAF250a, specific for the BAF complex (B) or two lentiviral shRNAs targeting BAF180, specific for the PBAF complex (C). Selected cells were treated with indicated titration of JQ1 for 18h. Representative data of at least three independent experiments (± SD) are shown for flow cytometry.
(D) Representative LM-PCR signal obtained from J-Lat A72 cells transduced with lentiviral shRNAs targeting BRG1 or BAF250a or scramble control.
(E) Area under the curve (AUC) for LM-PCR signal across three independent replicates (± SEM).
(F–H) Representative LM-PCR signal from J-Lat A72 cells transduced with scramble control shRNAs (F) or shRNAs targeting BRG1 (G) or BAF250a (H) treated with either DMSO or 625nM JQ1 for 18h.
(I) Fold change in AUC values for LM-PCR signals across three replicates (± SEM).
Previous research demonstrated opposing effects of two SWI/SNF remodeling complexes, PBAF and BAF, on HIV-1 transcription. While PBAF cooperates with Tat to enhance HIV transcription (Easley et al., 2010a; Mahmoudi et al., 2006; Treand et al., 2006), BAF promotes HIV-1 latency via enforcement of repressive nuc-1 positioning (Rafati et al., 2011). We found that knockdown of the BAF-specific BAF250a protein, but not of the PBAF-specific component BAF180, decreased JQ1 activity (Figure 3B, C), supporting a model in which JQ1 targets the repressive BAF complex at the latent HIV promoter. In addition, expression of stable components found in both complexes, BAF155 and BAF170, was also necessary for JQ1-mediated latency reversal (data not shown).
Next, we tested the effects of BAF knockdown on nuc-1 positioning using DNase I digestion and LM-PCR. Knockdown of BRG1 and BAF250a resulted in marked increases in accessibility of DNA sequences normally occupied by nuc-1 (Figure 3D), consistent with their designated roles in actively positioning nuc-1 during latency. Similar results were obtained in multiple independent experiments that were averaged by quantifying the area under the LM-PCR amplicon curve (Figure 3E). Importantly, while cells treated with control shRNAs showed increases in nuc-1 accessibility in response to JQ1 (Figure 3F), no further increase in nuc-1 accessibility was observed in BRG1 and BAF250a knockdown cells (Figures 3G—I), in line with the requirement of repressive BAF chromatin-remodeling activities for JQ1 activity in latency reversal.
Short BRD4 Binds SWI/SNF Complexes via Bromo- and ET Domains
We next hypothesized that BRD4S physically engages SWI/SNF complexes. We first mined published affinity purification/mass spectrometry data and found two studies examining interaction partners of BRD4S (Crowe et al., 2016; Rahman et al., 2011). Although not further validated in these studies, BRG1 was detected in both data sets with a mean z-score of 1 (Figure 4A), which was in a similar range as NSD3 (z=1.2), a validated interaction partner of BRD4S (Rahman et al., 2011; Shen et al., 2015). Interestingly, the second ATPase specific for SWI/SNF complexes, BRM, was not detected in either data set. Additional SWI/SNF family members, BAF155 and BAF170, were also weakly detected with mean z-scores of 0.07 and 0.002, respectively. As expected, bait protein BRD4S scored highly in both experiments (Figure 4A). These data implicate BRG1-containing SWI/SNF complexes as physical interaction partners of BRD4S.
Figure 4. Short BRD4 Interacts with BRG1 via Bromo- and ET Domains.

(A) Standard score plot of two N-terminal BRD4 IP/MS experiments (Rahman et al., 2011, Crowe et al., 2016) indicating BRD4 (bait), NSD3 (positive control, Rahman et al., 2011 and Shen et al., 2015), and SWI/SNF components BRG1, BAF155, and BAF170.
(B) Endogenous immunoprecipitation of N-terminal BRD4 from J-Lat A72 cells nuclear/chromatin extracts, followed by western blotting of BRD4 and SWI/SNF catalytic subunits BRG1 and BRM.
(C) Immunoprecipitation of FLAG-tagged BRD4 proteins from transfected 293T nuclear/chromatin extracts, followed by western blotting using FLAG and BRG1 antibodies.
(D) Immunoprecipitation of overexpressed FLAG-BRD4S domain deletions purified from transfected 293T nuclear/chromatin extracts, followed by western blotting using FLAG and BRG1 antibodies.
(E) Immunoprecipitation of overexpressed FLAG-BRD4 domains purified from transfected 293T nuclear/chromatin extracts, followed by western blotting using FLAG and BRG1 antibodies.
(F) Immunoprecipitation of recombinant GST, GST-BDS, or GST-ET from mixtures with recombinant BRG1, followed by western blotting using GST and BRG1 antibodies. Representative blots of three independent experiments with similar results are shown.
We confirmed this finding in co-immunoprecipitation experiments of endogenous BRG1 and BRD4 proteins in J-Lat T cells. Immunoprecipitation with antibodies directed against the N-terminus of BRD4 effectively recovered both BRD4 isoforms as well as endogenous BRG1, but not BRM, proteins (Figure 4B). Overexpression of FLAG-tagged BRD4 proteins showed that BRD4S binds endogenous BRG1 more efficiently than BRD4L (Figure 4C). Deletion of individual bromodomains (ΔBD1 or ΔBD2) or the ET domain (ΔET) in BRD4S did not abrogate BRG1 binding (Figure 4D). Individual FLAG-tagged proteins corresponding to the two bromodomains (BDS, aa 47–459) or the ET domain-containing C-terminus of BRD4S (aa 601–722) were sufficient to co-immunoprecipitate endogenous BRG1, while the interdomain space (IDS, aa 458–608) located between the bromo- and ET domains did not interact with BRG1 (Figure 4E). Pulldown of recombinant GST-BDS and GST-ET yielded efficient recovery of full-length recombinant BRG1, whereas pulldown of GST did not (Figure 4F), establishing a direct interaction between BRD4 and BRG1 mediated via BRD4 bromo- and ET domains.
Short BRD4 Recruits BRG1 to the HIV Promoter
Next, we examined BRD4 and BRG1 occupancy at the HIV-1 promoter by chromatin immunoprecipitation (ChIP) using N-terminal BRD4 and BRG1 antibodies followed by qPCR. Both factors were robustly detected at the latent promoter, and occupancy decreased upon JQ1 treatment (Figure 5A, B), consistent with the model that both BRD4 and SWI/SNF are dissociated from latent HIV-1 chromatin by JQ1. No effect of JQ1 treatment or BRD4 knockdown on overall BRG1 protein levels was observed (Figure S4). We also observed no significant increase in RNA polymerase II (Pol II) occupancy upon JQ1 treatment (Figure S5), as reported (Li et al., 2013), consistent with the finding that Pol II is paused at the latent HIV-1 promoter (Peterlin and Price, 2006).
Figure 5. Short BRD4 Recruits BRG1 to the Latent HIV-1 Provirus.

(A–B) ChIP-qPCR analysis with BRD4 (A) and BRG1 (B) antibodies at two positions downstream of the HIV TSS from J-Lat A72 cells treated with DMSO or 625nM JQ1 for 18h.
(C) Western blotting of BRD4 to assess knockdown efficiency in J-Lat A72 cells.
(D–E) ChIP-qPCR analysis with BRD4 (B) and BRG1 (C) antibodies at HIV nt +155 from J-Lat A72 cells transduced with scramble shRNA or BRD4-targeting shRNAs as schematized in Figure 1A. Nonspecific IgG values have been subtracted from all displayed ChIP data, with the average of three biological replicates ± SEM shown.
To test whether BRG1 occupancy at the latent HIV-1 promoter depends on BRD4, we performed ChIP experiments in cells transduced with shRNAs targeting BRD4L, BRD4S, or both isoforms (Figure 1A, Figure 5C). Treatment with individual BRD4-targeting shRNAs decreased corresponding BRD4 protein levels in western blot analysis, as expected (Figure 5C). All shRNAs markedly lowered BRD4 occupancy at the latent HIV-1 provirus (BRD4S+L>BRD4S>BRD4L, Figure 5D), indicating that both isoforms are present at latent HIV-1 chromatin. However, only depletion of BRD4S or both isoforms, and not BRD4L alone, markedly reduced BRG1 occupancy at the latent viral promoter (Figure 5E), supporting a model in which BRD4S is necessary to recruit repressive SWI/SNF chromatin remodeling complexes to the latent HIV promoter.
Genome-Wide Co-Occupancy of BRD4 and BRG1
To extend our findings beyond the HIV promoter, we performed high-throughput sequencing of ChIP DNA recovered using BRD4 and BRG1 antibodies. The analysis of these data revealed a quantitative relationship between BRD4 and BRG1 tag densities at all peaks identified (Pearson’s r=0.66; Figure 6A). Both BRD4 and BRG1 occupancies were diminished upon JQ1 treatment, with occupancies of BRD4 more significantly shifted than for BRG1 after drug treatment (Figure 6B, C). We subsampled BRD4 peaks to determine the percentage overlap with BRG1-bound peaks, and found that >80% of BRD4 peaks overlapped a BRG1 peak (Figure 6D). Upon JQ1 treatment, the percentage overlap between N-terminal BRD4 and BRG1 was reduced to 20–40% (Figure 6D), demonstrating that JQ1 treatment significantly disrupts the overlap between BRD4 and BRG1 occupancies.
Figure 6. BRD4 and BRG1 Exhibit Genome-Wide Co-Binding that is Disrupted upon BET Inhibition.

(A) Correlation of tag densities between N-terminal BRD4 and BRG1 across all peak regions. Red line is the line of best-fit.
(B, C) Tag density (DMSO) vs. fold change in ChIP signal between JQ1 (625nM) and DMSO treatments for N-terminal BRD4 (B) and BRG1 (C). Red line represents the moving average.
(D) Percentage overlap of subsampled BRD4 peaks and BRG1 peaks ±625nM JQ1 shown for two replicates.
We next performed unsupervised non-hierarchical clustering of all genomic regions on the basis of ChIP signal. We also included ChIP-seq data for Pol II and ATAC-seq data (Figure 2H). This analysis revealed nine distinct clusters, each partitioned by unique occupancy/accessibility profiles (Figure 7A). We annotated elements in each cluster for genomic feature, and calculated z-scores through permutation testing to quantify deviations from the average genomic distribution of each feature per cluster (Figure 7B).
Figure 7. BRD4:BRG1 co-binding across genomic features and endogenous LTR sequences.

(A) Heatmap corresponding to binned ChIP/ATAC signal across the genome with clusters displayed on the left.
(B) Heatmap of z-scores (range: −10 to +10) displaying enrichments of genomic features across all clusters. SVA – SINE/VNTR/Alu, SR – simple repeat, DNA_t – DNA transposons.
(C) Average profiles of ChIP/ATAC signal across LTR peaks (n=334).
Clusters self-segregated into two major classes based upon genomic feature (Figure 7B, dendrograms). Clusters 4–7 were enriched for coding sequences, containing promoter, exon, and transcription termination site sequences, while clusters 1–3, 8, and 9 were enriched for non-coding sequences containing intergenic, intronic, and class I (LINEs, SINEs, & LTRs) and class II (DNA) transposon sequences. Significant BRD4:BRG1 co-binding was observed at both coding (i.e., clusters 6 and 7) and non-coding elements (i.e., clusters 1 and 2), establishing that BRD4:BRG1 co-occupancy is not restricted to genes. Notably, clusters 1 and 2 harbor multiple endogenous LTR-containing sequences evolutionarily related to HIV-1.
To further interrogate the response of endogenous LTR sequences to BET inhibition, we performed average profiling of all focal LTR peaks defined in our combined ChIP-/ATAC-seq analyses and found striking similarities between these sequences and the latent HIV-1 LTR. Occupancies of BRD4 and BRG1 were diminished under JQ1 treatment (Figure 7C), as with the latent HIV-1 promoter (Figure 5C, D). Furthermore, polymerase occupancy was unchanged, if not modestly increased, similar to what we observed at the HIV LTR (Figure S5). Finally, transposition density decreased and modestly broadened over endogenous LTR sequences under BET inhibition (Figure 7C), as we had observed with latent HIV-1 chromatin (Figure 2H). These data identify similarities between ancestral LTR sequences and latent HIV with respect to BRD4:BRG1 co-occupancy and chromatin remodeling in response to JQ1 treatment.
DISCUSSION
Here we report an unexpected role of the short BRD4 isoform in transcriptional repression during HIV-1 latency. BRD4S is highly chromatin-engaged and recruits repressive BRG1-containing SWI/SNF complexes to latent HIV-1 chromatin. The BET inhibitor JQ1 efficiently displaces the BRG1 ATPase from viral chromatin, thereby relieving a repressive nucleosome structure to facilitate transcription and disrupt latency. We propose that JQ1-mediated release of BRD4S:BRG1 is a critical early derepressive step required to generate a local chromatin environment permissive to processive transcription. Subsequently, de novo Tat production and BRD4L displacement promotes unrestrained P-TEFb recruitment via Tat, as reported (Li et al., 2013; Zhu et al., 2012).
This model is based on the finding that knockdown of both BRD4 isoforms activates HIV transcription most efficiently (Figure 1A, S1A–B). Significant activation is also observed with knockdown of BRD4S alone, but not with knockdown of only BRD4L. This indicates that relief of the repressive function of BRD4S is necessary but not sufficient to maximally activate latent HIV transcription. We speculate that maximal latency reversal requires the additional dissociation of BRD4L to facilitate P-TEFb release in conjunction with Tat-dependent P-TEFb recruitment (Bartholomeeusen et al., 2012; Li et al., 2013; Zhu et al., 2012). As knockdown of BRD4L alone has minimal efficacy in activating latent HIV transcription, the P-TEFb-dependent activation step associated with BRD4L removal depends on concomitant derepression via BRD4S dissociation.
Such a model explains how BET inhibition reactivates HIV-1 latency before Tat is produced from the initially generated mRNA, resolving outstanding unanswered questions surrounding JQ1 activity in HIV-1 transcription (Boehm et al., 2013a; Boehm et al., 2013b). Furthermore, the newly described mechanism of JQ1 action may offer a molecular explanation for synergies observed with other LRAs, such as PKC agonists which function in latency reversal by increasing availability of active transcription factors, namely NF-κB (Jiang and Dandekar, 2015). The relief of repressive BRD4S:BRG1 complexes by JQ1 described herein delineate a transcriptional activation pathway fundamentally distinct from PKC-induced coactivator mobilization. We expect both pathways are highly synergistic in fully enhancing Pol II processivity on HIV chromatin.
Cooperation between BET proteins and SWI/SNF components, especially BRG1, has been previously reported in the literature, both via physical (Denis et al., 2006; Rahman et al., 2011; Shi et al., 2013) and functional interactions (Liu et al., 2014). The BET protein BRD2 interacts with BRG1 and BAF155 (Denis et al., 2006). We extended these findings to show that BRD4S interacts with BRG1 in T cells, and that this interaction is mediated directly by both conserved BET domains in BRD4. BRD4 recruits BRG1 to the Nanog locus in mouse embryonic stem cells to activate Nanog production (Liu et al., 2014). In the context of acute myeloid leukemia, Vakoc and colleagues proposed that BRD4 and BRG1 operate in parallel pathways despite overlapping target gene sets and chromatin occupancy profiles (Shi et al., 2013). While our data support a direct functional interaction between BRD4S and BRG1 in the regulation of HIV-1 transcription during latency (Figure 3, 5), we cannot exclude that BRD4 and BRG1 function in parallel, overlapping pathways at certain cellular genes.
In our genome-wide data set, we report extensive overlap in occupancies of BRD4 and BRG1, supporting a model in which many genes are potentially co-regulated by both factors. However, strikingly, only a small subset of mostly intergenic regions show characteristic BRD4 and BRG1 co-occupancy in conjunction with Pol II occupancy and chromatin accessibility in response to BET inhibition. The fact that LTR-containing elements are among these regions suggests that endogenous retroviral sequences possess a unique feature that recruits the repressive function of BRD4S:BRG1 complexes. It is currently unknown what this feature is, but we speculate that it involves an acetyl-lysine ligand, as JQ1 disrupts the bromodomain:acetyl-lysine interface. An interesting candidate is histone H3K27, which when acetylated binds the first bromodomain of BRD4 (Filippakopoulos et al., 2012) and co-occupies genomic regions of BRD4 binding in human CD4+ T cells (Zhang et al., 2012). H3K27 is also trimethylated by Polycomb complexes, and this residue, when unmethylated, enhanced the response of latent HIV to JQ1-mediated reactivation (Tripathy et al., 2015). Polycomb activities are classically antagonized by Trithorax chromatin modifiers, which include SWI/SNF chromatin remodelers as well as the Drosophila homolog of BRD4 and the only fly BET protein, fs(1)h (Digan et al., 1986). Genomic LTR sequences are known Polycomb targets (Ishak et al., 2016), and Polycomb is required to prevent endogenous murine leukemia virus element mobilization (Leeb et al., 2010). How BRD4S and SWI/SNF complexes act in concert with other silencing mechanisms such as Polycomb to transcriptionally restrict parasitic retroviral elements will be examined in future studies.
Multiple clinical trials with various BET inhibitors are currently in early stages specifically for applications in cancer (Jones et al., 2016; Shortt et al., 2017). In addition, combinatorial treatment with BET inhibitors and low dose PKC agonists is currently the most effective way to reactivate latent HIV from CD4+ T cells isolated from aviremic HIV+ individuals (Laird et al., 2015), rendering BET inhibitors attractive candidates in future clinical trials targeting HIV-1 latency. An important clinical implication of our data is that BET inhibitor treatment may stimulate expression of endogenous retroelements, which are otherwise transcriptionally dormant. It is likely, however, that multiple and perhaps redundant epigenetic (Martens et al., 2005) and posttranscriptional (Yang and Kazazian, 2006) restriction mechanisms remain in place to curb gene expression from these fossilized sequences under BET inhibition.
Collectively, our findings shed original light on the complex roles of BRD4 in HIV-1 transcription and latency, defining previously unknown isoform-specific functions of this factor and its cooperation with SWI/SNF chromatin remodelers. This work extends the known chromatin-associated functions of BRD4, such as its histone chaperone activity (Kanno et al., 2014), its reported histone acetyltransferase activity (Devaiah et al., 2016), and its ability to bind mononucleosomes (Larue et al., 2014). We anticipate that our data in the context of previous literature will inform future studies into the pleiotropic functions of this epigenetic regulator not only in HIV biology, but also in various noninfectious pathological conditions.
STAR ★ METHODS
Contact for Reagent and Resource Sharing
Requests for further information and reagents may be directed to the corresponding author and lead contact, Dr. Melanie Ott, at the Gladstone Institutes (mott@gladstone.ucsf.edu).
Experimental Model and Subject Details
Cell lines
293T cells were obtained from ATCC and cultured in DMEM supplemented with 10% FBS, 1% glutamine, and 1% penicillin-streptomycin at 37°C, 5% CO2. J-Lat cells were obtained from Dr. Eric Verdin (The Buck Institute for Research on Aging, Novato, CA) and cultured in RPMI supplemented with 10% FBS, 1% glutamine, and 1% penicillin-streptomycin at 37°C, 5% CO2 at an optimal seeding density of 500,000 cells/mL.
Bacterial strains
DH5α cells were obtained from Thermo, stored at −80°C, and grown in LB medium at 37°C. These cells were used exclusively to propagate plasmids.
METHODS DETAILS
Cell Fractionation and Western Blotting
Cell fractionation was performed via the Dignam & Roeder method (Carey et al., 2009a), with minor modifications. Cells were sedimented at 1000g for 5min at 4°C and washed with ice cold PBS. Pelleted cells were resuspended in 5 volumes of DR Buffer A (10mM HEPES-KOH, 10mM KCl, 1.5mM MgCl2, 0.5mM DTT, supplemented with 1X Halt) and incubated on ice for 10min. Cell suspensions were homogenized with 10 strokes of a Dounce tight pestle (Wheaton), and nuclei were pelleted at 10000rpm for 20min, 4°C. Supernantants were decanted, the resulting pellet was gently resuspended in DR Buffer C (20mM HEPES, 0.42M NaCl, 1.5mM MgCl2, 0.2mM EDTA, 25% w/v glycerol, 0.5mM DTT, 0.5mM PMSF, 1X Halt) at ~3×109 cells/mL and rotated for 1h at 4°C. The suspension was centrifuged at 10000rpm for 30min at 4°C, and supernatants were collected as the nuclear fraction. The remaining insoluable pelleted was resuspended in digestion buffer (10mM Tris-HCl pH=7.6, 10mM MgCl2, 50mM NaCl, 5mM CaCl2, 1mM DTT, 100µg/mL BSA, 0.1mM PMSF, 1X Halt) and incubated with MNase (60U/µL, NEB) for 20 minutes at room temperature. Samples were briefly spun down and sonicated (Cole Parmer Ultrasonic Processer) with 40% amplitude, pulsing 10s on, 2s off, for a total of 10 pulses, two times spinning down briefly in between sonication steps. Samples were spun down at maximum speed for 5 minutes at 4°C, with the resulting supernatant collected as the chromatin fraction. Protein concentrations of nuclear and chromatin fractions were determined using a Bradford assay (BioRad), and normalized among samples per experiment before analysis via western blotting using standard techniques.
ShRNA-Mediated Knockdown Experiments
Knockdown experiments were performed essentially as described (Boehm et al., 2013a). Knockdown data represent polyclonal cell lines obtained upon transduction and puromycin selection.
Flow Cytometry
All flow cytometry experiments were performed using the IntelliCyt HTFC with samples in each experiment analyzed in at least triplicate. Cell viability was estimated using forward and side scatter parameters. Roughly 5–20,000 live cells were analyzed per condition, gated such that ~1% of control samples were assigned as GFP+.
RNA Isolation, Reverse Transcription, and qPCR
RNA for qPCR experiments was isolated from approximately 106 J-Lat cells via RNA STAT-60 (AMSBIO) and Zymo-Spin IIC columns (Zymo Research) including on-column DNase I digestion. Isolated RNA primed with random hexamers (Thermo) was reverse transcribed using the AMV reverse transcriptase (Promega) according to manufacturer’s instructions. cDNA was analyzed via the 7500 Fast Real-Time PCR system (Applied Biosystems).
Plasmids
The majority of BRD4 plasmids were generous gifts from Dr. Eric Verdin (The Buck Institute for Research on Aging, Novato, CA). The BDS, IDS, and ET constructs (Figure 4D) were cloned into pFLAG-CMV-2 (Sigma) via standard restriction enzyme-dependent methods using primers listed in Table S2. SV40 NLS sequences were inserted using a linker to ensure proper localization of BRD4 truncations. All plasmids and corresponding sequence information are available upon request.
Accessibility of Nucleosomal DNA to Restriction Enzymes
Enzymatic digestion of chromatin was performed essentially as described (Verdin et al., 1993). 3–5×106 J-Lat cells (per condition) were spun down at 1000g for 5 minutes at 4°C and washed once with ice cold PBS. Cells were resuspended at 25×106 cells/mL in buffer A (10mM Tris-HCl pH=7.6, 10mM NaCl, 3mM MgCl2, 0.3M sucrose, 0.3mM spermidine, 1X Halt) and incubated on ice for 5min. An equal volume of buffer A supplemented with 0.2% NP-40 (Surfact Amps, Thermo) was added and suspensions were incubated on ice for 5min. Nuclei were pelleted by centrifugation at 300g for 5min at 4°C, and resuspended at 108 nuclei/mL in either buffer B (For DNase I digestion, 10mM Tris-HCl pH=7.6, 10mM MgCl2, 1mM CaCl2, 50mM NaCl, 1mM DTT, 100µg/mL BSA, 0.1mM PMSF, 1X Halt) or 1X CutSmart (for AflII digestion, NEB). Reactions were stopped with the addition of 2mM EDTA and 1% SDS, upon which proteinase K was added at a concentration of 400µg/mL and incubated at 55°C overnight. RNase was added at a concentration of 100µg/mL and incubated at 37°C for at least 1h. DNA was purified via QIAquick PCR purification kit (Qiagen).
To control AflII experiments, naked genomic DNA was incubated either with or without AflII and digested to completion for 20min at 37°C and heat inactivated at 65°C for 20mins. 10–80ng of DNA, normalized among samples, was subjected to qPCR using the 7500 Fast Real-Time PCR system (Applied Biosystems).
For ligation-mediated PCR, a standardized protocol (Carey et al., 2009b) was used with several adaptations, notably to amend the technique to fragment analysis using the 3730xl DNA Analyzer (Applied Biosystems, UC Berkeley DNA Sequencing Facility). For DNase I-treated DNA, first strand synthesis was performed on 1µg of control or in vivo digested DNA with Pfu II Ultra. Pre-annealed, unidirectional linkers were ligated to DNA overnight at room temperature using T4 DNA ligase (NEB). Ligated DNA was precipitated and amplified using a linker primer and a primer specific for the 5′-LTR. NEBNext High Fidelity 2X PCR MasterMix (NEB) and the following cycling conditions were used for amplification: 98°C 2min, 98°C 30s, 58°C 2min, 72°C 2min, steps 2–4 repeated 29x, 72°C 5 min, 4°C hold. A 6-FAM labeled probe was added and reactions were cycled for an additional three rounds of amplification: 98°C 2min, 98°C 1min, 62°C 2min, 72°C 10min, steps 2–4 3x, 4°C hold. Amplified, labeled DNA was precipitated and resuspended in 30µL HiDi Formamide. 1–5µL of DNA was mixed with 0.35µL GeneScan 1200 LIZ and analyzed on the 3730xl DNA Analyzer. Amplicon peaks were called using PeakScanner, and data was imported to GraphPad Prism for visualization and analysis by LOWESS regression.
Immunoprecipitations
Immunopreciptaitons were performed on nuclear/chromatin extracts from either J-Lat A72 (for endogenous complexes) or 293T cells (for domain mapping). Roughly 1.8×107 293T cells were transfected with 20µg of the indicated FLAG-BRD4 construct using PolyJet (Signagen) and harvested 48–72h post transfection. For endogenous IPs from J-Lat cells, roughly 2×107 cells were used per extraction.
Nuclear/chromatin extracts were prepared exactly as described for cell fractionation (see above), except upon resuspension in DR Buffer C, extracts were directly sonicated and cleared. 1–2mg of extract was diluted in FLAG-IP buffer (50mM Tris-HCl, 150mM NaCl, 1mM EDTA, 1% Triton-X-100, and 1X Halt) and ~2–4µg of indicated antibody was added (BRD4 N (Abcam, ab128874), IgG control (Santa Cruz, sc-2027) FLAG (Sigma, M2, F1804)). Dynabeads Protein A/G (Thermo, 50µL/IP) were washed in FLAG-IP buffer, added to samples, and rotated overnight at 4°C. IPs were washed 3x with FLAG-IP buffer before elution in 2X Laemmli buffer and processing via western blotting.
For immunoprecipitations of recombinant GST-tagged proteins, ~2µg GST (Abcam, ab70456), GST-BDS (Abcam, ab176941), or GST-ET (kind gift of Ming-Ming Zhou, Icahn School of Medicine at Mount Sinai, NY) were mixed with ~1µg recombinant full-length BRG1 (Abcam, ab82237), ~1µg of either nonspecific IgG (Santa Cruz, sc-2027) or anti-GST (Abcam, ab9085) antibodies, and Dynabeads Protein A (Thermo, 50µL/IP) diluted in FLAG-IP buffer. IPs were washed 3x with FLAG-IP buffer before elution in 2X Laemmli buffer and processing via western blotting using BRG1 and GST antibodies.
Chromatin Immunoprecipitation
Exponentially growing J-Lat A72 cells (~3×107 cells/IP) were fixed with 1% formaldehyde (Sigma-Aldrich) for 20 minutes at room temperature. Cross-linking was quenched with 125mM glycine for 5 minutes, after which cells were sedimented at 1000rpm for 5 minutes at 4°C and washed 3x with ice-cold PBS. Outer membranes were lysed with Lysis Solution #1 (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% Surfact Amps NP-40, 1X Halt) for 15min on ice and nuclei were spun down at 1000rpm. Supernantants were discarded, and the resulting pellet was washed once in MNase digestion buffer (10mM Tris-HCl pH=7.6, 10mM MgCl2, 50mM NaCl, 5mM CaCl2, 1mM DTT, 100µg/mL BSA, 1X Halt) and resuspended in the same buffer supplemented with 60U/µL MNase (NEB). Digestion reactions were rocked at room temperature for 20 minutes, stopped with 2mM EDTA, and centrifuged at 10000rpm for 5 minutes. Supernatants were discarded and the pellet was resuspended in Lysis Solution #2 (0.1% SDS, 10mM EDTA, 50mM Tris-HCl pH=8.0, 1X Halt). Samples were sonicated (Cole Parmer Ultrasonic Processer) with 40% amplitude, pulsing 10s on, 2s off, for a total of 10 pulses, two times spinning down briefly in between sonication steps. Samples were spun down for 5 minutes at 4°C, with the resulting supernatant collected as the chromatin fraction.
A small fraction (1/30) of isolated chromatin was used for quantification via reverse cross-linking with ~600mM NaCl, boiling, RNase-treatment, and DNA purification via QIAQuick PCR purification kit (Qiagen). Purified DNA was quantified and used to estimate chromatin concentration. DNA was also analyzed via agarose gel electrophoresis to ensure proper shearing of DNA to 200–1000bp. Chromatin concentrations were normalized among samples and pre-cleared with Dynabeads Protein A/G (Thermo, 20µL/IP). Approximately 30–100µg of chromatin was used for each IP, diluted in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2mM EDTA, 16.7mM Tris-HCl, 167mM NaCl, 1X Halt). Antibodies were added (BRD4 N (Abcam, ab128874): 3µg, BRG1 (Abcam, ab110641): 0.3µg, Pol II (Santa Cruz, N-20, sc-899): 4µg), along with the equivalent of 50µL of Protein A/G Dynabeads washed in ChIP dilution buffer. Immunoprecipitations were carried out overnight at 4°C and washed as follows: 3x in low salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris-HCl pH 8.0, 150mM NaCl), 1x in high salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris-HCl pH=8.0, 500mM NaCl), 1x in LiCl buffer (0.25M LiCl, 1% NP-40, 1% deoxycholic acid (sodium salt), 1mM EDTA, 10mM Tris-HCl pH=8.0), and 1x in TE buffer. DNA was eluted from beads 2x with elusion buffer (50mM NaHCO3, 1% SDS) at 65°C with shaking. NaCl was added to a final concentration of ~600mM and samples were reverse cross-linked overnight at 65°C. 10µg RNase A (Thermo) was added and samples were incubated for 20 minutes at 37°C, before purification of ChIP DNA on QIAquick columns. For qPCR experiments, ChIP DNA was analyzed using the 7500 Fast Real-Time PCR system (Applied Biosystems).
ChIP-seq
ChIP DNA (1–2 ng) was processed using the Ovation Ultralow Library Systems V2 (NuGEN) before quality control of libraries using BioAnalyzer 2100 (Agilent) and KAPA quantification. ChIP samples were sequenced on the HiSeq 2500 (Illumina) at the UCSF Institute for Human Genetics under the PE100 protocol. For mapping statistics, please see Table S1.
RNA-seq
For RNA-sequencing applications, RNA was isolated from exponentially growing J-Lat A72 cells via the RNeasy Plus Mini Kit (Qiagen) and was checked for quality on the BioAnalyzer 2100 (Aglient) before library preparation. Libraries were prepared using the Ovation RNA-Seq System V2 (NuGEN) and sequenced on the HiSeq 2500 (Illumina) at the UCSF Institute for Human Genetics under the PE100 protocol. For mapping statistics, please refer to Table S1.
ATAC-seq
ATAC-seq was performed essentially as described (Buenrostro et al., 2013), with the exception of outer membrane lysis prior to transposition. This step was removed from the original protocol to reduce contamination from mitochondrial DNA. Libraries were assessed for quality control on the BioAnalyzer 2100 (Aglient) to ensure nucleosomal phasing and complexity. Sequencing was performed on the HiSeq 2500 (Illumina) at the UCSF Institute for Human Genetics under the PE100 protocol. For mapping statistics, please see Table S1.
Quantification and Statistical Analysis
Data were processed and visualized using GraphPad Prism. Most quantified data represent mean ± SEM, as indicated. AflII accessibility was defined as (Ct,digested-Ct,undigested)nuclei/(Ct,digested-Ct,undigested)naked DNA, with Ct as the real-time qPCR threshold cycle number. LOWESS non-parametric regression analysis of raw LM-PCR data was performed in GraphPad Prism, with gDNA signal subtracted from that of DNA digested in vivo. Western blot band intensities were quantified using ImageJ.
Bioinformatic analysis
All reads were trimmed for adapters and quality using the fastq-mcf tool. ChIP-/ATAC-seq reads were then aligned to the hg19 human genome assembly using bowtie2 (Langmead and Salzberg, 2012), while RNA-seq reads were aligned with tophat2 (Kim et al., 2013). Tags that did not map to the genome uniquely (mapq≥30) were not kept. Separately, ATAC-seq data was aligned to the A72 HIV integrant (sequenced via primer walking, sequence available upon request) without removing non-uniquely mapping reads in order to allow alignment to homologous LTR regions.
To calculate tag density measurements for ATAC-seq, ChIP-seq, and RNA-seq data, reads were first processed in an experiment-specific manner, with the center point of each ChIP-seq fragment location being taken as the most likely position of factor binding, and ATAC-seq reads being shifted by 4bp from the edge of the fragment to more precisely indicate the location of transposition. To later estimate nucleosome occupancy, ATAC-seq fragments longer than the span of a nucleosome were kept and the center point of each fragment retained as the most likely position of a nucleosome for that fragment. After sample-specific processing of tags, tag densities were calculated. The genome was divided into 20bp bins and the number of tags that mapped to within 75bp of each 20bp bin was used as the tag count (tc) for each bin. Tag density was calculated using those counts as follows:
tagDensity = tc * #binsInGenome/#tagsTotal
and for ChIP data, input normalization was calculated for each bin as:
normalizedDensity = tagDensity(chip) - tagdensity(input)
Peaks were called for ChIP-seq and ATAC-seq by thresholding and merging bins with tag densities greater than 60. Replicate concordant peaks were identified as peaks that were observed in multiple replicates.
A region of the same size as each peak was taken from 10kb upstream of each peak to serve as a background region. The median peak score across all backgrounds regions for each sample was used to normalize the tag density values to correct for varying signal-to-noise ratios between experiments. All subsequent peak-based analyses were performed using the maximum normalized density score at each peak for each sample. Clustering of peaks was performed by first performing k-means clustering with 100 clusters, and then using HOPACH to collapse similar clusters. Peak overlap analysis was performed by sampling without replacement 1000 of the replicate concordant peaks and then observing the distribution of percent overlap with other factors. Genome feature enrichment was calculated by a permutation analysis in which the feature associations were shuffled 1000 times and the fraction of each peak cluster observed to map to each feature was observed. The mean and standard deviation these random associations served to model the significance of the enrichment of each peak cluster with each genomic feature. ChIP-/ATAC-seq average profiling was performed using deepTools.
Data and Software Availability
All software used in this study is listed in the Key Resource Table. GEO accession numbers can be found at GSE100266.
Supplementary Material
Acknowledgments
We thank all members of the Ott laboratory for sharing reagents and advice throughout the preparation of this manuscript. We also thank Eric Verdin, Danica Galonic Fujimori, and Hiten Madhani for guidance throughout the course of this work. We are grateful to Veronica Fonseca and John Carroll for administrative and graphical support, respectively. We gratefully acknowledge support from the NIH (R01 AI083139 and U19 AI096113 to M.O.). R.J.C. was supported in part by NIH Training Grant T32 GM007175. This publication was made possible with help from the University of California, San Francisco-Gladstone Institute of Virology & Immunology Center for AIDS Research (CFAR), an NIH-funded program (P30 AI027763), the amfAR Institute for HIV Cure Research (amfAR grant number 109301), and the James B. Pendleton Charitable Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental information includes Figures S1–S5 and Tables S1, S2 and can be found with their article online at ________.
AUTHOR CONTRIBUTIONS
R.J.C. performed the experiments with help from P.F., R.J.C and S.T. analyzed the data, H.S. first raised the idea of BRD4:BRG1 interactions, Q.Z. and M.M.Z. provided critical reagents, M.O. supervised, guided, and funded the research, R.J.C. and M.O. wrote the paper, with input from P.F. and S.T.
References
- Alsarraj J, Faraji F, Geiger TR, Mattaini KR, Williams M, Wu J, Ha NH, Merlino T, Walker RC, Bosley AD, et al. BRD4 Short Isoform Interacts with RRP1B, SIPA1 and Components of the LINC Complex at the Inner Face of the Nuclear Membrane. PLoS ONE. 2013;8(11):e80746. doi: 10.1371/journal.pone.0080746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012;92:1147–1154. doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012;287:36609–36616. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013a;12:452–462. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Conrad RJ, Ott M. Bromodomain proteins in HIV infection. Viruses. 2013b;5:1571–1586. doi: 10.3390/v5061571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, Smale ST. Dignam and Roeder nuclear extract preparation. Cold Spring Harb. Protoc. 2009a;2009 doi: 10.1101/pdb.prot5330. pdb prot5330. [DOI] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, Smale ST. In vivo DNase I, MNase, and restriction enzyme footprinting via ligation-mediated polymerase chain reaction (LM-PCR) Cold Spring Harb. Protoc. 2009b;2009 doi: 10.1101/pdb.prot5277. pdb prot5277. [DOI] [PubMed] [Google Scholar]
- Crowe BL, Larue RC, Yuan C, Hess S, Kvaratskhelia M, Foster MP. Structure of the Brd4 ET domain bound to a C-terminal motif from gamma-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. U.S.A. 2016;113:2086–2091. doi: 10.1073/pnas.1516813113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis GV, McComb ME, Faller DV, Sinha A, Romesser PB, Costello CE. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006;5:502–511. doi: 10.1021/pr050430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, Dey A, Ozato K, Singer DS. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016;23:540–548. doi: 10.1038/nsmb.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ, 3rd, Singer DS. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 2012;109:6927–6932. doi: 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Digan ME, Haynes SR, Mozer BA, Dawid IB, Gans M. Genetic and Molecular Analysis of Fs(1)H, a Maternal Effect Homeotic Gene in Drosophila. Dev. Biol. 1986;114:161–169. doi: 10.1016/0012-1606(86)90392-1. [DOI] [PubMed] [Google Scholar]
- Easley R, Carpio L, Dannenberg L, Choi S, Alani D, Van Duyne R, Guendel I, Klase Z, Agbottah E, Kehn-Hall K, et al. Transcription through the HIV-1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology. 2010a;405:322–333. doi: 10.1016/j.virol.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley R, Van Duyne R, Coley W, Guendel I, Dadgar S, Kehn-Hall K, Kashanchi F. Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim. Biophys. Acta. 2010b;1799:275–285. doi: 10.1016/j.bbagrm.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd SR, Pacold ME, Huang QY, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498 doi: 10.1038/nature12147. 246-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo RC. Shock and kill with caution. Science. 2016;354:177–178. doi: 10.1126/science.aaf8094. [DOI] [PubMed] [Google Scholar]
- Halper-Stromberg A, Lu CL, Klein F, Horwitz JA, Bournazos S, Nogueira L, Eisenreich TR, Liu C, Gazumyan A, Schaefer U, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wu G, Rusch M, Lukes L, Buetow KH, Zhang J, Hunter KW. Integrated cross-species transcriptional network analysis of metastatic susceptibility. Proc. Natl. Acad. Sci. U.S.A. 2012;109:3184–3189. doi: 10.1073/pnas.1117872109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell. 2016 doi: 10.1016/j.molcel.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jiang GC, Dandekar S. Targeting NF-kappa B Signaling with Protein Kinase C Agonists As an Emerging Strategy for Combating HIV Latency. AIDS Res. Hum. Retrov. 2015;31:4–12. doi: 10.1089/aid.2014.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC, Kornberg RD. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8538–8543. doi: 10.1073/pnas.95.15.8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nature Rev. Genet. 2016;17:630–641. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Science Adv. 2015;1:e1500447. doi: 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun HW, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D, et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014;21:1047–1057. doi: 10.1038/nsmb.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker-Scharff I, Darnell RB, Allis CD. BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nature Neurosci. 2015;18:1464–1473. doi: 10.1038/nn.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Invest. 2015;125:1901–1912. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue RC, Plumb MR, Crowe BL, Shkriabai N, Sharma A, DiFiore J, Malani N, Aiyer SS, Roth MJ, Bushman FD, et al. Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids. Res. 2014;42:4868–4881. doi: 10.1093/nar/gku135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010;24:265–276. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013;41:277–287. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Stein P, Cheng X, Yang W, Shao NY, Morrisey EE, Schultz RM, You J. BRD4 regulates Nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ. 2014;21:1950–1960. doi: 10.1038/cdd.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T, Parra M, Vries RG, Kauder SE, Verrijzer CP, Ott M, Verdin E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006;281:19960–19968. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- Martens JHA, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005;24:800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Ott M, Geyer M, Zhou Q. The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe. 2011;10:426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011;9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruelas DS, Greene WC. An Integrated Overview of HIV-1 Latency. Cell. 2013;155:519–529. doi: 10.1016/j.cell.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang E, Roe JS, Suzuki Y, Pappin DJ, Joshua-Tor L, Vakoc CR. NSD3-Short Is an Adaptor Protein that Couples BRD4 to the CHD8 Chromatin Remodeler. Mol. Cell. 2015;60:847–859. doi: 10.1016/j.molcel.2015.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortt J, Ott CJ, Johnstone RW, Bradner JE. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer. 2017;17:268. doi: 10.1038/nrc.2017.26. [DOI] [PubMed] [Google Scholar]
- Siliciano RF, Greene WC. HIV latency. Cold Spring Harb. Perspect. Med. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treand C, du Chene I, Bres V, Kiernan R, Benarous R, Benkirane M, Emiliani S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006;25:1690–1699. doi: 10.1038/sj.emboj.7601074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy MK, McManamy ME, Burch BD, Archin NM, Margolis DM. H3K27 Demethylation at the Proviral Promoter Sensitizes Latent HIV to the Effects of Vorinostat in Ex Vivo Cultures of Resting CD4+ T Cells. J. Virol. 2015;89:8392–8405. doi: 10.1128/JVI.00572-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan MJ, Pollard KS. Hybrid clustering of gene expression data with visualization and the bootstrap. J. Stat. Plan. Infererence. 2003;117:275–303. [Google Scholar]
- Van Duyne R, Guendel I, Narayanan A, Gregg E, Shafagati N, Tyagi M, Easley R, Klase Z, Nekhai S, Kehn-Hall K, et al. Varying modulation of HIV-1 LTR activity by Baf complexes. J. Mol. Biol. 2011;411:581–596. doi: 10.1016/j.jmb.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Verdin E. Dnase I-Hypersensitive Sites Are Associated with Both Long Terminal Repeats and with the Intragenic Enhancer of Integrated Human-Immunodeficiency-Virus Type-1. J. Virol. 1991;65:6790–6799. doi: 10.1128/jvi.65.12.6790-6799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Paras P, Jr, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Li Q, Helfer CM, Jiao J, You J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012;287:10738–10752. doi: 10.1074/jbc.M111.323493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell. 2013;49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H, Mo Y, Liao W, Zhang MQ. Genome-wide localization of protein-DNA binding and histone modification by a Bayesian change-point method with ChIP-seq data. PLoS Comput. Biol. 2012;8:e1002613. doi: 10.1371/journal.pcbi.1002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Kazazian HH., Jr L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat. Struct. Mol. Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJ, Thiessen N, Pettersson S, Jones SJ, Knapp S, et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J. Biol. Chem. 2012;287:43137–43155. doi: 10.1074/jbc.M112.413047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Nakamura T, Fu Y, Lazar Z, Spector DL. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nature Cell Biol. 2011;13:1295–1304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, Qu H, Walker BD, Elledge SJ, Brass AL. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012;2:807–816. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.