

Abstract
Lambert‐Eaton myasthenia (LEM) is a rare autoimmune disorder associated with debilitating muscle weakness. There are limited treatment options and 3,4‐diaminopyridine (3,4‐DAP) free base is an investigational orphan drug used to treat LEM‐related weakness. We performed a population pharmacokinetic/pharmacodynamic (PK/PD) analysis using 3,4‐DAP and metabolite concentrations collected from a phase II study in patients with LEM. The Triple Timed Up & Go (3TUG) assessment, which measures lower extremity weakness, was the primary outcome measure. A total of 1,270 PK samples (49 patients) and 1,091 3TUG data points (32 randomized patients) were included in the PK/PD analysis. A two‐compartment and one‐compartment model for parent and metabolite, respectively, described the PK data well. Body weight and serum creatinine partially explained the variability in clearance for the final PK model. A fractional inhibitory maximum effect (Emax) model characterized the exposure‐response relationship well. The PK/PD model was applied to identify a suggested dosing approach for 3,4‐DAP free base.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ LEM is a rare autoimmune disorder with debilitating muscle weakness and, although approved therapy exists in Europe, there is currently no US Food and Drug Administration approved therapy in the United States.
WHAT QUESTION DOES THIS STUDY ADDRESS?
☑ This study sought to perform a PK/PD analysis of 3,4‐DAP free base in patients with LEM using 3TUG as a measure of mobility.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This study shows that 3TUG is a useful measure to assess the improvement in disease‐related weakness in patients with LEM. This analysis identified potential sources of variability in the PK/PD of 3,4‐DAP free base and its dosing in patients with LEM.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study showed that 3TUG may be used as a useful measure of drug efficacy in patients with LEM. A concentration‐response relationship was observed with 3,4‐DAP free base in patients with LEM using the 3TUG assessment. The PK/PD model developed in this study helped us to identify a suggested dosing approach for 3,4‐DAP free base.
Lambert‐Eaton myasthenia (LEM) is a rare autoimmune disorder of the neuromuscular junction that results in debilitating muscle weakness and impaired mobility.1 The estimated incidence is reported to be 0.48 per million.2 In patients with LEM, autoantibodies are generated against the voltage‐gated calcium channels of the presynaptic neuronal membrane, which impairs calcium entry into the motor nerve terminal, and reduces the normal release of acetylcholine from synaptic vesicles.3, 4 In patients with LEM, weakness typically involves proximal weakness in the shoulders and hip girdle resulting in difficulty walking and climbing stairs, and falls. The rate of disease progression over 6 months is much higher in patients with small cell lung cancer‐LEM than in patients with nontumorous LEM.2, 5 The investigational orphan drug, 3,4‐diaminopyridine (3,4‐DAP) free base, is reported to improve LEM‐related muscle weakness.6 3,4‐DAP phosphate is currently approved by the European Medicines Agency for treatment of LEM, but there is currently no approved treatment for LEM in the United States.
3,4‐DAP blocks the presynaptic voltage‐gated potassium channels resulting in a prolonged action potential and increased influx of calcium, which facilitates the release of acetylcholine from the motor nerve terminal.7, 8 The dose of 3,4‐DAP is typically titrated to effect.9 3,4‐DAP free base is predominantly metabolized to its pharmacologically inactive metabolite, 3‐N‐acetyl‐3,4‐diaminopyridine (3‐Ac DAP), by N‐acetyl transferases and >80% of the administered drug is excreted through renal elimination as 3‐Ac DAP.10, 11 3,4‐DAP free base is reported to have a moderate steady–state volume of distribution (159 L), a high clearance (118 L/h), and a half‐life of ∼0.5–2 hours.12 After oral administration, peak clinical effects are seen within 1.5 hours and are maintained for 3–8 hours.13 In clinical practice, individualized dose titration is used to establish the effective dose for each patient and daily doses of ≤100 mg are typically used due to concerns that seizures could be related to higher doses. The optimal dosing regimen remains uncertain.
To date, five double‐blind randomized, controlled trials have favorably supported the effectiveness of 3,4‐DAP in patients with LEM.12, 14, 15, 16, 17 One of these studies sought to investigate whether the combination of 3,4‐DAP and pyridostigmine leads to an improvement in LEM symptoms.12 After i.v. administration of both drugs, given alone and in combination, to 9 patients with LEM, the pharmacokinetic (PK) and pharmacodynamic (PD) analyses demonstrated that 3,4‐DAP, but not pyridostigmine, resulted in a significant improvement in muscle strength and the electrophysiological measure‐compound muscle action potential. All of these studies suggest that 3,4‐DAP is usually well‐tolerated with minimal adverse events and results in a significant improvement in isometric muscle strength and LEM symptoms. Total daily doses of up to 100 mg were reported, although lower doses sufficed for some patients.14 Further investigations are needed to determine the concentration‐response relationship of oral 3,4‐DAP free base in a larger cohort of patients with LEM to evaluate whether any clinical covariates explain between‐subject variability (BSV) in PK/PD parameters, and to provide dosing recommendations.
In addition to the five studies noted above, a recent double‐blind, randomized, placebo‐controlled phase II study sought to evaluate the PK/PD profile of 3,4‐DAP free base in patients with LEM treated for at least 3 continuous months.18 Drug efficacy during gradual withdrawal of 3,4‐DAP was measured using the triple Time Up & Go test (3TUG). The TUG test has a long history of successful implementation in geriatric studies and measures a patient's ability to arise from the chair, walk for a short distance, return to the chair, and sit back down.19, 20, 21, 22, 23 The 3TUG, a modification in which the TUG is repeated three times without rest, was felt to be an ideal measure of LEM‐related weakness because it directly assesses proximal lower extremity muscle weakness, a typical and often debilitating feature of LEM. The primary study endpoint was a 30% deterioration in 3TUG following dose tapering.
The purpose of our analysis was to develop a population PK/PD model of orally administered 3,4‐DAP free base using data obtained from this phase II trial in patients with LEM. The final population PK/PD model was applied to perform simulations to evaluate the predicted improvement in 3TUG response with varied 3,4‐DAP free base doses.
MATERIALS AND METHODS
Patient population
The data used to develop the PK and PD models in this study were collected through a multicenter, inpatient, double‐blind, placebo‐controlled withdrawal study of 3,4‐DAP free base performed in patients with known LEM (clinicaltrials.gov no. NCT01511978). Patients enrolled in this study had an established diagnosis of LEM and were on continuous use of 3,4‐DAP free base provided by Jacobus Pharmaceutical Company through physician‐held investigational new drug protocols for at least 3 months (use of a minimum of 3 doses per day with no single dose <10 mg of 3,4‐DAP free base). Additional study details are available in Supplementary Information S1.
Study design, drug dosing, and 3TUG assessment
Patients meeting the inclusion criteria were either randomized to maintain their current dosing regimen (group A) or withdrawn from 3,4‐DAP free base treatment (group B). The primary endpoint of the study was a >30% deterioration in 3TUG from baseline following dose tapering.
The study consisted of three consecutive inpatient stages. Briefly, during stage 1, patients were admitted for 2½ days of testing on their stable prestudy treatment regimen to establish each patient's baseline (first half day for acclimation to the testing facility). During this stage, patients who were on their usual doses, and were unable to attain a target 3TUG response (as measured by ≥27% improvement compared to the 3TUG before the first morning of the 3,4‐DAP dose on 2 consecutive days or ≥30% improvement after the first morning, afternoon, or evening dose on day 1 plus ≥12% improvement after the first dose on day 2) following administration of 3,4‐DAP free base, were considered to be “screening failure” patients. During stage II, patients were randomized to continuation of their 3,4‐DAP free base treatment regimen or a tapered withdrawal. The double‐blind withdrawal stage lasted up to 3½ days. Throughout the study, the patients in group A continued to receive their stable individualized doses, as suggested by their own neurologists. A patient who was randomized to withdraw 3,4‐DAP free base (group B) received identical tablets on the same schedule and in the same number that they received during the baseline period, with sequentially lower doses of 3,4‐DAP free base and ending the series with placebo. During stage III, all patients had their prestudy treatment regimens reinstituted, and remained at the facility for up to half a day of monitoring and additional testing pending improvement or sufficient recovery on 3TUG. Whole blood samples for assessments of plasma drug concentrations were obtained at −30, +30, +90, and +150 minutes around each dose on the second day of stage I, including time points that correspond with 3,4‐DAP trough and peak measurements. During stage II and for the first dose of stage III, sampling was limited to −30 and +90 minutes around each dose of the day (Supplementary Figure S1).
The 3TUG was assessed as the average of three consecutive TUG laps without rest. For each lap, the patient is required to stand up from an armchair and then walk at normal pace to a line 3 meters (∼10 feet) from the front of the chair, turn around, return to the chair, and sit back down. The 3TUG was performed six times per day (prior to and about 2 hours after the first doses after 12 am, 12 pm, and 5 pm).
Analytical methods
PK samples were shipped to the University of Florida (Gainesville, FL) and were analyzed using a validated liquid chromatography‐tandem mass spectrometry method. The lower limit of quantification was 0.5 ng/mL and a nitrogen‐15 stable isotope labeled with 3,4‐DAP was used as an internal standard. Additional details of the analytical method are available in Supplementary Information S1.
Determination of acetylator status
Genomic DNA was isolated from lymphocytes in whole blood using the Qiagen DNA Blood Isolation Kit (Qiagen, Valencia, CA). The N‐acetyltransferase 2 (NAT2) single nucleotide polymorphisms (SNPs) were genotyped using TaqMan allelic discrimination (Applied Biosystems, Foster City, CA). Acetylator status was inferred using NAT2‐specific SNPs.24, 25 Additional details of the NAT2 SNPs evaluated are available in Supplementary Information S1.
Population PK/PD model development
A sequential population PK/PD model was developed using the plasma concentrations of 3,4‐DAP and 3‐Ac DAP, and the 3TUG measurements. The nonlinear mixed effects modeling software NONMEM version 7.3, was used (ICON Development Solutions, Ellicott City, MD). The first‐order conditional estimation method with interaction was implemented for all model runs. Run management was performed using Pirana version 2.9.2.26 Visual predictive checks (VPCs) and bootstrapping was performed using Perl‐speaks‐NONMEM version 3.7.6.27 Data manipulation and visualization was performed using the software R version 3.2.0 (R Foundation for Statistical Computing, Vienna, Austria) and RStudio version 0.99.442 (RStudio, Boston, MA); with the lattice, xpose4, and ggplot2 packages used for the latter.28, 29, 30 For PK analysis of below the quantification limit (BQL) samples, a zero value was used for both the parent and metabolite. Additionally, the final population PK model parameter estimates were evaluated after discarding the BQL data. The molecular weights of 3,4‐DAP and 3‐Ac DAP (109.13 g/mol and 151.18 g/mol, respectively) were used for conversion to nM such that concentrations in ng/mL are nM*109.13/1000 for 3,4‐DAP and nM*151.18/1000 for 3‐Ac DAP.
In the PK analysis, 3,4‐DAP and 3‐Ac DAP concentrations were modeled simultaneously. One and two‐compartment models were tested for both the parent and metabolite. The structural model was selected based on an assessment of the objective function value (OFV), precision of parameter estimates, diagnostic plots, and model stability. Given that 3,4‐DAP is largely converted to 3‐Ac DAP, the fraction of parent converted to metabolite (Fm) was fixed to 1 to obtain an identifiable model.11 Thus, all clearance and volume parameters for 3‐Ac DAP are relative to the Fm and the bioavailability of 3,4‐DAP (F), which we summarized as F3ACDAP (the product of Fm and F).31 Once the PK model met acceptance criteria, the individual PK estimates were fixed and the PD analysis was performed.
The PD component of the model was described by a fractional inhibitory maximum effect (Emax) model (Eq. (1)).
| (1) |
Where E0 is the baseline 3TUG in the absence of 3,4‐DAP, Emax is the fraction of maximum effect from the baseline, EC50 is the 3,4‐DAP concentration that produces 50% of Emax, and Cp is the predicted plasma concentration. BSV in PK/PD model parameters was assessed using an exponential relationship (Eq. (2)). An exception was that a logit transformation was used to estimate BSV for Emax while constraining individual Emax values between 0 and 1.32, 33
| (2) |
Where Pij denotes the estimate for parameter j in the ith individual; θPop,j is the population value for the parameter j; and ηij denotes the deviation from average population value for parameter j in the ith individual with a mean of zero and variance of ω2. Initially, BSV was estimated for all 3,4‐DAP and 3‐Ac DAP parameters, and then an assessment of between‐subject random effect (η)‐shrinkage and model stability was used to determine which BSV parameters should be retained in the model. The covariance between BSV parameters for clearance and volume of distribution was estimated and a correlation coefficient was calculated.32 A proportional residual error model was used for both 3,4‐DAP and 3‐Ac DAP, and separate parameters were estimated for 3,4‐DAP and 3‐Ac DAP. As the study consisted of three consecutive inpatient stages, we evaluated the interoccasion variability in clearance for 3,4‐DAP using the base and final models.
Covariate analysis
Covariates were analyzed for inclusion in the PK and PD components of the model. Visual inspection was performed using box and scatter plots (for categorical and continuous covariates, respectively) of the individual deviations from typical population values in the PK and PD parameters (ETAs) against covariates. In the population PK model, the following covariates were explored: genotype, total body weight (TBW), serum creatinine (SCR), age, and gender. In the population PD model, the following covariates were explored: acetylator status, TBW, SCR, age, gender, pyridostigmine comedication, and acetaminophen comedication. A forward inclusion (P < 0.05 and ΔOFV >3.8) and backward elimination (P < 0.01 and ΔOFV >6.6) approach was used to evaluate statistical significance for inclusion of covariates in the model. Descriptive statistics of the individual empirical Bayesian estimates (EBEs) were calculated.
The relationship between TBW and PK parameters specifically was evaluated using an allometric relationship for clearance parameters (systemic clearance (CL/F), intercompartmental clearance (Q/F), and metabolite clearance (CLm/F3ACDAP)) and a linear scale for volume parameters (V/F for one‐compartment model; central compartment volume (Vc/F) and peripheral compartment volume for 2‐compartment model; Eqs. (3) and (4)). Fixed exponents of 0.75 and 1 for clearance and volume parameters, respectively, were applied.34, 35
| (3) |
| (4) |
Where CLstd and Vstd denote the population estimates of CL/F and V/F, respectively, WTm denotes the median TBW of the evaluated patients, and WTi represents the TBW for the ith patient.
Other continuous and categorical covariates were tested using a power model and centered using the median covariate value for the sample (Eqs. (5) and (6)).
| (5) |
| (6) |
Where covi represents the individual covariate value; covm denotes the median covariate value of population; θcov is a parameter that denotes the covariate effect; and cat is a categorical variable that takes a value between 0 and 1 for the categorical covariates (comedication administered, genotype status, and assisted status) analyzed. For example, the categorical variable takes a value of 1 when comedication was administered and 0 when comedication was not administered.
Population PK/PD model evaluation and validation
Standard model diagnostic methods were used for the assessment of PK/PD model performance, including successful minimization, evaluation of diagnostic plots, plausibility and precision of parameter estimates, changes in the OFV and shrinkage values.36 Precision of population PK and PD parameters were evaluated using nonparametric bootstrapping (500 replicates) to generate 95% confidence intervals of the parameter estimates. The VPCs for the base and final models were performed by generating 1,000 Monte Carlo simulation replicates per time point. Additional details of the diagnostic and VPC plots generated are available in Supplementary Information S1.
Dosing simulations
The final PK/PD model was used to perform dosing simulations. A virtual population of 50 patients (49 patients in the current study plus 1 patient with a 70 kg standardized patient weight) was simulated 1,000 times for each dosing regimen evaluated. Based on the typical doses and dosing frequency used in the clinic, the following dosing regimens were simulated: 5, 10, 20, and 30 mg administered 3 times daily; 15, 20, and 30 mg administered 4 times daily; 15 and 20 mg administered 5 times daily; and 20 mg administered 6 times daily. The parameters obtained from PK simulations were used to simulate the concentration vs. 3TUG responses. The percentage of 3TUG improvement following steady‐state dosing was calculated for each virtual patient based on their respective model‐generated baseline (E0). The percentage of virtual patients that achieved a greater than 10%, 20%, 30%, and 50% improvement in 3TUG at any of the simulated time points (0.5, 1, 2, 3, 4, 5, 8, 10, 14, 16, 20, 22, and 24 hours) within the dosing was computed for each dosing regimen. Similarly, the number of patients that achieved a greater than 10%, 20%, 30%, and 50% sustained improvement in 3TUG at all the simulated time points was also computed for each dosing regimen.
RESULTS
Patient characteristics
A total of 1,270 samples collected from 49 patients receiving oral 3,4‐DAP were included in the population PK analysis. A summary of clinical and laboratory data for all patients is provided in Table 1. The median (range) single dose of 3,4‐DAP taken at baseline when entering the study was 20 mg (range, 10–30 mg). The 3TUG data was available from 32 patients who were randomized to continue treatment or taper 3,4‐DAP. All 1,091 available 3TUG data points for the 32 randomized patients were used for the PK/PD analysis. The median of 3TUG assessments per patient was 36 (range, 27–37). The median total daily 3,4‐DAP dose taken by the 32 randomized patients was 80 mg (range, 30–100 mg). A total of 26 (2%) PK samples were BQL for 3,4‐DAP and none for 3‐Ac DAP. The median number of PK samples per patient, including screening failures, was 29 (range, 4–59). Genotyping was performed on 11 (22%) study patients: 5 (10%), 5 (10%), and 1 (2%) were slow, intermediate, and rapid acetylators, respectively. For the remaining patients (38 (78%)), genotype data were not available.
Table 1.
Clinical data
| Covariate a | No. of patients b | Median (range) or count (%) |
|---|---|---|
| Age, years | 49 | 60.0 (23.0–83.0) |
| Weight, kg | 49 | 82.6 (45.8–131.5) |
| Serum creatinine, mg/dL | 49 | 0.8 (0.5–1.5) |
| BUN, mg/dL | 49 | 13.0 (3.0–23.0) |
| AST, U/L | 43 | 24.0 (10.0–50.0) |
| ALT, U/L | 43 | 22.0 (9.0–58.0) |
| Total bilirubin, mg/dL | 43 | 0.6 (0.2–1.9) |
| Direct bilirubin, mg/dL | 6 | 0.2 (0.1–0.5) |
| ALP, IU/L | 43 | 60.0 (38.0–215.0) |
| Albumin, g/dL | 43 | 4.0 (3.1–5.0) |
| Hemoglobin, g/dL | 49 | 14.2 (8.6–17.1) |
| Male sex | 49 | 23 (47%) |
| White race | 49 | 46 (94%) |
| Hispanic ethnicity | 49 | 1 (2%) |
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen.
aDescriptive statistics are calculated based on values at the time of the first recorded dose.
bNo. of patients signifies the number of patients with a measurement available for each respective variable. Forty‐nine and 32 patients contributed pharmacokinetic (PK) and pharmacodynamic data, respectively. The data for all 49 patients used for population PK model development are reported in this table.
Population PK model development and evaluation
The 3,4‐DAP and 3‐Ac DAP plasma concentration vs. time data are shown in Figure 1. A two‐compartment model for 3,4‐DAP and a one‐compartment model for 3‐Ac DAP described the PK data well. A summary of the covariate analysis for the PK model is presented in Supplementary Table S1. During the univariable analysis, TBW on CL/F, Vp, and CLm/F3ACDAP caused the highest drop of 11.6 points in the OFV, despite no additional parameters being estimated. Thus, all additional covariate relationships were evaluated after accounting for TBW on these parameters. In the multivariable analysis step, inclusion of SCR on CLm/F3ACDAP caused a further drop in OFV (8.5 points). Inclusion of the effect of SCR on CLm/F3ACDAP reduced the BSV on this parameter from 42.2% to 38.6%. This indicates that changes in renal function may explain a relatively small amount of the BSV in the clearance of 3‐Ac DAP for these study subjects. Thereafter, genotype on CL/F caused a further drop in the OFV; however, it was removed from the model in the backward elimination step due to a less than significant increase in the OFV (+5.0 points).
Figure 1.
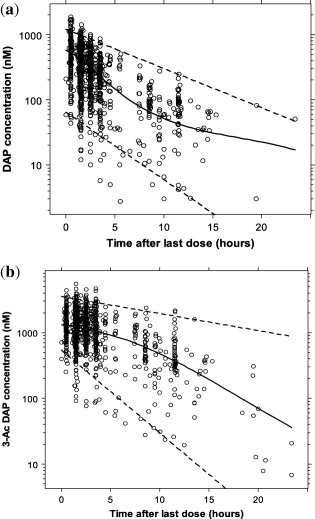
(a) 3,4‐diaminopyridine (3,4‐DAP) and (b) 3‐N‐acetyl‐3,4‐diaminopyridine (3‐Ac DAP) concentration (nM) vs. time after last dose (h) for all patients. The solid line represents the loess curve and the dashed lines represent the 2.5th and 97.5th percentiles of 3,4‐DAP and 3‐Ac DAP concentrations. The molecular weights of 3,4‐DAP and 3‐Ac DAP (109.13 g/mol and 151.18 g/mol, respectively) were used for conversion to nM such that concentrations in ng/mL are nM*109.13/1000 for 3,4‐DAP and nM*151.18/1,000 for 3‐Ac DAP.
The population PK parameter estimates and bootstrap analysis are summarized in Table 2. For the power function describing the effect of SCR on CLm/F3ACDAP , the observed exponent for SCR is positive; however, as implemented, the relationship predicts a decrease in CLm/F3ACDAP with SCR increases. The η‐shrinkage for KA, CL/F, Vc/F, Q/F, Vp/F, and CLm/F3ACDAP in the final model was 28%, 26%, 38%, 31%, 1%, and 5%, respectively. The ɛ‐shrinkage was 17%. The final population PK model parameters were estimated after discarding the BQL data and there were only minor changes in the parameter estimates. For the final model, the interoccasion variability in clearance was low (8%) and, thus, was not included in the final model.
Table 2.
Population PK parameter estimates and bootstrap analysis results for the final model
| Final model | Bootstrap b | ||||
|---|---|---|---|---|---|
| Parameter a | Estimate | RSE (%) | 2.5th percentile | Median | 97.5th percentile |
| KA, h−1 | 0.9 | 8.9 | 0.6 | 0.9 | 1.1 |
| CL/F, L/h | 90 | 7.4 | 71.6 | 88.0 | 107.7 |
| Vc/F, L | 24 | 33.3 | 1.2 | 16.8 | 72.6 |
| Q/F, L/h | 111 | 12.1 | 71.1 | 105.3 | 147.7 |
| Vp/F, L | 669 | 17.6 | 425.0 | 623.4 | 902.2 |
| CLm/F3ACDAP , L/h | 20.5 | 6 | 17.8 | 20.5 | 23.3 |
| Vm/F3ACDAP , L | 36 | 10.7 | 28.3 | 36.9 | 52.3 |
| Exponent for SCR on CLm/F3ACDAP c | 0.7 | 31.8 | 0.3 | 0.7 | 1.1 |
| Between subject variability (%CV) | |||||
| KA, % | 36.2 | 47.3 | 8.2 | 35.6 | 70.3 |
| CL/F, % | 48.1 | 28.9 | 33.5 | 47.6 | 61.6 |
| Vc/F, % | 182.4 | 36 | 100.7 | 208.5 | 401.1 |
| Q/F, % | 48.7 | 44.3 | 25.6 | 49.6 | 79.1 |
| Vp/F, % | 89.8 | 41.8 | 44.7 | 80.7 | 119.9 |
| CLm/F3ACDAP, % | 38.6 | 21.3 | 27.7 | 37.3 | 44.8 |
| CL/F‐Vc/F covariance d | 0.5 | 42.4 | −0.4 | 0.6 | 1.4 |
| Residual error | |||||
| 3,4‐DAP proportional error, % | 34.8 | 16.7 | 29.2 | 34.3 | 42.1 |
| 3‐Ac DAP proportional error, % | 20.1 | 12.7 | 17.8 | 19.9 | 23.3 |
3‐Ac DAP, 3‐N‐acetyl‐3,4‐diaminopyridine; 3,4‐DAP, 3,4‐diaminopyridine; CL/F, CLm/F3ACDAP , and Q/F, apparent oral clearance (L/h) for parent, metabolite, and intercompartment clearance (L/h), respectively; CV, coefficient of variation; F3ACDAP is the product of Fm and F; Fm, fraction of 3,4‐DAP converted to its metabolite (3‐Ac DAP); KA, absorption rate; RSE, relative standard error; SCR, serum creatinine; Vc/F, Vp/F and Vm/F3ACDAP, volume of distribution (L) for central compartment, parent, and metabolite respectively.
aPopulation estimates for CL/F, V/F, and CLm/F3ACDAP were scaled using the median weight of 82 kg: CL (L/h)=90*(WT/82)0.75;Vp/F(L)=669*(WT/82); CLm/F3ACDAP (L/h)=20.5*(WT/82)0.75*(0.8/SCR)0.7. WT is the body weight. bA nonparametric bootstrap analysis of the final model was performed with 500 replicates. cThe following mathematical relationship describes the exponent for SCR on CLm/F3ACDAP: CLm/F3ACDAP (L/h)=20.5*(WT/82)0.75*(0.8/SCR)0.7, where 0.8 is the median serum creatinine in mg/dL. dCorrelation coefficient for CL and Vc between subject variability parameters: 0.53.
The diagnostic plots for the final population PK model are shown in Figure 2 and Supplementary Figure S2. Some model misspecification is observed at the higher and lower concentration range of the 3,4‐DAP observations vs. individual predictions and population predictions plots. The VPCs indicated that the final model described the PK data adequately (Figure 2 c). In both VPCs, there was good agreement between the 2.5th and 50th percentile for the simulated and observed data, but the variability is slightly overestimated (as reflected by comparison of the 97.5th percentiles). The median values from the bootstrap analysis are close to the final model estimates, but the wide confidence intervals are possibly due to the small sample size. Individual EBEs of PK parameters obtained using the final populations PK model are summarized in Supplementary Table S2.
Figure 2.

(a) The 3,4‐diaminopyridine (3,4‐DAP) and (b) 3‐N‐acetyl‐3,4‐diaminopyridine (3‐Ac DAP) population and individual predictions vs. observations (nM). (c) Visual predictive check for 3,4‐DAP and 3‐Ac DAP using final population pharmacokinetic model. The solid black line in a and b is the line of unity. The shaded region in c denotes the 95% prediction interval.
Sequential PK/PD model development and evaluation
A fractional inhibitory Emax model characterized the exposure‐response relationship well. No covariates resulted in statistically significant reduction in the OFV. The final population PK/PD model parameters are shown in Table 3. The concentration‐response relationship is characterized according to the following equation: Effect (sec)=18.2*[1−(0.816*Cp)/(29.8+Cp)] with EC50 expressed in ng/mL. The corresponding EC50 value in nM is 273 nM. Diagnostic plots for the sequential PK/PD model are shown in Figure 3 a and Supplementary Figure S3. The model describes the data adequately, although some misspecification was observed for a limited number of average 3TUG values >20 seconds. The VPC indicated that the final PK/PD model described the data adequately (Figure 3 b); however, a comparison of the 97.5th percentiles for the observed and simulated data indicates overestimation of the variability. Thus, the results of the stochastic dosing simulations need to be interpreted with caution. The η‐shrinkage estimates for the BSV parameters on fractional Emax, EC50, and E0 were 20%, 30%, and 1%, respectively, and the ɛ‐shrinkage was 3%. Individual EBEs of PD parameters obtained using the final population PK/PD parameters are summarized in Supplementary Table S2.
Table 3.
Population PK/PD final model parameter estimates
| Final model | Bootstrap a | ||||
|---|---|---|---|---|---|
| Parameter | Estimate | RSE (%) | 2.5th percentile | Median | 97.5th percentile |
| Fractional Emax | 0.816 | 16 | 0.623 | 0.803 | 0.984 |
| EC50, ng/mL | 29.8 | 36 | 17.5 | 29.1 | 52.9 |
| E0, sec | 18.2 | 11.5 | 14.8 | 18.0 | 23.7 |
| Between subject variability, %CV | |||||
| Fractional Emax (%) b | 41 (2.93) | 71 | (0.35) | (2.84) | (12.85) |
| EC50, % | 88.3 | 63 | 16.4 | 84.0 | 358.5 |
| E0, % | 71.3 | 61 | 33.0 | 69.7 | 111.1 |
| Residual error | |||||
| Proportional error, % | 21.4 | 31 | 14.3 | 20.8 | 27.7 |
CV, coefficient of variation; EC50, the 3,4‐diaminopyridine (3,4‐DAP) concentration that produces 50% of Emax; E0, baseline Triple Timed Up & Go in the absence of 3,4‐DAP; Fractional Emax, the fraction of maximum effect from the baseline; RSE, relative standard error.
aA nonparametric bootstrap analysis of the final model was performed with 500 replicates. b%CV for Emax based on 1,000 simulations (SD(Emax,i)/mean(Emax,i)), the variance for the logit transform of Emax is shown in parentheses.33
Figure 3.
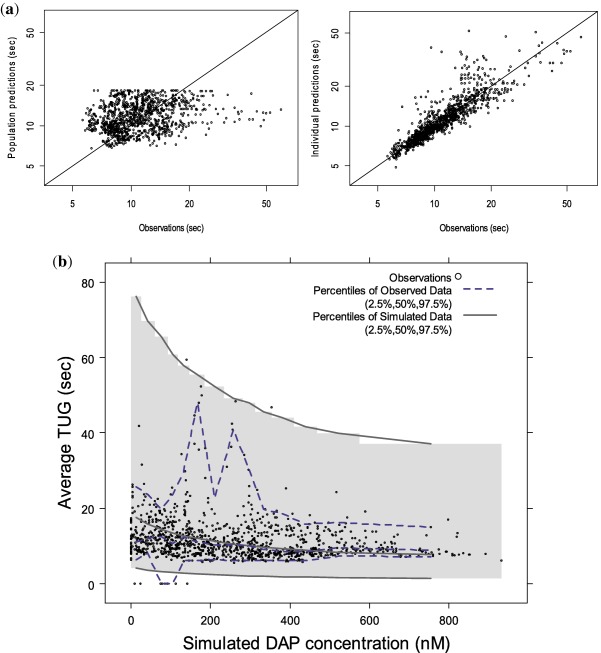
(a) Population and individual predictions vs. observations for observed average Triple Timed Up & Go (3TUG) (sec) and (b) visual predictive check for the final population pharmacokinetic/pharmacodynamic model. The shaded region in b denotes the 95% prediction interval. The molecular weights of 3,4‐diaminopyridine (3,4‐DAP; 109.13 g/mol) was used for conversion to nM such that concentrations in ng/mL are nM*109.13/1,000 for 3,4‐DAP. Observed 3TUG data points that were above 65 seconds (2 points (0.2%) of 1,091 points) were excluded in these figures for better visualization of the data.
Dosing simulations
Table 4 summarizes the percentage of virtual patients that achieved a greater than 10, 20, and 30% improvement in 3TUG with 3,4‐DAP dosing 3 to 6 times daily. The majority of virtual patients (>90%) had greater than 10% and 20% improvement in 3TUG times. A 20‐mg 5 or 6 times daily dosing regimen resulted in a 30% improvement in 3TUG in >88% of virtual patients. To achieve improvements of >50%, more frequent dosing would be required. Based on the simulations, there is an improvement in 3TUG in the majority of patients at one of the simulated time points, but a sustained improvement in this measure is observed in few virtual patients (Supplementary Table S3). The simulated sustained 3TUG percentage change from baseline seems to be higher during the first 2 hours after the last dose for the 20‐mg 4 times daily dosing regimen (Supplementary Figure S4). These findings support that some patients will need higher doses or more frequent dosing to achieve greater improvements in 3TUG from baseline.
Table 4.
Simulated percentage of patients with improvement in average 3TUG response
| Total oral daily dose | Improvement in 3TUG from baseline (therapeutic regimen) | >10% | >20% | >30% | >50% |
|---|---|---|---|---|---|
| 15 mg | 5 mg 3 times daily | 99.7% | 92.8% | 60.2% | 2.5% |
| 30 mg | 10 mg 3 times daily | 99.8% | 95.0% | 67.8% | 6.1% |
| 60 mg | 20 mg 3 times daily | 99.9% | 97.1% | 77.8% | 14.4% |
| 60 mg | 15 mg 4 times daily | 99.9% | 96.9% | 78.7% | 16.8% |
| 75 mg | 15 mg 5 times daily | 99.9% | 97.8% | 85.5% | 29.8% |
| 80 mg | 20 mg 4 times daily | 99.9% | 97.8% | 82.9% | 23.0% |
| 90 mg | 30 mg 3 times daily | 100.0% | 98.1% | 83.5% | 22.4% |
| 100 mg | 20 mg 5 times daily | 99.9% | 98.5% | 89.2% | 38.0% |
| 120 mg | 20 mg 6 times daily | 99.9% | 98.3% | 88.9% | 39.5% |
| 120 mg | 30 mg 4 times daily | 100.0% | 98.6% | 88.4% | 33.8% |
3TUG, Triple Timed Up & Go.
DISCUSSION
LEM is a rare autoimmune disorder characterized by impaired mobility and muscle weakness. Currently, there is no well‐defined efficacy measurement and different end points, such as compound muscle action potential, isometric muscle strength, and 3TUG, have been used to assess improvement in mobility of patients with LEM.12, 16 In clinical practice, 3,4‐DAP free base is titrated in each patient based on clinical response, but the optimum starting dose and dosing regimen for chronic treatment are unknown. Performing PK/PD studies in this patient population is challenging due to the limited number of patients with the condition, varying disease status, and lack of a well‐defined metric to assess drug efficacy. The current analysis investigates the effect of changing or tapering the dose and the impact of potential covariates on the PK/PD of 3,4‐DAP free base in the largest number of patients with LEM studied to date. The PK/PD model was applied to identify a suggested dosing approach for 3,4‐DAP free base in patients with LEM.
The population estimates obtained in our study (3,4‐DAP Vc/F and CL/F of 24 L and 90 L/h, respectively) are comparable to the previously published values for patients with LEM (34 L and 118 L/h).12 The PK component of the model included TBW and SCR as significant covariates that explain BSV in the PK of 3,4‐DAP and 3‐Ac DAP, respectively. Previous findings suggest that the majority of 3,4‐DAP is eliminated renally as 3‐Ac DAP and this explains the role of SCR as a predictor of metabolite clearance.11 However, a formal renal impairment study has not been performed and it is currently unknown whether dosing adjustments are needed. Additional covariates, such as age, gender, and genotype status, were tested and not found to be significant covariates in this population. In a previous study, NAT2 genotype was reported to play a role in the PK profile of 3,4‐DAP phosphate.11 In our study, genotype status was available for a limited number of patients (11/49) and this may explain the lack of an association in this study. Further studies are required to investigate the association of 3,4‐DAP free base and NAT2 genotype status. In the PD model, there was a lack of relationship between the average 3TUG effect and covariates, such as TBW, age, gender, SCR, and concomitant medications, which suggests that none of these clinical variables were important predictors of BSV in the 3TUG response.
Our results show that a concentration‐response relationship was achieved with 3TUG measurements. The 3TUG is a useful study endpoint in patients with LEM because it is a quick and noninvasive test that measures clinically relevant changes in patient's mobility and has been successfully demonstrated to be a primary outcome measure in geriatric studies and LEM.18, 19, 23 Consistent with our study findings, a concentration‐response relationship was observed in a previous study of 3,4‐DAP free base using dynamometry and compound muscle action potential.12
Different methods can be used to handle the BQL data. Although, in the current analysis, BQL samples were imputed to 0, we ran the final model after discarding the BQL samples and compared the parameter estimates. We found that the parameter estimates using both the methods were similar, which is consistent with the limited number of BQL samples in our data set (26 samples (2% of total samples)). Similarly, given that the majority of 3,4‐DAP is converted to 3‐Ac DAP, we fixed the Fm to 1, but also compared our results with fixing Fm to 0.81 based on previous findings and incorporating an additional clearance route.11 Based on the results, the population estimates and range of individual EBEs for PK/PD parameters, as well as the diagnostic plots and OFV, were comparable between the two models.
We performed simulations using multiple daily dosing regimens. Our simulation strategy corresponds with a clinically used approach for oral dosing of 3,4‐DAP free base starting with 5–10 mg doses administered 3 times daily to achieve >10 and 20% improvement in 3TUG and titrating the dose based on the observed response. A majority of the simulated patients (>90%) had 10% and 20% improvement in 3TUG times with all simulated dose regimens. Dose increases up to 20 mg 5 or 6 times or 30 mg 3 or 4 times daily resulted in the vast majority of virtual patients achieving a clinically relevant 30% improvement. The simulations show that more frequent dosing would be required to achieve greater 3TUG improvement from baseline. These findings are consistent with previous studies showing favorable effects in patients with LEM treated with 10 mg i.v. dose of 3,4‐DAP (suggested to be equivalent to ∼30 mg oral dose, administered 2–3 times).12, 37
CONCLUSIONS
A population PK/PD model was developed using data collected from a phase II study in patients with LEM. A concentration‐response relationship was observed with 3,4‐DAP free base in patients with LEM using the 3TUG assessment. Simulations showed that starting doses of 5 mg or 10 mg 3 times daily are likely to achieve a response in the majority of patients with LEM. Further dose increases to 20 mg up to 5 or 6 times daily or 30 mg 3 or 4 times daily may be warranted to optimize the response. The model predicts that even at total daily doses of 100 mg (maximum dose actually studied in DAPPER), 12% of patients with LEM may not experience a 30% improvement in 3TUG.
Supporting information
Additional Supplementary Information may be found in the online version of this article.
Supplementary Figure S1 Schematic representation of overall study design. (a) Different stages of the DAPPER trial. (b) Patients were randomized to either group A (continue active 3,4‐diaminopyridine (3,4‐DAP) or group B (withdrawal of 3,4‐DAP).
Supplementary Figure S2 Conditional weighted residuals (CWRES) vs. time (h) and population predictions (nM) for (a) 3,4‐diaminopyridine (3,4‐DAP) and (b) 3‐N‐acetyl‐3,4‐diaminopyridine (3‐Ac DAP).
Supplementary Figure S3 Conditional weighted residuals (CWRES) vs. time (h) and population predicted Triple Timed Up & Go (3TUG; sec).
Supplementary Figure S4 Simulated percent change in sustained Triple Timed Up & Go (3TUG) from baseline for 20 mg four times daily dosing regimen.
Supplementary Table S1 Summary of covariate analysis for population PK model development
Supplementary Table S2 Summary of individual empirical Bayesian parameter estimates for (a) PK model and (b) PK/PD model
Supplementary Table S3 Simulated percentage of patients with sustained improvement in average 3TUG response
Supplementary Information S1 Additional methods description
Supplementary Information S2 Final population PK and PK/PD model control streams
ACKNOWLEDGMENTS
DAPPER Study Group.
Duke University Medical Center: Vern C. Juel, MD (PI), Jeffrey T. Guptill, MD (co–I), Janice M. Massey, MD (co–I), Lisa D. Hobson–Webb, MD (co–I), Katherine Beck, RN (coordinator).
Baylor College of Medicine: Yadollah Harati, MD (PI), Ali Arvantaj, MD (co–PI), Cecile Phan, MD (co–I), Shima Mousavi, MD (co–I), Claire MacAdam, PT (coordinator).
University of Utah School of Medicine: A Gordon Smith, MD (PI), J. Rob Singleton, MD (co–I), Charles Latner (coordinator), Meg Lessard (coordinator).
Vanderbilt Medical Center: Amanda Peltier, MD MS (PI), Peter D. Donofrio, MD (co–I), Christopher Lee, MD, MPH (co–I), Kay Artibee, RN Med (coordinator).
Oregon Health and Science University: Jau–Shin Lou, MD, PhD (PI, presently at University of North Dakota School of Medicine & Health Science), Tessa Marburger, MD (PI), Alexandra Dimitrova, MD (co–I), Brent Burroughs, MD (co–I), Ghazaleh Jafari, MD (co–I), Julie Khoury, MD (co–I), Mananya Satayaprasert, MD (co–I), Diana Dimitrova, PhD (coordinator).
Indiana University: Robert M. Pascuzzi, MD (PI), Cynthia Bodkin, MD (co–I), John Kincaid, MD (co–I), Riley Snook, MD (co–I), Angela Micheels PT (coordinator), Sandra Guingrich (coordinator).
University of California, Davis: David P. Richman, MD (PI), Mark A. Agius, MD (co–I), Janelle Butters, RN (coordinator), Molly Lindsay, CRC (coordinator), Vardenick Khalatyan, CRC (coordinator).
Author Contributions
N.T. and D.G. wrote the manuscript. N.T., C.P., K.A., D.J., L.J., M.C.W., J.T.G., and D.G. designed the research. N.T., C.P., K.A., M.C.W., J.T.G., and D.G. performed the research. N.T. and D.G. analyzed the data.
Conflict of Interest
This study was funded by Jacobus Pharmaceutical Company. D.G. receives support for research from the National Institute for Child Health and Human Development (K23HD083465), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and from industry (Cempra, Inc. and Jacobus Pharmaceutical Company, Inc.) for drug development in adults and children. M.C.W. receives support for research from the National Institutes of Health (NIH; 1R01‐HD076676–01A1), the National Center for Advancing Translational Sciences of the NIH (UL1TR001117), the NIAID (HHSN272201500006I and HHSN272201300017I), the NICHD (HHSN275201000003I), the US Food and Drug Administration (1U01FD004858‐01), the Biomedical Advanced Research and Development Authority (HHSO100201300009C), the nonprofit Thrasher Research Fund (www.thrasherresearch.org), and from industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). J.T.G. receives support for research from the NIH (K23NS085049), the Myasthenia Gravis Foundation of America and from industry for drug development in adults (www.dcri.duke.edu/research/coi.jsp). The remaining authors have no conflicts of interest to disclose.
References
- 1. Eaton, L.M. & Lambert, E.H. Electromyography and electric stimulation of nerves in diseases of motor unit; observations on myasthenic syndrome associated with malignant tumors. J. Am. Med. Assoc. 163, 1117–1124 (1957). [DOI] [PubMed] [Google Scholar]
- 2. Titulaer, M.J. , Lang, B. & Verschuuren, J.J. Lambert‐Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 10, 1098–1107 (2011). [DOI] [PubMed] [Google Scholar]
- 3. Lang, B. , Newsom‐Davis, J. , Prior, C. & Wray, D. Antibodies to motor nerve terminals: an electrophysiological study of a human myasthenic syndrome transferred to mouse. J. Physiol. 344, 335–345 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lang, B. , Pinto, A. , Giovannini, F. , Newsom‐Davis, J. & Vincent, A. Pathogenic autoantibodies in the Lambert‐Eaton myasthenic syndrome. Ann. NY Acad. Sci. 998, 187–195 (2003). [DOI] [PubMed] [Google Scholar]
- 5. Titulaer, M.J. et al The Lambert‐Eaton myasthenic syndrome 1988–2008: a clinical picture in 97 patients. J. Neuroimmunol. 201–202, 153–158 (2008). [DOI] [PubMed] [Google Scholar]
- 6. Lundh, H. , Nilsson, O. & Rosén, I. Novel drug of choice in Eaton‐Lambert syndrome. J. Neurol. Neurosurg. Psychiatry 46, 684–685 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kirsch, G.E. & Narahashi, T. 3,4‐diaminopyridine. A potent new potassium channel blocker. Biophys. J. 22, 507–512 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molgó, J. , Lundh, H. & Thesleff, S. Potency of 3,4‐diaminopyridine and 4‐aminopyridine on mammalian neuromuscular transmission and the effect of pH changes. Eur. J. Pharmacol. 61, 25–34 (1980). [DOI] [PubMed] [Google Scholar]
- 9. Lindquist, S. & Stangel, M. Update on treatment options for Lambert‐Eaton myasthenic syndrome: focus on use of amifampridine. Neuropsychiatr. Dis. Treat. 7, 341–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ng F, Lee DC, Schrumpf LA, Mazurek ME, Lee Lo V, Gill SK, Maselli RA. Effect of 3,4–diaminopyridine at the murine neuromuscular junction. Muscle Nerve 55, 223–231 (2017). [DOI] [PubMed] [Google Scholar]
- 11. Haroldsen PE, Garovoy MR, Musson DG, Zhou H, Tsuruda L, Hanson B, O'Neill CA. Genetic variation in aryl N–acetyltransferase results in significant differences in the pharmacokinetic and safety profiles of amifampridine (3,4–diaminopyridine) phosphate. Pharmacol Res Perspect. 3, e00099 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wirtz, P.W. et al Efficacy of 3,4‐diaminopyridine and pyridostigmine in the treatment of Lambert‐Eaton myasthenic syndrome: a randomized, double‐blind, placebo‐controlled, crossover study. Clin. Pharmacol. Ther. 86, 44–48 (2009). [DOI] [PubMed] [Google Scholar]
- 13. Sanders, D.B. 3,4‐Diaminopyridine (DAP) in the treatment of Lambert‐Eaton myasthenic syndrome (LEMS). Ann. NY Acad. Sci. 841, 811–816 (1998). [DOI] [PubMed] [Google Scholar]
- 14. McEvoy, K.M. , Windebank, A.J. , Daube, J.R. & Low, P.A. 3,4‐Diaminopyridine in the treatment of Lambert‐Eaton myasthenic syndrome. N. Engl. J. Med. 321, 1567–1571 (1989). [DOI] [PubMed] [Google Scholar]
- 15. Sanders, D.B. , Massey, J.M. , Sanders, L.L. & Edwards, L.J. A randomized trial of 3,4‐diaminopyridine in Lambert‐Eaton myasthenic syndrome. Neurology 54, 603–607 (2000). [DOI] [PubMed] [Google Scholar]
- 16. Oh, S.J. , Claussen, G.G. , Hatanaka, Y. & Morgan, M.B. 3,4‐Diaminopyridine is more effective than placebo in a randomized, double‐blind, cross‐over drug study in LEMS. Muscle Nerve 40, 795–800 (2009). [DOI] [PubMed] [Google Scholar]
- 17. Oh, S.J. et al Amifampridine phosphate (Firdapse(®)) is effective and safe in a phase 3 clinical trial in LEMS. Muscle Nerve 53, 717–725 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Sanders, D. , Jacobus, L. , Ales, K. & Jacobus, D. Predicting responsiveness to study drug before randomization in the DAPPER trial of 3,4‐diaminopyridine (3,4‐DAP) in Lambert‐Eaton myasthenic syndrome (LEMS) (P7.066). Neurology 84 (suppl.), P7.066 (2015). [Google Scholar]
- 19. Huang, S.L. , Hsieh, C.L. , Wu, R.M. , Tai, C.H. , Lin, C.H. & Lu, W.S . Minimal detectable change of the timed “up & go” test and the dynamic gait index in people with Parkinson disease. Phys. Ther. 91, 114–121 (2010). [DOI] [PubMed] [Google Scholar]
- 20. Wall, J.C. , Bell, C. , Campbell, S. & Davis, J. The timed get‐up‐and‐go test revisited: measurement of the component tasks. J. Rehabil. Res. Dev. 37, 109–113 (2000). [PubMed] [Google Scholar]
- 21. Steffen, T.M. , Hacker, T.A. & Mollinger, L. Age‐ and gender‐related test performance in community‐dwelling elderly people: Six‐Minute Walk Test, Berg Balance Scale, Timed Up & Go Test, and gait speeds. Phys. Ther. 82, 128–137 (2002). [DOI] [PubMed] [Google Scholar]
- 22. Bischoff, H.A. et al Identifying a cut‐off point for normal mobility: a comparison of the timed ‘up and go’ test in community‐dwelling and institutionalised elderly women. Age Ageing 32, 315–320 (2003). [DOI] [PubMed] [Google Scholar]
- 23. Nordin, E. , Lindelöf, N. , Rosendahl, E. , Jensen, J. & Lundin‐Olsson, L. Prognostic validity of the Timed Up‐and‐Go test, a modified Get‐Up‐and‐Go test, staff's global judgement and fall history in evaluating fall risk in residential care facilities. Age Ageing 37, 442–448 (2008). [DOI] [PubMed] [Google Scholar]
- 24. Hein, D.W. & Doll, M.A. Accuracy of various human NAT2 SNP genotyping panels to infer rapid, intermediate and slow acetylator phenotypes. Pharmacogenomics 13, 31–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuznetsov, I.B. , McDuffie, M. & Moslehi, R. A web server for inferring the human N‐acetyltransferase‐2 (NAT2) enzymatic phenotype from NAT2 genotype. Bioinformatics 25, 1185–1186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keizer, R.J. , van Benten, M. , Beijnen, J.H. , Schellens, J.H. & Huitema, A.D. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput. Methods Programs Biomed. 101, 72–79 (2011). [DOI] [PubMed] [Google Scholar]
- 27. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 28. Jonsson, E.N. & Karlsson, M.O. Xpose–an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 29. Wickham, H. ggplot2 Elegant Graphics for Data Analysis (Springer, New York, NY, 2009). [Google Scholar]
- 30. Sarkar, D. Lattice: Multivariate Data Visualization with R (Springer, New York, NY, 2008). [Google Scholar]
- 31. Salman, S. et al Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob. Agents Chemother. 55, 5306–5313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Owen, J.S. & Fiedler‐Kelly, J. Introduction to Population Pharmacokinetic/Pharmacodynamic Analysis With Nonlinear Mixed Effects Models (John Wiley & Sons, Inc, Hoboken, NJ, 2014). [Google Scholar]
- 33. Trame, M.N. , Bergstrand, M. , Karlsson, M.O. , Boos, J. & Hempel, G. Population pharmacokinetics of busulfan in children: increased evidence for body surface area and allometric body weight dosing of busulfan in children. Clin. Cancer Res. 17, 6867–6877 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Wang, C. et al A bodyweight‐dependent allometric exponent for scaling clearance across the human life‐span. Pharm. Res. 29, 1570–1581 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mahmood, I. Interspecies scaling: predicting volumes, mean residence time and elimination half‐life. Some suggestions. J. Pharm. Pharmacol. 50, 493–499 (1998). [DOI] [PubMed] [Google Scholar]
- 36. Karlsson, M.O. & Savic, R.M. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 82, 17–20 (2007). [DOI] [PubMed] [Google Scholar]
- 37. Lundh, H. , Nilsson, O. , Rosén, I. & Johansson, S. Practical aspects of 3,4‐diaminopyridine treatment of the Lambert‐Eaton myasthenic syndrome. Acta Neurol. Scand. 88, 136–140 (1993). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supplementary Information may be found in the online version of this article.
Supplementary Figure S1 Schematic representation of overall study design. (a) Different stages of the DAPPER trial. (b) Patients were randomized to either group A (continue active 3,4‐diaminopyridine (3,4‐DAP) or group B (withdrawal of 3,4‐DAP).
Supplementary Figure S2 Conditional weighted residuals (CWRES) vs. time (h) and population predictions (nM) for (a) 3,4‐diaminopyridine (3,4‐DAP) and (b) 3‐N‐acetyl‐3,4‐diaminopyridine (3‐Ac DAP).
Supplementary Figure S3 Conditional weighted residuals (CWRES) vs. time (h) and population predicted Triple Timed Up & Go (3TUG; sec).
Supplementary Figure S4 Simulated percent change in sustained Triple Timed Up & Go (3TUG) from baseline for 20 mg four times daily dosing regimen.
Supplementary Table S1 Summary of covariate analysis for population PK model development
Supplementary Table S2 Summary of individual empirical Bayesian parameter estimates for (a) PK model and (b) PK/PD model
Supplementary Table S3 Simulated percentage of patients with sustained improvement in average 3TUG response
Supplementary Information S1 Additional methods description
Supplementary Information S2 Final population PK and PK/PD model control streams