

Abstract
Bacteria employ remarkable membrane-bound nanoarrays to sense their environment and direct their swimming. Arrays consist of chemotaxis receptor trimers of dimers that are bridged at their membrane-distal tips by rings of two cytoplasmic proteins, a kinase CheA and a coupling protein CheW. It is not clear how ligand binding to the periplasmic domain of the receptor deactivates the CheA kinase bound to the cytoplasmic tip ∼300 Å away, but the mechanism is thought to involve changes in dynamics within the cytoplasmic domain. To test these proposals, we applied solid-state NMR mobility-filtered experiments to functional complexes of the receptor cytoplasmic fragment (U–13C,15N-CF), CheA, and CheW. Assembly of these proteins into native-like, homogeneous arrays is mediated by either vesicle binding or molecular crowding agents, and paramagnetic relaxation enhancement is used to overcome sensitivity challenges in these large complexes. INEPT spectra reveal that a significant fraction of the receptor is dynamic on the nanosecond or shorter time scale, and these dynamics change with signaling state. The mobile regions are identified through a combination of biochemical and NMR approaches (protein truncations and unique chemical shifts). The INEPT spectra are consistent with an asymmetric mobility in the methylation region (N-helix mobility ≫ C-helix mobility) and reveal an increase in the mobility of the N-helix in the kinase-off state. This finding identifies functionally relevant dynamics in the receptor, and suggests that this N-helix segment plays a key role in propagating the signal.
Introduction
It is increasingly clear that protein dynamics play an important role in signaling pathways. Binding of a signaling molecule sometimes induces no change in the protein structure, but instead alters the protein dynamics.1 Bacterial chemotaxis receptors, an excellent system for investigating mechanisms of transmembrane signaling, are thought to undergo changes in dynamics as part of the signaling mechanism.2 Like most proteins that perform critical processes for the cell, chemotaxis receptors function within a multiprotein complex.3 Measurement of dynamics within functional complexes of chemoreceptors will provide insight into the mechanistic role of protein dynamics in transmembrane signaling.
Bacterial chemotaxis involves large (>100 MDa) multiprotein hexagonal arrays of transmembrane receptors that transmit signals into the cell to control swimming direction. These arrays, which have been observed in a wide range of chemotactic bacteria as well as in some Archaea,4 enable signaling with high sensitivity, cooperativity, and wide dynamic range. In native arrays in bacteria, the homodimeric helical receptors are arranged as hexagons of trimers of dimers, and these hexagons are bridged at their cytoplasmic tips by rings of two alternating proteins, a histidine kinase CheA, and a coupling protein CheW (Figure 1).5,6 In Escherichia coli, these receptors activate CheA phosphorylation, which is followed by phosphotransfer to response regulators CheB and CheY. CheY induces tumbling by interacting with the flagellar motor and changing its direction of rotation. Binding of attractant ligands inactivates the kinase, which suppresses tumbling until adaptation restores kinase activation and the basal tumbling level. Adaptation is mediated by methylation/demethylation of 4 glutamate residues on the receptor cytoplasmic domain, through the competing reactions of a methyltransferase CheR and a methylesterase CheB. After binding of attractant, the receptor methylation level increases because the kinase-off states is a better substrate for methylation by CheR, and because decreased phosphotransfer from CheA to CheB inhibits demethylation by CheB. The resulting increased level of methylation shifts the receptor back to the kinase-on state.
Figure 1.
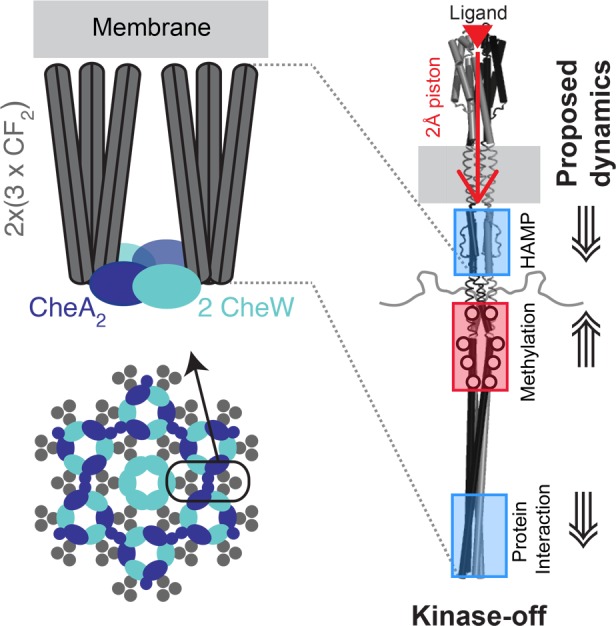
Overview of chemotaxis receptor array structure and dynamic changes proposed to propagate the signal. Cartoon of core unit (top left) formed by a pair of receptor trimer of dimers (gray rods) bridged by CheA kinase (blue) and CheW (cyan). Cartoon of the hexagonal array (bottom left) with receptor trimers of dimers (gray circles) at the vertices of the hexagons, connected by alternating CheA/CheW rings and CheW-only rings. Proposed signaling mechanism (right) begins with ligand binding to the periplasmic domain, causing a 2 Å downward piston of an α helix that extends through the transmembrane domain. The piston is proposed to cause changes in dynamics in the cytoplasmic domain to inactivate the kinase CheA bound to the membrane-distal tip: reduced dynamics in the HAMP and protein interaction regions (blue boxes) and increased dynamics in the methylation region (red box). All NMR samples in this study are native-like arrays of functional complexes of CheA, CheW, and the Asp receptor CF (cytoplasmic fragment), which begins near the junction of the HAMP/methylation regions. Array assembly is mediated by binding to membrane vesicles (left) or by molecular crowding agents (PEG 8000). Note that the CF lacks the periplasmic, transmembrane, and HAMP domains of the intact receptor.
Chemotaxis receptors of E. coli have been extensively studied to determine the mechanistic details of how ligand binding changes the structure and dynamics to control the activity of the kinase CheA that is bound to the cytoplasmic tip of the receptor ∼300 Å away.2 It is widely accepted that ligand binding causes a 2 Å downward piston motion of an α helix in the periplasmic and transmembrane domains (Figure 1, right), based primarily on the effects of engineered disulfide bonds on kinase and methylation activities, as well as a superposition of crystallographic structures of a receptor periplasmic fragment with and without ligand and site-directed NMR distance measurements in the periplasmic domain of the membrane-bound intact receptor.7,8 However, the mechanism of signal propagation through the cytoplasmic domain to control the kinase activity of CheA is not well understood. It has been proposed that changes in protein dynamics of the cytoplasmic domain play a role in this signaling, based on mutagenesis and disulfide cross-linking studies that suggest the methylation region is destabilized in the kinase-inhibited state, while the HAMP and signaling regions are stabilized in this state (Figure 1, right).9,10 EPR and hydrogen exchange mass spectrometry studies have begun to test these proposed mobility changes. Electron paramagnetic resonance (EPR) measurements of dynamics of spin labels incorporated at 15 positions of intact receptors in nanodiscs revealed that methylation decreased the dynamics of the methylation region, but ligand binding caused no changes in dynamics.11 Another EPR study of fusion proteins representing the kinase-on and kinase-off signaling states demonstrated inverse effects on the dynamics of the HAMP and protein interaction regions: in the kinase-off state, dynamics decreased in the HAMP region and increased in the protein interaction region, relative to the kinase-on state.12 These studies were done on receptors in the absence of CheA and CheW. Thus far, studies of receptor dynamics within functional complexes with CheA and CheW have been limited to hydrogen exchange mass spectrometry, with some initial observations of differences in exchange between samples representing the kinase-on and kinase-off signaling states.13 Additional studies of receptor dynamics within functional complexes are needed to investigate the role of dynamic changes in signaling.
This manuscript reports a solid-state nuclear magnetic resonance (NMR) study to detect receptor segments with mobility on the nanosecond or faster time scale within functional complexes. Biophysical studies of signal propagation in the cytoplasmic domain require homogeneous functional complexes of receptor, CheA, and CheW. We employ two methods for preparing native-like arrays, which are functional complexes of the E. coli Asp receptor cytoplasmic fragment (CF) that form extended arrays with native-like architecture containing the same 12 nm hexagonal spacing as observed for intact receptors.16 These CF array samples are more homogeneous than complexes of intact receptors prepared in nanodiscs17 or vesicles18 (with intact receptors inserted as a mixture in both directions in the membrane), or native membrane vesicles.16 CF arrays are also more amenable to NMR studies of the receptor,14 because of the 2-fold reduction in resonances from the 60 kDa intact receptor to the 33 kDa cytoplasmic fragment. One method for assembling CF arrays employs templating vesicles containing Ni-chelating lipid head groups that bind the N-terminal hexa-histidine tag of CF; CheA and CheW binding to the CF restores both kinase and methylation activities.19 Another method employs molecular crowding agents (PEG8000) to mediate assembly of complexes of CF with high kinase activity.20 These also form extended native-like arrays, but with a sandwich geometry: two CF layers overlap in the middle at the normally membrane-proximal ends, and each layer binds CheA and CheW on the exterior of the sandwich.21 Vesicle-mediated arrays can be prepared from both CF4E (the unmethylated CF) and CF4Q (which mimics the fully methylated CF); only CF4Q forms PEG-mediated arrays. Both vesicle-mediated and PEG-mediated arrays can be prepared in defined kinase-on and kinase-off signaling states for comparisons of the structure and dynamics of each state.
Magic angle spinning (MAS) solid-state NMR is an ideal tool for investigating the structure and dynamics of membrane proteins and supramolecular complexes. There has been remarkable progress in solid-state NMR methodology, enabling applications ranging from structure determination of a membrane-bound 35 kDa GPCR22 and of the micron-scale Type III secretion needle assembled from its 9 kDa protomer,23 to identification of dynamic segments in complex systems and samples such as histone proteins within nucleosome arrays,24 and both functional and disease-related amyloid fibrils.15–26 The approaches for detection of dynamic segments are applicable to the CF within its functional complexes and could reveal receptor dynamics involved in signal propagation. We have used complementary NMR methods to detect the mobile and rigid portions of U–13C,15N-CF4Q assembled with CheA and CheW into native-like functional arrays. These spectra demonstrate a change in NMR-detected mobility with signaling state. We have also constructed a truncated CF to assign the mobile segments. Comparison of refocused INEPT (insensitive nuclei enhanced by polarization transfer) spectra of functional arrays of CF4Q and CF4QΔ34 (lacking the C-terminal tail previously shown to be mobile)27 demonstrate the expected resonances of the mobile tail and detect an additional mobile segment, which is identified by chemical shifts of distinctive residues.
Experimental Methods
Plasmid Construction
PCR primers used for plasmid construction are listed in Table S1. The PCR reactions were done in a thermocycler (Eppendorf Master Cycler personal or BioRad MJ Mini) using reagents from New England Biolabs. Plasmids expressing TEV-cleavable His-tagged proteins were constructed for ease of purification and compatibility with the vesicle assembly of His-tagged CF. The CheY, CheA, and CheW-expressing regions were PCR-amplified from pT7 (CheY), pET6H-CheA, and pET6H-CheW plasmids, respectively. PCR products were separated in 1% Agarose gels, and each product with the expected length was gel-purified (QiAquick gel extraction Kit, Qiagen). The purified product was cloned into the pETite vector system using ExpressoR T7 cloning & Expression System (Lucigen Corporation, WI). PCR products and linearized pETite vectors were transformed into HI-Control 10G chemically competent cells, and recombinants were screened for kanamycin resistance. The resulting pTEVcheY, pTEVcheA, and pTEVcheW plasmids express each protein with a TEV-cleavable N-terminal His-tag. Cloning was confirmed by sequencing (Genewiz, South Plainfield, NJ).
A plasmid expressing CF4Q with 34 residues truncated from the C-terminus (pCF4QΔ34) was constructed to help assign the mobile segments of CF. A plasmid expressing CF4Q with A411 mutated to V was constructed to represent the kinase-off state.28 Site-directed mutagenesis was used to introduce either a new stop codon (pCF4QΔ34) or the A411V mutation (pCF4Q.A411V) into pHTCF4Q (pHTCF4Q expresses an N terminal His tag (MRGSHHHHHHGSP) appended to residues 257–553 of the E. coli Asp receptor).19 The PCR products were confirmed by gel electrophoresis in a 1% agarose gel, subjected to DpnI digestion, and transformed into DH5αF’ competent cells for plasmid purification. Sequences were verified for both plasmids (Genewiz).
Protein Purification
Since the CF expression plasmids do not contain lacIq (pHTCF4Q, pCF4QΔ34, pCF4Q.A411V, all encoding ampicillin resistance), all were cotransformed into BL21(DE3) with pCF430, which encodes lacIq and tetracycline resistance. Cells harboring CF expression plasmids were grown in M9 minimal media using U–13C-glucose and (15NH4)2SO4 as the carbon and nitrogen sources. A single colony (from cells grown on an LB/Agar plate containing 150 μg/mL ampicillin and 10 μg/mL tetracycline) was inoculated into 2 mL Luria–Bertani (LB) broth, and grown to OD600 ∼ 0.6 at 37 °C with 200 rpm shaking. This culture was used to inoculate 1 L of minimal media and grown overnight at 30 °C with 200 rpm shaking, followed by induction with 1 mM IPTG for at least 5 h at 25 °C. Cells were harvested by centrifugation and lysed with a microfluidizer at 16K psi; 1 mM PMSF was added every hour after cell lysis to inhibit proteolysis. CF proteins were purified by nickel-affinity chromatography as previously described.20 Unlabeled CheW, CheA, and CheY proteins were expressed in BL21(DE3) in LB broth with kanamycin (50 μg/mL). Cells were grown at 37 °C until OD600 ∼ 0.6, and then induced with 1 mM IPTG and grown overnight for 16 h at 15 °C (CheY) or 4 h at 30 °C for CheA and CheW, and then harvested by centrifugation. Each protein was purified by nickel affinity chromatography. Buffers contained 75 mM Tris-HCl pH 7.4 and 100 mM KCl (lysis), plus 10 mM imidazole (wash) or 250 mM imidazole (elution). Fractions containing protein were pooled; 5 mM EDTA was added before dialysis against lysis buffer. TEV cleavage to remove the His-tag was performed using a 1:40 ratio of TEV protease:substrate, incubated at 4 °C overnight, followed by 2–3 h at 25 °C. SDS-PAGE (12.5% acrylamide gel) was used to verify cleavage. The His-tagged TEV protease was then removed using nickel-affinity chromatography on a column equilibrated with lysis buffer. The digested sample was loaded on the column and the cleaved CF was collected in the flow-through. Protein concentrations were measured by BCA assay (Thermo Scientific), and purity was checked by SDS-PAGE.
Vesicle-Mediated Complex Assembly
DOGS-NTA-Ni2+ (1,2-dioleoyl-sn-glycero-3-[[N(5- amino-1-carboxy-pentyl) and DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) lipids (Avanti Polar Lipids) were combined in a 1:1.5 ratio in chloroform. Lipids were dried in a round-bottom flask in thin layer first with nitrogen gas and then placed under vacuum for 1 h, then resuspended in potassium phosphate kinase buffer (PPKB: 50 mM KxHxPO4, 50 mM KCl, 5 mM MgCl2, pH 7.5) to make a 3 mM stock solution containing 40% nickel-chelating lipid. After repeated vortexing of lipid in PPKB (30 s vortex, 5 min rest, repeated 3 times), the multilamellar vesicles were freeze–thawed (30 s in liquid nitrogen, 3 min at room temperature, 3 min at 37 °C, repeated 5 times) to make unilamellar vesicles. Vesicles were extruded, and complexes were assembled as previously described,29 except that excess CheW and CheA was used to reach maximum activity so that all of the CF would be in complexes.20 The 725 μM total lipid concentration (containing 290 μM DOGS-NTA) was chosen to provide sufficient surface area to accommodate all 30 μM CF in functional complexes with CheA and CheW, based on the known dimensions of the native arrays.5 In brief, arrays were assembled by combining (in order) autoclaved water, stock solutions of PPKB (5x), CheA, CheW, vesicles, CF, and PMSF. Final concentrations in the assembly were 12 μM CheA, 24 μM CheW, 725 μM lipids, 30 μM CF, and 1 mM PMSF. Complexes were incubated at 25 °C for 4 h or overnight before measuring sedimentation and kinase activity.
PEG-Mediated Complex Assembly
PEG 8000 and d-(+)-trehalose stocks were prepared as 20% and 40% (w/v) stock solutions, respectively, dissolved in Milli-Q water and passed through a 0.22 μm sterile filter. PEG-mediated arrays were assembled by combining (in order): autoclaved water, stock solutions of PPKB (5x), CheA, CheW, PEG, trehalose, CF, and PMSF. Final concentrations in the assembly were 12 μM CheA, 20 μM CheW, 4% (w/v) PEG 8000, 4% (w/v) D-(+)-trehalose, 50 μM CF and 1 mM PMSF. The mixture was incubated at 25 °C for 4 h or overnight before measuring the kinase activity.
Kinase, Binding, and Methylation Assays
The kinase activity was measured using an enzyme-coupled ATPase assay as previously described.20 For the protein binding assay (sedimentation assay), at least 35 μL of the assembled complex was centrifuged for 30 min at 125 000g in a Beckman ultracentrifuge (60 000 rpm in a TLA 120.2 Beckman rotor). The supernatant was carefully removed to another Eppendorf tube, and the pellet was resuspended in the original volume. Gel samples of the total (before centrifugation), supernatant, and resuspended pellet were separated by SDS-PAGE (12.5% acrylamide), and the gel bands were analyzed using ImageJ. The bound fraction of each protein was calculated as (intensity of pellet band)/(intensity of total band). To avoid distortion of gel bands due to high PEG concentrations, reduced sample volumes of 1.5–2 ul of PEG samples (compared to 5 μL for vesicle samples) were loaded onto 20% acrylamide gels. The protein concentrations for assembly were chosen to maximize activity and reach the 6:1:2 binding ratio of CF:A:W. Methylation of the CF was measured by assembling functional vesicle complexes as described above; final concentrations of 6 μM CheR and 10 μM SAM were added to the complexes to initiate methylation. Aliquots (20 μL) were removed after 10 min and 4.5 h, quenched by addition of gel sample buffer, and subjected to SDS-PAGE using a 10% polyacrylamide gel.19
NMR Sample Preparation
Each NMR sample was assembled as 4 mL of the complex mixture and incubated overnight at 25 °C in a circulating water bath. The activity and sedimentation were measured to ensure successful assembly before packing the NMR rotor. Complexes were collected by ultracentrifugation at 108 000g and 25 °C for 2 h (55 000 rpm in a TLA 120.2 Beckman rotor). To pack the 1.9 mm Bruker rotor, a 200 μL gel-loading pipet tip was prepared by slicing the narrow end to create a hole the size of the top cap of rotor, which was then flame-sealed. The jelly like pellet was transferred with a spatula to this pipet tip and then centrifuged down into the sealed tip (7–10s in benchtop microfuge at 14 000 rpm) using a 1.5 mL Eppendorf as a container. The sealed tip was cut and placed into the opening of a 1.9 mm rotor for an additional centrifugation to transfer the sample into the rotor. Approximately 14 mg of each pellet was packed to fill the volume of each 1.9 mm rotor. The mass of CF in each sample was calculated as (total volume of complex) × (CF concentration) × (packed sample weight)/(total pellet weight). The precision of the protein concentration measurements was ±2–4%. Due to reproducible assembly and packing conditions, all samples of each type (vesicle or PEG) contained very similar amounts of CF, as listed in Table 1.
Table 1. CF Amounts in NMR Samples.
| NMR sample: | vesicle CF4Q | vesicle CF4Q.A411V | vesicle CF4QΔ34 | PEG CF4Q | PEG CF4QΔ34 |
|---|---|---|---|---|---|
| nmol CF: | 79.7 | 81.0 | 78.9 | 120.2 | 122.3 |
NMR Spectroscopy
All NMR experiments were done on a 14.1 T Bruker Avance III spectrometer (1H = 600 MHz, 13C = 150 MHz, 15N = 60 MHz) in a 1.9 mm HNCF probe. 2D refocused insensitive nuclei enhanced by polarization transfer (INEPT) experiments on vesicle-mediated complexes were conducted at 40 kHz MAS with a 0.3 s recycle delay; 2D INEPT experiments on PEG-mediated complexes were conducted at 11.11 kHz MAS with a 1 s recycle delay. The 90° pulses were 3.2 μs, 5 μs, and 4.5 μs for 1H, 15N, and 13C, respectively, with SPINAL-64 decoupling at 78 kHz. INEPT parameters were chosen for 90 Hz JNH-couplings and 125 Hz JCH-couplings. Each 2D experiment contained 64 points in the indirect dimension (1200 scans per slice), for a total experiment time of approximately 7 h (vesicle-mediated complexes) and 21 h (PEG-mediated complexes). Proton T1 values were measured using a saturation-recovery pulse sequence.
Reported temperature values are estimated sample temperatures calibrated as follows. The heating due to MAS was determined by measuring the spin–lattice relaxation rate of KBr powder30 at the same MAS and gas flow rate as the experimental conditions. The heating due to decoupling was determined by measuring the proton chemical shift of TmDOTP,31 with DMPC vesicles in sample buffer for equivalent ionic strength. Measurements of heating due to MAS at various spin rates were used to choose a cooling/spinning protocol needed to maintain the sample temperature between 10 and 15 °C during stepwise increases of the MAS from 11 kHz to 40 kHz.
NMR Data Analysis
Topspin 3.2 was used for data processing. One-dimensional NMR data were processed with 100 Hz of exponential line broadening. Two-dimensional NMR data were processed using baseline correction (subtraction of the average intensity for the last quarter of the FID: BC-mod = quad and BCFW = 0.1 ppm in Topspin), linear prediction in the indirect dimension (16 points), cosine-bell multiplication, and zero filling in both dimensions. 13C chemical shifts were referenced to adamantane at 40.5 ppm (relative to DSS at 0 ppm).32 Indirect chemical shift referencing of 15N and 1H was calculated based on 13C, by SR(X) = [BF(13C) + SR(13C)] × (gX/gC) – BF(X) where, X is 15N or 1H, and g is the gyromagnetic ratio.33
Sparky (UCSF) was used for analysis and measuring peak volumes in 2D spectra. The lowest contour level was set to 15 times the estimated noise (2000 points) for the CF4Q arrays (Figure 3A for vesicle arrays and Figure S1-A for PEG arrays) and the same contour level was used for all other spectra of the same array type. The peak volumes for all resolved peaks were measured using the Gaussian fitting integration method (default settings plus fit baseline). The Cα region peak volume was estimated using the same Gaussian fit, removing the contribution of the overlapping Sβ peak by using the “subtract off fit peaks” option.
Figure 3.

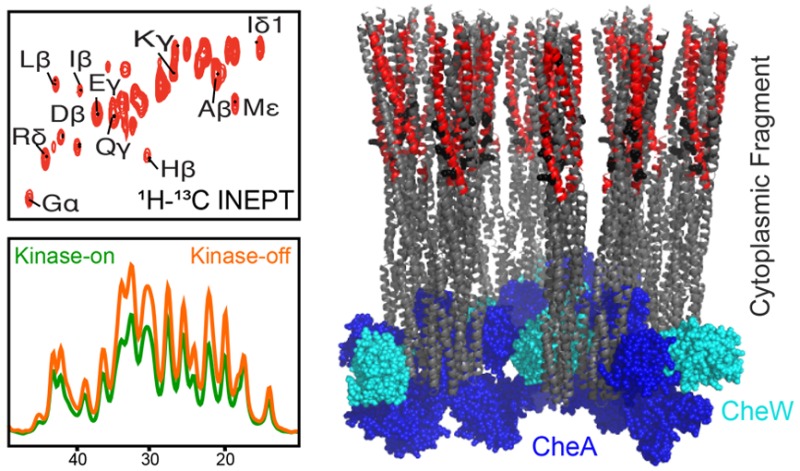
1H–13C INEPT of U–13C,15N-CF4Q and U–13C,15N-CF4QΔ34 in vesicle-mediated arrays with CheA and CheW. (A) CF4Q spectrum is compared to predicted resonance positions (black dots) for the C-terminal tail (red box), using average chemical shifts from the BMRB since this tail region is known to be mobile. Resonances are observed for all tail residues except Trp (gray). Several additional resonances are also observed (cyan arrows indicate aromatic His and Tyr, Gly Cα, His Cβ, and Met Cε) suggesting CF contains additional mobile region(s). (B) CF4QΔ34 spectrum is compared to predicted resonance positions (BMRB chemical shifts) for the most likely ∼50-residue mobile region (red box), corresponding to the sequence range indicated. Structural models are based on the 1qu7 crystal structure of a cytoplasmic fragment of the Ser receptor in the absence of CheA and CheW.38 The portion of the CF4Q extending beyond the end of the 1qu7 crystal structure is shown as a solid line (methylation region) and dotted line (HAMP region). Spectra are shown at comparable contour levels, demonstrating more intensity in the top spectrum for most of the peaks. The spectra were collected with 40 kHz MAS, ∼ 10 °C sample temperature, and a 0.3 s recycle delay (64 slices of 1200 scans/slice for a total experiment time of 7 h), on samples containing approximately 80 nmol CF (plus vesicles, CheA, and CheW).
Results and Discussion
NMR Detects Mobile Segments of Chemoreceptors in Functional Arrays
NMR methods for selective detection of rigid versus mobile residues reveal dynamic segments of the chemotaxis receptor cytoplasmic fragment within its vesicle-mediated functional complexes with CheA and CheW. Selective detection of rigid residues is achieved with cross-polarization (CP), which transfers magnetization via dipolar couplings that are large for rigid solids but averaged to zero by isotropic motion. Selective detection of mobile residues is achieved with refocused INEPT, which transfers magnetization via J couplings that are independent of dynamics. INEPT transfers occur during delays that serve as a relaxation filter: signals from rigid residues decay quickly due to large proton–proton dipolar couplings that result in short 1H T2 values,34 so segments with mobility on the ns or shorter time scale are detected in INEPT spectra.15 Thus, the rigid and mobile fractions can be detected with one-dimensional 13C CP and INEPT spectra, respectively. As shown in Figure 2 and Table 2, there is a significant mobile fraction (20–30% based on integrated intensities for the 10–80 ppm aliphatic region) detected in spectra of unfrozen samples of both the kinase-on state (U–13C,15N-CF4Q, blue) and kinase-off state (U–13C,15N-CF4Q.A411V, red). As anticipated for both states, all of the receptor becomes rigid upon freezing (zero intensity INEPT, data not shown). The correspondence between the sum of the INEPT (Figure 2A) and CP (Figure 2B) intensities for the unfrozen sample with the CP intensity for the frozen sample (Figure 2C) suggests that there are no segments with intermediate time scale motion that would not be detected by CP or INEPT (Table 2).
Figure 2.
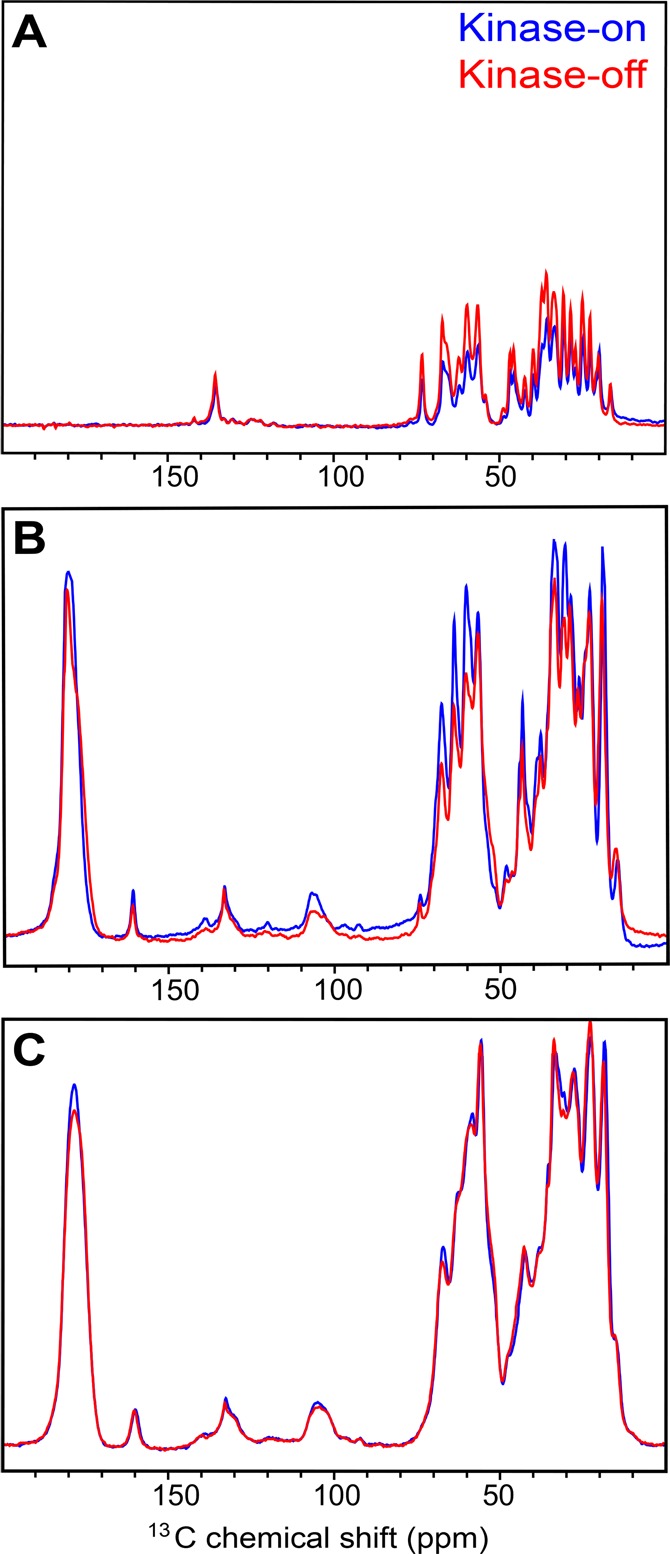
NMR spectra of U–13C,15N-CF in vesicle arrays with CheA and CheW reveal a mobile fraction in the kinase-on state (CF4Q, blue) and kinase-off state (CF4Q.A411V, red). 13C spectra of unfrozen arrays (15 °C) were collected using (A) refocused INEPT to observe the flexible segments and (B) CP to observe the rigid segments of the receptor in functional complexes. Integrated intensities of the aliphatic region (10–80 ppm) indicate that, at this temperature, the kinase-on receptor is approximately 20% flexible and 80% rigid, while the kinase-off receptor is approximately 30% flexible and 70% rigid. (C) 13C spectra of frozen samples at −25 °C show the expected loss of flexibility: INEPT spectra have zero intensity (not shown) and CP frozen spectra have intensity comparable to the sum of unfrozen INEPT + CP (see Table 2). Each spectrum was recorded with 2400 scans and a recycle delay of 1 s, at 11.11 kHz MAS.
Table 2. CP versus INEPT Integrated Intensitiesa for U–13C,15N-CF in Frozen and Unfrozen Samples of Kinase-On and Kinase-Off Vesicle Arrays with CheA and CheW.
| unfrozen INEPT | unfrozen CP | frozen INEPT | frozen CP | |
|---|---|---|---|---|
| CF4Q (kinase-on) | 0.19 | 0.79 | 0 | 1 |
| CF4Q.A411V (kinase-off) | 0.27 | 0.68 | 0 | 0.97 |
Integrated intensities from 10 to 80 ppm.
The extent of mobility is surprising, as the receptor is thought to be largely alpha helical with a mobile tail corresponding to only ∼10% of the CF.27 Furthermore, the mobility may be relevant to the signaling mechanism, since there is a mobility shift between the signaling states. The kinase-off state has a ∼ 10% increase in the INEPT intensity and a corresponding ∼10% decrease in the CP intensity relative to the kinase-on state. Since changes in dynamics of regions of the CF have been proposed to play a role in signaling,9,10 we pursued the identity of the mobile segments through a combination of biochemical and NMR approaches.
INEPT Spectra Identify a Mobile Segment in the Methylation Region
Sensitivity can be a challenge for two-dimensional NMR of CF in these native-like complexes, because CF is only 45% of the mass of the protein plus lipid components of the vesicle samples, resulting in only ∼80 nmol of labeled CF in the NMR rotor. NMR sensitivity was improved for vesicle-mediated array samples using paramagnetic relaxation enhancement.35 The Ni2+ bound to the DOGS lipid decreases the proton T1 to 129 ms (compared to 525 ms for PEG-mediated arrays), making it possible to use rapid recycling (0.3 s recycle delay). Furthermore, we observed greater INEPT intensities at higher MAS speeds (∼1.4-fold greater intensities in 1D HC INEPT spectra collected with 40 kHz MAS relative to 11 kHz MAS), most likely due to a longer proton T2 due to MAS averaging of proton dipolar couplings. Under these conditions, two-dimensional 1H–13C INEPT spectra containing 64 slices of 1200 scans each are collected in a total experiment time of 7 h.
Based on the known mobility27 of a 34-residue C-terminal tail that serves to tether the methyltransferase and methylesterase enzymes,36,37 we expected INEPT spectra to contain residues from this tail. Figure 3A shows the 1H–13C INEPT spectrum of U–13C,15N-CF4Q assembled into vesicle-mediated native-like arrays with CheA and CheW. Using average protein chemical shift values (black dots) tabulated in the Biological Magnetic Resonance Data Bank (BMRB), resonances were identified for all of the tail residues except Trp (gray). It is unclear why the Trp resonances are observed in 1H–15N INEPT spectra (Figure 4) but are absent in the 1H–13C INEPT spectra. Several resonances are also observed for residues not found in the tail, specifically for Tyr, His, Gly, and Met (cyan arrows). This suggests that CF in functional complexes contains additional mobile region(s).
Figure 4.

1H–15N INEPT spectra of U–13C,15N-CF4Q and U–13C,15N- CF4QΔ34 in vesicle-mediated functional arrays with CheA and CheW. Resonances are assigned to amino acid types based on average protein chemical shifts reported in the BMRB. (A) Spectra of CF4Q contain resonances expected for the flexible C-terminal tail, but also contain resonances for Gly, which is not present in the tail (cyan arrow). (B) Spectra of the tail-deleted CF4QΔ34 are consistent with removal of the tail (no Trp resonance), but indicate that another region of CF has high mobility. Same NMR conditions as Figure 3.
To determine what other CF region(s) are mobile, we constructed a plasmid encoding CF without the 34-amino acid C terminus, CF4QΔ34. Such a tail-truncated receptor has been previously shown to be functional in kinase assays in studies that demonstrated the role of this tail is to dock the methylation enzymes.36Figure 3B shows the 1H–13C-INEPT spectrum of the tail-deleted CF, U–13C,15N-CF4QΔ34, incorporated into functional vesicle-mediated arrays with CheA and CheW. This spectrum (normalized to the same amount of CF) has reduced intensity for all peaks except for those residues not present in the tail (cyan arrows in Figure 1). The absence of Phe ε,γ,Z and Pro δ peaks in the spectrum of the tail-deleted sample is consistent with removal of the tail, and indicates that these residues are not present in the receptor mobile region.
To identify the residue types observed in the CF4QΔ34 1H–13C INEPT spectrum, we used BMRB-tabulated chemical shift values. A number of resonances are uniquely identified in this spectrum for Ala β, Arg δ, Asp β, Glu γ, Gly α, His β,δ,ε1 Ile β,δ, Leu β, Lys ε, Met ε, Ser β, Thr β, Tyr δ,ε, and thus these residues are present in the mobile region(s) of CF4QΔ34. The presence of Tyr resonances (green in Figure 3B) is notable, as a single Tyr residue is found in the CF4QΔ34 sequence. If we assume that the INEPT spectrum comes from a single mobile region, the observed resonances suggest this is a region near the N terminus (red box in Figure 3B), that includes His (blue), Tyr (green), Met (brown), and Lys (orange). This mobile segment includes three of the four methylation sites (black spheres/underlined residues) in the receptor. Note that the N-terminal His-tag is anchored to the vesicle surface in these functional arrays, so mobility of this region is not simply a result of being near the truncated N-terminus of the protein.
Additional insights into the identity of the mobile region can be deduced from peak volume estimates tabulated in Table 3. Intensities were referenced to the Tyr Cδ peak intensity because there is a single Tyr in the CF. The Tyr Cε peak was judged to be less reliable, due to overlap with the nearby His Cδ2 peak. All peak volumes were first corrected for the small differences in nmol of CF in the samples, and then divided by half of the Tyr Cδ volume, to yield a Tyr peak volume of two, corresponding to two Cδ on the single Tyr residue in the CF. For clarity, the peak volumes were then divided by the number of unresolved correlations per residue (5 for Phe, 2 for Tyr Cδ, and 1 for everything else), so that the normalized volumes listed in Table 3 would correspond to the number of residues contributing to the peak. The volume for total Cα was calculated as the volume of the peaks above the horizontal brackets on the spectra (Figure 3) minus the volumes of the Ser β and Pro δ peaks.
Table 3. Peak Volumesa in 1H–13C and 1H–15N INEPT Spectra of 13C15NCF4Q and 13C15NCF4QΔ34 Assembled with CheA and CheW into Functional Native-Like Arrays on Vesiclesb.
| residue/s | ω1/1H | ω2/13C or 15N | CF4Q volume | CF4QΔ34 volume | mobile region residues | CF4Q – CF4QΔ34 = Δ34 volume | Δ34 (tail) residues |
|---|---|---|---|---|---|---|---|
| total Cα (no Gly) | 82.8 | 47.6 | 48 | 35.2 | 34 | ||
| Arg Nε | 7.3 | 84.9 | 5.7 | 2.8 | 3 | 2.9 | 3 |
| Asn Cβ + Tyr Cβ | 2.6 | 39.1 | 4.1 | 2.4 | 1 Asn, 1 Tyr | 1.7 | 2 Asn |
| Glu Cγ | 2.2 | 36.3 | 7.1 | 3.7 | 5 | 3.4 | 3 |
| Gly Cα | 4.0 | 45.4 | 2.8 | 2.8 | 3 | 0.0 | 0 |
| Gly N | 8.3, 8.2 | 108.7, 107.4 | 2.5 | 2.2 | 3 | 0.3 | 0 |
| His Cβ | 3.1 | 29.7 | 0.9 | 0.7 | 1 | 0.2 | 0 |
| His Cε1 | 7.8 | 138.4 | 0.9 | 0.8 | 1 | 0.1 | 0 |
| Ile Cβ | 1.8 | 38.7 | 3.2 | 1.9 | 2 | 1.3 | 1 |
| Ile Cδ1 | 0.8 | 13.0 | 3.3 | 2.0 | 2 | 1.3 | 1 |
| Leu Cβ | 1.6 | 42.2 | 4.3 | 2.5 | 3 | 1.7 | 2 |
| Met Cβ | 2.0 | 17.1 | 1.5 | 1.5 | 1 | 0.0 | 0 |
| Phe | 7.2 | 131.8 | 1.1 | 0 | 0 | 1.1 | 1 |
| Pro Cδ | 3.7 | 50.7 | 1.3 | 0 | 0 | 1.3 | 7 |
| Thr Cβ | 4.2 | 69.8 | 8.4 | 5.4 | 6 | 3.0 | 3 |
| Trp Nε | 10.0 | 129.1 | 1.0 | 0 | 0 | 1.0 | 1 |
| Tyr Cδ | 7.1 | 133.4 | 1.1 | 1.0 | 1 | 0.1 | 0 |
All peak volumes are corrected for differences in nmol of CF in the sample. HC peak volumes are then calibrated based on Tyr Cδ = 2 in the CF4QΔ34 vesicle sample, and then normalized to the number of unresolved correlations contributing to the peak (5 for Phe and 2 for Tyr Cδ) so that the reported volume should correspond to the number of residues. HN peak volumes are calibrated based on Trp Nε = 1 in the CF4Q vesicle sample.
Bold columns compare peak volumes to residues of the most probable 50-residue mobile region; italicized columns compare difference volumes to residues of the deleted 34-residue tail.
The difference between the normalized volumes of the CF4Q and CF4QΔ34 samples should correspond to the resonances of the Δ34 tail only. Table 3 shows fairly good agreement for the Δ34 tail (compare italicized columns for observed difference volumes and expected number of residues). The total Cα difference intensity of 35.2 is close to its predicted value of 34, and 11 individual 1H–13C INEPT peaks have difference intensities remarkably close to their predicted values when referenced to the volume of the Tyr Cδ peak. Only the Pro Cδ difference volume is much smaller (1.3) than predicted (7). Although we cannot explain the unexpectedly small Pro peak or the absence of the Trp peaks, the agreement for the rest of the peaks suggests that INEPT transfer efficiencies are similar for these residues, and that the normalized volumes of the INEPT spectra can provide a qualitative estimate of the number of residues in mobile regions. Since these are computed as differences between two samples, the agreement also suggests that our samples are consistent with each other, which is further corroborated by a similar subtraction performed with spectra of PEG-mediated arrays of CF4Q and CF4QΔ34 (see below and Table S2).
The agreement between the subtraction of the integrated intensities and the predicted intensities of the Δ34 residues validates the use of the INEPT integrals for CF4QΔ34 to gain further insight into the identity of the mobile region(s). The bold columns in Table 3 compare observed peak volumes for CF4QΔ34 to the expected number of residues in the likely ∼50-residue mobile region extending from His267 to Gln316, as shown in Figure 3B. The Cα intensity of 50.4 (47.6 total Cα plus 2.8 Gly Cα) is in reasonable agreement with the number of residues, as are all of the other 1H–13C INEPT peak intensities. The Lys peaks are not sufficiently resolved for accurate peak volume measurements (the overlap with Arg and Asp peaks is visible at a lower contour level for Lys Cε), but comparison of 1D slices for the Lys ε peak of CF4Q and CF4QΔ34 indicate that the mobile region contains at least one Lys.
1H–15N INEPT spectra (Figure 4) of the U–13C,15N-CF4Q and CF4QΔ34 vesicle-mediated array samples provide further evidence for a mobile region, and support the conclusions from the 1H–13C INEPT spectra shown in Figure 3. These spectra, labeled with the amino acid-type assignments based on average protein shifts tabulated in the BMRB, display the resonance of the single Trp NH located near the C-terminus of CF4Q (Figure 4A, red), which is absent in the CF4QΔ34 spectrum (Figure 4B). Other amino acids observed in the spectrum of CF4Q (Figure 4A) are also present in the mobile C-terminal tail, with the exception of glycine (cyan arrow). As in the 1H–13C INEPT spectra, the observed Gly resonances provide evidence that segments besides the C-terminal tail are mobile. This is confirmed by the 1H–15N INEPT spectrum of U–13C,15N- CF4QΔ34 arrays (Figure 4B), which detects a mobile region that contains G, N/Q, R, and S/T.
Peak volume measurements are listed in Table 3 only for the well-resolved peaks of the 1H–15N INEPT spectra: Gly N, Arg Nε, and Trp Nε. Intensities were referenced to that of the Trp Nε because there is a single Trp in the CF. After correction for the small differences in nmol of CF in the samples, all HN peak volumes were divided by the peak volume of the single Trp Nε, so that all volumes correspond to the number of residues. The results are consistent with the expected volumes for the deleted 34-residue tail (italicized columns in Table 3). Again, this empirical correlation between the difference peak volumes and the numbers of deleted residues suggests that the transfer efficiencies for Gly N, Arg Nε, and Trp Nε are similar enough that their peak volumes can provide a qualitative estimate of the numbers of these residues in the mobile region. The volumes in the CF4QΔ34 spectrum suggest that the INEPT-detectable mobile segment contains 3 Arg (volume = 2.8) and 3 Gly (volume = 2.5), as expected for the assigned mobile segment of the methylation region (bold columns in Table 3).
Thus, INEPT spectra demonstrate an asymmetric mobility property in the methylation region of the CF: the methylation N-helix is mobile on the nanoseconds time scale but the methylation C-helix is not. The N terminal side of this region (methylation N-helix), extending from His267 to Gln316, experiences mobility on the nanoseconds or shorter time scale (red block in Figure 5, top). This mobile segment spans the entire length of the methylation region: it begins immediately following the second HAMP domain helix (∼247–264, dashed line in Figure 3B and Figure 5) and includes all three methylation sites (295, 302, 309, black circles in Figure 5). Of course, it is also possible that multiple mobile segments contribute to the INEPT spectrum of CF4QΔ34. As illustrated in Figure 5, we considered the simplest possibility, that mobility is found in a pair of ∼25 amino acid mobile segments from opposite ends of the CF that are adjacent in the helical hairpin structure, for instance, segments of both the methylation N and C helices (red blocks in Figure 5, bottom). However, there are no such pairs of shorter segments adjacent in the structure that contain all of the rare amino acids (H,Y,M,K) identified in the INEPT spectrum.
Figure 5.
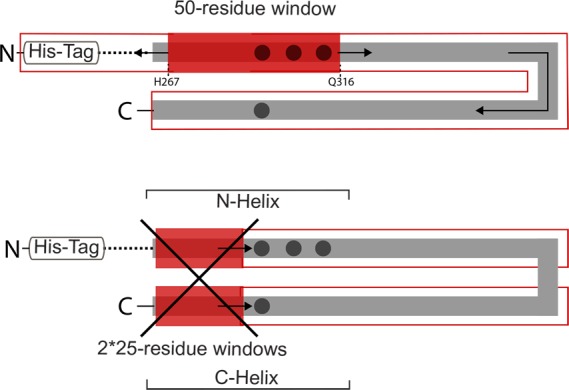
INEPT spectra indicate high mobility of the methylation N-helix in the CF in functional arrays. Black dots represent the methylation sites and dashed lines represent a portion of the HAMP region contained in the CF used in this study. Top: sliding a 50-residue window from N- to C-terminus of the CF sequence identifies the most probable mobile segment (red block, H267-Q316), corresponding to the N-helix side of the methylation region. No other contiguous 50-residue segment contains all of the amino acid residues observed in the INEPT spectra. Bottom: sliding a pair of 25-residue windows sequences adjacent in the structure along the length of the helix hairpin (within outlined red boxes) does not identify any positions at which the pair contain all of the amino acid residues observed in the INEPT spectra.
Mobile Methylation N-helix Is a Property of Functional Arrays
Similar 1H–13C INEPT and 1H–15N INEPT spectra are observed for U–13C,15N-CF in functional arrays with CheA and CheW, whether the arrays are assembled via His-tag binding to vesicles (vesicle-mediated arrays: Figures 3-4 and Table 3) or via molecular crowding (PEG-mediated arrays: Figures S1–S2 and Table S2). The differences in peak volumes in spectra of PEG-mediated arrays between 13C,15N-CF4Q and 13C,15N-CF4QΔ34 are close to those observed for vesicle-mediated arrays (compare Table 3 and Table S2, italicized columns), and again are qualitatively consistent with the deleted 34-residue tail. This again demonstrates the reproducibility of NMR spectra for different samples.
The spectra and peak volumes of 13C,15N-CF4QΔ34 also reveal a mobile segment in the methylation region of PEG-mediated arrays (Figure S1). Other than the appearance of PEG resonances, disappearance of the lipid resonance, and greater intensity of His resonances, the 1H–13C INEPT spectra of PEG-mediated arrays (Figure S1) are very similar to the spectra of vesicle arrays (Figure 3). This is further illustrated in Figure 6, by an expansion of the aromatic region of the 1H–13C INEPT spectra for PEG and vesicle samples, which shows the equivalence of the Phe resonances (Figure 6A), as well as the increased size of the His Cε1 resonance in 1D slices (Figure 6B). The 1H–15N INEPT spectra are also quite similar for PEG arrays (Figure S2) and vesicle arrays (Figure 4). Comparison of Table S2 (peak volumes for PEG array spectra) with Table 3 (peak volumes for vesicle array spectra) shows only small differences (≤1), except that the two histidine peak volumes increase by 4–5. The increased His intensity most likely comes from the N-terminal His-tag, which is the only region with more than 2 His residues close together. We conclude that without its anchor to the vesicles, the His-tag has some mobility in the PEG-mediated arrays, leading to an increased His intensity in the 1H–13C INEPT spectra of PEG arrays. Comparison of the INEPT spectra and peak volumes for PEG and vesicle-mediated CF arrays demonstrate that both contain a similar INEPT-detectable mobile region, suggesting that the mobile methylation N-helix is an intrinsic property of functional CF complexes.
Figure 6.
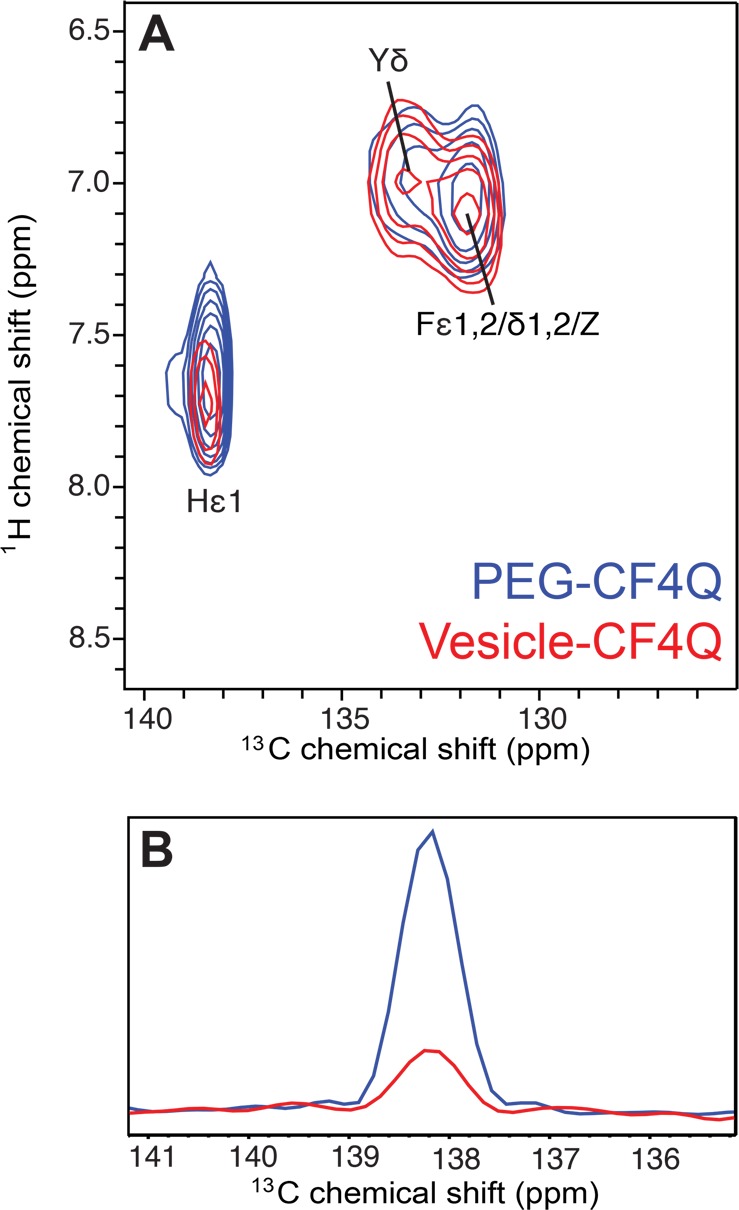
Comparison of 1H–13C INEPT spectra of U–13C,15N-CF4Q in PEG-mediated (blue) and vesicle-mediated (red) functional arrays with CheA and CheW. Spectra are each scaled to a peak volume of 1 for the single Tyr Cδ resonance. (A) Expansion of the aromatic region of the spectrum illustrates the similarity of the Phe resonances for both arrays, and the greater intensity of the His Cε1 resonance for the PEG array. (B) Comparison of one-dimensional slices for His Cε1 shows 4-fold larger peak intensity in the PEG array, which is consistent with limited mobility of the His-tag region. The PEG sample spectrum was collected with 11.11 kHz MAS, ∼ 10 °C sample temperature, and a 1 s recycle delay (64 slices of 1200 scans/slice for a total experiment time of 22 h), on a sample containing 120 nmol CF (plus PEG, CheA, and CheW). Vesicle sample spectrum collected as in Figure 3.
The asymmetric mobility identified by INEPT in the methylation region is consistent with results of EPR studies of intact receptors in nanodiscs and hydrogen exchange mass spectrometry (HDX-MS) studies of vesicle-mediated CF arrays with CheA and CheW. EPR measurements of site-directed spin label mobility have demonstrated that the mobility parameters of the N-helix are higher than those of the C-helix, and the N-helix mobility parameters are comparable to those of the unstructured mobile tail.39 Similar results have been reported for an HDX-MS study of vesicle-mediated CF arrays of functional complexes with CheA and CheW (comparable to the vesicle samples in our NMR study): both the methylation N-helix and the tail showed complete hydrogen exchange within 3 min, but the methylation C-helix exchanged more slowly.13 Although neither system is fully native, CF arrays lack half of the receptor and intact receptors in nanodiscs lack CheA and CheW, observation of the asymmetric mobility of the methylation region in both systems suggests that it is an intrinsic property of the receptor. Thus, a highly mobile methylation N-helix has been observed by three methods (HDX-MS, EPR, NMR) on multiple sample types (including intact receptors in nanodiscs, vesicle-mediated CF arrays, and PEG-mediated CF arrays), so this mobility is likely to be a property of the functional receptor.
Mobile Methylation N-Helix Undergoes Signaling-Related Mobility Changes
Neither the EPR nor the HDX-MS study observed a signaling-related change in the methylation N-helix mobility. There was no effect of ligand on the EPR mobility parameters,11 perhaps due to the absence of CheA and CheW, which can reduce coupling between the periplasmic and cytoplasmic domains (e.g., the methylation state alters receptor ligand affinity only for receptors in complexes with CheA and CheW).40 The fast exchange observed in the HDX-MS study (complete exchange at the first time point) makes it impossible to measure the exchange rate and detect differences between the kinase-on and kinase-off states.13 Thus, INEPT detection of this mobile segment within functional complexes with CheA and CheW makes it possible to investigate whether mobility changes with signaling state.
To prepare CF4Q arrays in the kinase-off state, we introduced the A411V mutation, previously shown to lock the intact receptor into a kinase-off state.28 As expected, CF4Q.A411V in vesicle and PEG arrays with CheA and CheW does not activate the kinase (Figure 7A). Because chemoreceptors have inverse kinase and methylation activities, a good mimic of the kinase-off state should have high methylation activity. Thus, methylation assays can be used to distinguish whether A411V is a kinase-off mutant or an inactive protein. Since CF4Q cannot be methylated, methylation assays were performed on CF4E proteins, which contain the methylatable Glu residue at all 4 methylation sites. CF4E can be assembled on vesicles under conditions that promote the kinase-on or kinase-off state.29 The CF4E.A411V mutant protein showed low kinase activity (Figure 7A) and high methylation activity (Figure 7B) when assembled under conditions that promote the kinase-on state, indicating that CF4E.A411V is locked in the kinase-off state of the receptor. This validates CF4Q.A411V as a kinase-off state.
Figure 7.

Kinase and methylation activity for vesicle and PEG-assembled complexes. (A) Kinase-activity of vesicle-assembled complex (black bars) and PEG-assembled complex (gray bars) indicate A411V is kinase-off (note that CF4E does not assemble in PEG). Error bars are ± one standard deviation of two replicates. (B) Gel resolving the methylated CF demonstrates that the kinase-on CF4E (left) is not methylated; under the same assembly conditions, CF4E.A411V (right) shows ∼50% methylation, which is comparable to methylation of CF4E assembled in the kinase-off state (not shown). Thus, A411V locks the receptor into the kinase-off, methylation-on state.
An overlay of the 1H–13C INEPT spectra of the kinase-on (blue, CF4Q) and kinase-off (red, CF4Q.A411V) signaling states (Figure 8), shows similar resonances for both states, which suggests that the C-terminal tail and methylation N-helix are dynamic in both states. The kinase-off spectrum shows increased intensities (an average of 1.4 ± 0.2-fold, Table 4) for resonances corresponding to the mobile methylation N-helix, but comparable intensities for resonances corresponding to the tail (Phe and Pro resonances in 1H–13C INEPT and Trp resonance in 1H–15N INEPT). Thus, we conclude that the C-terminal tail has similar mobility in both states, and the methylation N-helix has increased nanoseconds time scale dynamics in the kinase-off state, leading to increased INEPT intensity.
Figure 8.

1H–13C INEPT of the kinase-on (blue, CF4Q) and kinase-off (red, CF4Q.A411V) states of the uniformly 13C, 15N-labeled CF incorporated into vesicle-mediated functional complexes with CheA and CheW. Spectra are corrected for the difference in nmol of CF. The same resonances are observed for both signaling states, but the kinase-off state exhibits greater intensity for all peaks except peaks corresponding only to the C-terminal tail (Phe and Pro). Inset shows overlay of the 1D projection of each spectrum. Same NMR conditions as Figure 3.
Table 4. Comparison of INEPT Peak Volumes of Kinase-On (CF4Q) and Kinase-Off (CF4QA411V) Vesicle-Assembled Samples.
| residue/s | CF4Q volume | CF4Q.A411V volume | intensity ratio (kinase-on/kinase-off) |
|---|---|---|---|
| total Cα | 82.8 | 109.3 | 1.3 |
| Arg Nε | 5.7 | 7.4 | 1.3 |
| Asn Cβ + Tyr Cβ | 4.1 | 4.8 | 1.2 |
| Glu Cγ | 7.1 | 13.0 | 1.8 |
| Gly Cα | 2.7 | 4.6 | 1.7 |
| Gly N | 2.5 | 3.5 | 1.3 |
| His Cβ | 0.9 | 1.4 | 1.5 |
| His Cε1 | 1.5 | 1.9 | 1.3 |
| Ile Cβ | 3.2 | 4.6 | 1.4 |
| Ile Cδ1 | 3.3 | 4.7 | 1.4 |
| Leu Cβ | 4.3 | 6.1 | 1.4 |
| Met Cβ | 1.5 | 2.5 | 1.7 |
| *Phe | 1.1 | 1.1 | 1.0 |
| *Pro Cδ | 1.3 | 1.3 | 1.0 |
| *Trp Nε | 1.0 | 1.0 | 1.0 |
| Thr Cβ | 8.4 | 11.5 | 1.4 |
| Tyr Cδ | 1.1 | 1.3 | 1.1 |
| average (excluding tail-only residues*): | 1.4 ± 0.2 | ||
Conclusions
INEPT NMR experiments on native-like, functional bacterial chemoreceptor arrays reveal a remarkably dynamic segment of the receptor that undergoes changes in dynamics during signaling. The methylation N-helix, which connects the HAMP region to the protein interaction domain, exhibits dynamics on the nanosecond or shorter time scale, comparable to the dynamics of the unstructured C-terminal tail of the receptor. These results are consistent with previous HDX-MS13 and EPR39 results, and suggest that this segment of the receptor may not be helical and may not pack against the proximal methylation C-helix. This should be considered in the construction of structural models for the array, which have thus far have represented this region as helical.
Furthermore, this study of functional complexes of CF4Q with CheA and CheW tests current proposed models (see Figure 1) for the role of dynamics in propagating the signal that controls the activity of CheA bound at the membrane-distal cytoplasmic tip of the receptor. It has been proposed that methylation region dynamics increase in the kinase-off state.9,10 INEPT spectra support this idea for the methylation N-helix (which may not be helical). However, the less mobile methylation C-helix is not detected by INEPT (so its mobility is limited to the μs or longer time scale), and thus INEPT experiments cannot determine whether its dynamics increase. It has also been proposed that the dynamics of the protein interaction region increase in the kinase-on state.10 INEPT spectra demonstrate that any dynamics in the protein interaction region occur on the microsecond or longer time scale, and that this region is less dynamic than the methylation region in both kinase-on and kinase-off states.
Finally, we propose that the remarkably mobile methylation N-helix is a key element controlling signal propagation through the methylation region. This segment is likely to have more influence on signaling than the methylation C-helix: its conformational equilibrium is determined by the helix stutter in its connection to the HAMP region, and by the methylation of its three sites (compared to one site in the methylation C-helix). Increased fluctuations in this mobile segment in the receptor kinase-off state may destabilize important contacts with CheA to inhibit the kinase activity.
This study demonstrates the utility of mobility-filtered solid-state NMR experiments on native-like assemblies to detect functionally important dynamics. Such approaches are key to understanding the role of dynamics in signaling and in other critical processes that are mediated in cells by large multiprotein complexes.
Acknowledgments
The authors thank Aruni Karunanayake Mudiyanselage for construction of the plasmids expressing TEV-cleavable His-tagged CheA, CheW, and CheY, and Libbie Haglin for construction of the plasmid expressing CF4Q.A411V. We are grateful to Monifa Fahie for assistance with mutagenesis, to Weiguo Hu for advice on solid-state NMR experiments and data analysis, and to Jasna Fezno for assistance with NMR data processing. We also thank Libbie Haglin and Xuni Li for helpful comments on the manuscript. This research was supported by National Institutes of Health Grant R01-GM085288.
Glossary
Abbreviations
- BCA
bicinchonic acid
- BMRB
Biological Magnetic Resonance Bank
- CF
cytoplasmic fragment of Escherichia coli aspartate chemotaxis receptor
- CP
cross-polarization
- DMPC
1,2-Dimyristoyl-sn-glycero-3-phosphorylcholine
- DOGS-NTA-Ni2+
1,2-dioleoyl-sn-glycero-3- [N(5- amino-1-carboxy-pentyl)]
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- EPR
electron paramagnetic resonance
- GPCR
G-protein coupled receptor
- HAMP
histidine kinases, adenyl cyclases, MCPs, and some phosphatases
- HDX-MS
hydrogen–deuterium exchange mass spectrometry
- INEPT
insensitive nuclei enhanced by polarization transfer
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- LB
Luria–Bertani broth
- MAS
magic angle spinning
- NMR
nuclear magnetic resonance
- PEG
polyethylene glycol 8000
- PCR
polymer chain reaction
- PMSF
phenylmethylsulfonyl fluoride
- PPKB
potassium phosphate kinase buffer
- SAM
S-adenosyl methionine
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.7b06475.
1H–13C INEPT (Figure S1) and 1H–15N INEPT (Figure S2) spectra of U–13C,15N-CF4Q and U–13C,15N-CF4QΔ34 in PEG-mediated functional arrays with Che A and CheW; PCR primers used for plasmid construction (Table S1); peak volumes of INEPT spectra of U–13C,15N-CF4Q and U–13C,15N-CF4QΔ34 in PEG-mediated arrays (Table S2) (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of The Journal of Physical Chemistry virtual special issue “Recent Advances in Connecting Structure, Dynamics, and Function of Biomolecules by NMR”.
Supplementary Material
References
- Smock R. G.; Gierasch L. M. Sending Signals Dynamically. Science 2009, 324, 198–204. 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J. S.; Hazelbauer G. L.; Falke J. J. Signaling and Sensory Adaptation in Escherichia Coli Chemoreceptors. Trends Microbiol. 2015, 23, 257–266. 10.1016/j.tim.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B. The Cell as a Collection of Protein Machines: Preparing the Next Generation of Molecular Biologists. Cell 1998, 92, 291–294. 10.1016/S0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Gunsalus R.; Jensen G. J.; Briegel A.; Ortega D. R.; Huang A. N.; Oikonomou C. M. Structural Conservation of Chemotaxis Machinery across Archaea and Bacteria. Environ. Microbiol. Rep. 2015, 7, 414–419. 10.1111/1758-2229.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Li X.; Bilwes A. M.; Hughes K. T.; Jensen G. J.; Crane B. R. Bacterial Chemoreceptor Arrays Are Hexagonally Packed Trimers of Receptor Dimers Networked by Rings of Kinase and Coupling Proteins. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 3766–3771. 10.1073/pnas.1115719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Hu B.; Morado D. R.; Jani S.; Manson M. D.; Margolin W. Molecular Architecture of Chemoreceptor Arrays Revealed by Cryoelectron Tomography of Escherichia Coli Minicells. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E1481–E1488. 10.1073/pnas.1200781109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz S. A.; Falke J. J. Molecular Mechanism of Transmembrane Signaling by the Aspartate Receptor: A Model. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 2545–2550. 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke J. J.; Hazelbauer G. L. Transmembrane Signaling in Bacterial Chemoreceptors. Trends Biochem. Sci. 2001, 26, 257–265. 10.1016/S0968-0004(00)01770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain K. E.; Gonzalez M. A.; Falke J. J. Engineered Socket Study of Signaling through a Four-Helix Bundle: Evidence for a Yin-Yang Mechanism in the Kinase Control Module of the Aspartate Receptor. Biochemistry 2009, 48, 9266–9277. 10.1021/bi901020d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Ames P.; Parkinson J. S. Mutational Analyses of HAMP Helices Suggest a Dynamic Bundle Model of Input-Output Signalling in Chemoreceptors. Mol. Microbiol. 2009, 73, 801–814. 10.1111/j.1365-2958.2009.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelli N. L.; Hazelbauer G. L. Bacterial Chemoreceptor Dynamics: Helical Stability in the Cytoplasmic Domain Varies with Functional Segment and Adaptational Modification. J. Mol. Biol. 2016, 428, 3789–3804. 10.1016/j.jmb.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Borbat P. P.; Dzikovski B.; Freed J. H.; Crane B. R. Bacterial Chemoreceptor Dynamics Correlate with Activity State and Are Coupled over Long Distances. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2455–2460. 10.1073/pnas.1414155112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy S. S.; Li X.; Eyles S. J.; Weis R. M.; Thompson L. K. Hydrogen Exchange Differences between Chemoreceptor Signaling Complexes Localize to Functionally Important Subdomains. Biochemistry 2014, 53, 7755–7764. 10.1021/bi500657v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Wong M. L.; Hodges H. L.; Oikonomou C. M.; Piasta K. N.; Harris M. J.; Fowler D. J.; Thompson L. K.; Falke J. J.; Kiessling L. L.; et al. New Insights into Bacterial Chemoreceptor Array Structure and Assembly from Electron Cryotomography. Biochemistry 2014, 53, 1575–1585. 10.1021/bi5000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Hazelbauer G. L. Core Unit of Chemotaxis Signaling Complexes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9390–9395. 10.1073/pnas.1104824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sferdean F. C.; Weis R. M.; Thompson L. K. Ligand Affinity and Kinase Activity Are Independent of Bacterial Chemotaxis Receptor Concentration: Insight into Signaling Mechanisms. Biochemistry 2012, 51, 6920–6931. 10.1021/bi3007466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. J.; Struppe J. O.; Wylie B. J.; McDermott A. E.; Thompson L. K. Multidimensional Solid-State Nuclear Magnetic Resonance of a Functional Multiprotein Chemoreceptor Array. Biochemistry 2016, 55, 3616–3624. 10.1021/acs.biochem.6b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrout A. L.; Montefusco D. J.; Weis R. M. Template-Directed Assembly of Receptor Signaling Complexes. Biochemistry 2003, 42, 13379–13385. 10.1021/bi0352769. [DOI] [PubMed] [Google Scholar]
- Fowler D. J.; Weis R. M.; Thompson L. K. Kinase-Active Signaling Complexes of Bacterial Chemoreceptors Do Not Contain Proposed Receptor-Receptor Contacts Observed in Crystal Structures. Biochemistry 2010, 49, 1425–1434. 10.1021/bi901565k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A.; Ladinsky M. S.; Oikonomou C.; Jones C. W.; Harris M. J.; Fowler D. J.; Chang Y. W.; Thompson L. K.; Armitage J. P.; Jensen G. J. Structure of Bacterial Cytoplasmic Chemoreceptor Arrays and Implications for Chemotactic Signaling. eLife 2014, 3, e02151. 10.7554/eLife.02151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H.; Das B. B.; Casagrande F.; Tian Y.; Nothnagel H. J.; Chu M.; Kiefer H.; Maier K.; De Angelis A. A.; Marassi F. M.; et al. Structure of the Chemokine Receptor CXCR1 in Phospholipid Bilayers. Nature 2012, 491, 779–783. 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loquet A.; Sgourakis N. G.; Gupta R.; Giller K.; Riede D.; Goosmann C.; Griesinger C.; Kolbe M.; Baker D.; Becker S.; et al. Atomic Model of the Type III Secretion System Needle. Nature 2012, 486, 276–279. 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Nadaud P. S.; Bernier M. W.; North J. A.; Hammel P. C.; Poirier M. G.; Jaroniec C. P. Histone H3 and H4 N-Terminal Tails in Nucleosome Arrays at Cellular Concentrations Probed by Magic Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc. 2013, 135, 15278–15281. 10.1021/ja407526s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick K. K.; Debelouchina G. T.; Kayatekin C.; Dorminy T.; Jacavone A. C.; Griffin R. G.; Lindquist S. Distinct Prion Strains Are Defined by Amyloid Core Structure and Chaperone Binding Site Dynamics. Chem. Biol. 2014, 21, 295–305. 10.1016/j.chembiol.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S. A.; Bajakian T. H.; Soria M. A.; Falk A. S.; Service R. J.; Langen R.; Siemer A. B. Identification and Structural Characterization of the N-Terminal Amyloid Core of Orb2 Isoform A. Sci. Rep. 2016, 6, 1–11. 10.1038/srep38265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmus J. J.; Surewicz K.; Nadaud P. S.; Surewicz W. K.; Jaroniec C. P. Molecular Conformation and Dynamics of the Y145Stop Variant of Human Prion Protein in Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6284–6289. 10.1073/pnas.0711716105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelli N. L.; Hazelbauer G. L. Direct Evidence That the Carboxyl-Terminal Sequence of a Bacterial Chemoreceptor Is an Unstructured Linker and Enzyme Tether. Protein Sci. 2011, 20, 1856–1866. 10.1002/pro.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames P.; Parkinson J. S. Conformational Suppression of Inter-Receptor Signaling Defects. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9292–9297. 10.1073/pnas.0602135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besschetnova T. Y.; Montefusco D. J.; Asinas A. E.; Shrout A. L.; Antommattei F. M.; Weis R. M. Receptor Density Balances Signal Stimulation and Attenuation in Membrane-Assembled Complexes of Bacterial Chemotaxis Signaling Proteins. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 12289–12294. 10.1073/pnas.0802868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber K. R.; Tycko R. Measurement of Sample Temperatures under Magic-Angle Spinning from the Chemical Shift and Spin-Lattice Relaxation Rate of 79Br in KBr Powder. J. Magn. Reson. 2009, 196, 84–87. 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo C. S.; Metz K. R.; Sun Y.; Sherry A. D. NMR Temperature Measurements Using a Paramagnetic Lanthanide Complex. J. Magn. Reson. 1998, 133, 53–60. 10.1006/jmre.1998.1429. [DOI] [PubMed] [Google Scholar]
- Morcombe C. R.; Zilm K. W. Chemical Shift Referencing in MAS Solid State NMR. J. Magn. Reson. 2003, 162, 479–486. 10.1016/S1090-7807(03)00082-X. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Bigam C. G.; Yao J.; Abildgaard F.; Dyson H. J.; Oldfield E.; Markley J. L.; Sykes B. D. 1H, 13C, and 15N Chemical Shift Referencing in Biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- Andronesi O. C.; Becker S.; Seidel K.; Heise H.; Young H. S.; Baldus M. Determination of Membrane Protein Structure and Dynamics by Magic-Angle-Spinning Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 12965–12974. 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe N. P.; Parthasarathy S.; Jones C. R.; Bhardwaj C.; Long F.; Kotecha M.; Mehboob S.; Fung L. W.; Past J.; Samoson A.; et al. Nanomole-Scale Protein Solid-State NMR by Breaking Intrinsic 1H T1 Boundaries. Nat. Methods 2009, 6, 215–218. 10.1038/nmeth.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Li G.; Weis R. M. The Serine Chemoreceptor from Escherichia Coli Is Methylated through an Inter-Dimer Process. Biochemistry 1997, 36, 11851–11857. 10.1021/bi971510h. [DOI] [PubMed] [Google Scholar]
- Wu J.; Li J.; Li G.; Long D. G.; Weis R. M. The Receptor Binding Site for the Methyltransferase of Bacterial Chemotaxis Is Distinct from the Sites of Methylation. Biochemistry 1996, 35, 4984–4993. 10.1021/bi9530189. [DOI] [PubMed] [Google Scholar]
- Kim K. K.; Yokota H.; Kim S. Four-Helical-Bundle Structure of the Cytoplasmic Domain of a Serine Chemotaxis Receptor. Nature 1999, 400, 787–792. 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- Bartelli N. L.; Hazelbauer G. L. Differential Backbone Dynamics of Companion Helices in the Extended Helical Coiled-Coil Domain of a Bacterial Chemoreceptor. Protein Sci. 2015, 24, 1764–1776. 10.1002/pro.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Weis R. M. Covalent Modification Regulates Ligand Binding to Receptor Complexes in the Chemosensory System of Escherichia Coli. Cell 2000, 100, 357–365. 10.1016/S0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.