

Abstract
We report oriented immobilization of proteins using the standard hexahistidine (His6)-Ni2+:NTA (nitrilotriacetic acid) methodology, which we systematically tuned to give control of surface coverage. Fluorescence microscopy and surface plasmon resonance measurements of self-assembled monolayers (SAMs) of red fluorescent proteins (TagRFP) showed that binding strength increased by 1 order of magnitude for each additional His6-tag on the TagRFP proteins. All TagRFP variants with His6-tags located on only one side of the barrel-shaped protein yielded a 1.5 times higher surface coverage compared to variants with His6-tags on opposite sides of the so-called β-barrel. Time-resolved fluorescence anisotropy measurements supported by polarized infrared spectroscopy verified that the orientation (and thus coverage and functionality) of proteins on surfaces can be controlled by strategic placement of a His6-tag on the protein. Molecular dynamics simulations show how the differently tagged proteins reside at the surface in “end-on” and “side-on” orientations with each His6-tag contributing to binding. Also, not every dihistidine subunit in a given His6-tag forms a full coordination bond with the Ni2+:NTA SAMs, which varied with the position of the His6-tag on the protein. At equal valency but different tag positions on the protein, differences in binding were caused by probing for Ni2+:NTA moieties and by additional electrostatic interactions between different fractions of the β-barrel structure and charged NTA moieties. Potential of mean force calculations indicate there is no specific single-protein interaction mode that provides a clear preferential surface orientation, suggesting that the experimentally measured preference for the end-on orientation is a supra-protein, not a single-protein, effect.
Keywords: protein immobilization, multivalency, monolayers, molecular dynamics simulations, self-assembly
Proteins anchored on solid substrates play a crucial role in biomedical, bioanalytical, and biotechnological applications, biomaterials, and nanobiotechnological devices and surfaces.1−9 Specific properties of surface-based diagnostic assays and cell culture supports often depend on site-selective attachment of proteins to solid supports.10−12 Grafting a suitable binding motif to a specific site on the protein provides control over the orientation of proteins on solid supports,13−16 which, unlike nonspecific or non-site-selective anchoring, generates homogeneous surface coverage and, if well-considered, easy accessibility to the proteins’ active sites.10−12 Consequently, different types of bio-orthogonal reactions, both noncovalent and covalent, have been developed to site-specifically attach proteins to surfaces.10−12,17−31 While functional attachment of proteins to solid supports with some control over orientation has been achieved, firm structural evidence of uniformly oriented proteins is lacking. For example, Saavedra and co-workers studied, using emission anisotropy, the distribution of orientations of cytochrome c, which were site-selectively and covalently attached to substrates through disulfide bond formation between a single reduced cysteine residue on the proteins and the surface-exposed thiol groups of a self-assembled monolayer (SAM).32 A broad range (12° ± 33°) of heme orientations signified a disordered layer with a substantial fraction of nonspecifically adsorbed proteins.32 Taking advantage of a lipid bilayer to resist nonspecific protein adsorption yielded more oriented cytochrome c layers (41° ± 11°).33 Scoles and co-workers grafted metal-chelating thiols (specifically, nitrilotriacetic acid (NTA)-terminated thiols) into antifouling SAMs to capture hexahistidine (His6)-tagged antibodies with high affinity for specific epitopes on prion proteins.34 Oriented immobilization of prion proteins was topographically detected.34 Multivalent host–guest interactions have been used by us to immobilize light-harvesting protein LH2 complexes that were engineered with cysteine residues close to the C-terminus of each of the nine α-domains of LH2 and functionalized with adamantane guests.35 These positions ensured oriented, yet irreversibly bound, protein complexes upon binding to β-cyclodextrin host surfaces.35 In linear arrays of such oriented proteins characteristic energy migration could be observed.36 Tampé and co-workers attached a 20S proteasome to substrates functionalized with metal-chelating NTA complexes using His6-tags attached (randomly) to only one of the subunits, either the outer α- or inner β-subunit, of the 20S proteasome.37,38 Placing His6-tags on the outer α-subunits resulted in an end-on orientation of the barrel-shaped enzyme, while His6-tags attached to the inner β-subunits resulted in side-on immobilization.37,38 Proteolytic activity was determined for both orientations of the 20S proteasome on the surface and found to differ by a factor of 2.38 End-on immobilization of the proteasome demonstrated that one pore is sufficient for substrate entry and product release.38 Remarkably, end-on-oriented proteasomes could process only one substrate at a time, while in contrast, the side-on-immobilized proteasome could bind two substrates.38 These findings demonstrated clearly that orientational control over the immobilization of proteins influenced their efficiency and functionality. Yet, despite the fact that control over protein orientation and geometry has a strong influence on protein function when immobilized, protein immobilization has not as yet been demonstrated with tunable control over the orientation, binding strength, and reversibility of protein adsorption.
We have previously reported on a covalent disulfide lock between two ferrocenyl (guest)-modified fluorescent proteins to switch from monovalent to divalent interactions with the β-cyclodextrin (β-CD) host surface, yielding stable immobilized protein layers with homogeneous orientation.27 Here, using multivalency as a design principle,39,40 we study controlled immobilization based on a combination of site-directed mutagenesis, surface modification techniques, multivalent numerical models, and molecular simulations. Fluorescent proteins were engineered with His6-tag residues at specific positions on the proteins (Chart 1). These strategic positions ensured unique orientations and properties of proteins upon binding to Ni2+:NTA surfaces. Given the widespread use of His6-tag technology in protein diagnostics, isolation, surface-based devices, and bionanostructures, the results of this investigation should be of general interest.21,41−55 Isothermal calorimetry and stop-flow fluorescence studies in solution by Tampé and co-workers show that individual Ni2+:NTA-His2 complexes (forming one coordination pair) are of low affinity (Kd = 14 μM), two coordination pairs in Ni2+:NTA-His4 complexes are of moderate affinity (Kd = 0.27 μM), while high affinity (Kd = 20 nM) could be reached by incorporating three NTA moieties into a single multivalent chelator entity (forming three coordination pairs in Ni2+:NTA-His6 complexes).56 While not all of these values have been validated on surfaces, Szoka and co-workers found nanomolar affinities for immobilizing His6-tagged proteins on trivalent NTA chelators on gold chips, which depended on linker length and surface density of the chelator.62 Complex formation remains reversible upon addition of competitive binding moieties, which has been demonstrated in several other protein immobilization studies.21,47,48,55−64 On surfaces, dissociation of longer His10-tagged proteins could be inhibited by accumulated “patches” of multivalent NTA entities on surfaces.58,59 Analogously, we have established multivalent Ni2+:NTA surfaces anchored to β-cyclodextrin host surfaces employing a divalent adamantyl-NTA moiety, yet the binding affinities were estimated to be markedly lower when compared to solution data, due to incomplete complex formation.55,63 More precisely, on such β-cyclodextrin-anchored Ni2+:NTA surfaces about 60% of the His6-tagged proteins formed three coordination pairs with an estimated affinity of Kd = 1 μM taking advantage of the spatial arrangement of NTA ligands on the host surface, while the remaining 40% formed only two coordination pairs with an estimated affinity of Kd = 10 μM.55,63 Rant and co-workers employed an electric field to switch between horizontally and vertically oriented His6-tagged proteins on the Ni2+:NTA substrate.65 They also demonstrated that binding characteristics of His6-tagged proteins are largely affected by additional interactions in the local chemical environment between the protein and surface, yielding KD values ranging between 1 μM and 1 nM.65
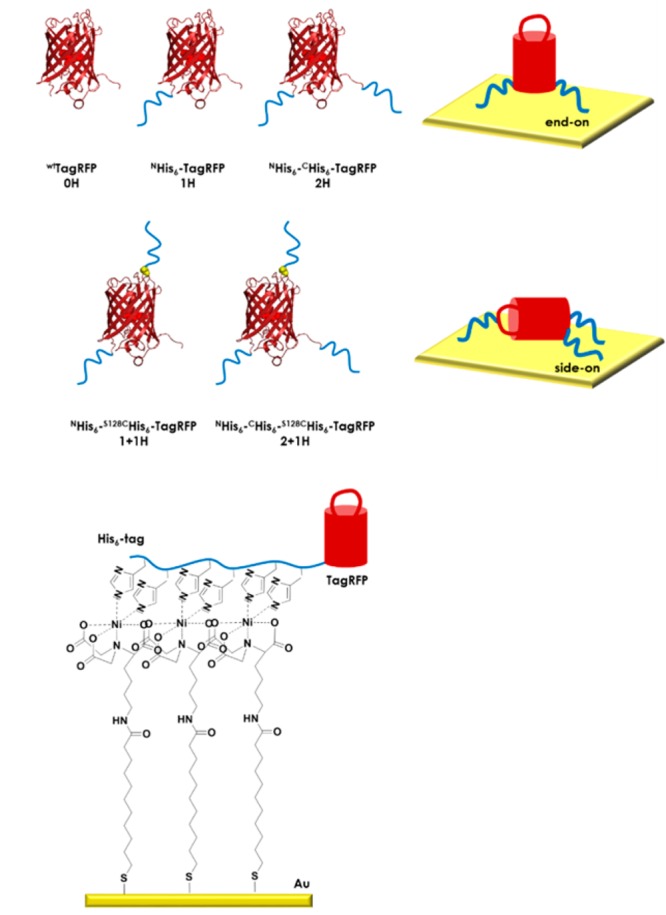
Chart 1. Schematic representation of the five TagRFP variants (0H, 1H, 2H, 1 + 1H, 2 + 1H) used in this study with different numbers of His6-tags located at the N- and/or C-terminus and/or at the serine-to-cysteine mutation site (S128C). End-on and side-on oriented TagRFP binding scenarios and the complexation of Ni2+:NTA SAMs (for preparation see Scheme S1) by a His6-tag are shown.
Here, we manipulate the orientation of histidine-tagged proteins on Ni2+:NTA functional surfaces without resorting to other substrate–protein interactions55,63 or external forces.65 The molecular-scale details of the protein–surface interface were monitored using fluorescence microscopy, IR spectroscopy, surface plasmon resonance, and time-resolved fluorescence anisotropy techniques supported by equilibrium and steered molecular dynamics computer simulations. Our results demonstrate that the orientation of proteins on surfaces can be controlled by strategic placement of His6-tags on the protein.
Results and Discussion
Engineering Protein Variants
The protein we employed is a red fluorescent protein (RFP), more specifically, an orange, monomeric variant of an RFP from the sea anemone Entacmaea quadricolor, TagRFP,66 and we trace the proteins at the surface using (time-resolved, polarized) spectroscopy, fluorescence microscopy, and surface plasmon resonance. Recombinant variants of TagRFP were made containing between zero and three His6-tags at different positions on the protein (Chart 1 and Methods). His6-tags were introduced at either one or both of the termini by cloning and/or after site-selective mutagenesis to create a single accessible cysteine residue by orthogonal conjugation. The studied proteins included a (wild-type) wtTagRFP (0H) that contains no His6-tags (Chart 1) and two mutants of wtTagRFP each with native cysteines C114 and C222 mutated to serine (C114S and C222S) to remove solvent-accessible cysteine residues. Both mutants have either one or two His6-tags, named NHis6-TagRFP (1H) and NHis6-CHis6-TagRFP (2H), respectively. The N- and C-termini in TagRFP are located at the base of its cylindrical structure, the so-called β-barrel, which places the two His6-tags of 2H at the same end of the β-barrel (Chart 1). Furthermore, two more mutants with two and three His6-tags were made, each containing three mutations, i.e., again C114S and C222S but now also serine S128 are mutated into a cysteine (S128C) to give a single, solvent-accessible cysteine residue. These two mutants were conjugated with a maleimide caproic acid modified hexahistidine tag (mic-His6). The conjugates were named NHis6-S128CHis6-TagRFP (1 + 1H) and NHis6-CHis6-S128CHis6-TagRFP (2 + 1H). The single accessible cysteine at position 128 is located in a flexible loop on the side of the β-barrel opposite the N- and C-termini. This means that conjugates 1 + 1H and 2 + 1H have the second or, respectively, the third of their His6-tags situated at the opposite end of their β-barrels with regard to the first or, respectively, the first two His6-tags (Chart 1 and Methods). The steady-state and time-resolved spectroscopic properties of all mutants and conjugates were verified to match those of the wild-type TagRFP (Table S1).
Demonstration of Reversible Binding of Protein Variants
For a qualitative assessment of the stability and reversibility of the immobilization, TagRFP variants 1H, 2H, 1 + 1H, and 2 + 1H were immobilized on bifunctional line patterns made by nanoimprint lithography (NIL; Methods and Scheme S2). Patterns consisted of wide Ni2+:NTA-terminated lines (varying width) and narrower (5 μm wide) poly(ethylene oxide) (PEG)-terminated lines. These patterned surfaces were incubated with solutions of the TagRFP variants for 30 min. After washing, no distinctive patterns were observed. By contrast, extended washing for 12 h in phosphate-buffered saline (PBS) with 5% Tween (PBST) yielded clean backgrounds and specifically immobilized proteins. Fluorescence micrographs (Figure 1) were recorded, and fluorescence intensity profiles measured across the lines (insets Figure 1). All variants were bound to Ni2+:NTA regions, and only faint signatures of nonspecific binding to the proteophobic PEG-terminated regions were observed, indicating site-selective binding of the His6-tag(s) to Ni2+:NTA-terminated surfaces. No binding was observed to the entire surface in the case of 0H, indicating the specificity of the binding of the proteins by the His6-tags. No significant differences in fluorescence intensities were observed between the different variants after 12 h of washing. However, after washing with PBST for 48 h the 1H line patterns had vanished (Figure 1b) and 2H and 1 + 1H patterns showed significantly reduced intensities, while 2 + 1H remained unchanged (Figure 1f,j,n). This indicates that the binding strength of a protein can be increased by increasing the number of binding motifs (multivalency).
Figure 1.

Fluorescence micrographs of NIL-patterned substrates with Ni2+:NTA- (broad lines) and PEG-terminated regions (narrow lines) after incubation with 1H (a–d), 2H (e–h), 1 + 1H (i–l), and 2 + 1H (m–p) and subsequent washing with PBS containing 5% Tween for 12 h (a, e, i, m), for 48 h (b, f, j, n), and for 2 h with PBS containing 5% Tween saturated with imidazole (c, g, k, o) or EDTA (d, h, l, p), and, subsequently, with PBS containing 5% Tween for 12 h (c, d, g, h, l, k, p, o). Insets show the corresponding intensity profiles perpendicular to the pattern. Imaging parameters, such as the exposure time of 2 s, were kept constant for all measurements.
Next, the reversibility of protein binding to the surface was tested by washing with imidazole, a monovalent ligand that competes to bind Ni2+:NTA, and ethylenediaminetetraacetic acid (EDTA) a hexadentated chelating agent for the (effectively irreversible) removal of Ni2+ ions from Ni2+:NTA (Figure 1). Washing with a large excess of monovalent imidazole resulted in nearly complete reversal of 1H immobilization (Figure 1c), while 2H and 1 + 1H showed only reduced intensities (Figure 1g, k) and 2 + 1H patterns remained essentially unchanged (Figure 1o) when compared with the surfaces after 12 h of washing in buffer (Figure 1a,e,i,m). These results support the idea that the higher the valency, the higher the resistance to replacement by the competitor, due to increased binding strength of the proteins with the surface. We relate these observations to multivalency, i.e., increased local concentration (see below), and not to cooperativity. While small differences in secondary structure can, in principle, exist between the different His6-tags on the protein due to conformation and location on the protein, our molecular dynamics results (see below) show that the conformations of the His6-tags appear to be random coil, as expected, and that the His6-tags point outward, as designed, away from the proteins, as can be seen from the structures in Figure S2, making us believe that multivalent effects are prevailing. The observations from the reversibility experiments are in agreement with literature showing reversible binding of single, short His6-tagged proteins and practically irreversible binding of longer His10-tagged proteins to high-valency Ni2+:NTA chelating groups on surfaces.55,56,58,59,63 By contrast, when washing with EDTA, the 2H and 1 + 1H patterns were much more severely reduced than when under treatment with imidazole (Figure 1h,l). As EDTA binds Ni2+, it removes the ion from the complex, preventing any further His6-tag (re)attachment. Hence, only the highest valency variant 2 + 1H can prevent the EDTA-mediated Ni2+ depletion to a significant degree (Figure 1p), indicating that 2 + 1H is, indeed, the most strongly bound variant.
Surface Binding Affinities
The qualitative findings from fluorescence microscopy were quantified using surface plasmon resonance (SPR). Figure 2 (left) shows maximum SPR responses after reaching thermodynamic equilibrium, for varying concentrations (10 pM to 20 μM) of each of the five variants binding to Ni2+:NTA-functionalized SPR sensors (Scheme S1) as well as their corresponding fits to a multivalency model (Table 1 and described below). Binding was clearly observed for all four His6-tagged proteins, while the control, 0H, shows only minimal binding even at a very high concentration (1 μM). These results confirm that the adsorption of TagRFP occurs through specific interaction between His6-tags and surface-bound Ni2+:NTA. Moreover, SPR corroborates the microscopy data, as the binding strength clearly increases, signified by the shifts of the inflection points of the curves of about 1 order of magnitude for each additional His6-tag (Figure 2, Table 1). In addition, SPR reveals information about the absolute amount of immobilized protein. The variants with His6-tag(s) on only one side of the β-barrel (1H and 2H) have a maximum attained coverage of approximately 3 ng/mm2, as estimated from the differential SPR angle shift (Δαmax) of around 300 millidegrees (see fitting below). By contrast, the remaining two variants (1 + 1H and 2 + 1H with His6-tags on opposite sides of the β-barrel) have a significantly lower maximum attained coverage of around 200 millidegrees. This difference in maximum attainable coverage, as well as the range of concentrations in which a particular mutant reaches a total surface coverage (Figure 2, right), suggests a difference in packing of the proteins on the surface. Each His6-tag contributes to the binding of the proteins to the surface (Table 1), which makes it plausible that protein variants with His6-tags on both sides of the β-barrel absorb in flat, side-on orientations on the surface (see Chart 1).
Figure 2.

(Left) Maximum response values of SPR titrations of TagRFP variants 0H (black, squares) 1H (red, circles), 2H (blue, triangles), 1 + 1H (green, pentagons), and 2 + 1H (orange, stars) at various concentrations binding to SPR sensors functionalized with Ni2+:NTA SAMs with their corresponding fits to the multivalency model (see text). (Right) Total surface coverage of the four His6-tagged TagRFP variants estimated from fitting each data point to the multivalency model, plotted as lines. Data represent single measurements that have been reproduced.
Table 1. Optimized Parameters Determined by Fitting the Experimental Data to Langmuir and Multivalency Modelsa.
| variant | KLM (M–1) | Kd,LM (nM) | Ceff (μM) |
|---|---|---|---|
| 1H | 2.7 × 106 | 370 | N/A |
| 2H | 3.8 × 107 | 26 | 1.8 |
| 1 + 1H | 4.7 × 107 | 21 | 5.7 |
| 2 + 1H | 2.7 × 108 | 3.7 | 12 |
KLM is the apparent overall binding association constant (Kd,LM is the related binding dissociation constant) obtained from the fit using a 1:1 Langmuir-type model for each variant. Ceff is the effective concentration obtained from a fit using a multivalency model (see text and Methods).
The observed difference in maximum coverage for the variants with binding motifs on only one side of the β-barrel suggests that those variants prefer to adopt a more upright, end-on orientation. This alternative orientation reduces their footprint to provide closer packing, which in turn allows higher surface coverages.
The SPR data were first fitted using a 1:1 Langmuir-type model,27 which assumes that each His6-tag interacts as a single entity with the Ni2+:NTA surface (Table 1). The resulting fits are in very good agreement with the experimental SPR data. As the Langmuir model yields only overall observable binding constants (KLM; Table 1), we employed a second more detailed model, based on the concept of multivalency and effective concentration (Ceff).67 This model provides deeper insight into the differences in binding conformations as a function of the number and position of the His6-tags of the different variants (Methods). Briefly, Ceff is a measure for the (much increased) probability, compared to a monovalent ligand, of a second (or third, etc.) binding moiety of a multivalent ligand attaching to the surface after the first moiety has bound. It takes the form of a concentration (number of molecules per unit volume), as it can be viewed as the number of binding sites the second (or third, etc.) binding moiety could reach within its probing volume (considering steric aspects). For fitting the SPR titration data for 1H, we used the maximum attained coverage Δamax and the intrinsic binding association constant of a single His6-tag (Ki,His6) as variables. We have treated the binding of an entire His6-tag as a single binding event throughout this analysis. We obtain a value for Ki,His6 = 2.8 × 106 M–1 (Kd = 0.36 μM), which is in very good agreement with the KLM value measured for 1H (Table 1), indicating that the histidine binding is equal to the overall binding constant. Our value is in agreement with literature values measured in solution for cases where two coordination bonds are formed between His6-tags and trivalent chelating NTAs55 and higher than literature values for cases of His6-tag binding to assembled Ni2+:NTA ligands on β-CD surfaces,57,63 indicating that, here, less than three coordination pairs (see Chart 1) between the His6-tag and surface are formed. Furthermore, the Langmuir fits show that variant 2 + 1H binds 1 order of magnitude stronger than the bivalent constructs 2H and 1 + 1H (Table 1), which is 1 order of magnitude stronger than literature values measured in solution for forming three coordination bonds with trivalent NTA chelating entities,56 indicating that more than three coordinating bonds were formed on our surfaces for 2 + 1H. For fitting the data of the other variants, Ki,His6 was fixed to the value found for 1H, while Δamax and Ceff were optimized. The resulting Ceff values of 1.8 μM (2H), 5.7 μM (1 + 1H), and 12 μM (2 + 1H) (Table 1) increase stepwise in magnitude and provide some insight into how the different TagRFP variants attach to the surface. It is worth mentioning at this point that typical Ceff values tend to be in the millimolar range,55,63,68 while here they are in the micromolar range. We attribute this fact to our use of N-hydroxy succinimide chemistry for the functionalization of the SPR sensors (Scheme S1), which resulted in low surface density of metal-chelating NTA units, which we, in turn, corroborated using X-ray photoelectron spectra (XPS) (Table S3). We did not include an explicit quenching of residual succinimidyl esters in our functionalization protocol because during intensive washing steps these esters are subject to hydrolysis.47 A low surface density of NTA puts a much lower number of binding sites in reach of the ligands and, thus, much lower Ceff than obtained previously with, for example, cyclodextrin-terminated SAMs.55,63,68 We emphasize that there is a significant difference between the His6-tag proteins interacting with NTA bound to a surface and with free NTA in solution. In solution NTA moieties are isolated from each other, while on a surface they are immobilized in close proximity to each other, allowing multivalent binding, here responsible for the observed enhanced binding of the multivalently tagged proteins. It is important to realize that any binding experiment in solution between monovalent NTA moieties and any His6-tag of the constructs will lead only to a determination of the monovalent binding event between one NTA moiety and (two) histidine residues. Therefore, we compared our binding constants on our surfaces with referenced solution data of Tampé, Piehler, and co-workers56,57 in which the interaction between a trivalent NTA moiety (i.e., three connected NTAs) with His-tags in solution was studied and also represents a case of multivalency where in solution multivalent interactions can occur between the three individual NTA moieties and the histidine residues.
As mentioned above, Ceff is directly proportional to the number of accessible binding sites, which in turn is directly proportional to the (accessible) surface area, with a square dependence on the linker length, r, linking the binding moieties. Furthermore, Ceff is inversely dependent on the probing volume, which has a cubic dependence on the linker length, i.e., Ceff ∼ area/volume ∼ r2/r3. With this in mind, it can be reasoned why 2 + 1H has the highest Ceff value of 12 μM. The conformational freedom of the third His6-tag is highly constrained in the divalently bound protein, and this reduction in flexibility reduces the accessible surface area, but more strongly the probing volume, resulting in the high Ceff for the trivalent complex. Interestingly, the Ceff values found for 2H and 1 + 1H clearly differ. Ceff is higher for 1 + 1H, the variant with the His6-tags on opposite sides of the β-barrel, than for 2H. This effect can be due only to the difference in position of the second His6-tag, which raises the possibility that site-specific attachment of binding motifs can provide different binding strengths even for complexes with the same number of binding motifs, evident from the separation between 2H and 1 + 1H data sets in the SPR titration plots in Figure 2. Intuitively, we expected that the necessary side-on orientational change needed to bind the second tag of 1 + 1H is more difficult than the search for a second NTA by the second tag of 2H. However, in the case of end-on binding of 2H, the two His6-tags compete for the same and limited NTA moieties, whereas this does not occur in the case of side-on binding of 1 + 1H. Moreover, in the case of side-on binding of 1 + 1H additional (possibly competing or repulsive) electrostatic interactions between a larger fraction of the β-barrel structure and charged NTA moieties could be possible. Taken together, the differences in KLM and Ceff for the different variants from SPR data, corroborated by our multivalency model, suggest that the binding affinity and orientation of the protein variants on the surface depend on both the position and number of binding motifs present.
Polarization-Resolved Lifetime Imaging on Immobilized Proteins
Polarization-resolved fluorescence lifetime imaging microscopy was performed to compare the anisotropy decay times of end-on and side-on oriented immobilized TagRFP. As concluded from SPR studies, two different types of surface coverages, i.e., packing, for each of which one representative variant was selected. As representative variants for this study, we used the proteins with the highest binding strengths (i.e., stability) per orientation, 2H (end-on) and 2 + 1H (side-on), as these samples would be most stable in terms of protein adsorption, as the measurements are very time-consuming (4 to 6 replicas). Representative plots of polarization anisotropy versus time can be found in Figure S15. The proteins were immobilized at the saturation levels determined from SPR (Figure 2). Anisotropy decay times were determined for 2H and 2 + 1H immobilized on various Ni2+:NTA/PEG line patterns on glass, measuring at different locations on different samples (see Methods for a detailed description of sample preparation, setup, and analysis). A plot with all fitted anisotropy lifetimes versus peak intensity is shown in Figure 3. Decay times cluster into two distinct groups (Figure 3) with decay times of 2H systematically shorter than those of 2 + 1H. The X-ray structure of TagRFP shows that the chromophore is oriented approximately perpendicular to the long axis of the barrel.69 The distribution of orientations of the chromophores is therefore expected to be the same for end-on and side-on orientation of the proteins on the surfaces, i.e., in both cases randomly oriented transition dipole moments parallel to the surface. The direction of detection is perpendicular to the surface. This means that there will be no difference in orientation dependence of the fluorescence of isolated chromophores (proteins) when comparing end-on and side-on adsorption. Differences in fluorescence intensity should therefore solely stem from differences in the density of proteins on the surface. From this it also follows that there should not be a difference in orientation dependence of polarization anisotropy decay between end-on and side-on orientations. Polarization anisotropy decay is strongly dependent only on the relative distance and orientation of two TagRFP chromophores that undergo energy transfer. Therefore, we believe that the loss of anisotropy could be caused by two conceivable mechanisms: energy transfer between the chromophores of neighboring molecules (homo-FRET) and freedom of movement or rotation. The loss of anisotropy is much faster for 2H, indicating that in this case proteins are either more closely packed, making for more efficient energy transfer between chromophores, or more mobile. When considering that relatively high, saturated, surface coverages were used for these measurements, homo-FRET seems the more likely explanation. The time-resolved anisotropy results, then, support the assumption of more close packing between end-on (2H) oriented TagRFP variants (2 + 1H can adsorb side-on; Chart 1). We note also that 2H samples all exhibited much higher fluorescence intensities than 2 + 1H samples (Table S2), which points to a higher surface coverage of 2H and thus a closer packing for end-on oriented 2H.
Figure 3.

Plot of the fitted anisotropy lifetimes τ versus the sum of their peak intensities for immobilized 2H (open squares) and 2 + 1H (solid squares). Data represent single measurements.
Polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS) was carried out on end-on (1H) and end-on (2 + 1H) oriented TagRFP immobilized on Ni2+:NTA-functionalized gold substrates at saturation levels (Figure 4). The immobilization of protein caused a marked increase in the proportion of the dichroic ratio coming from amide versus carbonyl groups when compared to bare Ni2+:NTA layers on gold, as monitored for the amide I band at 1660 cm–1 and the carbonyl stretch vibration of COOH groups at 1740 cm–1. The amide I to COOH ratio of unity on bare Ni2+:NTA rises to 4.5 for 1H but only to 2.3 for 2 + 1H, which is consistent with the existence of more closely packed protein in the case of the end-on binding mode.
Figure 4.
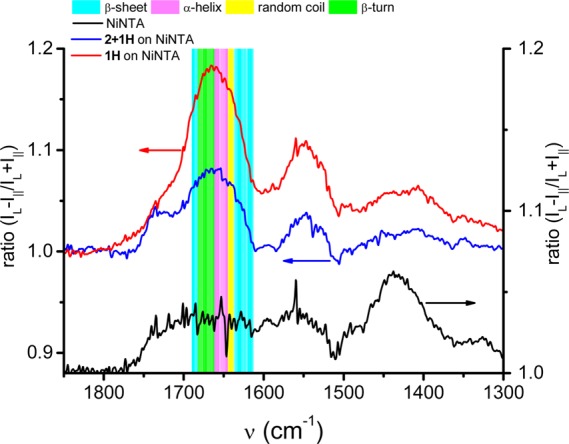
PM-IRRAS differential reflectance spectra of 1H (red line) and 2 + 1H (blue line) immobilized on Ni2+:NTA SAMs (the black line is the Ni2+:NTA surface prior to protein binding). Colored bands correspond to regions associated with specific secondary structural elements within the amide I region of the protein, and the changes in these bands are used to elucidate protein orientation. Intensities on the y-axis are given as dichroic ratios.
Furthermore, the amide I band between 1600 and 1700 cm–1 can be largely assigned to C=O stretch vibrations, and different regions of this band are correlated with different secondary structural elements of proteins.70 More specifically, the signal at 1654 cm–1 corresponds to α-helices (magenta) and the dichroic ratio at 1633 cm–1 to β-sheets (cyan).71 Ratios between the dichroic ratios for α-helix and β-sheet were determined to be 1.3 and 1.1 for 1H and 2 + 1H, respectively. Since 1H and 2 + 1H are structurally identical, apart from their number of His6-tags, this observation indicates a difference in orientation. From crystallographic data it can be seen that the only substantial α-helical structural element of TagRFP is oriented along the axis of the β-barrel. Therefore, the α-helical C=O stretch vibration, which is nearly parallel to the axis of the α-helix, should be observable in PM-IRRAS (for high angles of incidence of the polarized light on a conductive surface) only if the protein is oriented end-on. The differential reflectance spectrum of 1H, with its higher relative α-helix signal, indicates that the β-barrel and, thus, the α-helix are oriented more end-on, or normal to the surface, while the spectrum of 2 + 1H indicates that the β-barrel is oriented more side-on, or parallel to the surface. Taken collectively, all experimental data indicate a distinct preference for an end-on orientation for immobilized TagRFP variants with His6-tags placed on the same side of the β-barrel, in contrast with side-on orientation taken by alternative TagRFP variants with His6-tags placed on either end of the β-barrel.
Simulations of His6-Tagged Protein Complexation to Ni2+:NTA SAMs
In an effort to reveal the atomic-scale details of the effect that the number and positioning of His6-tags has on the strength of the binding interactions as well as the orientation of the protein upon binding to Ni2+:NTA SAMs, molecular dynamics (MD) simulations were performed (see Methods for a detailed description). There have been only a few reports of MD studies on the Ni2+:NTA-His6 system.72−74 Most notably, Zhang etal. performed molecular dynamics simulations on a His6-tag interacting with a single Ni2+:NTA complex and found that the residue pair His(2,3) was the most stable pair, while secondary structure analysis showed that the His6 structure has a high propensity for random coil conformations.74 While MD has been used to describe the behavior of fluorescent proteins in solution75−77 as well as the interactions of a green fluorescent protein with a silicon substrate,78 there have been, to the best of our knowledge, no MD studies on the immobilization of fluorescent proteins onto SAMs. Therefore, to better understand the atomic-scale structure of the protein–SAM interface, we modeled the immobilization of all our TagRFP proteins with a varying number of His6-tags (Figure S2) to Ni2+:NTA-modified SAMs. Top and side views of uniform Ni2+:NTA (control simulation) and mixed Ni2+:NTA/carboxylic acid SAMs are given in Figures S3 and S4. More ordered SAM structures were formed for the mixed monolayer,79 which represents our fabricated low NTA density SAMs. To study the protein–SAM interactions, the histidine residues of the TagRFP variants were placed near Ni2+:NTA in the SAM in positions where a complex might form. After equilibration, 20 ns of free dynamics was performed for each complex and showed that the magnitude of the computed protein–SAM interaction energy is directly correlated with the number of histidine residues complexed (Figures S5–S7). The final computed structures of the His6-tags binding to the uniform and mixed Ni2+:NTA SAMs are given in Figures S8–S13. In general, the N-terminal His6-tag has the largest number of NTA-complexed histidine residues, whereas the C-terminal His6-tag may have fewer complexed histidine residues because of repulsive interactions between the C-terminal region of the TagRFP and the NTA carboxylate oxygens. We estimated a tag–surface interaction energy (summed over electrostatic and van der Waals interactions) of −190 kcal/mol (Table S4) for the His6-tag with all six histidine residues bound. This gives an interaction energy per histidine of approximately −32 kcal/mol, which is similar to the value (−30 kcal/mol) calculated by Yang etal.80 using quantum mechanical simulations of the binding of a Ni2+ ion to one histidine. Computed interaction energies for the uniform and mixed SAMs with all TagRFP variants and orientations are given in Table S4. During simulations on both the uniform and mixed SAMs, the protein remains bound and maintains a near-constant, to within a few angstroms, center-of-mass height above the surface. Apart from the interactions at the His6-Ni2+:NTA binding sites, interactions between the SAM and the proteins are driven mostly by electrostatic interactions involving amino acids with charged side chains on the exterior of the protein and the charged terminal headgroups of the SAM. Also, the protein–SAM interaction energies are much larger for the uniform control SAM (Table S4) since the protein is in direct contact with more of the SAM. For the experimentally used mixed SAM, the interaction energy between the protein and the SAM is comparable to that of the interaction energy between the His6-tag and the SAM (Table S4). To investigate whether on balance the protein prefers a side-on or end-on orientation, the total interaction energy, including protein–water interactions as well as protein–SAM interactions, was evaluated. Increasing the number of His6-tags on the protein gives an increase in the total interaction energy, which agrees with experimental results. However, for both uniform and mixed SAMs, there are no large differences in the interaction energies for proteins immobilized in an end-on compared to a side-on orientation (Table S4). In fact, for both SAMs, the end-on oriented 1H has less favorable time-averaged interaction energies than 1H oriented side-on on the SAM. This indicates that the computed loss in protein–SAM interactions on standing up is greater than the gain in protein–water interactions. For 2H the time-averaged preference for the side-on orientation is smaller than in the case of 1H, but we still do not see a computed preference for the end-on orientation, at odds with the experimental observations. From these equilibrium simulations no strong preference in protein orientation can be deduced. We hypothesize that at experimental time scales proteins adopt an end-on orientation simply because it allows more proteins to be immobilized so the magnitude of the protein–SAM interaction energy per unit area is increased, as is the density of lateral protein–protein contacts. We tentatively propose an assembly mechanism analogous to the concentration-dependent assembly of alkanethiol SAMs on gold,81 which grow from sparse populations of horizontally oriented single or few-molecule clusters to tightly packed clusters of vertically oriented molecules with intermolecular forces becoming stronger than molecule–surface forces. The protein–surface contact area is approximately doubled when the protein is immobilized side-on, and so larger surface coverages are achieved for the end-on orientation, while the surrounding water density is comparable (Table S5). The protein conformational energy and radius of gyration values show little deviation between bulk solvated and SAM-bound (via the His6-tags) states (Table S6, less than 5% change in conformational stability and size). This indicates that the His6-tag method of immobilizing proteins allows the protein to be tethered to the SAM surface in its native conformation. This fits with the unchanged fluorescence properties of the proteins upon immobilization, which is known to be a sensitive probe of structural integrity for fluorescent proteins.
We used the computed MD protein–SAM structures as starting points for nonequilibrium steered MD (SMD) simulations to estimate the potential of mean force (PMF) required to remove an immobilized 2H His6-tagged TagRFP protein from the mixed Ni2+:NTA SAMs. The SMD simulation was used to pull the protein a distance of 11 nm away from the SAM over a time of 7 ns. Fifty-eight equally spaced configurations were extracted from the pulling trajectory, and umbrella sampling was performed for 10 ns on these to obtain the PMF. Figure 5 shows the PMF profiles that were calculated for detachment of 2H when immobilized in end-on and side-on orientations, while Figures 6 and 7 show key configurations along the unbinding pathways (starting configurations are shown in Figure S14). The two different binding orientations show different desorption paths. It costs approximately 18 kcal/mol to unbind 2H starting from the end-on orientation, in which only the His6-tags are in contact with the SAM, roughly half the energy required to unbind 2H starting from the side-on orientation (approximately 38 kcal/mol), in which the protein β-barrel is also in contact with the SAM.
Figure 5.
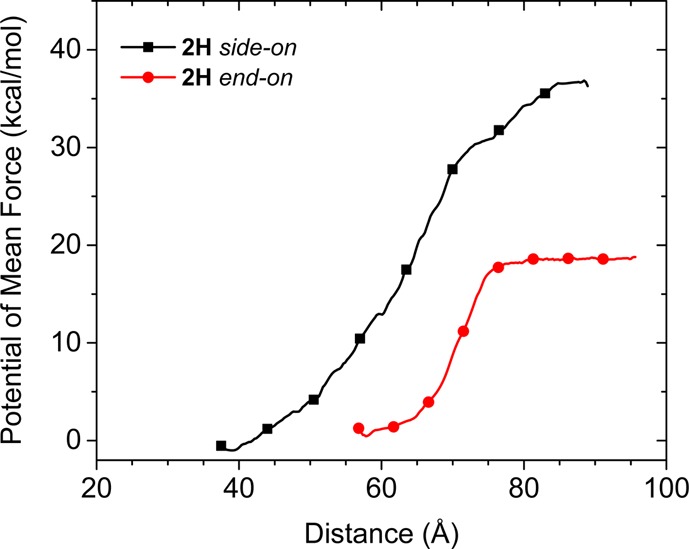
Computed potential of mean force profiles for 2H desorption in side-on (black squares) and end-on (red circles) orientations. The pulling force is applied to the main body of the protein excluding the His6-tags.
Figure 6.

2H desorption, starting from an end-on bound orientation. The structures correspond to distances of (a) 64 Å; (b) 65 Å; (c) 74 Å; and (d) 77 Å in the force profile (Figure 5, red curve).
Figure 7.

2H desorption, starting from a side-on bound orientation. The structures correspond to distances of (a) 48 Å; (b) 59 Å; (c) 70 Å; and (d) 71 Å in the force profile (Figure 5, black curve).
Figure 6a and b show configurations of end-on oriented 2H just before and after, respectively, desorbing the C-terminal His6-tag at around 65 Å on the horizontal axis, which agrees with the observation from the equilibrium MD simulations that the C-terminal His6-tag usually had a lower number of bound histidine residues, resulting in turn in a lower binding energy (Figure S8). Figure 6c shows the configuration of an intermediate state in which the N-terminal His6-tag was still immobilized and the C-terminal His6-tag was completely detached, and Figure 6d shows the configuration after both His6-tags were desorbed. The observation that the core protein barrel of end-on oriented 2H has practically zero interaction with the SAM (Figure 6) contrasts with the substantial protein–SAM interactions that must be broken to desorb the side-on oriented 2H (see also computed interaction energies in Table S4). For the side-on oriented 2H, the barrel desorbed from the SAM at a distance of 48 Å (Figure 7a) with a 3 kcal/mol magnitude loss in adsorption energy. Then, the C-terminal His6-tag desorbed from the SAM at 59 Å (Figure 7b), giving an additional 9 kcal/mol loss in energy. The N-terminal His6-tag desorbed at 71 Å (configurations shown in Figure 7c and d (before and after, respectively)), with an energy difference of 18 kcal/mol between the unique events where each of the two His6-tags were desorbing. However, these events were undetectable in the force profile for desorbing side-on oriented 2H (Figure 7), from which we conclude that it is, nonobviously, the protein pushing against water that contributes the most to the force. Protein adsorbed in the side-on orientation has a higher surface area in the direction of desorption (normal to the plane of the SAM) and also showed a much longer interaction length of 50 Å compared with 20 Å for end-on oriented 2H. Therefore, the PMF data quantify the energy required to disrupt water adlayers as the protein desorbs from the alternative bound conformations,82,83 which has been associated with a high entropic cost due to the disruption of the water hydrogen-bonding network.
The simulations then complement the experiments by suggesting a binding mechanism in which the protein approaches the surface end-on to minimize disruption to water adlayers (from the PMF calculations) then flips between approximately isoenergetic bound end-on and side-on states (from the equilibrium MD simulations) before (presumably) adopting an end-on orientation to maximize packing with other adsorbing proteins. Future work could involve explicit modeling of the adsorbed protein SAM, which would require sampling of the full range of possible protein–protein and protein–surface orientations, beyond the scope of the present work.
Conclusions
Proteins have been successfully engineered with strategically placed His6-tags on both N- and C-termini and a solvent-accessible loop of TagRFP. Steady-state and time-dependent spectroscopic properties of five mutants and conjugates matched those of the wild-type TagRFP. The binding strength of the variants increased with increasing numbers of binding motifs as qualitatively assessed by following fluorescent patterns in reversibility and competition experiments. These findings were corroborated by SPR studies signifying that each additional His6-tag increases the binding strength by 1 order of magnitude. With each His6-tag contributing to the binding of the proteins to the surface, additional information from SPR revealed that the absolute amount of immobilized proteins was reduced by a third when placing the binding motifs on opposite sides of the barrel. With binding motifs placed on the same side of the barrel a more upright, end-on orientation is adopted, while binding motifs placed at opposite sides of the barrel give a preference for a flat, side-on orientation. Measured binding affinities and binding modes calculated in the molecular simulations have shown that not all histidine residue pairs are involved in complexation, due to a combination of repulsive interactions between the protein and the solvated surface interactions and sterical hindrance at the His6-(Ni2+-NTA)3 sites. Time-resolved fluorescence anisotropy techniques verified that the orientation and thus coverage cause distinct, different fluorescent lifetime characteristics. Potential of mean force calculations coupled with calculated protein–surface adsorption energies suggest that the experimentally measured end-on orientation is a supra-protein effect driven by maximization of horizontal protein–protein interactions as the “footprint” of each individual protein is reduced.
The scientific challenge of this work was to provide evidence that protein immobilization occurs with envisioned control over orientation, surface coverages, affinities, and function. Firm structural evidence of uniformly oriented proteins is currently lacking, and firm quantification of binding characteristics by experiment and theory is lacking in most if not all protein immobilization studies. With this contribution we show evidence and discussion that tunable control over the orientation and binding strength of protein immobilization can be achieved by well-considered placement of multivalent binding motif(s) on the protein. We realize that in light of further development and understanding of protein immobilization and tag placement on the proteins it would be interesting to optimize the His6-tag NTA interaction by finding ways to fully involve each and every residue of all His6-tags in the formation of the maximum number of six coordination bonds with surface-bound NTA. We show by experiment and theory that this is not the case and that the solution to this challenge should not be sought in whether N- and C-termini or loops are chosen for tagging but rather in the spacing of the entire hexahistidine tag with respect to the protein and, equally important, the internal spacing of two histidine residues with respect to two other histidines to avoid steric crowding effects between the six histidine residues with the surface-bound NTA moieties.
The strict correlation between the intact tertiary structure (i.e., folding) and the unchanged fluorescence characteristics of fluorescent proteins makes them excellent model systems to probe the preservation of structural integrity of the protein upon immobilization. Any impairment of the structural integrity of the fluorescent protein would immediately lead to drastic changes in fluorescence characteristics, such as its intensity and lifetime, which can be observed by state-of-the-art fluorescence microscopy. A comparison of the relevant time traces showed no significant changes in fluorescence lifetimes between samples with proteins adsorbed side-on and end-on; only the anisotropy decay changed significantly. The realization of design of orientation of immobilized proteins not only is important for the fluorescent protein field itself but would be applicable to a range of much broader fields, such as the fundamental study of the protein dynamics and chemistry (folding, maturation, etc.) while being attached to an immobile surface. These developments are also highly relevant to fields to improve the performance of proteins when using His6-tag (or other tags) technology in diagnostic assays, purification protocols, and immobilization studies. Broad application of the results is foreseen when employing weak to moderate supramolecular interactions to immobilize proteins in applications where (reversible) control over orientation, binding strength, surface coverage, and function is required to optimize the performance of the assay, material, or device.
Methods
Materials
Maleimide caproic acid hexa(histidine) (mic-His6) was purchased from JPT Peptide Technologies, Germany. All solvents were of p.a. quality and purchased from Biosolve (Valkenswaard, The Netherlands). All other starting compounds were purchased from Acros (Geel, Belgium) or Sigma-Aldrich (Zwijndrecht, The Netherlands). All compounds were used as received unless stated otherwise. Deuterated solvents used for NMR spectroscopy were purchased from Cambridge Isotope Laboratories, and the water used was always of Milli-Q quality (Millipore, R = 18.2 MΩ·cm).
Preparation of NHis6-wtTagRFP
The following primers were used for PCR amplification of wtTagRFP using pTagRFP-C (Evrogen JSC) as DNA template: 5′-cgcggatccaatgagcgagctgattaaggagaacatgca-3′ containing a unique BamH I restriction site (underlined) and 5′-cgcgaattccttgtgccccagtttgctag-3′ containing a unique EcoR I restriction site (underlined). The PCR product was purified and digested with BamH I and EcoR I restriction enzymes (NEB) and ligated into pRSETB plasmid (Invitrogen), digested with the same restriction enzymes. pRSETB contains an N-terminal hexahistidine tag (NHis6-tag) for nickel-affinity purification and an enterokinase recognition site (DDDDK) to allow for the subsequent cleavage of the His6-tag. The resulting plasmid, pRSETB-wtTagRFP, was first transformed into E. coli (XL10 gold, Stratagene) using standard procedures in the presence of ampicillin (100 mg/L) for amplification and further mutagenesis. pRSETB-wtTagRFP plasmid was also transformed into E. coli BL21 pLysS using standard procedures in the presence of ampicillin (100 mg/L) and chloramphenicol (34 mg/L) for protein expression. Single-colony transformants were selected, and precultures were grown overnight at 37 °C. These precultures were each used to inoculate 2 L of LB medium containing ampicillin (100 mg/L) and chloramphenicol (34 mg/L) at 37 °C with shaking until an OD600 = 0.6 was reached. The cultures were cooled to 16 °C before protein expression was induced with isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM and incubated overnight at 16 °C. Cells were harvested by centrifugation at 4000g at 4 °C for 20 min. The resulting cell pellets were resuspended for 20 min in BugBuster reagent with benzonase nuclease (Novagen) according to the supplier’s instructions. The lysate was cleared by centrifugation at 16000g for 30 min at 4 °C. Ni2+:NTA agarose beads (QIAGEN) were added to the protein-containing supernatant at a 1:10 v/v ratio, respectively, and incubated at 4 °C for at least an hour with slow but continuous mixing. The agarose beads were filtered and washed with wash buffer (20 mM Tris buffer, 300 mM NaCl, 20 mM imidazole, pH 8.0), and the bound protein fraction was eluted with elution buffer (20 mM Tris buffer, 300 mM NaCl, 1 M imidazole, pH 8.0). The purified NHis6-wtTagRFP fractions (∼30 μM) were subsequently rebuffered using PD10 columns (GE Healthcare) into 0.1× PBS (0.8 mM phosphate buffer, 14.4 mM NaCl, 0.27 mM KCl, pH 7.4), aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C. The protein was characterized using SDS- and native PAGE (Figure S1), UV–vis, steady-state and time-resolved fluorescence spectroscopy, and MALDI-TOF mass spectrometry (see below).
Preparation of NHis6-CHis6-TagRFP
For the insertion of a second, C-terminal His6-tag (CHis6-tag), the following procedure was used regardless of the mutant. The TagRFP gene was amplified using 5′-cgcggatccaatgagcgagctgattaaggagaacatgca-3′ (BamH I restriction site is underlined) and 5′-gcggaattcttagtggtggtggtggtggtgcttgtgccccagtttgcta-3′ (EcoR I restriction site is underlined, encoding a His6-tag) as forward and reverse primers, respectively, and pRSETB-TagRFP as DNA template. After PCR purification, the gene product and pRSETB-TagRFP were digested sequentially, first with EcoR I, then BamH I, according to the manufacturer’s instructions. DNA ligations were performed using T4-ligase (NEB) at 16 °C overnight, and the resulting pRSETB-TagRFP-His6 plasmid was transformed into E. coli (XL10 gold, Stratagene) competent cells according to standard procedures in the presence of ampicillin (100 mg/L) for amplification and further mutagenesis. pRSETB-wtTagRFP-His6 plasmid was also transformed into E. coli BL21 pLysS in the presence of ampicillin (100 mg/L) and chloramphenicol (34 mg/L). Single-colony transformants were selected, and precultures were grown overnight at 37 °C. These precultures were each used to inoculate 2 L cultures of E. coli BL21 pLysS cells, which were grown at 37 °C to OD600 = 0.6 and cooled to 16 °C, and protein expression was induced with IPTG to a final concentration of 1 mM. The culture was incubated overnight at 16 °C. The cells were harvested by centrifugation (4000g, 20 min) and lysed using BugBuster (as described above). Protein purification and characterization were carried out as described above.
Preparation of NHis6-S128CTagRFP, NHis6-CHis6-S128CTagRFP, NHis6-TagRFP (1H), and NHis6-CHis6-TagRFP (2H)
By site-directed mutagenesis (QuikChange Multi kit, Stratagene Technologies), selected mutations were introduced into pRSETB-wtTagRFP (for both singly- and doubly-His6-tagged TagRFP) using the following primers: S128C forward 5′-ggtgaacttcccatgcaacggccctgtga-3′; reverse 5′-tcacagggccgttgcatgggaagttcacc-3′; C222S forward 5′-ggctgtggccagatactccgacctccc-3′; reverse 5′-gggaggtcggagtatctggccacagcc-3′; C114S forward 5′-gcctccaggacggctccctcatctacaac-3′; reverse 5′-gttgtagatgagggagccgtcctggaggc-3′.
Mutations S128C, C222S, and C114S yielded mutants “S128CTagRFP” containing a single accessible cysteine residue in a loop at the top of the β-barrel. Mutations C222S and C114S yielded mutants “TagRFP” with no accessible cysteine residues left. All mutant variants were expressed and purified under the same conditions as the wild-type, to yield NHis6-S128CTagRFP, NHis6-CHis6-S128CTagRFP, NHis6-TagRFP (1H), and NHis6-CHis6-TagRFP (2H). Characterization by SDS- and native PAGE (Figure S1), UV–vis, steady-state and time-resolved fluorescence spectroscopy, and MALDI-ToF mass spectrometry (see below) was carried out and confirmed the successful expression and purification of the respective proteins. The mutants showed no discernible differences regarding their photophysical properties from the wild-type variants NHis6-wtTagRFP and NHis6-CHis6-wtTagRFP, thus indicating that their overall structure and fluorescence properties upon site-directed mutagenesis are retained.
Preparation of wtTagRFP without His6-Tags (0H)
Removal of the His6-tag from the NHis6-wtTagRFP was accomplished by enterokinase digestion using enterokinase (Invitrogen) and its corresponding enterokinase removal kit (EKaway, Invitrogen) according to the manufacturer’s instructions. In short, enterokinase digestion of NHis6-wtTagRFP was carried out overnight at 37 °C using protein stock solution with varying amounts of enzyme, where undigested protein served as negative control. Cleavage was nearly complete using 4 units of enterokinase per 20 μg of NHis6-wtTagRFP. After digestion, enterokinase was removed by letting it bind to the removal kit’s enterokinase-binding resin, which was, in turn, removed by centrifugation (5000g, 2 min). Subsequently, residual, undigested protein was removed using nitrilotriacetatenickel (Ni2+:NTA)-affinity chromatography, resulting in a flow-through fraction of pure, His6-tag-free wtTagRFP (0H). Characterization by SDS-/native PAGE and Western blotting (Figure S1), UV–vis, and steady-state and time-resolved fluorescence spectroscopy (see below) was carried out and confirmed the successful expression and purification of 0H.
Preparation of NHis6-CHis6-S128CHis6-TagRFP (2 + 1H) and NHis6-S128CHis6-TagRFP (1 + 1H) Conjugation
NHis6-S128CTagRFP and NHis6-CHis6S128CTagRFP, which both possess a single accessible cysteine residue at position 128, were conjugated with a thiol-reactive maleimide-functionalized oligopeptide, maleimide caproic acid hexa(histidine) (mic-His6), to yield NHis6-S128CHis6-TagRFP (1 + 1H) and NHis6-CHis6-S128CHis6-TagRFP (2 + 1H), respectively. As negative and control NHis6-TagRFP and NHis6- CHis6-TagRFP were used, respectively. Conjugations were carried out by first reducing the cysteine residues using 10 mM phospate buffer (PB) containing 0.1 mM dithiothreitol (DTT) (pH 8.0) for 30 min and removing the DTT again (Zeba Spin, Thermo Scientific) and then by incubating a 10:1 mixture of maleimide/protein (∼20 μM) in 10 mM PB (pH 8.0) in the dark for 24 h at room temperature (RT). The reactions were subsequently quenched by adding a 10-fold molar excess (with respect to the maleimide) of DTT for 30 min at RT, after which the samples were rebuffered (Zeba Spin, Thermo Scientific) into 0.1× PBS at least three times to remove any residual peptide and DTT. Characterization was performed as described above, and conjugation yields were typically in the range 30–50% (see below).
Purification of the His6-tagged conjugates was conducted using an Äkta FPLC system and a 1 mL HisTrap HP column (GE Healthcare) according to the manufacturer’s instructions. Conjugates containing two and three His6-tags could be separated from the native proteins containing one and two His6-tags, respectively, and were isolated and characterized using SDS- and native PAGE (Figure S1) as well as MALDI-ToF MS (see below). The overall purity was found to be at least >80% after purification and rebuffering into 0.1× PBS using PD10 spin columns (GE Healthcare).
Surface Plasmon Resonance
SPR measurements were performed using instruments in Kretschmann configuration: either a Resonant Technologies GmbH (Germany) RT2005 SPR setup or an IBIS Technologies imaging SPR (iSPR) system (see below). In both cases glass substrates covered with a 50 nm gold layer were used. On the gold, Ni2+:NTA was self-assembled into a monolayer via Au–sulfide interactions (see below). In the case of the Resonant Technologies setup, these substrates were attached to a 70 μL volume microfluidic cell mounted on a prism, which in turn was mounted on a double goniometer head, with which the angle of incidence of the exciting laser on the prism (Schott, LaSFN9) could be controlled. Light from a 2 mW HeNe laser of 633 nm wavelength passed through the prism and onto the substrate. The intensity of the reflected light from the substrate was measured by a large-area photodiode. In both setups the gold-on-glass substrate was optically matched to the prism using index matching oil (Cargille; series B; nD25 = 1.700 ± 0.002). Imaging SPR measurements on the iSPR (IBIS Technologies), also set up in Kretschmann configuration, using a laser of 800 nm wavelength, were performed on the above type of substrates, and various regions of interest were assigned, for each of which SPR sensograms were determined, individually. Here too, the resonance angle was determined by continuously scanning through the surface plasmon resonance dip and finding the minimum, during binding experiments in a flow-cell. PBS with 0.01% Tween-20 was used as a running buffer in all experiments. All SPR experiments were performed at a continuous flow rate of 20 μL/min.
Steady-State and Time-Dependent Spectroscopy
Absorption spectra of all fluorescent proteins, their mutants, and conjugates were recorded using a Perkin Elmer LAMBDA850 UV/vis spectrophotometer. Fluorescence spectra and lifetime data of all fluorescent proteins, their mutants, and conjugates were recorded using a JobinYvon-Horiba Fluoromax4 fluorimeter including a TCSPC system for time-dependent measurements with pulsed LEDs for excitation at 561 nm. Results are summarized in Table S1.
Fluorescence Microscopy
Steady-state fluorescence microscopy images were recorded using an Olympus IX71 inverted microscope equipped with a U-RFL-T mercury burner lamp as light source and a digital Olympus DR70 color camera for image acquisition. Olympus filter cubes with appropriate band-pass or long-pass filters and dichroic mirrors (Semrock) were used. Fluorescence micrographs were acquired using a 20× Fluorplan objective from Olympus and exposure times of 2 s.
Fluorescence Anisotropy Measurements
Fluorescence anisotropy measurements were carried out on a custom-made microscope based on an Olympus IX71 body. Light from a Fianium SC 400-2-PP super continuum laser was passed through an acousto-optical tunable filter set to 515 nm and an additional band-pass filter (FF01-520/15, Semrock) into the microscope. The light was passed through a linear polarization filter (LPVISA100, Thorlabs) and focused on the sample with an Olympus UplanSApo 60× 1.2 NA water immersion objective. The generated fluorescence was filtered using a long-pass filter (LLP01-532R, Semrock) and a polarizer (LPVISB, Thorlabs). Detection was performed by an avalanche photodiode (PDM, Micro Photon Devices) that is connected to a Becker & Hickl SPC-830 counting card operating in time-tagged mode. For each measurement (all at equal exposure times) two sequential images of the same region were taken, one with the polarization parallel to the polarized excitation and one perpendicular; the results were checked to be independent of the order of acquisition. The anisotropy lifetime was obtained using a commercial software package (FluoFit v4.5, PicoQuant) by a fit of the observed time-dependent anisotropy using a single-exponential decay. Intensities were the sum of the peaks of the parallel and perpendicular time traces.
Mass Spectrometry
Mass spectrometry of small molecules and peptides was performed using a Waters micromass LCT ESI mass spectrometer. Mass spectrometry on all protein variants and conjugates was carried out using a MALDI ToF mass spectrometer (Voyager-DE-RP, Applied Biosystems) and sinapinic acid as matrix. Protein samples were desalted prior to spotting on the sample plate, and all measurements were carried out on several spots per sample with varying protein concentrations. For a summary of the mass spectrometry data of protein variants see Table S1.
Infrared Reflection Absorption Spectroscopy Measurements
Infrared spectra were recorded on monolayers of proteins immobilized on glass substrates covered with a thin layer of gold, which in turn was modified with a self-assembled thiol-monolayer terminated with Ni2+:NTA (see below), using a Thermo Fisher PM-IRRAS system under an angle of incidence of 81°. Protein immobilization was carried out according to the protocol below.
X-ray Photoelectron Spectroscopy
XPS were recorded using a Physical Electronics Quantera SXM (scanning XPS microprobe) spectroscope using the Al Kα line as monochromatic X-ray source at 1486.6 eV. For all samples one survey spectrum to identify regions for element analysis and 3 element scans per analyzed element were recorded in order to determine the relative abundance of carbon, sulfur, and, where present, nitrogen (Table S3). Spectra were recorded on 4 samples on 1 in. glass wafers covered with a 50 nm thick gold layer functionalized with monolayers (see below) of (1) 11-mercaptoundecanoic acid (MUA); (2) N-hydroxysuccinimide (NHS)-activated MUA; (3) Nα,Nα-bis(carboxymethyl)-l-lysine (NTA-NH2)-functionalized MUA; and (4) nickel(II) nitrilotriacetatenickel (Ni2+:NTA)-functionalized MUA.
Preparation of Ni2+:NTA Monolayers on Gold-Coated Substrates
SPR sensors (1 in., Ssense) were cleaned in a solution of H2SO4/H2O2, 3:1 (which should be handled with care, as it reacts violently with organic material), for 30 s, rinsed with water, dried in a flow of N2, and subsequently immersed in a solution of 2 mM MUA in 1:1 EtOH/water and left at least 16 h under ambient conditions to react. To remove any excess MUA, the samples were rinsed thoroughly with EtOH and dried in a flow of N2. The resulting carboxylic acid-terminated monolayers were activated using a freshly prepared solution of 300 mM NHS and 40 mM N-(dimethyl aminopropyl)-N′-ethylcarbodiimide (EDC) in 40 mM PB, pH 8.0, for at least 30 min under ambient conditions. After the reaction, the samples were rinsed with water and dried in a flow of N2, and the activated ester-terminated monolayers were immediately reacted with a 1 mM solution of Nα,Nα-bis(carboxymethyl)-l-lysine hydrate in 40 mM PB, pH 8.0, for 2 h under ambient conditions. Samples were then washed with water, incubated for 30 min in a 5 g/L aqueous solution of NiCl2·6H2O, washed with water, and dried in a flow of N2; the samples could be stored under dry N2 for a maximum of 4 weeks before use.
Preparation of Bifunctional NTA/PEG-Line-Patterned Glass Substrates Using Nanoimprint Lithography
Four-inch Borofloat (Schott, Borofloat 33) glass wafers were immersed in a solution of H2SO4/H2O2, 3:1 (which should be handled with care, as it reacts violently with organic material), for 10 min, rinsed with water, dried in a flow of N2, and subsequently spin-coated (Speedline, p6700) with a 6% w/v 350 kDa poly(methyl methacrylate) (PMMA) solution in toluene for 30 s at 3000 rpm. To remove residual solvent, the wafer was baked at 120 °C for 10 min. NIL was carried out using a Peltier temperature-controlled (Julabo FP 50) HP Specac NIL setup at 200 °C set to 1 ton for 10 min. As imprint master a 4 in. 1H,1H,2H,2H-perfluorodecyltrichlorosilane (AB111155, ABCR)-coated Si-wafer with 24 1 cm2 patterns of 10 to 25 μm wide ridges separated by 5 μm wide and ∼0.5 μm deep trenches was used. In detail, the patterned imprint master was brought into contact with the spin-coated PMMA layer on the Borofloat wafer, cushioned with Kunze heat-conducting cushioning foil (KU-TXE100) on one side and 4 layers of aluminum foil on the other side, and sealed with aluminum foil. This stack was heated to 200 °C, without applying pressure; then the pressure was carefully increased to the equivalent of 1 ton, and these conditions were kept constant for 10 min. Subsequently, the heating was switched off, while keeping the pressure constant. Upon reaching 100 °C, the pressure was left to decrease simultaneously with the temperature. Upon reaching 80 °C and complete pressure relief, the stack was removed from the NIL setup and disassembled and residual layer removal was carried out using an SPI Supplies Plasmaprep II oxygen-plasma cleaner. The progress of residual layer removal was monitored for each of the 1 cm2 patterns individually using an Olympus BH2 light microscope in transmission mode with a halogen lamp as light source. Upon completion of removing the residual layer (with only 5 μm wide ridges of PMMA remaining), the substrates were silanized by overnight chemical vapor deposition of N-[3-(trimethoxysilyl)propyl]ethylenediamine in vacuo. To remove any excess silane, the samples were rinsed thoroughly with ethanol. The resulting amine-terminated line patterns were reacted with phenyl diisothiocyanate (ITC). To this end, the samples were immersed in a 0.04 M solution of ITC in ethanol for 2 h at 50 °C under argon. After the reaction, the samples were rinsed with ethanol and dried in a flow of N2. The ITC-terminated line patterns were then functionalized with Nα,Nα-bis(carboxymethyl)-l-lysine (NTA-NH2). To this end, the samples were incubated in a 1 mM solution of NTA-NH2 in water at 50 °C for 2 h under argon. All remaining PMMA was then stripped from the Borofloat substrates by 30 min sonication in 1 L of acetone, rinsing with acetone, and drying with N2. Subsequently, the unfunctionalized (5 μm wide) lines were functionalized with a solution of 100 μL of [methoxy(polyethyleneoxy)propyl]trimethoxysilane (PEG)-silane (AB111226, ABCR) in 60 mL of dry toluene by leaving the samples to react overnight at room temperature under argon. After washing the samples with toluene they were dried under a flow of N2. The resulting NTA/PEG line patterns could be stored under N2 for a maximum of 4 weeks prior to use.
Protein Immobilization on (Patterned) Substrates
For protein immobilization NTA-terminated (patterned) layers were incubated, if necessary, with a 1 mM NiCl2·6H2O solution in water for 30 min, and the substrates were rinsed briefly with water. For the subsequent immobilization of proteins, substrates were incubated with the appropriate protein solution (of 1 μM concentration, unless stated otherwise) for 1 h (unless stated otherwise) in a humidity chamber. Protein solutions were then removed with a pipet and retained for further use, and samples washed on an orbital shaker (80 rpm) in the appropriate buffers overnight (unless stated otherwise). Samples were then rinsed with the appropriate buffer and subsequently with water and imaged using an Olympus IX70 inverted fluorescence microscope; see below.
Thermodynamic Multivalency Model for Fitting SPR Titration Data
We adapted a model previously reported in the literature41,63,67 in order to predict and determine the thermodynamic stability parameters of the protein–surface complexes to fit the SPR data obtained experimentally. We here consider each His6-tag to behave as a single motif, binding to three surface-bound Ni2+:NTA (NiNTA in formulas) units simultaneously, with an effective concentration value (Ceff) controlling the binding of consecutive His6-tags once the first one is bound. The model is summarized in the Supporting Information (eqs S1–S5) and solved using a spreadsheet approach.84
Molecular Dynamics
Molecular Langevin dynamics were performed using the NAMD program85 together with the CHARMM force field.86 Short-range nonbonded interactions were computed up to 1.2 nm distance. Ewald summation was used to calculate the electrostatic interactions, and a 2 fs time step was used for dynamics by constraining covalent bonds to hydrogen. The coordinates for TagRFP were taken from the PDB (ID: 3M22). The VMD87 mutate residue plugin was used to change residues Cys114, Cys222, and Ser128 to serine and cysteine, respectively. In order to run Molecular Dynamics simulations on the TagRFP-His6-Ni2+:NTA interface, it was necessary to parametrize the chromophore found in the center of the protein, the maleimide linker used to attach the third His tag to the protein, and the Ni2+:NTA molecule used in the SAM. Parametrization was carried out using the Paramchem88 Web tool, and the partial charges were mapped onto the CHARMM force field.89
A Au(111) slab of surface area 2800 Å2 was cut from bulk gold metal, and 384 Ni2+:NTA molecules were placed on one face. The SAM model was encased in a large water box with dimensions 20 × 20 × 10 nm, and periodic boundary conditions were applied. The SAM (31 872 atoms) was first relaxed using 6000 steps of steepest descent minimization and then allowed to equilibrate to a stable room-temperature structure over 1 ns of molecular dynamics and subjected to a further 7 ns of dynamics with a constant-density monolayer forming within 5 ns. The sulfur atoms were then weakly constrained at these equilibrated surface-bound positions to simulate the gold–sulfur bond that is formed in the experiments. A similar protocol was used to model the mixed SAM, with clusters of 5–10 Ni2+:NTA molecules surrounded by nonfunctionalized acid-terminated chains. Simulation input files and calculated SAM structures are available on request from D.T. To study the protein–SAM interactions, the TagRFP protein was placed near the SAM in two different orientations (end-on and side-on; see Chart 1) with a varying number of bound histidine residues, and the Avogadro program90 was used to position the histidine residues near the Ni2+ ions in the SAM in positions where a complex might form. The system was equilibrated with gradually loosening positional constraints on the histidine side chains for 8 ns. Free dynamics simulations were then run on the system for 20 ns, sampling every 100 ps. Image generation and Tcl script-based trajectory analysis were performed using the VMD program.87
For the steered MD simulations, two configurations were taken from the equilibrium MD simulations. They were chosen to have the same number of adsorbed histidine residues in order to be able to compare like with like as much as possible. For both configurations, SMD simulations were performed using GROMACS 5.1.191 and the CHARMM force field to pull the protein a distance of 110 Å over a time of 7 ns from the SAM. Fifty-eight configurations were then taken from these trajectories and subjected to 10 ns of umbrella sampling simulations. The PMF profile was calculated over the sampled windows using the gmx_wham command in GROMACS.
Acknowledgments
D.W. thanks the German Academic Exchange Service for financial support under postdoctoral research grant D/08/46093. Work by J.C.-D., E.T., and P.J. was funded by Project P2.02 OAcontrol of the research program of the BioMedical Materials Institute, cofunded by the Dutch Ministry of Economic Affairs. J.P. and V.S. acknowledge support from Stichting Technische Wetenschappen (STW) under the nanoscopy program (project no. 12149). The work by D.W. and P.J. was cofunded by Starting ERC Grant (259183 Sumoman). D.T. acknowledges financial support from the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant number 604530-2 (CellulosomePlus) and thanks Science Foundation Ireland (SFI) for financial support under Grant Number 15/CDA/3491 and for provision of computing resources at the SFI/Higher Education Authority Irish Centre for High-End Computing (ICHEC). The authors thank Dr. M. Koay from the University of Twente for discussions and comments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b03717.
Schemes S1, S2; Tables S1–S7; Figures S1–S15; and eqs S1–S5 (PDF)
Author Contributions
D.W. and P.J. conceived the project. D.W. engineered and together with E.T. produced the protein mutants. D.W., V.S., and P.J. analyzed the protein constructs. D.W. and J.C.-D. performed (patterned) protein immobilization studies. D.W., J.C.-D., and E.T. performed SPR binding studies. J.C.-D. applied the multivalent model, which was supervised by J.H. J.P. designed and together with D.W. performed fluorescence lifetime measurements, and J.P, D.W., and C.B. analyzed the data. C.B. and V.S. supervised polarization anisotropy setups. IRRAS measurements were done by D.W. and J.C.-D. S.O’M., P.-A.C., and D.T. performed and analyzed computer simulations, which was supervised by D.T. D.W., J.C.-D., J.P., S.O’M., P.-A.C., D.T., J.H., V.S., and P.J. were involved in the analysis of the data. D.W. and J.C.-D. prepared the manuscript. P.J. wrote the manuscript. P.J. supervised the entire project. All authors have given approval to the final version of the manuscript.
Author Contributions
# D. Wasserberg and J. Cabanas-Danés contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Dufva M.; Christensen C. B. V. Diagnostic and Analytical Applications of Protein Microarrays. Expert Rev. Proteomics 2005, 2, 41–48. 10.1586/14789450.2.1.41. [DOI] [PubMed] [Google Scholar]
- Terstappen G. C.; Schluepen C.; Raggiaschi R.; Gaviraghi G. Target Deconvolution Strategies in Drug Discovery. Nat. Rev. Drug Discovery 2007, 6, 891–903. 10.1038/nrd2410. [DOI] [PubMed] [Google Scholar]
- Weinrich D.; Jonkheijm P.; Niemeyer C. M.; Waldmann H. Applications of Protein Biochips in Biomedical and Biotechnological Research. Angew. Chem., Int. Ed. 2009, 48, 7744–7751. 10.1002/anie.200901480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli K.; Magliulo M.; Mulla M. Y.; Singh M.; Sabbatini L.; Palazzo G.; Torsi L. Printable Bioelectronics to Investigate Functional Biological Interfaces. Angew. Chem., Int. Ed. 2015, 54, 12562–12576. 10.1002/anie.201502615. [DOI] [PubMed] [Google Scholar]
- Mitchell A. C.; Briquez P. S.; Hubbell J. A.; Cochran J. R. Engineering Growth Factors for Regenerative Medicine Applications. Acta Biomater. 2016, 30, 1–12. 10.1016/j.actbio.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q.; Becherer T.; Angioletti-Uberti S.; Dzubiella J.; Wischke C.; Neffe A. T.; Lendlein A.; Ballauff M.; Haag R. Protein Interactions with Polymer Coatings and Biomaterials. Angew. Chem., Int. Ed. 2014, 53, 8004–8031. 10.1002/anie.201400546. [DOI] [PubMed] [Google Scholar]
- Tong Z.; Solanki A.; Hamilos A.; Levy O.; Wen K.; Yin X.; Karp J. M. Application of Biomaterials to Advance Induced Pluripotent Stem Cell Research and Therapy. EMBO J. 2015, 34, 987–1008. 10.15252/embj.201490756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Patel V. K.; Kant R.; Bhattacharya S. Surface Modification Strategies for Fabrication of Nano-biodevices: A Critical Review. RAA 2016, 4, 166–191. 10.7569/RAA.2016.097307. [DOI] [Google Scholar]
- Mendes P. M. Stimuli-Responsive Surfaces for Bio-Applications. Chem. Soc. Rev. 2008, 37, 2512–2529. 10.1039/b714635n. [DOI] [PubMed] [Google Scholar]
- Wong L. S.; Khan F.; Micklefield J. Selective Covalent Protein Immobilization: Strategies and Applications. Chem. Rev. 2009, 109, 4025–4053. 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- Rusmini F.; Zhong Z.; Feijen J. Protein Immobilization Strategies for Protein Biochips. Biomacromolecules 2007, 8, 1775–1789. 10.1021/bm061197b. [DOI] [PubMed] [Google Scholar]
- Jonkheijm P.; Weinrich D.; Schroeder H.; Niemeyer C. M.; Waldmann H. Chemical Strategies for Generating Protein Biochips. Angew. Chem., Int. Ed. 2008, 47, 9618–9647. 10.1002/anie.200801711. [DOI] [PubMed] [Google Scholar]
- Hackenberger C. P. R.; Schwarzer D. Chemoselective Ligation and Modification Strategies for Peptides and Proteins. Angew. Chem., Int. Ed. 2008, 47, 10030–10074. 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- Chen Y.-X.; Triola G.; Waldmann H. Bioorthogonal Chemistry for Site-Specific Labeling and Surface Immobilization of Proteins. Acc. Chem. Res. 2011, 44, 762–773. 10.1021/ar200046h. [DOI] [PubMed] [Google Scholar]
- Reyna K. V. L; Qing L. Bioorthogonal Chemistry: Recent Progress and Future Directions. Chem. Commun. 2010, 46 (46), 1589–1600. 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48, 2–27. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth B. P.; Xu J.; Taton T. A.; Guo A.; Distefano M. D. Site-Specific, Covalent Attachment of Proteins to a Solid Surface. Bioconjugate Chem. 2006, 17, 967–974. 10.1021/bc060125e. [DOI] [PubMed] [Google Scholar]
- Govindaraju T.; Jonkheijm P.; Gogolin L.; Schroeder H.; Becker C. F. W.; Niemeyer C. M.; Waldmann H. Surface Immobilization of Biomolecules by Click Sulfonamide Reaction. Chem. Commun. 2008, 32, 3723–3725. 10.1039/b806764c. [DOI] [PubMed] [Google Scholar]
- Christman K. L.; Broyer R. M.; Tolstyka Z. P.; Maynard H. D. Site-Specific Protein Immobilization Through N-Terminal Oxime Linkages. J. Mater. Chem. 2007, 17, 2021–2027. 10.1039/b618002g. [DOI] [Google Scholar]
- Hodneland C. D.; Lee Y.-S.; Min D.-H.; Mrksich M. Selective Immobilization of Proteins to Self-Assembled Monolayers Presenting Active Site-Directed Capture Ligands. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5048–5052. 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserberg D.; Nicosia C.; Tromp E. E.; Subramaniam V.; Huskens J.; Jonkheijm P. Oriented Protein Immobilization Using Covalent and Non-Covalent Chemistry on a Thiol-Reactive Self-Reporting Surface. J. Am. Chem. Soc. 2013, 135, 3104–3111. 10.1021/ja3102133. [DOI] [PubMed] [Google Scholar]
- Becker C. F. W.; Wacker R.; Bouschen W.; Seidel R.; Kolaric B.; Lang P.; Schroeder H.; Mueller O.; Niemeyer C. M.; Spengler B.; Goody R. S.; Engelhard M. Direct Readout of Protein-Protein Interactions by Mass Spectrometry from Protein-DNA Microarrays. Angew. Chem., Int. Ed. 2005, 44, 7635–7639. 10.1002/anie.200502908. [DOI] [PubMed] [Google Scholar]
- Kwon Y.; Coleman M. A.; Camarero J. A. Selective Immobilization of Proteins onto Solid Supports Through Split-Intein-Mediated Protein Trans-Splicing. Angew. Chem., Int. Ed. 2006, 45, 1726–1729. 10.1002/anie.200503475. [DOI] [PubMed] [Google Scholar]
- Hwang I.; Baek K.; Jung M.; Kim Y.; Park K. M.; Lee D.-W.; Selvapalam N.; Kim K. Noncovalent Immobilization of Proteins on a Solid Surface by Cucurbit[7]uril-Ferrocenemethylammonium Pair, a Potential Replacement of Biotin-Avidin Pair. J. Am. Chem. Soc. 2007, 129, 4170–4171. 10.1021/ja071130b. [DOI] [PubMed] [Google Scholar]
- Grunwald C.; Schulze K.; Reichel A.; Weiss V. U.; Blaas D.; Piehler J.; Wiesmuller K.-H.; Tampé R. In Situ Assembly of Macromolecular Complexes Triggered by Light. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6146–6151. 10.1073/pnas.0912617107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan P.; Wang Y.; Jiang Y.; Xu H.; Zhang X. Fabrication of Reactivated Biointerface for Dual-Controlled Reversible Immobilization of Cytochrome c. Adv. Mater. 2009, 21, 4362–4365. 10.1002/adma.200901237. [DOI] [PubMed] [Google Scholar]
- Yang L.; Gómez-Casado A.; Young J. F.; Nguyen H. D.; Cabanas-Danés J.; Huskens J.; Brunsveld L.; Jonkheijm P. Reversible and Oriented Immobilization of Ferrocene-Modified Proteins. J. Am. Chem. Soc. 2012, 134, 19199–19206. 10.1021/ja308450n. [DOI] [PubMed] [Google Scholar]
- Gutiérrez Sánchez C.; Su Q.; Schönherr H.; Grininger M.; Nöll G. Multi-Ligand-Binding Flavoprotein Dodecin as a Key Element for Reversible Surface Modification in Nano-Biotechnology. ACS Nano 2015, 9, 3491–3500. 10.1021/nn506993s. [DOI] [PubMed] [Google Scholar]
- Muller W.; Ringsdorf H.; Rump E.; Wildburg G.; Zhang X.; Angermaier L.; Knoll W.; Liley M.; Spinke J. Attempts to Mimic Docking Processes of the Immune System: Recognition-Induced Formation of Protein Multilayers. Science 1993, 262, 1706–1708. 10.1126/science.8259513. [DOI] [PubMed] [Google Scholar]
- Mrksich M.; Grunwell J. R.; Whitesides G. M. Biospecific Adsorption of Carbonic Anhydrase to Self-Assembled Monolayers of Alkanethiolates That Present Benzenesulfonamide Groups on Gold. J. Am. Chem. Soc. 1995, 117, 12009–12010. 10.1021/ja00153a029. [DOI] [Google Scholar]
- González-Campo A.; Eker B.; Gardeniers H. J. G. E.; Huskens J.; Jonkheijm P. A Supramolecular Approach to Enzyme Immobilization in Micro-Channels. Small 2012, 8, 3531–3537. 10.1002/smll.201200565. [DOI] [PubMed] [Google Scholar]
- Wood L. L.; Cheng S. S.; Edmiston P. L.; Saavedra S. S. Molecular Orientation Distributions in Protein Films 2. Site-Directed Immobilization of Yeast Cytochrome c on Thiol-Capped, Self-Assembled Monolayers. J. Am. Chem. Soc. 1997, 119, 571–576. 10.1021/ja9623673. [DOI] [Google Scholar]
- Edminston P. L.; Saavedra S. S. Molecular Orientation Distributions in Protein Films. 4. A Multilayer Composed of Yeast Cytochrome c Bound through an Intermediate Streptavidin Layer to a Planar Supported Phospholipid Bilayer. J. Am. Chem. Soc. 1998, 120, 1665–1671. 10.1021/ja972634k. [DOI] [Google Scholar]
- Sanavio B.; Scaini D.; Grunwald C.; Legname G.; Scoles G.; Casalis L. Oriented Immobilization of Prion Protein Demonstrated via Precise Interfacial Nanostructure Measurements. ACS Nano 2010, 4, 6607–6616. 10.1021/nn101872w. [DOI] [PubMed] [Google Scholar]
- Escalante M.; Zhao Y.; Ludden M. J. W.; Vermeij R.; Olsen J. D.; Berenschot E.; Hunter C. N.; Huskens J.; Subramaniam V.; Otto C. Nanometer Arrays of Functional Light Harvesting Antenna Complexes by Nanoimprint Lithography and Host–Guest Interactions. J. Am. Chem. Soc. 2008, 130, 8892–8893. 10.1021/ja802843m. [DOI] [PubMed] [Google Scholar]
- Escalante M.; Lenferink A.; Zhao Y.; Tas N.; Huskens J.; Hunter C. N.; Subramaniam V.; Otto C. Long-Range Energy Propagation in Nanometer Arrays of Light Harvesting Antenna Complexes. Nano Lett. 2010, 10, 1450–1457. 10.1021/nl1003569. [DOI] [PubMed] [Google Scholar]
- Dorn I. T.; Eschrich R.; Seemueller E.; Guckenberger R.; Tampé R. High-Resolution AFM-Imaging and Mechanistic Analysis of the 20S Proteasome. J. Mol. Biol. 1999, 288, 1027–1036. 10.1006/jmbi.1999.2714. [DOI] [PubMed] [Google Scholar]
- Hutschenreiter S.; Tinazli A.; Model K.; Tampé R. Two-Substrate Association with the 20S Proteasome at Single-Molecule Level. EMBO J. 2004, 23, 2488–2497. 10.1038/sj.emboj.7600262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M.; Choi S.-K.; Whitesides G. M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. . [DOI] [PubMed] [Google Scholar]
- Mulder A.; Huskens J.; Reinhoudt D. N. Multivalency in Supramolecular Chemistry and Nanofabrication. Org. Biomol. Chem. 2004, 2, 3409–3424. 10.1039/b413971b. [DOI] [PubMed] [Google Scholar]
- De M.; Rana S.; Rotello V. M. Nickel-Ion-Mediated Control of the Stoichiometry of His-Tagged Protein/Nanoparticle Interactions. Macromol. Biosci. 2009, 9, 174–178. 10.1002/mabi.200800289. [DOI] [PubMed] [Google Scholar]
- Schmitt J.; Hess H.; Stunnenberg H. G. Affinity Purification of Histidine-Tagged Proteins. Mol. Biol. Rep. 1993, 18, 223–230. 10.1007/BF01674434. [DOI] [PubMed] [Google Scholar]
- Hochuli E.; Dobeli H.; Schacher A. New Metal Chelate Adsorbent Selective for Proteins and Peptides Containing Neighbouring Histidine Residues. J. Chromatogr. A 1987, 411, 177–184. 10.1016/S0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- Young C. L.; Britton Z. T.; Robinson A. S. Recombinant Protein Expression and Purification: A Comprehensive Review of Affinity Tags and Microbial Applications. Biotechnol. J. 2012, 7, 620–634. 10.1002/biot.201100155. [DOI] [PubMed] [Google Scholar]
- Goodman R. P.; Erben C. M.; Malo J.; Ho W. M.; McKee M. L.; Kapanidis A. N.; Turberfield A. J. A Facile Method for Reversibly Linking a Recombinant Protein to DNA. ChemBioChem 2009, 10, 1551–1557. 10.1002/cbic.200900165. [DOI] [PubMed] [Google Scholar]
- Nieba L.; Nieba-Axmann S. E.; Persson A.; Hämäläinen M.; Edebratt F.; Hansson A.; Lidholm J.; Magnusson K.; Karlsson Å. F.; Plückthun A. BIACORE Analysis of Histidine-Tagged Proteins Using a Chelating NTA Sensor Chip. Anal. Biochem. 1997, 252, 217–228. 10.1006/abio.1997.2326. [DOI] [PubMed] [Google Scholar]
- André T.; Reichel A.; Wiesmueller K.-H.; Tampé R.; Piehler J.; Brock R. Selectivity of Competitive Multivalent Interactions at Interfaces. ChemBioChem 2009, 10, 1878–1887. 10.1002/cbic.200900001. [DOI] [PubMed] [Google Scholar]
- Alonso J. M.; Reichel A.; Piehler J.; del Campo A. Photopatterned Surfaces for Site-Specific and Functional Immobilization of Proteins. Langmuir 2007, 24, 448. 10.1021/la702696b. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Bilgin M.; Bangham R.; Hall D.; Casamayor A.; Bertone P.; Lan N.; Jansen R.; Bidlingmaier S.; Houfek T.; Mitchell T.; Miller P.; Dean R. A.; Gerstein M.; Snyder M. Global Analysis of Protein Activities Using Proteome Chips. Science 2001, 293, 2101–2105. 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- Selmi D. N.; Adamson R. J.; Attrill H.; Goddard A. D.; Gilbert R. J. C.; Watts A.; Turberfield A. J. 20. DNA-Templated Protein Arrays for Single-Molecule Imaging. Nano Lett. 2011, 11, 657–660. 10.1021/nl1037769. [DOI] [PubMed] [Google Scholar]
- Marin V. L.; Bayburt T. H.; Sligar S. G.; Mrksich M. Functional Assays of Membrane-Bound Proteins with SAMDI-TOF Mass Spectrometry. Angew. Chem., Int. Ed. 2007, 46, 8796–8798. 10.1002/anie.200702694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury P.; Escalante-Marun M.; Péter M.; Reinhoudt D. N.; Subramaniam V.; Huskens J. Creating Nanopatterns of His-Tagged Proteins on Surfaces by Nanoimprint Lithography Using Specific NiNTA-Histidine Interactions. Small 2007, 3, 1584–1592. 10.1002/smll.200700046. [DOI] [PubMed] [Google Scholar]
- Turchanin A.; Tinazli A.; El-Desawy M.; Großmann H.; Schnietz M.; Solak H. H.; Tampé R.; Gölzhäuser A. Molecular Self-Assembly, Chemical Lithography, and Biochemical Tweezers: a Path for the Fabrication of Functional Nanometer-Scale Protein Arrays. Adv. Mater. 2008, 20, 471–477. 10.1002/adma.200702189. [DOI] [Google Scholar]
- Hansen J. A.; Sumbayev V. V.; Gothelf K. V. An Electrochemical Sensor Based on the Human Estrogen Receptor Ligand Binding Domain. Nano Lett. 2007, 7, 2831–2834. 10.1021/nl071451l. [DOI] [PubMed] [Google Scholar]
- Cabanas-Danés J.; Dooms Rodrigues E.; Landman E.; van Weerd J.; van Blitterswijk C. A.; Verrips T.; Huskens J.; Karperien M.; Jonkheijm P. A Supramolecular Host-Guest Carrier System for Growth Factors Employing VHH Fragments. J. Am. Chem. Soc. 2014, 136, 12675–12681. 10.1021/ja505695w. [DOI] [PubMed] [Google Scholar]
- Lata S.; Reichel A.; Brock R.; Tampé R.; Piehler J. High-Affinity Adaptors for Switchable Recognition of Histidine-Tagged Proteins. J. Am. Chem. Soc. 2005, 127, 10205–10215. 10.1021/ja050690c. [DOI] [PubMed] [Google Scholar]
- Lata S.; Gavutis M.; Tampé R.; Piehler J. Specific and Stable Fluorescence Labeling of Histidine-Tagged Proteins for Dissecting Multi-Protein Complex Formation. J. Am. Chem. Soc. 2006, 128, 2365–2372. 10.1021/ja0563105. [DOI] [PubMed] [Google Scholar]
- Valiokas R.; Klenkar G.; Tinazli A.; Reichel A.; Tampe R.; Piehler J.; Liedberg B. Self-Assembled Monolayers Containing Terminal Mono-, Bis-, and Tris-nitrilotriacetic Acid Groups: Characterization and Application. Langmuir 2008, 24, 4959–4967. 10.1021/la703709a. [DOI] [PubMed] [Google Scholar]
- Lata S.; Piehler J. Stable and Functional Immobilization of Histidine-Tagged Proteins via Multivalent Chelator Headgroups on a Molecular Poly(ethylene glycol) Brush. Anal. Chem. 2005, 77, 1096–1105. 10.1021/ac048813j. [DOI] [PubMed] [Google Scholar]
- Tinazli A.; Tang J.; Valiokas R.; Picuric S.; Lata S.; Piehler J.; Liedber B.; Tampé R. High-Affinity Chelator Thiols for Switchable and Oriented Immobilization of Histidine-Tagged Proteins: A Generic Platform for Protein Chip Technologies. Chem. - Eur. J. 2005, 11, 5249–5259. 10.1002/chem.200500154. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Park J. I.; Watson D. S.; Hwang P.; Szoka F. C. Jr. Facile Synthesis of Multivalent Nitrilotriacetic Acid (NTA) and NTA Conjugates for Analytical and Drug Delivery Applications. Bioconjugate Chem. 2006, 17, 1592–1600. 10.1021/bc0602228. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Hwang P.; Watson D. S.; Cao L.; Szoka F. C. Jr. Tris-Nitrilotriacetic Acids of Subnanomolar Affinity Toward Hexahistidine Tagged Molecules. Bioconjugate Chem. 2009, 20, 1667–1672. 10.1021/bc900309n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludden M. J. W.; Mulder A.; Schulze K.; Subramaniam V.; Tampé R.; Huskens J. Anchoring of Histidine-Tagged Proteins to Molecular Printboards: Self-Assembly, Thermodynamic Modeling, and Patterning. Chem. - Eur. J. 2008, 14, 2044–2051. 10.1002/chem.200701478. [DOI] [PubMed] [Google Scholar]
- Zhen G.; Falconnet D.; Kuennemann E.; Vörös J.; Spencer N. D.; Textor M.; Zürcher S. Nitrilotriacetic Acid Functionalized Graft Copolymers: A Polymeric Interface for Selective and Reversible Binding of Histidine-Tagged Proteins. Adv. Funct. Mater. 2006, 16, 243–251. 10.1002/adfm.200500232. [DOI] [Google Scholar]
- Knezevic J.; Langer A.; Hampel P. A.; Kaiser W.; Strasser R.; Rant U. Quantitation of Affinity, Avidity, and Binding Kinetics of Protein Analytes with a Dynamically Switchable Biosurface. J. Am. Chem. Soc. 2012, 134, 15225–15228. 10.1021/ja3061276. [DOI] [PubMed] [Google Scholar]
- Merzlyak E. M.; Goedhart J.; Shcherbo D.; Bulina M. E.; Shcheglov A. S.; Fradkov A. F.; Gaintzeva A.; Lukyanov K. A.; Lukyanov S.; Gadella T. W.; Chudakov D. M. Bright Monomeric Red Fluorescent Protein with an Extended Fluorescence Lifetime. Nat. Methods 2007, 4, 555–557. 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- Huskens J.; Mulder A.; Auletta T.; Nijhuis C. A.; Ludden M. J.; Reinhoudt D. N. A Model for Describing the Thermodynamics of Multivalent Host-Guest Interactions at Interfaces. J. Am. Chem. Soc. 2004, 126, 6784–6797. 10.1021/ja049085k. [DOI] [PubMed] [Google Scholar]
- Mulder A.; Auletta T.; Sartori A.; Del Ciotto S.; Casnati A.; Ungaro R.; Huskens J.; Reinhoudt D. N. Divalent Binding of a Bis(adamantyl)-Functionalized Calix[4]arene to β-Cyclodextrin-Based Hosts: An Experimental and Theoretical Study on Multivalent Binding in Solution and at Self-Assembled Monolayers. J. Am. Chem. Soc. 2004, 126, 6627–6636. 10.1021/ja0317168. [DOI] [PubMed] [Google Scholar]
- Subach O. M.; Malashkevich V. N.; Zencheck W. D.; Morozova K. S.; Piatkevich K. D.; Almo S. C.; Verkhusha V. V. Structural Characterization of Acylimine-Containing Blue and Red Chromophores in mTagBFP and TagRFP Fluorescent Proteins. Chem. Biol. 2010, 17, 333–341. 10.1016/j.chembiol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey B. L.; Corn R. M. Covalent Attachment and Derivatization of Poly(L-lysine) Monolayers on Gold Surfaces as Characterized by Polarization-Modulation FT-IR Spectroscopy. Anal. Chem. 1996, 68, 3187–3193. 10.1021/ac9605861. [DOI] [Google Scholar]
- Goormaghtigh E.; Cabiaux V.; Ruysschaert J. M. Secondary Structure and Dosage of Soluble and Membrane-Proteins by Attenuated Total Reflection Fourier-Transform Infrared-Spectroscopy on Hydrated Films. Eur. J. Biochem. 1990, 193, 409–420. 10.1111/j.1432-1033.1990.tb19354.x. [DOI] [PubMed] [Google Scholar]
- Chen C.-W.; Liu H.-L.; Lin J.-C.; Ho Y. Molecular Dynamics Simulations of Metal Ion Binding to the His-tag Motif. J. Chin. Chem. Soc. 2005, 52, 1281–1290. 10.1002/jccs.200500185. [DOI] [Google Scholar]
- Watly J.; Simonovsky E.; Wieczorek R.; Barbosa N.; Miller Y.; Kozlowski H. Insight into the Coordination and the Binding Sites of Cu2+ by the Histidyl-6-Tag using Experimental and Computational Tools. Inorg. Chem. 2014, 53, 6675–6683. 10.1021/ic500387u. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lai C.-T.; Hinds B. J.; Schatz G. C. Kinetic Master Equation Modeling of an Artificial Protein Pump. J. Phys. Chem. C 2016, 120, 14495–14501. 10.1021/acs.jpcc.6b03076. [DOI] [Google Scholar]
- Nifosì R.; Tozzini V. Molecular Dynamics Simulations of Enhanced Green Fluorescent Proteins: Effects of F64L, S65T and T203Y Mutations on the Ground-State Proton Equilibria. Proteins: Struct., Funct., Genet. 2003, 51, 378–389. 10.1002/prot.10335. [DOI] [PubMed] [Google Scholar]
- Mitchell F. L.; Frank F.; Marks G. E.; Suzuki M.; Douglas K. T.; Bryce R. A. Molecular Dynamics Study of Chemically Engineered Green Fluorescent Protein Mutants: Comparison of Intramolecular Fluorescence Resonance Energy Transfer Rate. Proteins: Struct., Funct., Genet. 2009, 75, 28–39. 10.1002/prot.22218. [DOI] [PubMed] [Google Scholar]
- Demachy I.; Ridard J.; Laguitton-Pasquier H.; Durnerin E.; Vallverdu G.; Archirel P.; Lévy B. Cyan Fluorescent Protein: Molecular Dynamics, Simulations, and Electronic Absorption Spectrum. J. Phys. Chem. B 2005, 109, 24121–24133. 10.1021/jp054656w. [DOI] [PubMed] [Google Scholar]
- Nishiyama K.; Watanabe T.; Hoshino T.; Ohdomari I. Analysis of Interactions between Green Fluorescent Protein and Silicon Substrates Using Molecular Dynamics Simulations. Jpn. J. Appl. Phys. 2005, 44, 8210–8215. 10.1143/JJAP.44.8210. [DOI] [Google Scholar]
- Khassanov A.; Steinrück H.-G.; Schmaltz T.; Magerl A.; Halik M. Structural Investigations of Self-Assembled Monolayers for Organic Electronics: Results from X-ray Reflectivity. Acc. Chem. Res. 2015, 48, 1901–1908. 10.1021/acs.accounts.5b00022. [DOI] [PubMed] [Google Scholar]
- Yang Z. Y.; Zhao Y. P. QM/MM and Classical Molecular Dynamics Simulation of Histidine-Tagged Peptide Immobilization on Nickel Surface. Mater. Sci. Eng., A 2006, 423, 84–91. 10.1016/j.msea.2005.10.078. [DOI] [Google Scholar]
- Love J. C.; Estroff L. A.; Kriebel J. K.; Nuzzo R. G.; Whitesides G. M. Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem. Rev. 2005, 105, 1103–1170. 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437, 640–647. 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Höök F.; Rodahl M.; Kasemo B.; Brezezinski P. Structural Changes in Hemoglobin During Adsorption to Solid Surfaces: Effects of pH, Ionic Strength, and Ligand Binding. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 12271–12276. 10.1073/pnas.95.21.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huskens J.; Van Bekkum H.; Peters J. A. A Convenient Spreadsheet Approach to the Calculation of Stability Constants and the Simulation of Kinetics. Comput. Chem. 1995, 19, 409–416. 10.1016/0097-8485(95)00042-Q. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- CGenFF Home. Available at https://cgenff.paramchem.org/. Accessed: Sep 21, 2016.
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtolar T.; Schulz R.; Pall S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performace Molecular Simulations Through Multi-Level Parallelism From Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.