

Abstract
The rnhAB mutant E. coli, deficient in two RNase H enzymes that remove both R-loops and incorporated ribonucleotides (rNs) from DNA, grow slowly, suggesting accumulation of rN-containing DNA lesions (R-lesions). We report that the rnhAB mutants have reduced viability, form filaments with abnormal nucleoids, induce SOS and fragment their chromosome, revealing replication and/or segregation stress. R-loops are known to interfere with replication forks, and sensitivity of the double rnhAB mutants to translation inhibition points to R-loops as precursors for R-lesions. However, the strict specificity of bacterial RNase HII for RNA-DNA junctions indicates that R-lesions have rNs integrated into DNA. Indeed, instead of relieving problems of rnhAB mutants, transient inhibition of replication from oriC kills them, suggesting that oriC-initiated replication removes R-loops instead of compounding them to R-lesions. Yet, replication from an R-loop-initiating plasmid origin kills the double rnhAB mutant, revealing generation of R-lesions by R-loop-primed DNA synthesis. These R-lesions could be R-tracts, contiguous runs of ≥4 RNA nucleotides within DNA strand and the only common substrate between the two bacterial RNase H enzymes. However, a plasmid relaxation test failed to detect R-tracts in DNA of the rnhAB mutants, even though it readily detected R-patches (runs of 1-3 rNs). Instead, we detected R-gaps, single-strand gaps containing rNs, in the chromosomal DNA of the rnhAB mutant. Therefore, we propose that RNase H-deficient mutants convert some R-loops into R-tracts, which progress into R-gaps and then to double-strand breaks — explaining why R-tracts do not accumulate in RNase H-deficient cells, while double-strand breaks do.
Keywords: R-loops, R-lesions, double-strand DNA breaks, stable DNA replication, SOS-response
Graphical abstract
Introduction
Life not only faithfully transfers genetic information, but also preserves the chemistry of the information carrier. For example, ribonucleotides (rNs) are not tolerated in the DNA helix, as they affect its chemical stability [1-3] and alter its helical parameters from B-form to A-form [4-6]. DNA of wild-type cells has no detectable rNs; however, in vitro DNA synthesis in the presence of rNTPs and analysis of DNA isolated from mutants deficient in rN removal revealed that rNs incorporate into DNA at a significant frequency and by a variety of pathways [7,8]. Erroneous insertion by replicative or DNA-repair polymerases is the source for single rNs in DNA; this problem is accentuated by the fact that rNTPs are 10-100 fold overrepresented in the cellular nucleotide pools over dNTPs [9-12]. Extrapolations from in vitro measurements with purified enzymes, as well as from in vivo measurements suggest that replicative DNA polymerases incorporate single rNs frequently in bacteria, yeast and higher eukaryotes, with the total of, correspondingly, 2,000, >10,000 and 1,300,000 rNs misincorporated per replication round [13-15].
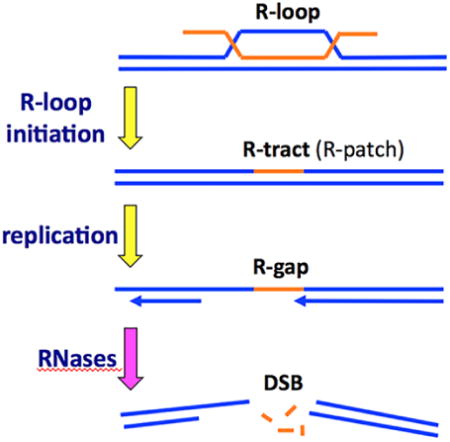
The 12 nt-long riboprimers of Okazaki fragments are introduced by RNA-primases during regular lagging strand replication (reviewed in [16,17]). A likely source of rN-runs longer than Okazaki primers are R-loops — the RNA:DNA hybrid structures formed by insertion of an RNA strand into the corresponding DNA duplex, with displacement of the identical DNA strand in a loop [18,19] (Fig. 1A). R-loops preferentially form at sites of active transcription — when an untranslated transcript inserts back into its cognate DNA — and, although lacking RNA-DNA junctions, are thought to be stable enough to lead to genome instability due to vulnerability of the exposed ssDNA and inhibition and breakage of replication forks [20-22]. R-loops also spawn both scheduled and unscheduled initiations of DNA replication, happening in small bacterial plasmids [23], during constitutive stable DNA replication (cSDR) in E. coli [24], in the 35S rRNA genes in yeast [25] and in mitochondrial DNA [26].
Fig. 1. Growth, morphology and viability of the double rnhAB mutants.
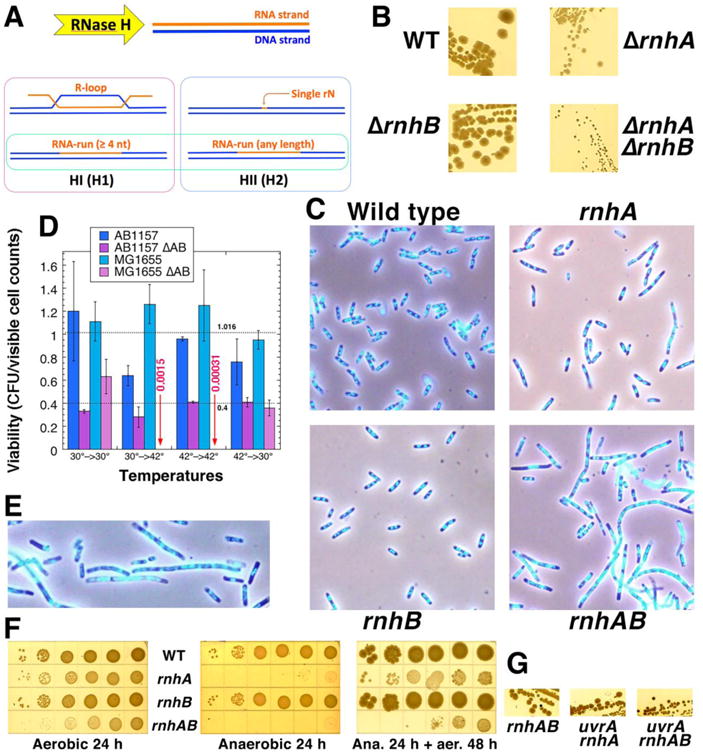
A. A scheme of in vivo substrates of the two RNase H enzymes. The common substrate, framed in bright green, is the RNA-run with at least four contiguous rNs, which we call “R-tract”. HI and H1, HII and H2 refer to RNase H enzymes of prokaryotes and eukaryotes accordingly. B. Colony size on LB agar, 37°C, 24 hours. Strains: WT, AB1157; ΔrnhA, L-413; ΔrnhB, L-415; ΔrnhAB, L-416. C. Images of rnh and wild type strains stained with DAPI and observed by Hiraga's fluo-phase combined method. Cells were grown at 37°C in LB. The strains are like in “B”. D. Viability of the strains, determined as the ratio of the colony forming units (CFUs) to the microscopic counts in the same volume of the culture. Overnight cultures grown at 30°C were diluted and grown at the temperature (indicated by the first number) to OD 0.2-0.3 (about 2 hours), then cultures were serially diluted and plated on LB plates developed for 16 hours at the temperature indicated by the second number in pairs. Average viability (± SEM) of the eight WT measurements and six measurements for the rnhAB mutant cells is shown (the low titers of the two MG1655 ΔrnhAB cultures at 42°C were not used in the calculation). Strains: AB1157, L-416, MG1655, L-419. E. An enlarged image of the rnhAB mutant cells (processed as in panel C), to show nucleoids of both filamenting and normal-looking cells in some detail. F. Anaerobic growth inhibition of the rnhA and anaerobic lethality of rnhAB strains. Dilution-spotting of strains (like in “B”) was done in an anaerobic chamber on LB plates. Plates were incubated at room temperature in the chamber for 24 hours, then shifted to 28°C aerobic conditions for another 48 hours. G. The uvrA defect further reduces the colony size of the rnhAB double mutant. Strains: rnhAB, L-416; uvrA rnhA, L-414; uvrA rnhAB, L-417.
One of the consequences of rN presence in DNA is inhibition of DNA replication fork movement. In vitro studies with both prokaryotic and eukaryotic replicative polymerases show that they pause at single rNs [13], bypassing them with various efficiencies. The bypass is impeded and becomes a complete block upon increase of a ribonucleotide stretch up to four consecutive rNs [14,27-29] (one paper reports an inefficient bypass of even a four-rN run [30]).
To keep genomic DNA rN-free, cells employ efficient ribonucleotide removal activities. Prokaryotes remove rNs from genomic DNA with two types of RNases H enzymes (reviewed in [31-33]). In E. coli these are RNase HI (type I) and RNase HII (type II) enzymes (Fig. 1A), encoded, correspondingly, by the rnhA and rnhB genes. RNase HI is the only enzyme from E. coli that attacks RNA:DNA hybrids in vitro; it has no sequence preference and endonucleolytically cleaves the RNA part in RNA:DNA hybrids, then further degrades it exonucleolytically [34]. The enzyme does not recognize single rN (or rNrN, or rNrNrN) in DNA; the RNA part must be at least four consecutive rNs for RNase HI to cleave it (Fig. 1A) [34-36].
Remarkably, as mentioned above, the same four consecutive rNs in the DNA template stall all tested DNA polymerases [28]. We call such ≥4 rN runs within otherwise duplex DNA “R-tracts”, while the ≤3 rN runs, including the most numerous single rNs, that are not recognized by RNase HI and do not stall DNA synthesis completely, are referred to as “R-patches”.
Although the main substrates of RNase HI in vivo are considered to be R-loops (Fig. 1A), this has never been tested in vitro (however, see our results), perhaps because of the general R-loop instability in linear DNA substrates [19]. Nevertheless, the ability of RNase H1 overproduction to suppress deficiency in topoisomerase I in E. coli, the main problem of which is accumulation of R-loops in DNA, is taken as sufficient evidence that RNase HI attacks R-loops in vivo [37,38].
The rnhA mutants in E. coli show several phenotypes: slower growth, synthetic lethality with various DNA metabolism defects (recBCD, rep, recG, topA) and initiation of unscheduled replication away from the origin and especially in the terminus region (the so-called “constitutive stable DNA replication”) [24,38-40]. Alternative initiations in the rnhA mutants are robust enough to support replication of the entire bacterial chromosome under conditions of blocking replication from oriC [41,42].
In contrast, bacterial RNase HII is a very different enzyme, as in the presence of the physiological cation Mg2+ it shows almost no (∼0.25%) RNase HI activity toward the RNA strand of RNA:DNA hybrids [43,44], but specifically cleaves the rN/dN transition within a DNA strand, equally attacking both R-patches and R-tracts and leaving a single rN on the 5′ end of the cleavage [45,46] (Fig. 1A). For this strict specificity, the bacterial RNase HII enzyme is referred to as the “junction ribonuclease” [46,47]. The ribonucleotide at the 5′-side of such cleavage has to be removed by nick translation activity of DNA polymerase I in E. coli [48]. Based on in vitro experiments and in vivo effects, nucleotide excision repair (NER) has also been implicated as a back up system to remove single rNs in rnhB mutants [49].
The biological importance of bacterial RNase HII, discovered in E. coli as a multicopy suppressor of the rnhA recBC lethality [44], is still obscure, as the corresponding rnhB single mutants behave essentially as WT cells (see below). The genomic DNA of the rnhB mutants in B. subtilis shows minor sensitivity to alkaline hydrolysis, indicative of low density of imbedded rNs, but no strong phenotypes of the rnhB mutants of B. subtilis are reported [13], suggesting that bacteria tolerate R-patches in their genomic DNA without difficulty.
The poor growth of mutants deficient in both RNases HI and HII in bacteria ([13], this work) suggests accumulation of hard-to-repair or even irreparable R-lesions (rN-containing DNA lesions). From in vitro experiments with eukaryotic type I and II RNase H enzymes, which both show robust and processive activity against RNA:DNA hybrids [50], the natural implication is that serious R-lesions must be exclusively R-loops, either still associated with transcription elongation complexes (TECs) that generated them or TEC-free R-loops [51-54]. There are even models proposing that TEC-free R-loops still stall and break replication forks, explaining the dramatic phenotypes of RNase H enzyme inactivation in eukaryotes [53,54].
However, at least three considerations make the TEC-free R-loops unlikely contributors to R-lesions. The first is the known gradual instability of model R-loops in vitro [19]. The second is the susceptibility of RNA:DNA hybrids to helicases: replicative helicases disassemble RNA:DNA hybrids (ensuring that a replication fork encountering a TEC-free R-loop always wins) [55], while a specialized DNA pump RecG and DNA helicase DinG disassemble lengthy R-loops [56,57]. Finally, the third argument against R-loops is the strict RNA-DNA junction specificity of the bacterial RNase HII enzymes [46,47]. R-loops have no RNA-DNA junctions, and yet, to inhibit growth severely, both RNase HI and RNase HII enzymes in bacteria have to be inactivated ([13], this work), suggesting that, in contrast to R-loops, growth-inhibiting R-lesions do have RNA-DNA junctions.
With the objective of learning the nature of R-lesions, we investigated the growth inhibition of the rnhAB double mutants in E. coli, seeking answers to the overarching question: does its poor growth result from a synergy between R-loops (substrates of RNase HI) and R-patches (substrates of RNase HII), or from the formation of a common substrate for the two enzymes, for example, R-tracts (Fig. 1A). To this end, we characterized the general growth parameters, the replication stress and various vulnerabilities of the double rnhAB mutants.
Results
The growth phenotypes: synergistic interactions of rnhA and rnhB mutations
To explore the effect of rN accumulation in the DNA of E. coli on cell growth and DNA metabolism, we created the rnhAB double mutant combining a complete deletion of the rnhB gene with an internal deletion of the rnhA ORF (364 of 468 nt removed). We then characterized the four-strain set comprising the rnhAB double mutant, the corresponding single rnh mutants and the WT strain in the AB1157 background. The size of colonies grown at 37°C on rich medium showed no difference between wild type and the rnhB mutant, smaller colonies for the rnhA mutant and the grossly inhibited rnhAB double mutant (Fig. 1B).
In liquid cultures, growth of the rnhAB double mutant was also severely inhibited. We compared cell shapes and nucleoid morphology in the early logarithmic cultures using DAPI staining and fluorescence-phase combined method [58] (Fig. 1C). Wild type and rnhB mutant cells were uniformly small with compact and well-defined nucleoids, while some rnhA mutant cells were elongated, but mostly with nucleoids of the same compact and bright morphology. Remarkably, the rnhAB double mutant had a mix of small WT-looking cells and grossly-elongated cells with obvious nucleoid defects (Fig. 1C): although the nucleoid material was generally distributed along the filamentous cells, the multiple nucleoids lacked compactness and looked like a continuous thin thread with periodic brighter sports (Fig. 1C,E and S1). Thus, in the absence of both RNase HI and HII enzymes, roughly half of all cells in liquid cultures reveal clear problems with both chromosome replication and segregation.
The grossly-inhibited growth of the double mutant could reflect its extremely low viability (∼5%, judging by the colony size (Fig. 1B)). In fact, the double rnhAB mutant in the MG1655 background was killed after only 2 hours at 42°C (Fig. 1D), confirming earlier reports of its temperature sensitivity [59,60]. Surprisingly, in contrast to the gross growth problems, the average viability of the rnhAB mutants measured by comparing direct cell count with the corresponding CFU titer was 40% in the AB1157 background at both 30°C and 42°C and in MG1655 background at 30°C (Fig. 1D). This unexpectedly high viability is consistent with the presence of a significant fraction of WT-looking cells in the rnhAB mutant cultures. This moderate defect in cell viability in combination with the severe growth inhibition suggests that the rnhAB double mutant cells frequently encounter R-lesions that require complex repair, yet most of these lesions are successfully mended.
Since irreparable lesions happen infrequently and independently of each other, Poisson distribution is appropriate to assess their frequency per generation. In particular, Poisson distribution with an average of one event has the zero class (no events) of ∼37% of the total. In other words, a viability of ∼40% indicates that irreparable R-lesions are formed in the rnhAB mutant cells, on the average, once per cell per generation. In WT cells, these R-lesions are either prevented or resolved by the RNase H enzymes.
We also report a novel phenotype for the rnhA mutants: the inability to grow and the loss of titer in anaerobic conditions (Fig. 1F). Both rnhA and rnhAB mutants were unable to form colonies on LB plates incubated anaerobically, but upon return to aerobic conditions, only the rnhAB double mutant took another 48 hours to form visible colonies and showed 1000-fold decrease in titer (Fig. 1F), suggesting formation of irreparable R-lesions. Overall, we confirm the lack of growth phenotype in a single rnhB mutant isogenic to WT strain, but in the rnhA mutant cells, the additional rnhB mutation confers a dramatic growth defect, suggesting R-lesions, in which rNs are incorporated into DNA.
The rnhAB mutants are SOS-induced and dependent on recombinational repair
Filamentation and the nucleoid problems in the rnhAB double mutants indicate chronic chromosome replication and segregation stress and predict induction of the SOS response. The SOS response to DNA damage in E. coli elevates expression of some 30 genes and decreases expression of some 20 genes [61,62], boosting the cell's capacity to repair or tolerate DNA damage [63]. We determined SOS induction levels by monitoring the activity of β-galactosidase in strains containing a sulA∷lacZ transcriptional fusion reporter with the operator sequence for the SOS repressor, LexA [64]. We found (Fig. 2A) that the level of β-galactosidase expression in the early logarithmic cultures of the rnhAB double mutant was ∼10 times higher than in wild type and rnhB mutant strains, and two times higher than in the rnhA single mutant (known to be SOS-induced [65]), as well as in two positive controls with the known elevated SOS response: dut-1 and the wild type cells growing in the presence of the DNA crosslinking agent Mitomycin C [66]. Thus, slow growth of the rnhAB mutant reflects an unusually high SOS induction.
Fig. 2. The rnhAB double mutants are induced for SOS, fragment their chromosome and depend on recombinational repair of double-strand breaks.
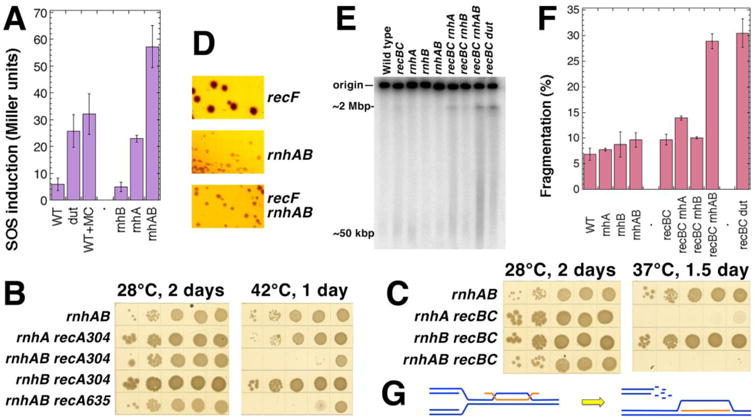
A. The level of SOS-induction in various strains: WT, AK43; dut-1, L-179 (positive control); WT grown in the presence of 100 μg/ml of mitomycin C (MC) (another positive control); rnhB, FK-7; rnhA, FK-6; rnhAB, FK-5. B. Synthetic lethality of rnhAB double defect with the recA defect. The double mutants with recA were built at 28°C in the presence of pEAK2 plasmid, which is RecA+, but ori(Ts). The plasmid is lost upon incubation at temperatures higher than 35°C, revealing the recA defect (all five strains lost pEAK2 with the same efficiency, data not shown.) Both recA304 and recA635 are complete deletions of different origin. The strains are: rnhAB, L-416; rnhA recA304, L-469; rnhAB recA304, L-470; rnhB recA304, L-471; rnhAB recA635, L-472. C. Synthetic lethality of rnhAB double defect with the recBC defect. The recBC defect is temperature-sensitive, fully defective at 37°C or higher. The strains are: rnhAB, L-416; rnhA recBC, L-465; rnhB recBC, L-466; rnhAB recBC, L-476. D. The recF defect does not reduce the colony size of the rnhAB double mutant. The strains are: recF, L-420; rnhA recF, L-431; rnhAB recF, L-435. E. PFGE analysis of chromosomal fragmentation in the rnh mutants. Chromosomal DNA of growing cells is labeled with 32P-orthophosphoric acid at 37°C for 5 hours, cells are lysed in agarose plugs before being subjected to PFGE. The dut recBC combination serves as a positive control for intense chromosome fragmentation. The strains are: WT, AB1157; recBC, SK129; recBC dut, AK107; rnhA, L-413; rnhB, L-415; rnhAB, L-416; rnhA recBC, L-465; rnhB recBC, L-466; rnhAB recBC, L-476. F. Quantification of chromosomal fragmentation from several gels like in “E”. Fragmentation is calculated as a percentage of radioactivity in a lane relative to the total radioactivity (the lane and the well counts). The values are means (n=3-6) ± SEM. G. A scheme of how replication fork collision with R-loop leads to replication fork disintegration (by an unspecified mechanism).
This high SOS induction in the rnhAB double mutants indicates formation of chromosomal lesions, mended by the RecA-dependent recombinational repair (SOS response is induced by RecA polymerization on chromosomal lesions [63]). Indeed, the rnhAB mutant is synthetic lethal in combination with recA inactivation (Fig. 2B, 42°C plate): the rnhAB ΔrecA strains fail to form colonies, whereas the rnhA ΔrecA and rnhB ΔrecA strains are viable.
In growing cells, chromosomal lesions are mostly generated by the process of replication [63]. Specifically, replication stress is associated either with disintegration of replication forks, resulting in double-strand breaks, or with inability to replicate through blocking lesions in the template DNA, resulting in formation of blocked single-strand gaps. The type of lesion could be inferred through genetic interactions with the recBCD versus recF mutations. In E. coli, double-strand breaks are mended by recombinational repair initiated by the RecBCD enzyme, whereas the RecFOR proteins are involved in repair of blocked single-strand gaps [63]. When combined with the recBC(Ts) allele, only the double rnhAB mutant shows synthetic lethality at 37°C (Fig. 2C), while the rnhA recBC(Ts) strain is strongly inhibited at 37°C on rich media (Fig. 2C), in agreement with the published results [44,65]. Our observation of the synthetic lethality of the rnhAB recBC mutant is compatible with the observation that synthetic lethality of rnhA recBC strain plated at 42°C on LB is suppressed by overexpressing rnhB+ gene [44]. These observations indicate RNase HII involvement in repair or prevention of R-lesions formed in the absence of RNase HI. In contrast, no synthetic lethality or even synthetic growth inhibition (smaller colony size) was observed for the rnhAB recF triple mutant (Fig. 2D). Overall, the synthetic lethality of the rnhAB mutations with both recA and recBC mutations suggests that double strand breaks are formed in the chromosome of the rnhAB mutants and are repaired by the RecABCD pathway.
Chromosomal fragmentation in the rnhAB double mutants
To detect these DSBs directly, we measured chromosomal fragmentation in the rnhAB recBC mutant conditions. As customary for this analysis in E. coli, we grew rnhAB recBC(Ts) mutants at 28°C (RecBC+ conditions) and then shifted them to 37°C (RecBC− conditions), to preserve the cumulative level of chromosomal fragmentation by simultaneously inhibiting both recombinational repair of broken DNA and linear DNA degradation [66,67]. Specifically, we radioactively labeled cultures growing at 28°C and then shifted them for 5 hours to 37°C (to inactivate the RecBCD(Ts) enzyme), prepared cells in agarose plugs and separated circular chromosomes from broken linear species in pulse-field gels (Fig. 2E). Two strains gave a statistically-significant increase in chromosomal fragmentation (Fig. 2EF): rnhAB recBC(Ts) had ∼20% increase over the background of the recBC(Ts) strain, while the rnhA recBC(Ts) strain showed a modest ∼5% increase of broken DNA. Some chromosomal fragmentation in the recBC(Ts)-version of the single rnhA mutant was expected, since the rnhA strain growth is slower at 37°C (Fig. 1B), the SOS response is elevated (Fig. 2A), and growth of the rnhA recBC(Ts) strain is grossly inhibited at 37°C (Fig. 2C). The rnhB single mutant again is not different from the WT strain in the chromosomal fragmentation test.
On the basis of these results, we propose that, during growth in the absence of both RNase HI and RNase HII enzymes, DSBs are formed continuously in the chromosomes of E. coli cells, presumably as a result of replication impairment at R-lesions, whose precise nature is unclear at the moment. The RecABCD recombinational repair pathway mends most of the double-strand breaks, whereas the observed level of lethality (∼40% viability) in the rnhAB (Rec+) strain indicates inability to repair at least one lesion per cell per generation.
It is commonly depicted [52-54] that R-loops, even without association with transcription complexes, represent strong enough impediments to replication forks that they could cause fork disintegration (Fig. 2G) — potentially explaining the chromosome fragmentation we observe in the rnhA mutants. This idea does not explain the dramatic effect of the additional rnhB inactivation in the rnhA mutant, though, as bacterial RNase HII enzymes do not act on RNA:DNA hybrids lacking rN-dN junctions [43,46,47] — and, therefore, cannot attack R-loops. To test the idea of the conflict between R-loops and replication forks, we measured how inhibition of translation, transcription or replication would affect the viability of the rnhAB double mutants.
The rnhA and rnhAB mutants are resistant to block of transcription initiation
Transcription makes R-loop formation possible, by producing the invading transcripts. We compared the effect of blocking transcription initiation with rifampicin on viability of the rnh mutants and the wild type cells, expecting that the rnhA mutants would show no negative effect with this antibiotic (in fact, their problems should be relieved, but the positive effect would be hard to detect). Rifampicin specifically binds DNA-dependent RNA polymerase and inhibits transcription at the level of initiation [68]. Concentrations up to 200 μg/ml are used to block transcription in vivo [69]. Unexpectedly, we observed that treatment with 100 μg/ml rifampicin for two hours reduced the titer of growing cultures of wild type and single rnhB mutants to ∼2%, while both rnhA and rnhAB mutants survived much better, around 25% (Fig. S2). We observed an even stronger rifampicin toxicity when we plated cells on LB supplemented with 2 μg/ml rifampicin (Fig. S3 and 3A), with the titer of the RnhA+ cells dropping 1,000 times, while that of rnhA mutants decreasing ten-fold (Fig. 3A, the orange bars). Interestingly, this unexpected toxicity of rifampicin is due to hydrogen peroxide accumulation in the medium, since supplementing plates with catalase eliminates the bulk of toxicity for all strains, resulting in ∼20% survival for the RnhA+ strains and ∼50% survival for the rnhA mutants (Fig. 3A, the blue bars, and Fig. S3). The synergy between rifampicin and hydrogen peroxide was noticed before [70].
Fig. 3. Response of rnh mutants to transcription and translation inhibition.
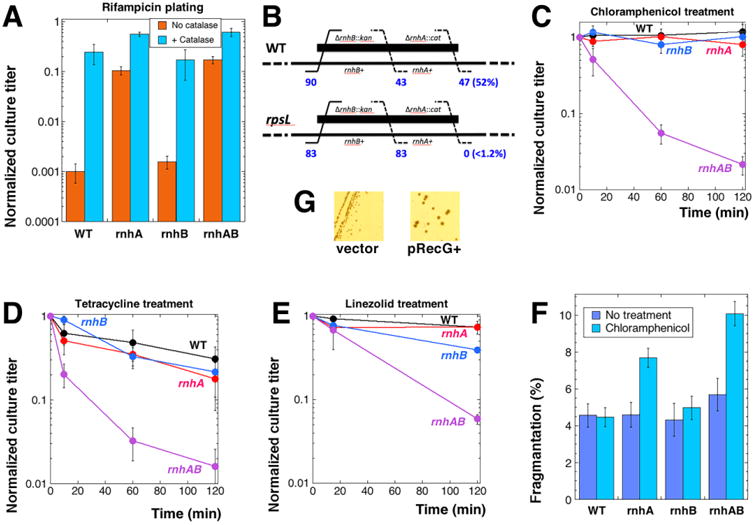
Four strains were used, unless indicated otherwise: WT, AB1157; rnhA, L-413; rnhB, L-415; rnhAB, L-416. Cultures in early exponential growth were exposed to the indicated antibiotics, samples of cultures were removed at the indicated times to determine culture titer. Fraction of survival was calculated after 24 hours of incubation at 30°C as a ratio of the culture titer at the particular time to the culture titer before the treatment was applied. In panels C-E, the mean of 5-6 independent measurements ± SEM are presented. A. Rifampicin resistance of rnhA and rnhAB strains. Cell cultures were plated on LB medium containing 2 μg/ml of rifampicin ± 150 U/ml of catalase. B. P1-cotransduction test for a synthetic lethal combination. The WT or rpsL mutant were P1 transduced with a double ΔrnhA∷cat ΔrnhB∷kan lysate (thick black line, strain L-418), the (leftmost number of) ΔrnhB∷kan transductants were selected on kanamycin-containing plates and then screened for resistance to chloramphenicol, signifying cotransduction of ΔrnhA∷cat (the rightmost number, followed by % cotransduction), or transduction of rnhB alone (the middle number). Strains: WT, AB1157: rpsL, JDW2525. C. Sensitivity to treatment with 100 μg/ml of chloramphenicol. D. Sensitivity to treatment with 50 μg/ml of tetracycline. E. Sensitivity to 50 μg/ml of linezolid. The strains in this case are: WT, L-437; rhnA, FK9; rnhB, FK10; rnhAB, L-438; all strains carry the acrB mutation in order to increase sensitivity to linezolid [103]. F. Chloramphenicol-induced chromosomal fragmentation. Cell cultures were treated with 100 μg/ml chloramphenicol at 28°C for two hours, and chromosomes were analyzed by PFGE. The values are means of four independent measurements ± SEM. The difference between the rnhA and rnhAB values is statistically significant. G. The slow growth of the rnhAB double mutant is improved by increased expression of RecG DNA pump. Strains: L-416 pSRK1 (recG+) and L-416 pBluescript (vector) were streaked on LB plates containing ampicillin and IPTG.
Clearly, the viability of rnhA mutants improves relative to the wild type cells by blocking the process that feeds generation of R-loops. Overall, the effect of rifampicin on rnhA(B) mutants is consistent with the idea that R-loops directly interfere with replication forks. The remaining minor rifampicin sensitivity of the RNase HI+ cells in the presence of catalase is curious and could be due to rifampicin blocking expression of an essential gene, for example via causing all transcription complexes within the gene to form R-loops, which RNase H1 dutifully destroys, effectively inhibiting expression of the gene. Under the circumstances, inactivation of R-loop removal in rnhA mutants could allow the rifampicin-treated cells to express the essential gene.
The rnhAB mutants are sensitive to inhibition of translation
If R-loops could indeed stall and break replication forks (Fig. 2G), then inhibiting translation elongation by interfering with ribosomes, either genetically or chemically, must be detrimental for both the rnhA single and rnhAB double mutants, as R-loop formation is amplified by availability of empty mRNA [71]. Enhancement of R-loop formation by slow translation was tested genetically, employing the E. coli S12 mutant (rpsL) with increased ribosome proofreading, which significantly reduces translation rate [72,73]. We noticed that we could not build rpsL rnhA strain by P1 transduction unless the rnhA+ gene was provided on a low copy number plasmid (pEAK39). To test the suspicion of synthetic lethality formally, we P1 co-transduced the ΔrnhA∷cat allele with the ΔrnhB∷kan (the two genes are about 30 kb apart) into the rpsL mutant strain. After selecting for the rnhB∷kan, we screened for the rnhA∷cat on plates. There were no rnhA co-transductants when the recipient was the rpsL mutant; at the same time, 52% of the rnhB transductants into the corresponding wild type strain also carried the rhnA mutation (Fig. 3B). Thus, rnhA rpsL strain is synthetic lethal, supporting the connection between a slow translation and R-loops.
We also completely blocked translation chemically, by treating growing cultures of the rnh mutants for up to 2 hours with translation inhibitors chloramphenicol, tetracycline or linezolid. In contrast to the rpsL synthetic lethality, the rnhAB double mutant was the only one showing the decrease in viability ranging from 15 to 100 fold with all the inhibitors, whereas wild type and the single mutants showed either no or minor loss of titer (Fig. 3CDE). Moreover, chloramphenicol treatment caused chromosomal fragmentation in the rnhAB mutants detectable by PFGE even in the RecABCD+ cells (Fig. 3F and S4). These observations support the argument that R-loops contribute to generation of R-lesions, but are inconsistent with R-loops being R-lesions themselves, — otherwise both the rnhA single and the rnhAB double mutants would be equally sensitive to translation inhibition. What this result indicates is that, in the rnhA mutants, RNase HII activity either prevents conversion of R-loops into R-lesions or repairs R-lesions.
Although translation inhibition could, in principle, kill rnhAB mutants by preventing SOS induction, transcription inhibition, which also blocks SOS, has the opposite effect on rnhAB cells, ruling out the SOS explanation and leaving the R-loop prevention or stimulation as the most relevant effects of the two treatments. We also tested the effect of overexpression of RecG, a dsDNA pump capable of dissociating R-loops [56,74], from a plasmid in the rnhAB double mutant. The RecG+ overproduction noticeably improves growth of the rnhAB double mutant (Fig. 3G). Overall, the results from experiments on translation inhibition are consistent with the idea that formation of R-loops is toxic for the rnhAB double mutant. However, the toxicity is indirect, rather than via direct R-loop conflict with replication, — otherwise the same toxicity would have been observed in the single rnhA mutants.
Inhibition of replication initiation from oriC exacerbates growth problems of the rnhAB mutants
The ultimate prediction of the scenario that envisions conflict between R-loops and replication forks (Fig. 2G) is that there should be no conflict without replication. In addition, there are suggestions that passage of replication forks through R-loops may also facilitate formation of R-lesions via re-priming from the nascent transcripts [75], leading to co-optation of transcript segments into DNA as R-tracts (Fig. 4A). The overall prediction of these two models is that the transient inhibition of replication should alleviate problems of both rnhA single and rnhAB double mutants.
Fig. 4. Effects of blocking oriC-dependent replication on growth of the RNase H deficient strains.
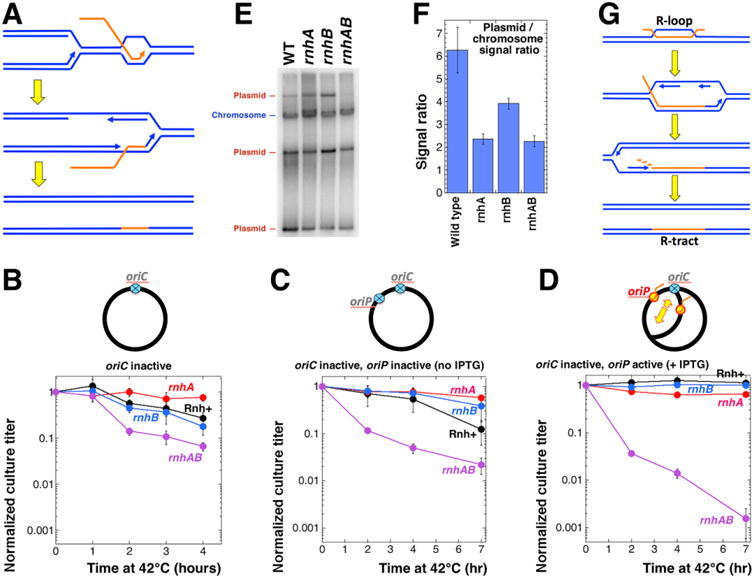
A. A model for R-tract formation due to co-directional transcript cooptation into DNA during replication in rnhAB strains. B-D All strains in these three panels are dnaA(Ts) mutants. In the chromosome schemes on the top of the corresponding graphs, inactive origins are shown as blue crossed circles, active origins are shown in red-and-yellow. The pEAK54 integration site (ColE1 origin under IPTG control) is marked as oriP. The direction of R-loop plasmid replication is identified by a bigger yellow arrow. Cultures were grown at 28°C in LB, serially diluted, spotted on LB plates (with or without IPTG) and incubated at 42°C for the indicated amount of time, then shifted to 28°C incubation for 16 hours to allow colony formation. The calculated titer of the culture for the indicated time of incubation at 42°C was normalized to the titer of the culture from the plate incubated at 28°C throughout. B. Survival of various rnh derivatives of the dnaA46(Ts) mutant after incubation at 42°C for up to 4 hours. Strains: rnh+, L-159: rnhA, L-483; rnhB, L-482; rnhAB, L-484. C. Survival of various rnh derivatives of the dnaA46(Ts) ColE1-ori mutant after incubation at 42°C for up to 7 hours without IPTG. Strains: rnh+, L-215; rnhA, L-486; rnhB, L-485: rnhAB, L-487. D. The experiment is done as in C with plates containing 1 mM IPTG for full ectopic origin induction. E. Determination of relative copy numbers of a ColE1 plasmid pAM34 in rnh strains. Total genomic DNA was isolated from the indicated strains transformed with pAM34 and grown in LB + 1 mM IPTG at 28°C to OD600 = 0.4. DNA was separated on a 1.1% agarose gel, transferred to a nylon membrane and hybridized to the pAM34 used as a probe with lacI-DNA hybridizing to the chromosome. The strains were: wild type, AB1157; rnhA, L413; rnhB, L-415; rnhAB, L-416. F. The ColE1 plasmid maintains a lower copy number per chromosome in the rnh mutants. Quantification of the relative ratio of the plasmid-to-chromosome signal from several gels like in “E”. G. A model for R-tract formation due to replication initiation from R-loop.
To test the effect of replication during RNase H deficiency we blocked initiation of chromosomal replication from oriC in either the dnaA(Ts) or dnaC(Ts) mutants for various amounts of time by incubating plates with serially diluted rapidly-growing cultures at the non-permissive temperature (42°C for dnaA46(Ts), 38°C for dnaC2(Ts)), but then returning plates to the permissive temperature to restore replication and to determine the surviving titer. The dnaC2(Ts) mutants turned out to be unsuitable for this test, as they rapidly lost viability after 2 hours at the non-permissive temperature independently of their rnh mutant status (Fig. S5, top). The dnaA46(Ts) mutants kept their viability during the first four hours of the initiation block (Fig. 4B), although later the cell titer would similarly collapse (Fig. S5, bottom).
Within these four hours of the relatively stable viability of the dnaA(Ts) mutants, there was no effect of the single rnhB defect on survival of the dnaA46(Ts) cells, while the rnhA dnaA(Ts) mutant survived even better than its RnhA+ progenitor (Fig. 4B), apparently due to the constitutive stable DNA replication (cSDR) initiated from transcripts at multiple locations along the chromosome [41,42]. At the same time, we observed that the dnaA(Ts) rnhAB strain lost at least an order of magnitude of its original titer (Fig. 4B). The result clearly shows that the problems of the rnhAB double mutant are distinct from those of the rnhA single mutant, because instead of improving, its viability worsens without replication. It looks like blocking replication from oriC in the absence of both RNase HI and RNase HII activities leads to accumulation of R-lesions (perhaps derived from R-loops), suggesting that replication forks arriving from oriC prevent and/or dilute them instead of aggravating them.
In the light of the dnaA(Ts) result that is numerically similar to our earlier inhibition of translation results (compare Fig. 3CDE with Fig. 4BC), it should be pointed out that translation inhibition also blocks initiation of new rounds of replication from oriC by blocking synthesis of replication initiator protein DnaA [76]. However, rifampicin does the same by blocking transcription, and yet the viability phenotype is the opposite, suggesting that the apparently similar dnaA and chloramphenicol results are due to distinct mechanisms.
Stable DNA replication generates R-lesions in rnhAB mutants
To further test the idea of the conflict between R-loops and replication forks in the rnhA mutants (Fig. 2G), we introduced a ColE1-type IPTG-dependent plasmid replication origin (oriP) of pAM34 [77] into the chromosome of the dnaA(Ts) rnh mutants. The plasmid replicon (that itself initiates via an R-loop-dependent mechanism [23]) maintains an increased copy number within the chromosome [78] and therefore should sensitize the rnhA mutants, due to increase in the conflict between R-loops and replication forks. We repeated our measurements of the viable culture titer on plates under conditions of either silent or IPTG-induced oriP. The “no IPTG” results (oriC inactive, oriP inactive) for the four strains looked generally similar to the previous strain set without oriP, except that the dnaA rnh+ and dnaA rnhB strains have improved their survival significantly between four and seven hours of incubation at 42°C (compare Fig. 4C with Fig. 4B and S5, bottom). However, upon plasmid origin induction by IPTG (oriC inactive, oriP active), the rnh+ and both single mutants showed complete recovery after seven hours of incubation at the non-permissive temperature, while the rnhAB double mutant lost almost three orders of magnitude of titer, indicating that replication initiated from R-loops is poisonous for the double mutant in the absence of replication from oriC (Fig. 4D).
Marker frequency analysis with probes on both sides of the plasmid integration locus confirmed that the IPTG-inducible ColE1 replicon in the chromosome fired with the expected polarity in both wild-type and rnhB mutant (Fig. S6). Interestingly, the polarity, but not the efficiency of replication from oriP, was lost for the rnhA mutants, while the induction was anemic in the rnhAB double mutant (Fig. S6). We estimated the replication potential of the same replicon as a free plasmid in rnhAB mutant by measuring copy number of plasmid monomers relative to the chromosomal DNA in DnaA+ strains. We found that the rnhA and rnhAB mutants have the plasmid copy number reduced to about one third of that in the wild-type strain (Fig. 4EF), indicating that this IPTG-inducible ColE1 replicon has problems operating in the rnhA mutants (natural ColE1 replicons also do [79]).
We interpret the inability of rnhAB mutants to recover after temporal block of replication from oriC, especially if additional initiations are induced via R-loops, as accumulation of the R-loop-derived R-lesions blocking chromosome replication. It is also apparent that RNase HII has a critical role in processing the R-lesions, while replication forks coming from oriC in the rnhAB mutant offer another possibility to dissolve the R-lesions.
Enzymatic and chemical hydrolysis of rNs in model substrates
An R-lesion derived from an R-loop could be the RNA remnant from the R-loop-initiation of cSDR (Fig. 4G), called “R-tract”, which we define as four or more consecutive rNs embedded in DNA. In fact, everything that we learned so far suggested that rnhAB mutants accumulate R-tracts derived from R-loops. What makes this idea even more appealing is the fact that R-tracts are the only R-lesions that should be recognized by both RNase H enzymes (Fig. 1A). To detect hypothetical R-tracts and estimate their density in the DNA of the rnhAB double mutants, we employed the in vitro plasmid relaxation assay with RNase HI or RNase HII enzymes. The specific activities of the RNase HI and RNase HII enzymes were verified in vitro with 52 bp-long dsDNA oligos containing either single rN (R-patch) or 5-rNs run (R-tract) in the same DNA sequence (Fig. 5A). To independently confirm the position and the number of rNs in the DNA oligos, we used high pH treatments, either 0.1 M NaOH, or sodium carbonate-sodium bicarbonate buffer with pH 9.3 (Fig. 5BC).
Fig. 5. Verification of RNase HI and RNase HII rN-DNA substrate specificity in vitro and the rN-density in DNA of the RNase H+ cells and rnh mutants.
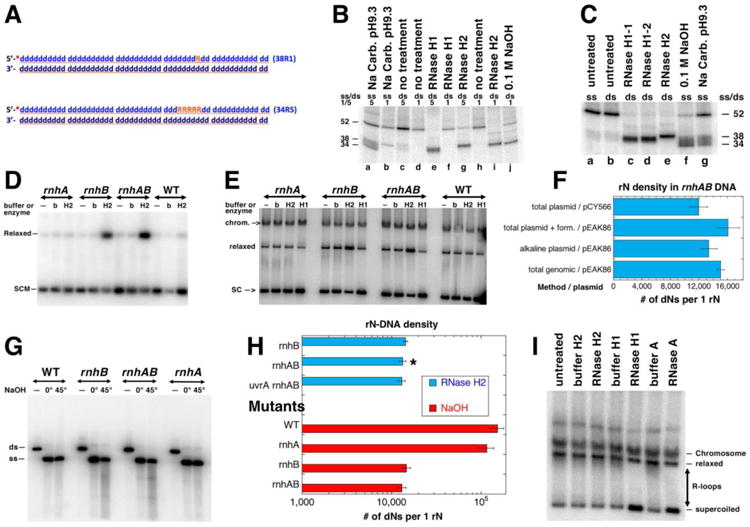
A. A scheme of the two double stranded oligo substrates: 38R1 (single rN) and 34R5 (five consecutive rN). The 32P label at the 5′ end is shown as a red asterisk. DNA nucleotides are shown as blue lower case “d”, ribonucleotides are orange uppercase “R”. B. Products of the rN-DNA substrate hydrolysis by E. coli RNase HI and RNase HII enzymes. The radiolabelled rN-containing dsDNA oligos (shown in A) were incubated with the RNase HI or RNase HII enzymes. “0.1 M NaOH” and “Na Carb. pH 9.3” refer to alkali conditions in which rN hydrolysis produces reference size products. Numbers “1” or “5” refer to 38R1 or 34R5 oligos (A); ss/ds refers to whether the substrate used in the reaction was single-stranded or double-stranded. RNase H1 and RNase H2 were the E. coli enzymes RNase HI and RNase HII. RNase H1-1 and RNase H1-2 were RNase HI enzymes from different producers. The numbers on the side of the gel represent the sizes of the substrate and cleavage products. The reaction products were analyzed in 18% urea-PAGE gel. C. Only 34R5 oligo was used as either ss or ds substrate. All designations are like in “B”. D. Treatment with RNase HII of the plasmid isolated by alkaline lysis protocol. SCM, supercoiled monomer; b, buffer; H2, RNase HII. Plasmid: pEAK86, plasmid isolation was done at 0°C. Strains for results shown in panels D-I were: WT, AB1157; rnhA, L-413; rnhB, L-415; rnhAB, L-416; uvrA rnhAB L-417. Product of the reactions were run in 1.1% agarose gel; autoradiogram of the representative Southern blot with the radiolabelled pEAK86 DNA as a probe is shown here and also in E and G. E. Treatment with either RNase HI or RNase HII enzymes of the plasmid isolated by the total genomic DNA protocol. SC, supercoiled plasmid; relaxed, relaxed plasmid; chrom., chromosomal DNA. Plasmid: pEAK86. Analysis of plasmid species was carried out as in D. F. Summary of quantification of the RNaseHII-revealed density of rNs in plasmid DNA isolated by various methods from the rnhAB double mutant. The density calculations are described in Methods. “Form.”, formamide. G. Alkali treatment analysis of rN-density. The plasmid DNA isolated by alkaline lysis at 0°C, was linearized and treated with NaOH. Treatment: “—”, no treatment; 0°, 0.2 M NaOH, 20 mM EDTA treatment on ice for 20 min; 45°, 0.3 M NaOH, 20 mM EDTA treatment at 45°C for 90 minutes. ds, linearized plasmid DNA, ss -single stranded plasmid. The samples were run in 1.1% agarose in TAE buffer, at 4°C. H. Summary of quantification of the rN-density determined by either RNase HII or by alkali treatments (from gels like in “G”). Various mutant comparison data are shown, pEAK86 was purified by alkaline lysis only, values are means of three independent measurements ± SEM. The star identifies the value already reported in panel “F”. I. R-loop removal by RNase HI or by RNase A. pAM34 isolated from rnhA (strain L-413) by the total genomic DNA protocol.
We confirmed that RNase HI hydrolyzes 20 nt-long RNA strand of an RNA/DNA hybrid (not shown), does not cleave a DNA strand with a single rN (Fig. 5B, compare lanes b, d, f and h), while cleaving the DNA strand with a run of five rNs in two positions around the middle of the run (Fig. 5B, compare lanes a, c, e and j; Fig. 5C, compare lanes b, c, d and f). At the same time, RNase HII enzyme cleaves both substrates, each at a single position at the rN-DNA junction (Fig. 5B lanes c, g, i and 5C, lanes b, e, g). We concluded that both RNase H enzymes can be used for in vitro plasmid relaxation assay to detect single rNs and R-tracts of at least five consecutive rNs.
Density of R-patches in the rnhAB mutants
Previously, we successfully used an in vitro plasmid relaxation assay with enzymes specific against modified nucleotides to measure the density of these modifications in E. coli DNA [80,81]. If the DNA of rnhB single mutants accumulates only R-patches (runs of 1-3 rNs), plasmids from rnhB cells would be relaxed in vitro only by RNase HII. At the same time, supercoiled plasmid DNA from the rnhAB double mutants, if it indeed contains additional R-tracts of at least five rNs, should be relaxed by both RNase HII (strongly, at both R-patches and R-tracts) and by RNase HI (weakly, only at R-tracts). We purified plasmid DNA from growing cultures of the WT and rnh mutant cells and digested it in vitro with the RNase HI and RNase HII enzymes.
For robust and unbiased measurements of rN-DNA density, we prepared plasmid DNA by three distinct protocols: 1) by alkaline lysis (Fig. 5D); 2) by the total plasmid DNA protocol with or without subsequent treatment with formamide (to remove potential R-loop species, which might affect the assay); 3) by the total genomic DNA protocol (Fig. 5E, I). Alkaline lysis procedure lyses cells and denatures DNA with 0.2 M NaOH, a short (∼1 minute) treatment that could potentially hydrolyze rNs in plasmid DNA. However, our control experiments demonstrated that the 52 bp-long dsDNA oligo (Fig. 5A) containing one ribonucleotide in the middle treated with up to 0.5 M NaOH (+20 mM EDTA) for up to five hours showed no signs of alkaline hydrolysis if the treatment was performed on ice (not shown). Therefore, plasmid DNA prepared by the alkaline lysis procedure can be used for ribodensity measurements as long as the samples were kept on ice during DNA isolation. To accommodate distinct replication initiation strategies, we used plasmids with either ColE1 replicon (replication initiates via R-loop formation) or pSC101 replicon (replication initiates by an initiator protein).
After the purified plasmid DNA was treated with either RNase HI or RNase HII, it was subjected to Southern analysis for quantification of the supercoiled-to-relaxed species conversion (Fig. 5 D, E and I). The fraction of the supercoiled DNA band remaining after the enzymatic treatment was calculated and considered to represent the zero class of the Poisson distribution, as before [80,81]. The average ribodensity in two different plasmid DNA, isolated from the rnhAB mutant by the three distinct protocols and determined by RNase HII treatment, was 1 rN per 14,098 nt (Fig. 5F). In the rnhB single mutant, the ribodensity was very similar, 1 rN per 13,619 nt (Fig. 5H). We also treated linearized plasmids from the same DNA samples with alkaline buffer at 45°C (Fig. 5G) and detected an average density of 1 rN per 13,895 nt, again with no significant difference in rN density between rnhB and rnhAB mutants (Fig. 5H). Thus, the ribodensity numbers determined by RNase HII and alkaline treatments are in a remarkable agreement (Fig. 5F vs. H). But there is also a difference: the in vitro RNase HII treatment yields no nicks in plasmids from WT and rnhA strains indicating no detectable ribodensity (Fig. 5DEI), whereas the alkaline buffer treatment “detects” 1 rN per 116,000-154,000 nt (Fig. 5H) — this is likely an artifact, but could reflect the presence of another alkaline-labile DNA modification. In conclusion: plasmid relaxation assays with RNase HII enzyme and alkali treatment do not reveal differences in the ribodensity in plasmid DNA between the rnhAB and rnhB strains and agree on the density of one R-patch in ∼14,000 nt of DNA.
No R-tracts in plasmid DNA from the rnhAB mutants
In contrast to the robust nicking of plasmids from rnhB and rnhAB mutants with RNase HII enzyme (Fig. 5DE), we observed no decrease of the supercoiled plasmid, or increase of the relaxed plasmid, by the RNase HI treatment regardless of the DNA purification procedure, plasmid type or mutant background (Fig. 5EI). At the same time, as a result of RNase HI treatment the smear between the bands of supercoiled and relaxed plasmid cleared, while the amount of the supercoiled plasmid noticeably increased as seen in ColE1-ori plasmid samples purified from the rnhA mutant by the total genomic DNA purification protocol (Fig. 5EI). We interpret this clearing by RNase HI or RNase A treatment accompanied by scDNA increase to mean that the species of apparently variable superhelicity that run as a smear between the bands of supercoiled and relaxed molecules in fact represent fully supercoiled molecules with R-loops of various sizes, which the RNase HI or RNase A treatment converts into the fully supercoiled species. This effect is another confirmation that the RNase HI enzyme is active in our tests. At the same time, RNase HII treatment has no effect on the “R-loop smear” (Fig. 5EI), consistent with reports that, under physiological conditions, RNase HII does not attack RNA:DNA hybrids lacking rN-dN junctions [43,46,47].
Since it has been proposed that the nucleotide excision repair (NER) system also participates in removal of rN from DNA [49], we constructed the ΔuvrA ΔrnhAB triple mutant strain in AB1157 background and indeed observed a slight reduction in colony size (Fig. 1G) and growth rate inhibition in liquid cultures, supporting NER contribution to rN-DNA removal. However, plasmid DNA purified from the uvrA rnhAB triple mutant showed no increase in the density of single rNs compared to the rnhAB double mutant (Fig. 5H), arguing against NER participation.
Thus, from our physical analysis of plasmid species after in vitro treatment with RNases H enzymes we conclude:
— The RNase HI treatment increases the fraction of supercoiled DNA, revealing R-loops in plasmids prepared by the total genomic DNA protocol.
— Since at the same time RNase HI fails to increase the amount of relaxed plasmid, the density of hypothesized R-tracts in the DNA of the rnhAB double mutant is below the sensitivity of our plasmid assay (calculated at 1 event per ∼100,000 nt [81]).
— The rnhB mutants accumulate a significant density of RNase HII-sensitive sites (R-patches, most of them likely single rNs) in their DNA (1 per 14,000 nt), but show no growth defects, meaning that single rNs at this density do not grossly interfere with DNA replication.
— Since the rnhAB double and uvrA rnhAB triple mutants accumulate the same R-patch density as the one in the rnhB mutants, neither RNase HI, nor NER contribute to removal of single rNs during regular growth.
RNase-induced breakage of the chromosomal DNA from rnh mutants
Puzzled by our inability to detect the expected R-tracts in plasmid DNA, we imagined that, in addition to their initial low density in the DNA of the rnhAB mutants, R-tracts (S1 in Fig. 6A) could be rapidly converted by DNA replication into R-gaps (ss-gaps in duplex DNA, in which the remaining strand contains one or more rNs), which would make them resistant to RNase HI attacks (S2 in Fig. 6A). At the same time, such R-gaps should still be substrates for both RNase HII and the non-specific RNase A enzymes (S2 in Fig. 6A, Table S1), which would convert them into double-strand breaks. To detect possible R-gaps in the chromosomal DNA of the rnh mutants isolated in agarose plugs, we treated the plugs with either RNase HI, RNase HII or RNase A (either low salt (LS) or high salt (HS) conditions), and subjected them to pulsed-field gel electrophoresis (Fig. 6B). RNase A(LS) attacks both ssRNA and dsRNA, as well as the RNA strand in the RNA:DNA hybrids; RNase A(HS) attacks only ssRNA [82]. Smearing DNA into the gel as a result of the RNase HII and A(HS) treatments in vitro would indicate formation of double-strand breaks, suggesting the presence of R-gaps in the chromosomal DNA.
Fig. 6. Chromosome fragmentation analysis by RNase HI, RNase HII and RNase A treatment in vitro.
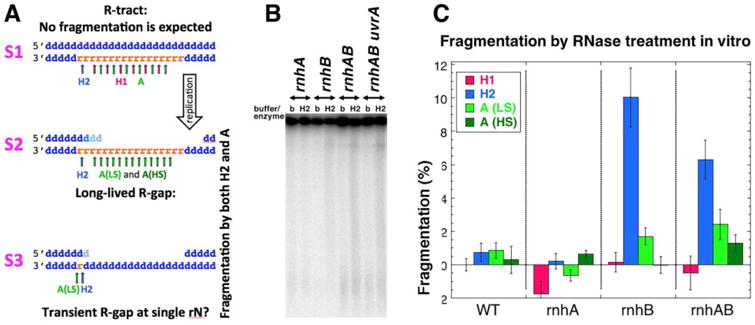
A. A scheme of various hypothetical R-lesions (R-tract, two types of R-gaps) with positions of cleavage by RNase HI, HII and A (in low salt (LS) and high salt (HS) conditions) shown with arrows of the corresponding color. Small blue “d” letters, dNs; small orange “r” letters, rNs. The strand polarity in a duplex is identified on the left. B. A representative pulsed-field gel detecting chromosomal fragmentation after RNase HII treatment. The lanes are marked either with “b” (buffer treatment control) or “H2” (RNase HII treatment). Strains: WT, AB1157; rnhA, L-413; rnhB, L-415; rnhAB, L-416; uvrA rnhAB, L-417. C. Quantification of the RNase treatment-induced fragmentation. The plotted values are means ± SEM from 3-6 independent measurements from gels like in “B”. For RNase A treatment, both low salt (LS) and high salt (HS) conditions are plotted. Since individual fragmentation values are differences between the enzyme and the buffer treatments, some values are negative.
As expected, we found no fragmentation after RNase HI treatment in any of the four strains (Fig. 6C) — because RNase HI has no DNA substrates that could be converted into double-strand breaks (Fig. 6A). In fact, none of the four treatments (RNase HI, HII, RNase A(LS), RNase A(HS)) fragmented chromosomes of the WT or rnhA single mutant cells (Fig. 6C), suggesting no rN residues near ss-gaps in this DNA. In contrast, there was a notable fragmentation of the chromosomal DNA from the rnhAB double mutant by RNase HII and a detectable fragmentation by RNase A (either LS or HS) (Fig. 6C), indicating R-gaps (S2 in Fig. 6A). In vitro, RNase HII enzyme of E. coli cleaves such partially single-stranded substrates normally [47]. Curiously, while there was no fragmentation of the chromosome from the single rnhB mutant with the RNase A(HS) treatment, there was a fragmentation similar to the double mutant levels upon RNase HII and RNase A(LS) treatments (Fig. 6C) — as if RNase A(LS) treatment can also (inefficiently) recognize single rNs in DNA (for which we have preliminary evidence (not shown)).
Because the single rnhB mutant, being RNase HI-proficient, is not expected to develop R-tracts in its chromosome, the observed HII or A-induced fragmentation most likely signifies transient ss-gaps across single rNs (S3 in Fig. 6A). Since the rnhB single mutant grows like WT, while the rnhAB double mutant is so much inhibited (Fig. 1B), we imagine that the rnhB single mutant accumulates only the innocuous transient ssDNA-gaps opposite single rNs (S3 in Fig. 6A), while the double rnhAB mutant also experiences long R-gaps (S2 in Fig. 6A), explaining its poor growth and low viability. Importantly, long R-gaps should be susceptible to various cellular RNases, converting them into double-strand breaks, which could explain high chromosomal fragmentation that we observe in the rnhAB recBC mutant (Fig. 2EF).
Discussion
Our genetic and physical comparison of the otherwise wild type E. coli deficient in RNase HI, RNase HII or in both enzymes revealed unexpected phenotypes of the double mutant. The double rnhAB mutant grows slowly, with a substantial fraction of highly filamentous cells in cultures that display abnormal nucleoid shapes. There is no obvious deficit of DNA in the elongated cells, though, suggesting no problem with gross DNA synthesis, while the peculiar shape of the nucleoids (like beads on the common string) suggests segregation problems, perhaps linked to random assortment of independent subnucleoids [83] due to multiple initiations away from the origin, expected in the rnhA mutants. We further report that, during regular growth, the rnhAB mutant is highly-induced for SOS-response to DNA damage, depends on recombinational repair of double-strand breaks for viability and develops high levels of chromosomal fragmentation when double strand break repair and linear DNA degradation are disabled by the recBC defect. The low levels of chromosomal breakage in the rnhAB rec+ cells, and their decreased viability are consistent with efficient DSB repair mending most of the lesions, while occasional irreparable DNA lesions causing the observed lethality.
Based on the specificity of RNase H enzymes broadly targeting RNA:DNA hybrids in which ribonucleotides are either integrated or not into a DNA strand, we propose “R-lesions”, a new class of DNA lesions containing rNs. The simplest R-lesion, in the current paradigm, is an R-loop, which is thought to be so stable as to block the progress of replication forks, causing their breakage [52-54]. In addition to R-loops (the structures in which the RNA strand is not contiguous with DNA strands), we considered two R-lesions in which rNs are integrated into DNA strands: R-patches (1-3 rN runs) and R-tracts (≥4 rN runs) — as contributors to the growth defect.
Our results with chemical inhibition of transcription and genetic inhibition of translation are consistent with the notion that R-loops contribute to the chromosomal problems in the rnhA(B) mutants and may even be the problem themselves. However, our results with chemical inhibition of translation, that kills the rnhAB double mutant, but not the rnhA single mutant, already suggest that R-loops cannot be the killing lesions themselves, as they lack rNs integrated into DNA duplex (the substrate of RNase HII). Finally, replication inhibition and induction of alternative initiations make it clear that regular replication from oriC prevents or removes R-lesions, rather than conflicting with them, while it is the R-loop-initiated replication from a plasmid origin in the chromosome that rapidly kills the double mutants, revealing the pathway for generation of R-lesions. The proposed pathway starts with R-loops, but then continues to RNA strand incorporation into DNA to create R-tracts (Fig. 4G).
Confusingly, we failed to detect R-tracts in the plasmid DNA from rnhAB double mutants (no relaxation of supercoiled plasmid DNA by RNase HI), even though we found indications of R-loops (increase in supercoiled plasmid by RNase HI) and of R-patches (supercoiled plasmid relaxation by RNase HII and linear DNA sensitivity to alkali). Of course, it could be that R-tracts do form in the chromosome readily, but only in a few specific locations, and therefore do not form in plasmids. It could be also that R-tracts in plasmid DNA are rapidly converted into a species resistant to RNase HI, and we indeed found that the chromosomal DNA from the rnhAB double mutant can be fragmented by in vitro treatment with RNase HII and non-specific RNase A, but not with RNase HI, suggesting additional types of R-lesions. The implications of this peculiar RNase sensitivity of genomic DNA will be discussed below.
The gross phenotypes of the rnhAB mutants are not due to R-patches
Bacteria in general and E. coli in particular offer a perfect model system to dissect the roles of two RNase H enzymes, since they have distinct substrate specificities in bacteria, attacking either RNA:DNA hybrids (RNase HI) or RNA-DNA junctions (RNase HII). Analogous eukaryotic enzyme RNase H2 is a heterotrimer with both activities residing in the same enzyme. For bacteria lacking both RNase H activities, temperature-sensitive growth, sensitivity to media and strain background, cell filamentation and high SOS response were documented before [13,59,60,84]. Yeast cells deficient for either one or both RNase H activities also exhibit replication stress, double strand DNA breaks and increased recombination [85-87].
Our in vitro (with RNase H enzymes) measurement of ribodensity in plasmid substrates isolated from the rnhB and rnhAB mutants show one R-patch in 14,000 dNs, which is also consistent with the chromosomal rN density in rnhB mutants estimated in alkaline sucrose gradients (Glen Cronan, personal communication), but no R-tracts. Similar to our results, in vitro sensitivity of genomic DNA, purified from RNase H2-mutant mouse cells, to RNase HII, but not to RNase HI hydrolysis, suggested the presence in DNA of one or two consecutive rNs with the density of 1 in 7,600 nt [15].
The essentially wild type behavior of the rnhB mutant cells combined with substantial density of R-patches in the DNA of rnhB mutants means that pausing of replisomes at single rNs in bacterial DNA does not result in negative consequences for DNA metabolism. At the same time, the severe growth inhibition of the rnhAB double mutant, which is at odds with a rather mild cell viability defect, suggests regular occurrence of replication-stalling lesions (not R-patches!) that, nevertheless, are mostly reparable.
The two types of R-loops
We report co-lethality of the slow-translation rpsL defect with the rnhA defect and sensitivity of the rnhAB double mutants to translation inhibitors, with the interpretation that the ribosome-free RNA-transcripts form R-loops with the template DNA. We base this inference on various conditions in E. coli that facilitate invasion of nascent untranslated transcripts into the cognate duplex DNA: increased negative supercoiling behind RNA polymerase in topA mutants [38,71] and availability of empty transcript due to the compromised transcription termination in rho or nusG mutants [60,88]. Synthetic lethality of rnhA topA and rnhA nusG double mutants is rationalized as R-loops persistance in the absence of RNase HI enzyme [60,88]. R-loops are considered to represent a strong barrier for replication forks, causing their breakage via still poorly-understood mechanisms [52-54]. In this respect, a simple interpretation of our results is that R-loops originate spontaneously, and if not removed by RNase HI, block replication forks, which we detect as increased chromosome fragmentation (Fig. 2G).
However, our finding of conditions in which the rnhA single mutant is fine, while the rnhAB double mutant dies (chemical inhibition of translation and blocking oriC initiation, induction of R-loop-initiated oriP) clearly shows that there is more to R-lesions than a simple replication fork conflict with R-loops. In fact, this difference in phenotypes between the single rnhA and the double rnhAB mutants indicates existence of at least two kinds of R-loops, with distinct outcomes for replication (Fig. 7A). The first kind is an R-loop forming behind the transcription-elongation complex (TEC) — which, according to the current consensus, is how most R-loops are initiated [52-54]. We propose to call this structure R-loop-aTEC (“R-loop-anchored transcription-elongation complex”) (Fig. 7A, left), to distinguish it from TEC-free R-loops (Fig. 7A, right) that remain after transcription termination or emerge by other processes [89,90]. Since transcription elongation complexes are extremely stable [91], their “anchoring” by the nascent short R-loops cannot terminate them. Instead, such R-loop anchoring effectively traps the negative supercoiling generated in the template DNA behind the advancing RNA-polymerase, channeling it into R-loop elongation (Fig. 7A, left). Such a growing R-loop anchor further strengthens the overall complex, making the resulting R-loop-aTEC a formidable obstacle in the path of a replication fork, capable of causing fork breakage [92,93].
Fig. 7. Two types of R-loops and a model of chromosomal fragmentation in the RNase H-deficient mutants.
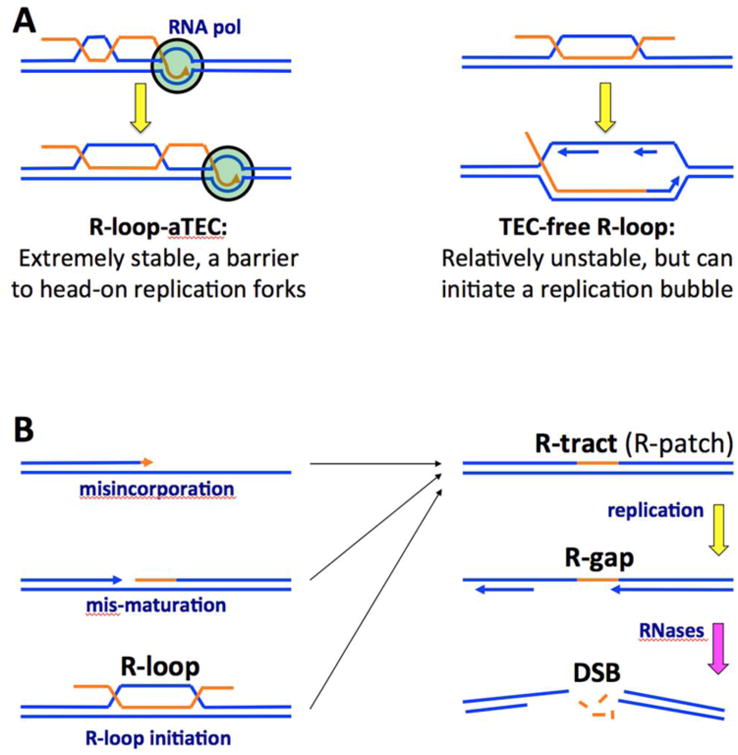
Blue lines, DNA strands; orange lines, RNA strands; arrowheads, direction of strand extension; smaller navy font, DNA metabolism processes; bigger black font, various R-lesions. A. Two types of R-loops in relation to replication. B. The model of chromosomal fragmentation in the absence of RNase H activities. Mis-maturation — incomplete maturation of Okazaki fragments. The last step with pink arrow — any cellular ssRNA endoribonuclease.
However, since this scenario of replication forks disintegrating at R-loop-aTEC works independently of the RNase HII status, the situation in the rnhAB mutants must be different. First, since RNA:DNA hybrids are poor substrates for RNase HII [43,46,47], there must be R-loop processing into R-tracts before RNase HII could act on these R-lesions. Second, the rnhAB mutants are especially poisoned by R-loop-initiated replication rounds, whereas R-loop-aTEC would not be able to initiate a replication fork because: 1) the 3′-end of the RNA transcript is not available for priming DNA synthesis; 2) the access for the priming activities to the potential replication fork structure is blocked by the RNA polymerase. Replication bubbles can only be initiated by a TEC-free R-loop [94] (Fig. 4G and 7A, right). However, TEC-free R-loops are relatively unstable, for the reasons given in the introduction. Remarkably, when TEC-free R-loops are “stabilized” by initiation of DNA replication, DNA synthesis around them should convert them into R-tracts (Fig. 4G). In other words, there are two types of R-loops inside the cell: 1) R-loop-aTECs that block replication fork progress due to their extreme stability; 2) TEC- free R-loops that are rather unstable, but could initiate replication, which should convert them into R-tracts that will block the next replication round.
R-loop initiations and chromosome fragmentation in the absence of RNase H
Recently, it was suggested in yeast rnh1 Δrnh201 strain that R-loop-primed replication can initiate break-induced replication (BIR) with lethal outcomes [85]. Our results demonstrate that during R-loop-primed replication, RNase HII enzyme plays an important role in processing the intermediates. In the absence of oriC-initiation, IPTG-induced-plasmid origin fired with reduced efficiency in the rnhAB mutants relative to its efficiency in rnhA mutants. At the same time, in contrast to the rnhA single mutant, the rnhAB double mutant was losing viability, which emphasizes a special role for RNase HII in processing R-loop-initiations (Fig. 4G), apparently preventing their conversion into irreparable R-lesions. Because of the substrate specificity of RNase HII toward RNA-DNA junctions, the most likely intermediates processed by RNase HII have such a junction. However, it should be noted that this thinking is solely based on the strict substrate specificity of bacterial RNase HII enzyme in vitro [43,46,47], and it is still formally possible that bacterial RNase HII has a limited in vivo activity against R-loops. Eukaryotic RNase H2 enzymes do attack RNA:DNA hybrids with no RNA-DNA junctions, representative of R-loops [95,96].
Since the poor growth of the rnhAB double mutant contrasted with normal / acceptable growth of single mutants, we did expect to find that the double mutant DNA accumulates a common substrate of the two RNase H enzymes, the R-tracts. We did not find R-tracts in plasmids, perhaps because the presumed R-tracts accumulate only at a few select sites on the chromosome that need to be identified first. Alternatively, it is possible that the RNase H-deficient mutants suffer from RNase HI-resistant and at the same time replication-blocking R-lesions, like R-gaps (Fig. 6A). Our observation of the RNase HII-induced DSBs in the chromosomal DNA from rnhB mutants is consistent with the formation of transient ssDNA gaps during pausing of replication across R-patches in the template DNA. Therefore, we would like to present a model of chromosomal fragmentation due to R-gap formation in the RNase H-deficient mutants (Fig. 7B). This model explains the poor growth of the RNase H-deficient mutants in bacteria and lower eukaryotes and the essentiality of RNase H enzymes in higher eukaryotes by a combination of frequent repairable lesions (R-tracts, R-gaps, double-strand breaks in replicated parts of the chromosome) and rare irreparable double-strand breaks in single-copy parts of the chromosome. The main difference from the previous models linking chromosome fragmentation with the conflict between R-loops and replication forks (Fig. 2G) is that R-loop formation is only the beginning in the pathway to generate real replication-stalling R-lesions (R-tracts and especially R-gaps), which eventually lead to chromosome fragmentation. The model invites experimental testing.
Material and Methods
Bacterial strains
E. coli K-12 strains and plasmids used are described in Table S2. Strain constructions were by P1 transduction [97] or by deletion-replacement method with the following removal of the antibiotic resistance by pCP20 [98]. The ΔrnhA∷cat deletion removes 35-154 aa of the ORF. The ΔrnhB∷kan mutant was from Keio collection [99] and along other such mutants was purchased from E. coli Genetic Stock Center. Deletions-replacements of the rnhA and rnhB genes were confirmed by PCR. The recA, recBCD, recF and uvrA mutants were confirmed by their characteristic UV-sensitivities. The dnaA and dnaC mutants were verified as unable to grow at 42°C. The acrB mutant was confirmed by sensitivity to 0.1% SDS on LB medium, the rpsL mutant was confirmed by decreased growth rate at 37°C and increased growth rate with 500 μg/ml streptomycin in LB medium at 37°C.
Plasmid constructions
pEAK39: 0. 619 kb PCR-amplified (primers #167, 168) chromosomal fragment carrying rnhA+ was first cloned into TOPO-vector giving rise to pEAK38, then the HindIII-XhoI fragment carrying rnhA+ gene was subcloned into HindIII and XhoI sites of pK80. pEAK84: the 1.89 kb PCR-amplified (primers #263, 264) chromosomal fragment carrying csdA+ gene was digested with Kpn I and Mlu I and ligated with KpnI, Mlu I sites of pZS*24-MCS-1. pEAK86: the 1.17 kb SacI-AatI fragment from pAM34 carrying bla+ was ligated with 4.8 kb SacI-AatI fragment of pEAK84.
Primers used for making deletion-replacements, PCR amplification, sequencing and verification of indicated chromosomal loci are listed in the Table S3 (Primers).
Media and growth conditions
Cells were grown in LB broth (10 g tryptone, 5 g yeast extract, 5 g NaCl per liter, pH 7.2, with NaOH) or on LB plates (15 g agar per liter of LB broth). The growth temperature was 28°C unless otherwise indicated in the description of experiments. When screening for mutations linked to antibiotic-resistant genes or when the cells were carrying plasmids, the media were supplemented with the required antibiotic: 100 μg/ml ampicillin, 50 μg/ml kanamycin, 10 μg/ml tetracycline or 10 μg/ml chloramphenicol. 1 mM IPTG (isopropyl-p-D-thiogalactopyranoside) was used when required for plasmid maintenance. In some experiments, liquid cultures were treated with 100 μg/ml rifampicin, 100 μg/ml chloramphenicol, 30 μg/ml tetracycline or 50 μg/ml linezolid.
Determination of viability
A fresh overnight culture was diluted 100-fold into 2 ml of LB and grown with shaking at either 28°C or 42°C until they reached OD600 = 0.4. Two 100 μl aliquots of the growing culture were taken at the same time: both were diluted 2-fold into 1% NaCl, and to one of them, 0.4 μl of 4 M NaOH was added to stop cell movement. The NaOH-treated aliquot was used to determine the titer by counting the cells under a microscope in a Petroff-Houser counting chamber and calculating the expected density of cells per 1 ml. The untreated aliquot was serially diluted in 1% NaCl, and the appropriate dilutions were plated on LB agar. The plate was incubated at either 28°C or 42°C overnight, and the colony-forming units were counted. The viability was expressed as the ratio of the number of colony-forming units and the titer of direct cell counts.
Determination of viability in various tests
Overnight cultures of tested strains were diluted 100-fold in the morning of the following day and grown in fresh LB media to OD600 ∼ 0.2-0.3. For liquid culture treatment, a specific amount of the agent was added directly to the growing culture. Five ten-fold serial dilutions of the culture were made in sterile 1% NaCl solution and spotted by 10 μl in one row on several square petri dishes with LB agar. Spots were dried, and the plates were treated according to the test.
For anaerobic sensitivity test, plates were prepared in advance and kept for 24 hours in the anaerobic chamber before used for spotting. Serial dilutions were applied on plates in the anaerobic chamber and incubated anaerobically at 37°C for 24 hours. Plates were moved to aerobic conditions and incubation at 37°C continued for 16, 24 or 48 hours.
For testing effects of dnaA46 or dnaC2 mutations, each plate was incubated either at 42°C for dnaA46 or at 38°C for dnaC2 for a time indicated in the figure and then transferred to 28°C incubation for 16-24 hours for colony development. Colonies were counted under a stereomicroscope. Survival was quantified by normalizing calculated culture titer from the plate developed at 42°C (with the subsequent shift to 28°C) to the titer of the culture from the plate developed only at 28°C.
Microscopic observation of cell morphology and nucleoids
We followed the “fluo-phase combined method” described in Hiraga et all [58]. Briefly: cells of growing cultures (OD600 = 0.4, 37°C) were centrifuged, washed once with sterile 1% NaCl and resuspended in 1% NaCl in the initial volume of the culture. 5 μl aliquots of the samples were placed on a slide glass (Superfrost Plus Microscope Slides, precleaned, Fisherbrand) and dried at room temperature. 5 μl of the methanol was applied per sample spot and was allowed to dry for 5 min. After fixation with methanol, the slide was briefly soaked six times in tap water. After washing, the slide was dried completely at room temperature. 10 μl freshly-prepared DAPI solution (NucBlue® Fixed Cell Stain ReadyProbes reagent purchased from Molecular probes by Life technologies) was applied to the sample spot and incubated for 5 min at room temperate. A clean glass cove slip was put on the solution drop, and the perimeter was sealed with clear polish nail liquid, to prevent the sample from drying out. Low fluorescence immersion oil was used on the cover slip for the sample observation through 100× of a Nikon microscope, with UV and phase contrast 2. The halogen lamp was dimmed to the optimal level to see fluorescent nucleoids in UV. Color images were taken with the microscope camera.
Measuring the SOS induction
The relative levels of the SOS induction, expressed in “Miller's units”, were determined in growing cultures by spectrophotometrically measuring the activity of β-galactosidase in the appropriate derivatives of AK43 according to the protocol of Miller [97] with modifications, as described [66].
Analysis of ribonucleotide incorporation by plasmid relaxation and alkali treatment assay
Strains transformed with plasmids were grown in 10 ml LB with ampicillin at 28°C to OD600 = 0.35-04. Cells from 10 ml cultures were collected by centrifugation, kept on ice and passed through one of the following protocols: total genomic DNA isolation [81], total plasmid DNA isolation [100] or small-scale alkaline lysis plasmid DNA isolation [101]. DNA concentrations were measured with Qubit 2.0 Fluorometer (Invitrogen).
Total genomic DNA isolation
Cells were resuspended in 1 ml LB, split into two tubes and collected by centrifugation. Cells in each tube were resuspended in 50 μl of 30% sucrose in TE buffer by vigorous vortexing, combined with 350 μl of 2% SDS in TE, mixed by inversion and incubated at 65°C until the lysate was clear (5 minutes). Lysate was chilled on ice for 5 minutes, after which the following extractions were performed, with removal of the organic phase: 400 μl phenol, 400 μl phenol/chloroform and 400 μl chloroform. The final aqueous phase was transferred into fresh tube and precipitated with 40 μl of 5M NaCl and 1 ml Ethanol. DNA was dissolved in 500 μl of TE overnight, reprecipitated with 20 μl of 5M NaCl and 1 ml of ethanol and dissolved in 100 μl of TE.
Total plasmid DNA purification
Cells were resuspended in 1 ml LB, split into two tubes and collected by centrifugation. Cells in each tube were resuspended in 50 μl of 30% sucrose in TE buffer (by vigorous vortexing), combined with 350 μl of 2% SDS in TE, mixed by inversion and incubated at 65°C until the lysate was clear (5 minutes). 100 μl of 5 M NaCl was added to slightly cooled tube, mix thoroughly by inversion and put on ice for one hour. The solution was centrifuged at 16,000 g for 20 minutes. Supernatant was transferred to the new tube and 1 ml of ethanol was added. After mixing the solution by inversion, the tube was centrifuged for 5 minutes at 16,000 g. The pellet was dissolved in 20 μl of TE. 30 μl of 6M LiCl was added to the solution, and after vortexing the tube was incubated on ice for 15 min. Precipitant was centrifuged down for 3 minutes at 16,000 g, and the supernatant was transferred to a new tube. The DNA was precipitated with 150 μl ethanol and then reprecipitated from 50 μl TE volume after addition of 5 μl of 5M NaCl and 150 μl of Ethanol. DNA was dissolved in 50 μl of TE.
Quantification of ribodensity in plasmids with RNase HI and RNase HII enzymes
50 or 500 ng of purified DNA (total plasmid, or alkaline lysis, or total genomic preparation) were incubated in 20 μl of 1× Thermopol buffer (NEB) containing either no enzyme or 1.5-2.5 units of RNase HII (NEB). DNA was incubated with 2.5 units of RNase HI (Ambion or ThermoScientific) in 20 μl of 1× RNase HI buffer (ThermoScientific): 20 mM Tris HCl (pH 7.8), 40 mM KCl, 8 mM MgCl2, 1 mM DTT. Incubation was at 37°C for 30 minutes. After incubation the reaction mixtures were chilled on ice and directly loaded on 1.1 % agarose gels and run in 1×TAE buffer.
To remove potential (RNA-DNA) intermediates formed in plasmids, the DNA samples were treated in 30 μl of 70% formamide at 65°C for 5 minutes, chilled on ice, precipitated with 500 mM NaCl and ethanol, dried on air and dissolved in water and aliquotted into three 20 μl reactions for treatment with RNase HI or RNase HII.
DNA species were analyzed by Southern hybridization, the radioactive membranes were scanned by PhosphorImager (FujiFilm FLA-3000, Fuji). For calculation of DNA representing zero class of the Poisson distribution by the enzymatic method, radioactivity in supercoiled monomer band was divided by the total radioactivity in the lane area between the relaxed monomer band and the supercoiled monomer band.
Quantification of ribodensity in plasmids using alkali treatment assay
Plasmid DNA purified by the Birnboim protocol, was linearized with MluI restriction enzyme, precipitated and dissolved in water. Treatment was done in 20 μl reactions containing 0.3 M NaOH and 20 mM EDTA at 45°C for 90 minutes. After the treatment the tubes were transferred to ice and the content diluted with water to 0.2 M NaOH. To denature double stranded DNA without RNA hydrolysis, DNA was kept in 20 μl reactions with 0.2 M NaOH, 20 mM EDTA on ice for 20 minutes. DNA samples were run in 1.0% agarose gel in cold 1×TAE electrophoresis buffer and analyzed by Southern with subsequent scanning of the radioactive membranes by PhosphorImager (FujiFilm FLA-3000, Fuji). For calculating ribodensity, radioactivity in ssDNA band was divided by the total radioactivity in the lane area from ssDNA band down to the botton of the gel.
Southern analysis
Before the transfer, the agarose gels were gently rocked for 40 minutes in two volumes of 0.25 M HCl, then for 45 minutes in two volumes of 0.5 M NaOH, then for 15 minutes in two volumes of 1 M Tris HCl, pH8.0. DNA from treated agarose gels was transferred either by vacuum or by capillary transfer to Hybond-N+ nylon hybridization membrane (GE Healthcare). DNA probes were labeled by random hexamer priming with Exo-Resistant Random Primers (ThermoScientific). Hybridization was performed as described [102].
Chromosomal fragmentation assay
Cultures were shaken overnight at 28°C in LB. Next morning, cultures were diluted 200 times into 2 ml of LB and were shaken at 28°C to OD600 = 0.1. The cultures were diluted two-fold in 2 ml LB supplemented with 5 μCi/ml 32P-orthophosphoric acid, shifted to 37°C and shaken for 2 hours to OD600 ∼ 0.2-0.7. Cultures were diluted again from 20 to 200 times, depending on the density and the genotype, and continued to grow at 37°C for another three hours. To make a single agarose plug, 0.5 ml of the culture with the OD600 = 0.35 was processed according to the protocol. The chromosomal DNA preparation in agarose plugs, conditions for pulse-field gel electrophoresis and quantification of chromosomal breakage were as described [66].
For the RNase sensitivity tests, the agarose plugs after incubation with the lysis buffer overnight at 60°C were washed at room temperature three times with gentle agitation in 1 ml TE buffer for 20 min each, followed by 30 min incubation on ice in 200 μl of the appropriate 1× reaction buffer: Thermopol (NEB) (for RNase HII); 20 mM Tris-HCl (pH 7.8), 40 mM KCl, 8 mM MgCl2, 1 mM DTT (for RNase HI); 10 mM Tris-HCl, pH 8.0, 1 mM EDTA or 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.3 M NaCl (for RNase A). Then half of the plug was treated in 50 μl of fresh 1× buffer and with 25 units RNase HI (Ambion) or 12.5 units RNase HII (NEB) or heat-treated 100 μg/ml RNase A (Boehringer) for 4 hours at 37°C. The second half of the plug was incubated with the corresponding buffer as control.
Alkali and enzymatic cleavage of ribonucleotide-containing DNA duplex substrates
The sequences of the oligonucleotides with lowercase letters representing rNMPs: 38R1 5′-GAC TAC GTA CTG TTA CGG CTC GAT CAA TAC GGC AAT CaA GGC AGATCT GCC-3′, 20r 5′-gac uac gua cug uua cgg cu-3′, 34R5 5′-GAC TAC GTA CTG TTA CGG CTC GAT CAA TAC GGC Caa uca AGG CCA GAT CTG CC.
The oligonucleotides (synthetized by Eurofins Genomics) were 5′ end-radiolabeled with [γ-32P]-ATP (PerkinElmer, Inc. Waltham., MA, USA) and T4 polynucleotide kinase (PNK) (New England Biolabs) in 25 μl of 1× Buffer (70 mM Tris-HCl, 10 mM MgCl2, 5 mM DTT, pH 7.6 at 25°C), 60 pmol of the oligo, 2.5 μl [γ-32P]-ATP (activity 6000 Ci/mmole or 4500 Ci/mmole) and 10 units of T4 PNK at 37°C for 50 minutes. After the reaction, the oligos were cleaned with the GE Health kit Illustra ProbeQuant G-50 micro column (GE Healthcare, UK Ltd), diluted with DEPC-treated water and kept at -20°C.
Labeled 20-mer RNA or 52-mer DNA-RNA-DNA oligonucleotides were annealed with 2 molar excess of unlabeled complementary 52-mer DNA: (20 pmole/40 pmole) in 20 μl STE buffer (50 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1 mM EDTA) at room temperature for 20 minutes or at 70°C for 5 minutes followed by slow cooling of the heating block to room temperature. Products of hybridization were analyzed on a 12% or 15% nondenaturing polyacrylamide gel to estimate the annealing efficiency.
RNaseHI and RNaseHII cleavage of the substrates was done in 20 μl reactions as described for the plasmid relaxation assay. After the reaction, the samples were precipitated with tRNA dissolved in TE, mixed with 2× loading dye (97% formamide, 10 mM EDTA, 0.1% xylene cyanol, 0.1% bromphenol blue), heated at 70°C for 3 minutes and analyzed on 18% polyacrylamide gel containing 8M urea. Patterns of bands migrating in the gel was compared to those produced by partial digestion of oligos.
Partial alkaline hydrolysis of labeled oligonucleotides was carried out in 0.1 M NaOH, 12.5 mM EDTA at 45°C for 10 minutes or in sodium carbonate-sodium bicarbonate mix (10 μl/90 μl of 0.1 M solution each (pH 9.2 at 20°C)) in which 1 μl of labeled oligo was diluted with 4 μl of the above mix and incubated at 92°C for 3 minutes before loading on the gel. The radiolabelled products were visualized after scanning the gels with PhosphorImager (FujiFilm FLA-3000, Fuji.)
Optimization of the alkali hydrolysis conditions with a single ribonucleotide-containing duplex DNA substrate
To find optimal temperature, molarity of NaOH and time of treatment to cleave a single ribonucleotide embedded in DNA, we treated 0.5 pmole of 5′ end-radiolabeled 38-R1 oligonucleotide annealed to the complementary DNA template substrate mixed with 6 ng of cold 2 kbp DNA oriC-fragment in either 10 or 20 μl reactions containing NaOH and 5 mM EDTA. The concentrations of NaOH varied from 0.1 to 0.5 M, the incubation time varied from 30 minutes to overnight, the tested temperatures were: 0, 4, 16, 25, 37, 45 and 55°C. After the reaction, half of the sample was analyzed on 12% native polyacrylamide gel (to test rNMP hydrolysis), while the other half was run on 0.7% agarose in 1×TAE gel followed by Southern analysis with the oriC-probe to test the stability of DNA. The polyacrylamide gel was directly scanned by PhosphorImager (FujiFilm FLA-3000, Fuji). The extent of hydrolysis was estimated by taking the ratio of radioactivity present in the cleaved product relative to the total signal (both the uncleaved and the cleaved products together).
Marker frequency analysis
Five strains (L-159, L-215, L-485, L-486 and L-487) were shaken overnight at 28°C, diluted 100-fold in the morning in 10 ml LB and shaken at 28°C until OD600= 0.2. At this point, the cultures were split in half, and one set of 5 ml halves was supplemented with IPTG (1 mM final concentration) and was moved to 42°C along with the other set of 5 ml “NO IPTG” cultures. Growth was continued at 42°C with shaking for 2 more hours. Total DNA isolation, DNA preparation for the dot-blot and data processing were performed as described [81]. Equal DNA samples purified from each strain grown either under “No IPTG, 42°C” or “1 mM IPTG, 42°C” conditions were applied to three membranes. The membranes were hybridized with Sfi29R, Sfi30 and oriC-probes, respectively. Location of probes is indicated in Fig. S6, at the borders of the pEAK54 integration site. Sfi29R/ori and Sfi 30/ori signal values from 42°C cultures were normalized to the respective values calculated for a saturated culture of AB1157 and taken as 1.0.
Supplementary Material
Fig. S1. Nucleoid morphology in the long rnhAB mutant cells. DAPI staining of rnhAB cells reveals defective nucleoids in filamentous cells: nucleoid transcends the cell as a thin thread with bright spots (red arrows). The WT-like rnhAB cells have regular compact brightly stained and well separated nucleoids (navy arrows).
Fig. S2. Sensitivity of rapidly-growing cultures to 100 μg/ml of rifampicin. The treatment was at 30°C in liquid cultures followed by plating on LB without antibiotic. Strains: WT, AB1157; ΔrnhA, L-413; ΔrnhB, L-415; ΔrnhAB, L-416.
Fig. S3. Rifampicin sensitivity of wt and rnhB strains, and its partial suppression by addition of catalase to the medium. Rifampicine resistence of strains deficient in RNase HI. LB plates were supplemented with 2 μg/ml rifampicin ± 150 U/ml catalase and incubated at 28°C. Strains: WT, AB1157; ΔrnhA, L-413; ΔrnhB, L-415; ΔrnhAB, L-416.
Fig. S4. A representative pulsed-filed gel documenting chloramphenicol-induced chromosomal fragmentation. Cell cultures were treated with 100 μg/ml chloramphenicol at 28°C for two hours and the state of the chromosomal DNA was analyzed by PFGE.
Fig. S5. Survival of the dnaC and dnaA mutants and their rnh derivatives at the non-permissive temperatures on plates. Survival of the rnhAB double mutant (DnaA+) is shown as a control. Dilutions of isogenic derivatives were spotted on LB plates and incubated at nonpermissive temperature and then returned to 28°C.
Top: Survival of the dnaC2(Ts) mutants at 38°C. Data are averages of 1-3 independent measurements.
Strains: dnaC, L-393; dnaC rnhA, L-502; dnaC rnhB, L-503; dnaC rnhAB, L-504; rnhAB, L-416.
Bottom: Survival of the dnaA46(Ts) mutants at 42°C. Strains: dnaA, L-159; dnaA rnhA, L-483; dnaA rnhB, L-482; dnaA rnhAB, L-484; rnhAB, L-416.
Fig. S6. Testing functionality of ColE1 replicon placed in the chromosome of dnaA(Ts) rnh mutants. Schematic orientation of positioning of the ColE1-type replicon relative to oriC in the left replicore of the E. coli chromosome. Designations: yellow line, integrated plasmid DNA; ovals, replication origins; black rectangles, the hybridization probes; the red arrow shows the transcription-initiated DNA synthesis from the ColE1 replicon.
The graphs show the copy number (relative to oriC) of two regions around the integrated plasmid. All tested strains are isogenic derivatives of the dnaA46(Ts) strain. The cultures were grown for 2 hours at 42°C either without or with 1 mM IPTG in the media before DNA was extracted from cells for dot-blotting analysis. Strains: wt, no oriect; L-159, wt, L-215; rnhB, L-485; rnhA, L-486 and rnhA, L-487. Values are means of at least three independent measurements ± standard errors. The rnhB data are the averages of two experiments.
Table S1. Predicted substrates to result in double-strand breaks in DNA upon RNase treatment of the chromosomes in agarose plugs.
Table S2. Strains and plasmids used in this study.
Table S3. Oligonucleotides and primers.
Highlights.
Slow growth of RNaseH-deficient rnhAB mutant E. coli cannot be due to only R-loops
rnhAB mutants filament, induce SOS and fragment their chromosome, showing DNA stress
Replication from oriC helps rnhAB mutants, while initiation from R-loops kill them
DNA of rnhAB mutants accumulates R-patches, R-gaps, but not the expected R-tracts
The proposed events in RNase H-deficient cells: R-loops -> R-tracts -> R-gaps -> DSBs
Acknowledgments
We would like to thank all members of this laboratory for enthusiastic discussion of our results, and Sharik Khan for strains and plasmids. Bill Metcalf (this department) generously helped us with fluorescent microscopy an offered to use his microscope. We are grateful to Bénédicte Michel for her interest in this work and for helpful suggestions to clarify the presentation. This work was supported by grant # GM 073115 from the National Institutes of Health.
Glossary
- R-lesions
ribonucleotide(rN)-containing DNA lesions
- R-patch
a run of 1-3 consecutive rNs in one strand of duplex DNA
- R-tract
a run of 4 or more consecutive rNs in one strand of duplex DNA
- R-gap
a ss-gap in duplex DNA, in which the contiguous strand contains rNs
- TEC-free R-loop
R-loop not associated with transcription-elongation complex
- R-loop-aTEC
R-loop-anchored transcription-elongation complex
Footnotes
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li Y, Breaker RR. Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2′-Hydroxyl Group. J Am Chem Soc. 1999;121:5364–5372. [Google Scholar]
- 2.Levene PA. The structure of the yeast nucleic acid: IV. Ammonia hydrolysis. J Biol Chem. 1919;40:415–424. [Google Scholar]
- 3.Oivanen M, Kuusela S, Lönnberg H. Kinetics and Mechanisms for the Cleavage and Isomerization of the Phosphodiester Bonds of RNA by Brønsted Acids and Bases. Chem Rev. 1998;98:961–990. doi: 10.1021/cr960425x. [DOI] [PubMed] [Google Scholar]
- 4.DeRose EF, Perera L, Murray MS, Kunkel TA, London RE. Solution structure of the Dickerson DNA dodecamer containing a single ribonucleotide. Biochemistry. 2012;51:2407–2416. doi: 10.1021/bi201710q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egli M, Usman N, Rich A. Conformational influence of the ribose 2′-hydroxyl group: crystal structures of DNA-RNA chimeric duplexes. Biochemistry. 1993;32:3221–3237. [PubMed] [Google Scholar]
- 6.Ban C, Ramakrishnan B, Sundaralingam M. A single 2′-hydroxyl group converts B-DNA to A-DNA. Crystal structure of the DNA-RNA chimeric decamer duplex d(CCGGC)r(G)d(CCGG) with a novel intermolecular G-C base-paired quadruplet. J Mol Biol. 1994;236:275–285. doi: 10.1006/jmbi.1994.1134. [DOI] [PubMed] [Google Scholar]
- 7.Dalgaard JZ. Causes and consequences of ribonucleotide incorporation into nuclear DNA. Trends Genet. 2012;28:592–597. doi: 10.1016/j.tig.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Williams JS, Kunkel TA. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair. 2014;19:27–37. doi: 10.1016/j.dnarep.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- 10.Buckstein MH, He J, Rubin H. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J Bacteriol. 2008;190:718–726. doi: 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bochner BR, Ames BN. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J Biol Chem. 1982;257:9759–9769. [PubMed] [Google Scholar]
- 12.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 13.Yao NY, Schroeder JW, Yurieva O, Simmons LA, O'Donnell ME. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc Natl Acad Sci U S A. 2013;110:12942–12947. doi: 10.1073/pnas.1309506110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundström EB, et al. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Nat Acad Sci USA. 2010;107:4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kornberg A, Baker TA. DNA Replication. New York: W.H. Freeman and Company; 1992. p. 931. [Google Scholar]
- 17.Corn JE, Berger JM. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acid Res. 2006;34:4082–4088. doi: 10.1093/nar/gkl363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White RL, Hogness DS. R loop mapping of the 18S and 28S sequences in the long and short repeating units of Drosophila melanogaster rDNA. Cell. 1977;10:177–192. doi: 10.1016/0092-8674(77)90213-6. [DOI] [PubMed] [Google Scholar]
- 19.Thomas M, White RL, Davis RW. Hybridization of RNA to double-stranded DNA: formation of R-loops. Proc Natl Acad Sci USA. 1976;73:2294–2298. doi: 10.1073/pnas.73.7.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahba L, Amon JD, Koshland D, Vuica-Ross M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell. 2011;44:978–988. doi: 10.1016/j.molcel.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell. 2003;12:711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Polisky B. ColE1 replication control circuitry: sense from antisense. Cell. 1988;55:929. doi: 10.1016/0092-8674(88)90235-8. [DOI] [PubMed] [Google Scholar]
- 24.Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stuckey R, García-Rodríguez N, Aguilera A, Wellinger RE. Role for RNA:DNA hybrids in origin-independent replication priming in a eukaryotic system. Proc Natl Acad Sci USA. 2015;112:5779–5784. doi: 10.1073/pnas.1501769112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clayton DA. Transcription and replication of mitochondrial DNA. Hum Reprod. 2000;15(2):11–17. doi: 10.1093/humrep/15.suppl_2.11. [DOI] [PubMed] [Google Scholar]
- 27.Watt DL, Johansson E, Burgers PM, Kunkel TA. Replication of ribonucleotide-containing DNA templates by yeast replicative polymerases. DNA Repair. 2011;10:897–902. doi: 10.1016/j.dnarep.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clausen AR, Murray MS, Passer AR, Pedersen LC, Kunkel TA. Structure-function analysis of ribonucleotide bypass by B family DNA replicases. Proc Natl Acad Sci U S A. 2013;110:16802–16807. doi: 10.1073/pnas.1309119110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair. 2013;12:121–127. doi: 10.1016/j.dnarep.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA. RNA-templated DNA repair. Nature. 2007;447:338–341. doi: 10.1038/nature05720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohtani N, Haruki M, Morikawa M, Kanaya S. Molecular diversities of RNases H. J Biosci Bioeng. 1999;88:12–19. doi: 10.1016/s1389-1723(99)80168-6. [DOI] [PubMed] [Google Scholar]
- 32.Tadokoro T, Kanaya S. Ribonuclease H: molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 2009;276:1482–1493. doi: 10.1111/j.1742-4658.2009.06907.x. [DOI] [PubMed] [Google Scholar]
- 33.Schroeder JW, Randall JR, Matthews LA, Simmons LA. Ribonucleotides in bacterial DNA. Crit Rev Biochem Mol Biol. 2015;50:181–193. doi: 10.3109/10409238.2014.981647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crooke ST, Lemonidis KM, Neilson L, Griffey R, Lesnik EA, et al. Kinetic characteristics of Escherichia coli RNase H1: cleavage of various antisense oligonucleotide-RNA duplexes. Biochem J. 1995;312:599–608. doi: 10.1042/bj3120599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogrefe HH, Hogrefe RI, Walder RY, Walder JA. Kinetic analysis of Escherichia coli RNase H using DNA-RNA-DNA/DNA substrates. J Biol Chem. 1990;265:5561–5566. [PubMed] [Google Scholar]
- 36.Haruki M, Tsunaka Y, Morikawa M, Kanaya S. Cleavage of a DNA-RNA-DNA/DNA chimeric substrate containing a single ribonucleotide at the DNA-RNA junction with prokaryotic RNases HII. FEBS Lett. 2002;531:204–208. doi: 10.1016/s0014-5793(02)03503-2. [DOI] [PubMed] [Google Scholar]
- 37.Baaklini I, Hraiky C, Rallu F, Tse-Dinh YC, Drolet M. RNase HI overproduction is required for efficient full-length RNA synthesis in the absence of topoisomerase I in Escherichia coli. Mol Microbiol. 2004;54:198–211. doi: 10.1111/j.1365-2958.2004.04258.x. [DOI] [PubMed] [Google Scholar]
- 38.Drolet M, Phoenix P, Menzel R, Masse E, Liu LF, et al. Overexpression of RNase H partially complements the growth defect of an Escherichia coli ΔtopA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc Natl Acad Sci USA. 1995;92:3526–3530. doi: 10.1073/pnas.92.8.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itaya M, Crouch RJ. A combination of RNase H (rnh) and recBCD or sbcB mutations in Escherichia coli K12 adversely affects growth. Mol Gen Genet. 1991;277:424–432. doi: 10.1007/BF00273933. [DOI] [PubMed] [Google Scholar]
- 40.Dimude JU, Stockum A, Midgley-Smith SL, Upton AL, Foster HA, et al. The Consequences of Replicating in the Wrong Orientation: Bacterial Chromosome Duplication without an Active Replication Origin. MBio. 2015;6:e01294–01215. doi: 10.1128/mBio.01294-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maduike NZ, Tehranchi AK, Wang JD, Kreuzer KN. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics. Mol Microbiol. 2014;91:39–56. doi: 10.1111/mmi.12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Massy B, Fayet O, Kogoma T. Multiple origin usage for DNA replication in sdrA (rnh) mutants of Escherichia coli K-12: initiation in the absence of oriC. J Mol Biol. 1984;178:227–236. doi: 10.1016/0022-2836(84)90141-4. [DOI] [PubMed] [Google Scholar]
- 43.Ohtani N, Haruki M, Muroya A, Morikawa M, Kanaya S. Characterization of ribonuclease HII from Escherichia coli overproduced in a soluble form. J BIochem. 2000;127:895–899. doi: 10.1093/oxfordjournals.jbchem.a022684. [DOI] [PubMed] [Google Scholar]
- 44.Itaya M. Isolation and characterization of a second RNase H (RNase HII) of Escherichia coli K-12 encoded by the rnhB gene. Proc Nat Acad Sci USA. 1990;87:8587–8591. doi: 10.1073/pnas.87.21.8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rydberg B, Game J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc Nat Acad Sci USA. 2002;99:16654–16659. doi: 10.1073/pnas.262591699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohtani N, Tomita M, Itaya M. Junction ribonuclease: a ribonuclease HII orthologue from Thermus thermophilus HB8 prefers the RNA-DNA junction to the RNA/DNA heteroduplex. Biochem J. 2008;412:517–526. doi: 10.1042/BJ20080140. [DOI] [PubMed] [Google Scholar]
- 47.Ohtani N, Tomita M, Itaya M. Junction ribonuclease activity specified in RNases HII/2. FEBS J. 2008;275:5444–5455. doi: 10.1111/j.1742-4658.2008.06673.x. [DOI] [PubMed] [Google Scholar]
- 48.Vaisman A, McDonald JP, Noll S, Huston D, Loeb G, et al. Investigating the mechanisms of ribonucleotide excision repair in Escherichia coli. Mutat Res. 2014;761:21–33. doi: 10.1016/j.mrfmmm.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaisman A, McDonald JP, Huston D, Kuban W, Liu L, et al. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet. 2013;9:e1003878. doi: 10.1371/journal.pgen.1003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Groh M, Gromak N. Out of balance: R-loops in human disease. PLoS Genet. 2014;10:e1004630. doi: 10.1371/journal.pgen.1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Costantino L, Koshland D. The Yin and Yang of R-loop biology. Curr Opin Cell Biol. 2015;34:39–45. doi: 10.1016/j.ceb.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 54.Sollier J, Cimprich KA. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015;25:514–522. doi: 10.1016/j.tcb.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin JH, Kelman Z. The replicative helicases of bacteria, archaea, and eukarya can unwind RNA-DNA hybrid substrates. J Biol Chem. 2006;281:26914–26921. doi: 10.1074/jbc.M605518200. [DOI] [PubMed] [Google Scholar]
- 56.Vincent SD, Mahdi AA, Lloyd RG. The RecG branch migration protein of Escherichia coli dissociates R-loops. J Mol Biol. 1996;264:713–721. doi: 10.1006/jmbi.1996.0671. [DOI] [PubMed] [Google Scholar]
- 57.Voloshin ON, Camerini-Otero RD. The DinG protein from Escherichia coli is a structure-specific helicase. J Biol Chem. 2007;282:18437–18447. doi: 10.1074/jbc.M700376200. [DOI] [PubMed] [Google Scholar]
- 58.Hiraga S, Niki H, Ogura T, Ichinose C, Mori H, et al. Chromosome partitioning in Escherichia coli: novel mutants producing anucleate cells. J Bacteriol. 1989;171:1496–1505. doi: 10.1128/jb.171.3.1496-1505.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Itaya M, Omori A, Kanaya S, Crouch R, Tanaka T, et al. Isolation of RNase H genes that are essential for growth of Bacillus subtilis 168. J Bacteriol. 1999;181:2118–2123. doi: 10.1128/jb.181.7.2118-2123.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leela JK, Syeda AH, Anupama K, Gowrishankar J. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc Natl Acad Sci U S A. 2013;110:258–263. doi: 10.1073/pnas.1213123110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernández de Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, et al. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- 63.Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage l. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ossanna N, Mount DW. Mutations in uvrD induce the SOS response in Escherichia coli. J Bacteriol. 1989;171:303–307. doi: 10.1128/jb.171.1.303-307.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kogoma T, Hong X, Cadwell GW, Barnard KG, Asai T. Requirement of homologous recombination functions for viability of the Escherichia coli cells that lacks RNase HI and exonuclease V activities. Biochimie. 1993;75:89–99. doi: 10.1016/0300-9084(93)90029-r. [DOI] [PubMed] [Google Scholar]
- 66.Kouzminova EA, Rotman E, Macomber L, Zhang J, Kuzminov A. RecA-dependent mutants in E. coli reveal strategies to avoid replication fork failure. Proc Natl Acad Sci USA. 2004;101:16262–16267. doi: 10.1073/pnas.0405943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu CW, Tweedy N. Mechanistic aspects of promoter binding and chain initiation by RNA polymerase. Mol Cell Biochem. 1982;47:129–149. doi: 10.1007/BF00229597. [DOI] [PubMed] [Google Scholar]
- 69.Reid P, Speyer J. Rifampicin inhibition of ribonucleic acid and protein synthesis in normal and ethylenediaminetetraacetic acid-treated Escherichia coli. J Bacteriol. 1970;104:376–389. doi: 10.1128/jb.104.1.376-389.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Humphrey TJ. The synergistic inhibition of Campylobacter jejuni by rifampicin and hydrogen peroxide. Lett Applied Microbiol. 1990;10:97–100. [Google Scholar]
- 71.Massé E, Drolet M. R-loop-dependent hypernegative supercoiling in Escherichia coli topA mutants preferentially occurs at low temperatures and correlates with growth inhibition. J Mol Biol. 1999;294:321–332. doi: 10.1006/jmbi.1999.3264. [DOI] [PubMed] [Google Scholar]
- 72.Siller E, DeZwaan DC, Anderson JF, Freeman BC, Barral JM. Slowing bacterial translation speed enhances eukaryotic protein folding efficiency. J Mol Biol. 2010;396:1310–1318. doi: 10.1016/j.jmb.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 73.Agarwal D, Gregory ST, O'Connor M. Error-prone and error-restrictive mutations affecting ribosomal protein S12. J Mol Biol. 2011;410:1–9. doi: 10.1016/j.jmb.2011.04.068. [DOI] [PubMed] [Google Scholar]
- 74.Fukuoh A, Iwasaki H, Ishioka K, Shinagawa H. ATP-dependent resolution of R- loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction-specific helicase. EMBO J. 1997;16:203–209. doi: 10.1093/emboj/16.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pomerantz RT, O'Donnell M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008;456:762–766. doi: 10.1038/nature07527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lark KG, Lark C. Regulation of chromosome replication in Escherichia coli: a comparison of the effects of phenethyl alcohol treatment with those of amino acid starvation. J Mol Biol. 1966;20:9–19. doi: 10.1016/0022-2836(66)90113-6. [DOI] [PubMed] [Google Scholar]
- 77.Gil D, Bouché JP. ColE1-type vectors with fully repressible replication. Gene. 1991;105:17–22. doi: 10.1016/0378-1119(91)90508-9. [DOI] [PubMed] [Google Scholar]
- 78.Khan SR, Mahaseth T, Kouzminova EA, Cronan G, Kuzminov A. Static and dynamic factors limit chromosomal replication complexity in Escherichia coli, avoiding dangers of runaway overreplication. Genetics. 2016;202:945–960. doi: 10.1534/genetics.115.184697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dasgupta S, Masukata H, Tomizawa J. Multiple mechanisms for initiation of ColE1 DNA replication: DNA synthesis in the presence and absence of ribonuclease H. Cell. 1987;51:1113–1122. doi: 10.1016/0092-8674(87)90597-6. [DOI] [PubMed] [Google Scholar]
- 80.Budke B, Kuzminov A. Production of clastogenic DNA precursors by the nucleotide metabolism in Escherichia coli. Mol Microbiol. 2010;75:230–245. doi: 10.1111/j.1365-2958.2009.06994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kouzminova EA, Kuzminov A. Fragmentation of replicating chromosomes triggered by uracil in DNA. J Mol Biol. 2006;355:20–33. doi: 10.1016/j.jmb.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 82.Edy VG, Szekely M, Loviny T, Dreyer C. Action of nucleases on double-stranded RNA. Eur J Biochem. 1976;61:563–572. doi: 10.1111/j.1432-1033.1976.tb10051.x. [DOI] [PubMed] [Google Scholar]
- 83.Kuzminov A. The Precarious Prokaryotic Chromosome. J Bacteriol. 2014;196:1793–1806. doi: 10.1128/JB.00022-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fukushima S, Itaya M, Kato H, Ogasawara N, Yoshikawa H. Reassessment of the in vivo functions of DNA polymerase I and RNase H in bacterial cell growth. J Bacteriol. 2007;189:8575–8583. doi: 10.1128/JB.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amon JD, Koshland D. RNase H enables efficient repair of R-loop induced DNA damage. Elife. 2016;5:e20533. doi: 10.7554/eLife.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O'Connell K, Jinks-Robertson S, Petes TD. Elevated Genome-Wide Instability in Yeast Mutants Lacking RNase H Activity. Genetics. 2015;201:963–975. doi: 10.1534/genetics.115.182725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang SN, Williams JS, Arana ME, Kunkel TA, Pommier Y. Topoisomerase I-mediated cleavage at unrepaired ribonucleotides generates DNA double-strand breaks. EMBO J. 2017;36:361–373. doi: 10.15252/embj.201592426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harinarayanan R, Gowrishankar J. Host factor titration by chromosomal R-loop as a mechanism for runaway plasmid replication in transcription termination-defective mutants of Escherichia coli. J Mol Biol. 2003;332:31–46. doi: 10.1016/s0022-2836(03)00753-8. [DOI] [PubMed] [Google Scholar]
- 89.Chattoraj DK, Stahl FW. Evidence of RNA in D loops of intracellular lambda DNA. Proc Natl Acad Sci U S A. 1980;77:2153–2157. doi: 10.1073/pnas.77.4.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wahba L, Gore SK, Koshland D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife. 2013;2:e00505. doi: 10.7554/eLife.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.von Hippel PH, Rees WA, Rippe K, Wilson KS. Specificity mechanisms in the control of transcription. Biophys Chem. 1996;59:231–246. doi: 10.1016/0301-4622(96)00006-3. [DOI] [PubMed] [Google Scholar]
- 92.Gan W, Guan Z, Liu J, Gui T, Shen K, et al. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011;25:2041–2056. doi: 10.1101/gad.17010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve-Grez H, et al. Topoisomeras I suppresses genomic instability by preventing interference between replication an transcription. Nat Cell Biol. 2009;11:1315–1324. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Asai T, Kogoma T. D-loops and R-loops: alternative mechanisms for the initiatio of chromosome replication in Escherichia coli. J Bacteriol. 1994;176:1807–1812. doi: 10.1128/jb.176.7.1807-1812.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pileur F, Toulme JJ, Cazenave C. Eukaryotic ribonucleases HI and HII generate characteristic hydrolytic patterns on DNA-RNA hybrids: further evidence that mitochondrial RNase H is an RNase HII. Nucleic Acids Res. 2000;28:3674–3683. doi: 10.1093/nar/28.18.3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, et al. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res. 2013;41:3130–3143. doi: 10.1093/nar/gkt027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. p. 466. [Google Scholar]
- 98.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006. doi: 10.1038/msb4100050. 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kuzminov A, Schabtach E, Stahl FW. Study of plasmid replication in Escherichia coli with a combination of 2D gel electrophoresis and electron microscopy. J Mol Biol. 1997;268:1–7. doi: 10.1006/jmbi.1997.0955. [DOI] [PubMed] [Google Scholar]
- 101.Birnboim HC. A rapid alkaline extraction method for the isolation of plasmid DNA. Methods Enzymol. 1983;100:243–255. doi: 10.1016/0076-6879(83)00059-2. [DOI] [PubMed] [Google Scholar]
- 102.Kuzminov A, Schabtach E, Stahl FW. c-sites in combination with RecA protein increase the survival of linear DNA in E. coli by inactivating ExoV activity of RecBCD nuclease. EMBO J. 1994;13:2764–2776. doi: 10.1002/j.1460-2075.1994.tb06570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xiong L, Kloss P, Douthwaite S, Andersen NM, Swaney S, et al. Oxazolidinone resistance mutations in 23S rRNA of Escherichia coli reveal the central region of domain V as the primary site of drug action. J Bacteriol. 2000;182:5325–5331. doi: 10.1128/jb.182.19.5325-5331.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Nucleoid morphology in the long rnhAB mutant cells. DAPI staining of rnhAB cells reveals defective nucleoids in filamentous cells: nucleoid transcends the cell as a thin thread with bright spots (red arrows). The WT-like rnhAB cells have regular compact brightly stained and well separated nucleoids (navy arrows).
Fig. S2. Sensitivity of rapidly-growing cultures to 100 μg/ml of rifampicin. The treatment was at 30°C in liquid cultures followed by plating on LB without antibiotic. Strains: WT, AB1157; ΔrnhA, L-413; ΔrnhB, L-415; ΔrnhAB, L-416.
Fig. S3. Rifampicin sensitivity of wt and rnhB strains, and its partial suppression by addition of catalase to the medium. Rifampicine resistence of strains deficient in RNase HI. LB plates were supplemented with 2 μg/ml rifampicin ± 150 U/ml catalase and incubated at 28°C. Strains: WT, AB1157; ΔrnhA, L-413; ΔrnhB, L-415; ΔrnhAB, L-416.
Fig. S4. A representative pulsed-filed gel documenting chloramphenicol-induced chromosomal fragmentation. Cell cultures were treated with 100 μg/ml chloramphenicol at 28°C for two hours and the state of the chromosomal DNA was analyzed by PFGE.
Fig. S5. Survival of the dnaC and dnaA mutants and their rnh derivatives at the non-permissive temperatures on plates. Survival of the rnhAB double mutant (DnaA+) is shown as a control. Dilutions of isogenic derivatives were spotted on LB plates and incubated at nonpermissive temperature and then returned to 28°C.
Top: Survival of the dnaC2(Ts) mutants at 38°C. Data are averages of 1-3 independent measurements.
Strains: dnaC, L-393; dnaC rnhA, L-502; dnaC rnhB, L-503; dnaC rnhAB, L-504; rnhAB, L-416.
Bottom: Survival of the dnaA46(Ts) mutants at 42°C. Strains: dnaA, L-159; dnaA rnhA, L-483; dnaA rnhB, L-482; dnaA rnhAB, L-484; rnhAB, L-416.
Fig. S6. Testing functionality of ColE1 replicon placed in the chromosome of dnaA(Ts) rnh mutants. Schematic orientation of positioning of the ColE1-type replicon relative to oriC in the left replicore of the E. coli chromosome. Designations: yellow line, integrated plasmid DNA; ovals, replication origins; black rectangles, the hybridization probes; the red arrow shows the transcription-initiated DNA synthesis from the ColE1 replicon.
The graphs show the copy number (relative to oriC) of two regions around the integrated plasmid. All tested strains are isogenic derivatives of the dnaA46(Ts) strain. The cultures were grown for 2 hours at 42°C either without or with 1 mM IPTG in the media before DNA was extracted from cells for dot-blotting analysis. Strains: wt, no oriect; L-159, wt, L-215; rnhB, L-485; rnhA, L-486 and rnhA, L-487. Values are means of at least three independent measurements ± standard errors. The rnhB data are the averages of two experiments.
Table S1. Predicted substrates to result in double-strand breaks in DNA upon RNase treatment of the chromosomes in agarose plugs.
Table S2. Strains and plasmids used in this study.
Table S3. Oligonucleotides and primers.