

Abstract
During filling, urinary bladder volume increases dramatically with little change in pressure. This is accomplished by suppressing contractions of the detrusor muscle that lines the bladder wall. Mechanisms responsible for regulating detrusor contraction during filling are poorly understood. Here we describe a novel pathway to stabilize detrusor excitability involving platelet-derived growth factor receptor-α positive (PDGFRα+) interstitial cells. PDGFRα+ cells express small conductance Ca2+-activated K+ (SK) and TRPV4 channels. We found that Ca2+ entry through mechanosensitive TRPV4 channels during bladder filling stabilizes detrusor excitability. GSK1016790A (GSK), a TRPV4 channel agonist, activated a non-selective cation conductance that coupled to activation of SK channels. GSK induced hyperpolarization of PDGFRα+ cells and decreased detrusor contractions. Contractions were also inhibited by activation of SK channels. Blockers of TRPV4 or SK channels inhibited currents activated by GSK and increased detrusor contractions. TRPV4 and SK channel blockers also increased contractions of intact bladders during filling. Similar enhancement of contractions occurred in bladders of Trpv4 −/− mice during filling. An SK channel activator (SKA-31) decreased contractions during filling, and rescued the overactivity of Trpv4 −/− bladders. Our findings demonstrate how Ca2+ influx through TRPV4 channels can activate SK channels in PDGFRα+ cells and prevent bladder overactivity during filling.
Introduction
Overactive bladder is a clinical syndrome accounting for over $12 billion in health care costs in the USA annually1. Current therapies to control this problem are less than adequate2. How the bladder remains mechanically stable during filling is a question lacking clear physiological explanation. We recently described a new class of interstitial cells in the murine bladder that are labeled specifically with antibodies against platelet-derived growth factor receptor-α (PDGFRα+ cells)3. We also identified a reporter strain of mice in which PDGFRα+ cells express a histone 2B-eGFP fusion protein driven off the endogenous promoters for Pdgfra (Pdgfrα/eGFP mouse)3. PDGFRα+ cells have also been identified in human and guinea pig detrusor muscles4. These cells are closely associated with varicose nerve processes in detrusor muscles (muscularis propria) suggesting that PDGFRα+ cells may be innervated and participate in purinergic regulation of bladder contraction5–7.
PDGFRα+ cells in detrusor muscles express small conductance Ca2+-activated K+ (SK3) channels5, and it is known that SK channel blockers and genetic deactivation of SK3 evokes bursts of action potentials in detrusor muscle preparations and increase contractions in response to stimulation of motor neurons8–11. PDGFRα+ cells display high current densities due to SK channels (i.e. whole cell currents averaging 13 pA/pF at −40 mV), but detrusor smooth muscle cells (SMCs) show very low SK current density (e.g. 0.5 pA/pF at + 10 mV)5. Thus stabilization of bladder excitability by SK channels appears to be due to the prominent expression of these channels in PDGFRα+ cells rather than in SMCs.
SK channels are activated by increased intracellular Ca2+ 12,13. In many cells Ca2+ influx occurs through activation of voltage-dependent Ca2+ channels. However, functional voltage-dependent Ca2+ conductances have not been resolved in PDGFRα+ cells5, raising the question of the source of Ca2+ for the regulation of SK channels in these cells. Transient receptor potential (TRP) channels are expressed by most cells and vanilloid TRP (TRPV) channels are relatively permeable to Ca2+ 14. Previous studies have shown Trpv4 is expressed in extracts of whole detrusor muscles that would have contained transcripts from SMCs and PDGFRα+ cells15. Trpv4 −/− mice displayed increased non-voiding contractions16,17. We tested the hypothesis that TRPV4 channels are expressed primarily by PDGFRα+ cells in detrusor muscles, and that Ca2+ entry via TRPV4 channels is linked to activation of SK channels. It is also important to note that TRPV4 channels can be mechanically activated14,18. Thus, it is possible that enhanced Ca2+ influx via TRPV4 channels during bladder filling increases the open probability of SK channels to dampen the excitability of detrusor muscles. Our results show that SK channel activation is closely linked to TRPV4-mediated Ca2+-influx and may be enhanced by direct interactions between TRPV4 and SK3 channels in PDGFRα+ cells.
Results
Expression of Trpv4 in PDGFRα+ cells
We investigated the expression of Trp genes in PDGFRα+ cells. We have shown previously that cells isolated enzymatically from bladders of Pdgfra tm11(EGFP)S ° r/J mice and sorted by fluorescence activated cell sorting (FACS) display significant enrichment in Pdgfrα transcripts and negligible expression of kit (ICC marker), Uchl1 (neuronal marker) and Myh11 (SMC marker)5. We isolated PDGFRα+ cells from detrusor muscles, purified these cells by FACS, and probed for expression of Trp genes. We found expression of Trpc1, Trpm5, Trpv2, Trpv3 and Trpv4 in PDGFRα+ cells. Trpc2, Trpc6, Trpm7 and Trpv2 transcripts were detected in SMCs (obtained from smMHC/Cre/eGFP mice; data not shown). Quantitative analysis of Trp transcripts from PDGFRα+ cells showed that Trpv4 (2.7 ± 0.2 fold) was highly expressed in PDGFRα+ cells vs. unsorted cells of the detrusor (n = 4, Fig. 1). Thus, we focused our investigations on the functional role of TRPV4 channels in PDGFRα+ cells since TRPC1 and TRPM5 channels are less permeable to divalent cations.
Figure 1.
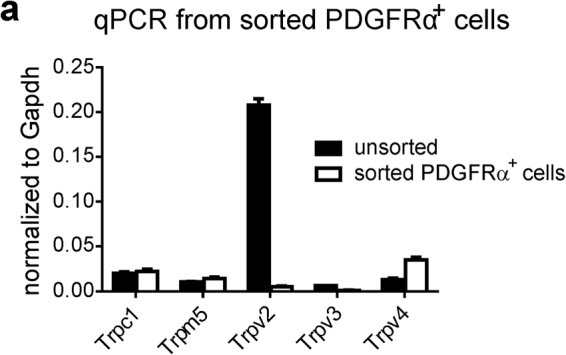
Quantitative analysis of Trp transcripts from sorted Pdgfrα+ cells. Quantitative analysis of Trp transcripts revealed Trpv4 is highly expressed in sorted PDGFRα+ cells (n = 4).
Effects of TRPV4 agonist and antagonists on PDGFRα+ cells
We tested the effects of TRPV4 agonist GSK1016790A (GSK)15 and antagonists on the generation of membrane currents and potentials in PDGFRα+ cells. Under whole-cell patch clamp conditions (cells dialyzed with Cs+-rich pipette solution; see Methods), GSK (100 nM) induced inward currents at holding potentials from −80, −60 and −40 mV (Fig. 2a,c,e; n = 12). When cells were depolarized with ramp protocols from −80 mV to + 80 mV (lower inset in Fig. 2b,d,f), negligible currents were evoked in control conditions (Fig. 2ba,da,fa). GSK activated non-selective cation currents with a reversal potential of −1.0 ± 0.4 mV (Fig. 2bb,db,fb). When cells were dialyzed with K+-rich pipette solution (see Methods), GSK (100 nM) activated inward current at a holding potential of −80 mV (n = 6, Fig. 2g), inward currents followed by outward current at −60 mV (n = 8, Fig. 2i), and outward currents at −40 mV (n = 35, Fig. 2k). The GSK-induced outward currents were voltage-independent and the whole cell reversal potential shifted toward the equilibrium potential for the K+ gradient (E K = ~−80 mV) (Fig. 2h,j,l).
Figure 2.

The effect of TRPV4 agonist on the inward currents and the outward currents in PDGFRα+ cells. GSK1016790A (GSK, 100 nM) activated inward currents at holding potentials −80 to −40 mV. Cells were dialyzed with Cs+-rich solutions (a,c,e). Expanded time scales (b,d,f) from each left panel during ramp depolarization at different holding potentials. a & b denote before and after GSK (100 nM), respectively. When cells were dialyzed with K+-rich solutions, GSK (100 nM) activated inward current at a holding potential of −80 mV (g). Expanded time scales (h) from panel g during ramp depolarization before (a) and after (b) GSK (100 nM) application, respectively. b-a denotes GSK-sensitive current. GSK (100 nM) activated inward current followed by outward current at holding potentials of −60 mV (i) and −40 mV (k). Expanded time scales (j,l) from panels i and k during ramp depolarization before (a) and after (b) GSK application, respectively. b-a denotes GSK-sensitive current.
TRPV4 channels can be activated by 4α-Phorbol 12,13-didecanoate (4α-PDD), swelling and mechanical stretch19–22. We examined whether activation of TRPV4 channels in PDGFRα+ cells by these alternative methods also led to activation of outward current. Cells were stretched using two patch electrodes: one to measure whole cell current and the other to elongate the cell23. After obtaining whole cell conditions with the first electrode, a second gigaseal was formed with the second electrode, and this was used to slowly stretch the cell by 1-2 µm. Mechanical stretch induced transient inward current followed by outward current (supplementary Fig. 2a,b). These effects were similar to the effects of GSK. In another series of experiments hypo-osmotic solution (200 mOsm) was used to swell cells. Exposure to hypo-osmotic solution induced inward current followed by reversal of the response to outward current (supplementary Fig. 2c,d). Finally, we also tested the effects of 4α-PDD, a non-selective TRPV4 agonist. Application of 4α-PDD induced inward current followed by outward current (supplementary Fig. 2e,f). Thus, all methods used to activate TRPV4 current (inward) resulted in secondary activation of an outward current as observed with GSK.
A selective TRPV4 antagonist, HC-067047 (1 μM, Fig. 3a,b)24 completely abolished the voltage-independent outward current evoked by GSK at −40 mV. In the same cells under current clamp (I = 0), GSK caused hyperpolarization averaging −38 ± 5 mV (n = 9). The average resting membrane potential was −35 ± 5.1 mV in control. HC-067047 (1 µM) abolished the GSK-induced hyperpolarization. Another selective TRPV4 antagonist, RN-173425 (10 μM, Fig. 3c,d) also inhibited GSK-activated currents under voltage clamp and abolished GSK-induced hyperpolarization under current clamp (I = 0, n = 7). These data suggest that activation of TRPV4 may result in Ca2+ influx and secondary activation of a Ca2+-sensitive K+ conductance.
Figure 3.

The effect of TRPV4 antagonist and SK channel blocker on the outward currents and membrane potentials in PDGFRα+ cells. In cells dialyzed with K+-rich solutions, GSK (100 nM) activated outward current at a holding potential of −40 mV in voltage clamp mode. In the same cell, GSK induced hyperpolarization under current clamp (I = 0). TRPV4 antagonist, HC-067047 (1 μM, HC, a) and RN-1734 (10 μM, RN, c) inhibited GSK-activated outward current and hyperpolarization. Expanded time scales (b,d) from panels a and c during ramp depolarization before (a) and after (b) GSK application, respectively. In next experiments, cells were dialyzed with K+-rich solutions and held at −40 mV. GSK (100 nM) activated outward current. Addition of an SK channel blocker, UCL1684 (UCL, 1 μM) inhibited the outward currents and revealed an inward current. TRPV4 antagonist, HC-067047 (1 μM, HC) inhibited the inward current activated by GSK (e). Expanded time scales (f) from panel e during ramp depolarization (see inset) before (a), after (b) GSK and additional application of HC (c). Under pretreatment of UCL, GSK activated only inward current at a holding potential of −40 mV. Addition of HC completely blocked the inward current (g). Expanded time scales (h) from panel g during ramp depolarization before (a) and after (b) GSK application in the presence of UCL. PDGFRα+ cells from Trpv4 −/− mice were dialyzed with K+-rich solutions (i). GSK (100 nM) did not activate outward current at a holding potential of −40 mV. In the same cell, an SK channel activator (SKA-31, 10 µM) activated outward currents (i). Expanded time scales (j) from panel i during ramp depolarization in control (a), and before (b) and after (c) SKA-31 application, respectively.
SK3 channels are highly expressed in PDGFRα+ cells5. The outward current activated by GSK had properties consistent with currents mediated by SK channels (i.e. voltage-independent; rectification at positive potentials5; see Fig. 2h,j,l). Therefore, we tested the effects of an SK channel blocker, UCL168426–28 on the outward current activated by GSK. Using K+-rich internal solution, cells were held at −40 mV. GSK activated outward current (Fig. 3e) and caused a negative shift in the reversal potential (Fig. 3fb). UCL1684 (1 µM) blocked the outward current activated by GSK and unmasked the inward current activated by GSK (172 ± 69 pA, n = 6, Fig. 3e) and shifted reversal potentials close to 0 mV (Fig. 3fc). The underlying inward current was completely blocked by HC-067047. We also tested the effects of GSK after pretreatment with UCL1684 (1 µM). GSK in the presence of UCL1684 (1 μM) activated inward current (122 ± 72 pA, n = 6), but no outward current, at a holding potential of −40 mV. The inward current reversed at −1.9 ± 0.6 mV and was blocked by HC-067047 (1 μM) (Fig. 3g,h; n = 5). These data suggest that Ca2+ influx via TRPV4 channels is sufficient to activate SK current and induce hyperpolarization of PDGFRα+ cells.
As a control for the effects of GSK, we tested the effects of this compound on PDGFRα+ cells from detrusor muscles of animals obtained by crossing PDGFRα/eGFP and Trpv4 −/− mice. GSK failed to activate inward or outward currents in these cells, but the SK channel activator (SKA-31) induced outward current (Fig. 3i,j). This observation suggests that GSK is selective for TRPV4 channels, and these channels are linked to activation of SK channels in detrusor PDGFRα+ cells.
Ca2+ influx through TRPV4 channels in PDGFRα+ cells
TRPV4 is a non-selective cation channel permeable to Ca2+, Mg2+, Na+ and K+ 29. In further tests of whether Ca2+ influx through TRPV4 can trigger SK channel currents, we utilized ion replacements. GSK activated small amplitude inward currents followed by outward current at a holding potential of −50 mV (Fig. 4a,c). When external Ca2+ was replaced by Mn2+ (Ca2+−0), the outward phase of the response to GSK disappeared leaving only a small amplitude inward current (Fig. 4a,b). The inward current was inhibited by HC-067047 (Fig. 4a). TRPV4 channels are also permeable to Na+. When external Na+ was replaced with equimolar NMDG and 0 mM Ca2+ (Ca2+−0/Na+−0), the inward current activated by GSK was abolished (Fig. 4c,d).
Figure 4.

The effects of cation replacement on the GSK-activated currents in PDGFRα+ cells. Cells were dialyzed with K+-rich solutions at a holding potential of −50 mV. GSK (100 nM) activated a small inward current followed by an outward current. Removal of external Ca2+ (Ca-0) inhibited the outward current and unmasked the inward current. Addition of HC-067047 (1 μM, HC) blocked the inward current activated by GSK (a). Expanded time scales (b) from panel a during ramp depolarization (see inset) before (a), after (b) GSK and Ca2+-free condition (c). GSK activated a small inward current followed by an outward current at a holding potential of −50 mV. External Ca2+-free solution (Ca-0) blocked the outward current and unmasked the inward current. Addition of external Ca2+ and Na+ free (Ca-0/Na-0) solution abolished the inward current (c). Expanded time scales (d) from panel c during ramp depolarization before (d), after (e) GSK and under Ca2+-free condition (f). After 10 min incubation with cyclopiazonic acid (CPA, 10 µM) GSK activated outward current at a holding potential of −40 mV (e). Expanded time scales (f) from panel e before (a) and after (b) GSK in the presence of CPA.
It is possible that Ca2+ influx via TRPV4 is amplified by Ca2+ release from intracellular stores. Experiments were performed on cells dialyzed with BAPTA (10 mM) in the presence of cyclopiazonic acid (10 µM) to test the role of Ca2+ stores in GSK responses. Under these conditions, GSK still activated outward currents (Fig. 4e,f), suggesting that Ca2+ influx through TRPV4 channels directly activates SK channels without initiating intracellular Ca2+ signaling (e.g by Ca2+ induced Ca2+ release).
TRPV4 current in smooth muscle cells
We also tested whether smooth muscle cells (SMCs) display functional expression of TRPV4, even though Trpv4 transcripts were not resolved in these cells (not shown). SMCs displayed voltage-dependent inward current during ramp depolarization when cells were dialyzed with Cs+-rich solution (supplementary Fig. 1a,b). GSK (100 nM) failed to evoke current responses in SMCs (n = 10). The effects of GSK were also tested on membrane potentials using K+-rich internal solution. GSK had no effect on membrane potential (supplementary Fig. 1c,d). These results are consistent with the transcript expression data, and demonstrate no role for TRPV4 channels in detrusor SMCs.
The effects of TRPV4 agonist and antagonist on detrusor muscle strips
It was previously reported that the TRPV4 agonist (GSK) increased spontaneous contractions of detrusor muscles with or without urothelium15. This result is contrary to our findings, because GSK, through activation of SK channels, would tend to exert inhibitory effects on contractions through activation of SK channels (see Figs 1–4). Therefore, we tested the effects of GSK (10 nM–30 µM) on contractions of detrusor muscle strips with urothelium removed. As predicted, GSK decreased the amplitude and frequency of spontaneous contractions and reduced basal tone in a concentration-dependent manner (Fig. 5a,b, n = 37 of 44 from 14 animals). The IC50 for the reduction in amplitude was 41.5 nM. A small number (7 of 44; 16%) of detrusor muscles showed a small increase in contractions at higher concentrations of GSK (≥1 µM). The TRPV4 agonist activated SK channels in PDGFRα+ cells, so the relaxation caused by the TRPV4 agonist might be due to activation of SK channels. To investigate this hypothesis, detrusor muscle strips were pretreated with apamin (Fig. 5c,d). In the presence of apamin GSK caused a concentration-dependent increase in contractions with an EC50 of 770 nM (Fig. 5d). The effects of a TRPV4 antagonist (RN-1734) on the contractility of detrusor muscle strips were also examined. RN-1734 (10 µM) increased contractions (area under curve; AUC) to 148 ± 13% compared to control (n = 7, P < 0.01, Fig. 5e–g). This observation is consistent with a link between TRPV4 channels and activation of SK channels. SKA-31 (10 µM, SK channel activator) reduced contractions caused by RN-1734 to 67 ± 15% of control (Fig. 5e–g). These findings suggest that the mechanical stretch (1 mN) applied to muscles under basal condition, enhanced activation of TRPV4 channels and produced basal activation of SK channels.
Figure 5.

The effects of TRPV4 agonist, antagonist and SK channel activator on phasic contractions in murine bladder strips. Concentration-response relationship for GSK shows inhibitory effect of this TRPV4 agonist on phasic contractions (a, upper panel). Excerpts in the record denoted by a–d are shown below and show contractions in the presence of 10−7, 10−6 and 3 × 10−6 M GSK (lower panel). Average concentration-response curve for GSK effects with contractile amplitudes normalized as a percentage of control (n = 37, b). Representative concentration-response showed that GSK increased the phasic contraction in the presence of apamin (c, upper panel) and expanded time scales (lower panel). Average concentration-response curve for GSK in the presence of apamin with contraction amplitudes normalized as a percentage of control (n = 9, d). Representative phasic contractions response to RN-1734 (RN, TRPV4 antagonist) and SKA-31 (SKA activator, e). Expanded time scales from panel f under control (a), RN-1734 (b) and SKA-31 (c). Summary of normalized area under curve (AUC) of phasic contractions under control, RN-1734 and SKA-31 (n = 7. g). **P < 0.01 and horizontal dotted lines in all panels denote baseline in control.
The effects of TRPV4 agonist and antagonist on ex vivo pressure-volume measurement
We also examined the pressure-volume relationships of intact bladders using ex vivo preparations to test the effects of GSK and apamin. In contrast to in vivo cystometry, ex vivo preparation exclude extrinsic neural regulation during filling, so this technique focuses on myogenic mechanisms regulating detrusor excitability during filling. Infusion of Krebs–Ringer bicarbonate buffer (KRB; 15 µl/min) initially induced a small increase in intravesical pressure (0.9 ± 0.1 cm H2O, n = 12) before pressure rose steeply to a threshold pressure (about 20 cm H2O) for voiding contractions (see Fig. 6 and Methods). GSK alone did not significantly affect the pressure-volume responses up to 20 cm H2O (n = 6, data not shown), but apamin (300 nM) increased transients contractions during filling (2.2 ± 0.3 cm H2O, n = 6, P < 0.001 compared with control, Fig. 6a,b,d,e,g,h). After apamin, GSK increased the amplitude (to 5.1 ± 0.3 cm H2O, P < 0.001, n = 6) of the transient contractions (Fig. 6c,f–h). These effects were significant (p < 0.0001) by ANOVA.
Figure 6.

The effects of apamin and TRPV4 agonist on spontaneous activity of murine bladder using ex vivo preparation. Ex vivo pressure-volume response for control (a), after apamin application (b) and after addition of GSK in the presence of apamin (c). Expanded time scales with adjustment of baseline under control (d), apamin (e) and apamin + GSK (f) from panels a–c (red box). Blue lines and dots denote detected responses to analyze the amplitude distribution (see panel g). Amplitude and frequency distribution of transient contractions (TC) in control, apamin, and apamin + GSK treatment were analyzed from data in panels d–f (g). Summarized amplitude (h) under control, after apamin and apamin + GSK application (n = 6). Horizontal dotted lines in panels d–f denote baseline (0 cm H20). *P < 0.001 by ANOVA.
TRPV4 antagonist, RN-1734 increased the transient contractions from 0.8 ± 0.1 cm H2O to 3.8 ± 0.4 cm H2O (n = 6, P < 0.001) during the filling phase (Fig. 7a–f). SKA (10 µM), in the presence of RN-1734, decreased the amplitude of transient contractions during filling (Fig. 7g–i). We also tested pressure responses in Trpv4 −/− mice, and responses were similar in bladders of these mice as in presence of the TRPV4 antagonist (Fig. 7j,k). SKA-31 (10 µM) abolished the transient contractions during filling (Fig. 7j,l).
Figure 7.

The effects of TRPV4 antagonist and SK channel activator in wild type and Trpv4 −/− bladder on ex vivo preparation. Representative ex vivo pressure-volume curves in control (a) and RN-1734 (b). Expanded time scales with adjustment of baseline from red box in (a and b) (c,d). Blue lines and dots denote detected transient contractions (TC). Frequency and amplitude distribution of TC in control and after RN-1734 (RN) were analyzed by data in panels c and d (e). Summarized amplitude (f) in control and after RN- application (n = 6). Horizontal dotted lines in panels c and d denote baseline (0 cm H20). Representative ex vivo pressure-volume curves in RN-1734 (g) and continuous application of SKA-31 (10 μM) in wild type animal. Expanded time scales after baseline adjustment under RN-1734 only (h) and combination with SKA-31 (i) from panel g red box. Gap (//) denotes 10 min incubation. (j) Representative ex vivo pressure response curve in control and after application of SKA-31 (10 μM) in Trpv4 −/− animal. Expanded time scales after baseline adjustment under control (k) after SKA-31 (i) from panel j (red box). Gap (//) denotes 10 min incubation. ***P < 0.001, **P < 0.01.
Protein-protein interaction between TRPV4 and SK3 channels by western blot & Proximity Ligation Assay
The specificity of the TRPV4 and SK3 antibodies were confirmed by immunoblots of detrusor membrane fractions (lane 2&5; Fig. 8a). TRPV4 was detected in the immunoblots of detrusor homogenates immunoprecipitated with the SK3 antibody (lane 3; Fig. 8a). The reverse co-immunoprecipitation (lane 6) shows that SK3 was detected in the immunoblots of detrusor homogenates immunoprecipitated with TRPV4 antibody. TRPV4 and SK3 were not detected in immunoblots of the detrusor membrane fraction incubated with non-immune rabbit IgG. These results suggest that there are protein-protein interactions between TRPV4 and SK3. Interactions between TRPV4 and SK3 in PDGFRα+ cells were investigated further by proximity ligation assays (PLA). PDGFRα+ cells were distinguished by the expression of eGFP (Fig. 8b,c). Incubation of the isolated detrusor PDGFRα+ cells with goat anti-TRPV4 and rabbit anti-SK3 antibodies followed by PLA detection show several sites of interaction present in these cells (Fig. 8d).
Figure 8.

Co-immunoprecipitation and proximity ligation assay of TRPV4 and SK3 proteins. Immunoblotting (IB) for TRPV4 or SK3 in SK3 or TRPV4 immunoprecipitates (IP) of detrusor membrane fraction (n = 4, a). Lane 1, protein standards; lane 2, IB for TRPV4 in detrusor membrane fraction (1 μg); lane 3 IB for TRPV4 in SK3 IP; lane 4, IB for TRPV4 in non-immune rabbit IgG IP of detrusor membrane fraction; lane 5, IB for SK3 in detrusor membrane fraction (1 μg); lane 6, IB for SK3 in TRPV4 IP; lane 7, IB for SK3 in non-immune rabbit IgG IP of detrusor membrane fraction (n = 4, b). Isolated detrusor PDGFRα+ cell morphology visualized by DIC microscopy (c). The same cell verified as PDGFRα+ by GFP expression. PLA analysis of the same cell using goat anti-TRPV4 and rabbit anti-SK3 antibodies (d).
Discussion
This study showed that expression of TRPV4 in detrusor smooth muscle tissues is due primarily to expression in PDGFRα+ cells. Patch clamp experiments confirmed that the TRPV4 conductance is functional in PDGFRα+ cells, because the TRPV4 agonist, GSK1016790A, activated inward current in these cells and little or no current in SMCs. Following the initial inward current, GSK also activated an outward current in PDGFRα+ cells when the cells were held positive to E K, but not when cells were held at E K. The outward current was due to activation of a conductance with properties consistent with it being due to SK channels, and UCL blocked the outward current activated by GSK. When the SK channels were blocked or when cells were held at E K, GSK activated only inward current. TRPV4 antagonists blocked both the inward and outward currents activated by GSK. TRPV4 has mechanosensitive properties14, and we hypothesized that detrusor excitability might be stabilized by activation of TRPV4 and subsequent activation of SK channels during bladder filling. The linkage between activation of TRPV4 and SK channels may be enhanced by protein-protein interactions between these channels, such that Ca2+ influx via TRPV4 channels activates SK channels. The functional relationship between TRPV4 channels and SK channels was verified in contractile and ex vivo bladder experiments. Inhibition of SK channels resulted in decreased compliance and increased transient contractions during filling. The same results were observed when TRPV4 channels were blocked or deactivated genetically, and an SK channel agonist reversed these effects. These findings describe a novel myogenic mechanism for controlling detrusor excitability during bladder filling and provide a rationale for the design of new therapies for overactive bladder.
TRPV4 channels (aka OTRPC4, VRL-2, VR-OAC, and TRP12) were first identified as a conductance activated by osmotic cell swelling30–33. Since this initial report, the properties of TRPV4 channels have been characterized extensively, and hypo-osmotic stress, mechanical stretch, temperature (30–43 °C) and phorbol ester derivatives are known to be modulators of TRPV4 activation19–22,34–37. TRPV4 channels have been linked to many cellular functions in various tissues throughout the body. For example, in vascular endothelium TRPV4 channels are linked to activation of IK and SK channels and dilation of resistance arteries38,39. These data suggested that Ca2+ influx through TRPV channels can directly regulate Ca2+-dependent K+ channels. Recently, we discovered cells distinguished by PDGFRα immunolabeling in detrusor muscles. These cells expressed SK3 channels but did not display functional voltage-dependent Ca2+ channels5. Thus, the present study sought to investigate a Ca2+ entry mechanism that might be linked to regulation of SK3 channels. With a pipette solution containing Cs+ to replace K+, inward current was activated by a TRPV4 agonist. However, when cells were dialyzed with a K+-rich solution, brief inward currents were followed by sustained outward current (e.g. holding potential −60 mV). TRPV4 channels are relatively Ca2+ selective with a P Ca/P Na permeability ratio of about 629,32. In our experiments, removal of Ca2+ from the external solution did not abolish the activation of inward current by GSK, but the outward current phase was completely blocked under these conditions. When cells were exposed to Na+-free and Ca2+-free solution, the inward and outward currents activated by GSK were completely abolished. These data are consistent with the idea that Ca2+ entry through TRPV4 channels (i.e. the inward current in response to GSK) activates outward current carried by SK3 channels.
Protein-protein interactions between TRPV4 and SK3 channels have been reported in rat mesenteric endothelial cells previously using co-immunoprecipitation and double immunolabeling methods38. This interactions between these proteins also appears to exist in detrusor muscles. However, it is possible that associations between proteins could develop in cell homogenates and involve proteins that are not naturally associated in situ. Therefore, we performed proximity ligation assays and found close associations between TRPV4 and SK3 channels in intact PDGFRα+ cells. Ca2+ concentrations at the intracellular mouths of activated TRPV4 channels may be far higher than in the greater cytoplasm, as shown by the development of sparklets and spatial decay of these events when TRPV4 channels are opened40. Thus, close associations between TRPV4 and SK3 may serve to directly regulate the open probability of SK3 channels.
Evidence for direct electrical coupling between PDGFRα+ cells and SMCs, as occurs in the gastrointestinal tract41, includes: (i) PDGFRα+ cells have high expression of SK3 channels and activation of these channels results in outward current and hyperpolarization of cells; SMCs have very low expression of SK channels and display no outward current or hyperpolarization response to SK channel activation at physiological holding potentials (−40 mV)5. (ii) Activation of SK conductance in intact detrusor muscles causes hyperpolarization and relaxation5. (iii) Blocking SK channels with apamin or genetically deactivating SK3 channels cause enhanced detrusor contractions8,10. (iv) P2Y1 receptors are expressed by PDGFRα+ cells and responses of these cells to purines include activation of SK channels; SMCs express only P2X-like inward currents in response to purines6. An inhibitory response to purines is manifest in whole detrusor muscles and is blocked by P2Y1 antagonists and in P2ry1 −/− mice6. (v) TRPV4 is expressed by PDGFRα+ cells and an agonist for these channels activates inward current and is secondarily coupled to activation of SK currents (current study). vi) SMCs have low expression of TRPV4, no response is activated in SMCs by the TRPV4 agonist, and detrusor muscles relaxed in a concentration-dependent manner to TRPV4 agonists unless SK channels were blocked by apamin (current study). For electrical responses transduced by PDGFRα+ cells to affect detrusor contractility, low resistance electrical coupling must exist between PDGFRα+ cells and SMCs.
A functional role for TRPV4 channels in regulating contractile behavior of the urinary bladder has been investigated previously with drugs to activate or block TRPV4 channels in wild-type mice and global Trpv4 −/− mice15,16. Previous studies reported that GSK enhanced contractions in strips of muscle with the urothelium intact or removed, and these responses were attributed to effects on SMCs15,42. We were unable to reproduce these results in the many experiments we performed using the same experimental conditions on detrusor muscles from the same strain of mice (C57BL/6). In fact, GSK caused concentration-dependent inhibition of contractions across the range of concentrations tested in 37 of 44 detrusor smooth muscle strips. In a small number of muscles (7 of 44) high concentrations of GSK caused slight restoration of contractions. The reasons for the differences in results are unclear, but with little or no TRPV4 expression in SMCs, no current responses activated by GSK in SMCs, molecular and functional expression of TRPV4 channels in PDGFRα+ cells, and linkage between Ca2+ entry via TRPV4 and activation of SK3 channels in PDGFRα+ cells, it is difficult to understand how GSK could elicit contractile responses in intact muscles. We also found that when muscles were treated with apamin to block SK channels, GSK activated small contractile responses. Contractions in the presence of apamin were likely due to the inward current and depolarization responses elicited in PDGFRα+ cells because expression of TRPV4 is extremely low in SMCs, and accordingly these cells displayed no activation of current in response to GSK. Finally, we also examined the effects of the TRPV4 antagonist (RN-1734). This compound increased detrusor contractions and the SK channel agonist abolished these responses. Taken together, these data support the concept that TRPV4 channels form a source of Ca2+ entry in PDGFRα+ cells that is linked directly to activation of SK channels. We detected no inward current activation attributable to TRPV4 in SMCs in any of our experiments.
Ex vivo preparation experiments were used to test myogenic mechanisms involved in regulation of TCs. These studies showed that TRPV4/SK3 coupling in PDGFRα+ cells provides an important means of moderating detrusor excitability during bladder filling. Apamin enhanced the frequency and amplitude of contractions during filling, confirming the idea that SK channels regulate detrusor excitability. In the presence of apamin, GSK increased the amplitude of contractions, suggesting that the inward currents activated in PDGFRα+ cells by GSK (see Fig. 4) can exert a depolarizing effect on detrusor muscles in intact bladders in the absence of SK channel activation. Finally, RN-1734, an antagonist of TRPV4 channels, increased contractions during filling. The enhanced contractions when TRPV4 channels were blocked by RN-1734 were reversed by SKA-31, an agonist for SK channels. These findings show that TRPV4 channels are coupled to stabilization of detrusor excitability through activation of SK channels. This is a previously unrecognized regulatory mechanism provided by PDGFRα+ cells and contributing to the integrated responses of detrusor muscles.
The role of TRPV4 was also tested by performing ex vivo preparation experiments on bladders from Trpv4 −/− mice. In a previous study Trpv4 −/−mice displayed decreased frequency in voiding contractions as measured by in vivo cystometry and voiding spot analysis16,17. Also apparent in the responses Trpv4 −/− mice, but not commented upon in previous studies, was a significant increase in non-voiding contractions16,17. The decrease in voiding contractions was thought to be due to reduced TRPV4 in the urothelium and thus an altered sensory phenotype16. In the urothelium TRPV4 expression has been postulated to regulate stretch-mediated release of ATP, a primary sensory mediator regulating the output of bladder afferent neurons16,43. However, the hypothesized urothelial functions of TRPV4 channels cannot explain the increase in non-voiding contractions observed in Trpv4 −/− mice. In addition to the proposed sensory role of TRPV4 in the bladder, our data suggest a novel role for these channels in myogenic regulation of detrusor excitability. TRPV4 channels are mechanosensitive and can be activated by stretch of cells29,30,32,36. Thus our findings suggest the following myogenic mechanism: as detrusor smooth muscle tissues are stretched during bladder filling, activation of TRPV4 channels provides Ca2+ entry into PDGFRα+ cells; the rise in [Ca2+]i (possibly localized near the cytoplasmic mouths of TRPV4 channels) activates SK3 channels that are highly expressed in PDGFRα+ cells5; enhanced outward current through SK3 channels causes hyperpolarization which conducts to electrically-coupled SMCs, restraining the excitability of these cells and reducing the tendency of SMCs to generate transient contractions. This non-neural, moment-to-moment mechanism for controlling excitability of detrusor SMCs and increasing bladder compliance is a novel physiological regulatory feature of normal bladders. Loss-of-function of this mechanism in genetic models such as Trpv4 −/− mice or after pharmacological block of TRPV4 channels or SK3 channels leads to enhanced transient contractions and symptoms experienced in overactive bladder syndrome. Finding a means to selectively regulate this pathway may provide novel therapeutic approaches to controlling overactive bladder.
Methods
Animal preparation and Cell Isolation of detrusor PDGFRα+ cells and SMCs
All experimental procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the animal use protocol, reviewed and approved by the Institutional Animal Use and Care Committee at the University of Nevada. C57BL/6, Pdgfra tm11(EGFP)S ° r/J (both purchased from Jackson Laboratory, Bar Harbor, ME, USA), smMHC/Cre/eGFP (donated by Dr. Michael Kotlikoff, Cornell University), and Trpv4 −/− mice (Donated by Dr. Kevin Thorneloe, GlaxoSmithKline LLC)15 were used. We used male only (8 to 12 weeks old) to exclude the effect of female steroid hormone on the ion channel expression. Dissection of detrusor smooth muscle was performed as previously described5. Briefly, Detrusor single SMCs and PDGFRα+ cells were isolated from detrusor tissues by enzymatic digestion using a combination of papain and collagenase5. Single SMCs and PDGFRα+ cells were used for patch clamp experiments within 12 h of isolation at 30 °C.
Cell purification and transcriptional analysis
Fluorescence-activated cell sorting (FACS; Becton Dickinson FACSAria using blue laser 488 nm and GFP emission detector; 530/30 nm) was used to purify PDGFRα+ cells and eGFP/SMCs. Tests for cell purity were described previously5. According to the manufacturer’s instructions, total RNA was isolated from PDGFRα+ cells and eGFP/SMCs using illustra RNAspin Mini RNA Isolation kit (GE Healthcare, Little Chalfont, UK), and first-strand cDNA was synthesized using SuperScript III (Invitrogen, Carlsbad, CA, USA). PCR products with specific primers (Supplementary Table S1) using Go-Taq Green Master Mix (Promega Corp., Madison, WI, USA) were analyzed on 2% agarose gels and visualized by ethidium bromide. The same primers as PCR using Fast Syber green chemistry (Applied Biosystems, Foster City, CA, USA) on the 7900HT Real Time PCR System (Applied Biosysytems) were used for Quantitative PCR (qPCR). Standard curves were generated by PCR Regression analysis of the mean values of three multiplex qPCRs for the log10 diluted cDNA. To give transcriptional quantification of each gene relative to the endogenous Gapdh standard after log transformation of the corresponding raw data, unknown amounts of messenger RNA (mRNA) were plotted relative to the standard curve for each set of primers which graphically plotted using Microsoft Excel and Graphpad Prism (v. 3.0, Graphpad Software Inc., San Diego, CA, USA) softwares.
Electrophysiological recordings
When filled with the pipette solution, tip resistances were 3-4 MΩs. Whole cell configuration was achieved in CaPSS bath solution (mM): NaCl 135, KCl 5, MgCl2 1.2, CaCl2 2, Glucose 10, HEPES 10, pH 7.4 with Tris-base. The MnPSS bath solution (also called Ca-0 solution) was replaced with equimolar Mn2+ in CaPSS solution. The Ca2+−0/Na+−0 solution was replaced with equimolar Mn2+ and NMDG+ in CaPSS solution. The K+-rich/Cs+-rich pipette solution contained (mM): KCl/CsCl 135, CaCl2 0.012, MgATP 3, Na2GTP 0.1, Creatine phosphate disodium 2.5, EGTA 0.1, Glucose 10, HEPES 10, pH 7.2 with Tris-base, respectively. Whole cell voltage- and current-clamp techniques were performed. Cells were placed in a 0.5 ml chamber mounted on an inverted microscope (Nikon, Japan). PDGFRα+ cells were identified by the fluorescence of eGFP in nuclei5. SMCs were easily identified by their morphology. An Axopatch 200B amplifier with a CV-4 headstage (Molecular Devices, Sunnyvale, CA, USA) was used. For mechanical stretch experiments, a gigaohm seal was formed with one pipette for recording whole cell currents, and the second pipette was cell-attached to a micromanipulator and used for stretching cells. All data were analyzed using pCLAMP software (Molecular devices, Sunnyvale, CA, USA) and Graphpad Prism. All recordings were made at ~30 °C.
Isometric Force Measurement
Contractions of strips of murine detrusor muscles were measured by standard organ bath techniques6. Briefly, muscle strips were attached to a fixed mount and an isometric force transducer (Fort 10, WPI, Sarasota, FL, USA). Muscles were immersed in organ baths perfused with oxygenated (95% O2 and 5% CO2) Krebs–Ringer bicarbonate buffer (KRB) solution and bath temperature was maintained at 37.5 ± 0.5 °C. The muscles were exposed to different concentrations of TRPV4 agonist, GSK (GSK1016790A) in the absence and presence of apamin and allowed to respond for at least 5 min to each concentration when stable responses were obtained. TRPV4 antagonist and SK channel agonist and antagonist were also tested. The effects of GSK were analyzed by calculating the average amplitude, frequency, and AUC (the area under curve) during control conditions and 5 min after addition of GSK. Contractile activity was recorded by computer running LabChart8 (AD Instruments, Colorado Springs, CO, USA) and measurements of peak amplitude and sustained amplitude were obtained.
Ex vivo preparation
Bladders were removed from animals and the ureters were ligated closed at the vesicoureteric junctions. A single catheter was placed in the urethral opening to fill the bladder and simultaneously measure the pressure. Intravesical pressure was recorded with reference to the air (i.e. atmospheric pressure = 0) by a water-filled pressure transducer placed level with the bladder and connected to a quad-bridge amplifier (AD Instruments). The infusion rate was 25 µl/min by a Genie Touch automated syringe pump (Kent Scientific) with KRB solution at 37 °C. Filling was stopped when the bladder pressure reached 40–45 cm/H2O to avoid over-distention which can induce permanent tissue damage. The effects of TRPV4 channel agonist and antagonist and SK channel agonist and antagonist were tested. Trpv4 −/− mice were also used for ex vivo volume-pressure evaluation. Ex vivo data were analyzed by Clampfit (Molecular Devices) with baseline adjustment to examine the frequency and amplitude of transient contractions occurring during the filling phase.
Co-immunoprecipitation
Detrusor smooth muscles from two mice were placed into 0.5 ml of ice cold lysis buffer (mM; 50 Tris HCl pH 8.0, 60 ß-glycerophosphate, 100 NaF, 2 EGTA, 25 Na-pyrophosphate, 1 DTT, 0.001% antifoam (Sigma) and protease inhibitor tablet (Roche, Indianapolis, IA, USA), and homogenized using a Bullet Blender (stainless steel bead, speed 7, 5 min) (Next Advance, Averill Park, NY, USA). The homogenates were centrifuged at 16,000 × g at 4 °C for 10 min, and the supernatants were then centrifuged at 100,000 × g at 4 °C for 1 hour. the 100,000 × g pellet (membrane fraction) was resuspended into lysis buffer 0.1% SDS, and analyzed for protein content with the Bradford assay. For the co-immunoprecipitation, 500 µg of pellet protein was pre-cleared with 25 µl of protein A/G-Dynabeads suspension (Thermo Fisher Scientific, Waltham, MA, USA) (rotating for 30 min at 4 °C). The rabbit TRPV4 (10 µg) (Abcam, Cambridge, MA, USA) or the rabbit SK3 antibody (10 µg) (Alomone Labs, Jeruselem, Israel) were each crosslinked to 25 μl of protein A/G-Dynabeads with DSS (disuccinimidyl suberate), using the Pierce Crosslink Immuno-precipitation Kit (Thermo Fisher Scientific, Waltham, MA, USA). The pellet protein samples were incubated with the protein A/G-Dynabeads suspension for 2 hours with rotation at 4 °C. The beads were placed on the magnet, the supernatants (flow-through) removed, and the beads washed three times with 200 µl lysis buffer. Immunoprecipitated proteins were eluted with low pH elution buffer, and then neutralized with the high pH buffer.
Automated capillary electrophoresis and chemiluminescent western blotting
Analysis of the IP eluates was performed using a Wes instrument (ProteinSimple (San Jose, CA, USA). The samples were mixed with fluorescent 5X Master Mix and incubated at 95 °C for 5 min. The samples were loaded into the Wes plate (Wes 12–230 kDa Pre-filled Plates with Split Buffer) along with biotinylated protein ladder, blocking reagent, primary antibodies, ProteinSimple HRP-conjugated anti-rabbit secondary antibody, luminol-peroxide, and washing buffer. The plates and capillary cartridges were loaded into the Wes for electrophoresis and chemiluminescence immunodetection imaging by a CCD camera using default settings: electrophoresis, 375 volts 25 min; blocking, 5 min; primary antibody, 30 min; secondary antibody, 30 min; camera exposure times, 1 sec to 120 sec. Compass software (ProteinSimple) was used to acquire and analyze the data, and to generate gel images and chemiluminescence signal intensity values. The digital lane views (bitmaps) of the immunodetected protein bands were generated by Compass software, with each lane corresponding to an individual capillary tube. The protein immunodetection figure was created from the digitized data using Corel PhotoPaint and Corel Draw × 4 (Corel Corp., Ottawa, Ontario, Canada).
Proximity Ligation Assay (PLA)
Enzymatically isolated detrusor PDGFRα+ cells were incubated overnight on slides coated with murine collagen (2.5 mg/ml, BD Falcon, Franklin Lakes, NJ, USA), fixed in 4% paraformaldehyde for 4 min, and then permeabilized and blocked with PBS containing 0.2% tween-20 and 1% bovine serum albumin for 10 min at room temperature. PLA was performed following the manufacturer’s instructions (Duolink Detect, Olink Bioscience, Sweden). The fixed and permeabilized cells were incubated with goat anti-TRPV4 specific antibody (1:200 dilution) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 hour at room temperature, washed 3 times with PBS and then incubated with the rabbit anti-SK3 antibody (1:200 dilution) (Alomone Labs, Jerusalem, Israel) for 1 hour at room temperature. The slides were washed 3 times with PBS and then incubated with the Duolink minus-anti rabbit IgG and plus-anti goat IgG secondary antibodies (1:5 dilution) at 37 °C for 1 hour, followed by the ligation and amplification reactions (Duolink detection kit red, ex.598/em.634). Finally, mounting medium with DAPI was used. The slides were examined using a LSM510 Meta (Zeiss, Jena, Germany) or Fluoview FV1000 confocal microscope (Olympus, Center Valley, PA, USA). Confocal micrographs are digital composites of the Z-series of scans (0.5 μm optical sections of 7 μm thick sections). Settings were fixed at the beginning of both acquisition and analysis steps and were unchanged. Final images were constructed using FV10-ASW 2.1 software (Olympus). In negative control, primary anti-TMEM16a and Myl9 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) antibodies were used.
Drugs
All drugs and reagents including GSK1016790A, HC-067047, RN-1734, SKA-31, UCL1684 and 4α-PDD were purchased from Sigma (Sigma-Aldrich, Co., Louis, MO, USA). Apamin was purchased from Santa Cruz Biotech (Santa Cruz Biotechnology, Inc., Dallas, Texas, USA). Dimethyl sulfoxide (DMSO) was used to make stock solutions. Final concentration of DMSO in the bath solution was less than 0.01%.
Statistical analyses
All data were expressed as means ± S.E.M. All statistical analyses were performed using Graphpad Prism. Student’s paired, unpaired t test or one-way ANOVA were used to compare groups of data and differences were considered to be significant at P < 0.05.
Electronic supplementary material
Acknowledgements
Support for this project was obtained from NIH/NIDDK R01 DK098388.
Author Contributions
Most of the patch data were collected and analyzed by H.L., B.H.K. and L.E.P. performed cell sorting and molecular study. R.D.C., N.E.G. and H.-T.L. performed ex-vivo preparation and organ bath experiments, respectively. Immunoblot and PLA were performed by B.P.B., Y.X. and B.A.P. H.L., T.C.C., K.M.S. and S.D.K. shared in the design of experiments, interpretation of the data and the writing of the manuscript. All authors approved the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-12561-7.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hu TW, et al. Estimated economic costs of overactive bladder in the United States. Urology. 2003;61:1123–1128. doi: 10.1016/S0090-4295(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 2.Jayarajan J, Radomski SB. Pharmacotherapy of overactive bladder in adults: a review of efficacy, tolerability, and quality of life. Res Rep Urol. 2013;6:1–16. doi: 10.2147/RRU.S40034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koh BH, et al. Platelet-derived growth factor receptor-alpha cells in mouse urinary bladder: a new class of interstitial cells. J Cell Mol Med. 2012;16:691–700. doi: 10.1111/j.1582-4934.2011.01506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monaghan KP, Johnston L, McCloskey KD. Identification of PDGFRalpha positive populations of interstitial cells in human and guinea pig bladders. J Urol. 2012;188:639–47. doi: 10.1016/j.juro.2012.03.117. [DOI] [PubMed] [Google Scholar]
- 5.Lee H, Koh BH, Peri LE, Sanders KM, Koh SD. Functional expression of SK channels in murine detrusor PDGFR+ cells. J Physiol. 2013;591:503–13. doi: 10.1113/jphysiol.2012.241505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee H, Koh BH, Peri LE, Sanders KM, Koh SD. Purinergic inhibitory regulation of murine detrusor muscles mediated by PDGFRalpha+ interstitial cells. J Physiol. 2014;592:1283–93. doi: 10.1113/jphysiol.2013.267989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H, et al. UTP activates small-conductance Ca2+-activated K+ channels in murine detrusor PDGFRalpha+ cells. Am J Physiol Renal Physiol. 2015;309:F569–74. doi: 10.1152/ajprenal.00156.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Afeli SA, Rovner ES, Petkov GV. SK but not IK channels regulate human detrusor smooth muscle spontaneous and nerve-evoked contractions. Am J Physiol Renal Physiol. 2012;303:F559–68. doi: 10.1152/ajprenal.00615.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashitani H, Brading AF. Ionic basis for the regulation of spontaneous excitation in detrusor smooth muscle cells of the guinea-pig urinary bladder. Br J Pharmacol. 2003;140:159–69. doi: 10.1038/sj.bjp.0705320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrera GM, et al. Urinary bladder instability induced by selective suppression of the murine small conductance calcium-activated potassium (SK3) channel. J Physiol. 2003;551:893–903. doi: 10.1113/jphysiol.2003.045914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parajuli SP, Hristov KL, Soder RP, Kellett WF, Petkov GV. NS309 decreases rat detrusor smooth muscle membrane potential and phasic contractions by activating SK3 channels. Br J Pharmacol. 2013;168:1611–25. doi: 10.1111/bph.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adelman JP, Maylie J, Sah P. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245–69. doi: 10.1146/annurev-physiol-020911-153336. [DOI] [PubMed] [Google Scholar]
- 13.Lee WS, Ngo-Anh TJ, Bruening-Wright A, Maylie J, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin: cell surface expression and gating. J Biol Chem. 2003;278:25940–6. doi: 10.1074/jbc.M302091200. [DOI] [PubMed] [Google Scholar]
- 14.Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol. 2010;103:2–17. doi: 10.1016/j.pbiomolbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Thorneloe KS, et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1 -piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther. 2008;326:432–42. doi: 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- 16.Gevaert T, et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J Clin Invest. 2007;117:3453–62. doi: 10.1172/JCI31766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshiyama M, et al. Functional roles of TRPV1 and TRPV4 in control of lower urinary tract activity: dual analysis of behavior and reflex during the micturition cycle. Am J Physiol Renal Physiol. 2015;308:F1128–34. doi: 10.1152/ajprenal.00016.2015. [DOI] [PubMed] [Google Scholar]
- 18.Liedtke W, et al. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2003;100(Suppl 2):14531–6. doi: 10.1073/pnas.2235619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278:27129–37. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- 20.Kung C. A possible unifying principle for mechanosensation. Nature. 2005;436:647–54. doi: 10.1038/nature03896. [DOI] [PubMed] [Google Scholar]
- 21.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci U S A. 2003;100:13698–703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe H, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–77. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 23.Koh SD, et al. TREK-1 regulation by nitric oxide and cGMP-dependent protein kinase. An essential role in smooth muscle inhibitory neurotransmission. J Biol Chem. 2001;276:44338–46. doi: 10.1074/jbc.M108125200. [DOI] [PubMed] [Google Scholar]
- 24.Everaerts W, et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A. 2010;107:19084–9. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vincent F, et al. Identification and characterization of novel TRPV4 modulators. Biochem Biophys Res Commun. 2009;389:490–4. doi: 10.1016/j.bbrc.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Campos Rosa J, et al. Synthesis, molecular modeling, and pharmacological testing of bis-quinolinium cyclophanes: potent, non-peptidic blockers of the apamin-sensitive Ca(2+)-activated K(+) channel. J Med Chem. 2000;43:420–31. doi: 10.1021/jm9902537. [DOI] [PubMed] [Google Scholar]
- 27.Malik-Hall M, Ganellin CR, Galanakis D, Jenkinson DH. Compounds that block both intermediate-conductance (IK(Ca)) and small-conductance (SK(Ca)) calcium-activated potassium channels. Br J Pharmacol. 2000;129:1431–8. doi: 10.1038/sj.bjp.0703233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strobaek D, Jorgensen TD, Christophersen P, Ahring PK, Olesen SP. Pharmacological characterization of small-conductance Ca(2+)-activated K(+) channels stably expressed in HEK 293 cells. Br J Pharmacol. 2000;129:991–9. doi: 10.1038/sj.bjp.0703120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plant, T.D. & Strotmann, R. Trpv4. Handb Exp Pharmacol, 189–205 (2007). [DOI] [PubMed]
- 30.Liedtke W, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–35. doi: 10.1016/S0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nilius B, Prenen J, Wissenbach U, Bodding M, Droogmans G. Differential activation of the volume-sensitive cation channel TRP12 (OTRPC4) and volume-regulated anion currents in HEK-293 cells. Pflugers Arch. 2001;443:227–33. doi: 10.1007/s004240100676. [DOI] [PubMed] [Google Scholar]
- 32.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 33.Wissenbach U, Bodding M, Freichel M, Flockerzi V. Trp12, a novel Trp related protein from kidney. FEBS Lett. 2000;485:127–34. doi: 10.1016/S0014-5793(00)02212-2. [DOI] [PubMed] [Google Scholar]
- 34.Guler AD, et al. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–14. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryskamp DA, et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci. 2011;31(19):7089–101. doi: 10.1523/JNEUROSCI.0359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc Natl Acad Sci USA. 2014;111:1316–21. doi: 10.1073/pnas.1319569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe H, et al. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277:47044–51. doi: 10.1074/jbc.M208277200. [DOI] [PubMed] [Google Scholar]
- 38.Ma X, et al. Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension. 2013;62:134–9. doi: 10.1161/HYPERTENSIONAHA.113.01500. [DOI] [PubMed] [Google Scholar]
- 39.Sonkusare SK, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan MN, Francis M, Pitts NL, Taylor MS, Earley S. Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Mol Pharmacol. 2012;82:464–72. doi: 10.1124/mol.112.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horiguchi K, Komuro T. Ultrastructural observations of fibroblast-like cells forming gap junctions in the W/W(nu) mouse small intestine. J Auton Nerv Syst. 2000;80:142–7. doi: 10.1016/S0165-1838(00)00089-8. [DOI] [PubMed] [Google Scholar]
- 42.Young JS, et al. The passive and active contractile properties of the neurogenic, underactive bladder. BJU Int. 2013;111:355–61. doi: 10.1111/j.1464-410X.2012.11300.x. [DOI] [PubMed] [Google Scholar]
- 43.Birder L, et al. Activation of urothelial transient receptor potential vanilloid 4 by 4alpha-phorbol 12,13-didecanoate contributes to altered bladder reflexes in the rat. J Pharmacol Exp Ther. 2007;323:227–35. doi: 10.1124/jpet.107.125435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.