

Abstract
IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. Additionally, patient clinical parameters (serum creatinine values and eGFRs) significantly correlated with Z scores derived from the expression profile of mesangial cell–positive standard genes. Among patients grouped according to Oxford MEST score, patients with segmental glomerulosclerosis had a significantly higher mesangial cell–positive standard gene Z score than patients without segmental glomerulosclerosis. By investigating mesangial cell proteomics and glomerular transcriptomics, we identified 22 common pathways induced in mesangial cells by gd-IgA, most of which mediate inflammation. The genes, proteins, and corresponding pathways identified provide novel insights into the pathophysiologic mechanisms leading to IgAN.
Keywords: IgA nephropathy, mesangial cells, transcriptional profiling
The glomerular mesangium is a key element for a number of human kidney diseases, including diabetic nephropathy, SLE, and IgA nephropathy (IgAN).1 However, knowledge of the role of mesangial cells in these diseases is not complete, partly due to difficulties in developing appropriate animal models2 and also because of the lack of mesangial cell–specific markers. IgAN is the most common GN in the world, and 30%–40% of the patients develop ESRD within 20–30 years after diagnosis.3 The diagnosis relies on histopathologic evaluation with findings of deposits of galactose-deficient IgA (gd-IgA) in the glomerular mesangial region in addition to mesangial cell proliferation and mesangial matrix expansion.4 Moreover, the amount of gd-IgA deposited is not correlated with mesangial cell damage or kidney injury.5 However, mesangial cell proliferation and inflammation induced by gd-IgA deposits are important for disease development.6–8 In our previous study,9 we showed that mesangial cells derived from patients with IgAN have increased susceptibility to gd-IgA compared with mesangial cells from control patients. To increase the understanding of the role of the mesangium in IgAN, we investigated IgAN from a broader perspective using both a transcriptomic and a proteomic approach. To achieve this, we conducted a human genome microarray analysis of glomeruli from patients with IgAN and healthy controls. The results were combined with proteomic data obtained from mass spectrometry (MS) analysis of mesangial cells treated with patient-derived gd-IgA.
In 2013, Ju et al.10 developed an iterative algorithm “in silico nanodissection” that predicts cell type–specific transcripts using large-scale datasets. On the basis of literature curation, they defined mesangial cell– or podocyte-positive standard genes as genes that are specifically expressed in mesangial cells or podocytes in the kidney. In this study, we have used these positive standard genes in our cohort to investigate their predictive and functional importance in IgAN disease onset and progression. We found that the mesangial standard genes play a dominant role in the disease and that mesangial standard gene expression correlates with patient clinical data, indicating their importance in IgAN pathophysiology. In addition, by integration of the profiling information from both microarray and MS analyses, we have found several common significantly differentially expressed pathways at both the transcriptomic and proteomic levels. Most of these are inflammatory pathways that might be critical in the understanding of the underlying mechanisms of IgAN.
Results
Statistics Reveal Important Transcriptomic Information about IgAN In Vivo
After quality check, normalized microarray data from 19 patients diagnosed with IgAN (one patient sample was run in duplicate in two different batches as an internal control) and 22 living donor controls were further analyzed (Table 1). A hierarchical clustering separated patient samples from the controls (Figure 1), indicating a distinct transcriptomic difference between patients and controls. Using the Significant Analysis of Microarray (SAM)11 method, we found 736 genes to be significantly differentially expressed between the groups (Supplemental Table 1). The significant genes were analyzed using the Ingenuity Pathway Analysis tool to investigate the corresponding significant pathways. 113 pathways were found to be significant with Fisher exact P<0.05, and most of the pathways were related to inflammation, cytokines, and growth factors. The top 25 differentially expressed pathways are listed in Table 2.
Table 1.
Clinical data for patients with IgAN
| Sex | Age, yr | Urine Albumin-to-Creatinine Ratio | Creatinine, μmol/L | eGFRa | CKD Stage | BP | BP Medication | Steroids | Immunosuppressive Therapy | Oxford Classification | Progressionb | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | E | S | T | |||||||||||
| Woman | 54 | 229 | 132 | 39.4 | 3 | 130/80 | Yes | No | No | 0 | 0 | 1 | 1 | −4.55 |
| Man | 46 | 323 | 155 | 45.6 | 3 | 150/90 | Yes | No | No | 1 | 1 | 1 | 1 | 0.60 |
| Woman | 45 | 155 | 132 | 40.5 | 3 | 140/80 | Yes | No | No | 1 | 0 | 1 | 1 | −3.05 |
| Woman | 41 | 189 | 165 | 33.0 | 3 | 130/80 | Yes | No | No | 0 | 1 | 1 | 1 | −5.37 |
| Man | 49 | 37 | 124 | 58.4 | 3 | 125/80 | No | No | No | 0 | 1 | 0 | 1 | −3.86 |
| Man | 48 | 65 | 139 | 51.3 | 3 | 125/85 | No | No | No | 0 | 0 | 1 | 1 | −18.99 |
| Man | 58 | 70 | 107 | 65.6 | 2 | 140/80 | Yes | No | No | 0 | 0 | 1 | 0 | −8.49 |
| Man | 35 | 14 | 103 | 80.7 | 2 | 120/80 | Yes | No | No | 0 | 0 | 1 | 0 | 2.40 |
| Man | 58 | 61 | 117 | 67.9 | 2 | 130/90 | Yes | No | No | 0 | 0 | 1 | 2 | −0.84 |
| Man | 34 | 5 | 124 | 64.9 | 2 | 126/85 | Yes | No | No | 0 | 0 | 1 | 1 | 1.18 |
| Man | 53 | 74 | 96 | 77.4 | 2 | 110/70 | No | Yesc | No | 0 | 1 | 0 | 0 | −27.74 |
| Man | 61 | 560d | 84 | 86.0 | 2 | 150/95 | Yes | No | No | 0 | 0 | 1 | 0 | −3.94 |
| Man | 65 | 490d | 93 | 74.0 | 2 | 155/95 | Yes | No | No | 0 | 0 | 1 | 1 | −1.88 |
| Woman | 17 | 15 | 71 | 108.2 | 1 | 130/80 | No | No | No | 1 | 1 | 1 | 0 | −2.24 |
| Man | 30 | 75 | 91 | 97.1 | 1 | 130/90 | Yes | No | No | 0 | 0 | 1 | 0 | 4.49 |
| Man | 20 | 1.2 | 76 | 124.8 | 1 | 100/60 | No | No | No | 0 | 0 | 0 | 0 | −6.94 |
| Woman | 23 | 0.6 | 68 | 109.3 | 1 | 110/65 | No | No | No | 0 | 0 | 0 | 0 | 3.92 |
| Man | 24 | 12 | 77 | 125.6 | 1 | 120/70 | No | No | No | 0 | 0 | 0 | 0 | −17.45 |
| Woman | 23 | 13 | 66 | 113.3 | 1 | 120/70 | No | No | No | 0 | 1 | 1 | 0 | 3.77 |
M, mesangial hypercellularity; E, endocapillary hypercellularity; S, segmental glomerulosclerosis; T, tubular atrophy/interstitial.
eGFR has been calculated using the CKD-EPI creatinine formula.bProgression is change in eGFR over time.
Before the biopsy.
Calculated from total urinary protein.
Figure 1.

Hierarchical clustering of patient and control samples. Clustering of the glomerular microarray data on the basis of whole-genome expression values using Euclidean distance with the Ward averaging method. Using whole-genome expression profiles, samples from patients with IgAN were clearly distinguishable from living donor controls.
Table 2.
Top 25 differentially expressed pathways from microarray data
| Ingenuity Canonical Pathwaysa | P Value | Ratio |
|---|---|---|
| Hepatic fibrosis/hepatic stellate cell activation | <0.001 | 30/181 |
| Phenylalanine degradation 4 (mammalian, via side chain) | <0.001 | 6/14 |
| Glycine betaine degradation | <0.001 | 5/10 |
| Acute-phase response signaling | <0.001 | 19/168 |
| LPS/IL-1–mediated inhibition of RXR function | <0.001 | 21/208 |
| LXR/RXR activation | <0.001 | 15/121 |
| Production of NO and reactive oxygen species in macrophages | <0.001 | 18/178 |
| Granulocyte adhesion and diapedesis | <0.001 | 17/165 |
| Atherosclerosis signaling | <0.001 | 14/121 |
| IL-8 signaling | <0.001 | 18/183 |
| Pathogenesis of multiple sclerosis | <0.001 | 4/9 |
| Methylglyoxal degradation 3 | <0.001 | 4/11 |
| Unfolded protein response | <0.001 | 8/53 |
| FXR/RXR activation | <0.001 | 13/125 |
| Regulation of cellular mechanics by calpain protease | 0.001 | 8/55 |
| Agranulocyte adhesion and diapedesis | 0.001 | 16/175 |
| Î2-alanine degradation 1 | 0.002 | 2/2 |
| ILK signaling | 0.002 | 16/181 |
| IL-6 signaling | 0.002 | 12/117 |
| Glutathione-mediated detoxification | 0.002 | 5/24 |
| eNOS signaling | 0.002 | 13/135 |
| Complement system | 0.002 | 6/36 |
| Dopamine receptor signaling | 0.003 | 9/77 |
| Sucrose degradation 5 (mammalian) | 0.003 | 3/8 |
| Inhibition of matrix metalloproteases | 0.003 | 6/38 |
Pathway P value is calculated using the Fisher exact method, and the ratio represents differentially expressed genes that map to the pathway divided by the total number of genes in the pathway. RXR, retinoid X receptor; LXR, liver X receptor; FXR, farnesoid X receptor; ILK, integrin-linked kinase; eNOS, endothelial nitric oxide synthase.
Using significant genes with adjusted q value <0.01 and unlogged fold changes >1.5 as upregulation and <0.67 as downregulation.
In an earlier study, it was shown that extracellular matrix genes play an important role in IgAN in vitro.9 To confirm these data in patient material, we investigated Gene Ontology (GO) terms using the significantly differentially expressed genes. As seen in Table 3, the top three enriched GO terms for biologic processes are extracellular matrix disassembly (GO:0022617), extracellular structure organization (GO:0043062), and extracellular matrix organization (GO:0030198). Additionally, considering GO terms of cellular components as seen in Table 3, extracellular matrix component (GO:0044420) and extracellular matrix (GO:0031012) were also found to be highly enriched along with basement membrane (GO:0005604) and cell-cell adherens junction (GO:0005913).
Table 3.
Top ten GO biologic processes and top ten GO cellular components
| GO Terms | Ref. List | Hits | Expected | Over/Under | Fold Enrichment | P Value |
|---|---|---|---|---|---|---|
| Top ten GO biologic processes | ||||||
| Extracellular matrix disassembly (GO:0022617) | 13 | 13 | 2.88 | + | 4.52 | 0.04 |
| Extracellular structure organization (GO:0043062) | 42 | 35 | 9.30 | + | 3.76 | <0.001 |
| Extracellular matrix organization (GO:0030198) | 42 | 35 | 9.30 | + | 3.76 | <0.001 |
| Bone development (GO:0060348) | 28 | 21 | 6.20 | + | 3.39 | <0.01 |
| Positive regulation of response to wounding (GO:1903036) | 34 | 25 | 7.53 | + | 3.32 | 0.002 |
| Regulation of B cell activation (GO:0050864) | 26 | 19 | 5.76 | + | 3.300 | 0.04 |
| Cell junction assembly (GO:0034329) | 40 | 29 | 8.86 | + | 3.27 | <0.001 |
| Negative regulation of nucleocytoplasmic transport (GO:0046823) | 37 | 26 | 8.19 | + | 3.17 | 0.002 |
| Negative regulation of MAP kinase activity (GO:0043407) | 30 | 21 | 6.64 | + | 3.16 | 0.03 |
| Protein folding (GO:0006457) | 36 | 24 | 7.97 | + | 3.01 | 0.01 |
| Top ten GO cellular components | ||||||
| Basement membrane (GO:0005604) | 21 | 18 | 4.65 | + | 3.87 | 0.002 |
| Endoplasmic reticulum lumen (GO:0005788) | 19 | 15 | 4.21 | + | 3.57 | 0.03 |
| Cell-cell adherens junction (GO:0005913) | 20 | 15 | 4.43 | + | 3.39 | 0.05 |
| Extracellular matrix component (GO:0044420) | 42 | 29 | 9.30 | + | 3.12 | <0.001 |
| Stress fiber (GO:0001725) | 30 | 20 | 6.64 | + | 3.01 | 0.02 |
| Actin filament bundle (GO:0032432) | 31 | 20 | 6.86 | + | 2.91 | 0.03 |
| Actomyosin (GO:0042641) | 35 | 22 | 7.75 | + | 2.84 | 0.02 |
| Proteasome complex (GO:0000502) | 46 | 28 | 10.18 | + | 2.75 | 0.002 |
| Proteinaceous extracellular matrix (GO:0005578) | 52 | 31 | 11.51 | + | 2.69 | 0.001 |
| Extracellular matrix (GO:0031012) | 159 | 94 | 35.20 | + | 2.67 | <0.001 |
MAP, mitogen-activated protein.
Mesangial Cell–Positive Standard Genes Have a Strong Effect in Patients with IgAN
On the basis of the algorithm presented in the study by Ju et al.,10 we investigated the expression profile of cell type–positive standard genes in the microarray data. In total, 35 positive standard genes were identified in mesangial cells, and 50 were identified in podocytes (for details of specific standard genes see Supplemental Table 2). As illustrated in Table 4, we found 77% (27 of 35) mesangial cell–positive standard genes in the dataset, and among these, 78% (21 of 27) were significantly differentially expressed (SAM q value <0.01). However, for the podocyte-positive standard genes, despite that 70% (35 of 50) were found in the dataset, only 43% (15 of 35) had a significantly changed expression (SAM q value <0.01).
Table 4.
Percentage of the standard genes in the microarray dataset
| Cells | Significant Genes (a) | Total Found (b) | Total Standard Genes (c) | Found (b/c), % | Significant (a/b), % |
|---|---|---|---|---|---|
| Mesangial | 21 | 27 | 35 | 77 | 78 |
| Podocyte | 15 | 35 | 50 | 70 | 43 |
To investigate the influence of the standard genes, we extracted the expression data using only the cell type–positive standard genes and conducted principal coordinate analysis12 to cluster the samples as shown in Figure 2. Using the mesangial cell–positive standard genes, samples from patients with IgAN were completely separated from the control samples (Figure 2, left panel), whereas the groups were overlapping when using podocyte-positive standard genes (Figure 2, right panel). The first eigenvalue (eig1) from the eigenvectors were 147.9 for mesangial cell and 48.6 for podocyte, which indicates approximately three times better separation of sample clusters using the mesangial cell–positive standard genes than the podocyte-positive standard genes. This result was confirmed using hierarchical clustering with the Ward averaging algorithm13 (Supplemental Figure 1).
Figure 2.

Principal coordinate analysis of the samples on the basis of the cell type–positive standard genes. Mesangial cell–positive standard genes could divide the samples into two separate groups (left panel). Podocyte-positive standard genes resulted in an overlapping population (right panel). Eig1 for mesangial cell–positive standards was 147.9, which was almost three times the eig1 for podocyte-positive standard genes (48.6). Eig1 represents the first value from eigenvector from the principal coordinate analysis.
Mesangial Cell–Positive Standard Genes Are Significantly Correlated with Patient Clinical Parameters
To determine how the cell type–positive standard genes are related to disease stage, the transformed patient Z scores were correlated with the patients’ clinical parameters. Patient Z score is calculated from the positive standard genes and represents the deviation of all of the positive standard genes from each individual patient (see Concise Methods). Patient serum creatinine values and eGFR (eGFR calculated by the Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI] creatinine equation14) significantly correlated with patient Z scores using the mesangial cell–positive standard genes (Figure 3, A and B), with Pearson correlation P values <0.05 (P=0.02 and P=0.03, respectively). However, using podocyte-positive standard genes, the Pearson correlation of the patient Z scores with clinical parameters was found to be NS (Figure 3, C and D). The patients were classified by a pathologist according to the Oxford mesangial hypercellularity, endocapillary hypercellularity, segmental glomerulosclerosis, and tubular atrophy/interstitial fibrosis (MEST) score for IgAN.3,15 The Oxford MEST scores were used to group the patients and then compared with their Z scores. The mesangial standard gene Z score was significantly higher in the group with a segmental glomerulosclerosis score of one compared with zero, whereas this was not found using the podocyte standard gene Z score (Figure 3, E and F). The other Oxford MEST scores (mesangial hypercellularity, endocapillary hypercellularity, and tubular atrophy/interstitial fibrosis) yielded no significant differences.
Figure 3.

Correlations of clinical data and patient Z scores. Patient Z score is calculated on the basis of the positive standard genes and represents the deviation of all of the positive standard genes from each individual patient compared with the rest of the patients. Using mesangial-positive standard genes, the patient Z score correlated significantly with (A) serum creatinine and (B) eGFR. However, patient Z score of podocyte-positive standard genes showed no significant correlation with either (C) serum creatinine or (D) eGFR. Grouping the patients according to their Oxford MEST score of segmental glomerulosclerosis (S; zero or one) showed a significant difference in (E) the patients’ mesangial standard gene Z score but not when (F) the patients’ podocyte standard gene Z score was used. Error bars represent SEM. *P<0.05.
Investigation of Mesangial Cells In Vitro
The results from the microarray analysis of glomeruli from patients with IgAN confirmed that mesangial cells play an important role in IgAN from a transcriptomic perspective. To investigate the effect of gd-IgA deposits on a cellular level, we investigated protein expression in cultured human mesangial cells. The cells were treated with IgA purified from patients with IgAN, who have a higher content of gd-IgA (Supplemental Figure 2). As a result, 5218 proteins were quantified using LC-MS/MS and then filtered for FDR 1%. Expression ratios were normalized on the basis of the average of all of the control values. Hierarchical analysis showed that IgA-treated samples were completely separated from the control untreated samples (Figure 4). After statistical analysis, we found 721 proteins to be significantly up- or downregulated in the gd-IgA–treated cells compared with untreated control cells (Supplemental Table 3). The top 25 differentially expressed pathways found using the significant proteins are listed in Table 5, and the pathways are predominantly involved in inflammation and cellular metabolism.
Figure 4.

gd-IgA treatment of human mesangial cells. Hierarchical clustering analysis of the cell proteomic data using the Ward averaging method. gd-IgA–treated cells were completely separated from the untreated cells.
Table 5.
Top 25 differentially expressed pathways from cell proteomic data
| Ingenuity Canonical Pathwaysa | P Value | Ratio |
|---|---|---|
| LXR/RXR activation | <0.001 | 29/121 |
| Superpathway of cholesterol biosynthesis | <0.001 | 14/27 |
| FXR/RXR activation | <0.001 | 26/125 |
| Acute-phase response signaling | <0.001 | 28/168 |
| Cholesterol biosynthesis 1 | <0.001 | 8/13 |
| Cholesterol biosynthesis 2 (via 24,25-dihydrolanosterol) | <0.001 | 8/13 |
| Cholesterol biosynthesis 3 (via desmosterol) | <0.001 | 8/13 |
| NRF2-mediated oxidative stress response | <0.001 | 21/177 |
| Superpathway of geranylgeranyldiphosphate biosynthesis 1 (via mevalonate) | <0.001 | 6/16 |
| Clathrin-mediated endocytosis signaling | <0.001 | 20/184 |
| Coagulation system | <0.001 | 8/35 |
| Aryl hydrocarbon receptor signaling | <0.001 | 16/135 |
| Mitochondrial dysfunction | <0.001 | 18/165 |
| Cell cycle control of chromosomal replication | <0.001 | 7/27 |
| Mevalonate pathway 1 | <0.001 | 5/12 |
| Oxidative phosphorylation | <0.001 | 13/104 |
| Superoxide radicals degradation | <0.001 | 4/8 |
| p53 Signaling | <0.001 | 12/98 |
| TR/RXR activation | <0.001 | 11/85 |
| Glutathione redox reaction 1 | <0.001 | 5/18 |
| Methylglyoxal degradation 3 | <0.001 | 4/11 |
| Thioredoxin pathway | <0.001 | 3/6 |
| Zymosterol biosynthesis | <0.001 | 3/6 |
| IL-12 signaling and production in macrophages | 0.001 | 13/131 |
| Vitamin C transport | 0.001 | 4/14 |
Pathway P value is calculated using the Fisher exact method, and the ratio represents the number of differentially expressed proteins that map the pathway from the total proteins in the pathway. LXR, liver X receptor; RXR, Retinoid X receptor; FXR, farnesoid X receptor; NRF2, nuclear factor (erythroid-derived 2)-like 2; TR, thyroid hormone receptor.
Using significantly regulated proteins with Benjamin–Hochberg-adjusted P value <0.05 and unlogged fold changes >1.15 as upregulation and <0.85 as downregulation.
gd-IgA–Treated Mesangial Cells Recapitulate Major Pathways Differentially Expressed in IgAN
To investigate the pathways affected in both tissue and mesangial cells, we used IPA to find common significantly differentially expressed pathways. Table 6 lists all of the common pathways with Fisher exact P<0.05. As a result, several inflammatory pathways were confirmed both in vivo and in vitro, such as acute-phase response signaling, atherosclerosis signaling, LXR/RXR activation, complement system, and cytokine/growth factor–related pathways (i.e., IL-12 signaling and production in macrophages and IL-8 signaling). Additionally, pathways related to iNOS/eNOS effects were also present. The genes and proteins involved in these common pathways are found in Supplemental Table 4.
Table 6.
Common significantly regulated pathways from both transcriptomic glomerular data and cell proteomic data
| Ingenuity Canonical Pathwaysa | P Value | Ratio | ||
|---|---|---|---|---|
| Array | Cell MS | Array | Cell MS | |
| Acute-phase response signaling | <0.001 | <0.001 | 19/168 | 28/168 |
| Agrin interactions at neuromuscular junction | 0.01 | 0.01 | 7/67 | 7/67 |
| Aryl hydrocarbon receptor signaling | 0.03 | <0.001 | 10/135 | 16/135 |
| Atherosclerosis signaling | <0.001 | 0.002 | 14/121 | 12/121 |
| Caveolar-mediated endocytosis signaling | 0.02 | <0.01 | 7/71 | 6/71 |
| Clathrin-mediated endocytosis signaling | 0.01 | <0.001 | 14/184 | 20/184 |
| Coagulation system | <0.01 | <0.001 | 5/35 | 8/35 |
| Complement system | 0.002 | <0.01 | 6/36 | 5/36 |
| FXR/RXR activation | <0.001 | <0.001 | 13/125 | 26/125 |
| Glioma invasiveness signaling | <0.01 | 0.02 | 7/57 | 6/57 |
| Glutathione redox reactions 1 | 0.03 | <0.001 | 3/18 | 5/18 |
| Glutathione-mediated detoxification | 0.002 | 0.01 | 5/24 | 4/24 |
| Hepatic fibrosis/hepatic stellate cell activation | <0.001 | <0.01 | 30/181 | 14/181 |
| IL-12 signaling and production in macrophages | 0.01 | 0.001 | 11/131 | 13/131 |
| IL-8 signaling | <0.001 | 0.02 | 18/183 | 13/183 |
| Inhibition of matrix metalloproteases | 0.003 | 0.05 | 6/38 | 4/38 |
| LXR/RXR activation | <0.001 | <0.001 | 15/121 | 29/121 |
| Methylglyoxal degradation 2 | <0.001 | <0.001 | 4/11 | 4/11 |
| NRF2-mediated oxidative stress response | 0.003 | <0.001 | 15/177 | 21/177 |
| Production of NO and reactive oxygen species in macrophages | <0.001 | <0.01 | 18/178 | 14/178 |
| Superpathway of citrulline metabolism | 0.02 | 0.01 | 3/14 | 3/14 |
| γ-Glutamyl cycle | 0.012 | 0.01 | 3/14 | 3/14 |
FXR, farnesoid X receptor; RXR, retinoid X receptor; LXR, liver X receptor; NRF2, nuclear factor (erythroid-derived 2)-like 2.
Pathway P value is calculated using the Fisher exact method, and the ratio represents (number of differentially expressed genes/proteins that map the pathway)/(total genes/proteins in the pathway).
Discussion
IgAN is the most common GN in the world, but the underlying mechanisms are not yet completely understood. The complexity of the disease has led to multiple hypotheses where the glycosylation pattern of the IgA molecule is considered to be important16 as well as a genetic component found in GWAS studies.17,18 To understand the development and progression of IgAN, the cellular mechanisms leading to IgAN must be understood. Therefore, we conducted a global human genome microarray analysis of glomeruli from patients with IgAN and healthy controls to investigate the pathophysiologic alterations of the disease. Using MS, we performed a global proteomic analysis on cultured human mesangial cells treated with patient-derived gd-IgA. By combining the results from the two approaches and using novel analytic methods, the aim was to broaden the understanding of the role of mesangial cells in IgAN pathophysiology.
From our microarray analysis, 736 genes were identified as significantly differentially expressed genes, even with a strict cutoff q value of <0.01. GO analyses revealed that many of these genes were related to extracellular events, which is consistent with previous studies.9,19–22 Deposition of gd-IgA–containing immune complexes in the mesangium of the patients is believed to induce mesangial cell proliferation by triggering release of extracellular cytokines and growth factors. In the end, this will lead to damage of the cells in the glomeruli, resulting in kidney injury.17 As suspected, in the pathway analysis, many of the top differentially expressed pathways were related to inflammatory processes, such as IL-8 signaling, IL-6 signaling, and LPS/IL-1–mediated inhibition of RXR function. Complement system pathways have been reported to be directly activated by IgA complexes in the mesangium of patients with IgAN,23,24 and this was consistent with our analysis. eNOS and iNOS signaling have been shown to correlate with the degree of glomerular injury in IgAN, and nitric oxide (NO) may be involved in disease progression.25,26 In the dataset, several pathways involved in NO signaling, such as production of NO and reactive oxygen species in macrophages and eNOS signaling, were identified.
Investigating the pathways and the GO terms identified using glomerular microarray data does not provide information on which cellular compartments of the glomeruli are affected. Ju et al.10 used an in silico approach to identify cell type–specific transcripts using human renal biopsy tissue in a genome-wide manner. These cell type–specific transcripts allow investigation of the involvement of the specific cell types in glomerular disease, such as IgAN, by examining tissue-derived transcriptomic data. In our study, we used both podocyte- and mesangial cell–positive standard genes defined by Ju et al.10 to investigate their function in IgAN. It has been discussed whether IgAN is a mesangial cell–dominant disease, because there is still no clear correlation between the IgA deposits and the degree of mesangial damage or kidney injury.5 Using the cell type–positive standard genes, we found that mesangial cell standard genes were significantly enriched in our tissue microarray data compared with podocyte standard genes. From a transcriptomic perspective, this indicates that mesangial cell–positive standard genes are more involved in IgAN pathogenesis than podocyte genes. Principal coordinate analysis showed that expression of mesangial cell–positive standard genes could clearly distinguish the patients with IgAN from the living donor controls, whereas the podocyte-positive standard genes failed to do so. This shows that the mesangial cell–positive standard genes could be useful for investigating and understanding disease development.
Several studies have shown that mesangial cell proliferation and inflammation may lead to kidney damage.7,8 Patient Z scores were calculated to represent the deviation of positive standard genes from different cell types. The patient Z score can explain how the changes of the positive standard genes influence the level of kidney injury. We used the patient Z score to correlate with patient clinical data. We found that eGFR and serum creatinine values at the time of biopsy were significantly correlated with mesangial cell–positive standard genes but not significantly correlated with positive podocyte standard genes. We also scored the patients according to the Oxford MEST score, a scoring system for IgAN.3,15 The MEST scores were used to group the patients, and then, the respective MEST scores were compared with the mesangial and podocyte Z scores. Dividing the patients according to their segmental glomerulosclerosis score (S1 or S0), a significantly higher mesangial standard gene Z score was found for the patients with S1 compared with for patients with S0. On the gene level, this shows that the expression changes of the mesangial cell–positive standard genes are significantly associated with the serum creatinine level at the time of renal biopsy. It is also interesting to note that, according to both their CKD status and their MEST scores (as shown in Table 1), the patient cohort in this study is in an early stage of the disease (CKD stages 1–3). In our cohort, only three patients have a mesangial hypercellularity score of one, indicating a low grade of mesangial hypercellularity, and this implies that the findings on the transcriptomic level are less likely to be due to an increased number of mesangial cells in the glomeruli. Despite the low CKD stages (1–3) of the patients, we can use the mesangial cell–positive standard gene Z score to discriminate between controls and patients and observe correlations with clinical data, illustrating the relevance of the mesangial standard genes in IgAN.
To investigate how gd-IgA specifically affects mesangial cells, an in vitro model was designed to simulate the cellular environment of IgAN by treating mesangial cells with patient-derived IgA. It is known that the levels of gd-IgA in the circulation of patients with IgAN differ, and there is evidence that the serum levels of gd-IgA are associated with disease progression.27 To standardize the experiments, pooled gd-IgA from three patients with IgAN was used to treat the cells with a specific amount of gd-IgA. The proteomics results from gd-IgA–treated mesangial cells showed that they could be distinguished from the untreated control cells by cluster analysis. Pathway analysis of the significantly differentially expressed proteins identified pathways involved in inflammation, metabolism, and oxidative stress. The LXR/RXR activation and FXR/RXR activation pathways are related to regulation of lipid metabolism and inflammation.28 Another inflammatory pathway is the acute-phase response signaling, which is a rapid inflammatory response pathway.29 These pathways are among the top differentially expressed pathways in the gd-IgA–treated mesangial cells and may be related to IgAN disease mechanisms. The pathway clathrin-mediated endocytosis signaling is also worth investigating, because it controls the internalization of molecules from the plasma membrane to intracellular compartments.30 The involvement of this pathway could explain how the mesangial cells attempt to internalize the gd-IgA for removal, triggering downstream signaling. The pathways involved in oxidative stress may be affected in IgAN due to imbalance of oxidation/antioxidation and advanced oxidation protein products, which are reported to be markers of proteinuria and disease progression.31
Combining the pathways from both transcriptomic and proteomic data gives us a new understanding of the pathophysiologic changes seen in the glomeruli from patients with IgAN that are a result of the mesangial response to gd-IgA. Nearly all common differentially expressed pathways are related to inflammation. The complement system is one of the inflammatory pathways that is found in both datasets, indicating that complement activation is induced in the mesangial cells by gd-IgA as discussed by other investigators.23
In summary, it was found that mesangial cells and their positive standard genes can be useful in investigating IgAN disease mechanisms. Additionally, the patient clinical parameters (serum creatinine values, eGFR and segmental glomerulosclerosis) significantly correlate with Z scores derived from expression profiles of mesangial cell–positive standard genes but do not correlate with those of podocyte standard genes. By investigating both mesangial cell proteomics and glomerular transcriptomics, we identified 22 common pathways, mainly related to inflammation. The genes, proteins, and their corresponding pathways identified in this study provide novel insights into the pathophysiologic mechanisms leading to progression of IgAN. The data clearly point toward a significant role for mesangial cellular events in the early stages of IgAN. Finally, the common regulated pathways confirmed both in vivo and in vitro constitute a solid base for further understanding of the mechanisms underlying IgAN.
Concise Methods
Patient and Control Biopsies
The study was conducted according to the Declaration of Helsinki, and the regional ethical board in Gothenburg approved the ethical application. All of the patients signed a written consent form before participating in the study. Whole-core patient biopsies were obtained as part of routine renal biopsy procedures, immediately placed in RNAlater (Ambion; Thermo Fisher Scientific, Waltham, MA) bedside, and stored at −80°C. Biopsies from patients diagnosed with IgAN were singled out for further experiments. Biopsies from healthy living kidney transplant donors were used as controls. These biopsies were taken after reperfusion in the kidney transplant recipient. In total, 25 patients with IgAN (age 43±15 years old, women-to-men ratio of 1:2) and 26 control samples (age 48±13 years old, women-to-men ratio of 1:1.27) were collected and included. Clinical data from the time of biopsy up to 4 years later were collected (in total, 15±7 serum creatinine values per patient). eGFR (milliliters per minute per 1.73 square meter per year) was calculated using the CKD-EPI formula. Renal disease progression was obtained by calculating the slope of the linear regression of the eGFR values over the timespan of the follow-up period.
Oxford Classification
Patients diagnosed with IgAN and used in the study for microarray analysis were scored according to the Oxford MEST scoring system by a renal pathologist in a blinded fashion.
RNA Isolation
On average, eight to ten glomeruli were microdissected by hand from the renal biopsies under a stereomicroscope, and RNA was extracted using the RNeasy Micro Kit (Qiagen, Venlo, The Netherlands) following standard protocols. The quality was checked using the HighSens Kit on Experion (Bio-Rad, Hercules, CA). NanoDrop 2000 (Thermo Fisher Scientific) was used to measure the RNA concentration of the samples.
Affymetrix Microarray of Glomeruli and Bioinformatics
RNA isolated from glomeruli was reversely transcribed to cDNA and amplified using the Ovation Pico WTA system (NuGen, San Carlos, CA). The cDNA was biotin labeled (NuGen) and used for microarray on the Affymetrix Human GeneChip U133 2.0 platform. The microarray experiments were conducted at five different time points. Data processing was done in R (University of Auckland, Auckland, New Zealand). The raw data were normalized using the Robust Multiarray Averaging32 algorithm in individual batches on the basis of the latest custom chip definition file (CDF file, version 19; BrainArray, Microarray Laboratory, Ann Arbor, MI). The sample quality was checked using a normalized unscaled SEM plot, a relative log expression plot, an RNA degradation plot, and a principal component analysis plot. A sample was considered an outlier if more than three of four of the plots showed poor quality. Outliers were excluded from the analysis, and the normalized data from each batch were then merged. The empirical Bayes algorithm33 was used to correct the batch effect, and microarray probes were filtered using a background signal threshold filtering technique as described in the work by Makarenkov et al.34 Data were clustered using the hierarchical Ward averaging13 method and principal coordinate analysis.12 The data were statistically analyzed using SAM11 as implemented in MultiExperiment Viewer (MeV; Dana-Farber Cancer Institute, Boston, MA). Significantly regulated genes were selected with SAM q value <0.01 and unlogged fold change <0.67 as downregulation and >1.5 as upregulation (equivalent logged fold change ±0.58). The strict q value was chosen to decrease the rate of false positive data.35 The fold change threshold was set to capture a robust differentiation in expression and an experimentally reliable validation using quantitative RT-PCR.36 Differentially expressed pathways were analyzed using Genomatix Genome Analyzer (Genomatix GmbH; https://www.genomatix.de/) and the Ingenuity Pathway Analysis tool (Ingenuity Qiagen). GO analysis was done using the online Gene Ontology Consortium tool (AmiGO; http://geneontology.org). The raw data are available at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE93798.
Taqman Quantitative PCR
Quantitative PCR was used to evaluate the quality of microarray data. RNA from glomeruli was reverse transcribed to cDNA using the High Capacity RNA to cDNA kit (Applied Biosystems, Waltham, MA) on a PTC-200 Peltier Thermal Cycler (MJ Research, Waltham, MA). Custom Taqman 384 low-density array cards were used to check the quantity of the mRNA level on the ViiA seven real-time PCR system (Applied Biosystems); 17 genes were selected, and GAPDH was used as the endogenous gene (Supplemental Figure 3).
Cell Type–Positive Standard Genes in Kidney Disease
Ju et al.10 developed an iterative algorithm (in silico nanodissection), which is an approach on the basis of machine learning to statistically define cell type–specific genes using high-throughput data. By applying the algorithm to kidney microarray datasets, the algorithm was crossvalidated with literature data and predicted cell type–positive standard genes for mesangial cells and podocytes for kidney diseases. The detailed lists of cell type–positive standards genes are in Supplemental Table 1.
Z-Score Transformation
To investigate how the patient clinical data are correlated with transcriptomic information, Z scores were calculated for each patient using the positive standard genes.
For each individual patient, the Z score was calculated as illustrated below:
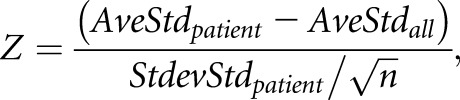 |
where AveStdpatient is the average value of the positive standard genes in each patient, AveStdall is the average value of the positive standard genes from all of the patients (as mean value for our patient population), StdevStdpatient is the SD of the positive standard genes in each patient, and n is the number of positive standard genes. The usage of Z scores has been described previously,37,38 and herein, the Z score for each patient represents how much the positive standard genes deviate in each patient compared with all of the other patients. The higher the Z score, the more the positive standard gene expression in the individual patient deviates from the cohort.
Pearson Correlation and P Value
The patient Z scores were correlated with the clinical data using Pearson correlation. The corresponding Pearson correlation P values were calculated on the basis of two-tailed distribution statistics. A P<0.05 was considered statistically significant.
IgA Purification
Sera from three patients diagnosed with IgAN and three healthy controls were collected and used for IgA1 purification using jacaline as reported elsewhere.39 The purified IgA from the two groups was pooled. The amount of gd-IgA purified from patients with IgAN compared with healthy controls was investigated using ELISA with Lectin Helix Aspersa (Sigma-Aldrich, St. Louis, MO). The concentration of the purified IgA was measured using nephelometry.
Cell Culture with IgA Treatment
Human mesangial cells commercially purchased from Lonza (Lonza, Basel, Switzerland) were cultured under standard conditions (5% CO2 and 37°C) with medium (DMEM F12 Ham medium [Lonza] + 20% serum [FBS; Hyclone, GE Healthcare, Little Chalfont, United Kingdom] + 1% antibiotics (Pen-Strep, BioWhittaker; Lonza) until 80% confluence. Before treatment, the cells were starved (DMEM Ham F12 medium + 0.5% FBS + 1% antibiotic) overnight. Purified IgA (100 μg/ml) from patients with IgAN was added to five 10-cm plates of mesangial cells for 48 hours of incubation.
Cells were washed twice with cold PBS (Hyclone; GE Healthcare) and subsequently lysed with lysis buffer (25 mM triethylammonium bicarbonate [Sigma-Aldrich] + 2% SDS [Sigma-Aldrich]) with PhosStop (Roche Life Science, Basel, Switzerland) and Protease Inhibitor (Roche Life Science). Protein concentration was measured using the Pierce BCA Kit (Thermo Fisher Scientific), and samples were stored at −80°C for further experiments.
MS
Thirty micrograms of protein was used for further analysis. Proteins were digested by trypsin using the filter-aided sample preparation method as described in the work by Wisniewski et al.40 The samples were reduced by dithiothreitol (Sigma-Aldrich) and diluted with 400 μl 8 M urea and 0.1 M TEAB. The samples were applied on Nanosep 30k Omega Filters (Pall Life Sciences, Portsmouth, United Kingdom), and 8 M urea and 0.1 M TEAB was used to repeatedly wash away SDS. Alkylation was performed with methylethane thiosulfonate diluted in 8 M urea and 0.1 M TEAB, and the filters were washed repeatedly. Trypsin (Sequencing Grade Modified Trypsin; Promega) was added at 1:100 dilution in the sample together with 1% sodium deoxycholate and 20 mM TEAB. pH was adjusted to approximately eight, and the samples were incubated at 37°C overnight. Another portion of trypsin was added, and incubation was continued. The peptides were collected by centrifugation, subjected to a unique isobaric mass tagging reagent, and combined according to the manufacturer’s instructions (Thermo Fisher Scientific). The sample was acidified with formic acid to a pH of approximately two to precipitate sodium deoxycholate; then, it was made basic with 1 M NH3 solution and fractionated with high-pH reversed-phase chromatography. The peptides were analyzed on an Orbitrap Fusion Tribrid Mass Spectrometer interfaced to an Easy-nLC1000 (Thermo Fisher Scientific). The raw data generated from LC-MS/MS were merged and identified using Proteome Discoverer, version 1.4 (Thermo Fisher Scientific). Database searches were performed by Mascot search engine (Matrix Science Ltd, London, United Kingdom) using SwissProt Homo sapiens protein database (Swiss Institute of Bioinformatics).
Statistical Analyses for the MS Data
Sample expression ratios were calculated on the basis of average quantity of all of the control samples. The IgA-treated sample expression ratios were compared with control expression ratios using t test statistics with Benjamin–Hochberg multivariable adjustment.41 A technical variance of 10% is observed using unique isobaric mass tagging reagent labeling in the Orbitrap Fusion Tribrid Mass Spectrometer. Thus, a 15% proportion change of the protein content is used for cutoff for the fold changes: fold change <0.85 indicated downregulation and >1.15 indicated upregulation (equivalent logged fold change ±0.2).42,43 Proteins were considered significantly differentially expressed at P<0.05. Differentially expressed pathways were analyzed using significantly regulated proteins in the IPA pathway and Genomatix Genome Analyzer pathway tools.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Alice Smith for providing the protocol for IgA purification and Helix aspersa–ELISA. We also thank Johan Mölne for mesangial hypercellularity, endocapillary hypercellularity, segmental glomerulosclerosis, and tubular atrophy/interstitial fibrosis scoring of the patients according to the Oxford classification for patients with IgAN, Elisabeth Rosqvist for excellent assistance, and Prof. Gerald F. DiBona for helpful discussion of the manuscript. The Swedish Medical Research Council grant 14764, the National Association for Kidney Diseases, the John and Brit Wennerström Research Foundation, the Inga-Britt and Arne Lundberg Research Foundation, and ALF grants from the Sahlgrenska University Hospital supported this study.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016101103/-/DCSupplemental.
References
- 1.Cove-Smith A, Hendry BM: The regulation of mesangial cell proliferation. Nephron, Exp Nephrol 108: e74–e79, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Hendry BM, Khwaja A, Qu QY, Shankland SJ: Distinct functions for Ras GTPases in the control of proliferation and apoptosis in mouse and human mesangial cells. Kidney Int 69: 99–104, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, D’Agati V, D’Amico G, Emancipator S, Emma F, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Leung CB, Li LS, Li PK, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H; Working Group of the International IgA Nephropathy Network and the Renal Pathology Society : The oxford classification of IgA nephropathy: Rationale, clinicopathological correlations, and classification. Kidney Int 76: 534–545, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Moresco RN, Speeckaert MM, Delanghe JR: Diagnosis and monitoring of IgA nephropathy: The role of biomarkers as an alternative to renal biopsy. Autoimmun Rev 14: 847–853, 2015 [DOI] [PubMed] [Google Scholar]
- 5.Yamaji K, Suzuki Y, Suzuki H, Satake K, Horikoshi S, Novak J, Tomino Y: The kinetics of glomerular deposition of nephritogenic IgA. PLoS One 9: e113005, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, Raska M, Renfrow MB, Julian BA, Novak J: The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front Immunol 7: 117, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasri H, Mubarak M: Extracapillary proliferation in IgA nephropathy; recent findings and new ideas. J Nephropathol 4: 1–5, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robert T, Berthelot L, Cambier A, Rondeau E, Monteiro RC: Molecular insights into the pathogenesis of IgA nephropathy. Trends Mol Med 21: 762–775, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Ebefors K, Liu P, Lassén E, Elvin J, Candemark E, Levan K, Haraldsson B, Nyström J: Mesangial cells from patients with IgA nephropathy have increased susceptibility to galactose-deficient IgA1. BMC Nephrol 17: 40, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju W, Greene CS, Eichinger F, Nair V, Hodgin JB, Bitzer M, Lee YS, Zhu Q, Kehata M, Li M, Jiang S, Rastaldi MP, Cohen CD, Troyanskaya OG, Kretzler M: Defining cell-type specificity at the transcriptional level in human disease. Genome Res 23: 1862–1873, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gower JC: Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53: 325–338, 1966 [Google Scholar]
- 13.Ward JH: Hierarchical grouping to optimize an objective function. J Am Stat Assoc 58: 236–244, 1963 [Google Scholar]
- 14.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) : A new equation to estimate glomerular filtration rate. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran DC, Coppo R, D’Agati V, D’Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H; Working Group of the International IgA Nephropathy Network and the Renal Pathology Society : The oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int 76: 546–556, 2009 [DOI] [PubMed] [Google Scholar]
- 16.Kiryluk K, Li Y, Moldoveanu Z, Suzuki H, Reily C, Hou P, Xie J, Mladkova N, Prakash S, Fischman C, Shapiro S, LeDesma RA, Bradbury D, Ionita-Laza I, Eitner F, Rauen T, Maillard N, Berthoux F, Floege J, Chen N, Zhang H, Scolari F, Wyatt RJ, Julian BA, Gharavi AG, Novak J: GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet 13: e1006609, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiryluk K, Novak J: The genetics and immunobiology of IgA nephropathy. J Clin Invest 124: 2325–2332, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyatt RJ, Julian BA: IgA nephropathy. N Engl J Med 368: 2402–2414, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Novak J, Julian BA, Mestecky J, Renfrow MB: Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin Immunopathol 34: 365–382, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, Monteiro RC: Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med 194: 417–425, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneko Y, Otsuka T, Tsuchida Y, Gejyo F, Narita I: Integrin α1/β1 and α2/β1 as a receptor for IgA1 in human glomerular mesangial cells in IgA nephropathy. Int Immunol 24: 219–232, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Kokubo T, Hiki Y, Iwase H, Tanaka A, Toma K, Hotta K, Kobayashi Y: Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol 9: 2048–2054, 1998 [DOI] [PubMed] [Google Scholar]
- 23.Floege J, Moura IC, Daha MR: New insights into the pathogenesis of IgA nephropathy. Semin Immunopathol 36: 431–442, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PH, Tamouza H, Jamin A, Bex-Coudrat J, Gestin A, Boumediene A, Arcos-Fajardo M, England P, Pillebout E, Walker F, Daugas E, Vrtosvnik F, Flamant M, Benhamou M, Cogné M, Moura IC, Monteiro RC: Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med 209: 793–806, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu X, Kong D, Zhang L, Sun Y, Na S, Han C, Jin X: Correlation analysis of angiotensin-converting enzyme, angiotensinogen, and endothelial nitric oxide synthase gene polymorphisms and the progression of immunoglobulin A nephropathy/membranous nephropathy. Hum Pathol 44: 2806–2813, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Furusu A, Miyazaki M, Abe K, Tsukasaki S, Shioshita K, Sasaki O, Miyazaki K, Ozono Y, Koji T, Harada T, Sakai H, Kohno S: Expression of endothelial and inducible nitric oxide synthase in human glomerulonephritis. Kidney Int 53: 1760–1768, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Suzuki Y, Matsuzaki K, Suzuki H, Okazaki K, Yanagawa H, Ieiri N, Sato M, Sato T, Taguma Y, Matsuoka J, Horikoshi S, Novak J, Hotta O, Tomino Y: Serum levels of galactose-deficient immunoglobulin (Ig) A1 and related immune complex are associated with disease activity of IgA nephropathy. Clin Exp Nephrol 18: 770–777, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desvergne B: RXR: From partnership to leadership in metabolic regulations. Vitam Horm 75: 1–32, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Jahangiri A: High-density lipoprotein and the acute phase response. Curr Opin Endocrinol Diabetes Obes 17: 156–160, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMahon HT, Boucrot E: Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12: 517–533, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Coppo R, Camilla R, Amore A, Peruzzi L: Oxidative stress in IgA nephropathy. Nephron Clin Pract 116: c196–c198, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Johnson WE, Li C, Rabinovic A: Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8: 118–127, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Makarenkov V, Zentilli P, Kevorkov D, Gagarin A, Malo N, Nadon R: An efficient method for the detection and elimination of systematic error in high-throughput screening. Bioinformatics 23: 1648–1657, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Dalman MR, Deeter A, Nimishakavi G, Duan ZH: Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinformatics 13[Suppl 2]: S11, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ju W, Nair V, Smith S, Zhu L, Shedden K, Song PX, Mariani LH, Eichinger FH, Berthier CC, Randolph A, Lai JY, Zhou Y, Hawkins JJ, Bitzer M, Sampson MG, Thier M, Solier C, Duran-Pacheco GC, Duchateau-Nguyen G, Essioux L, Schott B, Formentini I, Magnone MC, Bobadilla M, Cohen CD, Bagnasco SM, Barisoni L, Lv J, Zhang H, Wang HY, Brosius FC, Gadegbeku CA, Kretzler M; ERCB, C-PROBE, NEPTUNE, and PKU-IgAN Consortium : Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med 7: 316ra193, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheadle C, Cho-Chung YS, Becker KG, Vawter MP: Application of z-score transformation to affymetrix data. Appl Bioinformatics 2: 209–217, 2003 [PubMed] [Google Scholar]
- 38.Cheadle C, Vawter MP, Freed WJ, Becker KG: Analysis of microarray data using Z score transformation. J Mol Diagn 5: 73–81, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim MJ, McDaid JP, McAdoo SP, Barratt J, Molyneux K, Masuda ES, Pusey CD, Tam FW: Spleen tyrosine kinase is important in the production of proinflammatory cytokines and cell proliferation in human mesangial cells following stimulation with IgA1 isolated from IgA nephropathy patients. J Immunol 189: 3751–3758, 2012 [DOI] [PubMed] [Google Scholar]
- 40.Wiśniewski JR, Zougman A, Nagaraj N, Mann M: Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Benjamini Y, Hochberg Y: Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57: 289–300, 1995 [Google Scholar]
- 42.Kobayashi D, Kumagai J, Morikawa T, Wilson-Morifuji M, Wilson A, Irie A, Araki N: An integrated approach of differential mass spectrometry and gene ontology analysis identified novel proteins regulating neuronal differentiation and survival. Mol Cell Proteomics 8: 2350–2367, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karp NA, Huber W, Sadowski PG, Charles PD, Hester SV, Lilley KS: Addressing accuracy and precision issues in iTRAQ quantitation. Mol Cell Proteomics 9: 1885–1897, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.