

Abstract
P-glycoprotein (P-gp), an ATP-dependent efflux pump, is linked to the development of multidrug resistance in cancer cells. However, the drug-binding sites and translocation pathways of this transporter are not yet well-characterized. We recently demonstrated the important role of tyrosine residues in regulating P-gp ATP hydrolysis via hydrogen bond formations with high affinity modulators. Since tyrosine is both a hydrogen bond donor and acceptor, and non-covalent interactions are key in drug transport, in this study we investigated the global effect of enrichment of tyrosine residues in the drug-binding pocket on the drug binding and transport of P-gp. By employing computational analysis, 15 conserved residues in the drug-binding pocket of human P-gp that interact with substrates were identified and then substituted with tyrosine, including 11 phenylalanine (F72, F303, F314, F336, F732, F759, F770, F938, F942, F983, F994), two leucine (L339, L975), one isoleucine (I306), and one methionine (M949). Characterization of the tyrosine-rich P-gp mutant in HeLa cells demonstrated that this major alteration in the drug-binding pocket by introducing fifteen additional tyrosine residues is well tolerated and has no measurable effect on total or cell surface expression of this mutant. Although the tyrosine-enriched mutant P-gp could transport small to moderate size (<1000 Daltons) fluorescent substrates, its ability to transport large (>1000 Daltons) substrates such as NBD-cyclosporine A, Bodipy-paclitaxel and Bodipy-vinblastine was significantly decreased. This was further supported by the physico-chemical characterization of seventeen tested substrates, which revealed a negative correlation between drug transport and molecular size for the tyrosine-enriched P-gp mutant.
Keywords: ABC transporter, Cancer chemotherapy, Drug transport, Multidrug resistance, P-glycoprotein
Graphical abstract
1. Introduction
P-glycoprotein (P-gp, ABCB1) belongs to the large family of ATP-binding cassette (ABC) transporters [1]. It plays a crucial role in the efflux of a broad range of chemically dissimilar xenobiotics to the extracellular space [2]. Like many ABC transporters, P-gp utilizes the energy from ATP hydrolysis to actively pump substrates out of cells. Under normal physiological conditions, P-gp activity in the intestines, kidney, and liver facilitates secretion of harmful compounds into the feces, urine, and bile. Similarly, high expression of P-gp on the surface of endothelial cells of the blood-brain barrier significantly reduces penetration of toxic compounds and drugs into the brain [3, 4].
P-gp is reported to recognize and transport a vast array of chemically and structurally unrelated anti-cancer agents and confer multidrug resistance (MDR) to cancer cells. Expression of P-gp on tumor cell membranes limits intracellular drug accumulation and concentration, thus protecting cancer cells against chemotoxicity [5]. Considering P-gp’s crucial role in drug bioavailability and pharmacokinetics, there has been a keen interest in understanding the molecular mechanism of drug-binding and transport of P-gp. Understanding drug transport mechanism of this pump will allow the development of more potent and less toxic inhibitors. However, drug binding sites, substrate translocation pathways, substrate release, conformational transition, and the mechanism of drug transport by P-gp is not yet well characterized.
Structurally, P-gp consists of two transmembrane domains (TMDs) and two cytoplasmic nucleotide-binding domains (NBDs)[1, 2, 6–10]. Mutagenesis and biochemical studies suggest extensive conformational flexibility of P-gp, with two distinct conformations: an inward-facing or open (inverted V shape), and an outward-facing or closed (V shape) conformation (reviewed in [11]). These data also suggest that the transition between these conformations requires ATP hydrolysis [8, 12]. It is proposed that binding of amphipathic agents to the drug-binding pocket and ATP hydrolysis results in open to closed conformational switch and release of substrate into the extracellular space [13, 14].
While most of the P-gp substrates enhance ATP hydrolysis [1, 15, 16], a few third-generation modulators (zosuquidar, tariquidar, and elacridar) inhibit basal P-gp ATPase activity. By employing mutagenesis, we have recently reported the importance of drug-binding affinity for modulating inhibition of ATP hydrolysis. Our findings also suggested that hydrogen bond interactions are the key ligand-protein interactions controlling the binding affinity of some of the modulators to P-gp [17].
To specifically test the role of hydrogen bonds in ligand-protein interactions and P-gp function, we replaced fifteen key aromatic or hydrophobic amino acids known to interact with different substrates with tyrosine and generated what we termed the 15Y mutant P-gp. We characterized properties of the 15Y P-gp mutant by biochemical and functional analyses. Expression of 15Y mutant P-gp in HeLa cells by using Bac-Mam baculovirus demonstrated similar total and cell surface expression levels of this mutant when compared to wild type protein. For most of the substrates tested, 15Y mutant P-gp could efficiently transport them out of the cells. These results demonstrated that increasing the hydrogen bond potential by adding fifteen tyrosine residues has no major effect on the transport function of this transporter. However, three substrates- NBD-cyclosporine A, Bodipy-paclitaxel, and Bodipy-vinblastine, show little or no transport by this mutant. We found partial stimulation of ATPase activity of 15Y mutant P-gp by paclitaxel, suggesting that the observed decreased transport is probably due to a failure in substrate translocation and/or release, but not binding of paclitaxel. On the other hand, vinblastine didn’t stimulate or inhibit 15Y P-gp ATPase activity either in the presence or absence of verapamil, indicating lack of vinblastine binding to 15Y mutant P-gp. Physico-chemical characterization of seventeen tested substrates revealed a negative correlation between transport and substrate size for 15Y mutant.
2. Materials and methods
2.1 Chemicals
Cyclosporine A was purchased from the Alexis Corporation (Lausen, Switzerland). [125I]iodoarylazidoprazosin (IAAP) (2200 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). The fluorescent compounds tetramethylrosamine chloride (TMRC), Bodipy-Verapamil, BD-EDA, Bodipy-FL-4,4-difluoro-5,7-dimethyl-4-bora-3a,4a–diaza-s-indacene-3-propionyl ethylenediamine, hydrochloride. TMRE, tetramethylrhodamine ethyl ester perchlorate. DiOC2, 3,3’-Diethyloxancarbocyanine iodide. Cell tracker orange CMTMR, (5-(and-6)-(((4-chloromethy)benzyol)amino)tetramethylrhodamine). LDS-751, quinolinium, 6-(dimethylamino)-2-[4-[4-(dimethylamino)phenyl]-1-ethyl, perchlorate, SYTO 13, Bodipy-paclitaxel, Bodipy-forskolin, Calcein-AM, BD-prazosin, and dihydrorhodamine were purchased from Invitrogen (Carlsbad, CA). NBD-cyclosporine A was generously provided by Drs. Anika Hartz and Björn Bauer, University of Kentucky (Lexington, KY). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). The P-gp-specific monoclonal antibody C219 was provided by Fujirebio Diagnostic Inc. (Malvern, PA), and the MRK16 antibody used for flow cytometry studies was purchased from Kyowa Medex Company (Tokyo, Japan). FITC-labeled anti-mouse secondary antibody IgG2a was obtained from BD Biosciences (San Jose, CA).
2.2 Cell lines and culture conditions
HeLa cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and were maintained as described previously [18]. Briefly, cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 5 mM L-glutamine, 50 units/ml penicillin, and 50 µg/ml streptomycin at 37 °C.
2.3 Selection of residues for mutagenesis
Amino acids in the transmembrane region possibly interacting with substrates were identified based on previously published reports [10, 11, 19–21]. Key amino acids were identified based on their topological location in the homology model of human P-gp generated using the mouse P-gp crystal structure (4Q9H.pdb). Only those residues lining the drug-binding cavity in the transmembrane region were selected. Selected residues (F72, F303, F314, F336, F732, F759, F770, F938, F942, F983, F994, L339, L975, I306, and M949) were mutated to tyrosine using gene synthesis method as described previously [22] (GeneArt gene synthesis services, Life Technology, Carlsbad, CA).
2.4 Recombinant BacMam and baculovirus generation
The Bac-to-Bac Baculovirus Expression System (Life Technologies, Carlsbad, CA) was used to generate recombinant baculovirus and BacMam baculovirus [23, 24]. Briefly, the Gateway cloning technique was used to transfer WT and 15Y P-gp cDNA into pDest-008 and pDest-652 expression vectors for baculovirus expression and BacMam analysis, respectively. These expression vectors were then transformed into DH10Bac E. coli to allow site-specific transposition of the gene of interest into bacmid (the baculovirus shuttle vector). White clones were then selected and bacmid DNA was purified by alkaline lysis. Bacmid DNA was then transfected into Sf9 insect cells to generate BacMam baculovirus.
2.5 BacMam baculovirus transduction of HeLa cells and expression of WT and 15Y mutant P-gp
HeLa cells were transduced with the WT or 15Y mutant P-gp BacMam virus at a titer of 35–50 particles per cell as described previously [17]. Twenty-four hours’ post-transduction, the cells were harvested and analyzed for cell surface expression and transport function. Approximately 300,000 WT and 15Y mutant P-gp-expressing cells were analyzed for cell surface expression by 1-hour incubation at 37°C with control mouse IgG2a or human P-gp-specific MRK16 antibody (1 µg per 100,000 cells). These cells were then washed and incubated with FITC-labeled anti-mouse secondary antibody IgG2a (0.25 µg per 100,000 cells). The stained cells were analyzed by flow cytometry using a FACS CANTO II instrument, and the data were analyzed using FlowJo software (Tree Star, Inc. Ashland, OR)
2.6 Transport of fluorescent substrates
The transport function of WT and 15Y mutant P-gp was determined using flow cytometry, as previously described [17, 23]. Cells were incubated with fluorescent substrates; calcein-AM (0.5 µM) for 10 min; or TMRE (0.5 µM), TMR chloride (0.5 µM), BD-forskolin (0.5 µM), BD-verapamil (0.5 µM), BD-EDA (0.5 µM), DiOC2 (0.5 µM), SYTO 13 (0.5 µM), daunorubicin (4 µM), BD-paclitaxel (0.5 µM), BD-prazosin (0.5 µM), LDS-751 (0.5 µM), cell tracker orange (0.5 µM), rhodamine123 (1.3 µM), dihydrorhodamine 123 (1.3 µM), NBD-CsA (0.5 µM), and BD-vinblastine (0.5 µM) for 45 min. After incubation at 37°C with fluorescent substrates, cells were washed with cold PBS and re-suspended in cold PBS containing 1% BSA. The accumulation of substrates was measured with respect to the untransduced cells. The MFI of WT P-gp expressing cells is subtracted from untransduced cells and is taken as a 100% efflux for all transport experiments. Transport efficacy of 15Y is also calculated with same method and presented as percent of WT.
2.7 Efflux rate of NBD-CsA and BD-verapamil
HeLa cells (300,000/tube) expressing WT or 15Y mutant P-gp were incubated with 20 mM 2-deoxyglucose, and 5 mM sodium azide in 2 ml of DPBS without glucose for 10 min at 37 °C to deplete ATP from the cells. Then NBD-CsA or BD-verapamil (0.5 µM final concentration) was added to the same medium in each tube and incubated at 37 °C for another 20 min [25]. After incubation, cells were then washed with ice cold DPBS (PBS with 1mM MgCl2 and 0.1 mM CaCl2) and chilled on ice. The efflux of the substrates was initiated at 37°C by the addition of 2 ml of PBS containing 50 mM glucose, 0.1 mM CaCl2, and 1 mM MgCl2. The efflux was allowed to continue for 0, 1, 3, 6, 10, 20, and 30 min. The cells were then washed with cold PBS and analyzed after resuspending them in PBS with 1% BSA by flow cytometry. The MFI of untransduced cells for each time point is considered as 100%.
2.8 Preparation of total membranes from High-Five insect cells
High-Five insect cells (Invitrogen, Carlsbad, CA) were infected with recombinant baculovirus carrying WT or 15Y mutant P-gp with a 6XHis-tag and TEV site at the C-terminal end, as described previously [26]. Membrane vesicles were prepared by hypotonic lysis of P-gp-expressing High-Five insect cells and differential centrifugation as described previously [12].
2.9 Photo-crosslinking of WT and 15Y mutant P-gp with [125I]-Iodoarylazidoprazosin
Membranes of P-gp-expressing High-Five insect cells (60–80 µg/100 µl) were incubated in 50 mM MES-Tris buffer pH of 6.8 and 150 mM NaCl for 10 min. Then the samples were transferred to 4°C under subdued light and 5–7 nM IAAP was added. After 5 min incubation in ice water, samples were then photo-crosslinked with 365 nm UV light for 10 min. Protein bands were then separated by gel electrophoresis using 7% Tris-acetate gels and IAAP incorporation into P-gp bands quantified with a phosphorimager (GE Healthcare, Waldorf, MD), as reported earlier [27].
2.10 ATP hydrolysis measurements
ATP hydrolysis was measured in membranes of High-Five cells expressing WT or 15Y mutant P-gp. Membranes (100–200 µg protein/ml) were incubated in the presence or absence of sodium orthovanadate (0.3 mM) in a 50 mM MES-Tris pH 6.8 buffer containing 50 mM KCl, 10 mM MgCl2, 5 mM NaN3, 1 mM EGTA, 1 mM ouabain, and 2 mM DTT. Basal ATPase activity was measured in the presence of DMSO, while drug-modulated activity was measured in the presence of selected drugs. The reaction was started at 37°C by addition of 5 mM ATP and after 20 minutes’ incubation, it was stopped by addition of 2.5% SDS and the level of inorganic phosphate generated was quantified with a colorimetric method. The vanadate-sensitive ATPase activities were analyzed and plotted with Graph pad Prism software (version 7) as described previously [28].
2.11 Physico-chemical characterization of substrates and statistical analyses
All chemical characterizations, such as molecular weight, LogP3 value, H-bond donor/acceptor capacity, and surface area were collected from either the PubChem project website (https://pubchem.ncbi.nlm.nih.gov) or manufacturer’s data sheets. Multivariant analyses were performed by Microsoft Excel functions, and data were plotted as graphs using SigmaPlot (Systat, San Jose, CA).
3. Results
3.1 Substitution of fifteen amino acids with tyrosine in the P-gp drug-binding pocket
P-gp’s polyspecificity, along with lack of a high-resolution P-gp-substrate co-crystal structure, has hindered the identification of amino acids facilitating drug binding and transport [24, 29, 30]. So far, mutagenesis and molecular docking techniques are among the best approaches to identify the role of residues in substrate binding and transport. Co-crystal structure of mouse P-gp with several QZ-59 cyclopeptide inhibitor derivatives [21], and PBDE-100 pesticide [31] allowed identification of several key amino acids in the substrate or modulator binding pocket of mouse P-gp. We further used published bioinformatic and mutagenesis studies, and generated a list of key amino acids that interact with different substrates or modulators [11, 17, 20, 32–34]. Additionally, we generated a homology model of human P-gp based on the refined mouse P-gp crystal structure [10] and further selected amino acids based on their position and orientation with respect to the drug-binding pocket. Using this screening method, we identified fifteen key amino acid residues which appear to be critical for drug binding and transport (Table 1, Figure 1).
Table 1.
Conservation of the residues selected for mutagenesis across species
| Location | Residue | Rhesus monkey |
Cynomolgus monkey |
Mouse | Hamster | Rat | Chicken | C. elegans |
|---|---|---|---|---|---|---|---|---|
| TMH1 | F72 | ● | ● | ● | ● | ● | ● | |
| TMH5 | F303 | ● | ● | ● | ● | ● | ||
| TMH5 | I306 | ● | ● | ● | ● | ● | ||
| TMH5 | F314 | ● | ● | ● | ● | ● | ● | ● |
| TMH6 | F336 | ● | ● | ● | ● | ● | ● | |
| TMH6 | L339 | ● | ● | ● | ● | ● | ● | |
| TMH7 | F732 | ● | ● | ● | ● | ● | ● | ● |
| TMH8 | F759 | ● | ● | ● | ● | ● | ● | ● |
| TMH8 | F770 | ● | ● | ● | ● | ● | ||
| TMH11 | F938 | ● | ● | ● | ● | ● | ● | |
| TMH11 | F942 | ● | ● | ● | ● | ● | ● | |
| TMH11 | M949 | ● | ● | ● | ● | ● | ||
| TMH12 | L975 | ● | ● | ● | ● | ● | ||
| TMH12 | F983 | ● | ● | ● | ● | ● | ● | |
| TMH12 | F994 | ● | ● | ● | ● | ● | ● |
The residues in human ABCB1 (MDR1) were compared with isoforms in other species, which have higher sequence similarity. The filled circle denotes conservation. TMH, transmembrane helix.
Fig. 1. Schematic presentation of the location of fifteen residues mutated in 15Y mutant P-gp.

The homology model of human P-gp was built based on the mouse P-gp crystal structure (4Q9H.pdb). Schematic presentation of WT and 15Y mutant P-gps, generated by PyMOL. (A) Side view of 15Y P-gp protein (top). The N-terminal transmembrane domain and NBD1 and C-terminal transmembrane domain and NBD2 of P-gp are highlighted in green and cyan, respectively. The fifteen residues which are replaced by tyrosine are shown in sphere format in red. The expanded bottom view depicts location of 15 tyrosine residues in the transmembrane drug-binding pocket. (B) The residues in the transmembrane region of WT protein selected for substitution and after replacement with tyrosine residues in 15Y mutant are shown in stick format at top and bottom, respectively. The protein backbone is in grey color. Mutated residues and helices are labeled. Helices 2, 3, 4, 9, and 10 were deleted for clarity.
Comparing human P-gp amino acid sequence with different animal species, we found that the selected residues are highly conserved between species (Table 1). In fact, all fifteen of the residues we selected from human P-gp are 100% identical to Rhesus and Cynomolgus monkeys as well as to mouse and hamster species. The residues selected for substitution with tyrosine are located on different helices (Figure 1 A, B) but all are exposed to the lumen of the drug-binding pocket in the transmembrane region. The fifteen amino acids mutated to tyrosine include eleven phenylalanines (F72, F303, F314, F336, F732, F759, F770, F938, F942, F983, F994), two leucines (L339, L975), one isoleucine (I306), and one methionine (M949).
3.2 Cell surface and total expression of 15Y mutant P-gp
We have demonstrated high transduction efficacy (>98%) and high expression levels of ABC transporters using the BacMam baculovirus system in HeLa and other mammalian cells [23]. To test whether extensive tyrosine substitutions affect expression and/or function of this protein, we utilized the BacMam baculovirus system to express both WT and 15Y mutant P-gps in HeLa cells. HeLa cells, which don’t express detectable levels of P-gp, were transduced with BacMam baculovirus carrying WT or 15Y mutant P-gp, as described in the Materials and Methods section. The Western blot analysis of the whole cell lysate of the transduced HeLa cells demonstrated almost three-fold more expression of the mature form of 15Y mutant compared to WT P-gp (Fig. 2A, arrow). Consistent with the increased expression of 15Y mutant P-gp, it’s immature form is also seen (Fig. 2A, arrowhead)
Fig. 2. Characterization of expression and transport function of 15Y mutant P-gp in HeLa cells.

(A) Total expression: Whole cell lysates from untransduced HeLa cells, WT, 15Y BacMam Baculovirus-transduced HeLa cells were prepared and the lysate of equivalent number of cells (3×104 cells) was loaded per lane. The immunoblots were analyzed for P-gp using C219 monoclonal antibody (arrow and arrowhead indicate positions of mature and immature P-gps, respectively), and GAPDH as the loading control (bottom) (B) Cell surface expression of WT and 15Y mutant P-gps were compared in HeLa cells transduced with BacMam baculovirus carrying WT or 15Y mutant P-gp. After 24 h post-infection, the cells were harvested and stained with human P-gp-specific monoclonal antibody MRK-16 (compare black and red traces in the histogram, left) and quantification of expression is given in the bar graph (right). (C) WT and 15Y mutant P-gp expressing HeLa cells were incubated with fluorescent substrates and their respective mean MFI values were compared. Untransduced cells were used as control. Representative histograms show steady-state transport of four fluorescent substrates with most (TMRC, left) to least efficient (BD-vinblastine, right) by 15Y mutant P-gp in HeLa cells. (D) Transport efficacy of 15Y mutant P-gp for tested fluorescent substrates. MFI of WT P-gp expressing cells were subtracted from the one for untransduced cells and the difference was taken as 100% transport activity (dotted horizontal line). Transport efficacy of the 15Y mutant was calculated similarly and the values (mean ± SD) are presented in the waterfall bar graph. BD-Vinblastine (not shown) was not transported by the 15Y mutant. Data represent the mean ± SD of five independent experiments.
To determine the cell surface expression of the 15Y mutant, transduced HeLa cells were incubated with the human P-gp-specific monoclonal antibody MRK-16, which recognizes the extracellular loops of P-gp [35]. Consistent with the Western blot, flow cytometry analysis of the stained cells confirmed higher cell surface expression of 15Y mutant P-gp compared to WT (Fig. 2B). Quantitative analysis demonstrated almost 50% higher cell surface expression of 15Y mutant P-gp in HeLa cells compared to WT protein (Fig. 2B, right panel). As the expression of 15Y mutant at the cell surface and total cell level is higher, it is unlikely due to a change in the affinity for detection by either C219 or MRK-16 antibody (this is further supported by the increased level of the mutant protein even in the High-Five insect cell membranes, as shown in Fig. 6 A).
Fig. 6. 15Y mutant P-gp exhibits decreased incorporation of photoaffinity substrate IAAP.

Equal amounts of high five insect cell membranes expressing either WT or 15Y mutant P-gp (75 µg protein) were photo-crosslinked with IAAP (5–7 nM) as described in Materials and Methods. After 5 minutes, the samples were exposed to UV light for 10 min to crosslink bound IAAP to Pgp. Then the samples were prepared for electrophoresis. Labeled 30 µg protein was loaded per lane and the gels were stained for protein with colloidal blue dye, dried and scanned. Representative protein stained gel in (A) and an autoradiogram of the same gel in (B) is shown. The incorporation of IAAP in WT and 15 Y mutant P-gp was normalized to P-gp level in the stained gel (A) from three independent experiments (mean ± SD; bar graph on the right).
3.3 Transport efficacy of 15Y mutant P-gp with fluorescent substrates
For characterization of transport properties of the mutant transporter, we used established substrates with intrinsic fluorescence as well as several bodipy conjugated substrates including verapamil, vinblastine, paclitaxel, and prazosin [36, 37] and NBD derivative of cyclosporine A [38]. In addition, we used other fluorescently labeled compounds described by Strouse et. al. [39]. To assess the transport efficacy of 15Y mutant P-gp, we measured and compared the steady-state accumulation of seventeen fluorescent P-gp substrates in WT and 15Y mutant P-gp-expressing HeLa cells by flow cytometry (Fig. 2C and D). As expected, untransduced HeLa cells, which lack detectable levels of P-gp, have maximal accumulation of florescent substrates, while HeLa cells expressing WT P-gp actively pump out substrates and have significantly lower intracellular substrate accumulation. Figure 2C depicts histograms which represent four different efflux levels of the selected substrates, TMRC, rhodamine 123, NBD-CsA, and BD-vinblastine. For most of the tested substrates, we observed efficient transport (more than 75% of WT) by 15Y mutant P-gp (Table 2, TMRC, BD-verapamil, BD-EDA, TMRE, DiOC, BD-forskolin, calcein-AM, cell tracker orange, SYTO 13, and daunorubicin). BD-prazosin, LDS-751, rhodamine 123, and dihydrorhodamine show an intermediate level of transport by 15Y mutant. However, the intracellular level of BD-paclitaxel and NBD-CsA was slightly less than untransduced cells, demonstrating that 15Y mutant has decreased ability to transport these substrates. Notably, there was no detectable transport of BD-vinblastine in 15Y P-gp-expressing cells (Figure 2D and Table 2). It is quite possible that 15 mutations will affect the interaction with high-affinity modulators. For this reason, we focused on characterization of the transport function of the 15Y mutant using seventeen different substrates, and did not test the combination of a substrate and a modulator.
Table 2.
Steady-state transport function of 15Y mutant P-gp with seventeen fluorescent substrates
| Fluorescent substrate | % Transport (mean ± SD) | Transport efficiency |
|---|---|---|
| TMR Chloride | 121 ± 5 | Full transport (>75%) |
| BD-Verapamil | 116 ± 4 | |
| BD-EDA | 108 ± 9 | |
| TMRE | 100 ± 3 | |
| DiOC | 94 ± 3 | |
| BD-Forskolin | 93 ± 12 | |
| Calcein-AM | 88 ± 15 | |
| Cell Tracker Orange | 83 ± 6 | |
| SYTO 13 | 82 ± 5 | |
| Daunorubicin | 79 ± 11 | |
| BD-Prazosin | 71 ± 7 | Partial transport (30–75%) |
| LDS-751 | 70 ± 11 | |
| Rhodamine 123 | 66 ± 5 | |
| Dihydrorhodamine | 63 ± 12 | |
| BD-Paclitaxel | 29 ± 15 | Little to no transport (<30%) |
| NBD-CsA | 22 ± 9 | |
| BD-Vinblastine | No detectable transport |
Substrates were grouped into three categories based on their transport level when compared to WT; Full transport (>75%), Partial transport (30–75%), and little to no transport (<30%). The transport assays were carried out as described in the Materials and Methods section, and mean ± SD values are from four or more independent experiments. TMR, tetramethylrhosamine, BD-EDA, Bodipy-FL-4,4-difluoro-5,7-dimethyl-4-bora-3a,4a–diaza-s-indacene-3-propionyl ethylenediamine, hydrochloride. TMRE, tetramethylrhodamine ethyl ester perchlorate. DiOC2, 3,3’-Diethyloxancarbocyanine iodide. Cell tracker orange CMTMR, (5-(and-6)-(((4-chloromethy)benzyol)amino)tetramethylrhodamine). LDS-751, quinolinium, 6-(dimethylamino)-2-[4-[4-(dimethylamino)phenyl]-1-ethyl, perchlorate. NBD-CsA, [N-ε(4-nitrobenzofurzan-7-yl)-d-lys cyclosporine A.
In the transport assay described above, the fluorescence level of the cells at indicated steady-state time points is the net uptake and efflux of the tested substrate. Therefore, to specifically determine whether increased substrate accumulation is due to the lower efflux rate in 15Y mutant-expressing cells, but not increased passive or active influx, we depleted ATP from cells and then loaded them with substrate. Efflux was initiated by adding 50 mM glucose containing PBS buffer, but without the fluorescence substrate. For this purpose, we depleted ATP from untransduced, WT and 15Y–expressing cells by incubating the cells in 2-deoxyglucose and sodium azide, and added BD-verapamil (representing full transport substrate category) or NBD-CsA (representing little to no transport substrate category). Then the cells were washed and re-suspended in ice-cold DPBS containing glucose, and incubated at 37°C for different time points (Figure 3). Consistent with steady-state transport results, the 15Y mutant was not able to efflux NBD-CsA (Figure 3A), whereas WT was very efficient with t½ (50% efflux) of 2.17 min. However, BD-verapamil was transported at a comparable level to WT by 15Y P-gp-expressing cells (Figure 3B). The t1/2 for BD-verapamil efflux by WT was approximately 0.37 minutes compared to 0.73 minutes by 15Y mutant P-gp. These data suggest substrate-specific transport efficacy of 15Y mutant P-gp.
Fig. 3. Time course of the efflux of NBD-cyclosporine A and Bodipy-verapamil by WT and 15Y Mutant P-gp.

(A) HeLa cells expressing WT or 15Y mutant P-gp were first depleted of ATP by incubating with PBS containing 20 mM 2-deoxyglucose and 5 mM sodium azide for 20 min at 37°C and further incubated for 20 min with 0. 5 µM NBD-CsA (A) or BD-verapamil (B) in the same medium. The cells were then washed at 4°C with ice-cold PBS and incubated at 37°C with PBS supplemented with 50 mM glucose, 0.1 mM CaCl2, and 1 mM MgCl2 and the fluorescence intensity of cells was measured at different time points (0, 1, 3, 6, 10, 20, and 30 min) post-incubation. For each time point MFI of WT and 15Y was determined as percent of untransduced cells (which was taken as 100%). The time required for 50% loss of fluorescence (t½ min) values were calculated using GraphPad Prism version 7.0. Each data point represents the mean ± SD of four independent experiments.
3.4 Modulation of ATPase activity of 15Y mutant P-gp
ATP-binding and hydrolysis is known to provide energy for the conformational changes of P-gp required to transport substrates. Usually substrate-binding enhances the ATPase activity of P-gp. However, as noted earlier, this effect is substrate-specific and some modulators or substrates with higher binding affinity, such as zosuquidar, tariquidar, and elacridar, inhibit P-gp ATPase activity [17]. In addition to substrates, the P-gp microenvironment is also known to have an impact on the ATPase activity and transport ability of P-gp. We have recently described how the membrane and detergent micelle environment can modulate the ATPase activity of Pgp [40]. To evaluate P-gp ATPase function, we prepared membranes from High-Five insect cells expressing WT or 15Y mutant P-gps.
To test whether the transport efficacy of 15Y mutant P-gp is consistent with the rate of ATP hydrolysis, we measured the ATPase activity of WT and the 15Y mutant in the presence of different concentrations of selected substrates (Fig. 4). The basal ATPase activity of the 15Y mutant in the presence of the DMSO solvent alone was slightly higher (35–45 nmol Pi/mg protein/min) than WT P-gp (27–35 nmol Pi/mg protein/min). Comparing the ATPase activity of P-gp in the presence of TMRC, which is transported efficiently by the 15Y mutant, we observed a concentration-dependent substrate-mediated stimulation of ATPase activity of both WT and 15Y P-gps (Figure 4A). Similar but somewhat less stimulation of ATPase activity of 15Y mutant Pgp is also observed in the presence of verapamil (Figure 4B) and paclitaxel (Figure C and D). At a very low concentration of paclitaxel (0.01 µM), there is 50% stimulation of 15Y P-gp ATPase activity, which was not further increased by higher paclitaxel concentration (Figure 4D). On the other hand, as expected, increasing paclitaxel concentration further increases the ATPase activity of WT P-gp (Figure 4C). These findings are consistent with transport data (Figure 2D and Table 2). Both TMRC and verapamil, transported efficiently by the 15Y P-gp mutant, also stimulated the ATPase activity of this mutant.
Fig. 4. Effect of selected substrates on ATPase activity of WT and 15Y mutant P-gp.

High-Five insect cell membranes (100 µg protein/ml) expressing WT (filled circles) or 15Y mutant (unfilled squares) P-gp were incubated with DMSO (for basal activity), or various concentrations of the selected drugs, TMR chloride (A), verapamil (B), and paclitaxel (C). For clarity ATPase activity of paclitaxel at low concentrations is depicted separately in panel D. Substrates were selected based on their transport efficacy in Figure 2 and Table 2. TMRC and verapamil represent substrates which are efficiently transported, and paclitaxel represents the substrates with little to no transport. The ATPase activity was measured as described in the Materials and Methods section. Briefly, all drug stimulated ATPase activities were normalized to its vanadate control, and the basal ATPase activity was taken as zero. Each data point represents the mean ± SD. (three independent experiments). The data were fitted to the Michaelis-Menten curve using GraphPad Prism 7.0.
The lack of detectable transport of BD-vinblastine by 15Y P-gp mutant could be due to the inability of vinblastine to bind to the substrate-binding pocket and/or due to a defect in the translocation pathway before being released into the extracellular space. For this reason, we investigated the effect of vinblastine on P-gp ATPase activity. Additionally, we measured the effect of vinblastine on verapamil-stimulated ATPase activity of both WT and 15Y mutant P-gp (Figure 5). In the absence of verapamil (filled circles in Figure 5, A and B), we observed a concentration-dependent increase in ATPase activity of WT P-gp (A). However, the ATPase activity of 15Y mutant P-gp remains unchanged despite increasing the concentration of vinblastine (B). These findings correlate with WT and 15Y P-gp transport function (Fig. 2D and Table 2). In the presence of 5 µM verapamil (unfilled squares in Figure 5, A and B), which stimulates both WT and 15Y mutant P-gp ATPase activity (Figure 4B), vinblastine increases the ATPase activity of WT P-gp by 25%. However, increasing the concentration of vinblastine has no effect on verapamil-stimulated 15Y P-gp ATPase activity, which suggests that most likely the vinblastine does not bind to the 15Y mutant P-gp.
Fig. 5. Effect of vinblastine on ATPase activity of WT and 15Y P-gp mutant in the presence and absence of verapamil.

High-five insect cell membranes (100 µg protein/ml) were incubated with DMSO (for basal activity), or various concentrations of vinblastine (0, 0.5, 1, 2.5, 5, and 10 µM) in the presence (white squares) and absence of 5 µM verapamil (black filled circles). Vinblastine stimulated WT (A), but not 15Y mutant P-gp (B) ATPase activity in a concentration-dependent manner (compare filled black curves in A and B). We also measured the effect of vinblastine on verapamil-stimulated ATPase activity of both WT (A) and 15Y mutant P-gp (B) (compare the white squares). In both panels, the activities in the absence of vinblastine were taken as 100%. Each data point represents the mean ± SD. of three independent experiments. The data were fitted to the Michaelis-Menten curve using GraphPad Prism 7.0.
3.5 Fifteen tyrosine substitutions in the drug-binding pocket decreases the photolabeling of 15Y mutant P-gp with IAAP
Despite limited mechanistic insight, P-gp drug transport is known to be a multi-step process that includes drug-binding, ATP hydrolysis, conformational rearrangement, substrate translocation, substrate release, and resetting the cycle. Selective substrate transport observed with the 15Y mutant led us to evaluate whether the photolabeling with prazosin analog is affected.
Since extensive tyrosine substitutions might alter substrate binding sites, we investigated the drug binding ability of the 15Y mutant using an established photoaffinity labeled substrate. IAAP is a radiolabeled prazosin analog transported by P-gp [41]. The IAAP can be irreversibly crosslinked to P-gp by UV irradiation. Comparing the expression level of WT and 15Y mutant P-gp by colloidal blue gel staining, we observed higher level of 15Y mutant in High-Five insect cell membranes (Fig. 6A). However, photo labeling of WT and 15Y mutant P-gp with IAAP revealed a lower level of IAAP incorporation in 15Y mutant protein than WT P-gp (Fig. 6B, autoradiogram). Normalizing the IAAP incorporation to the protein expression level, we observed almost 60% lower IAAP labeling of 15Y mutant P-gp (Fig. 6B, right). These data demonstrate that similar to partial transport of Bodipy-prazosin (Table 2), photo labeling of this mutant with IAAP (another prazosin analog) is also decreased.
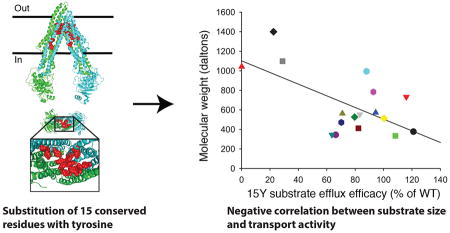
3.6 Physico-chemical properties and regression analyses revealed a negative correlation between substrate molecular weight and 15Y drug transport efficacy
Although the 15Y mutant was expressed well at the cell surface of HeLa cells, it was not able to transport a few of the substrates we tested. To understand the underlying reasons for this lack of transport, we analyzed the physico-chemical properties of the substrates used for transport studies. It is known that a larger polar surface area of a substrate increases the likelihood that it will act as a hydrogen bond acceptor. Regression analysis of these two known linked characteristics demonstrated the high positive correlation coefficient value of 0.90 (Fig. 7). Furthermore, comparing several key properties of substrates such as size, hydrogen bond acceptor and donor capacity, LogP3, and topological polar surface area, we found a negative correlation (correlation coefficient of −0.56) between the substrate size and transport efficacy of the 15Y mutant (Fig. 8). The fluorescent substrates with the highest molecular weight, such as BD-vinblastine (mol. wt. 1043 Da), NBD-CsA (mol. wt. ~1440 Da) and BD-paclitaxel (mol. wt. 1099 Da), were least efficiently transported by the 15Y mutant. In contrast, small substrates such as TMR chloride (mol. wt. 423 Da) and BP-EDA (mol. wt. 334 Da) were transported well by 15Y mutant P-gp (Fig. 8). The only exception was calcein-AM (mol. wt. 998 Da), which despite having a large size, is transported by the 15Y mutant.
Fig. 7. Positive correlation between the polar surface area of substrates and the total number of hydrogen bond acceptor of the substrates.

The positive correlation between polar surface area and total number of hydrogen bond acceptor (cross correlation coefficient of 0.90). Polar surface area and total hydrogen bond acceptor groups of each substrate were exported from the PubChem website and their correlations were calculated with Excel.
Fig. 8. Negative correlation between size of substrates and their transport by 15Y mutant P-gp.

Physico-chemical characterization of tested substrates revealed a negative correlation between molecular size and transport level of each substrate by 15Y mutant P-gp. The X axis of plot is the transport level (Fig. 2 and Table 2) and the Y axis is the molecular weight of the substrates. Correlation coefficient value is −0.56. Each shape represents one fluorescent substrate (list given on the right).
4. Discussion
Understanding the molecular mechanism of polyspecificity and transport pathway of Pgp could strongly enhance rational drug design to overcome resistance mediated by this transporter in cancer cells. Previously, we reported that substitution of three residues (Y307A, Q725A and Y953A) to alanine results in loss of binding of high-affinity modulators to their primary binding site, and we also demonstrated the importance of hydrogen bonds in inhibition of ATP hydrolysis [17].
Considering the lack of a high-resolution crystal structure of human P-gp, molecular modeling and mutagenesis are among the best approaches to study the role of residues in the drug-binding pocket in interactions with substrates and modulators. Since most of the fifteen selected residues were phenylalanine (11 out of 15), we replaced all residues with tyrosine, which is similar in structure to phenylalanine, causing subtle changes to P-gp structure while enhancing hydrogen bond formations. Remarkably, we observed that 15Y mutant P-gp was expressed at even higher level than WT in HeLa cells. The 15Y P-gp mutant retained its ability to transport many substrates efficiently. This observation further confirms the high molecular flexibility of P-gp [11, 42]. These data also suggest that a major alteration of the drug-binding pocket of P-gp is well-tolerated. To our knowledge, this is the first report demonstrating that human P-gp can be expressed well with fifteen substitutions in the transmembrane domains, especially in the drug-binding pocket.
We measured the electrostatic surface potential of the drug-binding pocket region of human P-gp (Fig. 9) using a plugin that integrates the molecular visualization program with Adoptive Poisson-Solver(APBS) [43]. Visual examination of the maps of WT and 15Y mutant P-gp drug-binding pockets clearly revealed a different charge potential landscape in areas where (non-polar) phenylalanine residues were replaced by polar, hydrophilic tyrosine (see areas denoted by circles in Figure 9). In these areas, 15Y mutant P-gp has tyrosine residues with hydroxyl oxygen providing two lone pairs of electrons available for hydrogen bond formation with H-donor groups of substrate molecules. The high probability of H-bond formation with large size substrates such as vinblastine (BD-vinblastine), cyclosporine A (NBD-CsA), and paclitaxel (BD-paclitaxel) supports the results of transport studies.
Fig. 9. Electrostatic surface potential of wild-type and 15Y mutant P-gp.

(A) Electrostatic surface potential of the N-terminal (TM5/Y310-side) (A) and C-terminal (TM11/Y953-side) (B) halves of WT and 15Y mutant P-gps’ drug-binding pocket. Homology models of human WT and 15Y mutant P-gps were built using the X-ray structure of mouse P-gp (4Q9H.pdb)[21] as a template. The TM5/Y310 side contains helices TM3, TM4, TM5, TM6, TM7, TM8 and NBD1, while the TM11/Y953 side contains helices TM1, TM2, TM9, TM10, TM11, TM12 and NBD2. The results of the calculations are shown as color-coded electrostatic surfaces with potential scale ranges from −8 KbT/ec (red) to 8 KbT/ec (blue). The central figure shows the surface of 15Y mutant P-gp in white, with red spots indicating the location of the hydroxyl-oxygen atoms of the tyrosine residues introduced in the mutant. The ovals indicate the areas where the introduced tyrosine residues significantly change the electrostatic landscape.
Furthermore, we found a negative correlation between substrate size and transport efficacy by 15Y mutant P-gp. Substrates with large molecular weight (>1000 Dalton) such as BD-Vinblastine, NBD-CsA, and BD-Paclitaxel, were not transported by 15Y mutant. Our in vitro ATPase assays further revealed more mechanistic insights into the limited transport ability of 15Y P-gp. Our data show that vinblastine, at a range of concentrations, had no effect on 15Y ATPase activity in the presence or absence of verapamil, suggesting vinblastine’s inability to bind to 15Y P-gp. However, paclitaxel could stimulate 15Y P-gp ATPase activity at very low concentrations, which suggests that paclitaxel binds to P-gp but does not progress efficiently through translocation pathway(s) and/or release from the 15Y mutant P-gp. To our knowledge, this is the first report demonstrating the role of residues in the drug-binding pocket in recognition of the size of substrates. As P-gp plays an important role in the pharmacokinetics and bioavailability of several classes of drugs in addition to anticancer agents, further studies with natural product and small molecule modulators are required.
Among the fifteen selected residues substituted with tyrosine in 15Y mutant P-gp, there are four phenylalanines (F303, F336, F732, F983), which have been previously reported to bind to different QZ-59 cyclopeptide derivatives [21]. Additionally, a recent co-crystal structure of mouse P-gp also reported two more conserved phenylalanine residues, F314 and F759, which are important for binding and transport of PBDE-100 pesticide [31]. Our findings suggest that replacing these six key phenylalanines (F303, F314, F336, F732, F759, and F983) with tyrosine has minimal effect on transport of small to medium size substrates. As described in the introduction section, there is a large body of published work describing the role of various residues in the drug-binding pocket by substitution of one or two of them at a time. The near normal function of 15Y mutant P-gp with a major alteration of the drug-binding pocket demonstrates that the effect of one individual residue is compensated by changes in other residues interacting with the substrates. The 15Y mutant has six mutated residues located in TMD1 and nine in TMD2. The generation of mutants with either six substitutions in TMD1 or nine in TMD2 will be useful to assess the role of each TMD of the transporter and to narrow the number of residues contributing to substrate size determination.
Taken together, these results provide new and important insight into the role of aromatic and hydrophobic residues in the drug-binding pocket for binding and transport of substrates by P-gp. Although the structural information gained from mutants such as 15Y P-gp will be very informative, further studies are needed to determine the specific residue(s) responsible for binding and transport of large substrates such as vinblastine, cyclosporine A and paclitaxel.
Acknowledgments
We thank George Leiman for editing the manuscript. We thank Drs. Kristen Pluchino and Suneet Shukla for the help with gene synthesis method and Ivannie Ortiz Rivera for help with ATP hydrolysis measurements. This research was funded by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, Center for Cancer Research. The high-performance computational capabilities of the Helix and Biowulf Systems at the National Institutes of Health, Bethesda, MD were used for building homology models and for comparing the electrostatic surface potentials of P-gps.
Non-standard abbreviations
- P-gp
P-glycoprotein
- ABC
ATP-binding cassette
- MDR
multidrug resistance
- NBD
nucleotide-binding domain
- TMD
transmembrane domain
- IAAP
[125I]iodoarylazidoprazosin
- TMRC
tetramethylrosamine chloride
- DMEM
Dulbecco’s modified Eagle’s Medium
- FBS
fetal bovine serum
- IMDM
Iscove's Modified Dulbecco's Medium
- MFI
mean fluorescence intensity
- BLAST
Basic Local Alignment Search Tool
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this work:
Calcein-AM (PubChem CID: 4126474)
Cyclosporine A (PubChem CID: 5284373)
Daunorubicin (PubChem CID: 30323)
3,3’-Diethyoxacyanine iodide (PubChem CID: 16204983)
Dihydrorhodamine 123 (PubChem CID: 105032)
Paclitaxel (PubChem CID: 36314)
Rhodamine 123 (PubChem CID: 65217)
Tetramethylrhodamine (PubChem CID: 155483)
Verapamil (PubChem CID: 62969)
Vinblastine (PubChem CID: 241903)
Conflict of interest
The authors declare that they have no conflict of interest with the content of this paper.
References
- 1.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 3.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 4.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U. S. A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 6.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 7.Dawson RJ, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 8.Sharom FJ. The P-glycoprotein multidrug transporter. Essays Biochem. 2011;50:161–178. doi: 10.1042/bse0500161. [DOI] [PubMed] [Google Scholar]
- 9.Choudhury HG, Tong Z, Mathavan I, Li Y, Iwata S, Zirah S, Rebuffat S, van Veen HW, Beis K. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U. S. A. 2014;111:9145–9150. doi: 10.1073/pnas.1320506111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chufan EE, Sim HM, Ambudkar SV. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): recent biochemical and structural studies. Adv. Cancer Res. 2015;125:71–96. doi: 10.1016/bs.acr.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerr KM, Sauna ZE, Ambudkar SV. Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J. Biol. Chem. 2001;276:8657–8664. doi: 10.1074/jbc.M010044200. [DOI] [PubMed] [Google Scholar]
- 13.Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, Xia D, Ambudkar SV, Subramaniam S. Cryo-EM Analysis of the Conformational Landscape of Human P-glycoprotein (ABCB1) During its Catalytic Cycle. Mol. Pharmacol. 2016;90:35–41. doi: 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verhalen B, Dastvan R, Thangapandian S, Peskova Y, Koteiche HA, Nakamoto RK, Tajkhorshid E, McHaourab HS. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature. 2017;543:738–741. doi: 10.1038/nature21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkadi B, Homolya L, Szakacs G, Varadi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiol. Rev. 2006;86:1179–1236. doi: 10.1152/physrev.00037.2005. [DOI] [PubMed] [Google Scholar]
- 16.Egido E, Muller R, Li-Blatter X, Merino G, Seelig A. Predicting Activators and Inhibitors of the Breast Cancer Resistance Protein (ABCG2) and P-Glycoprotein (ABCB1) Based on Mechanistic Considerations. Mol. Pharm. 2015;12:4026–4037. doi: 10.1021/acs.molpharmaceut.5b00463. [DOI] [PubMed] [Google Scholar]
- 17.Chufan EE, Kapoor K, Ambudkar SV. Drug-protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem. Pharmacol. 2016;101:40–53. doi: 10.1016/j.bcp.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapoor K, Bhatnagar J, Chufan EE, Ambudkar SV. Mutations in intracellular loops 1 and 3 lead to misfolding of human P-glycoprotein (ABCB1) that can be rescued by cyclosporine A, which reduces its association with chaperone Hsp70. J. Biol. Chem. 2013;288:32622–32636. doi: 10.1074/jbc.M113.498980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crowley E, Callaghan R. Multidrug efflux pumps: drug binding--gates or cavity? FEBS J. 2010;277:530–539. doi: 10.1111/j.1742-4658.2009.07484.x. [DOI] [PubMed] [Google Scholar]
- 20.Loo TW, Clarke DM. Mapping the Binding Site of the Inhibitor Tariquidar That Stabilizes the First Transmembrane Domain of P-glycoprotein. J. Biol. Chem. 2015;290:29389–29401. doi: 10.1074/jbc.M115.695171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, Doshi R, Urbatsch IL, Zhang Q, Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 2015;71:732–741. doi: 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pluchino KM, Hall MD, Moen JK, Chufan EE, Fetsch PA, Shukla S, Gill DR, Hyde SC, Xia D, Ambudkar SV, Gottesman MM. Human-Mouse Chimeras with Normal Expression and Function Reveal That Major Domain Swapping Is Tolerated by P-Glycoprotein (ABCB1) Biochemistry. 2016;55:1010–1023. doi: 10.1021/acs.biochem.5b01064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shukla S, Schwartz C, Kapoor K, Kouanda A, Ambudkar SV. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos. 2012;40:304–312. doi: 10.1124/dmd.111.042721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chufan EE, Kapoor K, Sim HM, Singh S, Talele TT, Durell SR, Ambudkar SV. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1) PLoS One. 2013;8:e82463. doi: 10.1371/journal.pone.0082463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ambudkar SV, Cardarelli CO, Pashinsky I, Stein WD. Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J. Biol. Chem. 1997;272:21160–21166. doi: 10.1074/jbc.272.34.21160. [DOI] [PubMed] [Google Scholar]
- 26.Ramachandra M, Ambudkar SV, Chen D, Hrycyna CA, Dey S, Gottesman MM, Pastan I. Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry. 1998;37:5010–5019. doi: 10.1021/bi973045u. [DOI] [PubMed] [Google Scholar]
- 27.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2000;97:2515–2520. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–514. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 29.Ecker GF, Csaszar E, Kopp S, Plagens B, Holzer W, Ernst W, Chiba P. Identification of ligand-binding regions of P-glycoprotein by activated-pharmacophore photoaffinity labeling and matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Mol. Pharmacol. 2002;61:637–648. doi: 10.1124/mol.61.3.637. [DOI] [PubMed] [Google Scholar]
- 30.Ambudkar SV, Kim IW, Sauna ZE. The power of the pump: mechanisms of action of P-glycoprotein (ABCB1) Eur. J. Pharm. Sci. 2006;27:392–400. doi: 10.1016/j.ejps.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Nicklisch SC, Rees SD, McGrath AP, Gokirmak T, Bonito LT, Vermeer LM, Cregger C, Loewen G, Sandin S, Chang G, Hamdoun A. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016;2:e1600001. doi: 10.1126/sciadv.1600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 33.Mittra R, Pavy M, Subramanian N, George AM, O’Mara ML, Kerr ID, Callaghan R. Location of contact residues in pharmacologically distinct drug binding sites on P-glycoprotein. Biochem. Pharmacol. 2017;123:19–28. doi: 10.1016/j.bcp.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Parveen Z, Stockner T, Bentele C, Pferschy S, Kraupp M, Freissmuth M, Ecker GF, Chiba P. Molecular dissection of dual pseudosymmetric solute translocation pathways in human P-glycoprotein. Mol. Pharmacol. 2011;79:443–452. doi: 10.1124/mol.110.067611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamada H, Tsuruo T. Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc. Natl. Acad. Sci. U. S. A. 1986;83:7785–7789. doi: 10.1073/pnas.83.20.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J. Membr. Biol. 2000;173:203–214. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]
- 37.Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Functional expression of human P-glycoprotein from plasmids using vaccinia virus-bacteriophage T7 RNA polymerase system. Methods Enzymol. 1998;292:456–473. doi: 10.1016/s0076-6879(98)92035-3. [DOI] [PubMed] [Google Scholar]
- 38.Schramm U, Fricker G, Wenger R, Miller DS. P-glycoprotein-mediated secretion of a fluorescent cyclosporin analogue by teleost renal proximal tubules. Am. J. Physiol. 1995;268:F46–52. doi: 10.1152/ajprenal.1995.268.1.F46. [DOI] [PubMed] [Google Scholar]
- 39.Strouse JJ, Ivnitski-Steele I, Waller A, Young SM, Perez D, Evangelisti AM, Ursu O, Bologa CG, Carter MB, Salas VM, Tegos G, Larson RS, Oprea TI, Edwards BS, Sklar LA. Fluorescent substrates for flow cytometric evaluation of efflux inhibition in ABCB1, ABCC1, and ABCG2 transporters. Anal. Biochem. 2013;437:77–87. doi: 10.1016/j.ab.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shukla S, Abel B, Chufan EE, Ambudkar SV. Effects of a detergent micelle environment on P-glycoprotein (ABCB1)-ligand interactions. J. Biol. Chem. 2017;292:7066–7076. doi: 10.1074/jbc.M116.771634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maki N, Hafkemeyer P, Dey S. Allosteric modulation of human P-glycoprotein. Inhibition of transport by preventing substrate translocation and dissociation. J. Biol. Chem. 2003;278:18132–18139. doi: 10.1074/jbc.M210413200. [DOI] [PubMed] [Google Scholar]
- 42.Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang WK, Gutierrez C, Zhang A, Shukla S, Madigan JP, Zhou T, Kwong PD, Ambudkar SV, Gottesman MM, Xia D. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017;292:446–461. doi: 10.1074/jbc.M116.755884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]