

Abstract
We present a three-dimensional nonlinear tumor growth model composed of heterogeneous cell types in a multicomponent-multispecies system, including viable, dead, healthy host, and extracellular matrix (ECM) tissue species. The model includes the capability for abnormal ECM dynamics noted in tumor development, as exemplified by pancreatic ductal adenocarcinoma, including dense desmoplasia typically characterized by a significant increase of interstitial connective tissue. An elastic energy is implemented to provide elasticity to the connective tissue. Cancer-associated fibroblasts (myofibroblasts) are modeled as key contributors to this ECM remodeling. The tumor growth is driven by growth factors released by these stromal cells as well as by oxygen and glucose provided by blood vasculature which along with lymphatics are stimulated to proliferate in and around the tumor based on pro-angiogenic factors released by hypoxic tissue regions. Cellular metabolic processes are simulated, including respiration and glycolysis with lactate fermentation. The bicarbonate buffering system is included for cellular pH regulation. This model system may be of use to simulate the complex interactions between tumor and stromal cells as well as the associated ECM and vascular remodeling that typically characterize malignant cancers notorious for poor therapeutic response.
Keywords: Nonlinear model, 3D simulation, cancer modeling, desmoplasia, vascular tumor, mathematical modeling, computational simulation
Graphical abstract
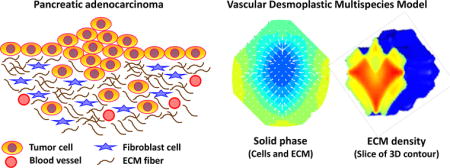
1. Introduction
Following the six hallmarks of cancer (Hanahan and Weinberg, 2000), carcinogenesis occurs when genetically defected cells acquire the ability to be self-sufficient in growth signals, insensitive to growth-inhibitory signals, have the ability to evade apoptosis, replicate with limitless potential, sustain angiogenesis, and ultimately invade surrounding tissue and metastasize. These acquired capabilities are supported by enabling characteristics (Hanahan and Weinberg, 2011) including the genomic instability in cancer cells (Hanahan and Weinberg, 2000; Lengauer et al., 1997; Luo et al., 2009; Negrini et al., 2010; Nowell, 1976) and tumor-promoting inflammation (Colotta et al., 2009), as well as reprograming of cellular energy metabolism and active evasion of immunosurveillance (Kroemer and Pouyssegur, 2008). The state of cancer cells is also characterized by the presence of DNA replication stress (Halazonetis et al., 2008), oxidative stress, mitotic stress, proteotoxic stress, and metabolic stress (Luo et al., 2009).
Genomic defects in a tumor cell alter its intrinsic cellular programs. Loss of cell cycle check control and programmed cell death mechanisms, along with modified differentiation and transformed metabolism, propel tumor cells to a hyper proliferating state. In the initial avascular growth phase, relying on diffusion of oxygen and nutrients from nearby existing blood vessels, tumor cells quickly outgrow the supply and reach a quiescent state with a hypoxic or necrotic core (Chaplain, 1996). In order to survive, hypoxic tumor cells upregulate an array of cytokines, growth factors, and proteases. The loss of appropriate balance in these molecules leads to the stimulation of new vessel growth, degradation of the extracellular matrix (ECM), and recruitment of immune cells. The resulting neovasculature provides additional oxygen and nutrients for the neoplastic growth. The destruction of normal ECM facilitates tumor angiogenesis and directed migration of invading tumor cells during metastasis. The infiltrating immune cells are subsequently coopted to promote tumorigenesis. Thus, advancing through the stages of normal, benign, malignant, and metastatic, cancer cells depend not only on changes inside the cell itself but also on what their environment is able to provide.
Since Paget’s seed-and-soil hypothesis (Paget, 1889) over a century ago, much has been understood about the importance of the tumor milieu on cancer growth and metastasis. A typical dynamic microenvironment in which tumors reside consists of cancer stem cells, highly proliferating neoplastic cells of different phenotypes, necrotic tumor cells, infiltrating innate and adaptive immune inflammatory cells, cancer-associated fibroblasts (CAF), ECM, blood vessels, endothelial cells (ECs), pericytes, host cells, and a variety of soluble molecules (de Visser and Coussens, 2006; Hanahan and Weinberg, 2000; Perez-Moreno, 2009; Tlsty and Coussens, 2006; Whiteside, 2008). The process of tumor progression is driven by the communication between the tumor cells and their surroundings. It is this tumor microenvironment that dictates the tumor progress from its unregulated neoplastic growth to eventual metastasis. An adequately and appropriately posed tumor model could be useful in predicting cancer behavior. Striving to mimic true biological aspects, we present here an attempt to model solid tumor growth with tumor-induced interactions in its heterogeneous milieu. The ultimate goal would be to predict tumor dynamics and treatment response so that good correspondence is achieved with in vivo or in situ tumor growth data.
Mathematical models of tumor growth have concentrated on simulating tumor behavior in response to certain stimuli in each of the stages of growth, including avascular and vascular conditions. These models generally fall into three categories: (i) continuum models (including single phase and multiphase/mixture mechanochemical approaches), (ii) discrete models, and (iii) hybrid models representing a combination of continuum and discrete approaches. In continuum models (see recent reviews (Andasari et al., 2011; Bachmann et al., 2012; Byrne, 2010; Chaplain, 2011; Cristini and Lowengrub, 2010; Deisboeck et al., 2011; Edelman et al., 2010; Frieboes et al., 2011; Kreeger and Lauffenburger, 2010; Lowengrub et al., 2010; Michor et al., 2011; Oden et al., 2015; Osborne et al., 2010; Preziosi and Tosin, 2009a; Rejniak and McCawley, 2010; Rejniak and Anderson, 2011; Roose et al., 2007; Tracqui, 2009; Vineis et al., 2010) and references therein), cell populations and molecular species that influence the cell cycle events are treated as continuous variables. These models typically make use of ODE or PDE approaches to describe an advection-diffusion-reaction system. For models which involve several cell types, tracking of the interfaces is necessary and may be accomplished using the level set method. Continuum multiphase/mixture mechanochemical models incorporate mechanical and chemical interactions between phases (cell types or species) (see (Araujo and McElwain, 2004; Astanin and Preziosi, 2008; Byrne et al., 2006; Graziano and Preziosi, 2007; Hatzikirou et al., 2005; Lowengrub et al., 2010; Preziosi and Tosin, 2009a; Quaranta et al., 2005; Roose et al., 2007; Tracqui, 2009) and associated references). Typical models of this approach introduce a stress tensor, pressure, and velocity for each phase by enforcing the mass, momentum, and energy balances (Ambrosi et al., 2002; Araujo and McElwain, 2005a; Araujo and McElwain, 2005b; Astanin and Preziosi, 2008; Bresch et al., 2010; Breward et al., 2002; Breward et al., 2003; Byrne and Preziosi, 2003; Byrne et al., 2003; Galle et al., 2009; Graziano and Preziosi, 2007; Klika, 2014; Preziosi and Tosin, 2009b; Preziosi and Vitale, 2011; Preziosi et al., 2010; Sciume et al., 2013). Related to the continuum multicomponent mixture models is the diffuse interface approach (Chen et al., 2014; Hawkins-Daarud et al., 2012; Oden et al., 2010). The square gradient theory can be used in this approach to describe the smooth transition within a thin interfacial region. The gradient contributes to the Helmholtz free energy, from which the component velocities, pressures, and diffusive terms are derived (Chen and Lowengrub, 2014; Wise et al., 2008). Continuum single- or multi-phase models that consider the effects of cell-cell and/or cell-ECM adhesion include among others (Ambrosi and Preziosi, 2009; Bearer et al., 2009; Chatelain Clément et al., 2011; Escher and Matioc, 2013; Frieboes et al., 2007; Frieboes et al., 2013; Kuusela and Alt, 2009), while in (Arduino and Preziosi, 2015; Gerisch and Chaplain, 2008; Preziosi and Tosin, 2009b; Psiuk-Maksymowicz, 2013; Sciume et al., 2014a; Sciume et al., 2014b; Wu et al., 2013), the ECM is represented as one of the key components of the tumoral tissue.
In this paper, we present a tumor growth model consisting of heterogeneous cell types in a multicomponent-multispecies system. Taken into consideration are the effects of metabolic molecules, tumorigenic factors, and desmoplastic reaction, coupled with tumor-induced angiogenesis. Since tumors may contain as many as 105 to 107 cells per mm3 (Fang et al., 2000; Fidler and Hart, 1982; Holmgren et al., 1995; Zheng et al., 2005), a continuum scale is thus appropriate to model tumor growth. Starting from a mixture system similar to Frieboes et al. (2010), we implement the diffuse interface approach, as derived in Wise et al. (2008), where thermodynamically consistent Darcy velocities and Fickian diffusive terms are determined from the energy variation. The square gradient model is used in the Helmholtz free energy equation (Cahn and Hilliard, 1958; Rowlinson, 1979; Yang et al., 1976) to describe interfaces arising from the adhesive properties of different cell components. Unlike Frieboes et al. (2010), continuous blood and lymphatic vessel densities here are modified from cell fluxes employed in Anderson and Chaplain (1998), Chaplain (1996), and Mantzaris et al. (2004), with different sprout initiation conditions included (Levine et al., 2000; Levine et al., 2001a; Levine et al., 2001b). We model the ECM as its own species to interact with the tumor cell species, and include an elastic energy that provides elasticity to the connective tissue. Stromal cells representing cancer-associated fibroblasts are modeled as principal contributors to the ECM remodeling. The stromal cells further support the tumor growth through the release of growth factors. For the tumor cells, we include the cellular metabolic processes of respiration and glycolysis with lactate fermentation, and a bicarbonate buffering system to simulate the cellular regulation of pH. Interactions between angiogenic factors, proteolytic enzymes, and ECM components described by Levine et al. (2001b) are also incorporated. Nutrients and waste products from cell metabolism are governed by fluxes and consumption/production rates modified from Casciari et al. (1992). We note that previous work has evaluated the important roles of glucose metabolism and microenvironmental acidity in tumor progression, including (Smallbone et al., 2008; Smallbone et al., 2005; Smallbone et al., 2007).
This paper develops as follows. An overview of relevant biology is outlined in Section 2 where the scope of the model biological hypotheses is described in detail. All components considered as well as interactions between each component are discussed. In Section 3, the model formulation is described, including the derivation of velocities, fluxes, and source terms. Governing equations are presented and the system of nonlinear partial differential equations is nondimensionalized. Numerical schemes for the solution are discussed in Section 4. In Section 5, simulation results of three-dimensional tumor cases are delivered and discussed. Finally, conclusions and the direction of future work are described in Section 6.
2. Biological Background and Hypotheses
The tumor system is a complex domain that includes heterogeneous cell types and stroma maintained by a vast interplay of signaling pathways. Enumerated in this section are key components on which we focused in modeling the neoplastic growth in the tumor microenvironment.
2.1. Tumor and Host Cells
Whether tumors originate from a single or multiple transformed cells, the resulting cancer cells from neoplastic progression within tumors display diverse phenotypes that may have varying proliferation and metastatic potentials (Fidler and Hart, 1982; Gupta et al., 2011; Heppner, 1984; Lobo et al., 2007). Although tumor heterogeneity is generally believed to be an outcome of genomic instability and selectivity (Grady and Markowitz, 2000; Heng et al., 2006), cancer stem-like cells (CSC) have been shown to play a role as well (Campbell and Polyak, 2007; Dontu et al., 2003; Marotta and Polyak, 2009). Multiple types of CSCs (Marotta and Polyak, 2009) may potentially arise from normal stem cells or transit-amplifying progenitor cells with oncogenic mutations (Clarke and Fuller, 2006; Lobo et al., 2007). Supported and protected by the CSC niche (Borovski et al., 2011), these stem-cell-like phenotypes retain their abilities to self-renew and differentiate (Clarke et al., 2006; Lobo et al., 2007), subsequently driving the tumor growth and metastasis (Borovski et al., 2011; Clarke and Fuller, 2006; Lobo et al., 2007; Marotta and Polyak, 2009). Cancer is also genetically related to autophagy malfunctions, a normally regulated cellular catabolic response to stress and nutrients deficiency in order to maintain homeostasis and facilitate cell survival (see refs (Kimmelman, 2011; Mathew et al., 2007; Mizushima et al., 2008; White, 2012; White et al., 2010)), which is cytoprotective and contributes to the survival of cancer cells in low nutrient environment and their resistance to anticancer treatments.
In this study, we make the simplifying assumption that there is only one viable tumor cell component, one dead tumor cell component, and a healthy host cell component: original viable phenotype (V), dead/necrotic tumor cell (D), and healthy host cell (H). Viable tumor species may undergo mitosis, apoptosis, and necrosis, whereas host cells are assumed to be homeostatic. Viable tumor species can also derive molecules like growth factors (refer to Section 2.5), angiogenic factors (Section 2.6), and matrix degrading enzymes (Section 2.7).
2.2. Stroma
In addition to the tumor cells, host cells, and infiltrating immune cells, a neoplastic tissue environment also consists of other resident cells such as fibroblasts and vascular cells that constitute the local blood and lymphatic vessels, ECM, interstitial fluid, as well as molecules secreted by cells. Among these secretions are ECM components, growth factors, cytokines, chemokines, proteases, and various metabolites. Note that stroma defined here encompasses all components stated above, excluding the tumor, host, and immune cells (which are accounted for in previous sections).
The ECM is a vital framework that plays a monumental role in tumor progression. In addition to providing cells with a mechanical scaffold for adhesion and migration, ECM also interacts with cells to directly or indirectly regulate developmental processes and control cellular activities such as cell differentiation, proliferation, and apoptosis (Aumailley and Gayraud, 1998).
Under homeostatic conditions, fibroblasts and vascular cells synthesize the appropriate amounts of ECM components (Bosman and Stamenkovic, 2003; Kalluri, 2003). Composition of ECM may vary considerably depending on the type of tissue and the location within the tissue. Furthermore, it may fluctuate to adapt to varying signals during normal developmental processes as well as pathological processes (Tlsty and Coussens, 2006). In general, the constitutive ECM components of most tissues include both fibrillar and nonfibrillar collagens, noncollagenous glycoproteins, and proteoglycans, which share common domains but have different physical and biochemical properties (Aumailley and Gayraud, 1998; Bosman and Stamenkovic, 2003; Oezbek et al., 2010; Whittaker et al., 2006). These components constitute the basement membrane and the interstitial matrix. Basement membrane is more compact and less porous than the interstitial matrix. It also consists of epithelial, endothelial, and stromal cells, keeping epithelium and endothelium separated from the stroma, and providing a scaffold for cell adhesion (Kalluri, 2003; Lu et al., 2012). Interstitial matrix is highly charged and hydrated. Rich in fibrillar collagens, proteoglycans, and various glycoproteins, it is also a main factor in the tensile strength of tissues (Egeblad et al., 2010).
The most abundant structural components of ECM are collagens, which have a propensity to form highly organized polymers (Aumailley and Gayraud, 1998). While the major component of basement membranes is collagen IV (Aumailley and Gayraud, 1998), roughly 80–90% of all collagenous proteins in soft tissues are type I collagen (Tlsty and Coussens, 2006). Type I collagen can release diffusible signaling molecules upon breakdown (Aumailley and Gayraud, 1998). It belongs to an ECM protein family that is crucial in maintaining the structural integrity of organs and tissues, and contributes to regulations of cell phenotype, polarity, survival, and migration (Vuorio and Decrombrugghe, 1990). Several classes of proteoglycans having various sizes and protein cores also constitute to the ECM. Proteoglycans function as a joint between various collagenous and glycoprotein networks by binding to other ECM molecules, thus regulating the structural arrangement and stabilizing the ECM architecture (Aumailley and Gayraud, 1998). Other than collagens and proteoglycans, noncollagenous glycoproteins also make up a prominent meshwork of the ECM. One of the most studied glycoproteins in the interstitial connective tissue is fibronectin. Fibronectins are known to initiate matrix assembly and form fibrils, but the polymerization is cell dependent and direct interactions with cell surface integrin receptors are required (Kadler et al., 2008; Singh et al., 2010). Embedded between endothelial and perivascular cells perivascular cells (Astrof and Hynes, 2009), it has also been shown to promote cell adhesion (Pierschbacher and Ruoslahti, 1984; Pierschbacher et al., 1984) and blood vessel development.
The diverse structural, biochemical, and biomechanical functions of the ECM components contribute to the regulation of essential cell behavior (Lu et al., 2012). The ECM provides cells with contextual biological information and a mechanical scaffold to respond appropriately following the onset of certain stimuli (Bissell et al., 2002; Howe et al., 1998; Weaver et al., 2002). Its physical features, such as rigidity, porosity, spatial arrangement, and orientation, allow it to support tissue architecture and integrity (Lu et al., 2012). The ECM is also a highly charged protein network that can directly initiate signaling events (Hynes, 2009; Lu et al., 2011), bind to a wide range of growth factors (Hynes, 2009), and transmit biochemical signals via cell-surface adhesion receptors (Hynes, 2002), thus controlling the accessibility, limiting the diffusive range, and dictating the signaling direction of ligands to their cognate receptors (Norton et al., 2005). Biomechanically, the elasticity of ECM governs how external forces are perceived by a cell (Gehler et al., 2009; Lopez et al., 2008; Paszek et al., 2005), guiding cellular behavior in response to environmental changes (DuFort et al., 2011; Fernandez-Gonzalez et al., 2009; Koelsch et al., 2007; Montell, 2008; Pouille et al., 2009; Solon et al., 2009), leading to the determination of cell differentiation and tissue function (Engler et al., 2006; Gilbert et al., 2010; Lutolf et al., 2009).
While ECM remodeling with a tightly regulated balance may be essential for maintaining tissue integrity (Bosman and Stamenkovic, 2003; Kalluri, 2003; Ruiter et al., 2002), abnormal ECM dynamics have been seen in tumor development (Bergers and Coussens, 2000; Cox and Erler, 2011; Egeblad and Werb, 2002; van Kempen et al., 2003). For instance, pancreatic cancer, especially pancreatic ductal adenocarcinoma (Pandol et al., 2009), exhibits dense desmoplastic reaction which is identified by a significant increase of interstitial connective tissue (Gress et al., 1995). Cancer-associated fibroblasts (CAFs, or myofibroblasts) are the main contributors of the ECM remodeling enzymes (Bhowmick et al., 2004; Orimo et al., 2005) in tumor tissues and are responsible for the synthesis, deposition, and remodeling of the ECM (Aboussekhra, 2011; Liu et al., 2011; Nakagawa et al., 2004; Östman and Augsten, 2009; Rasanen and Vaheri, 2010; Sirica et al., 2011) (refer to Section 2.4). To sustain neoplastic growth, it is essential for solid tumors to coopt fibroblasts, inflammatory, and vascular cells to upset the balance between ECM synthesis and degradation (Ruiter et al., 2002). Perturbation to the control mechanisms disorganizes the ECM and changes its architecture (Clarijs et al., 2003), facilitating oncogenic transformations (Levental et al., 2009) and upregulating signals that can promote cell survival and proliferation (Paszek et al., 2005; Wozniak et al., 2003). Deregulation of ECM modeling may also lead to apoptotic evasion due to the antiapoptotic effects exerted by fragments of various ECM components (Mott and Werb, 2004).
Another main component of the stroma is interstitial fluid. Increased peritumor interstitial fluid convection (Dafni et al., 2002) and higher lymphatic drainage to the sentinel lymph node (Harrell et al., 2007; Proulx et al., 2010) have been reported, suggesting an increased in interstitial flow within the tumor microenvironment. The hypoxia resulting from rapidly growing tumor fuses atypical angiogenesis, generating leaky tumor vessels that cause the accumulation of macromolecules in the neoplastic tissue. This condition, coupled with the mechanical stress from ECM remodeling (DuFort et al., 2011; Xu et al., 2009), leads to increased interstitial fluid pressure (IFP) within the tumor, ranging from 10–40 mmHg (Fukumura and Jain, 2007; Heldin et al., 2004). Tumor IFP may reach the levels of capillary pressure (Boucher et al., 1996) while normal tissue exhibits pressures ranging from −2 to 0 mmHg (Wiig, 1990).
In our model, stroma is assumed to be made up mainly by interstitial fluid (W) and the interstitial matrix (E). Here, we do not differentiate between various ECM components. Their secretions by viable cancer cells (V), ECs (B), LECs (L), and especially by myofibroblastic cells (F) are modeled. The decay of ECM involves proteolytic reactions with matrix degrading enzymes (refer to Section 2.7). Since ECM macromolecules diffuse very slowly (Levine et al., 2001b), we assume that the individual diffusive flux of various macromolecules within ECM are negligible. In our model, within the ECM component, we track the blood and lymphatic vessel densities (refer to Section 2.3), myofibroblastic cell density (Section 2.4). Concentrations of growth factors (Section 2.5), cytokines (Section 2.6), proteases (Section 2.7), and metabolites (Section 2.8) are also monitored within the interstitial fluid phase.
2.3. Blood and Lymphatic Vessels
According to Kerbel (2000), the term “tumor angiogenesis” was coined by Shubik in 1968 (Greenblatt and Shubi, 1968). From studies done by other investigators (Algire and Legallais, 1947; Algire et al., 1945; Greenblatt and Shubi, 1968; Ide et al., 1939; Warren and Shubik, 1966) and his own (Folkman, 1970; Folkman, 1972; Folkman, 1974; Folkman, 1976; Folkman and Gimbrone, 1971; Folkman et al., 1963; Folkman et al., 1966; Folkman et al., 1971), Folkman first hypothesized in 1971 that tumor growth is angiogenesis dependent and that angiogenesis could be a relevant target for tumor therapy. He suggested that tumor cells may produce a diffusible chemical signal to switch ECs from a resting state to a rapid growth phase, which in turns aids the otherwise diffusion-limited dormant tumor mass to expand relentlessly. These theories are now widely accepted with the discoveries of pro-angiogenic molecules, generally known as Tumor Angiogenic Factors (TAFs) (see Section 2.6).
Avascular tumor outgrowth is limited to 1–3 mm in diameter (Folkman et al., 1966; Macklin et al., 2009; Marmé and Fusenig, 2007). To sustain anabolic growth, tumor cells must recruit new blood vessels from the nearby pre-existing vasculature network. The first step of angiogenesis involves rearrangements and recruitments of ECs from the parental vessel to form new sprouts (Cliff, 1963; Paweletz and Knierim, 1989; Schoefl, 1963; Schoefl and Majno, 1964; Warren, 1970). The ECs also begin to secrete matrix degrading enzymes (see Section 2.7) to break down the surrounding ECM, making chemotactic migration up signal gradient possible (Mantzaris et al., 2004; Patel and Nagl, 2010). Proliferation of ECs occurs later, about 36 – 48 hours after the initial response (Sholley et al., 1977; Sholley et al., 1984; Warren et al., 1972), near the base of the sprout (Cliff, 1963; Schoefl, 1963; Schoefl and Majno, 1964). Following the development of lumina within solid strands of ECs formed in the ECM, neighboring sprouts join to form loops and enable circulation (Paweletz and Knierim, 1989).
Vascular tissue is composed of two cell types that interact with each other. While ECs line the inner wall of a vessel, pericytes (also known as Rouget cells, mural cells, or referred to as vascular smooth muscle cells VSMCs) embrace the abluminal endothelium wall and are embedded within the basement membrane. Microvessels are mainly consisted of ECs surrounded by a basal lamina loosely wrapped by single pericytes. Larger vessels are coated abluminally with multiple layers of smooth muscle cells and surrounded by collagenous fibers (Cleaver and Melton, 2003). On top of functioning as a scaffold, pericytes also communicate with ECs via gap junctions and adhesion plaques (Rucker et al., 2000). The cell-cell contact mechanisms may also be crucial in vessel maintenance and modulation of EC growth by pericytes (Orlidge and Damore, 1987).
In addition to angiogenesis, tumors also drive lymphangiogenesis in their microenvironment (Swartz and Lund, 2012). In many human tumors, increased lymphatic vessel density, along with high expressions of lymphangiogenic growth factors (refer to Section 2.8), are correlated with poor prognosis, invasion, and metastasis (Mumprecht and Detmar, 2009; Skobe et al., 2001; Tammela and Alitalo, 2010). Cancer cells are thought to first spread to a sentinel tumor-draining lymph node by recruiting lymphatic vessels and entering local lymphatic circulation (Swartz and Lund, 2012). From the lymphatic system, the malignant cells are transported to the blood circulation and subsequently spread via blood vessels to distal organs (Fujisawa et al., 1995; Taubert et al., 2004; Weiss and Ward, 1987).
Lymphatic flow plays an important role in circulation. Extravasated plasma leaking from blood capillaries, together with macromolecules and leukocytes, make their way through the interstitium and drains into local lymphatic vessels. The physiological function of lymphatic networks is to collect the lymph fluid at regional lymph nodes for immune surveillance and transfer them to the blood circulation (Pepper and Skobe, 2003; Stacker et al., 2002; Witte and Witte, 1987). Lymph flow from tumors has also been shown to be elevated (Dafni et al., 2002; Harrell et al., 2007) and increased lymph drainage has been correlated positively with metastasis (Pathak et al., 2006).
Tumors may go through a phase of lymphangiogenic switch. Similar to the angiogenic switch, it is likely to involve the over production of lymphangiogenic growth factors and downregulation of relevant inhibitors (Cao, 2005). A range of lymphangiogenic factors can be produced by tumor, inflammatory, and stromal cells (Cao et al., 2004; Chang et al., 2004; Veikkola and Alitalo, 2002), reflecting the presence of complex processes comparable to those involved in angiogenesis. Lymphatic Microvessels consist of an irregular wider lumen, contained by a single thin layer of overlapping non-fenestrated lymphatic endothelial cells (LECs), and either lack or have an incomplete basement membrane (Leak, 1971; Saharinen et al., 2004). The lymphatic capillaries are tethered to the ECM to ensure the patency of the vessels, and the capillary wall also exhibits valve-like structures that facilitate the uptake of fluid (Saharinen et al., 2004). Microvasculature found within a tumor environment is usually disorganized and leaky. Not surprisingly, studies found that peritumoral and intratumoral lymphatic microvessels are also disorganized and lack drainage function (Isaka et al., 2004; Padera et al., 2002). This structural irregularity might contribute to their susceptibility to invading malignant cells (Kim et al., 1988).
In our model, densities of blood (Bn) and lymphatic (Ln) vessels are tracked, represented by their respective endothelial cells, ECs and LECs. They secrete factors (refer to Section 2.5) that promote the growth of tumor cells and produce ECM macromolecules (Section 2.2). They also generate and uptake tumor angiogenic and lymphangiogenic factors (Section 2.6) as well as produce proteolytic enzymes (Section 2.7). The densities of these vessels also affect the supply of nutrients and the clearing of waste products (Section 2.8) within the tumor tissue.
2.4. Myofibroblastic Cells
It is well established that myofibroblasts play a vital role in wound healing and pathological ECM remodeling. In addition, the protagonistic involvement of myofibroblasts in the stroma reaction of epithelial tumors may stimulate cancer cell invasion (De Wever and Mareel, 2003).
Depending of the type of tissue and organ, myofibroblastic cells found responsible for the desmoplastic reaction in tumor stroma may come from a heterogeneous origin including residential fibroblasts or mesenchymal stem cells, vascular smooth muscle cells, pericytes, ECs, epithelial tumor cells via epithelial-mesenchymal transition, circulating fibrocytes or bone marrow–derived mesenchymal stem cells, adipocytes, hepatic stellate cells, and pancreatic stellate cells (PSCs) (De Wever et al., 2008; Hinz et al., 2007; Hinz et al., 2012; Östman and Augsten, 2009).
Under normal conditions in intact tissue, crosslinked ECM stress-shields fibroblastic cells, which produce little ECM and show few to no actin-associated cell-cell and cell-matrix contacts (Tomasek et al., 2002). After an event of tissue injury where the continuous remodeling of ECM disrupts the protective mechanical environment (Tomasek et al., 2002), or via coercion by malignant tumor cells, these fibroblastic cells undergo some phenotype changes and become myofibroblastic, the activated state. Myofibroblasts regulate connective tissue remodeling by synthesizing ECM components (Hinz, 2007), mediated by cytokines produced by inflammatory, resident (Werner and Grose, 2003), or malignant epithelial cells (De Wever and Mareel, 2003), and by exerting traction forces through their stress fibers (Tomasek et al., 2002) reminiscent of the contractile filaments on smooth muscle cells. In fact, the expression of α–smooth muscle actin (α–SMA) has been commonly used as a molecular marker for myofibroblast differentiation.
There are two stages to the formation of myofibroblasts. When fibroblasts experience mechanical tension, they acquire the proto-myofibroblast phenotype capable of generating contractile force, forming cytoplasmic actin-containing stress fibers that meet at fibronexus adhesion complexes, as well as expressing and organizing cellular fibronectin with ED-A splice variant at cell surface (Tomasek et al., 2002). In addition to extracellular stress, platelet-derived growth factor (PDGF, refer to Section 2.5) appears to be important in proto-myofibroblast formation (Lindahl and Betsholtz, 1998; Martin, 1997), where the absence of PDGF-A results in the lack of proto-myofibroblasts (Boström et al., 1996). Under persistent mechanical stress and the presence of both transforming growth factor–β1 (TGF–β1, refer to Section 2.5) and ED-A fibronectin (Desmouliere et al., 1993; Hinz et al., 2001; Serini et al., 1998), proto-myofibroblasts can further develop into differentiated myofibroblasts, distinguishable by their elevated ED-A fibronectin expression, amplified stress fibers–fibronexus assembly and complexity (Dugina et al., 2001; Vaughan et al., 2000), and most identifiably, their expression of α–SMA. In a tumor environment, myofibroblast differentiation is also induced by oxidative stress caused by reactive oxygen species (Toullec et al., 2010). After the extracellular tension has been resolved, as occurs after an event of tissue repair, the stress release leads to myofibroblast apoptosis (Grinnell et al., 1999).
CAFs also produce various soluble paracrine growth factors (refer to Section 2.5), such as epithelial growth factor, hepatocyte growth factor, or transforming growth factor-β (Kalluri and Zeisberg, 2006). These tumor-promoting growth factors are known to regulate cell proliferation, morphology, survival, and death (Tlsty and Coussens, 2006). Persistent DNA damage found in human precancerous lesions (Gorgoulis et al., 2005) and during the early stages of human tumorigenesis (Bartkova et al., 2005) has been reported to result in enhanced secretion of IL-6 and IL-8 by fibroblasts (Rodier et al., 2009). CAFs are also found to increase their secretion of cytokines and chemokines, including COX-2, CXCL1, CXCL2, IL–1β, IL-6, and CXCL14 (Augsten et al., 2009; Erez et al., 2010). Vascular endothelial growth factor (Dong et al., 2004) and fibroblast growth factor (Pietras et al., 2008) derived from CAFs are found to be crucial for tumor angiogenesis. A study with breast CAFs showed that their secretion of CXCL12 led to the recruitment of bone marrow-derived endothelial precursor cells into the tumor site (Orimo et al., 2005). In addition, CAFs are also able to buffer the acidity generated by tumor cells (Koukourakis et al., 2006), and most importantly, have a direct effect in promoting metastasis (Karnoub et al., 2007). The proliferation rate and taxis potential of activated PSCs, which are found in area of fibrosis and dispersed throughout the pancreatic tumor (Vonlaufen et al., 2008), have been shown to be upregulated by PDGF. Activated PSCs upregulate their production of ECM degrading enzymes (MMPs, refer to Section 2.7) and their inhibitors during pancreatic tissue remodeling (Phillips et al., 2003). Fibrosis by myofibroblastic cells is also induced by a hypoxic environment (Masamune et al., 2008). The behavior active PSCs exhibit and effects these cells exert in a tumor environment are similar to those of CAFs (see reviews (Apte and Wilson, 2012; Omary et al., 2007)).
Here, we neither distinguish between proto-myofibroblast and differentiated myofibroblast, nor the origins and types of myofibroblastic cells. All myofibroblastic cells are grouped under one species (F). Proliferation and migration of the myofibroblastic species are induced by tumor growth factors (Section 2.5). Myofibroblastic species in our model produces ECM (Section 2.2), tumor growth factors (Section 2.5), tumor angiogenic factors (Section 2.6), and upregulate their secretion of MDEs (Section 2.7). The species F is assumed to migrate towards sites of tumor, indicated by the presence of tumor growth factors. Their secretion of ECM macromolecules in the model is also set to be hypoxia induced.
2.5. Tumor Growth Factors and Growth Hormones
As one of the hallmarks, cancer can acquire the capability to sustain proliferation signaling which may be achieved in several ways (Hynes and MacDonald, 2009; Lemmon and Schlessinger, 2010; Perona, 2006; Witsch et al., 2010). Cancer cells can produce growth factors, and in an autocrine manner, respond by expressing their cognate receptors themselves. They may also signal stromal cells to secrete various paracrine mitogenic factors (Bhowmick et al., 2004; Cheng et al., 2008). High levels of receptor proteins as well as receptor molecules with altered structures may be expressed at the cancer cell surface, raising their responsiveness to growth factors (Hanahan and Weinberg, 2011).
In addition, cancer cells can avoid negative regulation of cell proliferation and evade apoptosis. Several families of growth factors can stimulate tumor cell proliferation and survival, including the epithelial growth factor (EGF) family, platelet-derived growth factor (PDGF), the insulin-like growth factor (IGF) family, hepatocyte growth factor (HGF), the fibroblast growth factor (FGF) family, and the transforming growth factor-β (TGF-β) family (Bhowmick et al., 2004; Lewis and Pollard, 2006; Siveen and Kuttan, 2009). Among these factors, HGF, EGF, TGF-α, FGF-2, FGF-7 (or keratinocyte growth factor, KGF), and FGF-10 are known to increase proliferation of tumor cells, while IGF-1, IGF-2, TGF-β1, TGF-β2, and TGF-β3 act as tumor cell mitogens and apoptosis inhibitors (Bhowmick et al., 2004). While CAFs are capable of secreting most of these growth factors, a number of them, such as EGF, PDGF, TGF-β1, and FGF-2, are also expressed by TAMs and are upregulated under hypoxic conditions (Goswami et al., 2005; Lewis and Murdoch, 2005; Osullivan et al., 1993; White et al., 2004).
EGF receptor (EGFR) belongs to the polypeptide growth factor receptor tyrosine kinase superfamily and its ligands belong to the EGF family, including EGF and TGF-α (Hynes and MacDonald, 2009; Tang et al., 1997). Elevated levels of EGFR and TGF-α have been implicated in malignant glioma and in the development of other solid tumors (Gullick, 1991; Holbro and Hynes, 2004; Nister et al., 1988; Schlegel et al., 1990; Yung et al., 1990), reflecting their growth-stimulatory functions involved in the carcinogenic process. In fact, studies have indicated that they are involved in a key autocrine loop in sustaining proliferation of human tumors (Sporn and Todaro, 1980; Tang et al., 1997), leading to unregulated neoplastic growth. In addition to production by fibroblasts, TGF-α is also expressed constitutively by cancer cells (Tang et al., 1997) and EGF is synthesized by TAMs in response to tumor-derived colony stimulating factor-1 (CSF-1) (Lewis and Pollard, 2006).
First isolated from platelets, PDGF was later found to also come from other cell types. Having mitogenic activity in connective tissue and glial cells, it is essential in wound healing and directs the chemotactic movements of fibroblasts, muscle cells, neutrophils, and monocytes (Perona, 2006). PDGF expressed by malignant skin cells may induce fibroblasts to express FGF-7 (Brauchle et al., 1994), which stimulates the proliferation of epithelial cells, leading to enhanced carcinogenesis (Yan et al., 1992).
The IGF family includes the polypeptides IGF-1 and IGF-2. Synthesized in the liver and are abundant in human infants, both IGFs and their receptors hold a key role in the regulation of malignant cell proliferation, inhibition of apoptosis, and tumor transformation (Perona, 2006). HGF is a stroma-derived paracrine growth factor (Bhowmick et al., 2004) predominantly produced by fibroblasts. However, its cognate receptor, c-Met, is mainly expressed by epithelia (Nakamura et al., 1997). Commonly observed in many cancers, the overexpression of c-Met may be caused by ligand-independent activation (Michieli et al., 2004) or increased sensitivity to physiological HGF levels (Pennacchietti et al., 2003).
Members of the TGF-β cytokine family exist as TGF-β1, TGF-β2, and TGF-β3. Elevated levels of plasma TGF-β1 are detected in cancer patients and linked to early metastasis (Shariat et al., 2001a; Shariat et al., 2001b; Tsushima et al., 2001). Nearly all human cell types are responsive to TGF-β (Massague, 2008), while most cell types are capable of both expressing and responding to the chemokine. Cellular sources of TGF-β in tumors vary and may include cancer cells, stromal fibroblasts, and inflammatory cells (Gold, 1999; Lewis and Pollard, 2006; Massague, 2008; Tlsty and Coussens, 2006). The presence of leukocytes, macrophages, bone marrow-derived endothelial, mesenchymal, and myeloid precursor cells in tumor milieu correlates to TGF-β secretion, suggesting that these cells are potential sources of tumor progressive TGF-β1 accumulating at the invasion front of tumors (Dalal et al., 1993; Yang et al., 2008).
The TGF-β family can impose both tumor suppressive and tumor promoting functions. This growth factor may act as a growth inhibitor in tumor suppression (Amendt et al., 1998; Bottinger et al., 1997; Gorska et al., 2003; Pierce et al., 1995; Tucker et al., 1984), its presence in the tumor microenvironment may also enhance pro-tumorigenic stroma, promote angiogenesis, and impair immunosurveillance (Bhowmick et al., 2004). Under normal conditions, tissue homeostasis is maintained by TGF-β via the regulation of cellular proliferation, differentiation, survival, adhesion, and environment (Massague, 2008). Malignant cells, however, can circumvent these suppressive effects either through inactivation of the TGF-β receptors or alter the signaling pathway downstream. In fact, increased expression of TGF-β in carcinoma cells is often detected along with loss of TGF-β sensitivity by the cells (Bhowmick et al., 2004). Pathological forms of TGF-β signaling permit loss of cell adherens junctions and activate a cellular program termed the epithelial-to-mesenchyme transition (EMT), awarding malignant cells with changes that favor invasion and metastasis (Akhurst and Derynck, 2001; Hanahan and Weinberg, 2011; Ikushima and Miyazono, 2010). Furthermore, TGF-β1 can inhibit cytotoxicity of TAMs (Ben-Baruch, 2006; Elgert et al., 1998) and may recruit other stromal cell types to generate a pro-tumorigenic microenvironment (Massague, 2008).
Tumor necrosis factor-α (TNF-α), a pivotal cytokine in inflammatory reactions, may be produced by epithelial tumor cells or stromal cells, including mononuclear phagocytes, neutrophils, activated lymphocytes, natural killer (NK) cells, ECs, mast cells, and TAMs (Balkwill, 2002; Borish and Steinke, 2003; Lewis and Pollard, 2006). TNF-α affects neoplastic growth directly by regulating the proliferation and survival tumor cells (de Visser et al., 2006). It controls the activation state and cellular localization of nuclear factor of kB (NF-kB) (Pikarsky et al., 2004), which is found to be constitutively activated in some types of cancer cell (Karin et al., 2002). The activated NF-kB is translocated into the cell nucleus (Senftleben et al., 2001), where it ignites a series of alterations involving immunoregulatory and inflammatory genes, antiapoptotic genes, positive cell proliferation regulating genes, and encoding genes for negative regulators of NF-kB (Karin et al., 2002), leading to changes in cell functions that promote proliferation and inhibit apoptosis (Beg and Baltimore, 1996; Liu et al., 1996; VanAntwerp et al., 1996; Wang et al., 1996). Activation of NF-kB is also linked to ECM destruction by cancer cells (Bond et al., 1998; Takeshita et al., 1999; Wang et al., 1999b) and anticancer drug or radiation treatment resistance (Wang et al., 1999a).
In our model, only one group of growth factors is currently included. Tumor growth factors (tgf) represent all the aforementioned factors and molecules which engage in autocrine or paracrine signaling. They are produced by the viable tumor cells (V), vascular cells (B, L), and myofibroblastic species (F). Another group of endocrine factors, termed tumor hormonal growth factors (h), which are carried by blood circulation and disseminated via microvessels in the tumor milieu, is not currently modelled and will be included in future studies. Both species diffuse through the tumor tissue with a certain rate of decay and are consumed by binding to tumor cell surface.
2.6. Tumor Angiogenic Factors
Avascular tumor can undergo angiogenic switch to attain vascular tumor development. The angiogenic switch induces the growth of neovasculature, subsequently increases the blood vessel density in tumor mass, enabling tumors to overcome growth restrictions imposed by insufficient oxygen and nutrients supplies. Potent inducers of angiogenic growth include fibroblast growth factors, vascular endothelial growth factors, and angiopoietins (Folkman and Kalluri, 2003).
The first angiogenic proteins to be isolated, basic fibroblast growth factor (bFGF or FGF-2) and acidic fibroblast growth factor (aFGF or FGF-1), are among the most potent angiogenic proteins in vivo (Folkman and Shing, 1992; Shing et al., 1984). The involvement of bFGF in tumorigenesis is evidenced by elevated levels of bFGF found in the serum, urine, and cerebrospinal fluid of cancer patients with different types of tumors (Li et al., 1994; Nguyen et al., 1994). Stored in ECM and synthesized by tumor cells and ECs in the tumor vasculature, these FGFs have high affinity for heparin and can stimulate EC mitosis and migration in vitro (Folkman and Kalluri, 2003). Some tumors are also known to recruit and activate macrophages to secrete bFGF, while others may attract mast cells to sequester bFGF due to their high heparin content (Schulzeosthoff et al., 1990). Under hypoxic condition, bFGF is upregulated by TAMs and tumor cells (Lewis and Murdoch, 2005). Tumors with elaborated bFGF levels may have heightened immune-tolerance attributable to the ability of bFGF to interfere with leukocyte adhesion to the endothelium (Melder et al., 1996).
Vascular endothelial growth factors (VEGFs) are a family of cytokines secreted by the majority of tumor cells and a wide variety of normal cells, including ECs and TAMs (Folkman and Kalluri, 2003; Lee et al., 2007; Lewis and Pollard, 2006; Maharaj et al., 2006). The VEGF gene family consists of VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor (PIGF) (Ferrara et al., 2003; Kim et al., 2002; Shibuya, 2006). Among them, VEGF-A (initially vascular permeability factor VPF) is the most important molecule that dictates blood vessel morphogenesis with known correlation between its expression and angiogenesis in tumors (Adams and Alitalo, 2007; Saharinen et al., 2011). It exists as five isoforms and two of its receptors are found predominantly on ECs (Devries et al., 1992; Terman et al., 1992). Shown to be an EC mitogen and motogen in vivo (Connolly, 1991; Dvorak et al., 1991), VEGF-A is also essential in the differentiation of endothelial precursor cells, assembly of ECs into vasculature, and vessel remodeling (Adams and Alitalo, 2007). In addition, it can bind to a receptor expressed on tumor cells. The autocrine secretion of VEGF-A by tumor cells facilitates the generation of a surface-bound VEGF-A gradient, leading to the chemotaxis of ECs to the tumor cells (Folkman and Kalluri, 2003). In several experimental systems, it was also shown to stimulate lymphatic growth (Hirakawa et al., 2005; Hong et al., 2004; Nagy et al., 2002). However, the mechanisms might be indirect and involve the recruitment of inflammatory cells and increased VEGF-C expression (Baluk et al., 2005; Cursiefen et al., 2004). In contrast to VEGF-A, VEGF-C binds to a receptor that is expressed predominantly on lymphatic endothelium (Chang et al., 2002). As a key regulator of lymphangiogenesis (Adams and Alitalo, 2007), it induces the proliferation and survival of LECs, hence, promoting the sprouting of lymphatic vessels (Karkkainen et al., 2004). Also showing lymphangiogenic activity is VEGF-D, though might not be crucial for lymphatic development (Baldwin et al., 2005), it has been linked with poor prognosis, invasion, and metastasis (Mumprecht and Detmar, 2009; Skobe et al., 2001; Tammela and Alitalo, 2010). Moreover, VEGF-C and VEGF-D participations have been indicated in angiogenesis under pathological conditions (Laakkonen et al., 2007). While VEGF-A expression by tumor cells is known to be upregulated by hypoxia and elevated near necrotic tumor areas (Bando et al., 2003; Folkman and Kalluri, 2003; Koong et al., 2000; Lal et al., 2001; Lewis and Pollard, 2006), hypoxic regulations of VEGF-C and VEGF-D remain unclear. While some studies showed that VEGF-C and VEGF-D are upregulated by hypoxia or correlates positively with hypoxia-inducible factor-1α (HIF-1α) (Currie et al., 2004; Daluvoy et al., 2009; Nilsson et al., 2004; Okada et al., 2005; Schoppmann et al., 2006; Simiantonaki et al., 2008; Tzao et al., 2008), others have reported contrarily (Enholm et al., 1997; Okada et al., 2005; Simiantonaki et al., 2008).
Another group of important angiogenic signaling molecules is the angiopoietins. Angiopoietin-1 (Ang1), Ang2, and Ang3/4 bind to the Tie1 and Tie2 receptors on ECs (Davis et al., 1996; Maisonpierre et al., 1997; Saharinen et al., 2010; Suri et al., 1996), hence are EC specific growth factors (Folkman and Kalluri, 2003). While VEGFs are involved in the initial assembly of the vasculature, the Ang-Tie system plays an essential role in the later stages of vascular development when the vessels remodel and recruit pericytes for coating (Saharinen et al., 2010). Ang1 is secreted and incorporated into the tumor-associated ECM, but Ang2 is not found to be ECM bound (Xu and Yu, 2001). While Ang1 can readily activate Tie2, Ang2 only induces Tie2 phosphorylation under certain conditions, such as in stressed ECs or at high levels of Ang2 (Daly et al., 2006; Kim et al., 2000). Ang1 is not an endothelial mitogen but induces ECs to recruit mural cells for the vessel wall (Folkman and Kalluri, 2003). Hence, overexpressing Ang1 leads to non-leaky vessels (Suri et al., 1998). Whereas Ang2 in the presence of VEGF-A may increase angiogenesis, Ang2 alone acts as an antagonist of Ang1, causing EC apoptosis, destabilizing vessels, and eventually regression of new microvasculature (Holash et al., 1999; Maisonpierre et al., 1997; Thurston et al., 1999). In the normal vasculature, Ang1 is produced mainly by periendothelial mural cells while Ang2 and Tie2 are expressed by ECs (Helotera and Alitalo, 2007; Saharinen et al., 2010). In activated endothelium found within a tumor microenvironment, Ang2 secretion exceeds that of Ang1, causing pericytes detachment and vessel regression, leading to hypoxia which drives the release of VEGFs and initiation of angiogenesis (Ahmad et al., 2001; Holash et al., 1999; Maisonpierre et al., 1997; Reiss et al., 2009; Yancopoulos et al., 2000).
In our model, the total effects of all TAFs are combined and modeled under one species (taf). We do not differentiate between angiogenic and lymphangiogenic growth factors, assuming that the growth factors affect ECs and LECs in a similar manner. Upregulated by hypoxia, production of the species is taken to come from viable tumor cells (V), host cells (H), ECs (B), LECs (L), and myofibroblastic cell species (F). The species, decaying at a certain rate, diffuses through the tumor tissue and is used by growing ECs and LECs. It is also involved in directing the chemotactic migrations of ECs and LECs.
2.7. Matrix Degrading Enzymes
Matrix metalloproteinases (MMPs) are matrix degrading enzymes (MDEs) involved in promoting the inflammatory response, tissue remodeling, wound healing, and angiogenesis. In a tumor environment, these proteinases are upregulated, leading to destruction of normal ECM (Bissell and Radisky, 2001). Degradation of ECM may indirectly enable the selection of apoptosis-resistant carcinoma cells and enhanced invasive potential (Mitsiades et al., 2001; Sethi et al., 1999). The ability of MMPs to bind to specific receptors on tumor cell surface, coupled with MMP retention on cell surface by ECM adhesion receptors, provide spatial control of its proteolytic activity and directional signals to invading tumor cells (Yu and Stamenkovic, 2000).
The MMP family of at 25 or more highly homologous, either secreted or plasma membrane-associated zinc-binding proteinases can be produced by nearly all cell types (Egeblad and Werb, 2002; Sternlicht and Werb, 2001). They are matrix degrading enzymes (MDEs) consisting of collagenase (MMP-1), gelatinase A (MMP-2), stromelysin (MMP-3), matrilysin (MMP-7), gelatinase B (MMP-9), and others (Siveen and Kuttan, 2009). Various studies have reported a correlation between elevated expression of MMPs in human malignant tissue and poor prognosis (Egeblad and Werb, 2002).
While some MMPs are secreted by ECs, the major source of the enzymes in both human and mouse cancer models is activated stromal cells, mainly innate immune cells and others, such as fibroblasts and vascular cells (Egeblad and Werb, 2002). MMPs can lead to the formation of distant metastases by remodeling cell-cell and cell-matrix adhesion molecules, as well as both soluble and insoluble ECM components. These restructurings relax the connective tissue supporting a tumor and alter intracellular signaling pathways, enabling malignant cells to detach from tumor mass and disseminate (Egeblad and Werb, 2002; Lamagna et al., 2006). Released by the proteolytic cleavage are bioactive cryptic protein fragments embedded within some ECM molecules (Egeblad and Werb, 2002; Kalluri, 2003). These protein fragments antagonize angiogenesis and could be used as potential drugs for tumor retardation.
Another involvement of MMPs in tumor growth is in angiogenesis. Highly expressed proangiogenic growth factors such as VEGFs, bFGF, and TNF have limited bioavailability because they are either bound to ECM molecules or tethered to cells (Bergers and Coussens, 2000). MMPs regulate the release of these growth factors, exposing them to their associated receptors on ECs, and promoting the development of neovasculature (Bergers and Coussens, 2000; Egeblad and Werb, 2002).
Hypoxia is known to upregulate TAM production of MMP-7 (Burke et al., 2003), while MMP-9 secretion by monocytic cell lines, blood monocytes, and brain macrophages can be elevated by macrophage chemoattractants CCL2 and CCL5 (Azenshtein et al., 2002; Cross and Woodroofe, 1999; Locati et al., 2002; Robinson et al., 2002). MMP-9 can mobilize ECM-sequestered VEGF (Bergers et al., 2000) and both MMP-2 and MMP-9 can activate latent TGF-β residing in the matrix (Yu et al., 2002), which lead to the proliferation of perivascular and ECs and stabilization of nascent microvessels (Jain, 2003; Jain, 2005). Being a major source of MMP-2 and MMP-9 (Lewis and Pollard, 2006; Siveen and Kuttan, 2009), TAMs also induce their productions by tumor cells in the presence of ET-1 and ET-2, stimulating the invasiveness of tumor cells (Grimshaw et al., 2002).
Here, we model the MMP family as a single MDE species (m). It is produced by viable tumor cells (V), ECs (B), LECs (L), and myofibroblastic cell species (F). The species diffuses through the tumor tissue with a certain rate of decay. It is involved in the degradation of ECM (E) and its expression can be upregulated by hypoxia. We acknowledge that diffusion-type MMP models can be problematic, as explored by (Mumenthaler et al., 2013) and (D’Antonio et al., 2013), since in reality MMPs are often membrane-bound or have very short diffusion distances. For simplicity, here we assume that diffusion is the main physical mechanism, even if over a short distance.
2.8. Nutrients and Waste Products
Specific to tumor cells is their altered metabolism first postulated by Nobel Laureate Otto H. Warburg. In papers published by Warburg and coworkers in the 1920s (Warburg, 1924; Warburg et al., 1924; Warburg et al., 1927), tumor tissues were shown to metabolize approximately tenfold more glucose to produce two orders of magnitude higher of lactic acid compared to normal tissues even under aerobic conditions (Koppenol et al., 2011). The use of tracer 2-[18F]fluoro-2-deoxy-D-glucose (FDG) with positron emission tomography (PET) (Gambhir, 2002) has been successfully used to identify many human cancers (Czernin and Phelps, 2002) and to show that primary and metastatic lesions display increased glucose uptake (Gatenby and Gillies, 2004).
The increase in glucose consumption as a carbon source for anabolic reactions and glycolytic ATP production facilitates tumor growth in several ways (Kroemer and Pouyssegur, 2008). By shifting towards glycolysis, tumor cells evade uncertainties in oxygen supply and mitigate their dependence on oxygen under hypoxic conditions. Lactic acid, a product of glycolysis, has been shown to suppress human T lymphocyte proliferation and its cytokine production up to 95%, leading to the reduction of its cytotoxicity by half (Fischer et al., 2007). Carbon dioxide released by cellular respiration is wetted in the interstitium to generate carbonic acid. Together with lactic acid, they contribute towards low extracellular pH levels that promote tumor growth and invasion (Swietach et al., 2007). The avoidance of acidic death is achieved by synchronized buffering between cancer and stromal cells. Lactate is pumped from cancer cell cytoplasm to the ECM and absorbed by stromal fibroblasts to use as fuel in pyruvate production. The excess pyruvate within fibroblasts is then exported to the ECM and taken up by cancer cells as fuel in lactate fermentation, ending in lactic acid production (Koukourakis et al., 2006). Furthermore, intermediates from the glycolytic pathways may also be deviated by cancer cells to participate in anabolic reactions that linked to cell growth and proliferation (Kroemer and Pouyssegur, 2008).
Another essential nutrient for most cancer cells is glutamine (Eagle, 1955). Cancer cell proliferation is also found to depend on glutamine, in which oxidative glutamine metabolism contributes to lipogenesis (Anastasiou and Cantley, 2012). Under hypoxia where HIF is stabilized and mitochondrial functions are impaired, malignant cells rely predominantly on glutamine to provide carbons for lipid production via reductive glutamine metabolism (Metallo et al., 2012; Mullen et al., 2012).
In our model, cellular metabolic processes considered are respiration and glycolysis with lactate fermentation. The bicarbonate buffering system is included for cellular pH regulation. Sodium and chloride ions are pumped across cell membranes. Oxidative and reductive glutamine pathways will be included in our future work. Nutrients and waste products involved in these reactions are glucose, oxygen, lactic acid, carbon dioxide, water, and carbonic acid. Water is assumed to exist in abundance and therefore is not a limiting factor. Both lactic and carbonic acids are also assumed to dissociate completely and exist only in ionic forms. We model the concentrations of glucose (g), oxygen (n), lactate ion (ℓ), carbon dioxide (w), bicarbonate ion (b), hydrogen ion (a), sodium (s), and chloride (r) ions.
3. Mathematical Model
The soft tissue in our model consists of a viable tumor cell species, a necrotic tumor cell species, and a healthy host cell species living within the stromal component (as defined in Sections 2.1). The stroma consists of mainly ECM and interstitial physiological fluid, with myofibroblastic cells (Section 2.4), vascular and lymphatic vessels (Section 2.2 and 2.3).
Variables of different cell components of the mixture are identified by the subscripts W, V, D, E, and H, representing interstitial fluid, viable tumor cells, necrotic tumor cells, ECM, and healthy host cells respectively. Whenever appropriate and necessary, we interchangeably denote the components in the order listed with the numeric subscripts 0 to 4:
-
0
– Interstitial physiological fluid (W)
-
1
− Viable tumor cells (V)
-
2
− Dead/necrotic tumor cells (D)
-
3
– Extracellular matrix (E)
-
4
– Healthy host cells (H)
The cell and ECM components are collectively considered as a solid phase and the interstitial component is taken to be an aqueous fluid phase, denoted by subscripts α and β respectively. Note that subscripts W, 0, and β are used interchangeably for the aqueous interstitial component.
Dependent variables in the continuum mixture model include the cell-ECM α phase pressure p, volume fraction ϕα, density ρα, and velocity vector uα; interstitial fluid β phase pressure q, volume fraction ϕβ, density ρβ, and velocity vector uβ. Within the solid cell-ECM α phase, component volume fractions are denoted as ϕα,1,⋯,ϕα,4, cell component densities as ρ1,⋯,ρ4, and velocities relative to stationary coordinates u1,⋯,u4.
Figure 1 presents a graphical overview of the main model components and their interactions. The exchange of key diffusible elements between these components, including tumor and angiogenic growth factors as well as nutrients and oxygen, drives the evolution of these components, and ultimately determines the net amount of viable tumor tissue and stroma at any given moment in time.
Figure 1.
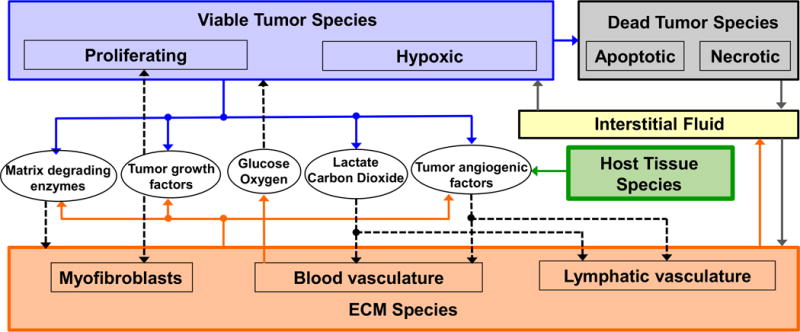
Graphical overview of the main model components and their interactions. Solid arrows indicate outputs from a component, while dashed arrows indicate particular inputs. Arrows penetrating into the boxes highlight specific recipients associated with particular input. For example, tumor growth factors are output by the viable tumor species and the stromal elements, and the growth factors in turn influence the behavior of proliferating tumor cells and myofibroblasts.
3.1. Basic Equations
The tumoral tissue is taken to be a mixture of solid cell-ECM phase α and interstitial fluid phase β. Assuming that there are no voids in the tissue, the saturation constraint implies that volume fractions
| (3.1.1) |
Assuming that the volume fractions are continuous in a tissue domain Ω, a mass balance equation is written for each phase:
| (3.1.2) |
where i = α,β. Sα and Sβ are the rates of production of phase α and β, respectively. These rates include mass exchange between the cell-ECM components and the interstitial fluid, mass gain or loss by biological processes, as well as external source/sink.
Following mixture theory, the mixture density is defined as
| (3.1.3) |
and the composite velocity of the mixture is defined as the weighted average of the phase velocities:
| (3.1.4) |
Summing Eq. (3.1.2) for α and β, and taking Eqs. (3.1.1), (3.1.3), and (3.1.4) into account, we get the mass conservation equation for the total mixture:
| (3.1.5) |
Assuming there are no external mass source and sink, the law of conservation of mass may be enforced by letting
| (3.1.6) |
and Eq. (3.1.5) is reduced to the equation of continuity for the mixture. Note that the circulatory systems are assumed to be co-located and the formation of edemas is excluded; these simplifications may not hold for some types of tumors, and deviations will be explored in future studies. The total mass flux, N, of each of the cell-ECM and interstitial phase is the resultant of the bulk tissue motion and the total nonadvective flux J:
| (3.1.7) |
where i = α,β. The nonadvective fluxes arise from movements associated with mechanical interactions between solid and fluid phase. Summing Eq. (3.1.7) over α and β yields
| (3.1.8) |
Now, assume that the solid phase is closely packed with all cells and ECM components, leaving no voids in the solid mixture. The saturation constraint implies that
| (3.1.9) |
where ϕα,i is the volume fraction of component i in phase α. The volume fractions are assumed to be continuous in the tissue domain Ω. Similarly, a mass balance equation of the following form can be written for each component in the solid phase:
| (3.1.10) |
and Sα,i are the source/sink terms that include interphase and external mass exchange, as well as mass gain/loss due to cellular progression.
Again, following mixture theory, the solid cell-ECM mixture density is defined as
| (3.1.11) |
and the composite velocity of the solid mixture is defined as the weighted average of the cell component velocities:
| (3.1.12) |
Summing Eq. (3.1.10) over all constituents and taking Eqs. (3.1.11), and (3.1.12) into account, we get the mass conservation equation for the solid (α) phase, appeared in Eq. (3.1.2), where
| (3.1.13) |
Also, the mass flux, Nα,i, of each cell component is the resultant of the bulk tissue motion and the total diffusive flux of the component:
| (3.1.14) |
where Jα,i are diffusive fluxes that arise from movements associated with mechanical interactions among cell-cell and cell-ECM components and biological driving forces such as chemotaxis and haptotaxis:
| (3.1.15) |
Summing Eq. (3.1.14) over all cells and ECM components and using Eqs. (3.1.11) and (3.1.12), we get
| (3.1.16) |
Therefore, Eqs. (3.1.6) and (3.1.16), together with Eq. (3.1.13), are enforced as consistency constraints for sources and diffusive fluxes. Letting ϕi = ϕαϕα,i and assuming constant ρi, Eq. (3.1.10) can be rewritten as
| (3.1.17) |
Expressions for velocities, diffusive fluxes, and source/sink terms for the cells and ECM components will be derived and discussed in Sections 3.2 and 3.3. The governing equations for dissolved species and vessels will be discussed separately in Sections 3.4 to 3.6.
3.2. Diffuse Interface Method
In diffuse interface theories, the sharp interface between phases is replaced by a diffuse interface of non-zero interface thickness, which arises from the finite range of molecular interactions. Cell-cell and cell-ECM interactions, combined with elastic effects and taxis potential, are considered and accounted for by a non-local contribution to the Helmholtz free energy (Cahn and Hilliard, 1958; Rowlinson, 1979; Yang et al., 1976), where the local free energy density depends on the both the local composition and the composition of the nearest surroundings.
We follow the energetic variational approach presented by Wise et al. (2008). The total Helmholtz free energy of the system is
| (3.2.1) |
Here, the tumoral tissue is considered an isothermal system. To construct the constitutive relations for velocities and fluxes that are consistent with the second law of thermodynamics, we begin with a generalized Helmholtz free energy equation of component interactions, with the added taxis potential term posed by Cristini et al. (2009). One way to model the elasticity is by evolving natural configurations and introducing the time derivative of the stress (Giverso and Preziosi, 2012; Giverso et al., 2015; Preziosi et al., 2010). Here, we opted to add the effects of elasticity to the free energy equation via an elastic energy term. Therefore, the generalized Helmholtz free energy equation can be expressed in the following form:
| (3.2.2) |
where E̅ is the Helmholtz free energy density of the system Fb and is the bulk free energy of components due to local interactions; the second term on the right hand side represents gradient energy due to interactions with nearest surroundings, κij > 0 are the strength of component interactions; the third term is the energy contribution due to the taxis of cell components, χil is the taxis coefficient of cell component i with respect to the chemical species l, and σl are the concentrations of taxis inducing species; the last term is the elastic free energy density contributed by component where is the infinitesimal strain tensor.
The time derivative of the total free energy is
| (3.2.3) |
where the boundary terms are dropped and omitted hereinafter. At equilibrium, the total Helmholtz free energy of the system has a minimum. By assuming that the volume fraction of the aqueous interstitial phase is constant , the volume fraction of the solid cell-ECM phase is reduced to . To impose the two constraints on the volume fractions, Lagrange multipliers p* and q, solid and aqueous pressure respectively, may be introduced. Using Eqs. (3.1.2) and (3.1.17), the time derivative in Eq. (3.2.3) is rewritten as
| (3.2.4) |
Using Gauss divergence theorem, it can be rewritten as
| (3.2.5) |
where new boundary terms are dropped and omitted hereinafter. Using Eq. (3.1.16), let and rewrite the solid pressure term as presented by Wise et al. (2008):
| (3.2.6) |
the time derivative of energy in Eq. (3.2.5) can be manipulated and rewritten as
| (3.2.7) |
Taking each term to be separately dissipative, constitutive relations that are thermodynamically consistent can be written for velocities and fluxes:
| (3.2.8) |
| (3.2.9) |
| (3.2.10) |
where kα and kβ are motilities of the solid and liquid phase respectively, and Mi is the mobility of the component. The constant volume fraction is absorbed into kβ.
Let the bulk energy of local interactions be the summation of potentials over the liquid and solid phases (Cogswell and Carter, 2011):
| (3.2.11) |
where is a constant and with positive as an energy scale for adhesion. Therefore the bulk energy of local interactions does not depend on the liquid component, ∂Fb/∂ϕβ = 0. Similar to the approach by Wise et al. (2008), we do not distinguish between the adhesive properties of various tumor cells (V and D):
| (3.2.12) |
As a start, we assumed the free energy term of a ternary system can be described by the following form, adapted from the one constructed by Kim and Lowengrub (2005):
| (3.2.13) |
where A1 to A5 are a set of constants. The collective tumor species T and healthy cell species H are immiscible, and the ECM species is more miscible with species H. For example, with A1 = 1000, A2 = 0.2, A3 = 0.4, A4 = 0.2, and A5 = 0.2, the free energy as stated in Eq. (3.2.13) has two minima, at (ϕT, ϕE, ϕH) = (−0.00002, 0.23485, 0.76517) and (0.63788, 0.36216, −0.00003), that fall slightly out of the Gibbs triangle. We note that the slight negative composition is not a concern here, since volume fractions of small negative values are taken to be zero, the combined tumor and ECM volume fraction (ϕT + ϕE) is generally maintained below 1, and the volume fraction of the healthy cell species is calculated post smoothing by ϕH = 1 − ϕT − ϕE.
Again using ϕT in Eq. (3.2.12) and the relation in Eq. (3.1.9) to eliminate ϕH, the gradient energy term in Eq. (3.2.2) can be rewritten as
| (3.2.14) |
where the adhesive flux of the liquid component is assumed negligible, κ0j = κi0 ≈ 0. The interface energy terms εi and εij are
| (3.2.15) |
where κij = κji, κiV = κiD = κiT, and κVD = κDV = κVV = κDD = κTT.
While the elastic energy contribution may take various forms, we currently adopt the generalized elastic energy density of the system following Leo et al. (1998) and Garcke (2005) given by
| (3.2.16) |
where is an energy scale for elastic effects εe, is an interfacial strain energy coefficient. The infinitesimal strain 𝛆, the elastic stiffness (a fourth order tensor) , and the stress-free strain (eigenstrain) 𝛆*(ϕ) are all symmetric tensors defined as the following:
| (3.2.17) |
| (3.2.18) |
| (3.2.19) |
where ud is the displacement vector with components , δij = 1 for i = j and δij = 0 for i ≠ j; 𝛆E* is the constant misfit tensor for the ECM component; we do not differentiate between cell types, and thus assume all cell components to have the same constant misfit tensor 𝛆C*. The cubic interpolation function for 0 ≤ γ ≤ 1 is given by Q3(γ) = 3γ2 − 2γ3. Assuming that all cell components have the same elastic properties, the two volume fraction dependent terms L1(ϕ) and L2(ϕ) used are given by
| (3.2.20) |
where , , and , are Lamé constants in regions of pure ECM and cells respectively. Hence 𝛆*(ϕ) is reduced to . Similarly, and are reduced to and respectively. The cell types are also not differentiated here. Furthermore, from Eq. (3.2.18), we define
| (3.2.21) |
therefore and are constants.
From Eqs. (3.2.1), (3.2.2), (3.2.11), (3.2.14), and (3.2.16), together with the Euler-Lagrange equation δE/δϕj = ∂E/∂ϕj −∇ · ∂E/∂∇ ϕj, the variational derivative of the energy with respect to each component is
| (3.2.22) |
From Eqs. (3.2.16) – (3.2.21), it is concluded that
| (3.2.23) |
where for j ≠ E. Assuming that the mechanical equilibrium is reached on a much faster time scale than mass diffusion and growth, a quasi-equilibrium is used for the displacement ud:
| (3.2.24) |
where is the symmetric stress tensor defined by
| (3.2.25) |
and Eq. (3.2.24) can be rewritten as a vector with the i component expressed below:
| (3.2.26) |
The displacement components , , and , determined from Eq. (3.2.26) and satisfying Eq. (3.2.24) above, are used to compute the elastic contribution via Eq. (3.2.23) to the potential of the ECM phase, given in Eq. (3.2.29) below.
Because there is no taxis χ0l = 0 and ∂∇ ϕ{T,E}/∂∇ ϕβ = 0, we get δE/δϕ0 = δE/δϕβ = 0. Eq. (3.2.8) is therefore reduced to
| (3.2.27) |
Since tumor cells are assumed to be not migratory, χVl = χDl = 0, we rewrite Eq. (3.2.22) for tumor cells as
| (3.2.28) |
There is no taxis of ECM macromolecules and healthy host cells, therefore χ6l = χ7l = 0. Eq. (3.2.22) can be written for ECM and host cells as
| (3.2.29) |
| (3.2.30) |
Substitute into Eq. (3.2.13) and subsequently into Eq. (3.2.11), we get
| (3.2.31) |
| (3.2.32) |
| (3.2.33) |
Let μT = δE/δϕT and μE = δE/δϕE. Eqs. (3.2.30) and (3.2.33) implies that δE/δϕH = 0. Substitute Eqs. (3.2.28), (3.2.29), (3.2.30), and (3.2.33) in Eq. (3.2.9), we get an expression for the solid phase velocity:
| (3.2.34) |
Assuming that the cell species densities are matched, ρi = ρ, and let Mi = Mϕiρ2, the diffusive fluxes of cell-ECM components given by Eqs. (3.2.10) and (3.1.16) become
| (3.2.35) |
where the mobility M is a positive constant.
3.3. Solid Tumor Cell Volume Fractions
Using fluxes given in Eq. (3.2.35) and by letting Sα,i/ρ = Si, the continuum multicomponent equations of change in Eq. (3.1.17) can be rewritten as
| (3.3.1) |
where chemical potentials μT and μE are given in Equations (3.2.28) and (3.2.29), respectively.
The source terms for solid species may be written as a combination of rates (r) related to biological processes. The viable tumor cell species (V) is subjected to mitotic gain (rM,V), apoptotic (rA,V) and necrosis loss (rN,V), as well as metastatic disseminations via the blood (rB,V) and lymphatic (rL,V) vessels. The viable tumor cell species is also assumed to potentially undergo autophagic degradation (rde,V). The dead tumor cell species, which accounts for both apoptotic and necrotic loss of viable tumor cells, undergoes lysis (rL,D) and is eventually released into the interstitium. Fibronectin may be secreted by viable tumor cells (rV,E), endothelial cells (rB,E, rL,E), and myofibroblastic cells (rF,E). The source term also includes its degradation (rde,E) by matrix degrading enzymes. The healthy host cell species is assumed to maintain homeostasis with negligible changes compared to the tumor and immune cell species, therefore SH = 0. We obtain the following expressions for species source terms:
| (3.3.2) |
Summing all the source terms in Eq. (3.3.2), and using the relations in Eqs. (3.1.6) and (3.1.13), we get the mass exchange terms between the solid cell and aqueous interstitial components:
| (3.3.3) |
As indicated in the expression above, ECM produced and mitotic gain of cells are assumed to come from aqueous interstitial components. Metastatic loss of tumor cells, degraded ECM, catabolized tumor cells due to autophagy, and lysed dead tumor cells are assumed to contribute to the interstitial component. We also assume that the phagocytosed tumor cells and degraded tumor cells by autophagy are processed and released to the interstitial space instantaneously.
The rate expressions used in source terms in Eq. (3.3.2) are given in Table 1. In the table of rate expressions, λM,V, λA,V, and λN,V are the mitosis, apoptosis, and necrosis rate constants, respectively, for viable tumor cell species V; λL,D is the lysis rate constant of the dead tumor cell species; λB,V and λL,V are the rate constants for metastatic dissemination via the blood and lymphatic vessels, respectively, for viable tumor cell species V; rde,V is the autophagic degradation of viable tumor species V whereas rde,E is the degradation of ECM by MDEs; ri,E is the rate of fibrosis contributed by cell species i; given by Bn and Ln, respectively, are the total EC and LEC concentrations in number of cells per volume tissue.
Table 1.
Rate expressions from Eq. (3.3.2).
|
|
|
|
|||
|
|
|
|
|||
|
|
|
|
|||
|
|
|
|
Tumor cells proliferate aggressively until a threshold level of hypoxia is reached. In areas where oxygen concentrations fall below the threshold level, tumor cells may stop proliferating and switch to anaerobic glycolysis for continuous energy production (Brown, 2000). Including the effects of mitogens and hypoxia, the adjustment factor for the mitosis rate constant of viable tumor cell species can be written as
| (3.3.4) |
where nh is the hypoxic threshold of oxygen level, gv,V is the glucose viability limit for viable tumor species V, is the effective factor of tumor growth factors on the mitosis rate of viable tumor species V, tgfsat is the saturation level of tgf species.
Taking into account the inhibition of apoptosis by tumor growth factors and desmoplasia, the adjustment factors for viable tumor species apoptosis rate may be given as
| (3.3.5) |
where and are the effective factors of tumor growth factors and ECM macromolecules, respectively, on the apoptosis rate of viable tumor species V.
Factors potentially affect the lysis rate constant of dead tumor cells are not considered here, hence, we let . We assume that necrosis in the viable tumor species can be triggered when one of the nutrients drops below their viable thresholds. Therefore, the adjustment factors for necrosis rates are taken to be
| (3.3.6) |
where nv,V is the oxygen viability limit and gv,V is the glucose viability limit for the viable tumor species V. Note that the effect of pH is not included here.
In experimental animals, tumor cells may appear in the circulation continuously after neovascularization of the primary tumor; the number of tumor cells shed has been shown to correlate positively with the density of blood vessels in the primary tumor and the number of metastases observed (Liotta et al., 1976; Liotta et al., 1980). The leaky fragmented basement membranes of proliferating capillaries facilitate the metastatic emigration of tumor cells (Dvorak et al., 1988; Liotta et al., 1976). High lactate levels in the primary tumor also correlate positively to the metastatic spread of carcinomas (Walenta et al., 2000). Therefore, we let the adjustment factors for the rates of metastatic dissemination via blood and lymphatic vessels be
| (3.3.7) |
| (3.3.8) |
where the threshold pressures pt,B and pt,L represent upper limits, above which blood and lymphatic vessels are considered crushed. The factors and , termed Metastatic Index here, act as indications of the likelihood of viable tumor cell dissemination via the blood and lymphatic vasculatures respectively. We let
| (3.3.9) |
| (3.3.10) |
where ℓsat is the maximum level of lactate, Bmax is the maximum density of blood vessels, and Lmax is the maximum level of lymphatic vessels.
Although pancreatic ductal adenocarcinoma is found to have elevated levels of autophagy even when nutrients are abundant (Yang et al., 2011), we let the onset of autophagy in our model be related to metabolic stress, hypoxia, and, potentially, growth factors:
| (3.3.11) |
where we assumed that the process occurs only when both oxygen and glucose levels fall between a certain range and the rate is decreasing as nutrient levels approach their viability limits; gde,v is the glucose upper limit for viable tumor species, below which autophagy may occur; is the effective factor of tumor growth factors on the autophagic degradation rate of viable tumor species.
The adjustment factors of ECM production and degradation are
| (3.3.12) |
| (3.3.13) |
where i = V, B, L, F, msat is the saturation level of MDEs in tissues, and are the effective factors of hypoxia in upregulating the production of ECM molecules by species i. Note that another expression for ECM degradation rate rde,E is discussed in Section 3.5.
3.4. Nutrients and Waste Products Concentrations
Molar concentrations of glucose (g), oxygen (n), lactate ion (ℓ), carbon dioxide (w), bicarbonate ion (b), hydrogen ion (a), as well as sodium (s) and chloride (r) ions in tissues follow the mass conservation equation given in Eq. (3.1.17). Nutrients and waste products diffuse through interstitial fluid space and may be permeable to the cellular membrane. Since the transport of these species in tissues is dominated by diffusion and takes place at time scales that are orders of magnitude shorter than cellular proliferation (seconds versus day or longer) (Wise et al., 2008), we use the following quasi-steady state governing equations for nutrients and waste transport in the interstitial space:
| (3.4.1) |
where σ = g, n, ℓ, w, b, a, s, and r. The molar flux and source term of species σ are given by Nσ and Sσ respectively. The molar flux term of uncharged species follow Fick’s law of diffusion:
| (3.4.2) |
where the diffusivity of specie σ in the tissue is Dσ. Following Casciari et al. (Casciari et al., 1992) for charged species, the flux is given by charged migration and diffusion:
| (3.4.3) |
where zσ and uσ are the charge and mobility of species σ, F is Faraday’s constant, and ψ is the electrical potential. Assuming dilute solution, the Nerst-Einstein equation links the mobility of species σ to its diffusivity by
| (3.4.4) |
where R and T are the gas constant and temperature respectively. Since there is no net current, electroneutrality gives
| (3.4.5) |
Hence, the sum of the charge fluxes due to the ionic species zℓJℓ + zbJb + zaJa + zsJs + zrJr = 0. Using this fact and Eq. (3.4.4), the molar flux of the charged species given by Eq. (3.4.3) can be rewritten as
| (3.4.6) |
where σ = ℓ, b, a, s, r. Assuming that all species diffusivities in the ECM Dσ,E and cell Dσ,C domains may be different, let
| (3.4.7) |
and the species diffusivity in the cell domain is given by
| (3.4.8) |
where Dσ,T and Dσ,H are species diffusivities in the tumor and host regions, respectively. The concentration of chloride ion is not modeled through the mass balance equation in Eq. (3.4.1), but via electroneutrality in Eq. (3.4.5).
While nutrients like oxygen and glucose are supplied via the vasculature network (rB,n, rB,q) and consumed by the cell species (rU,n, rU,g), waste products are produced (rW,w, rW,ℓ, rW,b, rW,a) and may leave through blood vessels (rB,w, rB,ℓ, rB,a). We let Sr = Ss = 0, and the remaining source terms of nutrients and waste products are
| (3.4.9) |
Although healthy host cells are assume to be at homeostasis with negligible nutrient consumption and waste production compared to tumor cells, their nutrient consumption terms are included in the model. Thus, the rates of supply and consumption of oxygen and glucose are
| (3.4.10) |
where σ = n, g. λB,σ is the transfer rate coefficient of species σ from capillaries, λU,i,σ is the uptake rate constant of σ by cell species i, σC is the concentration of σ in the capillaries, and are adjustment factors. Assuming that oxygen consumption is reduced during hypoxia and glucose glycolysis is increased, the adjustment factors are given as
| (3.4.11) |
| (3.4.12) |
where σ = n, g and i = V, H. Assuming that the nutrient transfer coefficients in the ECM λB,σ,E and cell λB,σ,C domains may be different, let
| (3.4.13) |
and
| (3.4.14) |
where λB,σ,T and λB,σ,H are the nutrient transfer coefficients in tumor and host regions, respectively.
Metabolic waste products such as CO2 and lactic acid may enter blood circulation, we let rB,a = rB,ℓ and
| (3.4.15) |
where σ = w,ℓ. λB,σ is the transfer rate coefficient of species σ to the capillaries and takes the form of Eqs. (3.4.13) & (3.4.14), and adjustment factors share the same expression as given in Eq. (3.4.11).
We consider the anaerobic and aerobic glycolysis, as well as the bicarbonate buffering system as presented by Casciari et al (Casciari et al., 1992):
| (3.4.16) |
| (3.4.17) |
| (3.4.18) |
Following the stoichiometry of the metabolic pathways given above, the production rate of lactate ion based on oxygen and glucose consumptions is
| (3.4.19) |
Similarly, the production rate of CO2, bicarbonate and hydrogen ions may be written as
| (3.4.20) |
where kf and kr are the forward and reverse reaction rate constants in Eq. (3.4.18).
3.5. Tumorigenic Species Concentrations
Essential in the neoplastic expansion of tumoral tissues are factors that facilitate cell growth and induce favorable environmental conditions. Tumorigenic factors considered in this model (refer to Sections 2.4 – 2.7) are the tumor growth factor, tumor angiogenic factor, matrix degrading enzyme, and myofibroblastic cell species. Molar concentrations of these molecules are represented by tgf, taf, m, and F respectively.
Similar to nutrients, tumor growth factors and angiogenic factors diffuses through tissues. Since their diffusions take place at much shorter time scales compared to cell proliferation, molar concentrations of these species follow Eqs. (3.4.1) and (3.4.2):
| (3.5.1) |
Since the diffusivity of matrix degrading enzymes is much smaller than that of oxygen (Macklin et al., 2009), the transient species concentrations obey:
| (3.5.2) |
The myofibroblastic cell species is assumed to reside only within the ECM component. The reduced weighted myofibroblastic cell concentration, F, per unit volume of tissue is defined as F = ϕEFE, where FE is the concentration of myofibroblastic cells within the ECM component, defined per unit volume of ECM. The governing equation for the ECM residing myofibroblastic cell species which taxis to tumor sites is
| (3.5.3) |
where SFE is the specific source term defined per unit volume of ECM and we also let ϕeSFE.
All species diffusivities Dσ are assumed to vary in the tumor Dσ,T, ECM Dσ,E, and host Dσ,H domains, therefore following the averaged diffusivity function given in Eqs. (3.4.7) & (3.4.8). Although myofibroblastic cells only reside in the ECM component, its diffusivity DF is also assumed to be affected by the surroundings.
The tumor growth factor species is produced by the viable tumor species (rV,tgf), as well as endothelial (rB,tgf, rL,tgf) and myofibroblastic (rF,tgf) cells. The source term also includes its degradation (rde,tgf) and uptake (rU,tgf) by tumor cells:
| (3.5.4) |
and
| (3.5.5) |
where λi,tgf is the production rate constant of tgf by species i, λde,tgf is the degradation rate constant of tgf, and λU,V,tgf is the tgf uptake rate constant of the viable tumor species. Adjustment factors are given as
| (3.5.6) |
where σ = B, L, F. Fmax is the maximum concentration of myofibroblastic cells in ECM. and are the effective factors of hypoxia and lactate level in upregulating the production of tgf by endothelial and myofibroblastic cells. ℓsat is the saturation level of lactate in tissues, and ℓtgf is a threshold lactate level that triggers tgf upregulation.
The tumor angiogenic factor species is produced by viable tumor (rV,taf), endothelial (rB,taf, rL,taf), and myofibroblastic (rF,taf) cells. The source term also includes its degradation (rde,taf) and uptake (rU,B,taf, rU,L,taf) by endothelial cells:
| (3.5.7) |
and
| (3.5.8) |
where λi,taf is the production rate constant of taf by species i, λde,taf is the degradation rate constant of taf. The uptake rates of taf will be discussed later in this section. To include VEGF upregulation by lactate accumulation (Kumar et al., 2007; Philp et al., 2005), adjustment factors are given as
| (3.5.9) |
where σ = B, L, F. and are the effective factors of hypoxia and lactate level in upregulating the production of taf by the viable tumor species. Similarly, and are effective factors of hypoxia and lactate level in upregulating the production of taf by endothelial and myofibroblastic cells. ℓsat is the saturation level of lactate in tissues, and ℓtaf is a threshold lactate level that triggers taf upregulation.
The matrix degrading enzyme species is produced by viable tumor (rV,m), endothelial (rB,m, rL,m), and myofibroblastic (rF,m) cells. The source term also includes a first order decay (rde,m):
| (3.5.10) |
and
| (3.5.11) |
where λi,m is the production rate constant of m by species i, λde,m is the decay rate constant of m, and msat is the saturation level of m in tissues. We defer the discussion of enzyme production rates by endothelial cells until later in this section. Adjustment factors are given by
| (3.5.12) |
where is the effective factor of hypoxia in upregulating the production of matrix degrading enzymes by the species F.
Source term of myofibroblastic cells includes the rates of mitosis (rM,F), apoptosis (rA,F), and necrosis (rN,F). Similar to tumor and immune cells, the source term and rate expressions with corresponding adjustment factors are shown below:
| (3.5.13) |
and
| (3.5.14) |
where the subscript i represents processes M, A, N. λM,FE, λA,FE, and λN,FE are rate constants of mitosis, apoptosis, and necrosis respectively. The adjustment factors are given as
| (3.5.15) |
where, is the effective factor of tgf on the mitosis rate of myofibroblastic cells. tgfFE is the trigger threshold of tgf, above which the mitosis rate of myofibroblastic cells is upregulated by tgf. nv,F and gv,F are the viability limits of oxygen and glucose, respectively. The effects of pH on the mitosis and necrosis rates of myofibroblastic cells are not considered here.
Tumor angiogenic factors bind to receptors on the endothelial cell wall, activating the secretion of proteolytic enzymes. We follow the mechanisms for the formation of matrix degrading enzymes and the degradation of ECM used in (Levine et al., 2001b):
| (3.5.16) |
where σ is Bn and Ln. Rσ denotes receptors on ECs and LECs. We let C and C′ be the concentrations of ECM macromolecules and degraded ECM macromolecules per volume of tissue. Assuming the number of moles of ECM macromolecules per unit volume of ECM is constant and represented by CE, therefore C = CEϕE and rde,E = −rde,C/CE.
Tumor angiogenic factors are assumed to bind to LEC in the same mechanism. The rate of production of the matrix degrading enzyme species may be expressed in relation to taf as
| (3.5.17) |
The rate of ECM degradation and angiogenic factor uptake can be expressed following mass action or Michaelis-Menten kinetics:
| (3.5.18) |
| (3.5.19) |
where σ = B,L, λU,σ,taf is the taf uptake rate constant by species σ, kcat,C and kcat,σ are rate constants of the reaction steps in Eq. (3.5.16), and Km,C and Km,σ are Michaelis constants. We also make the assumption that the number of receptors per endothelial cell remains constant and the cell receptor density is absorbed in the reaction rate kcat,σ.
3.6. Angiogenic and Lymphangiogenic Vessel Densities
Included in the model are new blood (Bn) and lymphatic (Ln) vessels densities defined per tissue volume. These vessels are assumed to reside within the ECM component in the model. The concentrations of new vessels within the ECM component, defined per unit volume of ECM, are and . The reduced weighted and specific vessel concentrations are related via and .
When an initial tumor mass outgrows the capacity of preexisting vasculature, angiogenic stimuli are secreted by hypoxic tumor cells to initiate angiogenesis. Endothelial cell rearrangement and migration are the first induced by angiogenesis (Paweletz and Knierim, 1989). Cell division is a secondary response and occurs a short distance behind newly formed sprout tips. A general mass conservation equation can be written for EC and LEC densities:
| (3.6.1) |
where σ is used for B (EC) and L (LEC) throughout this section. JσnE and SσnE are the fluxes and source terms, respectively, defined per ECM volume for neovessels. To account for the chemotactic response of ECs and LECs to angiogenic factors, as well as haptotaxis upgradient of ECM macromolecules, we let the flux terms be
| (3.6.2) |
where χche,σnE and χhap,σnE are the chemotactic and maximum haptotactic coefficients respectively. The last term represents the motility of vessels with diffusivity DσnE. Assuming that the EC and LEC diffusivities in the tumor DσnE,T, ECM DσnE,E, and host DσnE domains are different, DσnE is computed using Eqs. (3.4.7) & (3.4.8).
Adjustment factors for chemotactic rates are assumed to depend on the concentration of angiogenic factors following the receptor kinetic law (Anderson and Chaplain, 1998):
| (3.6.3) |
where kche,σnE are positive constants and is the fraction of σ vessel species that are sprouting. Assuming that the numbers of receptors on ECs and LECs stay constant and let the ratio of minimum to maximum taxis strength of σ vessel cells be ωσn, the adjustment factor for haptotaxis strength follows the functions given below:
| (3.6.4) |
where (ϕE)min,σn and (ϕE)max,σn are ECM volume fractions that correspond to the minimum and maximum haptotaxis strengths, respectively. The interpolation function is a quartic polynomial given by Q4(γ) = 16γ2 (1 − γ)2 for 0 ≤ γ ≤ 1.
Neovessels are remodeled (rde,σnE) by fibronectin and may be destroyed (rcrush,σnE) due to high pressure of the tumor tissue. Additionally, there are proliferative vessel (rm,σnE) sections located near growing sprout tips. Let the source terms be
| (3.6.5) |
The destruction rate of σ cells follow the functions given below:
| (3.6.6) |
| (3.6.7) |
The mitosis rate for σ species is given by
| (3.6.8) |
where λm,σnE is the maximum mitosis rate constant. To account for endothelial cell mitosis as a secondary response to angiogenesis, proliferation is assumed to occur only when a threshold concentration of angiogenic factors (tafσ) has been reached (Chaplain, 1996). Therefore, the adjustment factor is set to be
| (3.6.9) |
where is the maximum attainable density for the σ vessel species. If logistic growth is considered (Chaplain and Stuart, 1993; Sholley et al., 1984), the effective factor is set to 1. Otherwise, .
Note that the processes of anastomosis and secondary sprouting can be modeled via a periodic function in the vessel remodeling rate term (Chaplain, 1996). In which case, an additional term accounting for these two processes would be included in the remodeling rate:
| (3.6.10) |
| (3.6.11) |
where λde,σnE is the degradation rate constant of the vasculature due to remodeling by MDEs (Macklin et al., 2009) and λas,σnE is the periodic rate constant that accounts for the anastomosis and secondary sprouting of the σ neovessels. The periodic rate constant should be a function with zero average over a period. Therefore, the net gain due to anastomosis and the net loss due to secondary sprouting can be accounted equally.
3.7. Nondimensionalization of Governing Equations
Following the set of governing equations derived above, dependent variables (Table 2) and parameters (Table 3 – Table 6) are nondimensionalized accordingly. The resulting dimensionless independent variables, space and time , are obtained by scaling with the reaction-diffusion length of oxygen and the mitosis rate constant of viable tumor cell species:
| (3.7.1) |
| (3.7.2) |
Table 2.
Dimensionless Dependent Variables.
| Dimensionless Dependent Variable | Biological Representation | Scaling Factor (e.g. ) | ||
|---|---|---|---|---|
|
|
Viable tumor cells | (fixed solid volume fraction) | ||
|
|
Dead tumor cells |
|
||
|
|
Extracellular Matrix |
|
||
|
|
Healthy host cells |
|
||
|
|
Oxygen |
|
||
|
|
Glucose |
|
||
|
|
Carbon dioxide |
|
||
|
|
Lactate |
|
||
|
|
Bicarbonate |
|
||
|
|
Hydrogen ion |
|
||
|
|
Sodium ion |
|
||
|
|
Chloride ion |
|
||
|
|
Tumor growth factors |
|
||
|
|
Tumor angiogenic factors |
|
||
|
|
MDEs |
|
||
|
|
Myofibroblastic cells |
|
||
|
|
New blood vessels |
|
||
|
|
New lymphatic vessels |
|
||
|
|
Tumor cell potential |
|
||
|
|
Extracellular matrix potential |
|
||
|
|
Healthy host cell potential |
|
||
|
|
Solid phase tumor cell pressure |
|
||
|
|
Interstitial fluid phase pressure |
|
||
|
|
Interstitial fluid velocity |
|
||
|
|
Solid cell velocity |
|
Table 3.
Dimensionless Diffusivities.
| Dimensionless Parameter | Biological Representation | Scaling Factor* | Value Assigned | ||
|---|---|---|---|---|---|
|
|
Effective diffusivity of O2 | Dn,T | computed | ||
|
|
Diffusivity of O2 through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of O2 through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of O2 through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of glucose | Dn,T | computed | ||
|
|
Diffusivity of glucose through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of glucose through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of glucose through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of CO2 | Dn,T | computed | ||
|
|
Diffusivity of CO2 through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of CO2 through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of CO2 through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of lactate | Dn,T | computed | ||
|
|
Diffusivity of lactate through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of lactate through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of lactate through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of bicarbonate | Dn,T | computed | ||
|
|
Diffusivity of bicarbonate through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of bicarbonate through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of bicarbonate through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of H+ ions | Dn,T | computed | ||
|
|
Diffusivity of H+ through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of H+ ions through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of H+ ions through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of Na+ ions | Dn,T | computed | ||
|
|
Diffusivity of Na+ through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of Na+ ions through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of Na+ ions through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of Cl− ions | Dn,T | computed | ||
|
|
Diffusivity of Cl− through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of Cl− ions through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of CE ions through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of TGFs | Dn,T | computed | ||
|
|
Diffusivity of TGFs through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of TGFs through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of TGFs through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of TAFs | Dn,T | computed | ||
|
|
Diffusivity of TAFs through ECM regions | Dn,T | 1.0 | ||
|
|
Diffusivity of TAFs through tumor regions | Dn,T | 1.0 | ||
|
|
Diffusivity of TAFs through host regions | Dn,T | 1.0 | ||
|
|
Effective diffusivity of MDEs |
|
computed | ||
|
|
Diffusivity of MDEs through ECM regions |
|
0.05 | ||
|
|
Diffusivity of MDEs through tumor regions |
|
0.01 | ||
|
|
Diffusivity of MDEs through host regions |
|
0.01 | ||
|
|
Effective diffusivity of Myofibroblastic cells (MFC) |
|
computed | ||
|
|
Diffusivity of MFCs through ECM regions |
|
1.0 | ||
|
|
Diffusivity of MFCs through tumor regions |
|
0.0 | ||
|
|
Diffusivity of MFCs through host regions |
|
0.0 | ||
|
|
Effective diffusivity of ECS |
|
computed | ||
|
|
Diffusivity of ECs through ECM regions |
|
1.0 | ||
|
|
Diffusivity of ECs through tumor regions |
|
0.0 | ||
|
|
Diffusivity of ECs through host regions |
|
0.0 | ||
|
|
Effective diffusivity of LECs |
|
computed | ||
|
|
Diffusivity of LECs through ECM regions |
|
1.0 | ||
|
|
Diffusivity of LECs through tumor regions |
|
0.0 | ||
|
|
Diffusivity of LECs through host regions |
|
0.0 |
For example,
Most diffusivities are assumed to be on the same order of magnitude as Dn,T, hence the nondimensionalized values are set to 1. A wider range of values will be tested and analyzed in future work.
Table 6.
Dimensionless Constants.
| Dimensionless Constant | Biological Representation | Scaling Factors* | Value Assigned | ||
|---|---|---|---|---|---|
|
|
Interaction strength for tumor cells |
|
0.05 | ||
|
|
Interaction strength for ECM |
|
0.05 | ||
|
|
Interaction strength between tumor cells and ECM |
|
0.02 | ||
|
|
Strain energy coefficient |
|
0.001 | ||
|
|
Hypoxic level of O2 | n∞ | 0.3 | ||
|
|
O2 level in capillaries | n∞ | 1.0 | ||
|
|
Glucose level in capillaries | g∞ | 1.0 | ||
|
|
CO2 level in capillaries | n∞ | 0.0 | ||
|
|
Lactate level in capillaries | n∞ | 0.0 | ||
|
|
O2 viability limit of viable tumor cells | n∞ | 0.21 | ||
|
|
O2 viability limit of ECs | n∞ | 0.1 | ||
|
|
O2 viability limit of LECs | n∞ | 0.1 | ||
|
|
O2 viability limit of MFCs | n∞ | 0.21 | ||
|
|
H+ viability limit of viable tumor cells | n∞ | 0.7 | ||
|
|
H+ viability limit of MFCs | n∞ | 0.7 | ||
|
|
Glucose viability limit of viable tumor cells | g∞ | 0.1 | ||
|
|
Glucose viability limit of myofibroblast-like cells | g∞ | 0.1 | ||
|
|
Threshold level of glucose leading to the onset of autophagy for viable tumor cells | g∞ | 0.3 | ||
|
|
Saturation level of lactate in tissues | n∞ | 1.0 | ||
|
|
Threshold lactate level for tgf upregulation | n∞ | 0.8 | ||
|
|
Threshold lactate level for taf upregulation | n∞ | 0.8 | ||
|
|
Charge of a lactate ion | za | −1.0 | ||
|
|
Charge of a bicarbonate ion | za | −1.0 | ||
|
|
Charge of Na+ | za | 1.0 | ||
|
|
Charge of Cl− | za | −1.0 | ||
|
|
Threshold level of corresponding to the onset of the upregulation of myofibroblastic cell proliferation | tgfsat | 0.1 | ||
|
|
Threshold level of taf corresponding to the onset of EC proliferation | tafsat | 0.2 | ||
|
|
Threshold level of corresponding to the onset of LEC proliferation | tafsat | 0.2 | ||
|
|
Eigenstrain for the ECM component |
|
1.0 | ||
|
|
Eigenstrain for the cell components |
|
0.0 | ||
|
|
Lamé constants for ECM component |
|
1.0 | ||
|
|
Lamé constants for cell components |
|
1.0 | ||
|
|
Lamé constants for cell components |
|
1.0 | ||
|
|
Concentration of ECM macromolecules corresponding to the minimum EC haptotaxis strength |
|
0.2 | ||
|
|
Concentration of ECM macromolecules corresponding to the maximum EC haptotaxis strength |
|
0.8 | ||
|
|
Concentration of ECM macromolecules corresponding to the minimum LEC haptotaxis strength |
|
0.2 | ||
|
|
Concentration of ECM macromolecules corresponding to the maximum LEC haptotaxis strength |
|
0.8 | ||
|
|
Positive chemotaxis constant for ECs | tafsat | 1.0 | ||
|
|
Positive chemotaxis constant for LECs | tafsat | 1.0 | ||
|
|
Threshold pressure corresponding to the onset of blood vessel loss |
|
0.6 | ||
|
|
Threshold pressure corresponding to the onset of lymphatic vessel loss |
|
0.6 | ||
|
|
Threshold pressure corresponding to the maximum rate of neo-blood vessel loss |
|
0.8 | ||
|
|
Threshold pressure corresponding to the maximum rate of neo-lymphatic vessel loss |
|
0.8 |
For example, .
Nondimensionalized Eigenstrain and all Lamé constants are set to 1 as an initial value in this study. A wider range of values will be tested and analyzed in future work.
Other scaling factors are listed in Table 7. The dimensionless set of cell-ECM species equations is summarized below:
| (3.7.3) |
| (3.7.4) |
| (3.7.5) |
Table 7.
Scaling Factors
| Dimensional Scaling Factor | Biological Representation | Expression | ||
|---|---|---|---|---|
| ℒ | Characteristic length |
|
||
|
|
Characteristic time |
|
||
|
|
Characteristic cell pressure |
|
||
|
|
Characteristic fluid pressure |
|
||
| ℳ | Characteristic mobility |
|
||
|
|
Characteristic interaction strength |
|
||
|
|
Characteristic Strain |
|
||
|
|
Characteristic Myofibroblastic diffusivity |
|
The volume fraction of healthy cell species can be solved using the relation . The dimensionless chemical potentials are
| (3.7.6) |
| (3.7.7) |
| (3.7.8) |
where is the dimensionless bulk free energy given below:
| (3.7.9) |
| (3.7.10) |
Dimensionless elastic energy term involved in Eq. (3.7.7) can be written as
| (3.7.11) |
where
| (3.7.12) |
| (3.7.13) |
| (3.7.14) |
| (3.7.15) |
The dimensionless Lamé constants are , , , , , , and . Adjustment factors are given in Table 8. The nondimensional solid and interstitial fluid velocities are given by
| (3.7.16) |
| (3.7.17) |
where is a dimensionless cell adhesion parameter, for i = T,E. With fixed volume fractions and , Eq. (3.1.2), together with the mass conservation law given in Eq. (3.1.6), yield the continuity equations for the solid and fluid species:
| (3.7.18) |
| (3.7.19) |
where . Combining Eqs. (3.7.16), (3.7.17), (3.7.18), and (3.7.19), we obtain the Poisson equations for both pressure terms:
| (3.7.20) |
| (3.7.21) |
Table 8.
Adjustment Factors.
| From Eq. (3.7.22) |
|
|
| where |
| where |
| From Eq. (3.7.22) |
|
|
| From Eq. (3.7.22) |
|
|
| From Eqs. (3.7.26) |
|
|
| From Eq. (3.7.33) |
|
|
| From Eq. (3.7.33) |
|
|
| From Eq. (3.7.33) |
|
|
| From Eq. (3.7.33) |
|
|
| From Eqs. (3.7.37) |
|
|
| where the effect of TAF is not considered. |
| where |
| where the effect of TAF is not considered. |
| where |
| From Eqs. (3.7.38) |
|
|
The two velocities are calculated a posteriori in each iteration after the pressures have been computed. The nondimensional source terms are given below:
| (3.7.22) |
where adjustment factors are listed in Table 8.
The dimensionless governing equations for nutrients and waste products are
| (3.7.23) |
The chloride ion concentration is not modeled directly. It is calculated at the end of each iteration step by the relation given by electroneutrality, . The nondimensional flux terms for uncharged and ionic species are given below:
| (3.7.24) |
where σ = ℓ, b, a, and s. Diffusivities in the two equations above can be written in dimensionless form as
| (3.7.25) |
where . The nondimensional source terms of nutrients and waste products listed below:
| (3.7.26) |
where Rg,n = g∞/n∞ and adjustment factors are given in Table 8. The transfer rate coefficients , , , and follows the form
| (3.7.27) |
The governing equations of tumorigenic species shown in Eqs. (3.5.1) – (3.5.3) are nondimensionalized and presented below:
| (3.7.28) |
| (3.7.29) |
| (3.7.30) |
| (3.7.31) |
and , .
All diffusivity terms in Eqs (3.7.28) – (3.7.31) are written in the same form as Eq. (3.7.25). The source terms are expressed in the following dimensionless functions:
| (3.7.32) |
where Rtaf,m = tafsat/msat, Rλ = λU,V,n/λM,V, adjustment factors are given in Table 8, and the transfer rate coefficient takes the same form as depicted in Eq. (3.7.27). Additional rate expressions are nondimensionalized as
| (3.7.33) |
Adjustment factors are given in Table 8.
Neo–blood and neo–lymphatic vessels, expressed in dimensionless densities, are governed by the following equations:
| (3.7.34) |
| (3.7.35) |
Also, , , , and . The convective and diffusive flux terms for neovessels are nondimensionalized as
| (3.7.36) |
| (3.7.37) |
where diffusivities and are written as in Eq. (3.7.25) and adjustment factors are found in Table 8.
Dimensionless source terms are listed in the following:
| (3.7.38) |
where adjustment factors can be found in Table 8. Boundary conditions necessary to solve the set of governing equations listed here will be discussed in the following section.
3.8. Boundary Conditions
We model a tissue domain, Ω, bounded by an outer boundary, Σ. The tissue domain contains both tumor cell regions, ΩT, and healthy host regions, ΩH. A tumor region is defined by and is surrounded by a tumor boundary ΣT, taken to be here. Naturally, a healthy host region is therefore defined by .
Boundary conditions are not needed for the tumor boundary ΣT. For outer boundary Σ, we define the following Neumann conditions for the cell volume fractions and Dirichlet condition for solid cell pressure:
| (3.8.1) |
where n is the outward normal of the boundary. To allow cells to flow freely across the outer boundary, we set the conditions
| (3.8.2) |
For nutrients and waste products, Dirichlet conditions are imposed:
| (3.8.3) |
For tumorigenic species, Dirichlet conditions are imposed, except for myofibroblastic cells, where Neumann condition is imposed:
| (3.8.4) |
New vessels are assumed to be at and at all four boundaries.
The set of dimensionless governing equations listed here, combined with boundary conditions stated in the next section, will be solved using numerical methods described in Section 4.
4. Numerical Methods
The set of nondimensionalized governing equations are discretized implicitly in time using the Crank-Nicholson scheme as described in Wise et al. (2011). Spatial discretization methods employed are second order finite differences and central differences. In the computation of edge-centered approximation in the advection terms, both upwind donor-cell advection and third order upwind biased WENO scheme (Jiang and Shu, 1996; Liu et al., 1994) are used.
The discretized set is solved using a nonlinear full multigrid solver (Trottenberg et al., 2001) with self-adaptive mesh refinement. The nonlinear solver uses the full approximation scheme in V-cycles (Trottenberg et al., 2001; Wise et al., 2007), with Gauss-Seidel relaxations in red-black (odd-even) ordering. The block-structured composite Cartesian mesh consists of a hierarchy of levels with uniform grids that are increasing in their mesh spacings. In general, the composite mesh has a few adaptively refined finer levels sitting on top of several coarser global levels. Each global mesh level consists of one block that spans the entire domain. Each adaptively refined level may have one or more blocks generated during each time-step for areas where refinement is deemed beneficial. If the finest global level, also called the root level, is set as L0 with grid spacing h0, a subsequent coarser global level is set as L−1 with grid spacing h−1 = 2h0, followed by another coarser level L−2 with grid spacing h−2 = 2h−1, and so on. Above the finest global level sits the first adaptively refined level L1 with h1 = h0/2, followed by another adaptively refined level L2 with h2 = h1/2, and so on.
A total of five levels of refinement is used here for the tissue domain Ω = (0,40)3. The mesh spacings for L2 down to level L−2 are h2 = 40/128, h1 = 40/64, h0 = h1/32, h−1 = 40/16, and h−2 = 40/8, respectively. The root level has a grid size of 32×32×32. Several options for flagging cells for refinement are considered, ranging from the simple volume fraction test (Wise et al., 2008), undivided gradient test (Wise et al., 2007), and relative truncation error test (Trottenberg et al., 2001).
Simulations were performed on a node with 768 GB of RAM and 32 Intel Xeon 3.3GHz cores, running CentOS 6.7 ×86_64. The time step size was set to 1×10−2. At t = 1, it took approximately 2.6 hrs to complete 100 time steps; at t = 10, the solver completed 100 time steps in about 3.5 hrs; at t = 20, 100 time steps were completed in roughly 4.7 hrs. The computation time for t = 10 was approximately 1 day and the total computation time to reach t = 20 was about 3 days. The algorithms were partially parallelized using OpenMP, and we expect further detailed parallelization work in the future to yield higher performance.
5. Computational Results
To gently perturb the symmetry of the mathematical model and to test the model’s ability to simulate a morphologically asymmetric system, the fraction of blood vessel species ℱBn available for sprouting, remodeling, and taxis, as depicted in Eqs. (3.6.3), (3.6.4), (3.6.9), and (3.6.11), is set to a different constant for the eight equally-sized parts of the domain.
Figure 2 shows a sample of a desmoplastic tumor evolution in time through a cross-section in the index plane j = k = 58. The simulation parameters are as listed in Tables 2 through 6. Initially (t = 1), the tumor viable, dead, and ECM species are mostly symmetrical around the tumor center. There is a significant proportion of viable tissue that is hypoxic in the core (when the oxygen value < 0.3). The tumor essentially has a viable rim surrounding a mostly necrotic core (induced when oxygen value < 0.2). The ECM level is high, representing a fibrotic lesion. The pressure is high in the immediate surroundings of the lesion and significantly lower in the core in which there is little tissue proliferation. The diffusible substances driving the tumor evolution, including oxygen (O2), glucose (Glu), carbon dioxide (CO2), bicarbonate (Bic), lactate (Lac), and hydrogen ions (H+), reflect this symmetry in space. Oxygen and glucose experience a decreasing concentration into the core of the lesion, as their uptake by the viable tumor tissue depletes their concentration. The byproducts of glycolysis, carbon dioxide and bicarbonate, accumulate in the tumor tissue, with their diffusion outwards hindered by inadequate vascularization and adverse pressure gradients. Correspondingly, the concentration of lactate and hydrogen ions is significantly higher within the tumor tissue.
Figure 2.
Desmoplastic tumor evolution with regionally varied . Time progression from left to right for t = 1, 5, 10, 15, and 20, and plots are made for a cross section of the index plane j = k = 58. First row: tumor viable species , dead species , and ECM species . The overall tumor pressure is labeled by pα. Second row: diffusible substances driving the tumor evolution, including oxygen (O2), glucose (Glu), carbon dioxide (CO2), bicarbonate (Bic), lactate (Lac), and hydrogen ions (H+). Third row: Concentration of myfibroblasts (myF), tumor growth factors (TGF), and matrix degrading enzymes (MDE). Fourth row: corresponding density of blood vasculature (Bn), lymphatic vasculature (Ln), and tumor angiogenic factors (TAF).
The tumor growth factor (tgf) and myofibroblast (myF) concentrations are also higher within the tumor, while the matrix degrading enzyme (MDE) concentration, produced by the expanding viable tumor tissue as well as the vasculature in response to tumor angiogenic factors released by the hypoxic tumor tissue, is slightly higher and asymmetrical within the core of the tumor, which reflects the asymmetry in the vascular layout due to some regions having a higher probability of endothelial tissue proliferation. The hypoxic tissue engenders a high level of tumor angiogenic factors (TAF) that diffuse outward from the tumor into the surroundings, and in turn drive higher the concentration of both blood and lymphatic vasculature. After 20 days of growth, the tumor has evolved into a heterogeneous system resembling a typical pancreatic cancer, which is mostly fibrotic, hypoxic, acidic, and with a low density of viable tissue spread throughout.
For comparison, Figure 3 shows the first five days of evolution for a perfectly spherical tumor (cross-section in the index plane j = k = 58), for which the percentage of proliferating vasculature is the same everywhere. Although in this case the tumor also represents a hypoxic, fibrotic lesion, the symmetric vasculature enforces a correspondingly symmetric MDE concentration. In time, however, fluctuations in the blood and lymphatic vasculature density are expected to disturb this symmetry and lead to a heterogeneous system as in Figure 2.
Figure 3.
Desmoplastic tumor evolution with uniform . Time progression is from left to right for t = 1 – 5, and plots are made for a cross section of the index plane j = k = 58. First row: tumor viable species , dead species , and ECM species . The overall tumor pressure is labeled by pα. Second row: diffusible substances driving the tumor evolution, including oxygen (O2), glucose (Glu), carbon dioxide (CO2), bicarbonate (Bic), lactate (Lac), and hydrogen ions (H+). Third row: Concentration of myfibroblasts (myF), tumor growth factors (TGF), and matrix degrading enzymes (MDE). Fourth row: corresponding density of blood vasculature (Bn), lymphatic vasculature (Ln), and tumor angiogenic factors (TAF).
Figure 4 highlights the evolution of the velocity field along the pressure gradient in a sub-section of the x − z plane (at index j = 58). The velocity of solid phase components (viable and dead tumor, and ECM tissues) is represented by uα, while the velocity of aqueous phase component (interstitial fluid) is represented by uβ, along with the solid pressure p and and hydrostatic pressure q contours. Figure 5 shows the viable tissue and ECM contours corresponding to the scenario in Figure 4, and highlights the heterogeneous growth-up of these two species in time. In these two figures, clockwise starting from the lower left quadrant (i < 65 and k < 65), ℱBn is set to 0.8, 0.1, 0.2, and 0.15, respectively.
Figure 4.
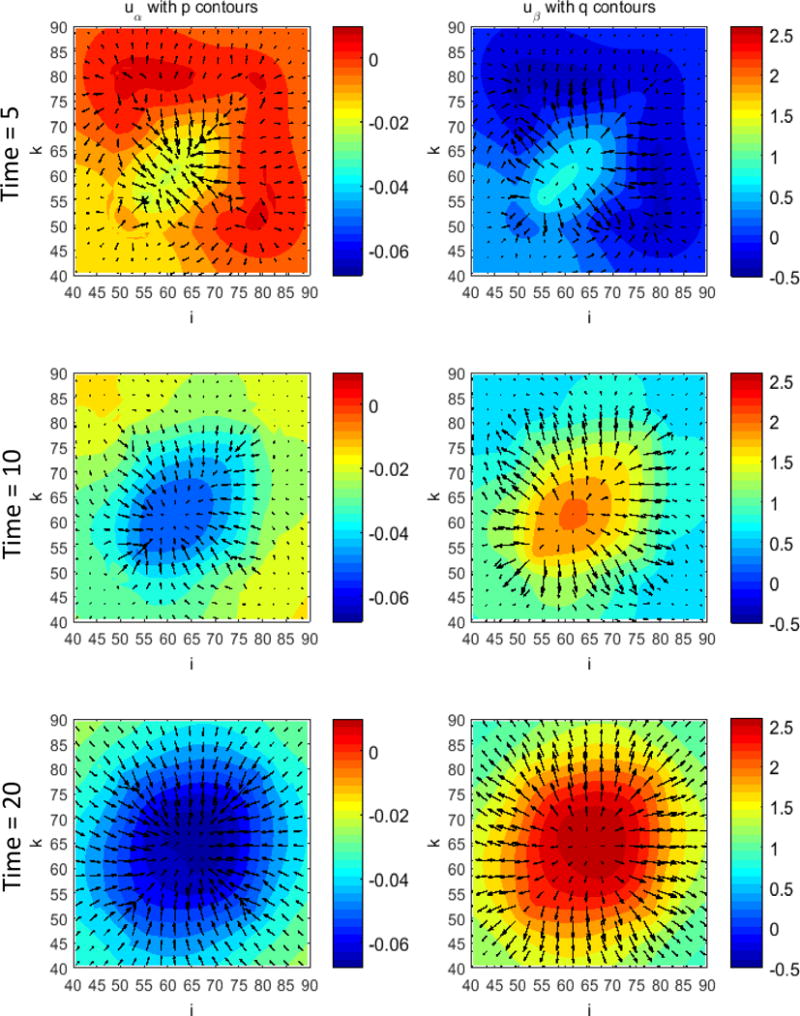
Evolution of the solid-ECM and interstitial fluid phase velocity fields along their corresponding pressure gradients. Plotted for t = 5, 10, and 20, in a sub-section of the x – z plane (at index j = 58). The arrows denote the velocity vectors, with longer arrows indicating a higher magnitude.
Figure 5.
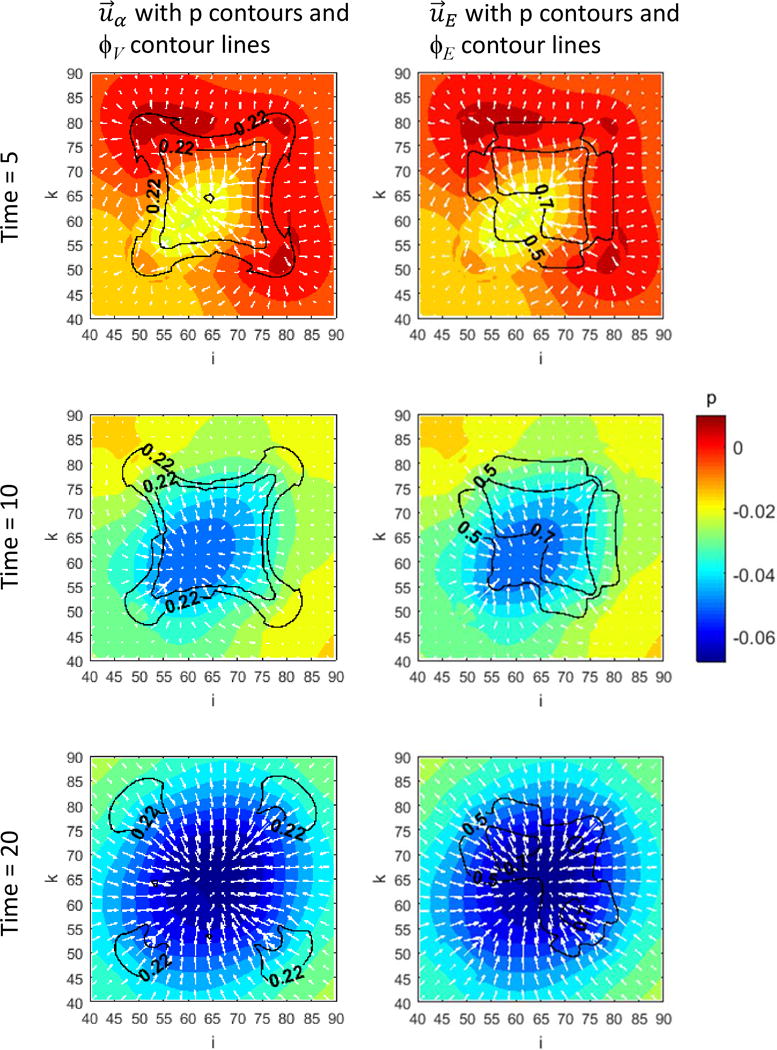
Evolution of viable tissue and ECM species (2D plot) with their corresponding velocity fields and pressure gradients. Plotted for t = 5, 10, and 20, in a sub-section of the x – z plane (at index j = 58). The arrows denote the velocity vectors, with longer arrows indicating a higher magnitude.
The evolution of the tumor in 3D space is presented in Figure 6. As time progresses the viable tissue tends to grow at the periphery, bulging out at the corners as has been previously reported with these types of mixture models (Frieboes et al., 2010; Wise et al., 2008). In the center of the tumor mass, oxygen and nutrient levels drop below the threshold necessary for viability, leading to tumor tissue becoming necrotic. Further, as TGF accumulates over time in the center due to its production by viable tumor tissue, thus drawing in myofibroblastic cells and upregulating their mitosis, more ECM is being secreted. As a result, the center of the tumor mass becomes denser with ECM (i.e., becomes fibrotic), while the viable tissue at the periphery, with access to higher nutrients levels from the surroundings, is able to continue growing. The dead tissue follows in structure the viable tissue, although in a smaller overall volume. The ECM species, initially mainly concentrated in the interior of the tumor, evolves non-uniformly to maintain a heterogeneous volume during the growth of the viable tissue. Figure 7 presents an alternate view of the viable, dead, and ECM species in 3D space (sliced through the center of the domain) at time t = 20 days, highlighting the heterogeneity of the structure in terms of the distribution of these species.
Figure 6.
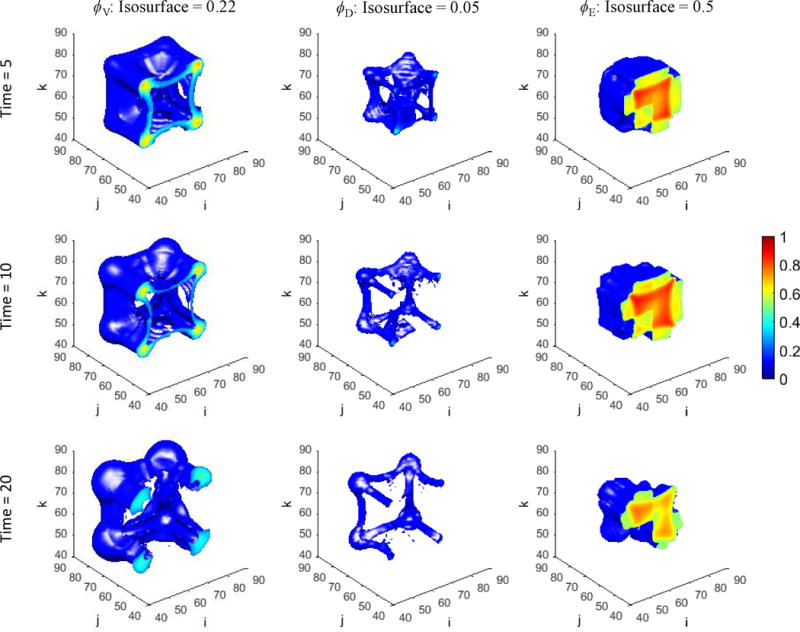
Evolution of the tumor viable, dead, and ECM species (3D plot) for t = 5, 10, and 20. Cross-sectional contours are on the x – z plane sliced at index j = 58. Highest density: red; lowest: blue.
Figure 7.
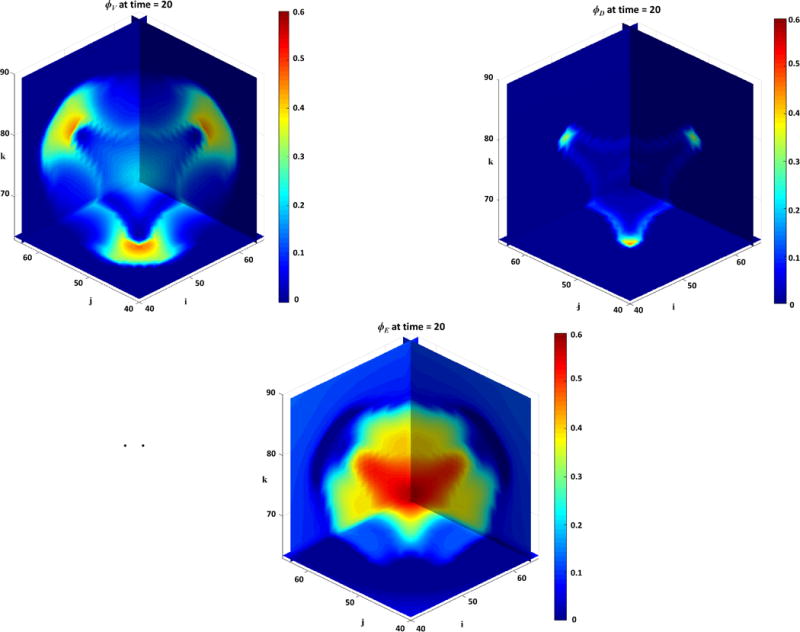
Tumor viable, dead, and ECM species at time t = 20 (3D plot). Cross-sectional contours are sliced at indices i = 58, j = 58, and k = 58. Highest density: red; lowest: blue.
The variation in the ECM species as a function of the elastic energy is shown in Figure 8. The interfacial strain energy coefficient εe as in Eqs. (3.2.16), (3.2.23), and (3.2.25), or the nondimensionalized form as in Eq. (3.7.11) is increased from for the base level ECM, to for 10 times the base level ECM, and to for 100 times the base level ECM. As the elastic energy contribution to the chemical potential increases over two orders of magnitude, the ECM species becomes more diffuse. For illustration, the ECM density at t = 2, initially with a high peak within the tumor region in the center of the x-dimension cross-section shown in the figure, decreases by about a third from the peak.
Figure 8.
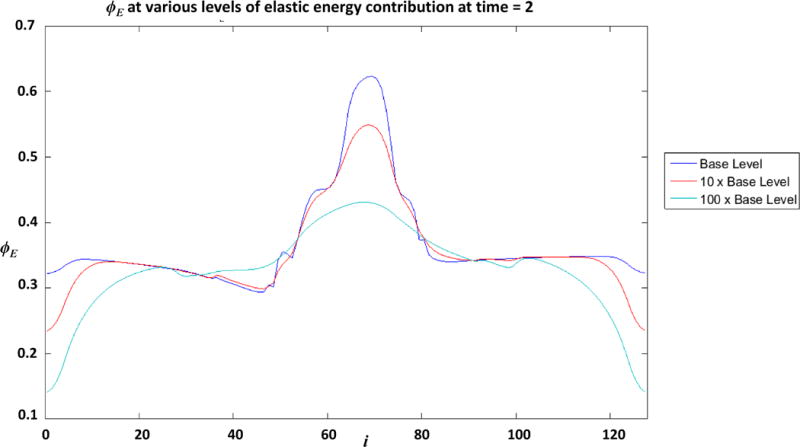
Variation in ECM species as a function of the elastic energy at t = 2. Blue: ; Red: ; Green: .
We performed a sensitivity analysis using the glucose uptake rate constant of viable tumor cells. Values of the rate constant used are 10, 1, and 0.1. Simulation results at t = 20 are given in Figure 9. Profiles of volume fractions, species concentrations, and vessel densities in the center column are from the base case shown in Figure 2, where . When the glucose uptake rate constant of viable tumor cells is increased to , the higher uptake causes the glucose levels in the center of the domain (where viable tumor cells are seeded) to drop below the glucose viability limit of 0.1 very early during the simulation. Viable tumor cells in the center of the domain become necrotic, turning into dead cells and eventually lysed. The low levels of viable tumor cells in the center also results in lower levels of TGF and TAF being produced. Lower levels of TGF lead to lower levels of myofibroblastic cells, hence less ECM being secreted. Similarly, lower TAF levels result in lower vessel densities. However, when the glucose uptake rate constant of viable tumor cells is decreased to , profiles of all volume fractions, species concentrations (except those involved in glycolysis), and vessel densities are the same as the base case. Since there is an abundance of glucose in the domain due to the low glucose uptake rate, oxygen is the limiting nutrient, as in the base case. Therefore, the system behavior is dictated by the same oxygen limitation and follows similar dynamics as in the base case.
Figure 9.
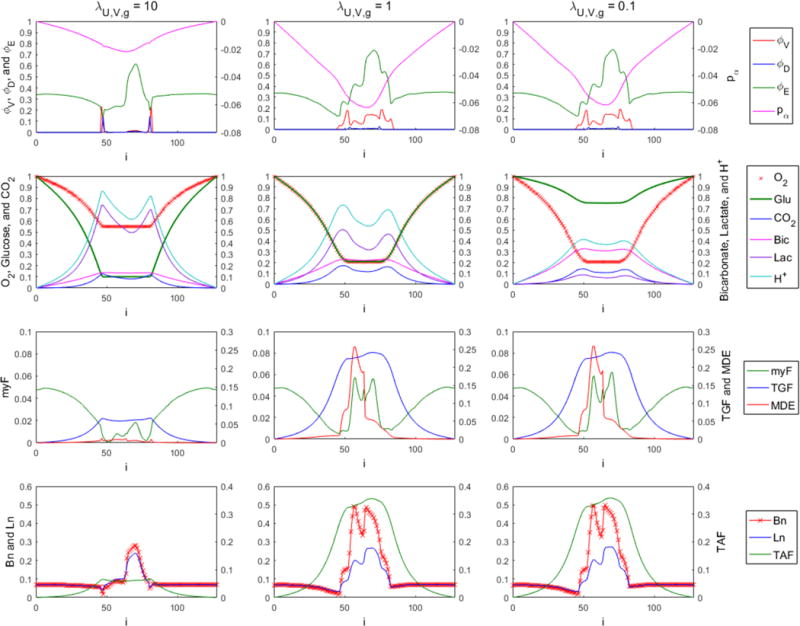
Desmoplastic tumor simulations at t = 20 for glucose uptake rate constants , 1 (the base case shown in Figure 1), and 0.1.
Lastly, we evaluated the effects of TGF on the tumor microenvironment. The levels of TGF are controlled through its secretion rate constant by viable tumor cells and degradation rate constant. Simulation results at t = 20 are plotted in Figure 10. Profiles of volume fractions, species concentrations, and vessel densities in the center column are from the base case shown in Figure 2, where and . To simulate a case with lower TGF levels compared to the base case, a higher degradation rate constant is used. In the nutrient-starved tumor mass center, viable tumor cells undergo necrosis, followed by lysing of the dead cells. Since the mitosis rate of viable tumor cells is not sufficiently upregulated by the lower TGF levels, viable cells are mostly absent in the low-nutrient center region at t = 20, and they are mostly present outside the edge of the hypoxic core where they still proliferate. The low TGF levels also do not sufficiently upregulate myofibroblastic cells in the tumor region, resulting in low levels of myofibroblastic cells, and therefore little ECM is being secreted. The lack of viable tumor cells results in a dip in the TAF profile, causing insignificant angiogenesis in the core.
Figure 10.
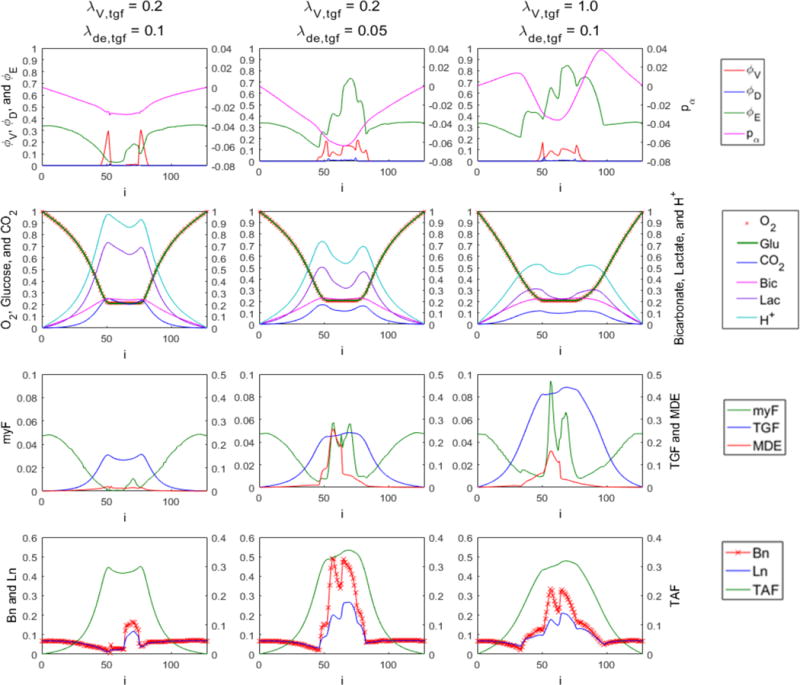
Desmoplastic tumor simulations at t = 20 for a decreased-TGF case ( and ), the base case shown in Figure 1 ( and ), and an increased-TGF case ( and ).
To achieve higher TGF levels compared to the base case, a higher production rate constant by viable tumor cells is used, accompanied by a higher degradation rate constant . The higher level of TGF upregulates the proliferation of myofibroblastic cells. The increased density of myofibroblastic cells leads to increased amount of ECM secreted, hence the ECM-rich tumor mass as seen in the upper right plot in Figure 10. Moreover, in the TGF rich environment, the mitosis rate of viable tumor cells within the nutrient-starved center and surrounding areas is upregulated, resulting in a wider viable tumor area. The increase in viable tumor mass might not be reflected in the one-dimensional profile, but it is shown clearly in Figure 11. The higher amount of proliferating viable tumor tissue is also responsible for the broader region of higher TAF in the tissue. As a result of the higher TAF levels, there is a broader region of elevated blood vessel growth, as also evidenced by the relatively more relaxed curves of oxygen, glucose, and metabolic byproducts.
Figure 11.
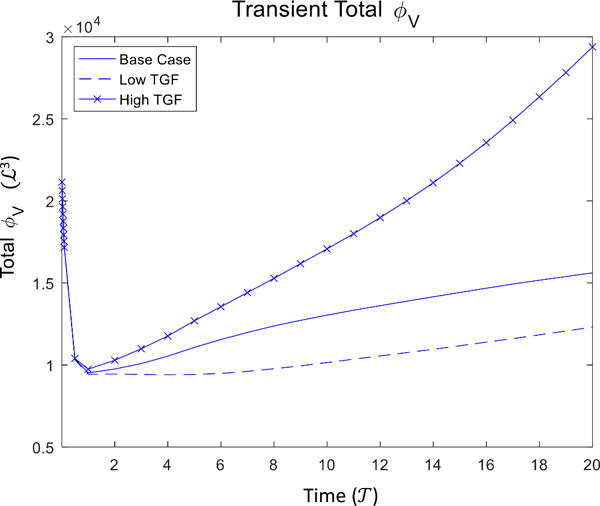
Transient total viable tumor volume corresponding to the three cases of different TGF levels in Figure 10.
6. Conclusion and Future Work
In this paper, we reviewed topics related to the study of the tumor microenvironment. We evaluated some of the key associated reactions and cell species, defined a scope that is sufficient for the purpose of simulating a vascular desmoplastic tumor microenvironment, and detailed the formulation of a mathematical model. We presented numerical simulations of symmetrical and nonsymmetrical desmoplastic tumor progression in 3D. We also showed that this model is capable of simulating an ECM-rich tumoral tissue and capturing complex morphological changes during growth of a tumor mass.
For future work, other forms of elastic energy contribution representing the mechanical behavior of biological tissues will be studied. The desmoplastic tumor growth could also include heterogeneous cell types such as mutated clones of tumor cells and cancer stem cells. To further approach a more complete tumor microenvironment, immune cell species could be included, as well as the tumor-associated inflammation response. Hormonal growth factors and chemoattractants can be incorporated in the model to drive the tumor growth and influence the migration of immune cells. We will obtain and make use of parameters and constants that are biologically appropriate. Reactions and responses in the model will also be studied in further detail, so that true dynamics can be captured in adjustment factors and modelled via empirical formulas obtained from experiments. As the model becomes more refined and well-tuned, the modelling of the transport and effects of anticancer drugs and nanovectors could be incorporated for evaluation of therapeutic efficacy.
Table 4.
Dimensionless Rate Constants.
| Dimensionless Parameter | Biological Representation | Scaling Factor * | Value Assigned | ||
|---|---|---|---|---|---|
|
|
Mitosis rate constant of viable tumor cells | λM,V | 1.0 | ||
|
|
Apoptosis rate constant of viable tumor cells | λM,V | 0.0 | ||
|
|
Necrosis rate constant of viable tumor cells | λM,V | 3.0 | ||
|
|
Metastasis rate constant of viable tumor cells via blood vessels | λM,V | 0.0 | ||
|
|
Metastasis rate constant of viable tumor cells via lymphatic vessels | λM,V | 0.0 | ||
|
|
Autophagy rate constant of viable tumor cells | λM,V | 0.0 | ||
|
|
Lysis rate constant of dead tumor cells | λM,V | 1.0 | ||
|
|
ECM rate of secretion by tumor viable cells | λM,V | 0.0 | ||
|
|
ECM rate of secretion by ECs |
|
0.0 | ||
|
|
ECM rate of secretion by LECs |
|
0.0 | ||
|
|
ECM rate of secretion by myofibroblastic cells |
|
5.0 | ||
|
|
Degradation rate of ECM | λM,V | 1.0 | ||
|
|
Degradation rate of ECM macromolecules catalyzed by MDEs |
|
5.0 | ||
|
|
Apparent transfer coefficient of O2 via capillary network | λU,V,n | computed | ||
|
|
Transfer coefficient of O2 via capillary network in ECM regions | λU,V,n | 0.1 | ||
|
|
Transfer coefficient of O2 via capillary network in tumor regions | λU,V,n | 0.001 | ||
|
|
Transfer coefficient of O2 via capillary network in host regions | λU,V,n | 0.01 | ||
|
|
Uptake rate constant of O2 by viable tumor cells | λU,V,n | 1.0 | ||
|
|
Uptake rate constant of O2 by healthy host cells | λU,V,n | 0.0001 | ||
|
|
Apparent transfer coefficient of glucose via capillary network | λU,V,n | computed | ||
|
|
Transfer coefficient of glucose via capillary network in ECM regions | λU,V,n | 0.1 | ||
|
|
Transfer coefficient of glucose via capillary network in tumor regions | λU,V,n | 0.001 | ||
|
|
Transfer coefficient of glucose via capillary network in host regions | λU,V,n | 0.01 | ||
|
|
Uptake rate constant of glucose by viable tumor cells | λU,V,n | 1.0 | ||
|
|
Uptake rate constant of glucose by healthy host cells | λU,V,n | 0.0001 | ||
|
|
Apparent transfer coefficient of CO2 via capillary network | λU,V,n | computed | ||
|
|
Transfer coefficient of CO2 via capillary network in ECM regions | λU,V,n | 1.0 | ||
|
|
Transfer coefficient of CO2 via capillary network in tumor regions | λU,V,n | 1.0 | ||
|
|
Transfer coefficient of CO2 via capillary network in host regions | λU,V,n | 1.0 | ||
|
|
Forward reaction rate of the dissolution of CO2 and H2O | λU,V,n | 1.0 | ||
|
|
Backward reaction rate of the dissolution of CO2 and H2O | λU,V,n/n∞ | 1.0 | ||
|
|
Apparent transfer coefficient of lactate via capillary network | λU,V,n | computed | ||
|
|
Transfer coefficient of lactate via capillary network in ECM regions | λU,V,n | 1.0 | ||
|
|
Transfer coefficient of lactate via capillary network in tumor regions | λU,V,n | 0.1 | ||
|
|
Transfer coefficient of lactate via capillary network in host regions | λU,V,n | 0.5 | ||
|
|
Production rate constant of TGFs by viable tumor cells | λU,V,n | 0.2 | ||
|
|
Production rate constant of TGFs by ECs | λU,V,n | 0.0 | ||
|
|
Production rate constant of TGFs by LECs | λU,V,n | 0.0 | ||
|
|
Production rate constant of TGFs by MFCs | λU,V,n | 0.0 | ||
|
|
Degradation rate constant of TGFs | λU,V,n | 0.05 | ||
|
|
Uptake rate constant of TGFs by viable tumor cells | λU,V,n | 0.0 | ||
|
|
Production rate constant of TAFs by viable tumor cells | λU,V,n | 0.2 | ||
|
|
Production rate constant of TAFs by ECs | λU,V,n | 0.0 | ||
|
|
Production rate constant of TAFs by LECs | λU,V,n | 0.0 | ||
|
|
Production rate constant of TAFs by MFCs | λU,V,n | 0.0 | ||
|
|
Degradation rate constant of TAFs | λU,V,n | 0.05 | ||
|
|
Uptake rate constant of TAFs by proliferating ECs | λU,V,n/Bmax | 0.0011574 | ||
|
|
Uptake rate constant of TAFs by proliferating LECs | λU,V,n/Lmax | 0.0011574 | ||
|
|
Rate constant for the loss of TAFs to the production of MDEs by proliferating ECs |
|
1.0 | ||
|
|
Rate constant for the loss of TAFs to the production of MDEs by proliferating LECs |
|
1.0 | ||
|
|
Michaelis constant for the uptake of TAF by proliferating ECs | tafsat | 1.0 | ||
|
|
Michaelis constant for the uptake of TAF by proliferating LECs | tafsat | 1.0 | ||
|
|
Production rate constant of MDEs by viable tumor cells | λM,V | 0.2 | ||
|
|
Production rate constant of MDEs by MFCs | λM,V | 0.0 | ||
|
|
Decay rate constant of MDEs | λM,V | 5.0 | ||
|
|
Mitosis rate constant of MFCs | λM,V | 0.1 | ||
|
|
Apoptosis rate constant of MFCs | λM,V | 0.1 | ||
|
|
Necrosis rate constant of MFCs | λM,V | 0.3 | ||
|
|
Maximum mitosis rate constant of ECs | λM,V | 1.0 | ||
|
|
Maximum degradation rate constant of new blood vessels due to cell pressure | λM,V | 1.0 | ||
|
|
Remodeling rate constant of new blood vessels by MDEs | λM,V/msat | 1.0 | ||
|
|
Anastomosis rate constant (periodic) of the new blood vessels | λM,V | 0.0 | ||
|
|
Maximum mitosis rate constant of LECs | λM,V | 1.0 | ||
|
|
Maximum degradation rate constant of new lymphatic vessels due to cell pressure | λM,V | 1.0 | ||
|
|
Remodeling rate constant of new lymphatic vessels by MDEs | λM,V | 1.0 | ||
|
|
Anastomosis rate constant (periodic) of the new lymphatic vessels | λM,V | 0.0 |
For example, .
Table 5.
Mobility, Motilities, and Taxis Coefficients
| Dimensionless Parameter | Biological Representation | Scaling Factor* | Value Assigned | ||
|---|---|---|---|---|---|
|
|
Mobility of cell species | ℳ | 0.1 | ||
|
|
Motility of the solid phase (cells) |
|
Computed† | ||
|
|
Motility of the tumor cell phase |
|
10.0 | ||
|
|
Motility of the ECM phase |
|
10.0 | ||
|
|
Motility of the healthy host cell phase |
|
10.0 | ||
|
|
Motility of the fluid phase (interstitial fluid) |
|
1.0 | ||
|
|
Chemotaxis coefficient of ECs |
|
1.0 | ||
|
|
Haptotaxis coefficient of ECs |
|
1.0 | ||
|
|
Minimum haptotaxis coefficient of ECs |
|
1.0 | ||
|
|
Chemotaxis coefficient of LECs |
|
1.0 | ||
|
|
Haptotaxis coefficient of LECs |
|
1.0 | ||
|
|
Minimum haptotaxis coefficient of LECs |
|
1.0 |
For example, .
The solid phase motility is computed from , , and using Eq. (3.7.25) by replacing with .
Nondimensionalized chemotaxis and heptotaxis coefficients are set to 1 as an initial value in this study. A wider range of values will be tested and analyzed in future work.
Highlights.
Created four-species model to simulate interactions between tumor and stromal cells
Cellular metabolic processes are simulated, including respiration and glycolysis
Growth-factor releasing fibroblasts are key contributors to abnormal ECM remodeling
An elastic energy is implemented to provide elasticity to the connective tissue
ECM remodeling simulates stromal desmoplasia in pancreatic ductal adenocarcinoma
Acknowledgments
HBF acknowledges partial support by the National Institutes of Health / National Cancer Institute (U54CA143907, R01CA180149, and R15CA203605), and by the Department of Bioengineering and the James Graham Brown Cancer Center at the University of Louisville. This work was conducted in part using the resources in the research computing group and the Cardinal Research Cluster (CRC) at the University of Louisville. The authors are grateful to Harrison Simrall for computing assistance and to Dylan Goodin for proofreading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboussekhra A. Role of cancer-associated fibroblasts in breast cancer development and prognosis. International Journal of Developmental Biology. 2011;55:841–849. doi: 10.1387/ijdb.113362aa. [DOI] [PubMed] [Google Scholar]
- Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- Ahmad SA, Liu WB, Jung YD, Fan F, Wilson M, Reinmuth N, Shaheen RM, Bucana CD, Ellis LM. The effects of angiopoietin-1 and-2 on tumor growth and angiogenesis in human colon cancer. Cancer research. 2001;61:1255–1259. [PubMed] [Google Scholar]
- Akhurst RJ, Derynck R. TGF-beta signaling in cancer – a double-edged sword. Trends in Cell Biology. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)82259-5. [DOI] [PubMed] [Google Scholar]
- Algire GH, Legallais FY. Growth Rate of Transplanted Tumors in Relation to Latent Period and Host Vascular Reaction. Cancer Res. 1947;7 [Google Scholar]
- Algire GH, Chalkley HW, Legallais FY, Park H. Vascular Reactions of Normal and Malignant Tissues In Vivo. I. Vascular Reactions of Mice to Wounds and to Normal and Neoplastic Implants. J Natl Cancer Inst. 1945;6:73–85. [Google Scholar]
- Ambrosi D, Preziosi L. Cell adhesion mechanisms and stress relaxation in the mechanics of tumours. Biomechanics and Modeling in Mechanobiology. 2009;8:397–413. doi: 10.1007/s10237-008-0145-y. [DOI] [PubMed] [Google Scholar]
- Ambrosi D, Preziosi L, Bellomo N. On the Closure of Mass Balance Models for Tumor Growth. Mathematical Models and Methods In Applied Sciences. 2002;12:737–754. [Google Scholar]
- Amendt C, Schirmacher P, Weber H, Blessing M. Expression of a dominant negative type II TGF-beta receptor in mouse skin results in an increase in carcinoma incidence and an acceleration of carcinoma development. Oncogene. 1998;17:25–34. doi: 10.1038/sj.onc.1202161. [DOI] [PubMed] [Google Scholar]
- Anastasiou D, Cantley LC. Breathless cancer cells get fat on glutamine. Cell Res. 2012;22:443–446. doi: 10.1038/cr.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andasari V, Gerisch A, Lolas G, South AP, Chaplain MAJ. Mathematical modeling of cancer cell invasion of tissue: biological insight from mathematical analysis and computational simulation. Journal of Mathematical Biology. 2011;63:141–171. doi: 10.1007/s00285-010-0369-1. [DOI] [PubMed] [Google Scholar]
- Anderson ARA, Chaplain MAJ. Continuous and discrete mathematical models of tumor-induced angiogenesis. Bulletin of Mathematical Biology. 1998;60:857–899. doi: 10.1006/bulm.1998.0042. [DOI] [PubMed] [Google Scholar]
- Apte MV, Wilson JS. Dangerous liaisons: Pancreatic stellate cells and pancreatic cancer cells. Journal of Gastroenterology and Hepatology. 2012;27:69–74. doi: 10.1111/j.1440-1746.2011.07000.x. [DOI] [PubMed] [Google Scholar]
- Araujo RP, McElwain DLS. A history of the study of solid tumour growth: The contribution of mathematical modelling. Bulletin of Mathematical Biology. 2004;66:1039–1091. doi: 10.1016/j.bulm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Araujo RP, McElwain DLS. A mixture theory for the genesis of residual stresses in growing tissues I: A general formulation. Siam Journal on Applied Mathematics. 2005a;65:1261–1284. doi: 10.1137/040607113. [DOI] [Google Scholar]
- Araujo RP, McElwain DLS. A mixture theory for the genesis of residual streses in growing tissues II: Solutions to the biphasic equations for a multicell spheroid. Siam Journal on Applied Mathematics. 2005b;66:447–467. doi: 10.1137/040607125. [DOI] [Google Scholar]
- Arduino A, Preziosi L. A multiphase model of tumour segregation in situ by a heterogeneous extracellular matrix. International Journal of Non-Linear Mechanics. 2015;75:22–30. doi: 10.1016/j.ijnonlinmec.2015.04.007. [DOI] [Google Scholar]
- Astanin S, Preziosi L. Multiphase Models of Tumour Growth. Birkhäuser Boston; Boston: 2008. [Google Scholar]
- Astrof S, Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis. 2009;12:165–175. doi: 10.1007/s10456-009-9136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augsten M, Hagglof C, Olsson E, Stolz C, Tsagozis P, Levchenko T, Frederick MJ, Borg A, Micke P, Egevad L, Ostman A. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3414–3419. doi: 10.1073/pnas.0813144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley M, Gayraud B. Structure and biological activity of the extracellular matrix. Journal of Molecular Medicine-Jmm. 1998;76:253–265. doi: 10.1007/s001090050215. [DOI] [PubMed] [Google Scholar]
- Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M, Wigler N, Keydar I, Ben-Baruch A. The CC chemokine RANTES in breast carcinoma progression: Regulation of expression and potential mechanisms of promalignant activity. Cancer research. 2002;62:1093–1102. [PubMed] [Google Scholar]
- Bachmann J, Raue A, Schilling M, Becker V, Timmer J, Klingmüller U. Predictive mathematical models of cancer signalling pathways. Journal of Internal Medicine. 2012;271:155–165. doi: 10.1111/j.1365-2796.2011.02492.x. [DOI] [PubMed] [Google Scholar]
- Baldwin ME, Halford MA, Roufail S, Williams RA, Hibbs ML, Grail D, Kubo H, Stacker SA, Achen MG. Vascular endothelial growth factor d is dispensable for development of the lymphatic system. Molecular and Cellular Biology. 2005;25:2441–2449. doi: 10.1128/mcb.25.6.2441-2449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine & Growth Factor Reviews. 2002;13:135–141. doi: 10.1016/s1359-6101(01)00020-x. [DOI] [PubMed] [Google Scholar]
- Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Yla-Herttuala S, Jackson DG, Alitalo K, McDonald DM. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. Journal of Clinical Investigation. 2005;115:247–257. doi: 10.1172/jci200522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bando H, Toi M, Kitada K, Koike M. Genes commonly upregulated by hypoxia in human breast cancer cells MCF-7 and MDA-MB-231. Biomedicine & Pharmacotherapy. 2003;57:333–340. doi: 10.1016/s0753-3322(03)00098-2. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anticancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bearer EL, Lowengrub JS, Frieboes HB, Chuang YL, Jin F, Wise SM, Ferrari M, Agus DB, Cristini V. Multiparameter Computational Modeling of Tumor Invasion. Cancer research. 2009;69:4493–4501. doi: 10.1158/0008-5472.can-08-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappa B in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Ben-Baruch A. Inflammation-associated immune suppression in cancer: The roles played by cytokines, chemokines and additional mediators. Seminars in Cancer Biology. 2006;16:38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Bergers G, Coussens LM. Extrinsic regulators of epithelial tumor progression: metalloproteinases. Current opinion in genetics & development. 2000;10:120–127. doi: 10.1016/s0959-437x(99)00043-x. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature cell biology. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Radisky D. Putting Tumours in Context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M, Fabunmi RP, Baker AH, Newby AC. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-kappa B. Febs Letters. 1998;435:29–34. doi: 10.1016/s0014-5793(98)01034-5. [DOI] [PubMed] [Google Scholar]
- Borish LC, Steinke JW. 2. Cytokines and chemokines. The Journal of allergy and clinical immunology. 2003;111:S460–75. doi: 10.1067/mai.2003.108. [DOI] [PubMed] [Google Scholar]
- Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: The place to be. Cancer Res. Cancer Research. 2011;71:634–639. doi: 10.1158/0008-5472.CAN-10-3220. [DOI] [PubMed] [Google Scholar]
- Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. Journal of Pathology. 2003;200:423–428. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C. PDGF-A Signaling Is a Critical Event in Lung Alveolar Myofibroblast Development and Alveogenesis. Cell. 1996;85:863–873. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor beta receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz- alpha -anthracene. Cancer research. 1997;57:5564–5570. [PubMed] [Google Scholar]
- Boucher Y, Leunig M, Jain RK. Tumor angiogenesis and interstitial hypertension. Cancer research. 1996;56:4264–4266. [PubMed] [Google Scholar]
- Brauchle M, Angermeyer K, Hubner G, Werner S. LARGE INDUCTION OF KERATINOCYTE GROWTH-FACTOR EXPRESSION BY SERUM GROWTH-FACTORS AND PRO-INFLAMMATORY CYTOKINES IN CULTURED FIBROBLASTS. Oncogene. 1994;9:3199–3204. [PubMed] [Google Scholar]
- Bresch D, Colin T, Grenier E, Ribba B, Saut O. COMPUTATIONAL MODELING OF SOLID TUMOR GROWTH: THE AVASCULAR STAGE. Siam Journal on Scientific Computing. 2010;32:2321–2344. doi: 10.1137/070708895. [DOI] [Google Scholar]
- Breward CJW, Byrne HM, Lewis CE. The role of cell-cell interactions in a two-phase model for avascular tumour growth. Journal of Mathematical Biology. 2002;45:125–152. doi: 10.1007/s002850200149. [DOI] [PubMed] [Google Scholar]
- Breward CJW, Byrne HM, Lewis CE. A multiphase model describing vascular tumour growth. Bulletin of Mathematical Biology. 2003;65:609–640. doi: 10.1016/s0092-8240(03)00027-2. [DOI] [PubMed] [Google Scholar]
- Brown JM. Exploiting the hypoxic cancer cell: mechanisms and therapeutic strategies. Molecular Medicine Today. 2000;6:157–162. doi: 10.1016/s1357-4310(00)01677-4. [DOI] [PubMed] [Google Scholar]
- Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, Lewis CE. Hypoxia-induced gene expression in human macrophages – Implications for ischemic tissues and hypoxia-regulated gene therapy. American Journal of Pathology. 2003;163:1233–1243. doi: 10.1016/s0002-9440(10)63483-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne H, Preziosi L. Modelling solid tumour growth using the theory of mixtures. Mathematical Medicine and Biology-a Journal of the Ima. 2003;20:341–366. doi: 10.1093/imammb20.4.341. [DOI] [PubMed] [Google Scholar]
- Byrne HM. Dissecting cancer through mathematics: from the cell to the animal model. Nat Rev Cancer. 2010;10:221–230. doi: 10.1038/nrc2808. , doi: http://www.nature.com/nrc/journal/v10/n3/suppinfo/nrc2808_S1.html. [DOI] [PubMed] [Google Scholar]
- Byrne HM, King JR, McElwain DLS, Preziosi L. A two-phase model of solid tumour growth. Applied Mathematics Letters. 2003;16:567–573. doi: 10.1016/s0893-9659(03)00038-7. [DOI] [Google Scholar]
- Byrne HM, Alarcon T, Owen MR, Webb SD, Maini PK. Modelling aspects of cancer dynamics: a review. Philosophical Transactions of the Royal Society a-Mathematical Physical and Engineering Sciences. 2006;364:1563–1578. doi: 10.1098/rsta.2006.1786. [DOI] [PubMed] [Google Scholar]
- Cahn JW, Hilliard JE. Free Energy of a Nonuniform System. I. Interfacial Free Energy. The Journal of Chemical Physics. 1958;28:258–267. [Google Scholar]
- Campbell LL, Polyak K. Breast tumor heterogeneity. Cell Cycle. 2007;6:2332–2338. doi: 10.4161/cc.6.19.4914. [DOI] [PubMed] [Google Scholar]
- Cao RH, Bjorndahl MA, Religa P, Clasper S, Garvin S, Galter D, Meister B, Ikomi F, Tritsaris K, Dissing S, Ohhashi T, Jackson DG, Cao YH. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004;6:333–345. doi: 10.1016/j.ccr.2004.08.034. [DOI] [PubMed] [Google Scholar]
- Cao Y. Opinion: Emerging Mechanisms of Tumour Lymphangiogenesis and Lymphatic Metastasis. Nature reviews. Cancer. 2005;5:735–43. doi: 10.1038/nrc1693. [DOI] [PubMed] [Google Scholar]
- Casciari JJ, Sotirchos SV, Sutherland RM. MATHEMATICAL-MODELING OF MICROENVIRONMENT AND GROWTH IN EMT6/RO MULTICELLULAR TUMOR SPHEROIDS. Cell Proliferation. 1992;25:1–22. doi: 10.1111/j.1365-2184.1992.tb01433.x. [DOI] [PubMed] [Google Scholar]
- Chang L, Kaipainen A, Folkman J. Lymphangiogenesis – New mechanisms. In: Rockson SG, editor. Lymphatic Continuum: Lymphatic Biology and Disease. Vol. 979. 2002. pp. 111–119. [DOI] [PubMed] [Google Scholar]
- Chang LK, Garcia-Cardena G, Farnebo F, Fannon M, Chen EJ, Butterfield C, Moses MA, Mulligan RC, Folkman J, Kaipainen A. Dose-dependent response of FGF-2 for lymphangiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11658–11663. doi: 10.1073/pnas.0404272101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplain MAJ. Avascular growth, angiogenesis and vascular growth in solid tumours: The mathematical modelling of the stages of tumour development. Mathematical and Computer Modelling. 1996;23:47–87. doi: 10.1016/0895-7177(96)00019-2. [DOI] [Google Scholar]
- Chaplain MAJ. Multiscale mathematical modelling in biology and medicine. IMA Journal of Applied Mathematics. 2011;76:371–388. doi: 10.1093/imamat/hxr025. [DOI] [Google Scholar]
- Chaplain MAJ, Stuart AM. A MODEL MECHANISM FOR THE CHEMOTACTIC RESPONSE OF ENDOTHELIAL-CELLS TO TUMOR ANGIOGENESIS FACTOR. Ima Journal of Mathematics Applied in Medicine and Biology. 1993;10:149–168. doi: 10.1093/imammb/10.3.149. [DOI] [PubMed] [Google Scholar]
- Chatelain Clément C, Ciarletta P, Ben Amar M. Morphological changes in early melanoma development: influence of nutrients, growth inhibitors and cell-adhesion mechanisms. Journal of Theoretical Biology. 2011;290:46–59. doi: 10.1016/j.jtbi.2011.08.029. [DOI] [PubMed] [Google Scholar]
- Chen Y, Lowengrub JS. Tumor growth in complex, evolving microenvironmental geometries: A diffuse domain approach. Journal of Theoretical Biology. 2014;361:14–30. doi: 10.1016/j.jtbi.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wise SM, Shenoy VB, Lowengrub JS. A stable scheme for a nonlinear, multiphase tumor growth model with an elastic membrane. International Journal for Numerical Methods in Biomedical Engineering. 2014;30:726–754. doi: 10.1002/cnm.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Transforming Growth Factor-beta Signaling-Deficient Fibroblasts Enhance Hepatocyte Growth Factor Signaling in Mammary Carcinoma Cells to Promote Scattering and Invasion. Molecular Cancer Research. 2008;6:1521–1533. doi: 10.1158/1541-7786.mcr-07-2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarijs R, Ruiter DJ, De Waal RMW. Pathophysiological implications of stroma pattern formation in uveal melanoma. Journal of Cellular Physiology. 2003;194:267–271. doi: 10.1002/jcp.10214. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Fuller M. Stem cells and cancer: Two faces of eve. Cell. 2006;124:1111–1115. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CHM, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer research. 2006;66:9339–44. doi: 10.1158/0008-5472.can-06-3126. [DOI] [PubMed] [Google Scholar]
- Cleaver O, Melton DA. Endothelial signaling during development. Nature Medicine. 2003;9:661–668. doi: 10.1038/nm0603-661. [DOI] [PubMed] [Google Scholar]
- Cliff WJ. Observations on Healing Tissue: A Combined Light and Electron Microscopic Investigation. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 1963;246:305–325. [Google Scholar]
- Cogswell DA, Carter WC. Thermodynamic phase-field model for microstructure with multiple components and phases: The possibility of metastable phases. Physical Review E. 2011;83 doi: 10.1103/PhysRevE.83.061602. [DOI] [PubMed] [Google Scholar]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-Related Inflammation, the Seventh Hallmark of Cancer: Links to Genetic Instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- Connolly DT. VASCULAR-PERMEABILITY FACTOR – A UNIQUE REGULATOR OF BLOOD-VESSEL FUNCTION. Journal of Cellular Biochemistry. 1991;47:219–223. doi: 10.1002/jcb.240470306. [DOI] [PubMed] [Google Scholar]
- Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Disease Models & Mechanisms. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristini V, Lowengrub J. Multiscale modeling of cancer: An integrated experimental and mathematical modeling approach. Cambridge University Press; Cambridge: 2010. [Google Scholar]
- Cristini V, Li X, Lowengrub J, Wise S. Nonlinear simulations of solid tumor growth using a mixture model: invasion and branching. Journal of Mathematical Biology. 2009;58:723–763. doi: 10.1007/s00285-008-0215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AK, Woodroofe MN. Chemokine modulation of matrix metalloproteinase and TIMP production in adult rat brain microglia and a human microglial cell line in vitro. Glia. 1999;28:183–189. doi: 10.1002/(sici)1098-1136(199912)28:3<183::aid-glia2>3.0.co;2-3. [PubMed] [Google Scholar]
- Currie MJ, Hanrahan V, Gunningham SP, Morrin HR, Frampton C, Han C, Robinson BA, Fox SB. Expression of vascular endothelial growth factor D is associated with hypoxia inducible factor (HIF-1 alpha) and the HIF-1 alpha target gene DEC1, but not lymph node metastasis in primary human breast carcinomas. Journal of Clinical Pathology. 2004;57:829–834. doi: 10.1136/jcp.2003.015644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cursiefen C, Chen L, Borges LP, Jackson D, Cao JT, Radziejewski C, D’Amore PA, Dana MR, Wiegand SJ, Streilein JW. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. Journal of Clinical Investigation. 2004;113:1040–1050. doi: 10.1172/jci200420465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czernin J, Phelps ME. Positron emission tomography scanning: Current and future applications. Annual Review of Medicine. 2002;53:89–112. doi: 10.1146/annurev.med.53.082901.104028. [DOI] [PubMed] [Google Scholar]
- D’Antonio G, Macklin P, Preziosi L. An agent-based model for elasto-plastic mechanical interactions between cells, basement membrane and extracellular matrix. Mathematical Biosciences and Engineering. 2013;10:75–101. doi: 10.3934/mbe.2013.10.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafni H, Israely T, Bhujwalla ZM, Benjamin LE, Neeman M. Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: Magnetic resonance imaging, confocal microscopy, and histological tracking of triple-labeled albumin. Cancer research. 2002;62:6731–6739. [PubMed] [Google Scholar]
- Dalal BI, Keown PA, Greenberg AH. IMMUNOCYTOCHEMICAL LOCALIZATION OF SECRETED TRANSFORMING GROWTH-FACTOR-BETA-1 TO THE ADVANCING EDGES OF PRIMARY TUMORS AND TO LYMPH-NODE METASTASES OF HUMAN MAMMARY-CARCINOMA. American Journal of Pathology. 1993;143:381–389. [PMC free article] [PubMed] [Google Scholar]
- Daluvoy SV, Avraham T, Kasten J, Mehrara BJ. Hypoxia induces upregulation of VEGF-C and lymphatic differentiation via Hif-1a pathway. Journal of the American College of Surgeons. 2009;209:S72–S72. [Google Scholar]
- Daly C, Pasnikowski E, Burova E, Wong V, Aldrich TH, Griffiths J, Ioffe E, Daly TJ, Fandl JP, Papadopoulos N, McDonald DM, Thurston G, Yancopoulos GD, Rudge JS. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15491–15496. doi: 10.1073/pnas.0607538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of Angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–1169. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Coussens LM. The Inflammatory Tumor Microenvironment and Its Impact on Cancer Development. Contributions to microbiology. 2006;13:118–37. doi: 10.1159/000092969. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nature reviews. Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. Journal of Pathology. 2003;200:429–447. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. International Journal of Cancer. 2008;123:2229–2238. doi: 10.1002/ijc.23925. [DOI] [PubMed] [Google Scholar]
- Deisboeck TS, Wang ZH, Macklin P, Cristini V. Multiscale Cancer Modeling. In: Yarmush ML, et al., editors. Annual Review of Biomedical Engineering. Vol. 13. Annual Reviews, Palo Alto; 2011. pp. 127–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. TRANSFORMING GROWTH-FACTOR-BETA-1 INDUCES ALPHA-SMOOTH MUSCLE ACTIN EXPRESSION IN GRANULATION-TISSUE MYOFIBROBLASTS AND IN QUIESCENT AND GROWING CULTURED FIBROBLASTS. Journal of Cell Biology. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. THE FMS-LIKE TYROSINE KINASE, A RECEPTOR FOR VASCULAR ENDOTHELIAL GROWTH-FACTOR. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- Dong JY, Grunstein J, Tejada M, Peale F, Frantz G, Liang WC, Bai W, Yu LL, Kowalski J, Liang XH, Fuh G, Gerber HP, Ferrara N. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. Embo Journal. 2004;23:2800–2810. doi: 10.1038/sj.emboj.7600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G, Al-Hajj M, Abdallah WA, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Proliferation. 2003;36:59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nature Reviews Molecular Cell Biology. 2011;12:308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugina V, Fontao L, Chaponnier C, Vasiliev J, Gabbiani G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. Journal of Cell Science. 2001;114:3285–3296. doi: 10.1242/jcs.114.18.3285. [DOI] [PubMed] [Google Scholar]
- Dvorak HF, Nagy JA, Dvorak JT, Dvorak AM. IDENTIFICATION AND CHARACTERIZATION OF THE BLOOD-VESSELS OF SOLID TUMORS THAT ARE LEAKY TO CIRCULATING MACROMOLECULES. American Journal of Pathology. 1988;133:95–109. [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF, Sioussat TM, Brown LF, Berse B, Nagy JA, Sotrel A, Manseau EJ, Vandewater L, Senger DR. DISTRIBUTION OF VASCULAR-PERMEABILITY FACTOR (VASCULAR ENDOTHELIAL GROWTH-FACTOR) IN TUMORS – CONCENTRATION IN TUMOR BLOOD-VESSELS. Journal of Experimental Medicine. 1991;174:1275–1278. doi: 10.1084/jem.174.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H. Nutrition Needs of Mammalian Cells in Tissue Culture. Science. 1955;122:501–504. doi: 10.1126/science.122.3168.501. [DOI] [PubMed] [Google Scholar]
- Edelman LB, Eddy JA, Price ND. In silico models of cancer. Wiley Interdisciplinary Reviews-Systems Biology and Medicine. 2010;2:438–459. doi: 10.1002/wsbm.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Reviews Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Current Opinion in Cell Biology. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: The macrophage connection. Journal of Leukocyte Biology. 1998;64:275–290. doi: 10.1002/jlb.64.3.275. [DOI] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Enholm B, Paavonen K, Ristimaki A, Kumar V, Gunji Y, Klefstrom J, Kivinen L, Laiho M, Olofsson B, Joukov V, Eriksson U, Alitalo K. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14:2475–2483. doi: 10.1038/sj.onc.1201090. [DOI] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappa B-Dependent Manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Escher J, Matioc AV. Analysis of a two-phase model describing the growth of solid tumors. European Journal of Applied Mathematics. 2013;24:25–48. doi: 10.1017/s0956792512000290. [DOI] [Google Scholar]
- Fang JM, Shing Y, Wiederschain D, Yan L, Butterfield C, Jackson G, Harper J, Tamvakopoulos G, Moses MA. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3884–3889. doi: 10.1073/pnas.97.8.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Simoes SdM, Roeper JC, Eaton S, Zallen JA. Myosin II Dynamics Are Regulated by Tension in Intercalating Cells. Developmental Cell. 2009;17:736–743. doi: 10.1016/j.devcel.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Hart IR. BIOLOGICAL DIVERSITY IN METASTATIC NEOPLASMS – ORIGINS AND IMPLICATIONS. Science. 1982;217:998–1003. doi: 10.1126/science.7112116. [DOI] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- Folkman J. The Intestine as an Organ Culture. In: Burdette WJ, editor. Carcinoma of the Colon and Antecedent Epithelium. CC Thomas, Springfield; Illinois: 1970. pp. 113–127. [Google Scholar]
- Folkman J. ANTI-ANGIOGENESIS – NEW CONCEPT FOR THERAPY OF SOLID TUMORS. Annals of surgery. 1972;175:409. doi: 10.1097/00000658-197203000-00014. -&, doi:10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. TUMOR ANGIOGENESIS FACTOR. Cancer research. 1974;34:2109–2113. [PubMed] [Google Scholar]
- Folkman J. VASCULARIZATION OF TUMORS. Scientific American. 1976;234:58–73. doi: 10.1038/scientificamerican0576-58. [DOI] [PubMed] [Google Scholar]
- Folkman J, Gimbrone MA. Perfusion of the Thyroid. In: Diczfalusy E, editor. Karolinska Symposia on Research Methods in Reproduction Endocrinology, 4th Symposium: Perfusion Techniques, Stockholm. 1971. pp. 237–248. [Google Scholar]
- Folkman J, Shing Y. ANGIOGENESIS. Journal of Biological Chemistry. 1992;267:10931–10934. [PubMed] [Google Scholar]
- Folkman J, Kalluri R. Chapter 11: Tumor Angiogenesis. In: Kufe D, editor. Holland-Frei Cancer Medicine. BC Decker; Hamilton, ON: 2003. [Google Scholar]
- Folkman J, Long DM, Jr, Becker FF. Growth and Metastasis of Tumor in Organ Culture. Cancer. 1963;16:453–67. doi: 10.1002/1097-0142(196304)16:4<453::aid-cncr2820160407>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Folkman J, Cole P, Zimmerman S. Tumor behavior in isolated perfused organs: in vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Annals of surgery. 1966;164:491–502. doi: 10.1097/00000658-196609000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Bach M, Rowe JW, Davidoff F, Lambert P, Hirsch C, Goldberg A, Hiatt HH, Glass J, Henshaw E. TUMOR ANGIOGENESIS – THERAPEUTIC IMPLICATIONS. New England Journal of Medicine. 1971;285:1182. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Frieboes HB, Chaplain MAJ, Thompson AM, Bearer EL, Lowengrub JS. Physical oncology: a bench-to-bedside quantitative and predictive approach. Cancer Research. 2011;71:298–302. doi: 10.1158/0008-5472.CAN-10-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieboes HB, Jin F, Chuang YL, Wise SM, Lowengrub JS, Cristini V. Three-dimensional multispecies nonlinear tumor growth-II: Tumor invasion and angiogenesis. Journal of Theoretical Biology. 2010;264:1254–1278. doi: 10.1016/j.jtbi.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieboes HB, Lowengrub JS, Wise S, Zheng X, Macklin P, Bearer EL, Cristini V. Computer simulation of glioma growth and morphology. NeuroImage. 2007;37(Supplement 1):S59–S70. doi: 10.1016/j.neuroimage.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieboes HB, Smith BR, Chuang Y-L, Ito K, Roettgers AM, Gambhir SS, Cristini V. An Integrated Computational/Experimental Model of Lymphoma Growth. Plos Computational Biology. 2013;9 doi: 10.1371/journal.pcbi.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T, Yamaguchi Y, Saitoh Y, Hiroshima K, Ohwada H. BLOOD AND LYMPHATIC VESSEL INVASION AS PROGNOSTIC FACTORS FOR PATIENTS WITH PRIMARY RESECTED NONSMALL CELL-CARCINOMA OF THE LUNG WITH INTRAPULMONARY METASTASES. Cancer. 1995;76:2464–2470. doi: 10.1002/1097-0142(19951215)76:12<2464::aid-cncr2820761210>3.0.co;2-u. doi: 10.1002/1097-0142(19951215)76:12<2464::aid-cncr2820761210>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Fukumura D, Jain RK. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. Journal of Cellular Biochemistry. 2007;101:937–949. doi: 10.1002/jcb.21187. [DOI] [PubMed] [Google Scholar]
- Galle J, Preziosi L, Tosin A. Contact inhibition of growth described using a multiphase model and an individual cell based model. Applied Mathematics Letters. 2009;22:1483–1490. doi: 10.1016/j.aml.2008.06.051. [DOI] [Google Scholar]
- Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nature Reviews Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- Garcke H. On a Cahn-Hilliard model for phase separation with elastic misfit. Annales De L Institut Henri Poincare-Analyse Non Lineaire. 2005;22:165–185. doi: 10.1016/j.anihpc.2004.07.001. [DOI] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature Reviews Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA, Keely PJ. Filamin A-beta 1 Integrin Complex Tunes Epithelial Cell Response to Matrix Tension. Molecular Biology of the Cell. 2009;20:3224–3238. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerisch A, Chaplain MAJ. Mathematical modelling of cancer cell invasion of tissue: Local and non-local models and the effect of adhesion. Journal of Theoretical Biology. 2008;250:684–704. doi: 10.1016/j.jtbi.2007.10.026. [DOI] [PubMed] [Google Scholar]
- Gilbert PM, Havenstrite KL, Magnusson KEG, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM. Substrate Elasticity Regulates Skeletal Muscle Stem Cell Self-Renewal in Culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giverso C, Preziosi L. Modelling the compression and reorganization of cell aggregates. Math Med Biol. 2012;29:181–204. doi: 10.1093/imammb/dqr008. [DOI] [PubMed] [Google Scholar]
- Giverso C, Scianna M, Grillo A. Growing avascular tumours as elasto-plastic bodies by the theory of evolving natural configurations. Mechanics Research Communications. 2015;68:31–39. [Google Scholar]
- Gold LI. The role for transforming growth factor-beta (TGF-beta) in human cancer. Critical Reviews in Oncogenesis. 1999;10:303–360. [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LVF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, DiTullio RA, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-b receptor exhibit impaired mammary development and enhanced mammary tumor formation. American Journal of Pathology. 2003;163:1539–1549. doi: 10.1016/s0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S, Sahai E, Wyckoff JB, Cammer N, Cox D, Pixley FJ, Stanley ER, Segall JE, Condeelis JS. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer research. 2005;65:5278–5283. doi: 10.1158/0008-5472.can-04-1853. [DOI] [PubMed] [Google Scholar]
- Grady WM, Markowitz S. Genomic instability and colorectal cancer. Current Opinion in Gastroenterology. 2000;16:62–67. doi: 10.1097/00001574-200001000-00012. [DOI] [PubMed] [Google Scholar]
- Graziano L, Preziosi L. Mechanics in Tumor Growth. In: Mollica F, et al., editors. Modeling of Biological Materials. Birkhäuser Basel. 2007. pp. 263–321. [Google Scholar]
- Greenblatt M, Shubi P. Tumor angiogenesis: transfilter diffusion studies in the hamster by the transparent chamber technique. Journal of the National Cancer Institute. 1968;41:111–24. [PubMed] [Google Scholar]
- Gress TM, Mullerpillasch F, Lerch MM, Friess H, Buchler M, Adler G. EXPRESSION AND IN-SITU LOCALIZATION OF GENES-CODING FOR EXTRACELLULAR-MATRIX PROTEINS AND EXTRACELLULAR-MATRIX DEGRADING PROTEASES IN PANCREATIC-CANCER. International Journal of Cancer. 1995;62:407–413. doi: 10.1002/ijc.2910620409. [DOI] [PubMed] [Google Scholar]
- Grimshaw MJ, Wilson JL, Balkwill FR. Endothelin-2 is a macrophage chemoattractant: implications for macrophage distribution in tumors. European Journal of Immunology. 2002;32:2393–2400. doi: 10.1002/1521-4141(200209)32:9<2393::AID-IMMU2393>3.0.CO;2-4. doi: 10.1002/1521-4141(200209)32:9<2393::aid-immu2393>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Zhu MF, Carlson MA, Abrams JM. Release of mechanical tension triggers apoptosis of human fibroblasts in a model of regressing granulation tissue. Experimental Cell Research. 1999;248:608–619. doi: 10.1006/excr.1999.4440. [DOI] [PubMed] [Google Scholar]
- Gullick WJ. PREVALENCE OF ABERRANT EXPRESSION OF THE EPIDERMAL GROWTH-FACTOR RECEPTOR IN HUMAN CANCERS. British Medical Bulletin. 1991;47:87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Fillmore CM, Jiang GZ, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harrell MI, Iritani BM, Ruddell A. Tumor-induced sentinel lymph node lymphangiogenesis and increased lymph flow precede melanoma metastasis. American Journal of Pathology. 2007;170:774–786. doi: 10.2353/ajpath.2007.060761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzikirou H, Deutsch A, Schaller C, Simon M, Swanson K. Mathematical modelling of glioblastoma tumour development: A review. Mathematical Models & Methods in Applied Sciences. 2005;15:1779–1794. doi: 10.1142/s0218202505000960. [DOI] [Google Scholar]
- Hawkins-Daarud A, van der Zee KG, Oden JT. Numerical simulation of a thermodynamically consistent four-species tumor growth model. International Journal for Numerical Methods in Biomedical Engineering. 2012;28:3–24. doi: 10.1002/cnm.1467. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – An obstacle in cancer therapy. Nature Reviews Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- Helotera H, Alitalo K. The VEGF Family, the Inside Story. Cell. 2007;130:591–592. doi: 10.1016/j.cell.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Heng HHQ, Stevens JB, Liu G, Bremer SW, Ye KJ, Reddy PV, Wu GS, Wang YA, Tainsky MA, Ye CJ. Stochastic cancer progression driven by non-clonal chromosome aberrations. Journal of Cellular Physiology. 2006;208:461–472. doi: 10.1002/jcp.20685. [DOI] [PubMed] [Google Scholar]
- Heppner GH. TUMOR HETEROGENEITY. Cancer research. 1984;44:2259–2265. [PubMed] [Google Scholar]
- Hinz B. Formation and function of the myofibroblast during tissue repair. Journal of Investigative Dermatology. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- Hinz B, Mastrangelo D, Iselin CE, Chaponnier C, Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. American Journal of Pathology. 2001;159:1009–1020. doi: 10.1016/s0002-9440(10)61776-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The Myofibroblast: One Function, Multiple Origins. The American journal of pathology. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbian G. Recent Developments in Myofibroblast Biology Paradigms for Connective Tissue Remodeling. American Journal of Pathology. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. Journal of Experimental Medicine. 2005;201:1089–1099. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- Holbro T, Hynes NE. ErbB receptors: Directing key signaling networks throughout life. Annual Review of Pharmacology and Toxicology. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- Holmgren L, Oreilly MS, Folkman J. DORMANCY OF MICROMETASTASES – BALANCED PROLIFERATION AND APOPTOSIS IN THE PRESENCE OF ANGIOGENESIS SUPPRESSION. Nature Medicine. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- Hong YK, Lange-Asschenfeldt B, Velasco P, Hirakawa S, Kunstfeld R, Brown LF, Bohlen P, Senger DR, Detmar M. VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha 1 beta 1 and alpha 2 beta 1 integrins. Faseb Journal. 2004;18:1111. doi: 10.1096/fj.03-1179fje. [DOI] [PubMed] [Google Scholar]
- Howe A, Aplin AE, Alahari SK, Juliano RL. Integrin signaling and cell growth control. Current Opinion in Cell Biology. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Current Opinion in Cell Biology. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Hynes RO. The Extracellular Matrix: Not Just Pretty Fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide AG, Baker NH, Warren SL. Vascularization of the Brown-Pearce Rabbit Epithelioma Transplant as Seen in the Transparent Ear Chamber. Am J Roentgenol. 1939;42:891–899. [Google Scholar]
- Ikushima H, Miyazono K. TGF beta signalling: a complex web in cancer progression. Nature Reviews Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- Isaka N, Padera TP, Hagendoorn J, Fukumura D, Jain RK. Peritumor lymphatics induced by vascular endothelial growth factor-C exhibit abnormal function. Cancer research. 2004;64:4400–4404. doi: 10.1158/0008-5472.can-04-0752. [DOI] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nature Medicine. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Jiang GS, Shu CW. Efficient implementation of weighted ENO schemes. Journal of Computational Physics. 1996;126:202–228. [Google Scholar]
- Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Current Opinion in Cell Biology. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nature Reviews Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature Reviews Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nature reviews. Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nature Immunology. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–U4. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. Tumor angiogenesis: past, present and the near future. Carcinogenesis. 2000;21:505–515. doi: 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- Kim BS, Chen J, Weinstein T, Noiri E, Goligorsky MS. VEGF expression in hypoxia and hyperglycemia: reciprocal effect on branching angiogenesis in epithelial-endothelial co-cultures. Journal of the American Society of Nephrology : JASN. 2002;13:2027–36. doi: 10.1097/01.asn.0000024436.00520.d8. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene. 2000;19:4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- Kim J, Lowengrub J. Phase field modeling and simulation of three-phase flows. Interfaces and Free Boundaries. 2005;7:435–466. [Google Scholar]
- Kim U, Park HC, Choi KH. DIFFERENTIAL PERMEABILITY OF LYMPHATIC AND BLOOD-VESSELS IN DETERMINING THE ROUTE OF METASTASIS AS DEMONSTRATED BY INDIRECT LYMPHOGRAPHY. Clinical & Experimental Metastasis. 1988;6:291–299. doi: 10.1007/bf01753576. [DOI] [PubMed] [Google Scholar]
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes & Development. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klika V. A Guide through Available Mixture Theories for Applications in Critical Reviews in Solid State and Material Science. Critical Reviews in Solid State and Materials Sciences. 2014;39:154–174. doi: 10.1080/10408436.2012.719132. [DOI] [Google Scholar]
- Koelsch V, Seher T, Fernandez-Ballester GJ, Serrano L, Leptin M. Control of Drosophila gastrulation by apical localization of adherens junctions and RhoGEF2. Science. 2007;315:384–386. doi: 10.1126/science.1134833. [DOI] [PubMed] [Google Scholar]
- Koong AC, Denko NC, Hudson KM, Schindler C, Swiersz L, Koch C, Evans S, Ibrahim H, Le QT, Terris DJ, Giaccia AJ. Candidate genes for the hypoxic tumor phenotype. Cancer research. 2000;60:883–887. [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer research. 2006;66:632–637. doi: 10.1158/0008-5472.can-05-3260. [DOI] [PubMed] [Google Scholar]
- Kreeger PK, Lauffenburger DA. Cancer systems biology: a network modeling perspective. Carcinogenesis. 2010;31:2–8. doi: 10.1093/carcin/bgp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Kumar VBS, Viji RI, Kiran MS, Sudhakaran PR. Endothelial cell response to lactate: Implication of PAR modification of VEGF. Journal of Cellular Physiology. 2007;211:477–485. doi: 10.1002/jcp.20955. [DOI] [PubMed] [Google Scholar]
- Kuusela E, Alt W. Continuum model of cell adhesion and migration. Journal of Mathematical Biology. 2009;58:135–161. doi: 10.1007/s00285-008-0179-x. [DOI] [PubMed] [Google Scholar]
- Laakkonen P, Waltari M, Holopainen T, Takahashi T, Pytowski B, Steiner P, Hicklin D, Persaud K, Tonra JR, Witte L, Alitalo K. Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer research. 2007;67:593–599. doi: 10.1158/0008-5472.can-06-3567. [DOI] [PubMed] [Google Scholar]
- Lal A, Peters H, St Croix B, Haroon ZA, Dewhirst MW, Strausberg RL, Kaanders J, van der Kogel AJ, Riggins GJ. Transcriptional response to hypoxia in human tumors. Journal of the National Cancer Institute. 2001;93:1337–1343. doi: 10.1093/jnci/93.17.1337. [DOI] [PubMed] [Google Scholar]
- Lamagna C, Aurrand-Lions M, Imhof BA. Dual role of macrophages in tumor growth and angiogenesis. Journal of Leukocyte Biology. 2006;80:705–13. doi: 10.1189/jlb.1105656. [DOI] [PubMed] [Google Scholar]
- Leak LV. PERMEABILITY OF LYMPHATIC CAPILLARIES. Journal of Cell Biology. 1971;50:300. doi: 10.1083/jcb.50.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. Genetic Instability in Colorectal Cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- Leo PH, Lowengrub JS, Jou HJ. A diffuse interface model for microstructural evolution in elastically stressed solids. Acta Materialia. 1998;46:2113–2130. doi: 10.1016/s1359-6454(97)00377-7. [DOI] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine HA, Sleeman BD, Nilsen-Hamilton M. A mathematical model for the roles of pericytes and macrophages in the initiation of angiogenesis. I. The role of protease inhibitors in preventing angiogenesis. Mathematical Biosciences. 2000;168:77–115. doi: 10.1016/s0025-5564(00)00034-1. [DOI] [PubMed] [Google Scholar]
- Levine HA, Sleeman BD, Nilsen-Hamilton M. Mathematical modeling of the onset of capillary formation initiating angiogenesis. Journal of Mathematical Biology. 2001a;42:195–238. doi: 10.1007/s002850000037. [DOI] [PubMed] [Google Scholar]
- Levine HA, Pamuk S, Sleeman BD, Nilsen-Hamilton M. Mathematical Modeling of Capillary Formation and Development in Tumor Angiogenesis: Penetration into the Stroma. Bulletin of Mathematical Biology. 2001b;63:801–863. doi: 10.1006/bulm.2001.0240. [DOI] [PubMed] [Google Scholar]
- Lewis C, Murdoch C. Macrophage Responses to Hypoxia: Implications for Tumor Progression and Anti-Cancer Therapies. The American journal of pathology. 2005;167:627–635. doi: 10.1016/s0002-9440(10)62038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer research. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- Li VW, Folkerth RD, Watanabe H, Yu CN, Rupnick M, Barnes P, Scott RM, Black PM, Sallan SE, Folkman J. MICROVESSEL COUNT AND CEREBROSPINAL-FLUID BASIC FIBROBLAST GROWTH-FACTOR IN CHILDREN WITH BRAIN-TUMORS. Lancet. 1994;344:82–86. doi: 10.1016/s0140-6736(94)91280-7. [DOI] [PubMed] [Google Scholar]
- Lindahl P, Betsholtz C. Not all myofibroblasts are alike: revisiting the role of PDGF-A and PDGF-B using PDGF-targeted mice. Current Opinion in Nephrology and Hypertension. 1998;7:21–26. doi: 10.1097/00041552-199801000-00004. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Kleinerman J, Saidel GM. SIGNIFICANCE OF HEMATOGENOUS TUMOR-CELL CLUMPS IN METASTATIC PROCESS. Cancer research. 1976;36:889–894. [PubMed] [Google Scholar]
- Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. METASTATIC POTENTIAL CORRELATES WITH ENZYMATIC DEGRADATION OF BASEMENT-MEMBRANE COLLAGEN. Nature. 1980;284:67–68. doi: 10.1038/284067a0. [DOI] [PubMed] [Google Scholar]
- Liu M, Xu JJ, Deng H. Tangled Fibroblasts in Tumor-Stroma interactions. International Journal of Cancer. 2011;129:1795–1805. doi: 10.1002/ijc.26116. [DOI] [PubMed] [Google Scholar]
- Liu XD, Osher S, Chan T. WEIGHTED ESSENTIALLY NONOSCILLATORY SCHEMES. Journal of Computational Physics. 1994;115:200–212. doi: 10.1006/jcph.1994.1187. [DOI] [Google Scholar]
- Liu ZG, Hsu HL, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappa B activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annual review of cell and developmental biology. 2007;23:675–99. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- Locati M, Deuschle U, Massardi ML, Martinez FO, Sironi M, Sozzani S, Bartfai T, Mantovani A. Analysis of the gene expression profile activated by the CC chemokine ligand 5/RANTES and by lipopolysaccharide in human monocytes. Journal of Immunology. 2002;168:3557–3562. doi: 10.4049/jimmunol.168.7.3557. [DOI] [PubMed] [Google Scholar]
- Lopez JI, Mouw JK, Weaver VM. Biomechanical regulation of cell orientation and fate. Oncogene. 2008;27:6981–6993. doi: 10.1038/onc.2008.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowengrub JS, Frieboes HB, Jin F, Chuang YL, Li X, Macklin P, Wise SM, Cristini V. Nonlinear modelling of cancer: bridging the gap between cells and tumours. Nonlinearity. 2010;23:R1–R91. doi: 10.1088/0951-7715/23/1/r01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. Journal of Cell Biology. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Takai K, Weaver VM, Werb Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harbor Perspectives in Biology. 2011;3 doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutolf MP, Gilbert PM, Blau HM. Designing materials to direct stem-cell fate. Nature. 2009;462:433–441. doi: 10.1038/nature08602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin P, McDougall S, Anderson ARA, Chaplain MAJ, Cristini V, Lowengrub J. Multiscale modelling and nonlinear simulation of vascular tumour growth. Journal of Mathematical Biology. 2009;58:765–798. doi: 10.1007/s00285-008-0216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj ASR, Saint-Geniez M, Maldonado AE, D’Amore PA. Vascular endothelial growth factor localization in the adult. American Journal of Pathology. 2006;168:639–648. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand S, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- Mantzaris NV, Webb S, Othmer HG. Mathematical modeling of tumor-induced angiogenesis. Journal of Mathematical Biology. 2004;49:111–187. doi: 10.1007/s00285-003-0262-2. [DOI] [PubMed] [Google Scholar]
- Marmé D, Fusenig N. Tumor Angiogenesis: Basic Mechanisms and Cancer Therapy. Springer; 2007. [Google Scholar]
- Marotta LLC, Polyak K. Cancer stem cells: a model in the making. Current opinion in genetics & development. 2009;19:44–50. doi: 10.1016/j.gde.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Martin P. Wound healing – Aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2008;295:G709–G717. doi: 10.1152/ajpgi.90356.2008. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF beta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melder RJ, Koenig GC, Witwer BP, Safabakhsh N, Munn LL, Jain RK. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nature Medicine. 1996;2:992–997. doi: 10.1038/nm0996-992. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Hiller K, Kelleher JK, Stephanopoulos G, Yang J, Iliopoulos O, Bell EL, Mattaini KR, Guarente L, Vander Heiden MG, Jewell CM, Johnson ZR, Irvine DJ. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michieli P, Mazzone M, Basilico C, Cavassa S, Sottile A, Naldini L, Comoglio PM. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 2004;6:61–73. doi: 10.1016/j.ccr.2004.05.032. [DOI] [PubMed] [Google Scholar]
- Michor F, Liphardt J, Ferrari M, Widom J. What does physics have to do with cancer? Nat Rev Cancer. 2011;11:657–670. doi: 10.1038/nrc3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer research. 2001;61:577–581. [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell DJ. Morphogenetic Cell Movements: Diversity from Modular Mechanical Properties. Science. 2008;322:1502–1505. doi: 10.1126/science.1164073. [DOI] [PubMed] [Google Scholar]
- Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Current Opinion in Cell Biology. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–U171. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumenthaler SM, D’Antonio G, Preziosi L, Macklin P. The Need for Integrative Computational Oncology: An Illustrated Example through MMP-Mediated Tissue Degradation. Front Oncol. 2013;3:194. doi: 10.3389/fonc.2013.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumprecht V, Detmar M. Lymphangiogenesis and cancer metastasis. Journal of Cellular and Molecular Medicine. 2009;13:1405–1416. doi: 10.1111/j.1582-4934.2009.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, Lawitts JA, Benjamin L, Tan XL, Manseau EJ, Dvorak AM, Dvorak HF. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. Journal of Experimental Medicine. 2002;196:1497–1506. doi: 10.1084/jem.20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Liyanarachchi S, Davuluri RV, Auer H, Martin EW, de la Chapelle A, Frankel WL. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23:7366–7377. doi: 10.1038/sj.onc.1208013. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Matsumoto K, Kiritoshi A, Tano Y. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: In vitro analysis of tumor-stromal interactions. Cancer research. 1997;57:3305–3313. [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic Instability – An Evolving Hallmark of Cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Watanabe H, Budson AE, Richie JP, Hayes DF, Folkman J. ELEVATED LEVELS OF AN ANGIOGENIC PEPTIDE, BASIC FIBROBLAST GROWTH-FACTOR, IN THE URINE OF PATIENTS WITH A WIDE SPECTRUM OF CANCERS. Journal of the National Cancer Institute. 1994;86:356–361. doi: 10.1093/jnci/86.5.356. [DOI] [PubMed] [Google Scholar]
- Nilsson I, Shibuya M, Wennstrom S. Differential activation of vascular genes by hypoxia in primary endothelial cells. Experimental Cell Research. 2004;299:476–485. doi: 10.1016/j.yexcr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Nister M, Libermann TA, Betsholtz C, Pettersson M, Claessonwelsh L, Heldin CH, Schlessinger J, Westermark B. EXPRESSION OF MESSENGER-RNAS FOR PLATELET-DERIVED GROWTH-FACTOR AND TRANSFORMING GROWTH FACTOR-ALPHA AND THEIR RECEPTORS IN HUMAN-MALIGNANT GLIOMA CELL-LINES. Cancer research. 1988;48:3910–3918. [PubMed] [Google Scholar]
- Norton WHJ, Ledin J, Grandel H, Neumann CJ. HSPG synthesis by zebrafish Ext2 and Extl3 is required for Fgf10 signalling during limb development. Development. 2005;132:4963–4973. doi: 10.1242/dev.02084. [DOI] [PubMed] [Google Scholar]
- Nowell PC. Clonal Evolution of Tumor-Cell Populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- Oden JT, Hawkins A, Prudhomme S. General Diffuse-Interface Theories and an Approach to Predictive Tumor Growth Modeling. Mathematical Models & Methods in Applied Sciences. 2010;20:477–517. doi: 10.1142/s0218202510004313. [DOI] [Google Scholar]
- Oden JT, Lima EABF, Almeida RC, Feng Y, Rylander MN, Fuentes D, Faghihi D, Rahman MM, DeWitt M, Gadde M, Zhou JC. Toward Predictive Multiscale Modeling of Vascular Tumor Growth: Computational and Experimental Oncology for Tumor Prediction. Archives of Computational Methods in Engineering. 2015 doi: 10.1007/s11831-015-9156-x. [DOI] [Google Scholar]
- Oezbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC. The Evolution of Extracellular Matrix. Molecular Biology of the Cell. 2010;21:4300–4305. doi: 10.1091/mbc.E10-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Osaki M, Araki K, Ishiguro K, Ito H, Ohgi S. Expression of hypoxia-inducible factor (HIF-1 alpha), VEGF-C and VEGF-D in non-invasive and invasive breast ductal carcinomas. Anticancer Research. 2005;25:3003–3009. [PubMed] [Google Scholar]
- Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. Journal of Clinical Investigation. 2007;117:50–59. doi: 10.1172/jci30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Orlidge A, Damore PA. INHIBITION OF CAPILLARY ENDOTHELIAL-CELL GROWTH BY PERICYTES AND SMOOTH-MUSCLE CELLS. Journal of Cell Biology. 1987;105:1455–1462. doi: 10.1083/jcb.105.3.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne JM, Walter A, Kershaw SK, Mirams GR, Fletcher AG, Pathmanathan P, Gavaghan D, Jensen OE, Maini PK, Byrne HM. A hybrid approach to multi-scale modelling of cancer. Philosophical Transactions: Mathematical, Physical and Engineering Sciences. 2010;368:5013–5028. doi: 10.1098/rsta.2010.0173. [DOI] [PubMed] [Google Scholar]
- Östman A, Augsten M. Cancer-associated fibroblasts and tumor growth – bystanders turning into key players. Current Opinion in Genetics & Development. 2009;19:67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Osullivan C, Lewis CE, Harris AL, McGee JO. SECRETION OF EPIDERMAL GROWTH-FACTOR BY MACROPHAGES ASSOCIATED WITH BREAST-CARCINOMA. Lancet. 1993;342:148–149. doi: 10.1016/0140-6736(93)91348-p. [DOI] [PubMed] [Google Scholar]
- Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ, Munn LL, Jain RK. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296:1883–1886. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- Paget S. THE DISTRIBUTION OF SECONDARY GROWTHS IN CANCER OF THE BREAST. The Lancet. 1889;133:571–573. doi: 10.1016/s0140-6736(00)49915-0. [DOI] [PubMed] [Google Scholar]
- Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of Pancreatic Ductal Adenocarcinoma. Clinical Gastroenterology and Hepatology. 2009;7:S44–S47. doi: 10.1016/j.cgh.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Patel MI, Nagl S. The role of model integration in complex systems modelling an example from cancer biology. Springer; Berlin; London: 2010. [Google Scholar]
- Pathak AP, Artemov D, Neeman M, Bhujwalla ZM. Lymph node metastasis in breast cancer xenografts is associated with increased regions of extravascular drain, lymphatic vessel area, and invasive phenotype. Cancer research. 2006;66:5151–5158. doi: 10.1158/0008-5472.can-05-1788. [DOI] [PubMed] [Google Scholar]
- Paweletz N, Knierim M. Tumor-Related Angiogenesis. Critical Reviews in Oncology/Hematology. 1989;9:197–242. doi: 10.1016/s1040-8428(89)80002-2. [DOI] [PubMed] [Google Scholar]
- Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Pepper MS, Skobe M. Lymphatic endothelium: morphological, molecular and functional properties. Journal of Cell Biology. 2003;163:209–213. doi: 10.1083/jcb.200308082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M. When Neighbourhood Matters: Tumour Microenvironment. Clinical & Translational Oncology. 2009;11:70–74. doi: 10.1007/s12094-009-0316-z. [DOI] [PubMed] [Google Scholar]
- Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clinical & translational oncology: official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2006;8:77–82. doi: 10.1007/s12094-006-0162-1. [DOI] [PubMed] [Google Scholar]
- Phillips PA, McCarroll JA, Park S, Wu MJ, Pirola R, Korsten M, Wilson JS, Apte MV. Rat pancreatic stellate cells secrete matrix metal loproteinases: implications for extracellular matrix turnover. Gut. 2003;52:275–282. doi: 10.1136/gut.52.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp A, Macdonald AL, Watt PW. Lactate – a signal coordinating cell and systemic function. Journal of Experimental Biology. 2005;208:4561–4575. doi: 10.1242/jeb.01961. [DOI] [PubMed] [Google Scholar]
- Pierce DF, Gorska AE, Chytil A, Meise KS, Page DL, Coffey RJ, Moses HL. MAMMARY-TUMOR SUPPRESSION BY TRANSFORMING GROWTH-FACTOR-BETA-1 TRANSGENE EXPRESSION. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:4254–4258. doi: 10.1073/pnas.92.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E. CELL ATTACHMENT ACTIVITY OF FIBRONECTIN CAN BE DUPLICATED BY SMALL SYNTHETIC FRAGMENTS OF THE MOLECULE. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- Pierschbacher MD, Hayman EG, Ruoslahti E. CELL ATTACHMENT TO FIBRONECTIN AND THE EXTRACELLULAR-MATRIX. In Vitro-Journal of the Tissue Culture Association. 1984;20:255–255. [Google Scholar]
- Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. Plos Medicine. 2008;5:123–138. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappa B functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Pouille P-A, Ahmadi P, Brunet A-C, Farge E. Mechanical Signals Trigger Myosin II Redistribution and Mesoderm Invagination in Drosophila Embryos. Science Signaling. 2009;2 doi: 10.1126/scisignal.2000098. [DOI] [PubMed] [Google Scholar]
- Preziosi L, Tosin A. Multiphase and Multiscale Trends in Cancer Modelling. Mathematical Modelling of Natural Phenomena. 2009a;4:1–11. doi: 10.1051/mmnp/20094301. [DOI] [Google Scholar]
- Preziosi L, Tosin A. Multiphase modelling of tumour growth and extracellular matrix interaction: mathematical tools and applications. Journal of Mathematical Biology. 2009b;58:4–5. doi: 10.1007/s00285-008-0218-7. [DOI] [PubMed] [Google Scholar]
- Preziosi L, Vitale G. A Multiphase Model of Tumor and Tissue Growth Including Cell Adhesion and Plastic Reorganization. Mathematical Models & Methods in Applied Sciences. 2011;21:1901–1932. doi: 10.1142/s0218202511005593. [DOI] [Google Scholar]
- Preziosi L, Ambrosi D, Verdier C. An elasto-visco-plastic model of cell aggregates. Journal of Theoretical Biology. 2010;262:35–47. doi: 10.1016/j.jtbi.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Proulx ST, Luciani P, Derzsi S, Rinderknecht M, Mumprecht V, Leroux JC, Detmar M. Quantitative Imaging of Lymphatic Function with Liposomal Indocyanine Green. Cancer research. 2010;70:7053–7062. doi: 10.1158/0008-5472.can-10-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psiuk-Maksymowicz K. Multiphase modelling of desmoplastic tumour growth. Journal of Theoretical Biology. 2013;329:52–63. doi: 10.1016/j.jtbi.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Quaranta V, Weaver AM, Cummings PT, Anderson ARA. Mathematical modeling of cancer: The future of prognosis and treatment. Clinica Chimica Acta. 2005;357:173–179. doi: 10.1016/j.cccn.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Experimental Cell Research. 2010;316:2713–2722. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- Reiss Y, Knedla A, Tal A, Schmidt MHH, Jugold M, Kiessling F, Burger AM, Wolburg H, Deutsch U, Plate KH. Switching of vascular phenotypes within a murine breast cancer model induced by angiopoietin-2. Journal of Pathology. 2009;217:571–580. doi: 10.1002/path.2484. [DOI] [PubMed] [Google Scholar]
- Rejniak KA, McCawley LJ. Current trends in mathematical modeling of tumor-microenvironment interactions: a survey of tools and applications. Experimental Biology and Medicine. 2010;235:411–423. doi: 10.1258/ebm.2009.009230. [DOI] [PubMed] [Google Scholar]
- Rejniak KA, Anderson ARA. Hybrid models of tumor growth. Wiley Interdisciplinary Reviews-Systems Biology and Medicine. 2011;3:115–125. doi: 10.1002/wsbm.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SC, Scott KA, Balkwill FR. Chemokine stimulation of monocyte matrix metalloproteinase-9 requires endogenous TNF-alpha. European Journal of Immunology. 2002;32:404–412. doi: 10.1002/1521-4141(200202)32:2<404::AID-IMMU404>3.0.CO;2-X. doi: 10.1002/1521-4141(200202)32:2<404::aid-immu404>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WAM, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nature cell biology. 2009;11:973–U142. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roose T, Chapman SJ, Maini PK. Mathematical models of avascular tumor growth. Siam Review. 2007;49:179–208. doi: 10.1137/s0036144504446291. [DOI] [Google Scholar]
- Rowlinson JS. TRANSLATION OF VANDERWAALS, JD THE THERMODYNAMIC THEORY OF CAPILLARITY UNDER THE HYPOTHESIS OF A CONTINUOUS VARIATION OF DENSITY. Journal of Statistical Physics. 1979;20:197–244. doi: 10.1007/bf01011513. [DOI] [Google Scholar]
- Rucker HK, Wynder HJ, Thomas WE. Cellular mechanisms of CNS pericytes. Brain Research Bulletin. 2000;51:363–369. doi: 10.1016/s0361-9230(99)00260-9. [DOI] [PubMed] [Google Scholar]
- Ruiter D, Bogenrieder T, Elder D, Herlyn M. Melanoma-stroma interactions: structural and functional aspects. Lancet Oncology. 2002;3:35–43. doi: 10.1016/s1470-2045(01)00620-9. [DOI] [PubMed] [Google Scholar]
- Saharinen P, Bry M, Alitalo K. How do angiopoietins Tie in with vascular endothelial growth factors? Current Opinion in Hematology. 2010;17:198–205. doi: 10.1097/MOH.0b013e3283386673. [DOI] [PubMed] [Google Scholar]
- Saharinen P, Tammela T, Karkkainen MJ, Alitalo K. Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. TRENDS IN IMMUNOLOGY. 2004;25:387–395. doi: 10.1016/j.it.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends in Molecular Medicine. 2011;17:347–362. doi: 10.1016/j.molmed.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Schlegel U, Moots PL, Rosenblum MK, Thaler HT, Furneaux HM. EXPRESSION OF TRANSFORMING GROWTH-FACTOR ALPHA IN HUMAN GLIOMAS. Oncogene. 1990;5:1839–1842. [PubMed] [Google Scholar]
- Schoefl GI. STUDIES ON INFLAMMATION. III. GROWING CAPILLARIES: THEIR STRUCTURE AND PERMEABILITY. Virchows Archiv fur pathologische Anatomie und Physiologie und fur klinische Medizin. 1963;337:97–141. doi: 10.1007/bf00963592. [DOI] [PubMed] [Google Scholar]
- Schoefl GI, Majno G. Regeneration of Blood Vessels in Wound Healing. Adv Biol Skin. 1964;5:173. [Google Scholar]
- Schoppmann SF, Fenzl A, Schindl M, Bachleitner-Hofmann T, Nagy K, Gnant M, Horvat R, Jakesz R, Birner P. Hypoxia inducible factor-1 alpha correlates with VEGF-C expression and lymphangiogenesis in breast cancer. Breast Cancer Research and Treatment. 2006;99:135–141. doi: 10.1007/s10549-006-9190-3. [DOI] [PubMed] [Google Scholar]
- Schulzeosthoff K, Risau W, Vollmer E, Sorg C. INSITU DETECTION OF BASIC FIBROBLAST GROWTH-FACTOR BY HIGHLY SPECIFIC ANTIBODIES. American Journal of Pathology. 1990;137:85–92. [PMC free article] [PubMed] [Google Scholar]
- Sciume G, Santagiuliana R, Ferrari M, Decuzzi P, Schrefler BA. A tumor growth model with deformable ECM. Physical Biology. 2014a;11 doi: 10.1088/1478-3975/11/6/065004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciume G, Gray WG, Hussain F, Ferrari M, Decuzzi P, Schrefler BA. Three phase flow dynamics in tumor growth. Computational Mechanics. 2014b;53:465–484. doi: 10.1007/s00466-013-0956-2. [DOI] [Google Scholar]
- Sciume G, Shelton S, Gray WG, Miller CT, Hussain F, Ferrari M, Decuzzi P, Schrefler BA. A multiphase model for three-dimensional tumor growth. New Journal of Physics. 2013;15 doi: 10.1088/1367-2630/15/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U, Cao YX, Xiao GT, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu YL, Fong A, Sun SC, Karin M. Activation by IKK alpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta 1. Journal of Cell Biology. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, Chilvers ER, Dransfield I, Donnelly SC, Strieter R, Haslett C. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nature Medicine. 1999;5:662–668. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- Shariat SF, Shalev M, Menesses-Diaz A, Kim IY, Kattan MW, Wheeler TM, Slawin KM. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. Journal of Clinical Oncology. 2001a;19:2856–2864. doi: 10.1200/JCO.2001.19.11.2856. [DOI] [PubMed] [Google Scholar]
- Shariat SF, Kim JH, Andrews B, Kattan MW, Wheeler TM, Kim IY, Lerner SP, Slawin KM. Preoperative plasma levels of Transforming growth factor beta(1) strongly predict clinical outcome in patients with bladder carcinoma. Cancer. 2001b;92:2985–2992. doi: 10.1002/1097-0142(20011215)92:12<2985::aid-cncr10175>3.0.co;2-5. doi: 10.1002/1097-0142(20011215)92:12<2985::aid-cncr10175>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. Journal of Biochemistry and Molecular Biology. 2006;39:469–478. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- Shing Y, Folkman J, Sullivan R, Butterfield C, Murray J, Klagsbrun M. HEPARIN AFFINITY – PURIFICATION OF A TUMOR-DERIVED CAPILLARY ENDOTHELIAL CELL-GROWTH FACTOR. Science. 1984;223:1296–1299. doi: 10.1126/science.6199844. [DOI] [PubMed] [Google Scholar]
- Sholley MM, Gimbrone MA, Cotran RS. CELLULAR MIGRATION AND REPLICATION IN ENDOTHELIAL REGENERATION – STUDY USING IRRADIATED ENDOTHELIAL CULTURES. Laboratory Investigation. 1977;36:18–25. [PubMed] [Google Scholar]
- Sholley MM, Ferguson GP, Seibel HR, Montour JL, Wilson JD. MECHANISMS OF NEOVASCULARIZATION – VASCULAR SPROUTING CAN OCCUR WITHOUT PROLIFERATION OF ENDOTHELIAL-CELLS. Laboratory Investigation. 1984;51:624–634. [PubMed] [Google Scholar]
- Simiantonaki N, Jayasinghe C, Michel-Schmidt R, Peters K, Hermanns MI, Kirkpatrick CJ. Hypoxia-induced epithelial VEGF-C/VEGFR-3 upregulation in carcinoma cell lines. International Journal of Oncology. 2008;32:585–592. [PubMed] [Google Scholar]
- Singh P, Carraher C, Schwarzbauer JE. Assembly of Fibronectin Extracellular Matrix. In: Schekman R, et al., editors. Annual Review of Cell and Developmental Biology. Vol. 26. Annual Reviews, Palo Alto; 2010. pp. 397–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirica AE, Campbell DJ, Dumur CI. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Current Opinion in Gastroenterology. 2011;27:276–284. doi: 10.1097/MOG.0b013e32834405c3. [DOI] [PubMed] [Google Scholar]
- Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunology Letters. 2009;123:97–102. doi: 10.1016/j.imlet.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Skobe M, Hamberg LM, Hawighorst T, Schirner M, Wolf GL, Alitalo K, Detmar M. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. American Journal of Pathology. 2001;159:893–903. doi: 10.1016/s0002-9440(10)61765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallbone K, Gatenby RA, Maini PK. Mathematical modelling of tumour acidity. J Theor Biol. 2008;255:106–12. doi: 10.1016/j.jtbi.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Smallbone K, Gavaghan DJ, Gatenby RA, Maini PK. The role of acidity in solid tumour growth and invasion. J Theor Biol. 2005;235:476–84. doi: 10.1016/j.jtbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol. 2007;244:703–13. doi: 10.1016/j.jtbi.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Solon J, Kaya-Copur A, Colombelli J, Brunner D. Pulsed Forces Timed by a Ratchet-like Mechanism Drive Directed Tissue Movement during Dorsal Closure. Cell. 2009;137:1331–1342. doi: 10.1016/j.cell.2009.03.050. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Todaro GJ. AUTOCRINE SECRETION AND MALIGNANT TRANSFORMATION OF CELLS. New England Journal of Medicine. 1980;303:878–880. doi: 10.1056/nejm198010093031511. [DOI] [PubMed] [Google Scholar]
- Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nature Reviews Cancer. 2002;2:573–583. doi: 10.1038/nrc863. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annual review of cell and developmental biology. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of Angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- Suri C, McClain J, Thurston G, McDonald DM, Zhou H, Oldmixon EH, Sato TN, Yancopoulos GD. Increased vascularization in mice overexpressing angiopoietin-1. Science. 1998;282:468–471. doi: 10.1126/science.282.5388.468. [DOI] [PubMed] [Google Scholar]
- Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nature reviews. Cancer. 2012;12:210–9. doi: 10.1038/nrc3186. [DOI] [PubMed] [Google Scholar]
- Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer and Metastasis Reviews. 2007;26:299–310. doi: 10.1007/s10555-007-9064-0. [DOI] [PubMed] [Google Scholar]
- Takeshita H, Yoshizaki T, Miller WE, Sato H, Furukawa M, Pagano JS, Raab-Traub N. Matrix metalloproteinase 9 Expression is induced by Epstein-Barr virus latent membrane protein 1 C-terminal activation regions 1 and 2. Journal of Virology. 1999;73:5548–5555. doi: 10.1128/jvi.73.7.5548-5555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammela T, Alitalo K. Lymphangiogenesis: Molecular Mechanisms and Future Promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- Tang P, Steck PA, Yung WKA. The autocrine loop of TGF-alpha/EGFR and brain tumors. Journal of Neuro-Oncology. 1997;35:303–314. doi: 10.1023/a:1005824802617. [DOI] [PubMed] [Google Scholar]
- Taubert H, Bhumke K, Bilkenroth U, Meye A, Kutz A, Bartel F, Lautenschlager C, Ulbrich EJ, Nass N, Holzhausen HR, Koelbl H, Lebrecht A. Detection of disseminated tumor cells in peripheral blood of patients with breast cancer: correlation to nodal status and occurrence of metastases. Gynecologic Oncology. 2004;92:256–261. doi: 10.1016/j.ygyno.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Terman BI, Doughervermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Bohlen P. IDENTIFICATION OF THE KDR TYROSINE KINASE AS A RECEPTOR FOR VASCULAR ENDOTHELIAL-CELL GROWTH-FACTOR. Biochemical and Biophysical Research Communications. 1992;187:1579–1586. doi: 10.1016/0006-291x(92)90483-2. [DOI] [PubMed] [Google Scholar]
- Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- Tlsty TD, Coussens LM. Tumor Stroma and Regulation of Cancer Development. Annual Review of Pathology: Mechanisms of Disease. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature Reviews Molecular Cell Biology. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, Richardson M, Rigaill G, Parrini MC, Lucchesi C, Bellanger D, Stern MH, Dubois T, Sastre-Garau X, Delattre O, Vincent-Salomon A, Mechta-Grigoriou F. Oxidative stress promotes myofibroblast differentiation and tumour spreading. Embo Molecular Medicine. 2010;2:211–230. doi: 10.1002/emmm.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracqui P. Biophysical models of tumour growth. Reports on Progress in Physics. 2009;72 doi: 10.1088/0034-4885/72/5/056701. [DOI] [Google Scholar]
- Trottenberg U, Oosterlee CW, Schüller A. Multigrid. Academic Press; San Diego: 2001. [Google Scholar]
- Tsushima H, Ito N, Tamura S, Matsuda Y, Inada M, Yabuuchi I, Imai Y, Nagashima R, Misawa H, Takeda H, Matsuzawa Y, Kawata S. Circulating transforming growth factor beta 1 as a predictor of liver metastasis after resection in colorectal cancer. Clinical Cancer Research. 2001;7:1258–1262. [PubMed] [Google Scholar]
- Tucker RF, Shipley GD, Moses HL, Holley RW. GROWTH INHIBITOR FROM BSC-1 CELLS CLOSELY RELATED TO PLATELET TYPE-BETA TRANSFORMING GROWTH-FACTOR. Science. 1984;226:705–707. doi: 10.1126/science.6093254. [DOI] [PubMed] [Google Scholar]
- Tzao C, Lee SC, Tung HJ, Hsu HS, Hsu WH, Sun GH, Yu CP, Jin JS, Cheng YL. Expression of hypoxia-inducible factor (HIF)-1 alpha and vascular endothelial growth factor (VEGF)-D as outcome predictors in resected esophageal squamous cell carcinoma. Disease Markers. 2008;25:141–148. doi: 10.1155/2008/468323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kempen L, Ruiter DJ, van Muijen GNP, Coussens LM. The tumor microenvironment: a critical determinant of neoplastic evolution. European Journal of Cell Biology. 2003;82:539–548. doi: 10.1078/0171-9335-00346. [DOI] [PubMed] [Google Scholar]
- VanAntwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappa B. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta 1 promotes the morphological and functional differentiation of the myofibroblast. Experimental Cell Research. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- Veikkola T, Alitalo K. Dual role of Ang2 in postnatal angiogenesis and lymphangiogenesis. Developmental Cell. 2002;3:302–304. doi: 10.1016/s1534-5807(02)00231-9. [DOI] [PubMed] [Google Scholar]
- Vineis P, Schatzkin A, Potter JD. Models of carcinogenesis: an overview. Carcinogenesis. 2010;31:1703–1709. doi: 10.1093/carcin/bgq087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, Apte MV. Pancreatic Stellate Cells: Partners in Crime with Pancreatic Cancer Cells. Cancer research. 2008;68:2085–2093. doi: 10.1158/0008-5472.can-07-2477. [DOI] [PubMed] [Google Scholar]
- Vuorio E, Decrombrugghe B. THE FAMILY OF COLLAGEN GENES. Annual Review of Biochemistry. 1990;59:837–872. doi: 10.1146/annurev.biochem.59.1.837. [DOI] [PubMed] [Google Scholar]
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, Mueller-Klieser W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer research. 2000;60:916–921. [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-kappa B. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang CY, Cusack JC, Liu R, Baldwin AS. Control of inducible chemoresistance: Enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappa B. Nature Medicine. 1999a;5:412–417. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- Wang WX, Abbruzzese JL, Evans DB, Chiao PJ. Overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma is regulated by constitutively activated RelA. Oncogene. 1999b;18:4554–4563. doi: 10.1038/sj.onc.1202833. [DOI] [PubMed] [Google Scholar]
- Warburg O. Verbesserte Methode zur Messung der Atmung und Glykolyse. Biochem Zeitschr. 1924;152:51–63. [Google Scholar]
- Warburg O, Posener K, Negelein E. Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschr. 1924;152:309–344. [Google Scholar]
- Warburg O, Wind F, Negelein E. THE METABOLISM OF TUMORS IN THE BODY. The Journal of General Physiology. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren BA. The ultrastructure of the microcirculation at the advancing edge of Walker 256 carcinoma. Microvascular research. 1970;2:443–53. doi: 10.1016/0026-2862(70)90037-3. [DOI] [PubMed] [Google Scholar]
- Warren BA, Shubik P. The growth of the blood supply to melanoma transplants in the hamster cheek pouch. Laboratory investigation; a journal of technical methods and pathology. 1966;15:464–78. [PubMed] [Google Scholar]
- Warren BA, Greenbla M, Komminen Vr. TUMOR ANGIOGENESIS – ULTRASTRUCTURE OF ENDOTHELIAL CELLS IN MITOSIS. British Journal of Experimental Pathology. 1972;53:216. [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JCR, Giancotti F, Werb Z, Bissell MJ. beta 4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss L, Ward PM. LYMPHOGENOUS AND HEMATOGENOUS METASTASIS OF LEWIS LUNG-CARCINOMA IN THE MOUSE. International Journal of Cancer. 1987;40:570–574. doi: 10.1002/ijc.2910400423. [DOI] [PubMed] [Google Scholar]
- Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiological Reviews. 2003;83:835–870. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Reviews Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Current Opinion in Cell Biology. 2010;22:212–217. doi: 10.1016/j.ceb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, Beard GL, Mundy CR, Naylor S. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics. 2004;83:1–8. doi: 10.1016/s0888-7543(03)00215-5. [DOI] [PubMed] [Google Scholar]
- Whiteside TL. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker CA, Bergeron KF, Whittle J, Brandhorst BP, Burke RD, Hynes RO. The echinoderm adhesome. Developmental Biology. 2006;300:252–266. doi: 10.1016/j.ydbio.2006.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig H. EVALUATION OF METHODOLOGIES FOR MEASUREMENT OF INTERSTITIAL FLUID PRESSURE (PI) – PHYSIOLOGICAL IMPLICATIONS OF RECENT PI DATA. Critical Reviews in Biomedical Engineering. 1990;18:27–54. [PubMed] [Google Scholar]
- Wise S, Kim J, Lowengrub J. Solving the regularized, strongly anisotropic Cahn-Hilliard equation by an adaptive nonlinear multigrid method. Journal of Computational Physics. 2007;226:414–446. [Google Scholar]
- Wise SM, Lowengrub JS, Cristini V. An adaptive multigrid algorithm for simulating solid tumor growth using mixture models. Mathematical and Computer Modelling. 2011;53:1–20. doi: 10.1016/j.mcm.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise SM, Lowengrub JS, Frieboes HB, Cristini V. Three-dimensional multispecies nonlinear tumor growth – I – Model and numerical method. Journal of Theoretical Biology. 2008;253:524–543. doi: 10.1016/j.jtbi.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witsch E, Sela M, Yarden Y. Roles for Growth Factors in Cancer Progression. Physiology. 2010;25:85–101. doi: 10.1152/physiol.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte MH, Witte CL. LYMPHATICS AND BLOOD-VESSELS, LYMPHANGIOGENESIS AND HEMANGIOGENESIS – FROM CELL BIOLOGY TO CLINICAL MEDICINE. Lymphology. 1987;20:257–266. [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. Journal of Cell Biology. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Frieboes HB, McDougall SR, Chaplain MAJ, Cristini V, Lowengrub J. The effect of interstitial pressure on tumor growth: Coupling with the blood and lymphatic vascular systems. Journal of Theoretical Biology. 2013;320:131–151. doi: 10.1016/j.jtbi.2012.11.031. doi: http://dx.doi.org/10.1016/j.jtbi.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Boudreau A, Bissell MJ. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer and Metastasis Reviews. 2009;28:167–176. doi: 10.1007/s10555-008-9178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yu Q. Angiopoietin-1, unlike angiopoietin-2, is incorporated into the extracellular matrix via its linker peptide region. Journal of Biological Chemistry. 2001;276:34990–34998. doi: 10.1074/jbc.M103661200. [DOI] [PubMed] [Google Scholar]
- Yan GC, Fukabori Y, Nikolaropoulos S, Wang F, McKeehan WL. HEPARIN-BINDING KERATINOCYTE GROWTH-FACTOR IS A CANDIDATE STROMAL TO EPITHELIAL-CELL ANDROMEDIN. Molecular Endocrinology. 1992;6:2123–2128. doi: 10.1210/me.6.12.2123. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- Yang AJM, Fleming PD, Gibbs JH. MOLECULAR THEORY OF SURFACE-TENSION. Journal of Chemical Physics. 1976;64:3732–3747. doi: 10.1063/1.432687. [DOI] [Google Scholar]
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Mosesl HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Wang XX, Contino G, Liesa M, Sahin E, Ying HQ, Bause A, Li YH, Stommel JM, Dell’Antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes & Development. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes & Development. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Woessner JF, McNeish JD, Stamenkovic I. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes & Development. 2002;16:307–323. doi: 10.1101/gad.925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung WKA, Xin Z, Steck PA, Hung MC. DIFFERENTIAL AMPLIFICATION OF THE TGF-ALPHA GENE IN HUMAN GLIOMAS. Cancer Communications. 1990;2:201–205. doi: 10.3727/095535490820874416. [DOI] [PubMed] [Google Scholar]
- Zheng X, Wise SM, Cristini V. Nonlinear simulation of tumor necrosis, neo-vascularization and tissue invasion via an adaptive finite-element/level-set method. Bulletin of Mathematical Biology. 2005;67:211–259. doi: 10.1016/j.bulm.2004.08.001. [DOI] [PubMed] [Google Scholar]