

Abstract
Paramagnetic relaxation enhancement is an NMR technique that has yielded important insight into the structure of folded proteins, although the perturbation introduced by the large spin probe might be thought to diminish its usefulness when applied to characterizing the structural ensembles of intrinsically disordered proteins (IDPs). We compare the computationally generated structural ensembles of the IDP amyloid-β42 (Aβ42) to an alternative sequence in which a nitroxide spin label attached to cysteine has been introduced at its N-terminus. Based on this internally consistent computational comparison, we find that the spin label does not perturb the signature population of the β-hairpin formed by residues 16–21 and 29–36 that is dominant in the Aβ42 reference ensemble. However, the presence of the tag induces a strong population shift in a subset of the original Aβ42 structural sub-populations, including a sevenfold enhancement of the β-hairpin formed by residues 27–31 and 33–38. Through back-calculation of NMR observables from the computational structural ensembles, we show that the structural differences between the labeled and unlabeled peptide would be evident in local residual dipolar couplings, and possibly differences in homonuclear 1H-1H nuclear Overhauser effects (NOEs) and heteronuclear 1H-15N NOEs if the paramagnetic contribution to the longitudinal relaxation does not suppress the NOE intensities in the real experiment. This work shows that molecular simulation provides a complementary approach to resolving the potential structural perturbations introduced by reporter tags that can aid in the interpretation of paramagnetic relaxation enhancement, double electron-electron resonance, and fluorescence resonance energy transfer experiments applied to IDPs.
Introduction
Intrinsically disordered proteins (IDPs) are an important class of proteins that play a significant role in cellular functions as well as a deleterious role in diseases (1, 2, 3). They confound the structure-function paradigm since they do not have a single dominant tertiary structure but instead sample between multiple tertiary conformations in solution. Paramagnetic relaxation enhancement (PRE) experiments are a widely used NMR technique employed to provide information about long-range order in both folded proteins (4, 5, 6) and more recently in IDP structural ensembles (7, 8, 9, 10, 11, 12, 13, 14, 15, 16). It requires the introduction of a nitroxide spin label into the peptide of interest via a covalent bond to cysteine residues, a technique commonly known as site-directed spin labeling (17). If the protein doesn’t have a cysteine residue, then cysteine is added to the sequence either as a point mutation or as an additional residue. The benefit of adding the label is that the unpaired electron spin in the PRE tag causes additional dipole-dipole interactions that result in line broadening in the NMR spectrum, from which information about distances up to 25 Å between the probe and a desired site can be extracted.
Of course, the PRE tag attached to the cysteine residue is a relatively large side-group perturbation relative to the original sequence, whose structural consequences must be carefully quantified to extract useful structural information about the unlabeled system. For well-folded proteins, most available sequence sites are able to accommodate the addition of the spin label without any significant structural changes (4, 5, 18, 19, 20). On the other hand, IDPs are remarkably different from well-folded proteins because of their structural plasticity, such that it might be expected that point mutations, an addition of a single residue, or introduction of a spin label, might bring about extensive structural changes in the IDP ensemble. At present, however, most experiments on IDPs are analyzed under the assumption that the probe has only a localized effect on structure and dynamics, and that the corresponding long-range features of the untagged ensemble are minimally perturbed. The motivation behind this work is to have a better understanding of the effect of probe attachment to an IDP based on a comparison of its structural ensemble to the same IDP structural ensemble without a label, so that the PRE experiments on IDPs are interpreted correctly.
Here, we study the effects of the PRE spin label on the structural dynamics of the amyloid-β peptide (Aβ42), a 42-residue peptide that is the major molecular player in Alzheimer’s disease (21, 22). It has been widely studied both experimentally (11, 12, 13, 21, 23, 24, 25) and computationally (26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37) and the results show that the monomeric form of Aβ42 is classified as an IDP, sampling among an extensive set of conformations in solution. We chose Aβ42 for our study because our previous computational work has characterized its structural ensemble extensively (34, 35, 36) and we have made thorough comparisons to the experimental and computational work of others (23, 38, 39, 40). In this work, we have combined two different sampling techniques—replica-exchange molecular dynamics (41) (REMD) and temperature cool walking (42, 43) (TCW) to generate structural ensemble averages with error bars that take into account sampling uncertainty. We have performed two independent simulations of the untagged monomeric Aβ42 peptide and a tagged version of the Aβ42 peptide in which we add a cysteine residue to the beginning of the N-terminus to which we attach the methanethiosulfonate spin label (MTSL) (Fig. 1), a commonly used spin label for PRE experiments. Any structural differences between the computationally generated ensembles of Aβ42 and MTSL-Cys-Aβ42 peptides that falls outside sampling uncertainty would then allow us to determine whether the spin label changes the IDP structural characteristics relative to the reference ensemble in any significant way.
Figure 1.
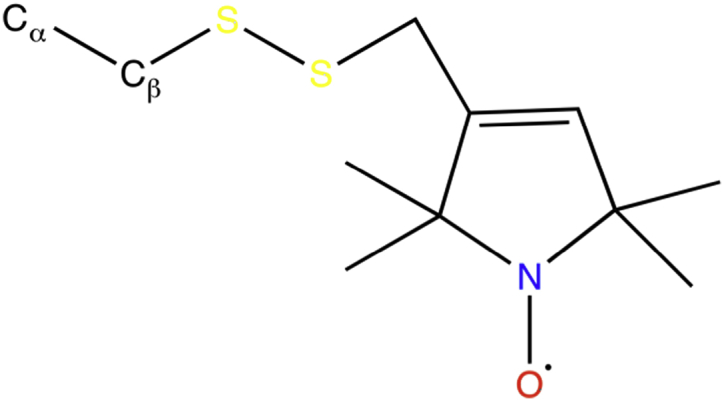
Schematic of MTSL. MTSL is a commonly used PRE tag because of its sulfhydryl specificity and side-chain flexibility. The oxygen atom is the paramagnetic center. To see this figure in color, go online.
From this internally consistent computational comparison we find that most of the important structural features of the untagged Aβ42 ensemble have been preserved, such as the dominant β-hairpin between the central hydrophobic cluster (CHC) comprising residues 16–21 and the C-terminal residues 29–36 (34, 35, 36), which is likely critical for subsequent fibril formation. However, the spin label introduces strong population shifts toward greater enhancement of what were originally lightly populated β-hairpins in the C-terminal region, including a sevenfold enhancement of the β-hairpin formed by residues 27–31 and 33–38 that is largely driven by the hydrophobic character of the MTSL label. Through back-calculation of many types of NMR observables, our computational results would be supported by differences in residual dipolar couplings, and possibly differences in homonuclear 1H-1H nuclear Overhauser effects (NOEs) and heteronuclear 1H-15N NOEs, if the paramagnetic contribution to the longitudinal relaxation does not suppress the NOE intensities of these nuclei in the real experiment. We conclude that PRE experiments can be used to answer structural and mechanistic questions regarding IDP ensembles, when carefully interpreted with the aid of additional experiments combined with high-quality molecular simulations.
Methods
Here, we report briefly on the simulation protocol and analysis methods; further details can be found in previous publications (34, 35, 36, 42).
REMD simulation protocol
All simulation setup and production runs utilized Amber14 (44). For addition of the PRE-tag, a Cys residue was first added to the N-terminus of the Aβ42 sequence, and the MTSL spin label was then attached through a disulfide bond at the Cβ position of Cys0 of the Cys-Aβ42 peptide. The Cys residue was introduced in the N-terminus to be consistent with the PRE experimental protocol reported by Fawzi et al. (11). The peptides were modeled using the Amberff99sb force field (45, 46) and the water molecules using the TIP4P-Ew water model (47). The parameters for the MTSL tag were generated using the CHAMBER module of Amber by Xue and Skrynnikov (48), a protocol that yields a consistent force field with the standard unlabeled peptide.
We created two extended configurations for Aβ42 and the MTSL-Cys-Aβ42 peptides, and the peptides were subsequently minimized. The initial simulation boxes were ∼700 nm3 in volume, and 3 and 4 Na+ atoms were added to the simulation box to neutralize the peptide for the untagged Aβ42 and MTSL-Cys-Aβ42, respectively. The starting configurations were heated to 287 K at constant volume and equilibrated for 2 ns under constant pressure of 1 bar, to achieve the correct density. This was followed by a 2 ns high-temperature simulation at 500 K to obtain a more collapsed peptide. Two distinct collapsed states were chosen for each peptide to start two independent REMD simulations. The peptides were resolvated to obtain cubic simulation boxes of 239 and 257 nm3 for Aβ42 and MTSL-Cys-Aβ42 peptides, respectively. The final numbers of water molecules for the Aβ42 and MTSL-Cys-Aβ42 boxes were 7800 and 8372, respectively.
The REMD module of the Amber14 MD package was used to generate the structural ensembles. Fifty-eight replicas were used in the temperature range 287–450 K with an exchange probability of 18–22% and an exchange attempt of 0.5 ps. The production runs were performed in NVT ensemble using a 1 fs time step and with constraints on the heavy-atom hydrogen covalent bonds. A Langevin thermostat was used to regulate the temperature. Long- range electrostatic forces were calculated using particle-mesh Ewald with a 9.0 Å cutoff for the real-space electrostatics and Lennard-Jones forces. The total length of each simulation was 100 ns, with the first 50 ns being discarded as equilibration; the final Boltzmann weighted ensemble consisted of 10,000 structures from the final 50 ns of the two independent equilibrated simulations at 287 K.
TCW simulation protocol
TCW is a non-equilibrium alternative to REMD, using only two temperature replicas to generate a Boltzmann-weighted ensemble (43). Structures from the high temperature are sequentially cooled to the low temperature and detailed balance is satisfied based on corresponding heating runs that together comprise a complete Metropolis acceptance criterion. TCW has been shown in a previous publication to converge faster for small peptides such as alanine dipeptide, met-enkephalin, and also the Aβ42 peptide (42). The same equilibrated starting structures for REMD simulations were chosen for the TCW method. The high- and low-temperature replicas were at 456.2 and 287 K with 50 intermediate annealing steps. Temperature was maintained using an Andersen thermostat (49). Long-range electrostatic forces were calculated using Ewald, with a cutoff of 9.5 Å for the real-space electrostatics and Lennard-Jones forces. The frequency of exchange attempt was 500 fs. Similar to the REMD simulations, two independent simulations were performed for 100 ns with only the last 50 ns being used for analysis.
Trajectory analysis
Both the cpptraj module of Amber and our own in-house codes for evaluating NMR observables were used for analyzing the structural ensemble properties of Aβ42 and MTSL-Cys-Aβ42. For the analysis, the cysteine residue of the modified MTSL-Cys-Aβ42 peptide will be referred to as Residue 0. The radius of gyration (Rg) was calculated using backbone heavy atoms for residues Asp1–Ala42; secondary structures were assigned using the Dictionary of Secondary Structure Prediction criterion; and hydrogen bonds were calculated using distance cutoffs of 3.5 and 4.0 Å, respectively, between heavy atoms and a 60° angle cutoff for both.
Calculation of NMR observables
Full details for back calculation of NMR observables have been reported in previous publications, including chemical shifts from ShiftX2 (50), J-coupling constants (51), residual dipolar couplings (RDCs) based on local (52) and global alignments (53), and 1H-1H and 1H-15N NOEs (34, 35) from the 287 K structural ensembles and MD correlation (34, 35) times for both the tagged and untagged peptide. We give a brief summary here for homonuclear and heteronuclear NOE intensities.
We performed short NVE simulations to calculate the spectral density functions (which are related to the NOE peak intensity). The normalized correlation function for each pair of hydrogen atoms in the Aβ peptides is calculated using the equation
| (1) |
where r(t) is the vector between each hydrogen-atom pair at time t, χt,t + τ is the angle between the r(t) and r(t + τ) vectors, and P2 is the second-order Legendre polynomial. Each correlation function is then fitted to a multi-exponential form over a 5 ns range to obtain τ.
| (2) |
with N = 1, 2, 3, or 4, and
The resulting time correlation function is then Fourier-transformed,
| (3) |
to obtain
| (4) |
where τi corresponds to the correlation time constant, τi.
The final step in homonuclear NOE calculation is to obtain the relaxation matrix R, composed of diagonal elements,
| (5) |
and off-diagonal elements,
| (6) |
The intensity obtained for each nucleus in the NOE spectra directly relates to the magnitudes in the matrix R.
A method similar to that employed above was used to calculate the spectral density function for each pair of H-N atoms for the H-N backbone bond for each residue in Aβ42. The steady-state NOE enhancement factor of the 15N spin by the 1H NOE was then estimated using the equation
| (7) |
where γH and γN are the gyromagnetic ratios of 1H and 15N, respectively. The 1H-15N cross-relaxation rate constant is defined as
| (8) |
and the 15N self-relaxation as
| (9) |
where ω0,H is the Larmor frequency of 1H and ω0,N is the Larmor frequency of 15N. The constant factor K is given as
| (10) |
where is Planck’s constant and μ0 is the permeability of free space.
Results
The characterization of an IDP structural ensemble is a highly underdetermined problem when compared to that of the folded-protein class, and thus, it is paramount to quantify the errors in computational reproducibility through independent ensemble calculations of the same IDP sequence. This is necessary to ensure that a fair comparison is made as to whether there are meaningful structural ensemble differences between the IDP Aβ42 and MTSL-Cys-Aβ42 peptides that arise from perturbations due to the spin label versus what arises from intrinsic uncertainties due to sampling limitations of the IDP ensemble. Although errors in force fields may contribute to direct agreement with experiment, use of the same protein and water force fields (Amberff99sb force field (45, 46) and TIP4P-Ew water model (47)) provides an internally consistent comparison among the computationally generated ensembles.
We therefore have performed two independent de novo MD simulations of the Aβ42 and MTSL-Cys-Aβ42 peptides using standard REMD (54, 55, 56) and also two independent TCW simulations of the two peptides in this work. Together, these independent ensembles provide a measure of reproducibility between computationally generated IDP ensembles, such that the standard deviation among them defines the uncertainty due to the sampling protocol. With the calculated model uncertainties thus defined as a standard deviation in all properties of the Aβ42 reference ensemble, any structural ensemble differences that are larger than this intrinsic sampling error when comparing against the MTSL-Cys-Aβ42 ensemble would thus define a genuine perturbation to the structural sub-populations due to the spin label.
Fig. 2 shows the normalized probability density distributions of the radius of gyration (Rg) in the Aβ42 and MTSL-Cys-Aβ42 ensemble. The mean and STD values in the Rg for the Aβ42 averaged ensembles and the MTSL-Cys-Aβ42 ensembles are 12.1 ± 2.1 and 11.6 ± 1.3 Å, respectively. All untagged Aβ42 ensembles are qualitatively the same in the sense that each exhibits a longer tail in the Rg distribution compared to the tagged peptide. The MTSL-Cys-Aβ42 ensemble has slightly more compact distribution than the Aβ42 ensemble, but there is enough uncertainty in the Rg estimates that we need to consider other types of analysis of their structural characteristics to quantify the effect of the spin label on the observed sub-populations of structure.
Figure 2.
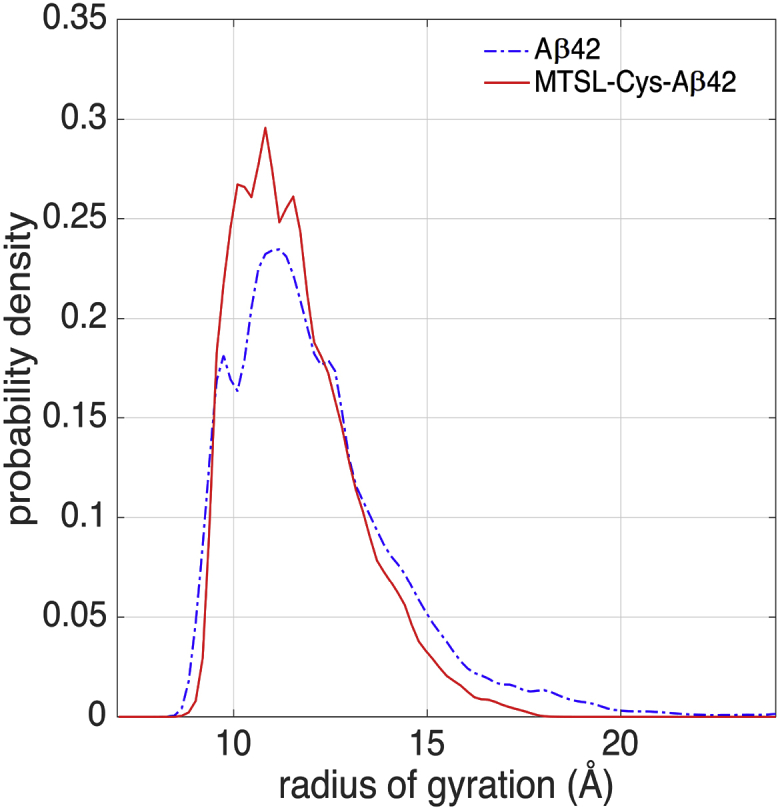
Probability distribution of the radius of gyration of the simulated Aβ42 (dash-dotted line) and MTSL-Cys-Aβ42 (solid line) ensembles. To see this figure in color, go online.
Fig. 3 shows the propensities by residue for each peptide to form helical structures (α-helix, 3–10 helix, and π-helix), β-structure (intramolecular β-bridges, β-hairpins, or β-sheets), and localized turns (with and without hydrogen bonds), as averages over their conformational ensembles. Table 1 provides the subset of secondary-structure classifications in Fig. 3 that are supported by internal hydrogen bonds (according to the Dictionary of Secondary Structure Prediction (57)), which would likely comprise secondary-structure sub-populations that are typically more populated due to greater Boltzmann weighting. It is important to note we have combined populations of turns and helices at the same sequence positions since any helical structure is typically localized to only one turn.
Figure 3.

Percentage of simulated Aβ42 (dotted line) and MTSL-Cys-Aβ42 (solid line) ensembles involved in different types of secondary structure: α-helix (top), β-bridges or β−strands (middle), and turns (bottom). Standard deviations are based on differences between the two ensembles generated from REMD and TCW sampling. To see this figure in color, go online.
Table 1.
Comparison of Important Secondary Structures Present in the Aβ42 and MTSL-Cys-Aβ42 Ensemble
| Sequence Region | Secondary Structure % | Aβ42 | MTSL-Cys-Aβ42 |
|---|---|---|---|
| Localized secondary structure (helices and turns) for N-terminal residues 1–15 | turn 1–2/turn 2–3∗ | 4.4 ± 1.5∗ | 23.3 ± 2.3∗ |
| turn 3–4/turn 5–6 | 7.6 ± 5.3 | 10.0 ± 5.0 | |
| turn 7–8/turn 8–9 | 36.0 ± 17.4 | 32.6 ± 25.0 | |
| turn 9–10/turn 10–11 | 14.4 ± 4.7 | 15.6 ± 0.2 | |
| turn 11–12 | 7.4 ± 4.0 | 7.4 ± 1.1 | |
| turn 12–13/turn 13–14 | 12.2 ± 1.2 | 12.6 ± 4.7 | |
| turn/helix 14–15 | 24.4 ± 11.4 | 9.3 ± 3.4 | |
| turn 17–18 | 3.6 ± 0.6 | 12.8 ± 13.8 | |
| turn 21–22/turn 22–23 | 5.0 ± 2.7 | 11.6 ± 2.5 | |
| Localized secondary structure (helices and turns) for central residues 16–30 | turn 23–24/turn 24–25 | 10.7 ± 2.0 | 22.8 ± 9.5 |
| turn/helix 25–26 | 21.0 ± 4.4 | 21.2 ± 15.7 | |
| turn/helix 26–27 | 22.8 ± 4.7 | 12.8 ± 4.4 | |
| turn 29–30 | 10.8 ± 5.0 | 4.6 ± 2.4 | |
| Localized secondary structure (helices and turns) for C-terminal residues 31–42 | turn 30–31/turn 31–32∗ | 15.8 ± 1.6∗ | 3.8 ± 0.2∗ |
| turn 32–33∗ | 7.3 ± 1.3∗ | 45.6 ± 1.6∗ | |
| turn 33–34∗ | 9.3 ± 1.8∗ | 3.2 ± 2.2∗ | |
| helix 32–33/helix 33–34 | 6.8 ± 1.1 | 12.3 ± 8.1 | |
| helix 34–35 | 6.4 ± 1.9 | 8.4 ± 0.8 | |
| helix 35–36 | 3.2 ± 1.1 | 6.1 ± 0.6 | |
| turn 36–37 | 6.6 ± 0.5 | 5.2 ± 0.1 | |
| Residues involved in β-strand pairings | turn 37–38 | 6.6 ± 1.9 | 9.3 ± 6.7 |
| β-strands 3–6 and 31–41 | 0.5 ± 0.7 | 6.6 ± 8.6 | |
| β-strands 16–21 and 29–36 | 8.7 ± 6.6 | 17.8 ± 13.8 | |
| β-strands 27–31 and 33–38∗ | 5.0 ± 4.3∗ | 38.0 ± 6.9∗ | |
| β-strands 34–36 and 39–40 | 2.2 ± 1.5 | 5.4 ± 6.8 |
The asterisks indicate the structural features whose populations differ in the untagged and tagged ensembles by a larger standard deviation than that observed for REMD and TCW ensembles of the Aβ42 ensemble.
When Table 1 and Fig. 3 are considered together, it is evident that many of the secondary-structure sub-populations are quantitatively preserved between the Aβ42 and MTSL-Cys-Aβ42 ensemble. The reference peptide has a large number of highly populated turns and helices throughout the sequence (Fig. 3), whose population percentages are almost completely explained by their stabilization due to internal hydrogen bonds (Table 1). The most prominent β-hairpin structure in the original Aβ42 ensemble is formed by β-strand pairings between the CHC and residues 29–36, sometimes stabilized by turns in the residue region Ala21–Gly25, such that the overall percentage of this β-hairpin averaged across all untagged Aβ42 ensembles is ∼9% (34, 35, 36). Although we find that the MTSL-Cys-Aβ42 structural ensemble stabilizes this β-hairpin at close to 18%, this population of β-hairpin is largely preserved within statistical uncertainty compared to the reference ensemble. Even so, this β-hairpin is not present in the simulated Aβ40 ensemble, and so the fact that it is retained in the labeled Aβ42 peptide is encouraging for preserving one of the primary structural differences between the two Aβ peptides.
However, there are some substantial shifts from the sub-population percentages seen for the unlabeled peptide in the C-terminal regions of the sequence due to the spin label. The reference Aβ42 ensemble exhibits two additional small sub-populations, 1) an anti-parallel β-strand pairing between residues 27–31 and 33–38 in ∼5% of the ensemble, and 2), an N-terminal turn 1–2/2–3 present in ∼4% of the ensemble. The percentages within these sub-populations shift dramatically in the presence of the MTSL-Cys addition to the N-terminus. Although the perturbation in the turn population in the N-terminus might be expected, what is the most striking effect of the spin label is the greater enhancement in the population of the β-hairpin population (∼38%), stabilized by β-turn 32–33. (Fig. S1). There is a simultaneous decrease in the turn content in the neighboring residues (turn 30–31/31–32/33–34) to accommodate the increased β-turn propensity of residues 32–33. This increase in secondary-structure content is largely driven by the hydrophobic interactions of the spin label with many residues along the chain (Fig. 4), which introduces a new long-range interaction between the N- and C-terminus of the MTSL-Cys-Aβ42 peptide that contributes to a more collapsed ensemble, corroborated by the Rg data.
Figure 4.
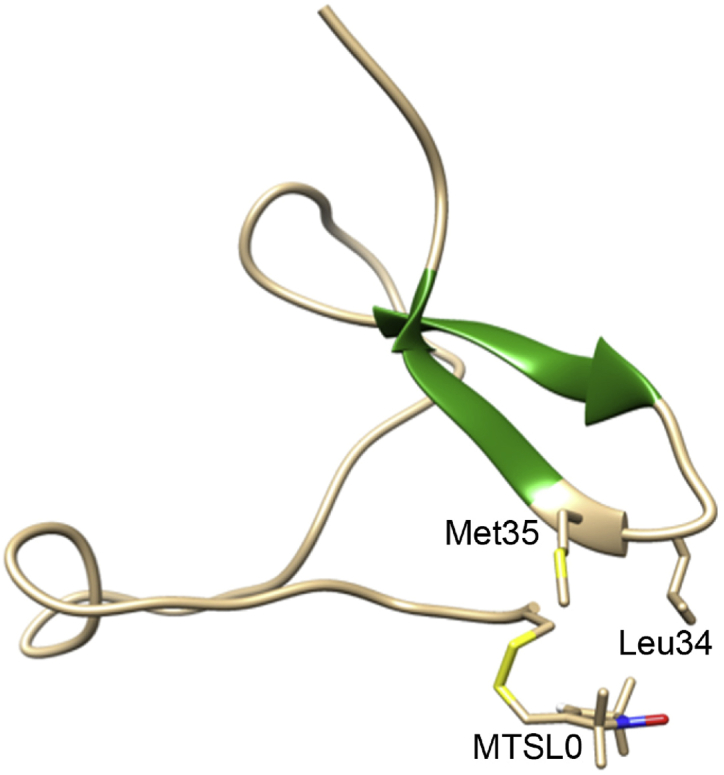
Representative configuration of the MTSL-Cys-Aβ42 ensemble, showing increased side-chain interactions between MTSL-Cys and Leu34 and Met35, which contributes to a more collapsed structural ensemble and β-strand pairings (darker shade) between residues 29–32and 36–38. This new β-structure is present in negligible amounts in the untagged peptide. Chimera ((73); University of California, San Francisco) was used to generate the molecular graphics. To see this figure in color, go online.
Fig. 5 shows a heavy-atom contact map that averages over the Aβ42 and MTSL-Cys-Aβ42 structural ensembles; although the overall pattern of residue-residue contacts are similar in the two sequences, the MTSL-Cys residue is involved in long-range heavy-atom contacts with residues 9–42 in >90% of the ensemble that explain these population shifts (Fig. S2). Considering the hydrophobic nature of the tag, a basic intermolecular interaction that is captured more than adequately by the force fields used here, the differences between the Aβ42 and MTSL-Cys-Aβ42 cannot be attributed wholesale to non-optimized force-field parameters but instead would be expected to give rise to the changes observed here. Thus, our observations of strong population shifts arising in the MTSL-Cys-Aβ42 ensemble can be tested experimentally.
Figure 5.

Heavy-atom contact map of the simulated ensembles of Aβ42 (top) and MTSL-Cys-Aβ42 (bottom). The contact maps portray the probability of interactions between each pair of residues in the simulated monomer ensembles. A contact is represented between two residues if any of their heavy atoms are <5 Å apart, and it is averaged over the ensemble to represent it with a probability ranging from 0 (black) to 1 (white). To see this figure in color, go online.
However, we require a set of NMR observables that might reveal their structural differences. We have previously found that chemical shifts, J-couplings, and global RDCs are not sufficiently discriminating NMR observables to differentiate between IDP ensembles for Aβ40 versus Aβ42, and Figs. S3–S5 confirm that these observables are not particularly helpful for discerning differences between Aβ42 and MTSL-Cys-Aβ42. However, we have previously found that local RDCs introduced by the Forman-Kay group (58), as well as homonuclear 1H-1H NOEs and heteronuclear 1H-15N NOEs, were found to better aid in the discrimination between different ensembles for Aβ40 versus Aβ42 (35). Fig. 6 shows the local 1DNH RDCs back-calculated from the untagged and tagged ensembles. It is evident that the local 1DNH values are larger on average in the presence of the spin label, where the RDC differences between the untagged and tagged peptide are larger than the experimental error of 0.1–0.6 Hz reported for Aβ42. Thus, we would predict that the 1DNH RDCs would have enough signal above the highly averaged conformational background to detect these β-hairpins.
Figure 6.

Calculated local residual dipolar couplings for Aβ42 and MTSL-Cys-Aβ42. Results are generated based on local alignments using the ENSEMBLE software package (58).
Fig. 7 compares the 15N NOE enhancement factor for REMD simulations of the Aβ42 and MTSL-Cys-Aβ42 ensembles. Although the trend is similar in both the ensembles, the MTSL-Cys-Aβ42 ensemble has higher NOE intensity values, indicating that the backbone is more ordered, essentially over the entire peptide. The experimental uncertainty for Aβ42 15N NOE enhancement varies between 0.006 and 0.08 based on data reported by Yan and Wang (23). Thus, we expect 15N NOE experiments to capture the differences in the tagged and untagged peptide observed in the simulations. In our simulated homonuclear NOE peaks for the Aβ42 and MTSL-Cys-Aβ42 ensembles, we find that the additional structure in the C-terminal region manifests as additional NOEs in the spin-labeled peptide (Fig. S6). That said, it should be noted that the unpaired electron in the PRE tag present in the MTSL-Cys-Aβ42 ensemble has a paramagnetic contribution to the longitudinal relaxation of the nuclei of interest, and typically dominates the relaxation process such that the underlying NOEs are very diminished in the real experiment (19). Since we do not consider the paramagnetic relaxation component for the NOE calculations for the MTSL-Cys-Aβ42 ensemble, further experimental judgment is required regarding whether the 1H and 15N NOE features would have sufficient strength to be observed.
Figure 7.

Comparison of the simulated 1H-15N NOE enhancement for Aβ42 (dotted line) and MTSL-Cys-Aβ42 (solid line) from REMD simulations. Higher values in the tagged peptide support a more ordered backbone. To see this figure in color, go online.
Discussion
Our results showing that structural perturbations are inevitable due to the addition of a spin label are supported by a number of previous PRE studies on folded proteins as well as IDPs. Although for well-folded proteins most sites are able to accommodate the addition of a paramagnetic tag like MTSL without any significant structural changes (18, 59, 60), both computational (20) and experimental studies (4) on the folded state of cytochrome c did show a slight decrease in protein helicity after probe attachment. In the study by Kjaergaard et al. (14) on the disordered activator for thyroid hormone and retinoid receptors peptide, the authors observed a 3–7% increase in the populations of existing helical segments after introduction of the MTSL spin label, although they considered the perturbations not strong enough to influence their conclusions about long-range interactions between the targeted set of helical domains. MTSL and TEMPOL, a different PRE tag, have been shown to have high affinity toward hydrophobic surfaces (61, 62), and the increased surface interaction in turn induces more aggregation in the labeled peptide (12). Finally, another way of conducting PRE experiments is to introduce small sequence motifs that bind to paramagnetic metal ions like Cu2+, Ni2+, or lanthanide ions, but these methods are not popular for IDP experiments, because they require larger sequence modification if the motif is not present in the original protein and would certainly perturb the underlying structural ensemble significantly (63).
However, the question is what can be learned from PRE experiments when applied to IDPs in solution, despite the inevitable perturbations due to the tag. In light of the simulation results presented here, PRE experiments aimed at looking into the kinetics of fibril or oligomer-growth mechanisms of Aβ42 (or Aβ40 peptide (11)), or interactions with membranes (64), should be analyzed with caution. Fibrils usually elongate by a faster “dock” and slower “lock” mechanism (65), and the tagged peptide might take longer to properly lock because of the introduction of free-energy barriers associated with the new/enhanced β-structure assemblies. For smaller oligomeric intermediates, the paramagnetic tag will possibly introduce more structured, compact conformations and increase the aggregation rates. The enhanced structure formation appears to arise from excessive hydrophobicity of the MTSL label that could be reduced by using a modified MTSL tag designed to be more hydrophilic (66, 67). This is a welcome addition given the importance of experiments such as PRE that can probe long-range order in IDP structures in solution, although a similar study to this one would be needed to help quantify the structural ensemble perturbations introduced by the more hydrophilic label.
Conclusion
The most widely used method for gaining detailed structural insights into IDPs is to combine experimental techniques such as NMR, infrared spectroscopy, and small-angle x-ray scattering with computational techniques like MD, Monte Carlo simulations, or knowledge-based methods (36, 68, 69, 70). The experimental observables are typically used to either validate IDP structural ensembles that are generated from Boltzmann-weighted MD simulations (26, 30, 34, 35, 36), to select for conformations from computationally generated IDP (typically random coil) ensembles (58, 68, 69), to provide structural restraints during guided MD simulations (7, 8, 68, 71), or to use in Bayesian analysis (72). Here, we have utilized molecular simulations to predict the perturbations caused by addition of the commonly used MTSL tag in PRE experiments when applied to the disordered Aβ42 peptide.
We have employed several independent calculations of the structural ensemble of the native Aβ42 ensemble to provide estimates of uncertainty due to sampling protocol to distinguish real structural perturbations measured from the average of two independently generated ensembles of the labeled MTSL-Cys-Aβ42 peptide. To sum up the effects of the spin label, a majority of the main structural features of the peptide, including the dominant β-hairpin formed by residues 16–21 and 29–36 (34, 36), are able to withstand the perturbations introduced by the tag. Nevertheless, the tag causes population enhancements in the C-terminus involving the β-hairpin formed by residues 27–31 and 33–38. If 1H and 15N NOE experiments can be performed without the paramagnetic spin label dominating the relaxation mechanisms, experiments would point toward a more ordered backbone and new NOE peaks arising from the enhanced/new β-strand contacts. Our results will also be supported by RDC experiments in which local alignments are considered (58), which will show increased 1DNH values resulting from higher β-structure propensities, especially in the C-terminus.
In conclusion, PRE experiments, fluorescence resonance energy transfer, and double electron-electron resonance experiments can probe long-range order in polypeptide structures in solution, although the requirement of a reporter tag will likely perturb the underlying structural ensemble to some degree (7, 8, 63). Furthermore, the nature of the structural perturbation will vary with peptide size, sequence, tag type and position, and whether it is being used to probe a folded or disordered structural ensemble. Since quantifying the exact extent of the perturbations caused by the spin label can be difficult to determine experimentally, we believe that MD simulation is a powerful and complementary approach to resolving the potential structural perturbations and that it will suggest experiments that can confirm the theoretical predictions.
Author Contributions
T.H.-G. proposed the project and approach. S.S. and J.L. performed the simulations. S.S., J.L., and T. H.-G. analyzed the data. S.S. and T.H.-G. wrote the manuscript.
Acknowledgments
S.S. thanks Dr. Aurelia K. Ball and Eugene Yedvabny for help in setting up the de novo REMD simulations and in using the T.H.-G. lab in-house NMR analysis tools.
T.H.-G. acknowledges support of this work by National Institutes of Health (NIH) grant U01GM121667-01. J.L. acknowledges partial support from an NIH Molecular Biophysics training grant, T32-GM008295. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
Editor: Wendy Shaw.
Footnotes
Six figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(17)30758-0.
Supporting Material
References
- 1.Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 2012;37:509–516. doi: 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 3.Dunker A.K., Silman I., Sussman J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Qu K., Vaughn J.L., Fetrow J.S. Kinetics and motional dynamics of spin-labeled yeast iso-1-cytochrome c: 1. Stopped-flow electron paramagnetic resonance as a probe for protein folding/unfolding of the C-terminal helix spin-labeled at cysteine 102. Biochemistry. 1997;36:2884–2897. doi: 10.1021/bi962155i. [DOI] [PubMed] [Google Scholar]
- 5.Parigi G., Bertini I., Piva A. Paramagnetic constraints in structure-determination programs. J. Inorg. Biochem. 2001;86 507–507. [Google Scholar]
- 6.Bertini I., Donaire A., Poggi L. Paramagnetism-based versus classical constraints: an analysis of the solution structure of Ca Ln calbindin D9k. J. Biomol. NMR. 2001;21:85–98. doi: 10.1023/a:1012422402545. [DOI] [PubMed] [Google Scholar]
- 7.Dedmon M.M., Lindorff-Larsen K., Dobson C.M. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 8.Allison J.R., Várnai P., Vendruscolo M. Determination of the free energy landscape of α-synuclein using spin label nuclear magnetic resonance measurements. J. Am. Chem. Soc. 2009;131:18314–18326. doi: 10.1021/ja904716h. [DOI] [PubMed] [Google Scholar]
- 9.Ganguly D., Chen J. Structural interpretation of paramagnetic relaxation enhancement-derived distances for disordered protein states. J. Mol. Biol. 2009;390:467–477. doi: 10.1016/j.jmb.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 10.Wu K.-P., Baum J. Detection of transient interchain interactions in the intrinsically disordered protein α-synuclein by NMR paramagnetic relaxation enhancement. J. Am. Chem. Soc. 2010;132:5546–5547. doi: 10.1021/ja9105495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fawzi N.L., Ying J., Clore G.M. Kinetics of amyloid β monomer-to-oligomer exchange by NMR relaxation. J. Am. Chem. Soc. 2010;132:9948–9951. doi: 10.1021/ja1048253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fawzi N.L., Ying J., Clore G.M. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature. 2011;480:268–272. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu L., Liu C., Guo Z. Structural insights into Aβ42 oligomers using site-directed spin labeling. J. Biol. Chem. 2013;288:18673–18683. doi: 10.1074/jbc.M113.457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iešmantavičius V., Jensen M.R., Kjaergaard M. Modulation of the intrinsic helix propensity of an intrinsically disordered protein reveals long-range helix-helix interactions. J. Am. Chem. Soc. 2013;135:10155–10163. doi: 10.1021/ja4045532. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs E.B., Cook E.C., Showalter S.A. Application of NMR to studies of intrinsically disordered proteins. Arch. Biochem. Biophys. 2017 doi: 10.1016/j.abb.2017.05.008. Published online May 11, 2017. [DOI] [PubMed] [Google Scholar]
- 16.Konrat R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014;241:74–85. doi: 10.1016/j.jmr.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hubbell W.L., Gross A., Lietzow M.A. Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 18.Koteiche H.A., Mchaourab H.S. Folding pattern of the α-crystallin domain in αA-crystallin determined by site-directed spin labeling. J. Mol. Biol. 1999;294:561–577. doi: 10.1006/jmbi.1999.3242. [DOI] [PubMed] [Google Scholar]
- 19.Bertini I., Luchinat C., Parigi P. Elsevier; Amsterdam, the Netherlands: 2001. Solution NMR of Paramagnetic Molecules, vol. 2: Applications to Metallobiomolecules and Models. [Google Scholar]
- 20.Murzyn K., Rog T., Froncisz W. Influence of the disulfide bond configuration on the dynamics of the spin label attached to cytochrome c. Proteins. 2006;62:1088–1100. doi: 10.1002/prot.20838. [DOI] [PubMed] [Google Scholar]
- 21.Barrow C.J., Yasuda A., Zagorski M.G. Solution conformations and aggregational properties of synthetic amyloid β-peptides of Alzheimer’s disease. analysis of circular dichroism spectra. J. Mol. Biol. 1992;225:1075–1093. doi: 10.1016/0022-2836(92)90106-t. [DOI] [PubMed] [Google Scholar]
- 22.Goedert M., Spillantini M.G. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 23.Yan Y., Wang C. Aβ42 is more rigid than Aβ40 at the C terminus: implications for Aβ aggregation and toxicity. J. Mol. Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 24.Roche J., Shen Y., Bax A. Monomeric Aβ (1–40) and Aβ (1–42) peptides in solution adopt very similar Ramachandran map distributions that closely resemble random coil. Biochemistry. 2016;55:762–775. doi: 10.1021/acs.biochem.5b01259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vivekanandan S., Brender J.R., Ramamoorthy A. A partially folded structure of amyloid-β(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sgourakis N.G., Yan Y., Garcia A.E. The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fawzi N.L., Phillips A.H., Head-Gordon T. Structure and dynamics of the Aβ(21–30) peptide from the interplay of NMR experiments and molecular simulations. J. Am. Chem. Soc. 2008;130:6145–6158. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam A.R., Teplow D.B., Urbanc B. Effects of the Arctic (E22→G) mutation on amyloid β-protein folding: discrete molecular dynamics study. J. Am. Chem. Soc. 2008;130:17413–17422. doi: 10.1021/ja804984h. [DOI] [PubMed] [Google Scholar]
- 29.Melquiond A., Dong X., Derreumaux P. Role of the region 23–28 in Aβ fibril formation: insights from simulations of the monomers and dimers of Alzheimer’s peptides Aβ40 and Aβ42. Curr. Alzheimer Res. 2008;5:244–250. doi: 10.2174/156720508784533330. [DOI] [PubMed] [Google Scholar]
- 30.Sgourakis N.G., Merced-Serrano M., Garcia A.E. Atomic-level characterization of the ensemble of the Aβ (1–42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J. Mol. Biol. 2011;405:570–583. doi: 10.1016/j.jmb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu C., Shea J.-E. The structure of intrinsically disordered peptides implicated in amyloid diseases: insights from fully atomistic simulations. In: Dokholyan N., editor. Computational Modeling of Biological Systems: From Molecules to Pathways. Springer; 2012. pp. 215–227. [Google Scholar]
- 32.Lin Y.S., Pande V.S. Effects of familial mutations on the monomer structure of Aβ42. Biophys. J. 2012;103:L47–L49. doi: 10.1016/j.bpj.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenman D.J., Connors C.R., García A.E. Aβ monomers transiently sample oligomer and fibril-like configurations: ensemble characterization using a combined MD/NMR approach. J. Mol. Biol. 2013;425:3338–3359. doi: 10.1016/j.jmb.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ball K.A., Phillips A.H., Head-Gordon T. Homogeneous and heterogeneous tertiary structure ensembles of amyloid-β peptides. Biochemistry. 2011;50:7612–7628. doi: 10.1021/bi200732x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ball K.A., Phillips A.H., Head-Gordon T. Differences in β-strand populations of monomeric Aβ40 and Aβ42. Biophys. J. 2013;104:2714–2724. doi: 10.1016/j.bpj.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ball K.A., Wemmer D.E., Head-Gordon T. Comparison of structure determination methods for intrinsically disordered amyloid-β peptides. J. Phys. Chem. B. 2014;118:6405–6416. doi: 10.1021/jp410275y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran L., Ha-Duong T. Exploring the Alzheimer amyloid-β peptide conformational ensemble: a review of molecular dynamics approaches. Peptides. 2015;69:86–91. doi: 10.1016/j.peptides.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Kim W., Hecht M.H. Sequence determinants of enhanced amyloidogenicity of Alzheimer Aβ42 peptide relative to Aβ40. J. Biol. Chem. 2005;280:35069–35076. doi: 10.1074/jbc.M505763200. [DOI] [PubMed] [Google Scholar]
- 39.Morimoto A., Irie K., Shirasawa T. Analysis of the secondary structure of β-amyloid (Aβ42) fibrils by systematic proline replacement. J. Biol. Chem. 2004;279:52781–52788. doi: 10.1074/jbc.M406262200. [DOI] [PubMed] [Google Scholar]
- 40.Lim K.H., Collver H.H., Kenney J.M. Characterizations of distinct amyloidogenic conformations of the Aβ (1–40) and (1–42) peptides. Biochem. Biophys. Res. Commun. 2007;353:443–449. doi: 10.1016/j.bbrc.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 41.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 42.Lincoff J., Sasmal S., Head-Gordon T. Comparing generalized ensemble methods for sampling of systems with many degrees of freedom. J. Chem. Phys. 2016;145:174107. doi: 10.1063/1.4965439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown S., Head-Gordon T. Cool walking: a new Markov chain Monte Carlo sampling method. J. Comput. Chem. 2003;24:68–76. doi: 10.1002/jcc.10181. [DOI] [PubMed] [Google Scholar]
- 44.Case D.A., Babin V., Gohlke H. University of California; San Francisco: 2014. AMBER 14. [Google Scholar]
- 45.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wickstrom L., Okur A., Simmerling C. Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys. J. 2009;97:853–856. doi: 10.1016/j.bpj.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horn H.W., Swope W.C., Head-Gordon T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004;120:9665–9678. doi: 10.1063/1.1683075. [DOI] [PubMed] [Google Scholar]
- 48.Xue Y., Skrynnikov N.R. Motion of a disordered polypeptide chain as studied by paramagnetic relaxation enhancements, 15N relaxation, and molecular dynamics simulations: how fast is segmental diffusion in denatured ubiquitin? J. Am. Chem. Soc. 2011;133:14614–14628. doi: 10.1021/ja201605c. [DOI] [PubMed] [Google Scholar]
- 49.Andersen H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980;72:2384–2393. [Google Scholar]
- 50.Neal S., Nip A.M., Wishart D.S. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J. Biomol. NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 51.Karplus M. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 1959;30:11–15. [Google Scholar]
- 52.Marsh J.A., Baker J.M.R., Forman-Kay J.D. Calculation of residual dipolar couplings from disordered state ensembles using local alignment. J. Am. Chem. Soc. 2008;130:7804–7805. doi: 10.1021/ja802220c. [DOI] [PubMed] [Google Scholar]
- 53.Zweckstetter M., Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 2000;122:3791–3792. [Google Scholar]
- 54.Hansmann U.H. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997;281:140–150. [Google Scholar]
- 55.Marinari E., Parisi G. Simulated tempering: a new Monte Carlo scheme. Europhys. Lett. 1992;19:451–458. [Google Scholar]
- 56.Hukushima K., Nemoto K. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 1996;65:1604–1608. [Google Scholar]
- 57.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 58.Krzeminski M., Marsh J.A., Forman-Kay J.D. Characterization of disordered proteins with ENSEMBLE. Bioinformatics. 2013;29:398–399. doi: 10.1093/bioinformatics/bts701. [DOI] [PubMed] [Google Scholar]
- 59.Matthews B.W. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 1995;46:249–278. doi: 10.1016/s0065-3233(08)60337-x. [DOI] [PubMed] [Google Scholar]
- 60.Mchaourab H.S., Lietzow M.A., Hubbell W.L. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 61.Deschamps M.L., Pilka E.S., Boyd J. Probing protein-peptide binding surfaces using charged stable free radicals and transverse paramagnetic relaxation enhancement (PRE) J. Biomol. NMR. 2005;31:155–160. doi: 10.1007/s10858-004-7912-6. [DOI] [PubMed] [Google Scholar]
- 62.Showalter S.A., Bruschweiler-Li L., Brüschweiler R. Quantitative lid dynamics of MDM2 reveals differential ligand binding modes of the p53-binding cleft. J. Am. Chem. Soc. 2008;130:6472–6478. doi: 10.1021/ja800201j. [DOI] [PubMed] [Google Scholar]
- 63.Clore G.M., Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yagi-Utsumi M., Kameda T., Kato K. NMR characterization of the interactions between lyso-GM1 aqueous micelles and amyloid β. FEBS Lett. 2010;584:831–836. doi: 10.1016/j.febslet.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 65.Sasmal S., Schwierz N., Head-Gordon T. Mechanism of nucleation and growth of Aβ40 fibrils from all-atom and coarse-grained simulations. J. Phys. Chem. B. 2016;120:12088–12097. doi: 10.1021/acs.jpcb.6b09655. [DOI] [PubMed] [Google Scholar]
- 66.Fawzi N.L., Fleissner M.R., Clore G.M. A rigid disulfide-linked nitroxide side chain simplifies the quantitative analysis of PRE data. J. Biomol. NMR. 2011;51:105–114. doi: 10.1007/s10858-011-9545-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Card P.B., Erbel P.J., Gardner K.H. Structural basis of ARNT PAS-B dimerization: use of a common β-sheet interface for hetero- and homodimerization. J. Mol. Biol. 2005;353:664–677. doi: 10.1016/j.jmb.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 68.Marsh J.A., Forman-Kay J.D. Ensemble modeling of protein disordered states: experimental restraint contributions and validation. Proteins. 2012;80:556–572. doi: 10.1002/prot.23220. [DOI] [PubMed] [Google Scholar]
- 69.Schneider R., Huang J.-R., Blackledge M. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol. BioSys. 2012;8:56–68. doi: 10.1039/c1mb05291h. [DOI] [PubMed] [Google Scholar]
- 70.Bhowmick A., Brookes D.H., Head-Gordon T. Finding our way in the dark proteome. J. Am. Chem. Soc. 2016;138:9730–9742. doi: 10.1021/jacs.6b06543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.López-Méndez B., Güntert P. Automated protein structure determination from NMR spectra. J. Am. Chem. Soc. 2006;128:13112–13122. doi: 10.1021/ja061136l. [DOI] [PubMed] [Google Scholar]
- 72.Brookes D.H., Head-Gordon T. Experimental inferential structure determination of ensembles for intrinsically disordered proteins. J. Am. Chem. Soc. 2016;138:4530–4538. doi: 10.1021/jacs.6b00351. [DOI] [PubMed] [Google Scholar]
- 73.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.