

Abstract
Regulatory and signaling networks coordinate the enormously complex interactions and processes that control cellular processes (such as metabolism and cell division), coordinate response to the environment, and carry out multiple cell decisions (such as development and quorum sensing). Regulatory network inference is the process of inferring these networks, traditionally from microarray data but increasingly incorporating other measurement types such as proteomics, ChIP-seq, metabolomics, and mass cytometry. We discuss existing techniques for network inference. We review in detail our pipeline, which consists of an initial biclustering step, designed to estimate co-regulated groups; a network inference step, designed to select and parameterize likely regulatory models for the control of the co-regulated groups from the biclustering step; and a visualization and analysis step, designed to find and communicate key features of the network. Learning biological networks from even the most complete data sets is challenging; we argue that integrating new data types into the inference pipeline produces networks of increased accuracy, validity, and biological relevance.
I. Introduction
Regulatory networks (RNs) can provide global models of complex biological phenomena, such as cell differentiation or disease progression. Knowledge of the underlying RNs has been key to understanding the functioning of diseases such as certain cancers (Carro et al., 2010; Suzuki et al., 2009), the creation of biofuels, and understanding the functioning of newly sequenced organisms (Bonneau et al., 2007). Although some cancers can be traced to a single causative mutation, many cancers are much more functionally complex, requiring simultaneous mutations in multiple genes that result in aberrations in the functions of multiple signaling pathways. Elucidation of the global RN allows for the study of disease-associated mutations in their global context. Biological regulation is a process that inherently occurs on multiple levels, such as transcription, translation, phosphorylation, and metabolism that span varying temporal and physical scales. Effective methods for RN inference must likewise integrate multiple types and scales of data – transcriptomic, proteomic, metabolomic – in order to most accurately recapitulate the complex underlying RNs. Our work, as described in this chapter, focuses on methods that can integrate multi-level data to elucidate an RN-scale view of complex biological processes.
The current explosion in the quantity, quality, and availability of high-throughput, genome-scale measurements provides powerful new tools to understand complex processes. Such measurements are now becoming available at different biological levels (e.g., transcriptomics and proteomics) for the same cell types or disease processes. At present, the most readily available genome-wide data type is micro-array data, capturing the “transcriptomic state” of the cell. We first discuss this data type in the context on network inference, then discuss other equally relevant data types. Microarray data provide genome-wide measurements of the abundance of mRNA for every transcript for which there is a probe on the microarray (typically thousands). Online compendia such as the Gene Expression Omnibus (Edgar et al., 2002) and Microbes online (http://www.microbesonline.org/) contain many thousands of such microarrays spanning many species and diseases, making this the most complete data available for the purpose of RN inference. Since these data are collected on the transcript level, they allow for the interrogation of only transcriptional effects. The mediators of these effects are transcription factors (TFs), which are proteins that bind the DNA and modulate the transcript abundance of their downstream targets, and environmental factors (EFs), which are environmental cues that modify the transcriptional program.
Inferring accurate, global RNs from such data remains a challenge for a multitude of reasons. The error (or variance in replicate measurements) in the measurement of transcript abundance is proportional to the expression level that is being measured (more expression means more error in the measurement) and many statistical methods do not properly account for this heteroskedasticity. Additionally, many of the data compendia used contain experiments from different laboratories and can thus contain batch effects (changes in expression that are mostly due to variations in experimental procedure from different labs). Even if a data compendium were to be normalized for batch effects and the other types of noise, the best data set would contain many more variables (genes) than data points that can be used for inferring regulatory interactions (conditions), leading to a computationally underdetermined problem. Finally, as transcriptomics data only capture one level of regulatory interactions, it provides an inherently biased and incomplete view of the underlying RN. Despite these caveats, it has been shown by us and others that novel biological interactions can be elucidated from these data.
A. Experimental Design
No technique or technology can provide a “one size fits all” solution to network inference that is optimal with respect to the completeness or accuracy of the learned network topology or the ability of the model to describe system behavior. Further, careful experimental design is needed to balance the biological goals of any given systems biology effort (what cell processes are of interest to the effort as a whole, what biology is interesting to the graduate person doing the work). In general, microarray experimental designs fall into two broad categories: (1) steady-state experiments, and (2) time-series experiments. Balancing steady-state measurements following perturbations (both genetic and environmental) with time-series experiments that provide measurements of the system in action (capturing key changes post-perturbation and providing a means of characterizing system dynamics) is key to the success of efforts to elucidate biological networks. In steady-state experiments, a perturbation (i.e., drug or genetic perturbation such as knock-down of a gene by RNAi) is introduced for a period of time, presumably until the system has reached a steady state, at which point the state of the system is assayed via microarray. We refer to these experiments as “steady state” even though the system may not have achieved a true steady state when measured. In a time-series experiment, a perturbation is introduced, and the response of the system is measured at multiple time points. Although these time-series experiments are more costly, this type of information can aid in resolving causality, and inferring not only topological structure but also the degree of regulation (i.e., kinetic parameters).
However, these types of data cover only the transcriptional level of regulation, and even state-of-the-art methods (Greenfield et al., 2010; Huynh-Thu et al., 2010; Pinna et al., 2010; Prill et al., 2010) still make a significant number of false predictions. The accuracy of these predictions can be improved with the addition of other data types. Recently, chromatin immunoprecipitation followed by sequencing (ChIP-seq) has become a widely used method for collecting direct TF–DNA binding data. These data can be used to help infer direct binding events. Note that these data typically contain many false positives as many binding events are non-functional. Another approach involves using single nucleotide polymorphism (SNP) data in conjunction with mRNA expression data to learn the extent to which each mutation can have a functional effect (Lee et al., 2009). Such an approach can be used to uncover the TFs that are more likely to have a phenotypically important effect. Current approaches to learning RNs combine (1) binding data (from ChIP-seq and scans using well-characterized binding sites), (2) priors on network structure from known/validated regulatory interactions, (3) perturbation/genetic data, and (4) expression/proteomics data to triangulate regulatory interactions. We discuss our pipeline in the context of expression data (to describe our core model and prior work) and then develop a method that can integrate these four sources of information (Chen et al., 2008; Christley et al., 2009; De Smet and Marchal, 2010; Friedman and Nachman, 1999; Geier et al., 2007; Gevaert et al., 2007; Husmeier and Werhli, 2007; Huynh-Thu et al., 2010; Ideker et al., 2001; Lee et al., 2009).
B. Estimating Co-regulated Genes Prior to Network Inference
If only mRNA expression data are available, as is the case in many processes/diseases of interest, other steps can be taken to improve the quality of the final output network. One means of dealing with the ambiguities of direct inference from micro-array data is to reduce the complexity of the problem by estimating co-regulated groups via clustering or biclustering, which we discuss here specifically in the context of RN inference. Automatic learning of genetic RNs from microarray data presents a severely under-constrained problem: even in the most complete data set, the number of genes is greater than the number of experimental conditions. This is traditionally addressed by applying dimensionality reduction techniques to reduce the number of genes, for example, eliminating genes based on signal-to-noise ratio, or clustering genes based on similarity of expression.
Clustering methods, when applied correctly, can reflect the known biological property that many gene products work together in functional modules under identical regulatory control, forming components of tightly conserved pathways or molecular machines. Thus, applying a clustering method prior to network inference serves not only as a dimensionality reduction technique, but also as an additional method to capture the relevant underlying biology. Standard clustering groups together genes that show common expression across all experimental conditions (referred to as co-expression). However, co-expression may not extend across all conditions, particularly as the number of conditions in a data set increases. A subset of genes may be co-expressed over only a subset of conditions; or a gene may participate in multiple processes, and therefore be co-expressed with several different subsets of genes across different subsets of conditions.
Biclustering (Cheng and Church, 2000) refers to simultaneous clustering of both genes and conditions, and can account for these more complex patterns of co-expression (Cheng and Church, 2000; Lazzeroni and Owen, 1999). Both genes and conditions can belong to multiple biclusters, and each bicluster’s subset of genes and conditions represents a putative functional module reflecting the organization of known biological networks into modules (Singh et al., 2008). Early works (Morgan and Sonquist, 1963) introduced the idea of biclustering as “direct clustering” (Hartigan, 1972), node deletion problems on graphs (Yannakakis, 1981), and biclustering (Mirkin, 1996). More recently, biclustering has been used in several studies to address the biologically relevant condition dependence of co-expression patterns (Ben-Dor et al., 2003; Bergmann et al., 2003; Cheng and Church, 2000; DiMaggio et al., 2008; Gan et al., 2008; Kluger et al., 2003; Lu et al., 2009; Supper et al., 2007; Tanay et al., 2004). Biclustering also provides another advantage relating to increasing the signal (relative to the noise) of microarray data. These data are noisy due to both random noise (e.g., fluctuations in the scanner’s laser) and systematic effects (e.g., sequence-specific differences in performance of probes or PCR amplification), as well as inherent biological noise, all of which occur per-gene. When genes are combined into modules, the average expression of the module is used, and thus the per-gene noise is averaged out, and the expression of the signal (relative to the noise) is increased.
Traditional biclustering is based solely on microarray data. Additional genome-wide data (such as association networks and TF binding sites) greatly improves the performance of these approaches (Elemento and Tavazoie, 2005; Huttenhower et al., 2009; Reiss et al., 2006; Tanay et al., 2004). Examples include the most recent version of SAMBA, which incorporates experimentally validated protein–protein and protein–DNA associations into a Bayesian framework (Tanay et al., 2004), and cMonkey (Reiss et al., 2006), an algorithm we recently introduced. Bicluster inference has also been extended to detect conservation of modules across multiple species (Waltman et al., 2010). These integrative biclustering methods provide more accurate and biologically relevant biclusters, and provide a template for the use of integrative methods in network inference.
Biclusters are of particular interest for network inference: inference on these putative functional modules is both more tractable and more easily interpreted than inference on individual genes. The regulation of biclusters (or individual genes) by the relevant TFs and EFs in the system can be learned in a variety of ways. Common difficulties for any network inference method include determining the direction of a regulatory relationship (does gene a regulate gene b or does gene b regulate gene a), and separating direct from indirect regulatory relationships (does gene a regulate gene b directly, or does gene a regulate gene c which then regulates gene b) (Marbach et al., 2010). The ability to resolve the directionality of a regulatory relationship can be improved by using microarray data collected from time-series or genetic-knockout experiments, as such data allow for causal inferences to be made (Chaitankar et al., 2010; Madar et al., 2010; Marbach et al., 2009b; Pinna et al., 2010; Schmitt et al., 2004; Yip et al., 2010). However, it is still difficult to distinguish direct interactions from indirect: again, data such as ChIP-seq and ChIP-chip help resolve this ambiguity, and would ideally be available for the construction of an accurate, global RN. Even in cases where multiple, putatively complementary data types are available (i.e., microarray and ChIP-seq), validation of the output RN and comparison of RNs generated by different methods is a challenging task. For example, many top-performing methods are likely to involve data-integration methods that may integrate data with complex relationships and co-dependencies. Also, as the full integrated data set used for network inference becomes more complex, generating leave-out test sets that are completely separate from the integrated inference data becomes a research problem of its own.
C. Validation of Network Inference Methods is Key to Progress
The plethora of different methodologies available for RN inference makes the comparison of the RNs produced by different algorithms a challenging problem. Until recently, a group developing an RN inference algorithm would generate a long list of hypothesis, experimentally validate their first few predictions, and consider their method successful. This tradition of validating top predictions makes good sense when one considers that biologists may only have the capacity to follow up on a limited number of top predictions. This focus on top predictions, however, is insufficient for comparing network inference methods and assessing their relative strengths and weaknesses, as a typical RN inference method generates thousands of predictions. For such comparisons, a gold-standard RN inference data set is needed in which the topology of the underlying network is unambiguously and completely determined (Marbach et al., 2009c, 2010; Prill et al., 2010; Stolovitzky et al., 2007). Databases for model organisms that collate thousands of validated regulatory interactions (such as Transfac, RedFly, and RegulonDB) are also critical to developing and validating RNs. A fully complete RN gold standard, however, cannot currently be obtained from real biological experiments, as known biological networks, even in the simplest organisms, are both extremely complex and considered to be incomplete.
In an effort to standardize the comparison and assessment of algorithms for RN inference, the Dialogue in Reverse Engineering Assessments and Methods (DREAM) has posed a set of challenges to the network inference community in a double-blind fashion (Marbach et al., 2009c, 2010; Prill et al., 2010; Stolovitzky et al., 2007). Participating groups only see the microarray data (either synthetically generated, or from real data compendia) and not the underlying topology. Likewise the evaluators only see the predictions from each group (in the form of ranked lists of regulatory interactions), and not the method that was used to generate them. When such gold standards exist, metrics such as area under the precision recall curve (AUPR) and area under the receiver operator curve (AUROC) (Davis and Goadrich, 2006) can be used to assess an algorithm’s performance. However, when applying RN inference techniques to mammalian data, relatively little of the true underlying topology is known, and AUPR and AUROC are not nearly as informative as for simpler systems.
D. Visualization
Analysis of inferred RNs for such systems presents a difficult set of problems. RNs have an intuitive visual representation as graphs consisting of nodes connected by directed or undirected edges, and programs such as Cytoscape (Shannon et al., 2003) provide a straightforward means of rendering these graphs and annotating them with manifold types of associated information. This visual representation can be used by a researcher with domain knowledge of the underlying biological problem to extract the most meaningful and interesting parts of the network. Unfortunately, for networks larger than tens of nodes connected by at most hundreds of edges, this straightforward visualization becomes too dense to comprehend as a whole, presenting visually as the familiar network “hairball.” While this dense representation contains much valuable information that can be interpreted by a researcher who has spent days or weeks investigating it, to the uninitiated it is essentially meaningless. Dimensionality reduction (e.g., via biclustering) can reduce visual complexity, but imposes other issues: a gene name is unambiguous, but how best to label a collection of genes and conditions? Whether inference is performed directly on all genes or on a reduced set of TFs and biclusters, the challenges are the same: to tease out the meaningful information contained in the network, and to convey this information effectively to other researchers not intimately familiar with the overall network. Thus, a set of visualization and analysis tools is necessary to query the network in an intuitive, meaningful, and easily accessible manner. Below, we describe one coordinated visualization system that allows users to explore biclusters, networks, and annotations.
II. Overview of Model/Algorithm
Our pipeline for network inference (Fig. 1) consists of three main steps: (1) inference of co-regulated modules using cMonkey, (2) RN inference using the Inferelator (Inf) pipeline, and (3) network analysis and visualization using software tools connected by the Gaggle (Fig. 2). Here, we present an overview of biclustering methods, RN inference methods, and a detailed overview of each step of our pipeline.
Fig. 1.
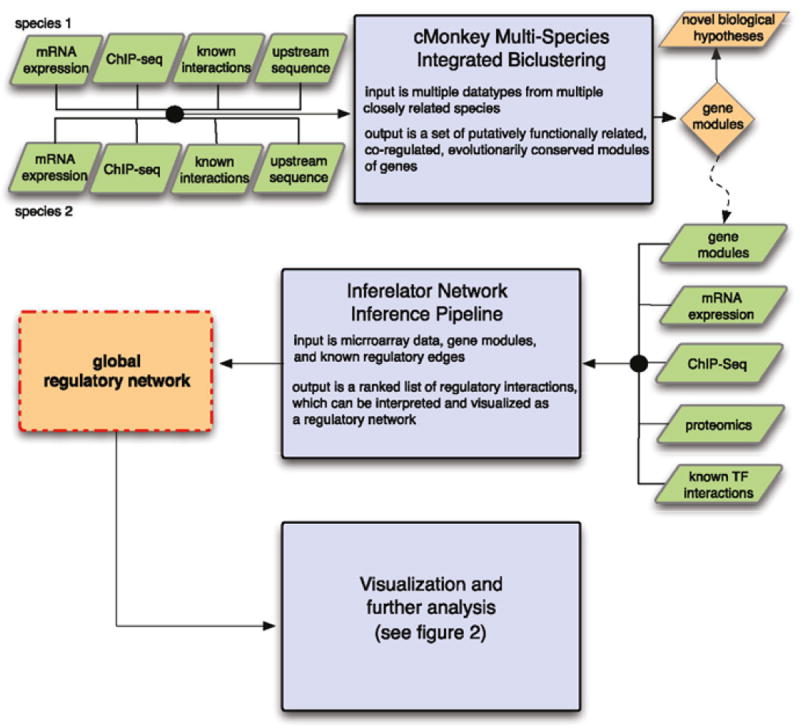
Overall inference pipeline. Our inference pipeline is composed of three main steps: (1) inference of biclusters, which are putative functionally related, co-regulated modules of genes, by Multi-species cMonkey (MScM); (2) inference of the regulation of these biclusters by transcription factors (TFs) via our Inferelator inference pipeline; and (3) analysis and visualization using a collection of Gaggle-connected tools. The input to cMonkey consists of mRNA expression data, known as interactions (some of which come from ChIP-seq), and upstream sequence information from two or more species. The output of MScM is biclusters that are conserved between multiple species. These biclusters can be used for hypothesis generation, and also serve as the input to the Inferelator network inference pipeline. Along with biclusters, the Inferelator also uses mRNA expression data, known interactions between relevant TFs and their targets, proteomics data, and ChIP-seq data. The output of the Inferelator inference pipeline is a set of regulatory interactions between the biclusters and TFs. This putative regulatory network can be visualized and analyzed by the Gaggle-connected set of tools shown in Fig. 2. (For color version of this figure, the reader is referred to the web version of this book.)
Fig. 2.
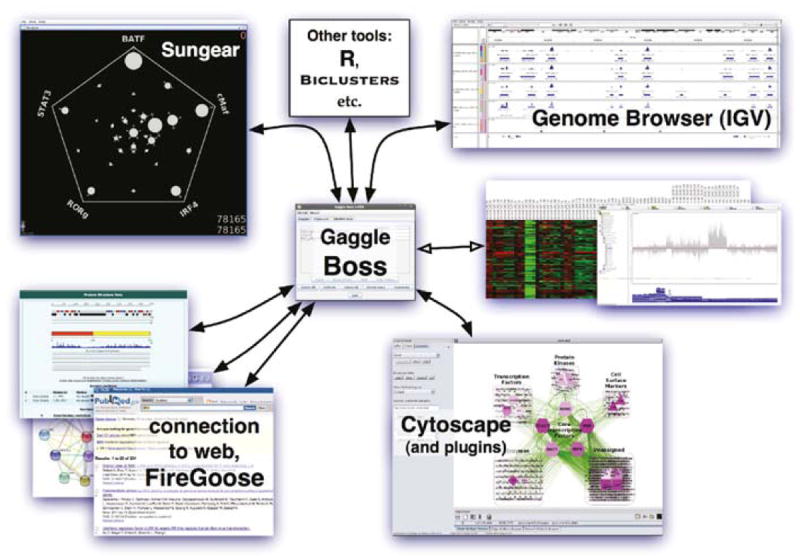
Gaggle visualization and analysis framework. The Gaggle Boss, shown in the center, coordinates communication among the various member tools (geese), removing the need for file import/export and format translation. Also shown is a subset of geese, including two – Cytoscape and Sungear – that are used as part of the analysis discussed in Biological Insights section and Methods section. Each of the geese can both send to and receive from the Boss, which permits an iterative workflow: for example, a small set of genes from Sungear can be sent to Cytoscape, analyzed to find its 1-hop network, then sent back to Sungear for further analysis. In addition, several geese provide extensible means to connect to a larger set of tools: Cytoscape and Sungear via plug-in frameworks, FireGoose via its connections to other websites, and R via its downloadable packages. (For color version of this figure, the reader is referred to the web version of this book.)
A. Biclustering
Biclustering methods can be broken into three categories, which we will refer to as co-expression, co-regulation, and conserved co-regulation. Some methods, such as that of Cheng and Church (2000), rely solely on gene expression data to find groups of genes that are co-expressed. More recently, algorithms such as cMonkey (Reiss et al., 2006; Waltman et al., 2010), COALESCE (Huttenhower et al., 2009), and the most recent version of SAMBA (Tanay et al., 2004) consider additional types of data such as common binding motifs, protein–DNA binding, and protein–protein interaction networks. These integrative techniques infer modules that are co-regulated rather than simply co-expressed. This distinction is of particular importance for RN inference, as genes in co-regulated biclusters are more likely to exhibit shared transcriptional control. Finally, several techniques (Bergmann et al., 2003; Waltman et al., 2010) extend the integrative approach by searching for conserved biclusters across different species.
The biclustering method cMonkey was designed to produce putatively co-regulated biclusters that are optimal for network inference. In addition to microarray expression data, cMonkey also incorporates upstream sequences and interaction networks into the biclustering process. Upstream sequences are used to find putative common binding motifs among genes in a bicluster, providing additional evidence for possible co-regulation. Co-regulated genes are also more likely to share other functional couplings, which will be reflected as an above-average number of connections between genes within a bicluster according to databases of known interactions such as BIND (Bader et al., 2003) and DIP (Salwinski et al., 2004) – in other words, these genes form small, highly connected sub-networks within these larger networks. Compared to other methods, cMonkey generates biclusters that are “tighter” (have lower variance across bicluster gene expression values) yet include more experimental conditions.
Multi-species cMonkey (MScM) (Waltman et al., 2010) is an extension of the cMonkey method to allow discovery of modules conserved across multi-species datasets. Recent work (Ihmels et al., 2005; Tirosh and Barkai, 2007) shows significant conservation of co-regulated modules across species. Therefore, biclusters that are highly conserved between organisms are most likely to be biologically relevant. In addition, by pairing a well-studied model organism such as yeast or mouse with a closely related but less well-studied organism, MScM is more likely to find meaningful biclusters in the other organism. Even pairing well-studied organisms may be beneficial as different processes may be better elaborated in each organism. The regulation of these putative conserved functional modules of genes can be inferred using the Inf-based inference pipeline.
B. Regulatory Network Inference
The key question that RN inference aims to answer is which EFs and TFs regulate which genes? In other words, given a set of observations (e.g., expression data), what is the underlying network responsible for observed data? Furthermore, can predictions be made from the output network? In order for quantitative predictions to be made about the response of the system to new perturbations, the dynamics of the system must be learned from time-series data. A multitude of inference methods exist, using varying underlying assumptions and modeling principles. We limit ourselves to the discussion of the following broad groups of methods: (1) Bayesian methods, (2) mutual information (MI)-based methods, and (3) ordinary differential-equation (ODE)-based methods. We briefly describe each grouping, and then proceed with a description of our network inference method. Here, we focus on methods that scale to systems with thousands of interactions.
A Bayesian network is defined as a graphical model that represents a set of random variables and their conditional dependencies. Such a framework naturally applies to RN inference, as RNs can intuitively be though of as directed graphs. The observed data are used to compute the model whose probability of describing the data is the highest, and such methods have resulted in several notable works (Friedman et al., 2000; Friedman and Nachman, 1999; Husmeier and Werhli, 2007; Sachs et al., 2002; Sachs et al., 2005; Segal et al., 2003). Bayesian methods also allow for the incorporation of priors such as sparsity constraints and structured priors (Geier et al., 2007; Gevaert et al., 2007; Mukherjee and Speed, 2008). However, Bayesian methods have difficulty in explicitly handling time-series data. Additionally, many Bayesian methods suffer from the identifiability problem: multiple network topologies produce equally high probabilities. In this situation, it is unclear which topology is best.
Differential-equation-based methods for RN inference attempt to learn not only the topology of the network but also the dynamical parameters of each regulatory interaction. RN models resulting from these methods can be used to predict the system-wide response to previously unseen conditions, future time points, and the effects of removing system components. A drawback of these methods is that they generally require time-series data and more complete datasets than many alternative methods. Typically these methods are based on ordinary differential equations (ODEs) due to several assumptions that improve the computational cost for parameterizing these models. ODE-based methods model the rate of change in the expression of a gene as a function of TFs (and other relevant effectors) in the system. Differential-equation-based methods differ in their underlying functional forms, how the system of equations is solved or parameterized (coupled or uncoupled solution, optimization procedures, etc.), and how structured priors and sparsity constraints are imposed on the overall inference procedure. For example, several methods have been proposed that use complex functional forms (Mazur et al., 2009) and solve a coupled system (Madar et al., 2009; Mazur et al., 2009), while other methods solve a simplified linear system of ODEs (Bansal et al., 2006; Bonneau et al., 2007; Bonneau et al., 2006; di Bernardo et al., 2006; di Bernardo et al., 2005). Several methods have been developed that are able to incorporate structured priors into network inference (Christley et al., 2009; Yong-a-poi et al., 2008).
A number of methods for detecting significant regulatory associations are based on similarity metrics derived from information theory, such as MI (Shannon, 1948). The MI between two signals (in this case the expression of a TF and its target) is calculated by subtracting the joint entropy of each signal from the sum of their entropies. It is similar to correlation (higher values connote stronger relationships), but is more generally applicable as it assumes neither a linear relationship between two signals nor continuity of signal. At their core, methods that rely on MI generally infer undirected interactions, as the MI between two variables is a symmetric quantity (Butte and Kohane, 2000; Faith et al., 2007; Margolin et al., 2006); however, modifications can be made that allow for the inference of direction (Chaitankar et al., 2010; Liang and Wang, 2008; Madar et al., 2010).
Each RN inference method has its own simplifying assumptions, biases, and data requirements. Recently, there has been much interest and progress in combining methods that use multiple different data types and modeling algorithms into RN inference pipelines. For example, it has been demonstrated by us and others (Greenfield et al., 2010; Pinna et al., 2010; Prill et al., 2010; Yip et al., 2010) that the response of a system to a genetic knockout is a very powerful data type for uncovering the topology of the underlying RN. Methods that take this into account performed very well in the DREAM3 and DREAM4 network inference challenges (Greenfield et al., 2010; Pinna et al., 2010; Yip et al., 2010).
It has also been shown that when multiple network inference methods, or ensembles of networks generated by the same method, are combined, the overall performance is better than that of any individual method (Greenfield et al., 2010; Marbach et al., in press, 2009a; Prill et al., 2010). This improvement in performance due to combining multiple methods is an important technique that can be applied to complex biological problems where complete knockout data are not available. In such cases it is also important to supplement microarray data with other available data types. The Encyclopedia of DNA Elements Consortium (ENCODE) has been compiling a vast database of high-sequence data such RNA-seq, ChIP-seq, and genome-wide distribution of histone modifications. These data can be used in many ways to influence the confidence that a network inference algorithm assigns to a regulatory interaction. We have incorporated these ideas into our network reconstruction methods in two forms: (1) topology dominated, where evidence from different data types is combined to rank interactions by converting all regulatory hypothesis derived from each data type into p-values or ranks, then combining them to form an overall p-value or rank for all regulatory interactions (Greenfield et al., 2010; Marbach et al., in press), and (2) model dominated, where information from different data types is used as structure priors during the network inference step (described below).
Our inference pipeline is built on three core principles: (1) combining multiple methods and data types in a mutually reinforcing manner, (2) using time-series information to infer putative causal, directed relationships (as opposed to undirected associations), and (3) inferring sparse models of regulation using model selection. The input to our method is a microarray dataset consisting of multiple types of experiments. All data sets include steady-state data (in response to perturbation), time-series data is often available; and in the best-case scenario, genetic-knockout steady-state data are available as well. The core of our inference pipeline comprises two methods that work in tandem: time-lagged context likelihood of relatedness (tlCLR) and the Inferelator 1.0. tlCLR computes a prediction of the RN that is further refined and optimized by the Inferelator 1.0. The output of tlCLR is the input to Inf, and we refer to the combined method as tlCLR-Inf. tlCLR-Inf uses all available microarray data and treats all steady-state data the same (making no distinction between knockout perturbations and any other perturbations). tlCLR-Inf takes advantage of the time-series data to learn putatively causal, directed edges, and assign dynamical parameters (see Methods).
tlCLR (Greenfield et al., 2010; Madar et al., 2010) is based on the well-known RN inference algorithm context likelihood of relatedness (CLR) (Faith et al., 2007). CLR uses MI followed by background correction to calculate the confidence in the existence of any regulatory interaction. tlCLR uses the same CLR strategy of MI followed by background correction, but takes advantage of the time-series data to learn the direction of the regulatory interaction. This method is described in detail in the Methods section. The output of this method is a set of predicted regulators for each target, and is used by the Inf to remove the least likely regulatory interactions and improve accuracy and computational efficiency.
The Inf models the network as a system of linear ODEs. The rate of change for each gene is modeled as a function of the known regulators in the system. This function can take many different functional forms, and can be easily modified to capture biologically relevant behaviors. For example, it is common in biological systems that two TFs must act in tandem in order to affect their target. The core Inf model allows for these non-linear combinatorial interaction terms. Additionally, it is known that the activation of a target by its regulator follows a hill-type curve (multiple functions with a roughly sigmoidal shape can be used to model biologically relevant activation thresholds, cooperation, and saturation of TF-target response). This can be incorporated into the core Inf model by approximating this behavior via sigmoidal functions compatible with efficient learning methods, such as constrained logistic regression. Once a functional form is chosen, the parameters for each regulatory interaction are calculated using least angle regression (LARS) (Efron et al., 2004) which is a constrained linear-regression approach that imposes an l1 constraint on the model parameters. This constraint ensures that sparse models are learned (in concordance with the known properties of TF RNs). Importantly, we have modified this core model selection algorithm, LARS, such that we can influence the degree to which a predictor is incorporated into or removed from a model. Using this modification, we can incorporate structured priors (derived from validated interactions, literature search algorithms, or alternate data types) into our network inference approach. We have shown that using a simple linear model with (and also without) interaction terms performs well in recovering the topology of the network.
C. Network Visualization and Analysis
RNs often consist of hundreds or thousands of nodes connected by thousands or more of regulatory edges. Analysis methods for networks of this scale generally fall into two categories that we will refer to as “network-centric” and “gene-centric,” with some techniques bridging the two. Network-centric (or “holistic”) techniques accumulate statistics about the network as a whole that can provide a sense of the validity of the overall network (e.g., by comparing statistics with those of validated biological networks) or guide further exploration (e.g., by pointing out the existence of highly connected nodes or densely inter-connected sub-networks). The simplest of these network-centric techniques is simply to count each node’s in- and out-degrees, that is, its incoming and outgoing regulatory edges, respectively. Analysis of node out-degree will highlight network “hubs”: those TFs that regulate many more genes than average. Examination of the distribution of node in- and out-degree also provides valuable information. Biological networks, such as metabolic networks (Jeong et al., 2000), as well as many other types of complex networks, tend to be “scale free” networks: the probability of a node having k in or out edges is described by P(k) ≈ k−γ. This is considerably different from random networks generated according to the classical Erdös-Rényi model, where any two nodes in a graph have an equal probability of being connected: such graphs are characterized by a Poisson distribution that peaks strongly at the average number of connections (Jeong et al., 2000). Average shortest-path length (the “small world” property), average clustering coefficient distribution (Ravasz et al., 2002), and many other have metrics have been shown or theorized to have biological relevance. Zhou et al. (2010) provide an example of using general network statistics to characterize and differentiate between ecological networks under different conditions. Cytoscape plug-ins such as NetworkAnalyzer (http://med.bioinf.mpi-inf.mpg.de/netanalyzer/index.php), the R packages sna (Butts, 2008), and igraph (http://cneurocvs.rmki.kfki.hu/igraph/) are designed to perform these and many other types of network analysis.
In general, and particularly with inferred networks, these network-centric metrics act as a guide to suggest areas of further exploration – such as network hubs – rather than an explicit measure of network plausibility. Gene-centric (or “constructive”) analysis techniques tend to follow a “find and connect” approach. They start with a small set of nodes – such as a set of genes of interest, a small sub-network of known function, a bicluster with significant functional annotations, or a set of network hubs identified through network-centric analysis – then gradually add connected nodes to grow the size of the network. The most basic approach is to start with a single gene, then examine its “1-hop” sub-network within the full network: the genes directly connected to it, that is, its direct targets and regulators. One can also make a 1-hop network for multiple genes that is simply the union of the 1-hop networks of the individual genes. These sub-networks can be expanded to an arbitrary number of “hops,” with each additional step adding all nodes directly connected to those already in the sub-network. Typically, the hope is that the small 1- or 2-hop networks will include some known regulatory edges (as a “sanity check” of the inference process) as well as some plausible novel edges that bear further investigation.
Another gene-centric approach is to find, for some set of genes or biclusters, the smallest sub-network that includes all these genes or biclusters of interest (the phrase “gene-centric” is used generically to refer to network consisting of genes or biclusters). This sub-network may resemble a known functional module (another “sanity check”); it may connect known genes or biclusters in a novel way; and it may include unknown or unexpected genes in an otherwise well-described functional module. While no Cytoscape plug-in provides this functionality directly, the Subgraph Creator (http://metnet.vrac.iastate.edu/MetNet_fcmodeler.htm) plug-in can be used to find the sub-network with a given number of directed hops for a set of starting genes, and so iteratively find a sub-network containing all the desired genes.
1. Gaggle Tools
We supplement the network viewer-based approaches above by providing a collection of tools that provide analysis of different types of information at varying scales. The Gaggle (Shannon et al., 2006) connects together many independent tools into a cohesive framework where the component tools (geese) can exchange information such as lists of genes directly without the need for intermediate files or format conversion. A key aspect of this type of approach is that it enables iterative exploration across multiple tools, where results are repeatedly sent from one tool to the next and further refined with each step in this process. Gaggle-enabled tools include
Network viewers Cytoscape (Shannon et al., 2003) and nBrowse (http://www.gnetbrowse.org)
Firegoose (Bare et al., 2007), a Firefox plug-in that provides data exchange with external web resources such as STRING (Snel et al., 2000; Szklarczyk et al., 2011)
The Comparative Microbial Module Resource (CMMR) (Kacmarczyk et al., 2011), a comprehensive bicluster visualization and analysis tool
The Data Matrix Viewer (DMV) (http://gaggle.systemsbiology.net/docs/geese/dmv.php), a data matrix exploration tool
MultiExperiment Viewer (MeV) (Saeed et al., 2003), a sophisticated analysis tool for microarray data
Sungear (Poultney et al., 2007), a set analysis and exploration tool
The Integrative Genomics Viewer (IGV) (Robinson et al., 2011), a browser for associating annotations and other data with chromosomal locations
The statistical programming language R (http://www.R-project.org).
III. Biological Insights
In this section, we will focus mainly on ways of extracting potential insights or points for further investigation. The networks discussed here were chosen to show a range of inference and analysis techniques across different network scales. For details of the methods used to create these networks, see the Computational Methods section.
Fig. 3 shows a subset of a larger network inferred on biclusters derived from the Immunological Genome Project (IMMGEN) (Painter et al., 2011) mouse immune cell data set and human immune cell experimental data from GEO (see Methods for details). This sub-network has been chosen to show the subset of biclusters from the full network that are most strongly linked to various hallmarks of cancer (Hanahan and Weinberg, 2000, 2011). An immediately striking feature of this network is that different hallmarks separate naturally into sub-regions of the network, joined by the TF MTA2. This is not a deliberate design feature of this sub-network, but rather an intriguing consequence of choosing the set of biclusters with high-confidence connections (via significant GO terms) to hallmarks of cancer. The top region, whose regulators include FOXM1 and MYC, includes all biclusters annotated with the hallmark “limitless replicative potential” (blue icon). The bottom region includes all biclusters annotated with the hallmark “self-sufficiency in growth signals” and all but one bicluster annotated with the hallmark “insensitivity to anti-growth signals.” The few biclusters annotated with “evading apoptosis” are spread evenly between the network clusters.
Fig. 3.
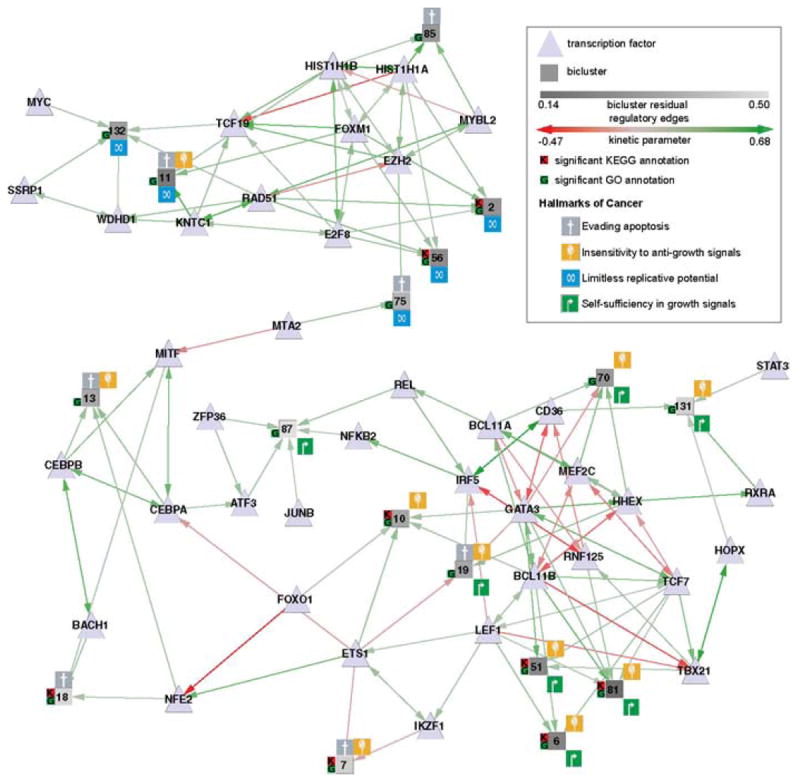
Hallmarks of cancer shown overlaid on a sub-network of biclusters and transcriptions factors (TFs). Biclusters are shown as squares, with shading indicating the bicluster residual (variance in gene expression values). Surrounding icons indicate the putative hallmarks of cancer. A small K or G to the bicluster left indicates particularly significant enrichment for one or more KEGG or GO terms, respectively. TFs are shown as triangles, with regulatory edges to biclusters and other TFs. Green edges indicate upregulation, and red edges downregulation. Four of the six original hallmarks are represented in the network: biclusters associated with self-sufficiency in growth signals and insensitivity to anti-growth signals are clustered together, as are those associated with limitless replicative potential; biclusters inferred to be involved in evading apoptosis are spread through the network. (See color plate)
Figs. 4-6 illustrate a comparative analysis of two different cell lines: normal human breast epithelial tissue (MCF-10) and invasive, metastatic breast cancer (MDA-MB-231) (see Methods for details). In all three figures, a blue-to-yellow continuum is used to indicate relative specificity of a gene, gene product, or regulatory edge to MDA-MB-231 (blue) or MCF-10A (yellow), with the intermediate gray denoting neutrality. Fig. 4 illustrates a typical network “hairball”: with 1866 nodes and 4822 regulatory edges, it is useful mostly for giving a general sense of the proportion of edges more active in MDA-MB-231 (blue) and MCF-10 (yellow), as well as the abundance of proteomics data (nodes colored yellow, blue, or dark gray).
Fig. 4.
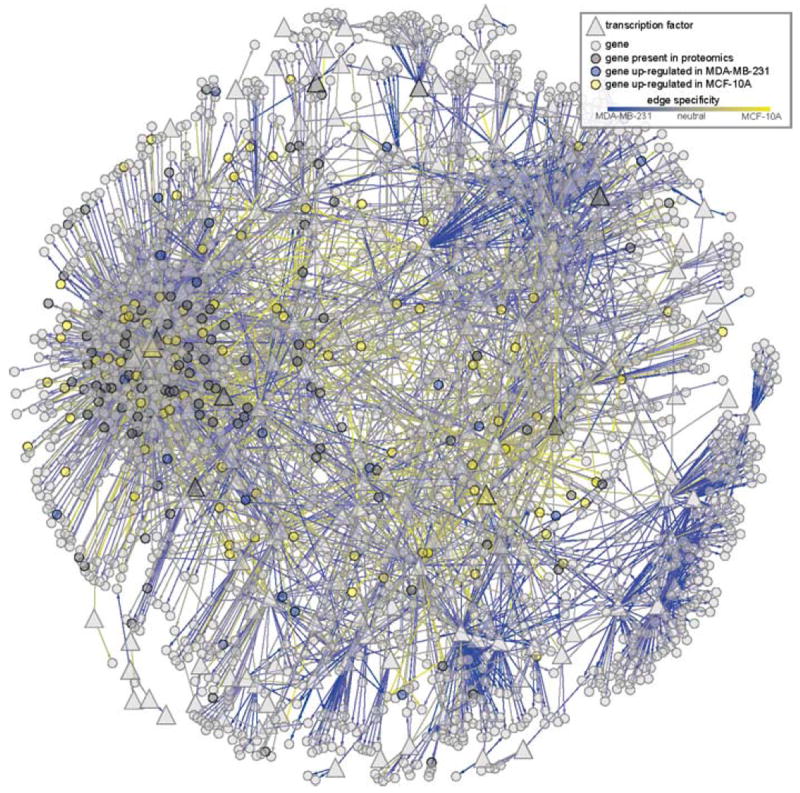
Breast cancer network with the top 4822 edges ranked by combined confidence from the two cell line inference runs. Edge color denotes differential inferred regulation on a yellow-to-blue gradient from MCF-10A (yellow) to MDA-MB-231 (blue). Nodes are rendered semi-transparent so that the distribution of cell-line-specific regulatory edges can be clearly seen. Proteomics data from MCF-10A/MDA-MB-231 comparison are also shown using node colors: differential expression in MCF-10A is shown in yellow, and MDA-MB-231 in blue. Genes present but not differentially expressed are shown in darker gray. (See color plate)
Fig. 6.
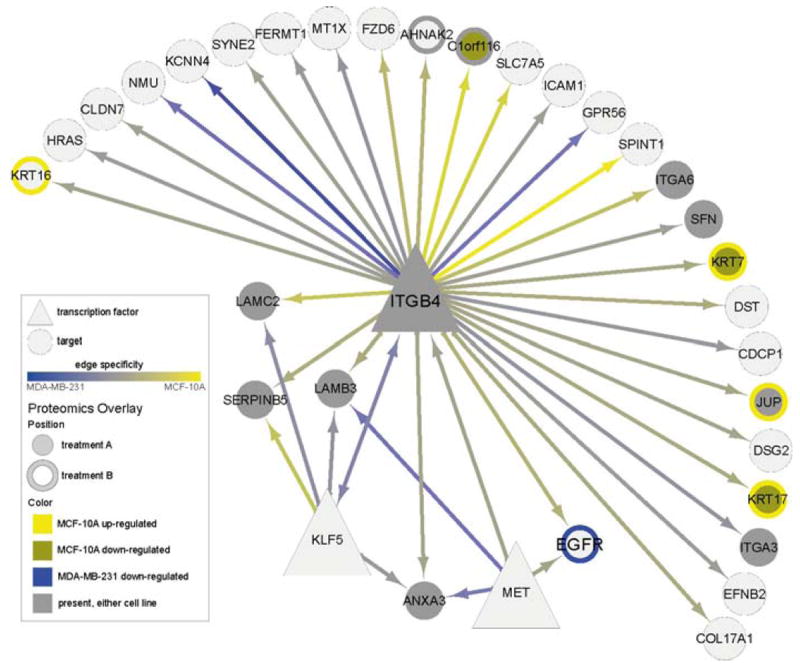
A sub-network extracted from the cell line comparison network illustrating all interactions with ITGB4 along with overlays of experimental proteomics (SILAC) data. Shown is the 1-hop network from gene ITGB4 along with differential expression in two experimental conditions, referred to as treatment A and treatment B. ITGB4 was identified a priori as a gene of interest, and is inferred to regulate gene of interest EGFR and several Laminins. Differential expression in treatment A is shown using node center, and in treatment B using node border, as follows: bright yellow denotes upregulation in MCF-10A, bronze denotes downregulation in MCF-10A, and blue denotes downregulation in MDA-MB-231. Gray denotes proteins that were present in either cell line but that did not meet the differential expression cutoff. Therefore, KRT17 (bottom right) is downregulated in MCF-10A with treatment A but upregulated in MCF-10A with treatment B, while EGFR is downregulated in MDA-MB-231 with treatment B. Edge colors are as in Fig. 5. (See color plate)
Fig. 5 is designed to present a summary of Fig. 4 that allows much more intuitive identification of features of interest. It represents the largest connected sub-network of TFs (142 of 220 total TFs in the original network). The number of regulatory targets of each TF is represented by the size of the node, while the node color denotes the ratio of regulatory edges strongly active in one cell type or another on a gradient from blue (MDA-MB-231) to yellow (MCF-10A). This “summary” representation of targets and regulatory edges allows the removal of all non-TF targets and their corresponding regulatory edges so that hubs such as CSDA, COPS2, IKZF1, and FBN1 are easily spotted: they are large and brightly colored. This constitutes a powerful use of simple network-centric techniques to simplify network visualization and analysis.
Fig. 5.
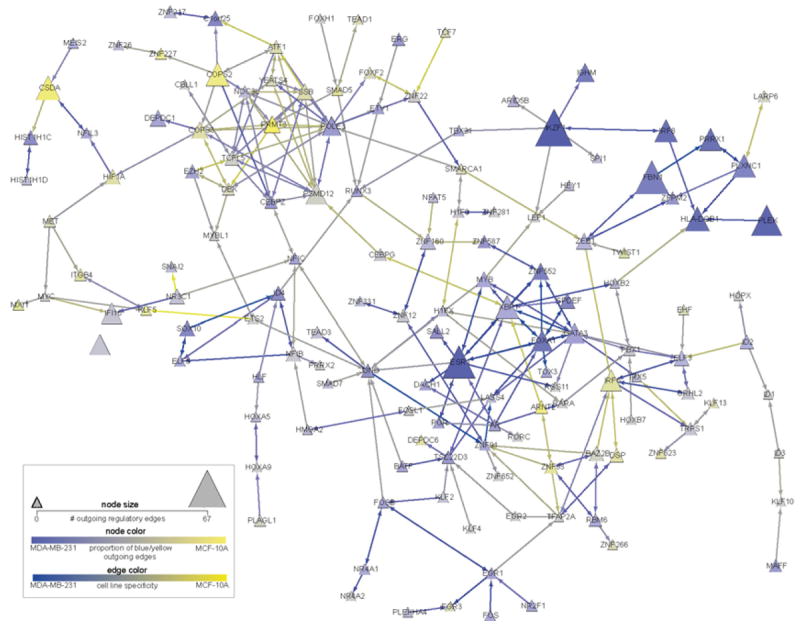
Largest connected sub-network of transcription factors (TFs) from the overall cell line comparison network. A “summary” of the entire network is provided by (a) hiding all targets of the shown TFs that are not themselves TFs, and (b) setting the size and color of each remaining TF node to reflect its number and proportion of cell-line-specific edges. Node size shows the number of edges in the master network that were above a cutoff for specificity to either cell line. Larger nodes have more cell-line-specific edges; the largest, IKZF1, has 67 edges above the threshold. Node color is determined by the ratio of above-cutoff edges specific to MCF-10Aversus MDA-MB-231, with yellow denoting more MCF-10A edges and blue more MDA-MB-231 edges. Nodes with many edges specific to one cell line or the other are therefore large and brightly colored, such as IKZF1 or COPS2. Edges are colored on a yellow-to-blue gradient based on the inferred confidence of the edge in the MCF-10A cell line (yellow) or MDA-MB-231 cell line (blue). (See color plate)
Fig. 6 shows a putative sub-network involved in cell motility. Our data set includes differential proteomics data for two conditions, shown in this network using node center and border colors. An analysis of sub-networks containing all differentially expressed proteins in both conditions found a sub-network centered on ITGB4 – identified a priori as a protein of interest involved in cell matrix, cell–cell adhesion, and motility – that contained an unusual number of differentially expressed proteins given the relatively small number of differentially expressed proteins in the network (ρ = 5 × 10−7 via hypergeometric distribution). Among the genes inferred to be regulated by ITGB4 are two members of the laminin family also thought to be involved in motility, providing a degree of “sanity check” as mentioned earlier. The presence of JUP in this sub-network is particularly interesting because of (a) its differential expression in one of the proteomics conditions, and (b) its known participation in c-MET and EGFR signaling cascades (Guo et al., 2008).
IV. Open Challenges
Combining multiple data types in the inference of RNs is still in its beginning stages, and many questions remain to be answered. Among these are the integration of additional data types into both the biclustering and inference processes, integrating across multiple temporal and physical scales, validation of inferred networks, using multiple-species datasets, and visualization of networks that are multi-scale and change across time and conditions.
A. Integrating New Data Types
New types of experimental data are becoming available that will be informative to the network inference process. Metabolomic data can provide detailed measurements of changes in hundreds of metabolite levels in response to changing cell state or environment. Techniques such as surface plasmon resonance imaging (SPRi) (Smith and Corn, 2003) can provide additional high-throughput data on protein-binding constants via measurements of association and dissociation rates, potentially providing small but high-accuracy interaction networks. Mass cytometry can provide single-cell measurements of phosphorylation on a very fine time scale (Bendall et al., 2011). New data types can be added to cMonkey fairly easily since its basic model is already integrative (see Methods). The network inference pipeline can accommodate some of these data, such as SPRi-derived interaction networks, by using them to influence the likelihood that a regulatory interaction is incorporated into the model. However, other data types – particularly those that are on different time scales, like mass cytometry –pose a more difficult challenge for network inference. Even integrating proteomics data – which may superficially resemble microarray data – into the inference pipeline, rather than simply overlaying it on inferred microarray-derived networks, poses new challenges. Proteomics measurements still produce sparser data sets than microarrays, and techniques such as SILAC (Ong et al., 2002) will be systematically biased against certain proteins. A more serious issue is that TFs tend to have low expression values, and proteomics techniques do a poor job of capturing proteins expressed at low levels.
B. Validation
Validation of inferred networks of biclusters and genes is a key issue that we address explicitly in Methods. It should be emphasized, however, that new data types as discussed above will not only improve the quality of the inferred biclusters and networks, but will aid validation as well. Bicluster enrichment analysis already provides an example of using independent data types (KEGG and GO pathways) for validation as the annotations used for enrichment analysis are independent from those used in bicluster inference: because of this independence, significant enrichment provides one indicator of bicluster quality. After such enrichment analysis, it is crucial for experts with domain knowledge to highlight their most interesting genes and pathways. With the thousands of predictions that are made in a single run of our pipeline and the lack of a true gold-standard data set, such biological expertise is crucial to fully realize the hypothesis-generating potential of our methods.
C. Visualization
One issue that needs to be addressed with current visualization tools concerns displaying per-gene measurements, like the proteomics overlays in Fig. 6, in networks consisting mostly of biclusters like the hallmarks network in Fig. 3 – in other words, what is the best way to indicate differential expression of a small subset of genes contained in one or more biclusters? This may only be relevant until overlays of data from other sources are replaced by integration of these data into the inference pipeline, but for now the issue of overlaying single-gene data on biclustered networks remains open.
A larger issue is that network visualizations such as those produced by Cytoscape show a single view of a network as it might exist at one point in time. This network view may also represent a superset of the RNs that produced the data: any regulatory interaction with enough support across the various conditions is reproduced in the final network. But networks change over time, as is shown in many cancers; and different parts of any network will be active under different conditions. In other words, what is currently shown might be called a union or average of many potentially valid inferred networks. As inference tools and data availability improve, what is really desired is a tool (or set of tools) that can be used to explore this multiplicity of possible networks. This will probably require tools that can display changes in networks, in real time and in interpretable fashion, extending the “network-centric/gene-centric” metaphor introduced earlier: network-centric techniques would summarize the possible network changes over time and/or condition with the goal of steering the user to interesting features of the data, gene-centric techniques would create network sets from one or more networks of interest, and hybrid techniques might answer questions posed by the user about specific alterations in the network.
V. Computational Methods
A. cMonkey Integrative Biclustering
The steps below describe the cMonkey algorithm. Examples of data sources used are those for Escherichia coli in a multi-species biclustering by Kacmarczyk et al. (2011). For further details on cMonkey and MScM see Reiss et al. (2006) and Waltman et al. (2010).
1. Data Preparation
cMonkey uses three main data types: microarray expression, upstream sequences, and networks of associations or interactions. Data preparation and translation into a format that cMonkey can use is a key and non-trivial part of running cMonkey. Specifics of this process will be addressed in the section for each data type. The overall data preparation process involves (a) finding appropriate expression data, (b) determining upstream sequence information for the relevant organism(s), (c) downloading the association and interaction network data to be used, and (d) processing network data as necessary to reduce it to a list of interacting pairs of genes. A crucial issue across all these steps is determining a single-gene naming convention across all input data types and converting as necessary. cMonkey uses the Global Translator goose for this (http://err.bio.nyu.edu/cytoscape/bionetbuilder/translator.php).
2. Expression Data
Expression data for cMonkey is given in matrix form, where rows represent genes and columns represent experimental conditions. Expression data are row-normalized to have mean = 0, SD = 1. E. coli expression data for the multi-species biclustering by Kacmarczyk et al. were comprised of 507 conditions covering 16 projects from the Many Microbe Microarrays Database (M3D) (Faith et al., 2008).
We denote the expression levels of the genes by x = (x1, …, xNg)T. We store the C observations of these Ng genes in an Ng × C matrix, X, where the columns correspond to the experimental observations. For a given bicluster k, if p(xij) is defined as the normally distributed likelihood of the expression value xij within bicluster k, then the co-expression p-value rik for gene i relative to bicluster k is rik = Σj∈Jk p (xij) where Jk indexes the conditions in bicluster k. The co-expression p-value rjk for condition j is defined similarly.
3. Sequence Data
Methods for obtaining and processing upstream sequence data depend on the organism. Generally the regulatory sequence analysis tools (RSAT) (van Helden, 2003) are used to extract upstream cis-regulatory sequences: sequence length depends on whether the organisms are archaea, bacteria, or eukaryotes, and additional processing may be required to account for the presence of operons (see Reiss et al., 2006, for details). E. coli data for the Kacmarczyk et al. biclustering were obtained using RSATas above and adding network edges between genes known to share operons to the network data (see below).
For a given bicluster k, MEME (Bailey and Elkan, 1994) is used to determine a set of motifs common to some or all of the upstream sequences of the genes in that cluster. MAST (Bailey and Gribskov, 1998) is then used to calculate a motif value sik for each gene i relative to bicluster k (motif values for conditions are set to zero).
4. Network Data
Network data are the most varied, generally comprising multiple network types for a given biclustering analysis. These data break down into two types: association and metabolic networks, such as Prolinks (Bowers et al., 2004) and Predictome (Mellor et al., 2002); and interaction networks, such as DIP (Salwinski et al., 2004) and BIND (Bader et al., 2003). While data sources such as DIP provide pairs of interacting proteins directly, others must be processed to generate these lists of interacting pairs. For example, KEGG (Kanehisa and Goto, 2000) metabolic pathways are examined for pairs of genes that participate in a reaction sharing one or more ligands (excluding water and ATP). Network data are also the most species-dependent as different network data types are available for different organisms. This is reflected in the number and diversity of network data types in the Kacmarczyk et al. E. coli biclustering: operon edges between genes known to lie on the same operon; metabolic edges from KEGG as described above; gene neighbor, phylogenetic profile, and gene cluster edges from Prolinks; and COG-code edges from COG (Tatusov et al., 2000).
For a given bicluster k, gene i, and network N, the network association p-value is computed using a hypergeometric distribution based on the number of connections between gene i and bicluster k, connections between gene i and genes not in bicluster k, and connections within and between genes in k and not in k. This metric assigns better p-values to densely connected sub-networks of genes that are likely to participate in common functional modules.
5. cMonkey Bicluster Model
cMonkey determines biclusters by iteratively (a) updating the conditional probability of each bicluster based on its previous state, and (b) further optimizing the bicluster by adding or dropping genes and/or conditions. This constitutes a Markov chain process where the probabilities in the optimization step depend only on the previous state of the bicluster. Additions and deletions are made by sampling from the conditional probability distribution using a Monte Carlo procedure. The component contributions to the conditional probability come from the expression, sequence, and network p-values described above, which are combined into a regression model. Denoting an arbitrary gene or condition by i, we define the vector gik as the projection into one dimension of the space defined by rik, sik, and qik, as follows:
| (1) |
The use of r̃ik instead of rik denotes that the log(rik) values have been normalized, for each bicluster, to have mean = 0, SD = 1; the same applies for s̃ik and , placing all three on the same scale for each bicluster. The likelihood of any gene or condition i belonging to bicluster k is then
| (2) |
The parameters β0, β1 determine the conditional probability of membership of gene or condition i in bicluster k. The importance of each evidence type can be adjusted using the “mixing parameters”r0, s0, .
A cMonkey run starts with “seeding” of initial biclusters, with each bicluster randomly seeded according to one of several algorithms. After seeding, each iteration (a) updates the bicluster motifs, (b) recalculates the probabilities πik described above for each gene or condition i, and (c) preferentially adds or drops genes or conditions according to their probability of membership using a simulated annealing protocol. Unlikely moves (additions or deletions) are permitted according to an annealing temperature T that is decreased over time. Mixing parameters r0, s0, are also varied according to a set schedule: s0 starts small early in the process, when biclusters are unlikely to have coherent motifs, and is gradually ramped up until its influence is equivalent to that of r0. Values of follow a schedule that depends on the networks involved.
6. Multi-Species cMonkey
MScM is similar to single-species cMonkey as described above, with a few additional steps. The overall MScM process, assuming a two-species run, is to (1) find orthologous genes between the two species; (2) perform the cMonkey Markov Chain Monte Carlo procedure, using orthologous gene pairs identified in step 1 instead of individual genes, to produce biclusters of “orthologous core” genes; (3) for each organism, elaborate these orthologous core biclusters by adding and dropping individual genes (instead of orthologous gene pairs) using the normal single-species cMonkey process, with the restriction that no orthologous core genes are dropped; and optionally (4) perform separate single-species cMonkey runs on the remaining genes for each species. Orthologous genes are identified using existing tools, such as the Mouse Genomics Informatics database (Bult et al., 2008) or the InParanoid algorithm (Remm et al., 2001). Determination of biclusters in step 2 begins by calculating values for each species U and V, and as in Eq. (1). These are combined to produce a likelihood of an orthologous pair i belonging to cluster k similarly to Eq. (2)
| (3) |
7. Enrichment Analysis
Analysis and validation of biclusters is a key component of the cMonkey design. As a post-biclustering step, biclusters are analyzed for significant enrichment according to standard annotations such as GO (Ashburner et al., 2000), KEGG, and COG (Tatusov et al., 2000). These annotations provide a standard way to assign putative functions to biclusters, somewhat resolving the issue of giving meaningful names to biclusters in inferred networks. With the exception of the shared-ligand network derived from KEGG, these annotations are separate from the data used to infer the biclusters, so enrichment analysis also provides a means of assessing bicluster quality (see the Validation section below).
8. Integrating New Data Types
Integration of new data types into cMonkey is relatively straightforward. Additional network types are easily added as additional terms. New data, such as relative expression levels from proteomics experiments, could be incorporated as a fourth major data type (in addition to microarray expression, sequence, and networks) and added to the calculation of gik. In both cases, an appropriate annealing schedule for the weight given to the new network or data type would have to be determined.
B. Inferelator Pipeline
We have applied our network inference pipeline to a variety of different data sets (synthetic, prokaryotic, yeast, human white blood cells). We have developed several closely related variants of the core pipeline, which is composed of two core methods: (1) tlCLR, and (2) the Inferelator 1.0. A coarse prediction of the topology is made using tlCLR, which is further refined by the Inf. This pipeline of tlCLR followed by Inf is repeated for multiple permutations of the data set (resampling), resulting in an ensemble of predicted RNs, which is then combined into one final network. Here we present a brief description of tlCLR (for a more detailed description we refer to the reader to Greenfield et al., 2010 and Madar et al., 2010). Additionally, we present a modification to the core Inf method that allows for the incorporation of a priori known regulatory edges.
1. Problem Setup
As in the description of cMonkey, we denote the expression levels of the genes by x = (x1, …, xNg)T. We store the C observations of these Ng genes in an Ng × C matrix, where the columns correspond to the experimental observations. These observations can be of two types: time-series data (Xts), and steady-state data (Xss). Since we make explicit use of the time-series data in the description of our inference procedure, we denote time-series conditions by t1, t2, …, tk, (i.e., x(t1), x(t2), …, x(tk) are the k time-series observations that constitute the columns of Xts). Our inference procedure produces a network in the form of a ranked list of regulatory interactions, ranked according to confidence. We refer to the final list of confidences as an Ng × Np matrix Zfinal, where Np denotes the possible predictors. Element i,j of Zfinal represents our confidence in the existence of a regulatory interaction between xi and xj.
2. Core Method 1: Time-Lagged Context Likelihood of Relatedness
tlCLR (Greenfield et al., 2010; Madar et al., 2010) is a MI-based method that extends the original CLR algorithm (Faith et al., 2008) to take advantage of time-series data. MI (Shannon, 1948) is an information theory metric of mutual dependence between any two random variables. The original formulation of CLR was unable to learn directionality of regulatory edges as MI is a symmetric measure. In the tlCLR algorithm, we make explicit use of the time-series data to learn directed regulatory edges. We describe, in brief, three main steps: (1) model the temporal changes in expression as an ODE, (2) calculate the MI between every pair of genes, and (3) apply a background correction (filtering) step to remove least likely interactions. We refer the reader to Greenfield et al. (2010) and Madar et al. (2010) for a thorough description of this method.
We assume that the temporal changes in expression of each gene can be approximated by the linear ODE:
| (4) |
where αi is the first-order degradation rate of xi and the βij s are a set of dynamical parameters to be estimated. Note that the functional form presented above treats the rate of change of the response (xi) as linear function of the predictors (xjs). Here, we describe only this linear form for simplicity, but in several applications we employ more complex functional forms. The value of βij describes the extent and sign of the regulation of target gene xi by regulator xj. We store the dynamical parameters in a N × P matrix β, where N is the number of genes, and P is the number of possible regulators. Note that β is typically sparse, that is, most entries are 0 (reflecting the sparsity of transcriptional RNs). Later, we describe how to calculate the values βij by a constrained linear-regression scheme. First, we briefly describe how to use the time-series data in the context of improving the calculation of MI values between a gene xi and its potential regulator xj.
We first apply a finite approximation to the left-hand side of Eq. (4), for each xi, i = 1, …, Ng and rewrite it as a response vector yi, which captures the rate of change of expression in xj. We pair the response yi with a corresponding explanatory variable xj, j = 1 … Np. Note each xj is time-lagged with respect to the response yi, that is, xj(tk) is used to predict yj(tk+1). For more details of this transformation, we refer the reader to Greenfield et al. (2010). As a measure of confidence for a directed regulatory interaction between a pair of genes (xj → xi), we use MI, I(xi, xj), where a pair that shows a high MI score (relative to other pairs) is more likely to represent a true regulatory interaction. Note that I(yi, xj) ≠ I(yj, xi), making one regulatory direction more likely than the other. We refer to the MI calculated from I(yi, xj) as dynamic MI, as it takes advantage of the temporal information available from time-series data (distinguishing time-series data from steady-state data). As described above, we calculate I(xi, xj) and I (yi, xj) for every pair of genes and store the values in the form of two Ng × Np matrices Mstat and Mdyn, respectively. Note that Mstat is symmetric, while Mdyn is not. We now briefly describe how tlCLR integrates both static and dynamic MI to produce a final confidence score for each regulatory interaction. For a more detailed explanation, we refer the reader to Greenfield et al. (2010) and Madar et al. (2010).
For each regulatory interaction xj → xi, we compute two positive Z-scores (by setting all negative Z-scores to zero): one for the regulation of xi by xj based on dynamic-MI (i.e., based on Mdyn), , where σi is the standard deviation of the entries in row i of Mdyn; and one for the regulation of xi by xj based on both static and dynamic MI, where σj is the standard deviation of the entries in column j of Mstat. We combine the two scores into a final tlCLR score, . Note that some entries in ZtlCLR are zero, that is, ZtlCLR is somewhat sparse. The output of tlCLR, ZtlCLR, is used as the input to Inf, as only the highest ranked predictors from row i of ZtlCLR are considered as possible predictors for gene i
3. Core Method 2: Inferelator 1.0
We use Inf to learn a sparse dynamical model of regulation for each gene xi. As potential regulators of xi, we consider only the P highest confidence (non-zero) regulators (i.e., the Pi most-highly ranked regulators from row i of ZtlCLR). Accordingly, for each gene, xi, we denote this subset of potential regulators as xi. We then learn a sparse dynamical model of regulation for each xi as a function of the potential regulators xi using Inf. We assume that the time evolution in the xis is governed by , i = 1, …, N which is exactly Eq. (4) with our constraint on the number of regulators. LARS (Efron et al., 2004) is used to efficiently implement l1 constrained regression to determine a sparse set of the parameters β. This is done by minimizing the following objective function, amounting to a least-square estimate based on the ODE in Eq. (4) under an l1-norm penalty on regression coefficients,
| (5) |
where βols are the values of β determined by ordinary least squares regression (ols), and si, the shrinkage parameter. This parameter is in the range [0,1], and controls the sparsity of the model, with si = 0 amounting to a null model, and si = 1 amounting the full ols model. We select the optimal values of si by 10-fold cross validation. After applying this l1 regression, we have β, an Ng × Np matrix of dynamic parameters βij for each regulatory interaction xj → xi. We use the percentage of explained variance of each parameter βij as confidences in these regulatory interactions, as described in Greenfield et al. (2010). We store these confidences in ZInf. We combine these confidences in a rank-based way such that each method is weighted equally, as described in Greenfield et al. (2010), to generate ZtlCLR–Inf, which represents our confidence in each regulatory interaction after running our pipeline one time. We now describe how we resample our network inference pipeline to generate an ensemble of predicted networks (i.e., lists of confidences for each regulatory interaction).
4. Using Resampling to Improve Network Inference
To further improve the quality of our ranked list, we apply a resampling approach to the pipeline described above to generate an ensemble of putative RNs. We denote the matrix of response variables yi, i = 1, …, Ng by Y. Similarly we denote the matrix of predictor variables xj, j = 1, …, Np by X. We sample with replacement from the indices of the columns of Y, generating a permutation of the indices, c*. We use this permutation c* to permute the columns of Y and X, generating Y* and X*, respectively. Note that (1) c* is typically picked to be the number of conditions in the dataset (i.e., we sample from all experimental conditions), and (2) the columns of Y match the columns of X in the sense that the time-lagged relationship between the response and the predictors is preserved. We generated ZtlCLR, ZInf, and ZtlCLR–Inf as described before, with the only difference being that we use the response and explanatory vectors from Y* and X* instead of Y and X. We repeat this procedure B times. This generates an ensemble of B predicted RNs. The final weight we assign to each regulatory interaction is the median weight assigned to that interaction from each of the B networks. Thus, the final weight can be considered an “ensemble vote” of the confidence the ensemble of networks has in that edge: .
5. Incorporating Prior Information Directly into Network Inference
Our tlCLR–Inf pipeline is capable of inferring not only topology but also dynamical parameters, which can be used to predict the response of the system to new perturbations (Greenfield et al., 2010). Our predictions, like those of any network inference method, contain false-positive interactions. One way to improve the performance of network inference is to constrain the model selection procedure to incorporate regulatory interactions that are known a priori, as many databases of known regulatory interactions exist (Aranda et al., 2010; Bader et al., 2003; Ceol et al., 2010; Chautard et al., 2011; Croft et al., 2011; Goll et al., 2008; Knox et al., 2011; Lynn et al., 2008; Michaut et al., 2008; Prieto and Rivas, 2006; Razick et al., 2008; Stark et al., 2011). However, if one is studying a particular process (e.g., lymphoma) not all of the known interactions will be relevant in lymphoma. Thus, a method is needed that incorporates a known edge only if it is supported by the given data. We do so by solving Eq. (4) subject to the following constraint:
| (6) |
which is exactly Eq. (5) with the parameter θij (Yong-a-poi et al., 2008; Zou, 2006). This parameter is referred to as the adaptive weight, and regulates the degree to which βij is shrunk out of the model. If it is known from an external data type (e.g., literature mining, ChIP-seq, etc.) that xj regulates xi, then this knowledge can be incorporated by setting θij < 1, which will make it less likely that βij will be shrunk (removed from the model) by LARS. If there exists negative prior knowledge (i.e., knowledge that xj does not regulate xi), this can be incorporated by setting θij > 1. The exact values of θij that are needed to incorporate an a priori known interaction vary from dataset to dataset and must be chosen heuristically. This behavior is similar to that of many other methods for incorporating priors, including Bayesian methods, which require a heuristically chosen hyper-parameter to determine the shape of the prior (Mukherjee and Speed, 2008). In our method, once an informed choice of θij is made, an edge is incorporated only if it is supported by the data. Even if θij is set to a very low value (approaching zero, reflecting strong belief in the existence of this edge), the corresponding parameter, βij, will be non-zero only if there is support from the data set. This is exactly the desired behavior when we are given a priori knowledge that may or may not be completely relevant for our data sets.
C. Analysis and Visualization
Given the wide range of network properties, features of interest, and intended audiences, there is no “silver bullet” approach to visualizing biological networks. The most effective visualizations come from detailed analysis of the network, followed by a careful linking of important network properties to visual features such that interesting properties are immediately and intuitively obvious. The steps below show how Figs. 3-6 were created, and are intended to provide an arsenal of examples and tools to arrive at an effective combination of analysis and representation.
Fig. 3 uses publicly available mouse and human microarray data from GEO. The mouse data consisted of 508 conditions from the IMMGEN (Painter et al., 2011) data set of experiments on characterized mouse immune cell lineages (GEO accession number: GSE15907). Human microarray data were gathered from 23 different experiment sets measuring the response of human immune cells to different stimuli. In an attempt to mirror the conditions of the IMMGEN data set, only the control conditions from the different experiment sets were used, yielding a total of 140 conditions. The network was generated from a full run of the MScM and Inf pipelines on the data described above as follows:
Run MScM to generate a collection of 176 mouse and human biclusters.
Perform enrichment analysis over all biclusters using generic GO slim, GO, and KEGG.
Run the Inf pipeline using the mouse biclusters and known TFs for mouse alone to produce a preliminary mouse-specific network. Although the human biclusters are not used directly, their presence in the MScM run should improve the biological relevance of the mouse biclusters as discussed above.
Remove low-confidence edges (Inf z-score < 3.5, or |β| < 0.1) to produce a refined preliminary network.
Find biclusters with significantly enriched GO slim terms and label them with hallmarks of cancer associated with these terms. This results in 17 biclusters with hallmark annotations.
Reduce the network to the smallest possible network containing all 17 biclusters identified above along with their regulators, giving the final Fig. 3 network.
Further analysis of this network would begin with further investigation of the biclusters to obtain a better sense of the function represented by each bicluster. The Annotation Viewer (http://gaggle.systemsbiology.net/docs/geese/anno.php) is a Gaggled tool that allows browsing of arbitrary gene or bicluster annotations – in this case, bicluster GO and KEGG annotations. The CMMR provides more detailed examination of all facets of the biclusters: genes, conditions, residuals, etc. When bicluster functions are better understood, one can then ask whether the inferred regulatory interactions make sense, and investigate the significance of the observed separation of cancer hallmarks into two different clusters.
The next three examples (Figs. 4-6) use breast cell lines from normal breast epithelial tissue (MCF-10A) (Soule et al., 1990) and invasive, metastatic breast cancer tumor tissue (MDA-MB-231) (Cailleau et al., 1978). Data for each cell line were gathered from a total of eight GEO data sets, giving a total of 103 MCF-10A conditions and 121 MDA-MB-231 conditions covering roughly 12,000 genes. Proteomics data consisting of genes up- and downregulated in each cell line under two treatments were also provided. Here, we infer regulation of individual genes directly, instead of regulation of biclusters, so that we can overlay the proteomics data on the corresponding genes in the resulting network. As a result, we are unable to take advantage of the dimensionality reduction and noise reduction provided by cMonkey, and used the following heuristic approach instead. From the initial 12,000 genes, we selected those genes whose standard deviation across experiments was at the 75th percentile or better, then added the 2000 genes with the most differential expression according to significance analysis of microarrays (SAM) (Tusher et al., 2001). The final input set for inference on each cell line consisted of 4619 genes, 289 of which were TFs. Network inference proceeded as follows:
Perform separate Inf pipeline runs on each cell line to produce a ranked list of putative regulatory edges for each cell line.
Combine separate network edges from the individual runs into a single “differential network” where edge color shows the likelihood of each regulatory edge being active in one cell type or another. The rank of each edge in this differential network is determined by using Stouffer’s method to combine the Inf scores from the individual networks. Specificity to one cell line or the other is calculated as the log ratio of individual edge ranks.
Retain the top-ranked 5000 edges of the differential network; remove those with |β| < 0.05 for a final total of 4822 edges and 1866 nodes. Overlay proteomics data from two experimental conditions to produce the network are shown in Fig. 4.
Starting with the network in Fig. 4, find the largest connected sub-network of TFs using the Subgraph Creator Cytoscape plug-in. Map node out-degree to node size. Map node color to fraction of edges specific to one cell line or the other, counting only those edges with absolute value rank ratio above 4. This provides the network “summary” shown in Fig. 5.
Given lists of genes up- and downregulated in each cell line in the two proteomics experiments, load these lists into Sungear. Send each gene list to Cytoscape using the Gaggle and annotate genes according to cell line, experimental condition, and up- or downregulation.
Find the smallest sub-network for each condition that includes all differentially expressed genes. This identifies ITGB4 as a likely key gene involved in motility as discussed in Biological Insights.
Find the 1-hop network around ITGB4. Distinguish up- and downregulated genes for each treatment using the annotations assigned earlier to the Sungear-derived gene lists, producing Fig. 6.
Further analysis of the network in Fig. 6 might proceed as follows: send the set of differentially regulated genes back to Sungear to look for interesting intersections, such as over-representation within a particular intersection of conditions; or broadcast ITGB4 and some of its targets to the Firegoose, then from there to EMBL STRING to look for additional evidence for the inferred edges or grow the network further.
D. Validation
Validation of inferred networks of genes or biclusters (i.e., of predicted regulatory topology and kinetic models) is a critical challenge that has not been well resolved. In all cases, the best validation is of course follow-up experimentation to verify the computational results, but this approach is inherently limited by available time and resources.
Bicluster validation is currently a more tractable problem than network validation. Several metrics used to compare biclustering methods (Waltman et al., 2010) can also be used to assess the quality of individual biclusters and biclustering runs. Each bicluster has a residual score that shows the variance in expression data within the bicluster; lower residuals mean higher coherence in expression values. Significant bicluster enrichment implies that a cluster contains co-functional genes. High coverage of the input expression matrix, in terms of fraction of overall genes and conditions included in the overall set of biclusters, is favorable, as is a low degree of overlap between biclusters. For multi-species biclustering, the degree of conservation between species in a bicluster is also important.
Inferred networks present a more significant challenge, especially when no gold standard is available. A simple approach is to calculate some of the network statistics mentioned earlier: for example, compare the distribution of node in- and out-degrees to the expected power-law curves. However, the inference technique itself, as well as any means used to filter results or select sub-networks of interest, may skew these distributions: for example, the Inf limits the in-degree of any gene to a user-defined threshold. Another means of network validation is to determine the degree to which the inferred network recapitulates known network edges. However, this can become circular when known edges are used to provide priors for inference. Recent work by the DREAM consortium incorporated the prediction of multiple methods by different research groups into one “community” prediction, and experimentally validated the top-ranked predictions. Ideally, such integrative methods will continue to be developed and shed light on previously unknown biology. Although the notion of “community predictions” is novel and exciting, such vast resources not always exist. Even in such cases, methods that are integrative in terms both the algorithms and data types used show great promise in building global, predictive RNs of complex biological phenomena.
References
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K, Hermjakob H. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–D530. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake J, ABotstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Betel D, Hogue CW. BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Int Conf Intell Syst Mol Biol; Proceedings /.. International Conference on Intelligent Systems for Molecular Biology; ISMB; 1994. pp. 28–36. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- Bansal M, Gatta GD, di Bernardo D. Inference of gene regulatory networks and compound mode of action from time course gene expression profiles. Bioinformatics. 2006;22:815–822. doi: 10.1093/bioinformatics/btl003. [DOI] [PubMed] [Google Scholar]
- Bare JC, Shannon PT, Schmid AK, Baliga NS. The Firegoose: two-way integration of diverse data from different bioinformatics web resources with desktop applications. BMC Bioinformat. 2007;8:456. doi: 10.1186/1471-2105-8-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Dor A, Chor B, Karp R, Yakhini Z. Discovering local structure in gene expression data: the order-preserving submatrix problem. J Comput Biol. 2003;10:373–384. doi: 10.1089/10665270360688075. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann S, Ihmels J, Barkai N. Iterative signature algorithm for the analysis of large-scale gene expression data. Phys Rev E Stat Nonlin Soft Matter Phys. 2003;67:031902. doi: 10.1103/PhysRevE.67.031902. [DOI] [PubMed] [Google Scholar]
- Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, Thorsson V, Shannon P, Johnson MH, Bare JC, Longabaugh W, Vuthoori M, Whitehead K, Madar A, Suzuki L, Mori T, Chang D-E, Diruggiero J, Johnson CH, Hood L, Baliga NS. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, Thorsson V. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7:R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers PM, Pellegrini M, Thompson MJ, Fierro J, Yeates TO, Eisenberg D. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol. 2004;5:R35. doi: 10.1186/gb-2004-5-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult CJ, Eppig JT, Kadin JA, Richardson JE, Blake JA. The Mouse Genome Database (MGD): mouse biology and model systems. Nucleic Acids Res. 2008;36:D724–D728. doi: 10.1093/nar/gkm961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte AJ, Kohane IS. Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. 2000:418–429. doi: 10.1142/9789814447331_0040. [DOI] [PubMed] [Google Scholar]
- Butts CT. Social network analysis with sna. J Stat Softw. 2008;24:1–51. doi: 10.18637/jss.v024.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailleau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. in vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol A, Aryamontri AC, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–D540. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitankar V, Ghosh P, Perkins EJ, Gong P, Zhang C. Time lagged information theoretic approaches to the reverse engineering of gene regulatory networks. BMC Bioinformat. 2010;11(Suppl 6):S19. doi: 10.1186/1471-2105-11-S6-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chautard E, Fatoux-Ardore M, Ballut L, Thierry-Mieg N, Ricard-Blum S. MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res. 2011;39:D235–D240. doi: 10.1093/nar/gkq830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh Y-H, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung W-K, Clarke ND, Wei C-L, Ng H-H. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Church GM. Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol. 2000;8:93–103. [PubMed] [Google Scholar]
- Christley S, Nie Q, Xie X. Incorporating existing network information into gene network inference. PloS One. 2009;4:e6799. doi: 10.1371/journal.pone.0006799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D’Eustachio P, Stein L. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–D700. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Goadrich M. The relationship between precision-recall and ROC curves. Proceedings of the 23rd International Conference on Machine Learning – ICML’06; 2006. pp. 233–240. [Google Scholar]
- De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nature reviews. Microbiology. 2010;8:717–729. doi: 10.1038/nrmicro2419. [DOI] [PubMed] [Google Scholar]
- di Bernardo D, Bansal M, Gatta GD. Inference of gene regulatory networks and compound mode of action from time course gene expression profiles. Bioinformatics. 2006;22:815–822. doi: 10.1093/bioinformatics/btl003. [DOI] [PubMed] [Google Scholar]
- di Bernardo D, Thompson MJ, Gardner TS, Chobot SE, Eastwood EL, Wojtovich AP, Elliott SJ, Schaus SE, Collins JJ. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat Biotechnol. 2005;23:377–383. doi: 10.1038/nbt1075. [DOI] [PubMed] [Google Scholar]
- DiMaggio PA, Jr, McAllister SR, Floudas CA, Feng XJ, Rabinowitz JD, Rabitz HA. Biclustering via optimal re-ordering of data matrices in systems biology: rigorous methods and comparative studies. BMC Bioinformat. 2008;9:458. doi: 10.1186/1471-2105-9-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Hastie T, Johnstone I, Tibshirani R. Least angle regression. Ann Statist Data. 2004:407–451. [Google Scholar]
- Elemento O, Tavazoie S. Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol. 2005;6:R18. doi: 10.1186/gb-2005-6-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Driscoll ME, Fusaro VA, Cosgrove EJ, Hayete B, Juhn FS, Schneider SJ, Gardner TS. Many Microbe Microarrays Database: uniformly normalized Affymetrix compendia with structured experimental metadata. Nucleic Acids Res. 2008;36:D866–D870. doi: 10.1093/nar/gkm815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:54–66. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Linial M, Nachman I. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- Friedman N, Nachman I. Learning Bayesian Network Structure from Massive Datasets: The “Sparse Candidate” Algorithm. UAI; San Fransisco, CA: 1999. pp. 206–215. [Google Scholar]
- Gan X, Liew AW, Yan H. Discovering biclusters in gene expression data based on high-dimensional linear geometries. BMC Bioinformat. 2008;9:209. doi: 10.1186/1471-2105-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier F, Timmer J, Fleck C. Reconstructing gene-regulatory networks from time series, knock-out data, and prior knowledge. BMC Syst Biol. 2007;11 doi: 10.1186/1752-0509-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevaert O, Van Vooren S, De Moor B. A framework for elucidating regulatory networks based on prior information and expression data. Ann N Y Acad Sci. 2007;1115:240–248. doi: 10.1196/annals.1407.002. [DOI] [PubMed] [Google Scholar]
- Goll J, Rajagopala SV, Shiau SC, Wu H, Lamb BT, Uetz P. MPIDB: the microbial protein interaction database. Bioinformatics. 2008;24:1743–1744. doi: 10.1093/bioinformatics/btn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield A, Madar A, Ostrer H, Bonneau R. DREAM4: combining genetic and dynamic information to identify biological networks and dynamical models. PLoS ONE. 2010;5:e13397. doi: 10.1371/journal.pone.0013397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, Wang Y, MacNeill J, Mitchell J, Gygi SP, Rush J, Polakiewicz RD, Comb MJ. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–697. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hartigan JA. Direct clustering of a data matrix. J Amer Statist Assoc. 1972;67:123–129. [Google Scholar]
- Husmeier D, Werhli AV. Bayesian integration of biological prior knowledge into the reconstruction of gene regulatory networks with Bayesian networks. Computational systems bioinformatics/Life Sciences Society. Comput Syst Bioinformat Conf. 2007;6:85–95. [PubMed] [Google Scholar]
- Huttenhower C, Mutungu KT, Indik N, Yang W, Schroeder M, Forman JJ, Troyanskaya OG, Coller HA. Detailing regulatory networks through large scale data integration. Bioinformatics. 2009;25:3267–3274. doi: 10.1093/bioinformatics/btp588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE. 2010;5:e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Ranish Ja, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science (New York, NY) 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Ihmels J, Bergmann S, Berman J, Barkai N. Comparative gene expression analysis by differential clustering approach: application to the Candida albicans transcription program. PLoS Genet. 2005;1:e39. doi: 10.1371/journal.pgen.0010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Kacmarczyk T, Waltman P, Bate A, Eichenberger P, Bonneau R. Comparative Microbial Modules Resource: Generation and Visualization of Multi-species Biclusters. PLoS computational biology. 2011;7:e1002228. doi: 10.1371/journal.pcbi.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger Y, Basri R, Chang JT, Gerstein M. Spectral biclustering of microarray data: coclustering genes and conditions. Genome Res. 2003;13:703–716. doi: 10.1101/gr.648603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39:D1035–D1040. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzeroni L, Owen A. Plaid models for gene expression data. Statistica Sinica. 1999;12:61–86. [Google Scholar]
- Lee S-I, Dudley AM, Drubin D, Silver PA, Krogan NJ, Pe’er D, Koller D. Learning a prior on regulatory potential from eQTL data. PLoS Genet. 2009;5:e1000358. doi: 10.1371/journal.pgen.1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K-C, Wang X. Gene regulatory network reconstruction using conditional mutual information. EURASIP J Bioinformat Syst Biol. 2008;2008:253894. doi: 10.1155/2008/253894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Huggins P, Bar-Joseph Z. Cross species analysis of microarray expression data. Bioinformatics. 2009;25:1476–1483. doi: 10.1093/bioinformatics/btp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, Gardy JL, Roche FM, Chan THW, Shah N, Lo R, Naseer M, Que J, Yau M, Acab M, Tulpan D, Whiteside MD, Chikatamarla A, Mah B, Munzner T, Hokamp K, Hancock REW, Brinkman FSL. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol Syst Biol. 2008;4:218. doi: 10.1038/msb.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar A, Greenfield A, Ostrer H, Vanden-Eijnden E, Bonneau R. The inferelator 2.0: A scalable framework for reconstruction of dynamic regulatory network models. IEEE Eng Med Biol Soc Conf; Conference proceedings:.. Annual International Conference of the IEEE Engineering in Medicine and Biology Society; 2009. pp. 5448–5451. [DOI] [PubMed] [Google Scholar]
- Madar A, Greenfield A, Vanden-Eijnden E, Bonneau R. DREAM3: network inference using dynamic context likelihood of relatedness and the inferelator. PLoS ONE. 2010;5:e9803. doi: 10.1371/journal.pone.0009803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Costello J, Küffner R, Vega N, Prill R, Camacho D, Allison K, Consortium TD, Kellis M, Collins J, Stolovitzky G. Wisdom of crowds for gene network inference. doi: 10.1038/nmeth.2016. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Mattiussi C, Floreano D. Combining multiple results of a reverse-engineering algorithm: application to the DREAM five-gene network challenge. Ann N Y Acad Sci. 2009a;1158:102–113. doi: 10.1111/j.1749-6632.2008.03945.x. [DOI] [PubMed] [Google Scholar]
- Marbach D, Mattiussi C, Floreano D. Replaying the evolutionary tape: biomimetic reverse engineering of gene networks. Ann N Y Acad Sci. 2009b;1158:234–245. doi: 10.1111/j.1749-6632.2008.03944.x. [DOI] [PubMed] [Google Scholar]
- Marbach D, Prill RJ, Schaffter T, Mattiussi C, Floreano D, Stolovitzky G. Revealing strengths and weaknesses of methods for gene network inference. Proc Natl Acad Sci U S A. 2010;107:6286–6291. doi: 10.1073/pnas.0913357107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Schaffter T, Mattiussi C, Floreano D. Generating realistic in silico gene networks for performance assessment of reverse engineering methods. J Comput Biol. 2009c;16:229–239. doi: 10.1089/cmb.2008.09TT. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera RD, Califano A. ARACNE: an algorithm for the reoncstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformat. 2006;15:1–15. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur J, Ritter D, Reinelt G, Kaderali L. Reconstructing nonlinear dynamic models of gene regulation using stochastic sampling. BMC Bioinformat. 2009;10:448. doi: 10.1186/1471-2105-10-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor JC, Yanai I, Clodfelter KH, Mintseris J, DeLisi C. Predictome: a database of putative functional links between proteins. Nucleic Acids Res. 2002;30:306–309. doi: 10.1093/nar/30.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaut M, Kerrien S, Montecchi-Palazzi L, Chauvat F, Cassier-Chauvat C, Aude J-C, Legrain P, Hermjakob H. InteroPORC: automated inference of highly conserved protein interaction networks. Bioinformatics. 2008;24:1625–1631. doi: 10.1093/bioinformatics/btn249. [DOI] [PubMed] [Google Scholar]
- Mirkin BG. Mathematical classification and clustering. Kluwer Academic Publishers; Dordrecht, the Netherlands: 1996. [Google Scholar]
- Morgan JN, Sonquist JA. Problems in the analysis of survey data, and a proposal. J Amer Stat Assoc. 1963:415–434. [Google Scholar]
- Mukherjee S, Speed TP. Network inference using informative priors. Proc Natl Acad Sci U S A. 2008;105:14313–14318. doi: 10.1073/pnas.0802272105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Painter MW, Davis S, Hardy RR, Mathis D, Benoist C. Transcriptomes of the B and T lineages compared by multiplatform microarray profiling. J Immunol. 2011;186:3047–3057. doi: 10.4049/jimmunol.1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinna A, Soranzo N, de la Fuente A. From knockouts to networks: establishing direct cause-effect relationships through graph analysis. PloS ONE. 2010;5:e12912. doi: 10.1371/journal.pone.0012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poultney CS, Gutiérrez Ra, Katari MS, Gifford ML, Paley WB, Coruzzi GM, Shasha DE. Sungear: interactive visualization and functional analysis of genomic datasets. Bioinformatics (Oxford, England) 2007;23:259–261. doi: 10.1093/bioinformatics/btl496. [DOI] [PubMed] [Google Scholar]
- Prieto C, Rivas JDL. APID: agile protein interaction dataanalyzer. Nucleic Acids Res. 2006;34:W298–W302. doi: 10.1093/nar/gkl128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prill RJ, Marbach D, Saez-Rodriguez J, Sorger PK, Alexopoulos LG, Xue X, Clarke ND, Altan-Bonnet G, Stolovitzky G. Towards a rigorous assessment of systems biology models: the DREAM3 challenges. PloS ONE. 2010;5:e9202. doi: 10.1371/journal.pone.0009202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- Razick S, Magklaras G, Donaldson IM. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformat. 2008;9:405. doi: 10.1186/1471-2105-9-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformat. 2006;7:280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs K, Gifford D, Jaakkola T, Sorger P, Lauffenburger DA. Bayesian network approach to cell signaling pathway modeling. Science’s STKE: signal transduction knowledge environment. 2002;2002:pe38. doi: 10.1126/stke.2002.148.pe38. [DOI] [PubMed] [Google Scholar]
- Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science (New York, NY) 2005;308:523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt WA, Jr, Raab RM, Stephanopoulos G. Elucidation of gene interaction networks through time-lagged correlation analysis of transcriptional data. Genome Res. 2004;14:1654–1663. doi: 10.1101/gr.2439804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Shannon C. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379–423. [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon PT, Reiss DJ, Bonneau R, Baliga NS. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformat. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AH, Wolf DM, Wang P, Arkin AP. Modularity of stress response evolution. Proc Natl Acad Sci U S A. 2008;105:7500–7505. doi: 10.1073/pnas.0709764105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EA, Corn RM. Surface plasmon resonance imaging as a tool to monitor biomolecular interactions in an array based format. Appl Spectrosc. 2003;57:320A–332A. doi: 10.1366/000370203322554446. [DOI] [PubMed] [Google Scholar]
- Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442–3444. doi: 10.1093/nar/28.18.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Stark C, Breitkreutz B-J, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Auken KV, Wang X, Shi X, Reguly T, Rust JM, Winter A, Dolinski K, Tyers M. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D700. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovitzky G, Monroe D, Califano A. Dialogue on reverse-engineering assessment and methods: the DREAM of high-throughput pathway inference. Ann N Y Acad Sci. 2007;1115:1–22. doi: 10.1196/annals.1407.021. [DOI] [PubMed] [Google Scholar]
- Supper J, Strauch M, Wanke D, Harter K, Zell A. EDISA: extracting biclusters from multiple time-series of gene expression profiles. BMC Bioinformat. 2007;8:334. doi: 10.1186/1471-2105-8-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Forrest ARR, van Nimwegen E, Daub CO, Balwierz PJ, Irvine KM, Lassmann T, Ravasi T, Hasegawa Y, de Hoon MJL, Katayama S, Schroder K, Carninci P, Tomaru Y, Kanamori-Katayama M, Kubosaki A, Akalin A, Ando Y, Arner E, Asada M, Asahara H, Bailey T, Bajic VB, Bauer D, Beckhouse AG, Bertin N, Björkegren J, Brombacher F, Bulger E, Chalk AM, Chiba J, Cloonan N, Dawe A, Dostie J, Engström PG, Essack M, Faulkner GJ, Fink JL, Fredman D, Fujimori K, Furuno M, Gojobori T, Gough J, Grimmond SM, Gustafsson M, Hashimoto M, Hashimoto T, Hatakeyama M, Heinzel S, Hide W, Hofmann O, Hörnquist M, Huminiecki L, Ikeo K, Imamoto N, Inoue S, Inoue Y, Ishihara R, Iwayanagi T, Jacobsen A, Kaur M, Kawaji H, Kerr MC, Kimura R, Kimura S, Kimura Y, Kitano H, Koga H, Kojima T, Kondo S, Konno T, Krogh A, Kruger A, Kumar A, Lenhard B, Lennartsson A, Lindow M, Lizio M, Macpherson C, Maeda N, Maher CA, Maqungo M, Mar J, Matigian NA, Matsuda H, Mattick JS, Meier S, Miyamoto S, Miyamoto-Sato E, Nakabayashi K, Nakachi Y, Nakano M, Nygaard S, Okayama T, Okazaki Y, Okuda-Yabukami H, Orlando V, Otomo J, Pachkov M, Petrovsky N, Plessy C, Quackenbush J, Radovanovic A, Rehli M, Saito R, Sandelin A, Schmeier S, Schönbach C, Schwartz AS, Semple CA, Sera M, Severin J, Shirahige K, Simons C, St Laurent G, Suzuki M, Suzuki T, Sweet MJ, Taft RJ, Takeda S, Takenaka Y, Tan K, Taylor MS, Teasdale RD, Tegnér J, Teichmann S, Valen E, Wahlestedt C, Waki K, Waterhouse A, Wells CA, Winther O, Wu L, Yamaguchi K, Yanagawa H, Yasuda J, Zavolan M, Hume DA, Arakawa T, Fukuda S, Imamura K, Kai C, Kaiho A, Kawashima T, Kawazu C, Kitazume Y, Kojima M, Miura H, Murakami K, Murata M, Ninomiya N, Nishiyori H, Noma S, Ogawa C, Sano T, Simon C, Tagami M, Takahashi Y, Kawai J, Hayashizaki Y. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet. 2009;41:553–562. doi: 10.1038/ng.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Sharan R, Kupiec M, Shamir R. Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc Natl Acad Sci U S A. 2004;101:2981–2986. doi: 10.1073/pnas.0308661100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Barkai N. Comparative analysis indicates regulatory neofunctionalization of yeast duplicates. Genome Biol. 2007;8:R50. doi: 10.1186/gb-2007-8-4-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J. Regulatory sequence analysis tools. Nucleic Acids Res. 2003;31:3593–3596. doi: 10.1093/nar/gkg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltman P, Kacmarczyk T, Bate AR, Kearns DB, Reiss DJ, Eichenberger P, Bonneau R. Multi-species integrative biclustering. Genome Biol. 2010;11:R96. doi: 10.1186/gb-2010-11-9-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannakakis M. Node-deletion problems on bipartite graphs. SIAM J Comput. 1981;10:310–327. [Google Scholar]
- Yip KY, Alexander RP, Yan K-K, Gerstein M. Improved reconstruction of in silico gene regulatory networks by integrating knockout and perturbation data. PLoS ONE. 2010;5:e8121. doi: 10.1371/journal.pone.0008121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong-a-poi J, Someren EV, Bellomo D, Reinders M. Adaptive least absolute regression network analysis improves genetic network reconstruction by employing prior knowledge. Commun Theory. 2008:1–14. [Google Scholar]
- Zhou J, Deng Y, Luo F, He Z, Tu Q, Zhi X. Functional molecular ecological networks. mBio. 2010;1:e00169–00110. doi: 10.1128/mBio.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H. The adaptive lasso and its oracle properties. J Amer Statis Assoc. 2006;101:1418–1429. [Google Scholar]