

Abstract
Apoptosis is a cell death pathway that is activated in ischemic stroke. The interaction between Fas and its ligand (FasL) initiates a complex pattern of intracellular events involving the recruitment of specific adaptor proteins and the development of apoptosis. We recently reported that dynamin is increased after experimental stroke, and its inhibition improves neurological outcome. Dynamin has been shown to transport Fas from the endoplasmic reticulum to the cell surface where it can be bound by its ligand, FasL. Hypothermia has been shown to improve outcome in numerous stroke models, and this protection is associated with reduced apoptosis and Fas expression. To explore the contribution of dynamin to hypothermic neuroprotection, we subjected mice to distal middle cerebral artery occlusion (dMCAO) and applied one of two cooling paradigms: one where cooling began at the onset of dMCAO (early hypothermia) and another where cooling began 1 hour later (delayed hypothermia), compared with normothermia (Norm). Both cooling paradigms reduced numbers of apoptotic cells, as well as Fas and dynamin compared with Norm. Fas and dynamin were co-expressed in neurons. Neuronal cultures were exposed to oxygen glucose deprivation. Hypothermia decreased dynamin as well as surface expression of Fas, and this correlated to reduced cell death. The results of this study suggest that dynamin may participate in the Fas-mediated apoptotic pathway, and its reduction may be linked to hypothermic neuroprotection.
Keywords: : hypothermia, ischemic stroke, apoptosis, dynamin, Fas
Introduction
Apoptosis has long been recognized to contribute to brain cell death after stroke in experimental models (Ferrer and Planas, 2003). There are two main apoptotic pathways: the intrinsic pathway, which occurs within the cell's mitochondria (Green and Reed, 1998), and the extrinsic pathway, which is triggered via a cell surface receptor (Ashkenazi and Dixit, 1998). Although much of the focus on apoptosis in experimental stroke has centered around the intrinsic or mitochondrial pathway, it is also recognized that the extrinsic or receptor-mediated cell death occurs as well. The most well known of these receptors is Fas (also referred to as CD95 or APO-1). The binding of Fas to its extracellular ligand (FasL) begins the formation of a so-called “death complex.” This binding leads to the recruitment of Fas-associated death domain (FADD) protein and its cytoplasmic adaptor protein (Lavrik and Krammer, 2012) along with procaspase-8 (Love, 2003). This FasL-Fas-FADD-procaspase-8 complex is often referred to as a death-inducing signaling complex (DISC) that, ultimately, leads to the generation of activated caspase-8 and apoptosis via effector caspases such as caspase-3. The Fas pathway has been shown to participate in several experimental models of neurological conditions, including ischemic stroke, traumatic brain injury, neuroinflammation, and neurodegeneration (Padosch et al., 2003).
Dynamin is a guanine triphosphatase that is responsible for endocytic functions (Ferguson and De Camilli, 2012). Dynamin-1 is expressed exclusively in neuronal cells of the brain (Li et al., 2015). Accumulated evidence in several laboratories demonstrated that dynamin has been implicated in numerous membrane trafficking events, including phagocytosis (Gold et al., 1999), trafficking to and from late endosomes and trans-Golgi membranes (Jones et al., 1998), and endocytosis-mediated pathogen entry into human cells (Meier and Greber, 2004). Dynamin has recently been shown to translocate Fas protein from the Golgi apparatus to the cell surface where it can be bound by FasL (Ivanov et al., 2006). However, dynamin has not been studied extensively in the brain, and it has been studied even less in brain ischemia.
Hypothermia has long been recognized as a potent neuroprotectant. Experimental studies (Krieger and Yenari, 2004) and clinical trials (Bernard et al., 2002) have shown that hypothermia protects the brain from ischemia. Hypothermia has been shown to slow metabolism, prevent loss of cellular energy stores, and inhibit release of excitotoxins, including glutamate. However, these mechanisms do not fully account for this robust protective effect, as even small decreases in temperature lead to similar brain protection. Thus, other processes may contribute to this phenomenon. Several labs, including ours, have reported that early hypothermia reduces infarct size and markedly improves neurological recovery after experimental stroke. Protection by hypothermia is not only associated with the inhibition of apoptosis via the intrinsic or mitochondria-dependent pathway (Inamasu et al., 2000; Xu et al., 2002) but also appears to decrease apoptosis via the extrinsic or receptor-mediated pathways (Liu et al., 2008). We previously showed that therapeutic hypothermia was associated with the inhibition of FasL cleavage, as levels of soluble FasL were decreased by cooling (Liu et al., 2008). Decreased soluble FasL presumably inhibits Fas ligation and, in turn, reduces caspase-8 activation (Liu et al., 2008).
We recently identified dynamin as a profoundly suppressed protein in HSP70-overexpressing mice subjected to experimental stroke. Further, HSP70 is known to protect the brain from stroke, and the inhibition of dynamin does the same (Kim et al., 2016). Since hypothermia has been viewed as a “gold standard” of neuroprotection, we explored whether dynamin suppression may be similarly linked to neuroprotection by hypothermia. We employ a paradigm identical to that used in our previously published report where hypothermia led to improved neurological deficits and smaller infarct size (Kawabori et al., 2013) as a model of hypothermic neuroprotection.
Materials and Methods
All experimental manipulations in mice were in accordance to protocols approved by the San Francisco VA Medical Center's Institutional Animal Care and Use Committee (IACUC), and they were within NIH guidelines.
Stroke model
Male C57/B6 mice (Simonson Labs), weighing 20–25 g and 8–12 weeks old, were anesthetized with isoflurane (5% for induction, 2% for maintenance via facemask) in an air/oxygen mixture (3:1) and subjected to permanent distal middle cerebral artery occlusion (dMCAO) as previously described with slight modifications (Kawabori et al., 2013). A small craniotomy was made above the proximal segment of the middle cerebral artery (MCA), and the MCA was exposed after the dura was opened and retracted. The MCA was occluded by coagulation with a bipolar forceps at the MCA segment just proximal to the olfactory branch, which was consistently present. The duration of anesthesia was the same in all experimental groups. At the time of recovery from anesthesia, animals were assessed for neurological deficits by using a Bederson score (Zheng et al., 2008). Those with a score of 0 or no deficit were removed from the experiment. Animals were euthanized at 1, 3, and 7 days after ischemia; brains were prepared for subsequent studies by transcardial perfusion with normal saline, followed by cryoprotection in 20% sucrose and fixation in 4% paraformaldehyde.
Hypothermia paradigms
Mice were randomized into three groups: early hypothermia (eHypo), delayed hypothermia (dHypo), and normothermia (Norm). Cooling to a rectal temperature of 29.5–30.5°C was achieved within 5 minutes of turning off the heating blanket and spraying the body with 70% alcohol. Rewarming was achieved within 5–10 minutes by turning on the heating blanket and placing a lamp over the animal (Tang et al., 2013). In the eHypo group, cooling began at the time of dMCAO; whereas in the dHypo group, cooling began 1 hour after dMCAO. In both paradigms, cooling was maintained for 2 hours. In the Norm group, rectal temperature was maintained in the normal range (36.5–37.5°C) throughout the experiment. We previously showed that rectal temperatures of 37°C and 30°C corresponded to brain temperatures of 38°C and 33°C, respectively (Yenari et al., 2000; Tang et al., 2013).
Assessing in situ detection of cells with fragmented DNA (TUNEL stain)
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) using the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon) was performed as previously described by our group (Kim et al., 2014). Briefly, cryosections in the coronal plane were incubated in terminal deoxynucleotidyl transferase (TdT) enzyme and digoxigenin-labeled dUTP at 37°C for 1 hour, followed by reacting with 3,3′-diaminobenzidine (DAB) substrate. Sections were counterstained with cresyl violet (CV). Negative control sections were stained in the same way, but with TdT eliminated, whereas the lactating mammary gland was used as a positive control. TUNEL-positive cells were counted from five random nonoverlapping fields within the cortex adjacent to the outer boundary of the infarct. The number of TUNEL-positive cells was expressed as the percentage of total cells counted from comparable anatomic brain regions of sham animals.
Immunohistochemistry and immunofluorescence
Brain sections were immunostained for dynamin and Fas. Cryosections were incubated sequentially in 0.1% hydrogen peroxidase (3 minutes), 2.5% goat serum blocking buffer (Vector Laboratories) for 30 minutes, and mouse anti-dynamin-1 (1:1000; Santa Cruz) or rabbit anti-Fas (1:100; Santa Curz) antibodies overnight at 4°C. Immunoreactivity was amplified and detected with biotinylated anti-mouse IgG (1:1000; Vector Laboratories) or anti-rabbit IgG (1:200; Sigma), peroxidase-conjugated avidin (ABC Elite, Vector Laboratories), and DAB substrate. Sections were counterstained with CV. Colabeling for dynamin and Fas was performed by dual-fluorescence immunostaining. After blocking in normal serum for 30 minutes, brain sections were incubated in antibodies against dynamin and Fas diluted in blocking solution overnight at 4°C. After three washes with phosphate buffered saline (PBS), sections were incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:200; Invitrogen) and Alexa Fluor 588-conjugated goat anti-mouse IgG (1:1000; Invitrogen) for 2 hours at room temperature. After three washes in PBS, sections were mounted and coverslipped.
In vitro model of ischemia-like injury
The immortalized mouse neuron cell line, Neuro2A, was purchased from the American Type Culture Collection. Cultures were grown and maintained in Dulbecco's modified eagle medium (Cellgro) supplemented with 10% Fetal Bovine Serum Defined (Hyclone) and antibiotics penicillin and streptomycin at 100 U/mL (Gibco). Under a humidified 5%, CO2 95% air atmosphere and at 37°C, cells were plated in 25 cm2 cell culture flasks (Corning). Media were changed 2–3 days after seeding, and flasks were split twice a week. For experiments, cells were plated on 12-well dishes (1 × 106 cells/well).
Cells were exposed to ischemia-like injury by subjecting them to oxygen glucose deprivation (OGD) as previously described (Kim et al., 2015). Cultures were incubated inside an anoxia chamber for 2 hours at 37°C (33°C for cultures in the hypothermia group), and oxygen tension was maintained at <0.001% (Coy Laboratories). Media were removed and cultures were washed three times with balanced salt solution (BSS0) lacking serum, glucose, and oxygen. Control cultures were incubated at similar temperatures (humidified, 5% CO2) in BSS containing 5.5 mM glucose (BSS5.5) at normoxia. After 2 hours of OGD, glucose was added to each well to a final concentration of 5.5 mM, and plates were incubated at normoxia for 24 hours at 5% CO2, at 37°C (33°C for hypothermia groups).
Cell death and viability were assessed by morphological assessment by light microscopy, vital staining (trypan blue; Sigma), and the tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) assay.
Immunoblotting
Neuro2A cells were homogenized and solubilized in radioimmunoprecipitation assay (RIPA) lysis buffer with protease inhibitor mixture (Complete; ROCHE Applied Science). Twenty-microgram aliquots of proteins were subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, transferred to polyvinylidinene fluoride membranes (Millipore), and probed with mouse anti-dynamin-1 (1:1000; Santa Cruz) followed by corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (1:1000; Bio-Rad). Blots were visualized by using the ECL system (Amersham) according to the manufacturer's instructions, and they were imaged by using ChemiDoc (Bio-Rad). Equal protein loading was confirmed by re-probing actin, which was used as a housekeeping protein.
Flow cytometry
After OGD, 1 × 106 N2a cells were washed once in PBS and blocked with Flow Cytometry Staining Buffer (eBioscience) for 10 minutes. Nonpermeabilized cells were then stained with mouse anti-Fas (1:1000; Santa Cruz) antibody for 1 hour, Alexa Fluor 488-conjugated goat anti-mouse IgG (1:1000; Invitrogen) for 20 minutes and normal mouse IgG-PE (Santa Cruz) was used as an isotype control for 10 minutes at 4°C in darkness. N2a cells were sorted on FACSCalibur (BD Biosciences), and data were analyzed by using CellQuest (BD Biosciences).
Statistical analysis and rigor
For in vivo studies, animals were assigned to experimental groups in a randomized fashion. For in vitro studies, a given run was carried out at the same temperature for a given day, since the anoxia chamber and incubator had to be re-adjusted to the different temperatures. However, experiments were carried out in triplicate from different aliquots of cells, and they were repeated three times on different days. All data analysis was performed by investigators who were blinded to the experimental conditions.
All data were analyzed with standard statistical methods (T-test for two groups and analysis of variance (ANOVA) for more than two groups; Systat Software, Inc., San Jose, CA). p Values of ≤0.05 were considered significant. Quantitative data are presented as the mean ± standard error.
Results
A total of 103 mice were subjected to dMCAO; none were excluded from the analysis as all animals had an obvious deficit at the time of recovery from anesthesia and had a visible ischemic lesion on histology. Four of the animals died prematurely, with three in the Norm group and one in the dHypo group.
Hypothermia decreases TUNEL staining after ischemic stroke
Brain sections from mice subjected to dMCAO followed by 3 days of survival were stained with TUNEL to identify cells with DNA damage, consistent with apoptosis. The brains from the Norm group had numerous TUNEL-positive cells in the ischemic cortex compared with the sham group. In contrast, both hypothermia-treated groups had significantly reduced numbers of TUNEL-positive cells (Fig. 1).
FIG. 1.

Hypothermia reduces numbers of TUNEL-positive cells 3 days after experimental stroke. (a) Under conditions of normothermia (Norm), increased numbers of TUNEL-positive cells are present in the ischemic cortex. These numbers are reduced when compared with when cooling was delayed for 1 hour (dHypo) or instituted immediately after occlusion (eHypo). (b) Graphs of these counts show TUNEL cells normalized to total cell counts from sham uninjured sections stained with cresyl violet. There are fewer TUNEL-positive cells in brains from both hypothermic groups compared with the Norm group, and significantly fewer positive cells among the eHypo compared with the dHypo (n = 6/group; *p < 0.01, **p < 0.05, scale bar = 50 μm). TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Hypothermia decreases expression of dynamin-1 and Fas protein after ischemic stroke
Very little dynamin-1 or Fas could be seen in uninjured sham brains, but both proteins were increased after stroke. Marked increases in dynamin-1 and Fas were observed at 3 and 7 days. Expression of both proteins in eHypo brains was lower than that observed in dHypo, but both hypothermic groups had lower counts compared with the Norm group (Fig. 2).
FIG. 2.

(a, b) Dynamin-1 is increased in neuron-like cells after dMCAO compared with uninjured shams. One, 3, and 7 days post-dMCAO, dynamin-1 is increased more in Norm brains, compared with the hypothermia groups (dHypo, eHypo). (c, d) Fas expression is similarly increased in ischemic brains compared with uninjured shams, and Fas staining is decreased in dHypo and eHypo brains (n = 7–8/group; *p < 0.01, **p < 0.001).
To determine whether dynamin-1 and Fas were expressed in the same cells, we colabeled for both proteins. We found that, indeed, both proteins colocalized to neuron-like cells and were increased in Norm brains compared with eHypo and dHypo (Fig. 3). Numbers of colabeled cells were lower in the hypothermic brains, whereas the pattern of colocalization did not change.
FIG. 3.
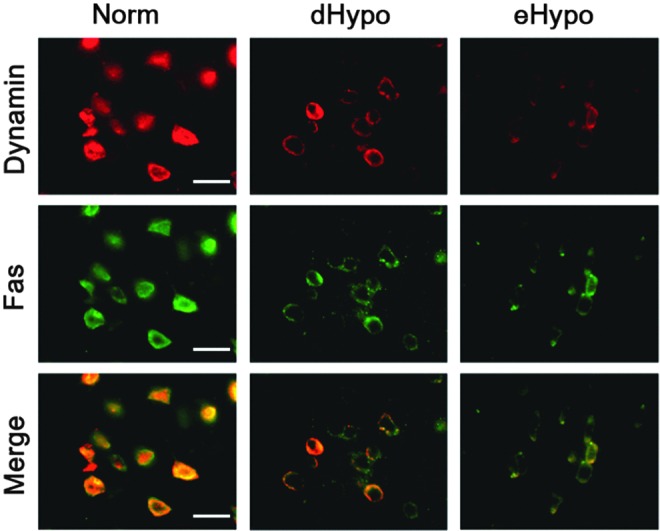
Dynamin-1 and Fas colocalize in neuron-like cells 3 days after experimental stroke. Immunofluorescent stains of dynamin-1 (red) and fas (green) colocalize in brains of mice subjected to dMCAO. A merged image (Merge) shows that many of these cells express both proteins (yellow). Expression of both Fas and dynamin-1 was reduced in hypothermic brains (dHypo and eHypo) compared with normothermic brains (Norm) (scale bar = 10 μm). dMCAO, distal middle cerebral artery occlusion.
Hypothermia decreases dynamin-1 and surface expression of Fas in a cell culture model of ischemia-like injury
To further explore the mechanism of dynamin in hypothermic protection, we turned to a well-established model of in vitro ischemia. OGD-exposed N2a cells under hypothermic conditions (Hypo) survived better and suffered less cell death compared with the Norm group (Fig. 4a, b). To better quantitatively assess dynamin expression, Western blot analysis showed that OGD increased dynamin-1 expression nearly twofold compared with uninjured control cultures, and this was blunted by hypothermia (Fig. 4c, d). Since dynamin is believed to traffick Fas to the surface where it can be ligated by FasL, we surface labeled (non-permeabilizing conditions) N2a cells with antibodies against Fas and estimated the numbers of Fas-positive neurons by flow cytometry. OGD increased numbers of Fas-positive cells, which was decreased by ∼38% by Hypo (Fig. 4e, f).
FIG. 4.

Hypo protects cultured neurons from ischemia-like insults and similarly decreases dynamin-1 and expression of Fas on the cell surface. Cultures of N2a cells were subjected to OGD for 2 hours followed by 24 hours of reperfusion at either normothermia (Norm, 37°C) or hypothermia (Hypo, 33°C). Cells not exposed to OGD but placed in similar media containing 5.5 mM glucose were studied in parallel (Cont). MTT (a) and trypan blue stains (b) showed increased cell viability and decreased cell death, respectively, with Hypo. Immunoblots of N2a cells exposed to OGD showed increases in dynamin-1 expression, whereas Hypo reduced this expression. (c, d) Quantification of the immunoblots shows a significantly greater reduction of dynamin-1 by Hypo compared with Norm. (e, f) Nonpermeabilized N2a cells were subjected to flow cytometry after labeling with anti-Fas antibody. OGD increased numbers of cells that expressed Fas on the surface, and this was decreased by Hypo (All experiments represent pooled results from cultures studied in triplicate and repeated on three separate days. *p < 0.01). Hypo, hypothermia; OGD, oxygen glucose deprivation.
Discussion
We show here that dynamin is upregulated after dMCAO, and this is associated with increased Fas expression. We detected both proteins in the same cells, and both were similarly reduced under conditions of therapeutic hypothermia. Because dynamin is believed to traffick Fas to the cell surface (Ivanov et al., 2006), increased dynamin could conceivably increase apoptosis and neuronal death. We further show that in an in vitro model, ischemia-like injury increased the expression of Fas on the cell surface, which was also reduced by hypothermia. These observations may indicate that one mechanism by which hypothermia protects is by inhibiting dynamin expression and, thus, downregulating Fas expression on the cell's surface.
Hypothermia has been shown to inhibit both apoptotic pathways, but more studies have focused on its effect on the intrinsic pathway (Yenari and Han, 2012). In models of global cerebral ischemia, hypothermia has been shown to inhibit mitochondrial cytochrome c release, a major trigger of the intrinsic apoptotic pathway, and subsequent caspase activation. Hypothermia also differentially alters the expression of Bcl-2 family members. The pro-apoptotic family member Bax decreases with cooling, whereas the anti-apoptotic member Bcl-2 increases. Hypothermia has been studied less with respect to the extrinsic pathway, but as previously mentioned, both FasL and caspase-8 are reduced with cooling.
Relevant to neurological disease, dynamin-1 deficiency has been linked to deficits in γ-amino butyric acid transmission and epilepsy (Ferguson and De Camilli, 2012). Although mutations in dynamin-2 have been the best characterized in the inherited neuropathy, Charcot-Marie-Tooth disease, intact dynamin has also been shown to have detrimental results. Dynamin has also been linked to Alzheimer's disease pathology, where dynamin was shown to influence amyloid generation through beta-site amyloid precursor protein-cleaving enzyme-1 (Zhu et al., 2012). Its inhibition or deficiency led to a reduction in Alzheimer's pathology and neuronal degeneration (Cirrito et al., 2008). Recently, a proteomics-based approach showed that dynamin-1 was upregulated 6 hours poststroke (Chen et al., 2014). This is consistent with our own observations of dynamin in our stroke model, and that it can be downregulated by the neuroprotective chaperone protein Hsp70 (Kim et al., 2016).
There are scant reports of dynamin in the ischemia literature, but there are a few reports of dynamin-related protein-1 (Drp1) and its role in mitochondrial fission. In cardiac and renal ischemia, the inhibition of Drp1 was found to prevent mitochondrial apoptosis (Brooks et al., 2009). Therapeutic hypothermia was also shown to suppress Drp1 dephosphorylation and to maintain mitochondrial morphology and myocardial function (Sharp et al., 2014). There are a few reports characterizing Drp1 in experimental stroke (Liu et al., 2012), and Drp1 inhibition also decreased apoptosis (Grohm et al., 2012). However, the site and the mechanism of action of Drp-1 and dynamin-1 appear distinct.
The precise relationship between dynamin and hypothermia is completely unknown, and to our knowledge, has never been reported earlier. Considered together, the data presented here suggest that dynamin contributes negatively to brain cell survival after ischemia, and it is reduced by hypothermia. Based on our own experiments and those in related literature, potential links between dynamin and hypothermia include the regulation of dynamin at the transcriptional level. For instance, the promoter region of dynamin contains a sequence similar to that recognized by nuclear factor kappa B (NF-kB) (Han et al., 2003). In prior work, our lab showed that hypothermia inhibited the transcriptional activity of NF-kB by directly binding NF-kB or interfering with its inhibitory kinase (Han et al., 2003). Thus, it is possible that hypothermia interferes with dynamin at the level of mRNA induction. It is also possible that hypothermia may directly inhibit dynamin-dependent Fas translocation by attenuating dynamin-1 in the cytosol through cooling, thereby preventing the trafficking of Fas by dynamin. The work presented here is correlative and does not prove causation. Although our prior study showed that dynamin inhibition led to improved neurological outcome in this same stroke model, our data here do not establish whether dynamin is definitively responsible for the observed neuroprotection by hypothermia (Kim et al., 2016). Future studies might address whether hypothermia fails to protect when dynamin is overexpressed, and functional studies should clarify whether dynamin is regulated at the transcriptional or post-translational stage in brain ischemia. It also bears mention that the hypothermia paradigms here are used as a model of neuroprotection to understand stroke pathophysiology. They do not necessarily translate directly to clinical scenarios where different circumstances such as longer durations of cooling may be necessary, and where there are other challenges regarding the induction of hypothermia.
Conclusion
Our findings here identify a previously unappreciated correlation between hypothermia and its anti-apoptotic function via suppression of dynamin. Taken in context with the current dynamin literature, these results suggest that dynamin may be an unappreciated link in the extrinsic apoptotic pathway in stroke, and they shed light on one possible mechanism underlying hypothermia-mediated neuroprotection. This work also suggests that dynamin may be a viable therapeutic target.
Acknowledgments
This study was funded by grants from the National Institutes of Health (RO1 NS40516), the Department of Defense, and the Veteran's Merit Award (I01 BX000589) to M.A.Y.; the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1D1A1B03933017) to J.Y.K.; and a National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT & Future Planning (NRF-2016M3C7A1905098) to J.E.L. Grants to M.A.Y. were administered by the Northern California Institute for Research and Education, and they were supported by resources of the Veterans Affairs Medical Center, San Francisco, California.
Author Disclosure Statement
No competing financial interests exist.
References
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998;281:1305–1308 [DOI] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 2002;346:557–563 [DOI] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009;119:1275–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Chiang YH, Ma HI. Analysis of spatial and temporal protein expression in the cerebral cortex after ischemia-reperfusion injury. J Clin Neurol 2014;10:84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 2008;58:42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 2012;13:75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol 2003;62:329–339 [DOI] [PubMed] [Google Scholar]
- Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 1999;190:1849–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–1312 [DOI] [PubMed] [Google Scholar]
- Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ 2012;19:1446–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HS, Karabiyikoglu M, Kelly S, Sobel RA, Yenari MA. Mild hypothermia inhibits nuclear factor-kappaB translocation in experimental stroke. J Cereb Blood Flow Metab 2003;23:589–598 [DOI] [PubMed] [Google Scholar]
- Inamasu J, Suga S, Sato S, Horiguchi T, Akaji K, Mayanagi K, Kawase T. Postischemic hypothermia attenuates apoptotic cell death in transient focal ischemia in rats. Acta Neurochir Suppl 2000;76:525–527 [DOI] [PubMed] [Google Scholar]
- Ivanov VN, Ronai Z, Hei TK. Opposite roles of FAP-1 and dynamin in the regulation of Fas (CD95) translocation to the cell surface and susceptibility to Fas ligand-mediated apoptosis. J Biol Chem 2006;281:1840–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Howell KE, Henley JR, Cao H, McNiven MA. Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science 1998;279:573–577 [DOI] [PubMed] [Google Scholar]
- Kawabori M, Hokari M, Zheng Z, Kim JY, Calosing C, Hsieh CL, Nakamura MC, Yenari MA. Triggering receptor expressed on myeloid cells-2 correlates to hypothermic neuroprotection in ischemic stroke. Ther Hypothermia Temp Manag 2013;3:189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Ho H, Kim N, Liu J, Tu CL, Yenari MA, Chang W. Calcium-sensing receptor (CaSR) as a novel target for ischemic neuroprotection. Ann Clin Transl Neurol 2014;1:851–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim N, Zheng Z, Lee JE, Yenari MA. 70-kDa heat shock protein downregulates dynamin in experimental stroke: a new therapeutic target? Stroke 2016;47:2103–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Yenari MA, Lee JE. Regulation of inflammatory transcription factors by heat shock protein 70 in primary cultured astrocytes exposed to oxygen-glucose deprivation. Neuroscience 2015;286:272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger DW, Yenari MA. Therapeutic hypothermia for acute ischemic stroke: what do laboratory studies teach us? Stroke 2004;35:1482–1489 [DOI] [PubMed] [Google Scholar]
- Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ 2012;19:36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Chen XN, Fan XX, Zhang YJ, Gu J, Fu XW, Wang ZH, Wang XF, Xiao Z. Upregulated dynamin 1 in an acute seizure model and in epileptic patients. Synapse 2015;69:67–77 [DOI] [PubMed] [Google Scholar]
- Liu L, Kim JY, Koike MA, Yoon YJ, Tang XN, Ma H, Lee H, Steinberg GK, Lee JE, Yenari MA. FasL shedding is reduced by hypothermia in experimental stroke. J Neurochem 2008;106:541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Tian F, Kurata T, Morimoto N, Abe K. Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J Neurosci Res 2012;90:1183–1189 [DOI] [PubMed] [Google Scholar]
- Love S. Apoptosis and brain ischaemia. Prog Neuropsychopharmacol Biol Psychiatry 2003;27:267–282 [DOI] [PubMed] [Google Scholar]
- Meier O, Greber UF. Adenovirus endocytosis. J Gene Med 2004;6 Suppl 1:S152–S163 [DOI] [PubMed] [Google Scholar]
- Padosch SA, Popp E, Vogel P, Bottiger BW. Altered protein expression levels of Fas/CD95 and Fas ligand in differentially vulnerable brain areas in rats after global cerebral ischemia. Neurosci Lett 2003;338:247–251 [DOI] [PubMed] [Google Scholar]
- Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 2014;28:316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XN, Liu L, Koike MA, Yenari MA. Mild hypothermia reduces tissue plasminogen activator-related hemorrhage and blood brain barrier disruption after experimental stroke. Ther Hypothermia Temp Mang 2013;3:74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Yenari MA, Steinberg GK, Giffard RG. Mild hypothermia reduces apoptosis of mouse neurons in vitro early in the cascade. J Cereb Blood Flow Metab 2002;22:21–28 [DOI] [PubMed] [Google Scholar]
- Yenari MA. Pathophysiology of acute ischemic stroke. Cleve Clin J Med 2004;71 Suppl 1:S25–S27 [DOI] [PubMed] [Google Scholar]
- Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischemia. Nat Rev Neurosci 2012;13:267–278 [DOI] [PubMed] [Google Scholar]
- Yenari MA, Onley D, Hedehus M, deCrespigny A, Sun GH, Moseley ME, Steinberg GK. Diffusion- and perfusion-weighted magnetic resonance imaging of focal cerebral ischemia and cortical spreading depression under conditions of mild hypothermia. Brain Res 2000;885:208–219 [DOI] [PubMed] [Google Scholar]
- Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab 2008;28:53–56 [DOI] [PubMed] [Google Scholar]
- Zhu L, Su M, Lucast L, Liu L, Netzer WJ, Gandy SE, Cai D. Dynamin 1 regulates amyloid generation through modulation of BACE-1. PLoS One 2012;7:e45033. [DOI] [PMC free article] [PubMed] [Google Scholar]