

Abstract
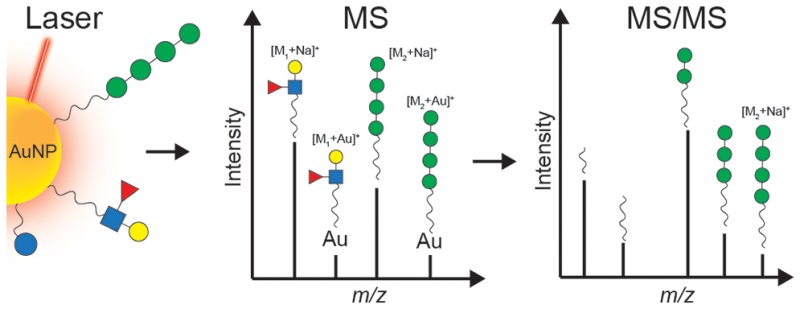
Biofunctionalized gold nanoparticles (AuNPs) enable innovative translational research and development in biomedicine. Biomolecules such as peptides, proteins, lipids, and carbohydrates can be assembled onto AuNPs to yield nanomaterials with unique properties for applications in imaging, photothermal therapy, vaccination strategies, and drug delivery. The characterization of functionalized AuNPs still remains an analytical challenge that normally requires the combination of multiple techniques. Laser desorption/ionization (LDI) and matrix-assisted LDI (MALDI) have been applied successfully in combination with time-of-flight (TOF) mass spectrometry (MS) for the analysis of the surface chemistry of AuNPs functionalized with synthetic ligands, however only for ligands with a molecular mass limited to 1000 Da. TOF-MS-based approaches in addition exhibit limited performance in terms of mass resolution and MS/MS possibilities. To overcome these limitations, we designed an approach for the analysis of AuNPs based on ultrahigh resolution Fourier transform ion cyclotron resonance (FTICR) MS and a combination of LDI and MALDI. To illustrate the performance of the method, we present a comprehensive characterization of the surface chemistry of AuNPs conjugated via a thiol-ending linker to either the ovalbumin peptide (OVA 323-339), the Lewis X antigen (Galβ1-4[Fucα1-3]GlcNAcβ1) trisaccharide, the tetramannoside Manα1-2Manα1-2Manα1-3Manα1, or a mixture of both carbohydrates. Collision-induced dissociation (CID) was used to characterize the structure of pseudomolecular ions generated by LDI/MALDI in-depth. These included [M + H]+ and [M + Na]+, and importantly also [M + Au]+ and [M + 2Au–H]+ ions. This first observation of gold-containing pseudomolecular ions provides direct evidence for the Au-conjugation of ligands. In addition, we show the applicability of the method to monitor proteolytic cleavage of peptides that are conjugated to the AuNP surface. The presented LDI/MALDI–FTICR–MS and MS/MS approach will be applicable to the characterization of a wide range of functionalized AuNPs.
Keywords: gold nanoparticles, mass spectrometry, surface functionalization, MALDI−FTICR−MS, surface chemistry, biofunctionalized nanomaterial
Gold nanoparticles (AuNPs) are versatile nanomaterials with a vast field of applications.1,2 The possibility to tailor specific biological, physical, and chemical properties through particle size and via surface modification makes AuNPs one of the most promising and studied tools in nanotechnology-enabled biomedicine. AuNPs display a high adsorption capacity due to a large surface-to-volume ratio, they are relatively nontoxic, can be functionalized with a variety of biomolecules, and they strongly interact with light by a physical phenomenon called surface plasmon resonance. These characteristics have stimulated the exploration of AuNPs in medical applications including imaging, photothermal therapy, vaccination, and drug delivery.3−6 Development of functional nanoparticles that carry multiple (bio)molecules will further extend the field of (biomedical) applications.2
AuNPs in the 2–100 nm size range are typically synthesized in solution by redox reactions and can be functionalized by introducing molecules containing a thiol group to form a strong nanoparticle–sulfur bond.7,8 Structure characterization of the surface of nanomaterials with regard to conjugation chemistry, ligand definition, and overall quality control is a key-factor in the development of functionalized AuNPs. A suitable method for the characterization of AuNPs must provide reliable information on the composition of the AuNP surface with respect to ligand structure, density, and stoichiometry. Analytical techniques should also allow the detection of possible modifications induced by the exposure to enzymes and other reactants in a biological environment such as the bloodstream or target organs. Clearly, characterization of biofunctional AuNPs is challenging, and may require a multimethod approach, including complementary analytical techniques such as UV–vis, IR, and NMR spectroscopy, enzyme-linked immunosorbent assay, X-ray crystallography, and mass spectrometry (MS).9
MS has proved to be a powerful tool for the characterization of functionalized AuNPs by providing information with regard to the mass of the ligands.10 While ESI-MS and ESI-MS/MS have been used for the characterization of gold nanoclusters,11,12 laser desorption/ionization (LDI) MS has been successfully applied in the mass analysis of various AuNPs ligands;10,13−16 however, only small synthetic ligands (i.e., <1000 Da) could be analyzed.17 This limitation is caused by a low desorption/ionization efficiency of specific ligands in commercial matrix assisted LDI-time of flight (MALDI-TOF) platforms and a decreased sensitivity of TOF mass analyzers at higher m/z. The combination of LDI–MS with MALDI allowed an enhancement of the detection of the ligands of AuNPs in biological samples.18 LDI/MALDI methods rely on the fact that AuNPs absorb laser irradiation resulting in the release and ionization of ligands that are subsequently detected by MS. For many applications, mass measurements by LDI/MALDI–MS and tandem mass spectrometry (MS/MS) may be required for full structural identification of complex ligands such as lipids, peptides, and carbohydrates which are of interest for biomedical applications and often have a mass higher than 1000 Da. Unfortunately, LDI/MALDI–MS analyses on AuNPs have hitherto been performed on time-of-flight (TOF) MS instruments with limited MS/MS possibilities.
To overcome the limitations of LDI-TOF–MS instruments with respect to mass range, mass resolution, accuracy, and MS/MS possibilities for the comprehensive analysis of biomolecular functionalization of AuNPs, we here apply ultrahigh resolution Fourier transform ion cyclotron resonance (FTICR) MS and a combination of LDI and MALDI. FTICR MS provides the highest resolving power, mass accuracy, and precision among the different mass analyzers and provides a higher dynamic range than TOF-MS.19,20 To illustrate the applicability of the FTICR MS method, we characterize ∼2 nm AuNPs conjugated via a thiol-ending linker to either the ovalbumin peptide (OVA 323-339), the Lewis X antigen (Galβ1-4[Fucα1-3]GlcNAcβ1) trisaccharide, the tetramannoside Manα1-2Manα1-2Manα1-3Manα1, or both carbohydrates. In addition, LDI/MALDI–FTICR MS using 1,5-diaminonaphthalene (1,5-DAN) as a matrix and collision-induced dissociation (CID) fragmentation studies on pseudomolecular ions, such as [M + H]+, [M + Au]+, and [M + 2Au–H]+, allowed unequivocal proof of attachment and composition of ligands to the AuNP surface.
Results and Discussion
OVA 323-339 peptide, Lewis X, and Tetramannoside AuNPs
Peptide- and carbohydrate-functionalized AuNPs find increased application in various biomedical research areas.21 For the current study, we synthesized AuNPs conjugated to the immunogenic OVA 323-339 peptide, and two different carbohydrate-coupled AuNPs. The OVA peptide was previously explored in the context of AuNPs as a T-cell epitope-containing carrier for immunization with S. pneumonia antigens.22 Carbohydrate AuNPs were constructed of a tetramannoside, recently also used to mimic HIV gp120 oligomannose clusters,23,24 and of the immunomodulatory Lewis X antigen.25 In each case a relatively long bifunctional thiol-ending linker (i.e., SCN-TEG-C11-SH) was used to couple the functional carbohydrate ligand through a thiourea bond as previously described.26 A glucose thiol-ending short derivative (i.e., -C5-SH) was used to modulate the density of the functional carbohydrate ligands on the AuNPs while still allowing them to protrude from the AuNP surface. A schematic of the AuNPs used in this study is depicted in Figure 1.
Figure 1.
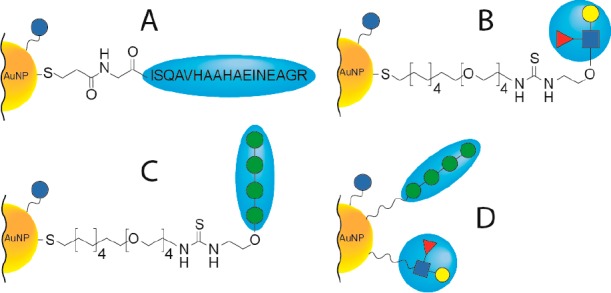
Gold nanoparticles characterized in this study. AuNPs were all functionalized with glucose-based ligands (indicated with solid blue circles) and in addition with various ligands (one-pot procedure described in Methods). Four different types of AuNPs were obtained: (A) AuNP functionalized with an ovalbumin (OVA 323-339) peptide-based ligand; (B) AuNP functionalized with a Lewis X ligand; (C) AuNP functionalized with a tetramannoside ligand; (D) AuNP functionalized with both Lewis X and tetramannoside ligands.
Analysis of OVA 323-339 peptide AuNPs by LDI-, LDI/MALDI–FTICR MS and MS/MS
In this study, we aimed to analyze AuNPs functionalized with biomolecules with a mass larger than 1000 Da. First, AuNPs of ∼2 nm conjugated to a modified OVA 323-339 peptide (monoisotopic mass = 1917.912 Da) were analyzed by both LDI- and LDI/MALDI–FTICR MS. As depicted in Figure 2A, LDI results in a pseudomolecular ion of the OVA peptide ligand ([M + Na]+), implying a release from the surface of the AuNPs upon ionization. In combination with the 1,5-DAN matrix (Figure 2B) the spectrum changes dramatically: the OVA peptide ligand is now observed as a protonated pseudomolecular ion at m/z 1918.933 at a peak intensity about 80 times higher compared to LDI. The 1,5-DAN MALDI matrix exhibits a high hydrogen donor capacity which in general enhances the signal compared to LDI-measurements (see Supporting Information, Figure S1). LDI-TOF MS did not yield any ions of the OVA peptide ligand, whereas LDI/MALDI-TOF MS resulted in a low quality spectrum compared to LDI/MALDI–FTICR MS. Thus, we conclude that the mass limitation of 1000 Da reported earlier by Vachet and co-workers17 can be overcome by LDI/MALDI-TOF MS, however for confident characterizations LDI/MALDI–FTICR MS which offers superior mass resolution and fragmentation capacity is required.
Figure 2.

Relevant parts of the FTICR mass spectra of AuNPs with OVA 323-339 peptide-based ligand obtained from LDI only (A) and LDI/MALDI (B). The use of 1,5-DAN as a MALDI matrix resulted in the detection of pseudomolecular ions [M + H]+, [M + Au]+, and [M + 2Au–H]+, highlighted with three different colors. Additional pseudomolecular ions observed were (a) [M–S–H + 1,5-DAN]+ at m/z 2043.030, (b) [M–H + 1,5-DAN]+ at m/z 2074.998, and (c) pseudomolecular ion of coupled glucose-based and OVA-based ligands at m/z 2199.038 (as explained in the Results and Discussion section). Further structure characterization of [M + Au]+, [M + H]+, and [M + 2Au–H]+ was performed by collision-induced dissociation (CID) and as an example the tandem mass spectrum of the first species is shown in C (the other two can be found in Figure S2). The obtained high sequence coverages (D) allowed to determine the preferred coordination site(s) of Au+ by evaluating all gold-containing fragment ions (indicated with yellow circles).
Interestingly, LDI/MALDI–FTICR MS allowed the detection of the OVA peptide ligand as [M + Au]+ and [M + 2Au–H]+ at m/z 2114.892 and m/z 2310.859, respectively (Figure 2B). To our knowledge, although Au+ ions have been detected in LDI spectra of AuNPs,18,27 the generation of gold-cluster ions linked to the ligands of functionalized AuNPs by LDI-MS has never been reported. Upon using AuCl3 in combination with alpha-cyano-4-hydroxycinnamic acid, gold ion-angiotensin peptide complexes (i.e., [M + Au(I)]+ and [M + 2Au(I)–H]+) have been observed in MALDI-spectra, with preferred coordination sites for gold being Cys, His, and Arg as determined from tandem-MS analysis.28 In our study, no single Au+ ions or Au+ clusters were detected and both the synthesis protocol (i.e., using 21 equiv of reducing agent) and the purification procedures (i.e., extensive dialysis) rule out the presence of free Au+ in solution. Thus, we rationalize that the observed [M + Au]+ and [M + 2Au–H]+ species result from a detachment of ligands together with Au+ ions from the nanoparticle surface.
An advantage of LDI/MALDI–FTICR MS, essential to the analysis of AuNPs with complex ligands, is the possibility of combining accurate ion mass determination with tandem mass spectrometry, using, for example, CID. The MS/MS spectra with assignments of the [M + Au]+, and [M + H]+and [M + 2Au–H]+ species of the OVA peptide-based ligand are depicted in Figure 2C and Figure S2, respectively. The CID of [M + H]+ resulted in fragments representing nearly all peptide bond cleavages (sequence coverage 94%, Figure S2B), fully confirming the OVA 323-339 amino acid sequence. In general, for this species, b-type fragmentation was favorable compared to y-type (a scheme of b- and y-type fragmentation is reported in Figure S3), although the most abundant fragment ion was a y-type ion detected at m/z 659.347 generated from the cleavage between Glu11 and Ile12. Full sequence coverage was obtained for [M + Au]+ (Figure 2D) and, as for [M + H]+, the most abundant CID fragment was the protonated y-type ion detected at m/z 659.347 (cleavage at Glu11-Ile12) suggesting that Au+ is not localized on the C-terminal part of the peptide. The detection of both protonated (more abundant) and gold-linked (less abundant) fragment ions for the same fragmentation site did not allow an unambiguous localization of Au+. However, the data indicate that both the terminal sulfur and His9 are preferred coordination sites of Au+. The MS/MS analysis of [M + 2Au–H]+ (Figure S2C,D) showed that the fragmentation between Glu11 and Ile12 was not predominant and while y-type ions carried one Au+ (main species), b-type ions carried two gold atoms. This suggests that both His6 and the terminal sulfur atom are involved in the coordination of Au+. Internal fragmentation (see inset Supporting Information, Figure S2C) resulted in low-intensity fragment ions containing His6 and gold. These data obtained for [M + Au]+ and [M + 2Au–H]+ species are indicative of the affinity of His6 and His9 for Au+ in the gas phase only, since the structure of b- and y-type fragments is an important factor for the coordination of cations. In contrast, on the surface of AuNPs a gold–histidine bond is energetically unfavorable compared to the strong gold–sulfur bond. In fact, Aldeek et al. have shown that proteins expressing an N-terminus polyhistidine tag self-assembled onto AuNPs only when NPs were partially capped with thiol-terminated ligands or were stabilized with a weakly binding cap such as citrate groups.29
As previously reported, LDI of AuNPs functionalized through an Au–S bond can lead to loss of SH2 from the ligand resulting in the formation of a double bond between adjacent carbons.13,18 As depicted in the inset in Figure 2B, this loss was observed at m/z 1884.945. In addition, due to the high hydrogen-donor capacity of the 1,5-DAN matrix, the reduced form [M–S + H]+ of [M–SH2 + H]+ was detected at m/z 1886.960. The structural characterization of these species by CID allowed confirmation of the presence/absence of a double bond in the structure of the linker (Supporting Information, Figure S4).
The OVA peptide AuNPs were synthesized using the glucose ligand and the OVA peptide ligand at a concentration ratio of about 9:1. The glucose ligand (theoretical m/z 282.1137) was hardly detected by both LDI- and LDI/MALDI–FTICR MS. Interestingly, the coupled glucose- and OVA peptide-ligand attached to each other via an S–S bond was detected at m/z 2199.038 (Figure 2B). Earlier, Yan et al. reported the formation of S–S bonds between ligands from monolayer-protected AuNPs induced by LDI.13 The CID spectrum of the glucose-OVA peptide species is reported in Figure S5. The most abundant fragment ions were b-type ions generated after the loss of the glucose molecule (i.e., fragment ion at m/z 2037.000), which is in line with the well-known lability of glycosidic bonds in CID of protonated glycoconjugates.30 In-source decay of the OVA peptide ligand was observed in both LDI- and LDI/MALDI–FTICR MS analyses. Preliminary data suggest that the use of sinapinic acid as the MALDI matrix instead of 1,5-DAN can drastically reduce in-source decay (Figure S6).
The detection of different forms of the OVA peptide ligand shows the potential of our strategy for the analysis of ligands with molecular weight higher than 1000 Da. In our previous study, we have shown that MALDI–FTICR MS allows the measurements at isotopic resolution of proteins up to about m/z 17 000.31 From this, we rationalize that the analysis of proteins linked to AuNPs is limited to similar sizes.
It is known from surface plasmon resonance studies that AuNPs with different particle sizes have different light absorption properties (e.g., from a laser). The here reported strategies apply to AuNPs with a diameter of approximately 2 nm. An additional and independent evaluation would be needed in case larger particles were of interest, or other metal or metal oxide were considered.
Enzymatic Digestion of OVA Peptide AuNPs and LDI/MALDI–FTICR MS
In situ activation of AuNPs, for example by the proteolytic cleavage of bioactive peptides or enzymatic release of fluorescent molecules for visualization allows innovative applications of AuNPs in biomedicine.32,33 Therefore, it is essential to develop MS-methods for the characterization of biofunctionalized AuNPs after modification or activation, providing information about the changes of surface chemistry and composition. Here, we studied the surface chemistry of the OVA peptide AuNPs after enzymatic digestion. To this end, the OVA peptide AuNPs were partially digested using endoproteinase Glu-C and then characterized by LDI/MALDI–FTICR MS and MS/MS. The undigested intact OVA peptide ligand was observed at m/z 1918.949 as major species in the spectrum (Figure 3B). The OVA peptide contains two theoretical cleavage sites at Glu11 and Glu14 (Figure 3A). The shorter OVA peptide ligand generated from the cleavage at Glu11 ([M1 + H]+ in Figure 3B) was detected at m/z 1278.607 while the ligand generated from the cleavage at Glu14 and missed cleavage at Glu11 ([M2 + H]+ in Figure 3B) was detected at m/z 1634.786 at lower peak intensity. As for the intact OVA peptide ligand, also the shorter M1 peptide was detected bound to one or two Au+, the glucose ligand and the 1,5 DAN matrix. The species generated from the loss of either SH2 or S were also detected. The MS/MS spectra and sequence coverage generated from the analysis of [M1 + H]+, [M1 + Au]+, and [M1 + 2Au–H]+ are reported in Supporting Information Figure S7, and Figure 3. While b-type fragmentation was preferred for [M1 + H]+, y-type fragments were more abundant for the gold-complexed species. The characterization of [M1 + Au]+ suggests that Au+ is coordinated at His6 while the second Au+ of [M1 + 2Au–H]+ is localized on the terminal sulfur of the linker. CID analysis also confirmed the structure of the M1 ligand linked to the glucose ligand via an S–S bond detected at m/z 1558.715 (Supporting Information Figure S7).
Figure 3.

(A) Enzymatic digestion of the OVA 323-339 peptide-based ligand attached to a gold nanoparticle. (B) Resulting LDI/MALDI–FTICR MS spectrum after Glu-C digestion. The most abundant product is derived from the cleavage at Glu11 and was detected as [M1 + H]+ at m/z 1278.607, [M1 + Au]+ at m/z 1474.571 and [M1 + 2Au–H]+ at m/z 1670.536. A second product is derived from the cleavage at Glu14 (with a missed cleavage at Glu11) and was detected as [M2 + H]+ at m/z 1634.786. Other assignments are (a)[M1–S–H + 1,5-DAN]+ at m/z 1402.703. (b)[M–S–H + 1,5-DAN]+ at m/z 2043.052. (c)[M1–H + glucose ligand]+ at m/z 1558.715. (d)[M–H + glucose ligand]+ at m/z 2199.038. (C) Sequence coverage obtained from CID analysis of [M1 + Au]+ (mass spectra of [M1 + H]+, [M1 + Au]+, and [M1 + 2Au–H]+ are shown in Figure S7). The data suggests that the preferred coordination sites of Au+ are Histidine 6 and the terminal sulfur. Yellow circles = [fragment + Au]+. Double yellow circles = [fragment + 2Au–H]+.
Together these data confirm the successful proteolysis by Glu-C of the OVA peptide at the surface of the AuNPs and show the potential of this method for monitoring of in vitro or in vivo ligand modifications. Our strategy allowed to detect enzymatic modifications of the AuNPs on the particles themselves in contrast to the detection of cleavage products, released from the particles.
Analysis of Lewis X and Tetramannoside AuNPs by LDI/MALDI–FTICR MS and MS/MS
AuNPs functionalized with two different carbohydrate ligands were analyzed by LDI/MALDI–FTICR MS, namely the Lewis X ligand (monoisotopic mass = 993.4749 Da) and the tetramannoside ligand (monoisotopic mass = 1130.4961 Da). The LDI/MALDI–FTICR spectrum of Lewis X AuNPs is reported in Figure 4. Here, the most abundant species detected at m/z 982.484 was identified as [M–SH2 + Na]+. MS/MS analysis (Supporting Information, Figure S8) confirmed the loss of SH2 generated from the terminal thiol group of the linker (Figure 4B). Interestingly, a double loss of SH2 was observed at m/z 948.495. This double loss generated from both the terminal thiol group and the thiourea group of the linker. These losses were not observed in the MALDI–FTICR–MS analysis of the purified Lewis X ligand (i.e., not linked to AuNPs; Supporting Information, Figure S9) which indicates that the sulfur at the thiourea group may also form a bond with gold. Contrary to what was observed for the OVA peptide, the [M–S + Na]+ was not detected. In-source decay of [M–SH2 + Na]+ led to the loss of fucose from the carbohydrate moiety (i.e., species detected at m/z 836.424).
Figure 4.
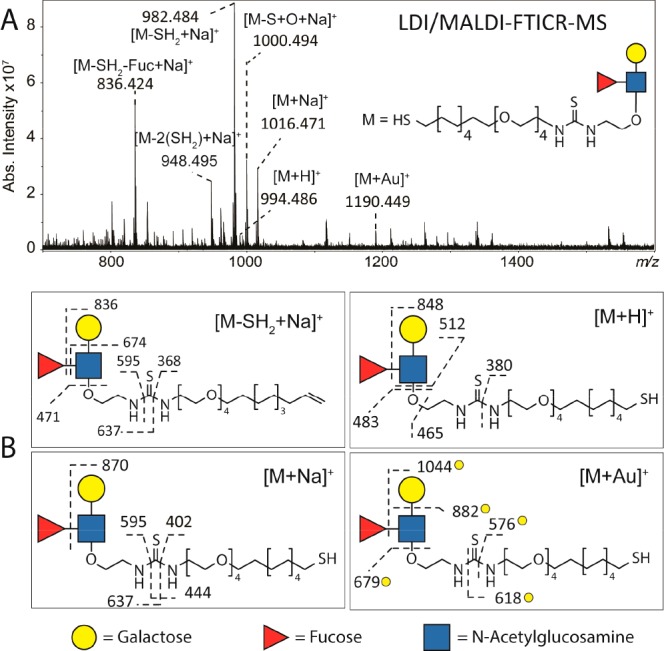
(A) LDI/MALDI–FTICR MS spectrum of AuNP functionalized with Lewis X-based ligand. The use of NaCl in the spotting procedure led to the detection of the intact Lewis X ligand at low intensity as [M + H]+, [M + Na]+, and [M + Au]+, and the detection of fragmented species at higher intensity such as [M–SH2 + Na]+ and [M–SH2–Fuc + Na]+. (B) From structural characterization performed by CID a double bond resulting from the loss of SH2 was confirmed. Furthermore, MS/MS data of three different pseudomolecular ions provided structure confirmation of the Lewis X-based ligand. The analysis of the [M + Au]+ suggests the terminal sulfur as the preferred coordination site of Au+. Yellow circles indicate gold-linked fragment ions.
Fragmentation of carbohydrates by CID provides detailed structural information from glycosidic linkage cleavages as well as cross-ring cleavages.34 The CID spectra from Lewis X and tetramannoside ligands were evaluated based on the identification of glycosidic cleavages. The characterization of [M + Na]+, [M + H]+ [M + Au]+ by CID resulted in different fragmentation patterns (Supporting Information Figure S8 and Figure 4) and provided complementary information on the structure of the ligand. In fact, the larger ion generated from the fragmentation of the Gal(β1-4)-GlcNAc bond was only detected in the MS/MS spectrum of [M + Au]+ at m/z 882 and not in the MS/MS spectra of [M + H]+ and [M + Na]+. Other examples of fragment ions providing complementary information are those detected in the CID spectrum of [M + H]+ at m/z 465 and 512 and those in the spectrum of [M + Na]+ at m/z 595 and 637. The presence of gold-clustered fragments indicated that Au+ was not localized on the carbohydrate part but rather in the proximity of the terminal sulfur of the linker. Further evidence for this localization was provided by the detection of the gold-clustered fragment at m/z 576. The species detected at m/z 1000.494 was identified as a Lewis X ligand with urea instead of a thiourea in the structure of the linker.
The LDI/MALDI–FTICR MS analysis of the tetramannoside AuNPs led to the detection of ion species similar to those detected for the Lewis X AuNPs (see Figures 4 and 5). Also in this case, the most abundant peak in the spectrum was identified as [M–SH2 + Na]+. The fragmentation patterns of [M + Na]+ and [M + Au]+ were clearly different with [M + Na]+ providing more information on the structure of the linker (i.e., fragments at m/z 444, 774, and 732) while [M + Au]+ provided more information on the structure on the carbohydrate part (i.e., fragments at m/z 841, 1003, 1165) (Figure 5B). The detection of gold-clustered fragments indicates that the Au+ is localized on the terminal sulfur of the linker (Supporting Information Figure S10 and Figure 5B). As for the Lewis X AuNPs, the detection of the fragment at m/z 576 in the MS/MS spectrum of [M + Au]+ provided additional evidence for this localization. Interestingly, when NaCl was not used in combination with the 1,5-DAN matrix, the tetramannoside ligand was detected at high intensity as [M + H]+, [M + Au]+, and [M + 2Au–H]+ (Supporting Information Figure S11).
Figure 5.
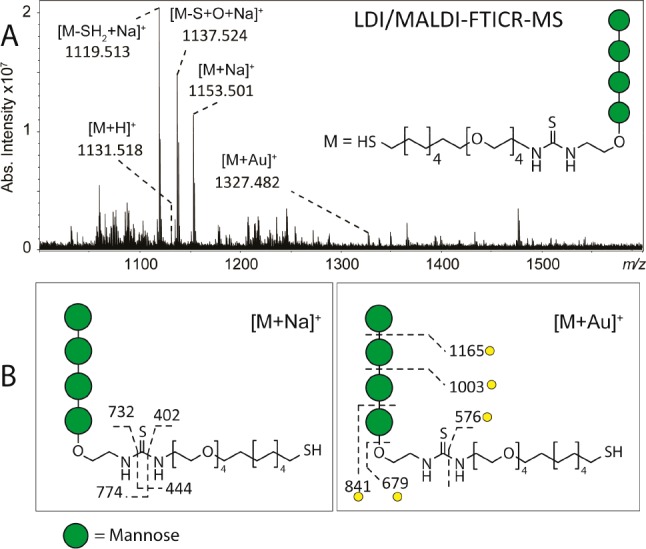
(A) LDI/MALDI–FTICR MS spectrum of AuNP functionalized with tetramannoside-based ligand. The tetramannoside-based ligand was detected as [M + H]+, and [M + Au]+ at low intensity, and detected as [M + Na]+, [M–SH2 + Na]+ and [M–S + O + Na]+ at higher intensities. (B) Structure characterization performed by CID confirmed the identity of these species. Fragmentation of [M + Au]+ allowed characterization of the carbohydrate moiety of the ligand, whereas CID analysis of [M + Na]+ provided more information on the structure of the linker. Yellow circles indicate gold-linked fragment ions.
To explore the applicability of LDI/MALDI–FTICR MS for the analysis of AuNPs with mixed functional ligands, we analyzed AuNPs carrying both Lewis X and tetramannoside. The ligand ratio anticipated on the basis of reaction stoichiometry and measured by 1H NMR spectroscopy was approximately 1:2, and the ratio between these ligands as detected by LDI/MALDI–FTICR MS and determined as ([M1–SH2 + Na]+ + [M1–S + O + Na]+ + [M1 + Na]+)/ ([M2–SH2 + Na]+ + [M2-S + O + Na]+ + [M2 + Na]+) was ∼3.0 (see Figure 6 and Figures S13–S15). Although it is clear that these results may be biased due to different ionization efficiencies for the different ligand species, it shows the potential of this approach to determine ligand ratios after synthesis of multicomponent AuNPs or for example after dynamic covalent exchange, as previously reported using LDI-MS-based methods.13,14
Figure 6.
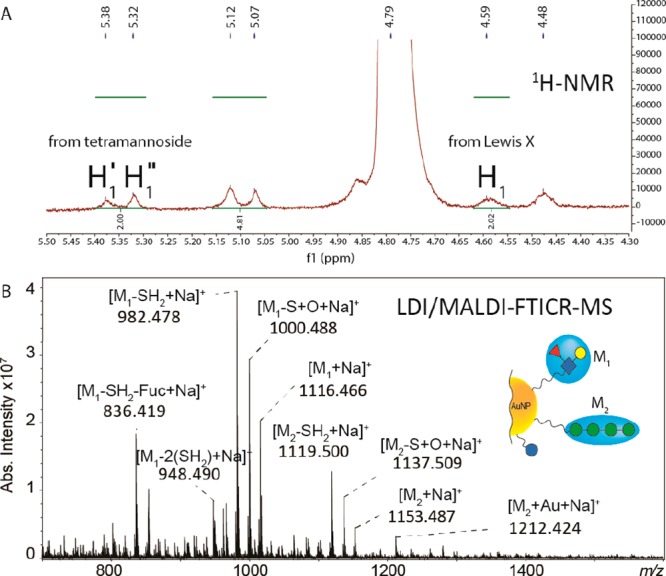
(A) 1H NMR spectrum of AuNPs functionalized with both Lewis X and tetramannoside ligands in D2O at 500 MHz. A detailed description of the NMR spectrum is reported in Supporting Information Figure S13. (B) LDI/MALDI–FTICR MS spectrum of AuNPs functionalized with both Lewis X and tetramannoside ligands.
Conclusions
We performed detailed structural characterization of peptide and carbohydrate functionalized AuNPs of ∼2 nm using a combination of LDI/MALDI–FTICR MS and CID. The here described strategy allows the structural analysis of AuNPs with ligands larger than 1000 Da by generating spectra of a much higher quality than spectra obtained with previously described LDI-TOF MS methods, including fragmentation spectra. All analyzed AuNPs were functionalized with either the OVA 323-339 peptide, the Lewis X antigen, or the tetramannoside, or a combination of the latter two ligands. The MS analysis of these AuNPs allowed for the detection of the ligands as pseudomolecular species, such as [M + H]+, [M + Na]+, [M + Au]+, and [M + 2Au–H]+ and the detection of modified ligands, such as [M–SH2 + Na]+, which were formed during ionization. We showed that LDI/MALDI-CID-FTICR MS allows for the in-depth structural characterization of these species providing comprehensive information on the surface chemistry of biofunctionalized AuNPs. In fact, MS/MS analysis allowed identification of the ligands present on the surface of AuNPs and an understanding of how these ligands can be linked to or interact with gold atoms. The identification and characterization of different [M + nAu]+ species could be used, in general, to confirm AuNP surface attachment of thiol coupled ligands. To our knowledge, the generation of these gold-containing ions directly from AuNPs has never been reported.
In addition, using the endoproteinase Glu-C, we showed that the OVA peptide AuNPs can be enzymatically modified and that surface modifications can be monitored by LDI/MALDI–FTICR MS, further indicating the potential of the approach in the field of AuNP research and development.
Methods
Conjugation of AuNPs with OVA Peptide, Lewis X Antigen and Tetramannoside
Four different types of AuNPs functionalized with thiol-ending derivatives of OVA 323-339 peptide, Lewis X, and tetramannoside were prepared as previously described (Figure 1).22,26 For each AuNP a thiol-ending derivative of glucose (i.e., 5-(mercapto)pentyl β-d-glucopyranoside) was used as the inner component to allow the incorporation of defined proportions of ligands, controlling their loading on the AuNPs (i.e., 10% of total surface load). To this end, a 9:1 mixture of inner component-ligand was used for the synthesis. Thus, AuNPs were functionalized with a thiol-ending conjugate of either a tetramannoside (Manα1-2Manα1-2Manα1-3Manα) or a Galβ1-4[Fucα1-3]GlcNAcβ1 (Lewis X antigen) or a 1:2 mixture of the two carbohydrates (i.e., ligands), as previously described.22,26,35 The OVA 323-339 peptide with an additional glycine and mercapto-propionic acid linker at the N-terminus HS(CH2)2C(O)GISQAVHAAHAEINEAGR was obtained from GenScript Corp (Piscataway, NJ, USA,); AuNPs carrying 10% of OVA 323-339 peptide and 90% of inner component were prepared as previously described.22 Briefly, a water solution of tetrachloroauric acid (HAuCl4, Strem Chemicals, 0.025 M, 1 equiv) was added to a 0.012 M (3 equiv) MeOH solution of the mixture of 10% thiol-ending conjugates and 90% of glucose-conjugate. A solution of NaBH4 1 M (21 equiv) was then added, and the black suspension was shaken for 2 h at room temperature. Thus, obtained AuNPs were washed with EtOH and MeOH, dissolved in a minimal volume of nanopure water and purified by dialysis (3500 Da MWCO), 5 times with 5 L during 72 h. Transmission electron microscopy showed a mean gold core diameter of approximately 2 nm for all the prepared AuNPs. Detailed experimental conditions for the synthesis of each NP and supporting structural information are reported in Supporting Information pages S13–S15. For the analysis of the ligand ratio on the AuNPs, proton nuclear magnetic resonance (1H NMR) was used as previously described.22,36 Before MALDI–FTICR–MS analysis AuNPs were washed with nanopure water (3 times with 500 μL) on 30 kDa centrifugal filters and AuNPs were suspended in nanopure water to a final concentration of 1–2 mg/mL.
Enzymatic Digestion of OVA Peptide AuNPs
Endoproteinase Glu-C with enzymatic activity ≥ 15 units/mg protein was purchased from Sigma-Aldrich. This solution was diluted 1 to 1000 in water, and 1 μL was mixed with 10 μL of 1 mg/mL solution of OVA 323-339 AuNPs. This mixture was then incubated for 4 h at 37 °C. Digested OVA 323-339 AuNPs were analyzed by MS without any further purification.
LDI and MALDI Spotting
For LDI analysis, each solution of functionalized AuNPs was spotted (1 μL) onto a ground-steel MALDI-target plate either undiluted or after a 1:10 dilution with solution A (50% ACN, 49.95% water, and 0.05% formic acid). For LDI/MALDI analysis, 1 μL of the same solutions was spotted onto a ground-steel MALDI-target together with a saturated solution of either 1,5 diaminonaphthalene (1,5-DAN) or sinapinic acid (SA), both prepared in solution A. One microliter of a NaCl solution at 1 mg/mL was mixed on the MALDI-target plate with each AuNPs solution and MALDI matrixes.
LDI- and LDI/MALDI–FTICR MS and MS/MS
All LDI- and LDI/MALDI-ESI-FTICR MS experiments were performed on a 15T solariX XR FTICR mass spectrometer controlled by ftmsControl software and equipped with a CombiSource and a ParaCell (Bruker Daltonics, Bremen, Germany). A Bruker Smartbeam-II Laser System was used for irradiation at a frequency of 500 Hz using the “medium” predefined shot pattern. The instrumental parameters were tuned for optimal sensitivity in the m/z-ranges between 92 and 5000 and 966.1–5000. The number of laser shots, laser power, and number of acquired scans were optimized for each type of functionalized AuNP. MALDI–FTICR spectra were generated with 1 M data points. Precursor ions were isolated through a quadrupole (Q), accumulated in the hexapole collision cell, and fragmented by collision-induced dissociation (CID). Accumulation times and collision energies were optimized for each precursor ion. DataAnalysis Software 4.2 (Bruker Daltonics) was used for the visualization and data analysis of both LDI- and LDI/MALDI-(CID)-FTICR spectra.
Acknowledgments
F.C. acknowledges NWO–CW Veni Grant No. 722.014.008 for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b03402.
Further MS and MS/MS spectra and peptide sequence coverage from biofunctionalized AuNPs analyzed in this study; detailed experimental conditions for the synthesis of each NP and supporting structural information (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dreaden E. C.; Alkilany A. M.; Huang X.; Murphy C. J.; El-Sayed M. A. The Golden Age: Gold Nanoparticles for Biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. 10.1039/C1CS15237H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C.-C.; Chompoosor A.; Rotello V. M. The Biomacromolecule-Nanoparticle Interface. Nano Today 2007, 2, 34–43. 10.1016/S1748-0132(07)70085-3. [DOI] [Google Scholar]
- Kailasa S. K.; Wu H.-F. Nanomaterial-Based Miniaturized Extraction and Preconcentration Techniques Coupled to Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry for Assaying Biomolecules. TrAC. TrAC, Trends Anal. Chem. 2015, 65, 54–72. 10.1016/j.trac.2014.09.011. [DOI] [Google Scholar]
- Niikura K.; Matsunaga T.; Suzuki T.; Kobayashi S.; Yamaguchi H.; Orba Y.; Kawaguchi A.; Hasegawa H.; Kajino K.; Ninomiya T.; Ijiro K.; Sawa H. Gold Nanoparticles as a Vaccine Platform: Influence of Size and Shape on Immunological Responses in Vitro and in Vivo. ACS Nano 2013, 7, 3926–3938. 10.1021/nn3057005. [DOI] [PubMed] [Google Scholar]
- Zong J.; Cobb S. L.; Cameron N. R. Peptide-Functionalized Gold Nanoparticles: Versatile Biomaterials for Diagnostic and Therapeutic Applications. Biomater. Sci. 2017, 5, 872–886. 10.1039/C7BM00006E. [DOI] [PubMed] [Google Scholar]
- Dykman L. A.; Khlebtsov N. G. Immunological Properties of Gold Nanoparticles. Chem. Sci. 2017, 8, 1719–1735. 10.1039/C6SC03631G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkinen H. The Gold-Sulfur Interface at the Nanoscale. Nat. Chem. 2012, 4, 443–55. 10.1038/nchem.1352. [DOI] [PubMed] [Google Scholar]
- Pensa E.; Cortés E.; Corthey G.; Carro P.; Vericat C.; Fonticelli M. H.; Benítez G.; Rubert A. A.; Salvarezza R. C. The Chemistry of the Sulfur–Gold Interface: In Search of a Unified Model. Acc. Chem. Res. 2012, 45, 1183–1192. 10.1021/ar200260p. [DOI] [PubMed] [Google Scholar]
- Qian H.; Eckenhoff W. T.; Zhu Y.; Pintauer T.; Jin R. Total Structure Determination of Thiolate-Protected Au38 Nanoparticles. J. Am. Chem. Soc. 2010, 132, 8280–8281. 10.1021/ja103592z. [DOI] [PubMed] [Google Scholar]
- Unnikrishnan B.; Chang C.-Y.; Chu H.-W.; Anand A.; Huang C.-C. Functional Gold Nanoparticles Coupled with Laser Desorption Ionization Mass Spectrometry for Bioanalysis. Anal. Methods 2016, 8, 8123–8133. 10.1039/C6AY02378A. [DOI] [Google Scholar]
- Fields-Zinna C. A.; Sampson J. S.; Crowe M. C.; Tracy J. B.; Parker J. F.; deNey A. M.; Muddiman D. C.; Murray R. W. Tandem Mass Spectrometry of Thiolate-Protected Au Nanoparticles Naxau25(Sc2h4ph)18–Y(S(C2h4o)5ch3)Y. J. Am. Chem. Soc. 2009, 131, 13844–13851. 10.1021/ja905787y. [DOI] [PubMed] [Google Scholar]
- Negishi Y.; Chaki N. K.; Shichibu Y.; Whetten R. L.; Tsukuda T. Origin of Magic Stability of Thiolated Gold Clusters: A Case Study on Au25(Sc6h13)18. J. Am. Chem. Soc. 2007, 129, 11322–11323. 10.1021/ja073580+. [DOI] [PubMed] [Google Scholar]
- Yan B.; Zhu Z.-J.; Miranda O. R.; Chompoosor A.; Rotello V. M.; Vachet R. W. Laser Desorption/Ionization Mass Spectrometry Analysis of Monolayer-Protected Gold Nanoparticles. Anal. Bioanal. Chem. 2010, 396, 1025–1035. 10.1007/s00216-009-3250-6. [DOI] [PubMed] [Google Scholar]
- della Sala F.; Kay E. R. Reversible Control of Nanoparticle Functionalization and Physicochemical Properties by Dynamic Covalent Exchange. Angew. Chem., Int. Ed. 2015, 54, 4187–4191. 10.1002/anie.201409602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.-J.; Ghosh P. S.; Miranda O. R.; Vachet R. W.; Rotello V. M. Multiplexed Screening of Cellular Uptake of Gold Nanoparticles Using Laser Desorption/Ionization Mass Spectrometry. J. Am. Chem. Soc. 2008, 130, 14139–14143. 10.1021/ja805392f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S.; Sikora K. N.; Tang R.; Liu Y.; Lee Y.-W.; Kim S. T.; Jiang Z.; Vachet R. W.; Rotello V. M. Quantitative Differentiation of Cell Surface-Bound and Internalized Cationic Gold Nanoparticles Using Mass Spectrometry. ACS Nano 2016, 10, 6731–6736. 10.1021/acsnano.6b02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B.; Kim S. T.; Kim C. S.; Saha K.; Moyano D. F.; Xing Y.; Jiang Y.; Roberts A. L.; Alfonso F. S.; Rotello V. M.; Vachet R. W. Multiplexed Imaging of Nanoparticles in Tissues Using Laser Desorption/Ionization Mass Spectrometry. J. Am. Chem. Soc. 2013, 135, 12564–12567. 10.1021/ja406553f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsico A. L. M.; Elci G. S.; Moyano D. F.; Yesilbag Tonga G.; Duncan B.; Landis R. F.; Rotello V. M.; Vachet R. W. Enhanced Laser Desorption/Ionization Mass Spectrometric Detection of Gold Nanoparticles in Biological Samples Using the Synergy between Added Matrix and the Gold Core. Anal. Chem. 2015, 87, 12145–12150. 10.1021/acs.analchem.5b02985. [DOI] [PubMed] [Google Scholar]
- Nikolaev E. N.; Kostyukevich Y. I.; Vladimirov G. N. Fourier Transform Ion Cyclotron Resonance (FT ICR) Mass Spectrometry: Theory and Simulations. Mass Spectrom. Rev. 2016, 35, 219–58. 10.1002/mas.21422. [DOI] [PubMed] [Google Scholar]
- Nicolardi S.; Bogdanov B.; Deelder A. M.; Palmblad M.; van der Burgt Y. E. M. Developments in FTICR-MS and Its Potential for Body Fluid Signatures. Int. J. Mol. Sci. 2015, 16, 27133–27144. 10.3390/ijms161126012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marradi M.; Chiodo F.; Garcia I.; Penades S. Glyconanoparticles as Multifunctional and Multimodal Carbohydrate Systems. Chem. Soc. Rev. 2013, 42, 4728–45. 10.1039/c2cs35420a. [DOI] [PubMed] [Google Scholar]
- Safari D.; Marradi M.; Chiodo F.; Th Dekker H. A.; Shan Y.; Adamo R.; Oscarson S.; Rijkers G. T.; Lahmann M.; Kamerling J. P.; Penades S.; Snippe H. Gold Nanoparticles as Carriers for a Synthetic Streptococcus Pneumoniae Type 14 Conjugate Vaccine. Nanomedicine (London, U. K.) 2012, 7, 651–62. 10.2217/nnm.11.151. [DOI] [PubMed] [Google Scholar]
- Di Gianvincenzo P.; Chiodo F.; Marradi M.; Penades S. Gold Manno-Glyconanoparticles for Intervening in HIV gp120 Carbohydrate-Mediated Processes. Methods Enzymol. 2012, 509, 21–40. 10.1016/B978-0-12-391858-1.00002-2. [DOI] [PubMed] [Google Scholar]
- Marradi M.; Di Gianvincenzo P.; Enriquez-Navas P. M.; Martinez-Avila O. M.; Chiodo F.; Yuste E.; Angulo J.; Penades S. Gold Nanoparticles Coated with Oligomannosides of HIV-1 Glycoprotein gp120 Mimic the Carbohydrate Epitope of Antibody 2g12. J. Mol. Biol. 2011, 410, 798–810. 10.1016/j.jmb.2011.03.042. [DOI] [PubMed] [Google Scholar]
- Tundup S.; Srivastava L.; Norberg T.; Watford W.; Harn D. A Neoglycoconjugate Containing the Human Milk Sugar Lnfpiii Drives Anti-Inflammatory Activation of Antigen Presenting Cells in a CD14 Dependent Pathway. PLoS One 2015, 10, e0137495. 10.1371/journal.pone.0137495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Avila O.; Hijazi K.; Marradi M.; Clavel C.; Campion C.; Kelly C.; Penades S. Gold Manno-Glyconanoparticles: Multivalent Systems to Block HIV-1 gp120 Binding to the Lectin Dc-Sign. Chem. - Eur. J. 2009, 15, 9874–88. 10.1002/chem.200900923. [DOI] [PubMed] [Google Scholar]
- Li Y.-J.; Tseng Y.-T.; Unnikrishnan B.; Huang C.-C. Gold-Nanoparticles-Modified Cellulose Membrane Coupled with Laser Desorption/Ionization Mass Spectrometry for Detection of Iodide in Urine. ACS Appl. Mater. Interfaces 2013, 5, 9161–9166. 10.1021/am4025824. [DOI] [PubMed] [Google Scholar]
- Lee J.; Jayathilaka L. P.; Gupta S.; Huang J. S.; Lee B. S. Gold Ion-Angiotensin Peptide Interaction by Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2012, 23, 942–51. 10.1007/s13361-011-0328-0. [DOI] [PubMed] [Google Scholar]
- Aldeek F.; Safi M.; Zhan N.; Palui G.; Mattoussi H. Understanding the Self-Assembly of Proteins onto Gold Nanoparticles and Quantum Dots Driven by Metal-Histidine Coordination. ACS Nano 2013, 7, 10197–10210. 10.1021/nn404479h. [DOI] [PubMed] [Google Scholar]
- An H. J.; Froehlich J. W.; Lebrilla C. B. Determination of Glycosylation Sites and Site-Specific Heterogeneity in Glycoproteins. Curr. Opin. Chem. Biol. 2009, 13, 421–426. 10.1016/j.cbpa.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolardi S.; Switzar L.; Deelder A. M.; Palmblad M.; van der Burgt Y. E. M. Top-Down MALDI-in-Source Decay-FTICR Mass Spectrometry of Isotopically Resolved Proteins. Anal. Chem. 2015, 87, 3429–3437. 10.1021/ac504708y. [DOI] [PubMed] [Google Scholar]
- Wang J.; Yue Y.; Chen G.; Xia J. Protease-Promoted Drug Delivery Using Peptide-Functionalized Gold Nanoparticles. Soft Matter 2011, 7, 7217–7222. 10.1039/c1sm05242j. [DOI] [Google Scholar]
- Chanana M.; Rivera Gil P.; Correa-Duarte M. A.; Liz-Marzan L. M.; Parak W. J. Physicochemical Properties of Protein-Coated Gold Nanoparticles in Biological Fluids and Cells before and after Proteolytic Digestion. Angew. Chem., Int. Ed. 2013, 52, 4179–83. 10.1002/anie.201208019. [DOI] [PubMed] [Google Scholar]
- Domon B.; Costello C. E. A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS/MS Spectra of Glycoconjugates. Glycoconjugate J. 1988, 5, 397–409. 10.1007/BF01049915. [DOI] [Google Scholar]
- Arosio D.; Chiodo F.; Reina J. J.; Marelli M.; Penadés S.; van Kooyk Y.; Garcia-Vallejo J. J.; Bernardi A. Effective Targeting of Dc-Sign byA-Fucosylamide Functionalized Gold Nanoparticles. Bioconjugate Chem. 2014, 25, 2244–2251. 10.1021/bc500467u. [DOI] [PubMed] [Google Scholar]
- Chiodo F.; Marradi M.; Park J.; Ram A. F.; Penades S.; van Die I.; Tefsen B. Galactofuranose-Coated Gold Nanoparticles Elicit a Pro-Inflammatory Response in Human Monocyte-Derived Dendritic Cells and Are Recognized by DC-SIGN. ACS Chem. Biol. 2014, 9, 383–389. 10.1021/cb4008265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.