

Abstract
Objective
The role of hemoglobin and myoglobin in the cardiovascular system is well established, yet other globins in this context are poorly characterized. Here, we examined the expression and function of cytoglobin (CYGB) during vascular injury.
Approach and Results
We characterized CYGB content in intact vessels and primary vascular smooth muscle (VSM) cells and used two different vascular injury models to examine the functional significance of CYGB in vivo. We found that CYGB was strongly expressed in medial arterial VSM and human veins. In vitro and in vivo studies indicated that CYGB was lost upon VSM cell de-differentiation. In the rat balloon angioplasty model, site-targeted delivery of adenovirus encoding shRNA specific for CYGB prevented its re-expression and decreased neointima formation. Similarly, four weeks after complete ligation of the left common carotid, Cygb knockout mice displayed little to no evidence of neointimal hyperplasia in contrast to their wild-type littermates. Mechanistic studies in the rat indicated that this was primarily associated with increased medial cell loss, TUNEL staining, and caspase-3 activation, all indicative of prolonged apoptosis. In vitro, CYGB could be re-expressed upon VSM stimulation with cytokines and hypoxia and loss of CYGB sensitized human and rat aortic VSM cells to apoptosis. This was reversed upon antioxidant treatment or NOS2 inhibition.
Conclusions
These results indicate that CYGB is expressed in vessels primarily in differentiated medial vascular smooth muscle cells where it regulates neointima formation and inhibits apoptosis after injury.
Keywords: smooth muscle, restenosis, hemoglobin, myoglobin, cytoglobin, nitric oxide, programmed cell death, apoptosis, angioplasty, arteriovenous fistula, caspase
Introduction
Maladaptive vessel remodeling is a characteristic feature of many vascular disorders including atherosclerosis, vasculitis, post-angioplasty restenosis, vein graft stenosis, and arteriovenous fistula (AVF) failure. Although the etiology of these conditions may differ, they all involve the coordinated destruction and synthesis of vascular wall components in response to mechanical and inflammatory injuries, changing flow, and altered tensile forces. Stress-response pathways and programed cell death such as apoptosis and necroptosis contribute to these processes.
The oxygen-carrier proteins hemoglobin (HB) and myoglobin (MB) are expressed in the vascular wall and regulate endothelial and smooth muscle functions, primarily through nitric oxide (NO) and nitrite (NO2-) signaling 1, 2. In addition to HB and MB, the mammalian globin family includes neuroglobin (NGB), cytoglobin (CYGB), and androglobin (ADGB) 3-7. The functions of the latter 3 are poorly defined but their intracellular concentrations and localization in non-muscle cells would suggest roles independent of molecular oxygen (O2) transport 8. Given some similarities with other globins, as well as its unique terminal extensions, heme-hexacoordination and ligand pocket, a variety of functions have been proposed for CYGB. These include electron transfer, O2 sensing, NO detoxification, or a combination of the above 9. In vivo studies have defined a cytoprotective role for CYGB during liver and kidney fibrosis and carcinogenesis, potentially related to its capacity to regulate oxidative stress 5, 10. Cytoglobin is a candidate tumor suppressor gene in upper aerodigestive tract squamous cancers11. It is also upregulated in activated myogenic progenitor cells and proliferating myoblasts, where it serves anti-apoptotic functions and contributes to muscle regeneration12. A number of studies have highlighted anti-apoptotic functions associated with CYGB in response to various stressors including hypoxia, high glucose, and oxidants13-15. In the cardiovascular system, CYGB is expressed in the heart and increases during hypoxia-induced cardiac hypertrophy in the mouse 16. CYGB mRNA and protein are also found in human and rodent vascular smooth muscle (VSM) 17 and recent evidence for a role for CYGB as a regulator of NO homeostasis in vivo has been provided18. However, the functional significance of CYGB expression in the context of vascular remodeling is unknown.
Because past reports suggest that CYGB is involved in cellular stress response12, 19, 20, we tested the hypothesis that CYGB regulates cell survival in the vessel wall during vascular injury. We found that CYGB is expressed in contractile VSM cells and its abundance is decreased in de-differentiated VSM cells. Overall, our results suggest that CYGB is critical for VSM cell survival and regulate apoptosis during vascular injury through effector caspase regulation. This is the first example of a globin regulating vascular injury through modulation of pro-apoptotic signals.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Cytoglobin is preferentially expressed in contractile vascular smooth muscle cells and downregulated during smooth muscle cell de-differentiation
In light of past studies indicating some level of expression of myoglobin (MB) and cytoglobin (CYGB) in vessels and to better understand the conditions associated with CYGB expression 2, 17, 18, 21, we first compared CYGB mRNA levels to those of myoglobin (MB) using quantitative real-time polymerase chain reaction (qPCR). Overall, human aortic samples showed statistically significantly higher mRNA levels for CYGB than MB (Figure 1A). We also evaluated CYGB expression in a de-identified cohort of discarded vein trimmings obtained from patients undergoing arteriovenous fistula (AVF) placement for hemodialysis or at revision of the AVF. Placement vessels expressed low levels of CYGB mRNA, in amounts not different from those of MB (Figure 1B). In contrast, vessels obtained at revision due to late failure of the AVF showed a statistically significant increase in CYGB gene expression with no change for MB. Western blotting of CYGB in protein lysates from placement and revision vessels recapitulated these findings (Figure I in the online-only Data Supplement). In mouse and rat abdominal aortas and carotids, Cygb mRNA transcripts were expressed at least in a hundred fold excess over Mb (Figure 1C and D). In contrast, lysates derived from mouse and rat hearts indicated levels of Mb mRNA in large excess to those of Cygb (Figure 1C and D).
Figure 1. Cytoglobin is expressed in human and rodent vessels.
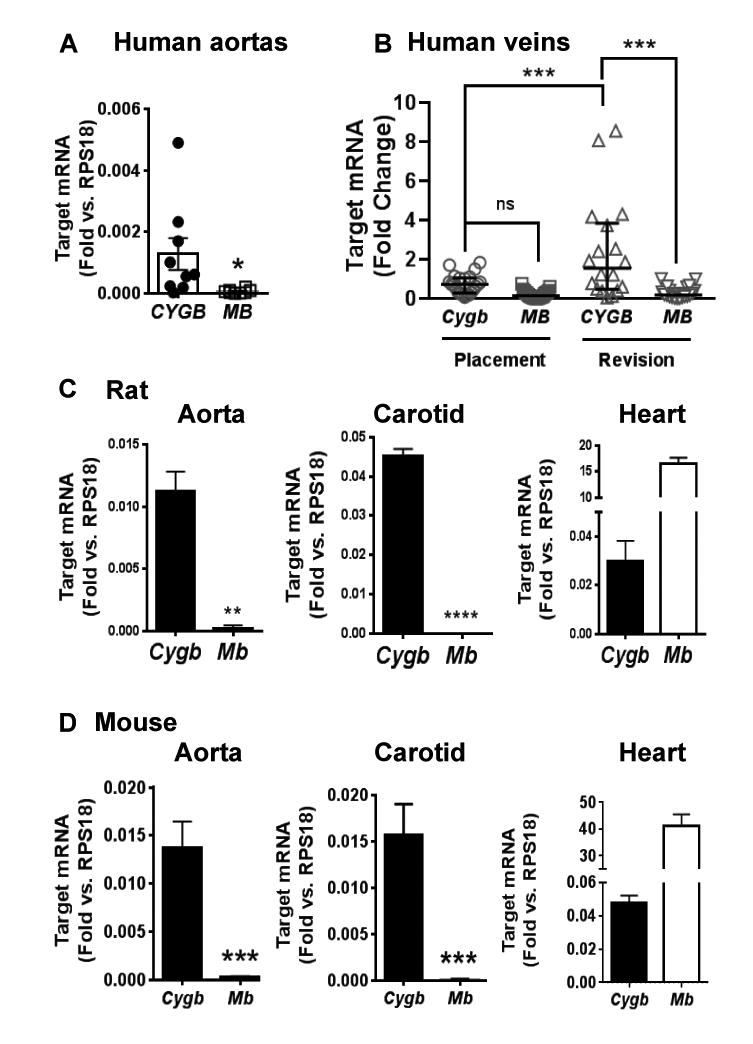
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) results showing relative mRNA levels for cytoglobin (CYGB) and myoglobin (MB) in human aortas (n = 9, A), and human veins derived from vessels trimmings obtained from patients undergoing arteriovenous fistula (AVF) placement (n=26) or at revision (n=20) of failed AVFs (B). Levels of Cygb and Mb mRNA transcripts in mouse aorta (n=11), carotid (n = 6), and heart (n= 4, C); and rat aorta (n=5), carotid (n=3), and heart (n=6; D). Values represent the mean±SEM; *p≤0.05, compared to CYGB; **p≤0.01, compared to Cygb; ***p≤0.001, compared to Cygb. ****p≤0.0001, compared to Cygb.
To expand on these observations, we analyzed Cygb mRNA in contractile (freshly isolated medial layers) and subcultured synthetic rat aortic VSM cells (Figure 2A). Similar to VSM contractile markers Myh11 and Lmod1, we found that Cygb transcript levels were dramatically reduced in sub-cultured de-differentiated SMC. Double immunofluoresence staining of tissue sections showed that CYGB was primarily expressed in medial VSM cells in intact carotids, based on colocalization with the VSM contractile marker calponin (CNN1) (Figure 2B) and similar results were obtained rat aortas (Figure II in the online-only Data Supplement) and in tissue sections derived from cephalic veins from patients undergoing AVF placement or at revision (Figure III in the online-only Data Supplement). Western blotting analysis indicated that medial rat aortic VSM cell dispersions progressively lost CNN1 and alpha smooth muscle actin (ACTA2) protein expression upon 120 h of primary culture and subsequent passage (Figure 2C). This was accompanied with a decrease in CYGB protein expression indicating that acute phenotypic modulation of VSM cells in vitro includes a mark decrease in CYGB protein content. To establish whether CYGB protein expression could be altered in vivo during vascular injury, we examined changes in CYGB protein levels after balloon injury in the rat carotid. A time course analysis by Western blot (Figure 2D) showed maximal decrease in CYGB expression peaking at 3 days post-injury with partial recovery in the latter stages such that levels of CYGB amounted to approximately 50% of the Sham conditions. Immunofluorescence staining of injured vessels at 14 days revealed co-localization of CYGB with CNN1 in the media with limited expression of CYGB in the neointima and limited to no CYGB expression in the adventitia (Figure 2E). Overall, these results indicate CYGB expression in medial differentiated VSM cells and downregulation after balloon injury in the rat that precedes neointima formation and might be concurrent with medial VSM cell de-differentiation.
Figure 2. Cytoglobin is expressed in medial SMCs and downregulated during de-differentiation in vitro and in vivo in the rat.
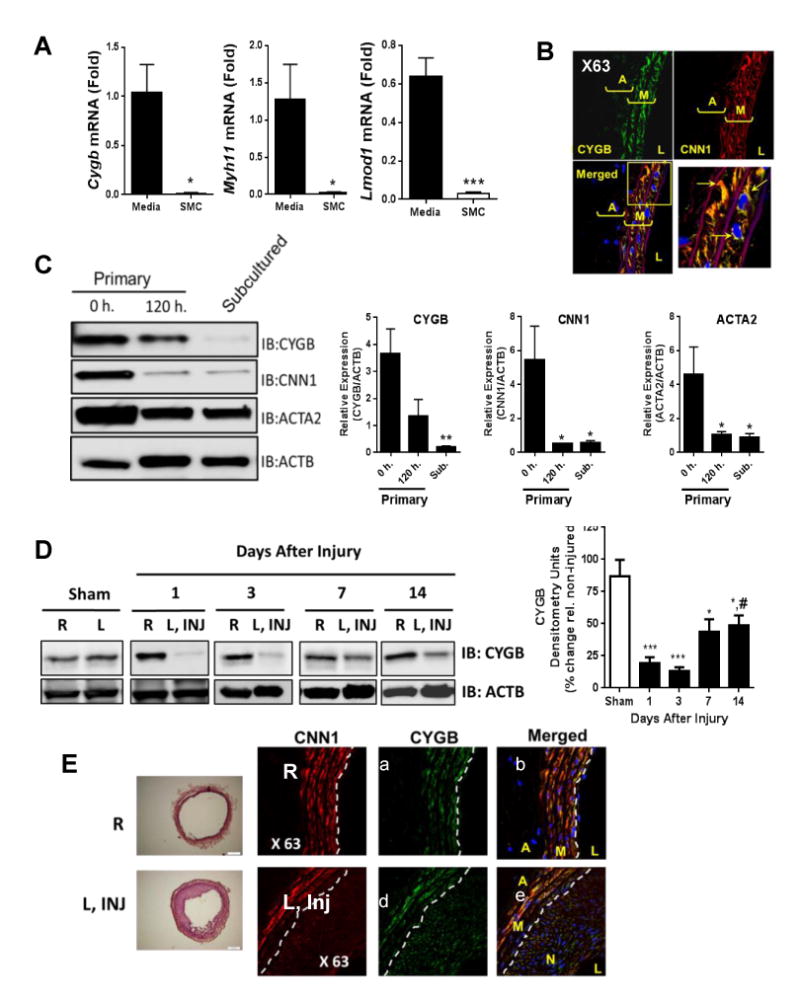
A) qRT-PCR showing relative mRNA levels for Cygb, Myh11, and Lmod1 in the media of rat aorta (n=5) and subcultured medial SMCs (average from 4 different dispersions). B) Double immunofluorescence studies of rat carotids. We used antibodies against CYGB (green) and CNN1 (red). Blue staining is DAPI., yellow inset is shown at higher magnification on the lower right panel and shows colocalization (arrows) of CNN1 and CYGB in the media. A = adventitia, M = media, L = lumen. Representative of 3 independent experiments. C) WB analysis from medial rat aortic VSM cell at 0 or 120 hours after initial dispersion, and from subcultured RAVSMCs. Primary antibodies against CYGB, CNN1, and ACTA2 were used. Right panels, Densitometric analysis of results shown in left panel. Mean±SEM (n=3), *p≤0.05 and **p≤0.01 compared to 0 h. D) WB analysis showing the expression of CYGB in rat carotids following balloon injury in right uninjured (R) and left injured (L, INJ) carotids. Right panel, densitometric analysis. Values are mean ± SEM (n = 5-7). *P<0.05 compared with Sham, ***P<0.001 as compared to Sham, #P<0.05 as compared to 3 days. E) Left panels represent H&E staining of right uninjured (R) and left injured (L, INJ) carotids. Panel a, b, c, d, e, and f, double immunofluorescence staining of rat carotids, 14 days following balloon angioplasty injury. We used antibodies against CYGB (green) and CNN1. Blue staining is DAPI. A = adventitia, M = media, L = lumen. White dashed lines indicate the internal elastic lamina. Representative of 3 experiments.
Loss of cytoglobin prevents neointima formation in vivo
To investigate the effect of a loss of Cygb in the rat balloon injury model, an adenovirus encoding a short hairpin targeting Cygb was infused in the carotid lumen at the time of injury. This treatment resulted in a 65% decrease in CYGB expression over the non-injured right carotid, 7 days after endoluminal denudation (Figure 3A). At this time point and upon silencing of CYGB, neointimal area was decreased by 80% with no change in medial or luminal cross-sectional areas (Figure 3B and C).
Figure 3. Loss of Cytoglobin inhibits neointima formation in rodents.
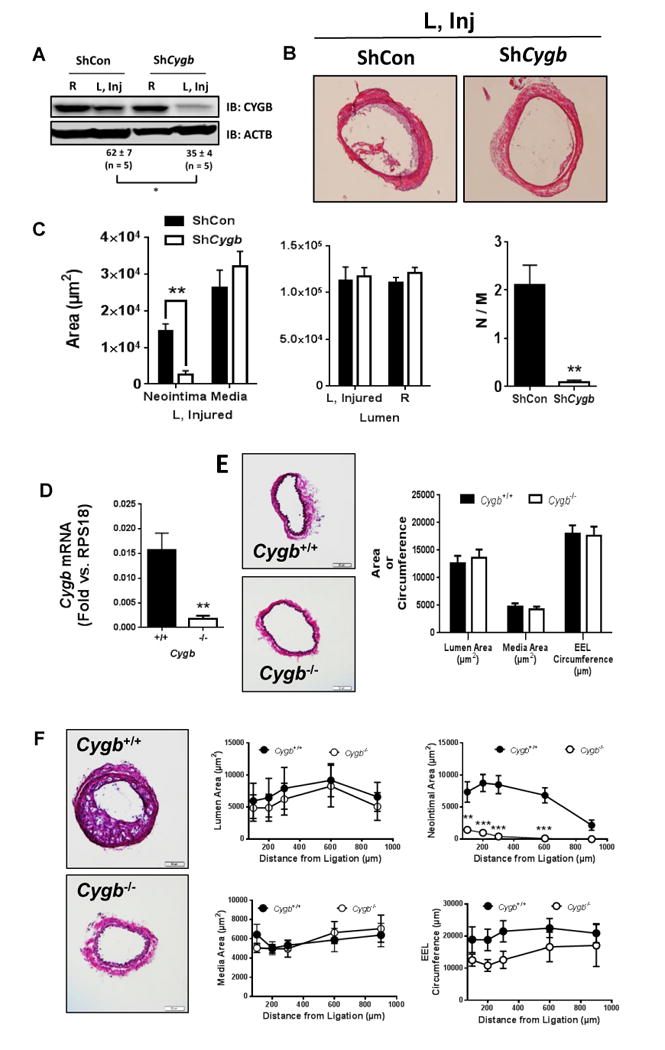
A) Western blot analysis showing the expression of CYGB in rat carotids from balloon-injured rat carotids treated with ShCon or ShCygb adenoviruses, 7 days post-injury and targeted adenoviral treatment. R indicates right uninjured carotid and L, Inj indicates left injured carotid. Values represent the mean ± SEM of the percentage change in CYGB immunoreactivity in left injured carotids relative to right non-injured carotids using ACTB as an internal reference; *P<0.05, n=5. B) representative H&E staining of left injured (L, Inj) carotids, 7 days after injury and targeted adenoviral delivery. Data shown is representative of 6 animals for each group. C) cross-sectional areas of the neointima, media, and lumen in rat carotids, 7 days after injury and targeted adenoviral delivery. Values shown represent the mean ± SEM (n=6). D) qRT-PCR showing relative mRNA levels for Cygb in mouse carotids of Cygb+/+ and Cygb-/- mice. Bars represent the Mean ± SEM (**P<0.01; n=5 and 6 for Cygb+/+ and Cygb-/- , respectively). E) Representative H&E staining of cross-sections from Cygb+/+ and Cygb-/- mouse carotids. Rigt panel, morphological characterization of results shown in left panel. Bars represent the Mean ± SEM (n = 5). There was no statistically significant differences between Cygb+/+ and Cygb-/- mice. F) Neointima formation was induced by carotid artery ligation in Cygb+/+ and Cygb-/- mouse littermates and analyzed 4 weeks after ligation; Left panels are representative images of H&E staining of cross sections obtained from Cygb+/+ and Cygb-/- mice and morphological characterization are shown on the right panels, including medial, neointimal, and luminal areas, as well as external elastic lamina (EEL) circumference. There was no difference between Cygb+/+ and Cygb-/- mice except for neointimal areas. Points represent Mean ± SEM (n = 7, **P<0.01; ***P<0.001 vs. Cygb+/+).
To evaluate a broader role for CYGB in vascular remodeling, we studied carotid artery sections from Cygb-/- and Cygb+/+ mice subjected to disturbed flow and increased shear stress as a result of carotid ligation. We first verified by qRT-PCR (Figure 3D) and Western blotting (Figure IV in the online-only Data Supplement) that CYGB was absent in Cygb-/- compared to Cygb+/+ mice vessels. There were no changes in the level of two smooth muscle cell markers (CNN1 and ACTA2, Figure IV in the online-only Data Supplement). We observed a significant decrease in systolic aortic internal diameter in the Cygb-/- mice (Figure V in the online-only Data Supplement) but all other morphological characteristics of aortas and carotids cardiac output, and heart rate were unchanged between genotypes (Figure 3E, Figure V in the online-only Data Supplement). Most significantly, four weeks after complete ligation of the left common carotid ligation, Cygb-/- mice displayed little to no evidence of neointimal hyperplasia in contrast to the Cygb+/+ mice (Figure 3F). No measurable differences in EEL circumference, luminal and medial area were observed (Figure 3F).
Suppression of CYGB protein expression increases medial cell apoptosis and caspase-3 activation
Loss of medial VSM cells is a characteristic feature of models of vascular injury that involve intraluminal endothelial denudation and distention of the vessel. In the case of the rat carotid injury model, the number of medial apoptotic cells peaks within a few hours and is rapidly superseded by increased VSM migration and proliferation that drive the hyperplastic response generally observed 22. We reasoned that if CYGB serves cytoprotective functions, adenoviral-mediated silencing of CYGB might prolong the onset of de-cellularization beyond the few hours usually observed post-injury in the rat carotid balloon angioplasty model. We found that enumeration of nucleus showed a decrease in medial cell density associated with silencing of CYGB, 7 days after balloon angioplasty (Figure VI in the online-only Data Supplement). At the four-day time point, protein levels of CYGB in the injured vessels with targeted delivery of shCYGB were approximately half of the levels found in the injured vessels with shCON (Figure 4A). Under these conditions, there was no change in cell proliferation in the injured vessels as indicated by proliferating cell nuclear antigen (PCNA) protein expression, whether CYGB was silenced or not (Figure 4B). In contrast, we found an increase in the abundance of terminal uridine nick-end labeling (TUNEL) in situ in the media, indicative of increased apoptosis (Figure 4C). Caspase-3 activation was dramatically increased upon CYGB silencing as shown by the approximately 6-fold increase in the ratio of cleaved-caspase to procaspase (Figure 4D, right panel). Cleavage of poly-ADP-ribose polymerase (PARP), a downstream target of caspase-3 activity was also evident (Figure 4E). All together, these results suggest that the loss of CYGB potentiates medial cell apoptosis through an increase in caspase-3 activation in the absence of an apparent decrease in cell proliferation.
Figure 4. Loss of Cytoglobin exacerbates apoptosis in the injured vessel.
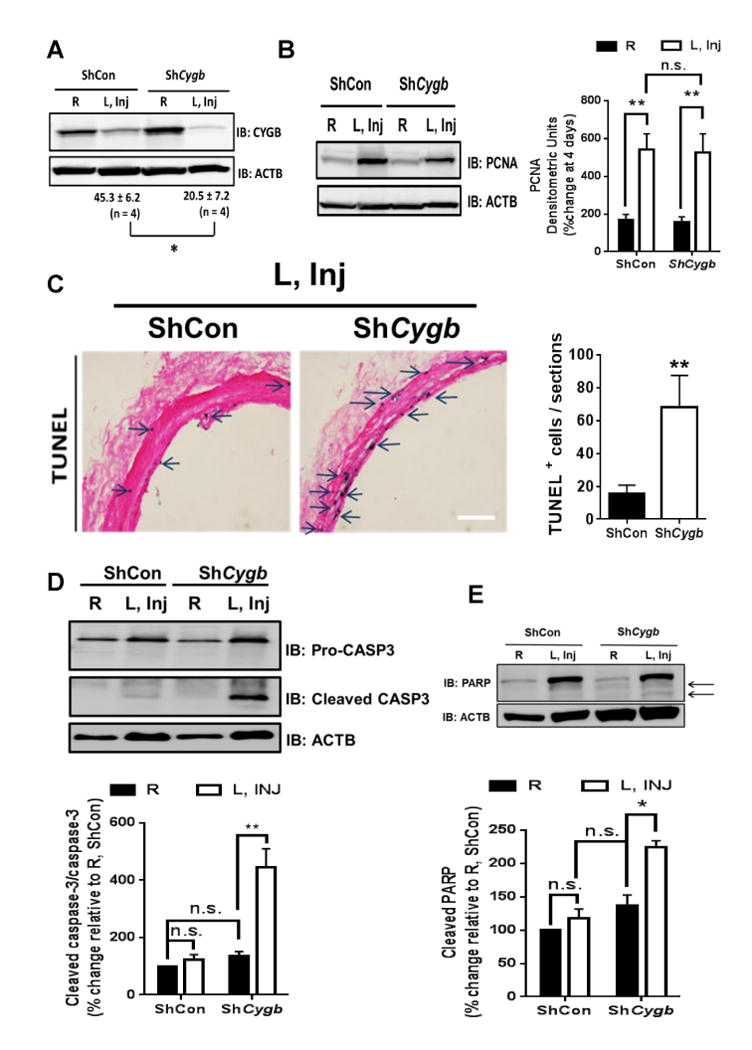
A, WB showing the expression of CYGB in rat carotids from balloon-injured rat carotids treated with ShCon or ShCygb adenoviruses 4 days post-injury and targeted adenoviral treatment; n=4, *P<0.05. B, WB analysis showing the expression of PCNA in rat carotids from balloon-injured rat carotids, 4 days post-injury and targeted adenoviral treatment. Right panel, n.s. = not statistically significant. C, Representative results from immunohistochemistry analysis of carotids harvested from conditions as described in (A). Sections were stained for TUNEL. Arrows indicate occurrence of purple staining indicative of TUNEL; Analysis is shown on the right panel; n=4, *P<0.05 compared to shCon . D, and E, WB analysis showing the expression of pro- and cleaved caspase-3 (Panel D, n=7), and PARP (Panel E, arrows indicate occurrence of cleaved PARP, n = 4) in rat carotids harvested from conditions as described in (A). Graphs shown below each WB represent the mean ± SEM values derived from the densitometric analysis of WB. *P<0.01, **P<0.01, as determined by single or paired sample t-test with Bonferroni correction.
Cytokine-mediated CYGB expression protects synthetic VSM cells from NOS2-dependent cytotoxicity
We next sought to establish whether CYGB effects on SMC death was independent of other cell types or other factors that may prevail in vivo. To this end, we first established conditions that may be conducive to CYGB re-expression in sub-cultured VSM cells. VSM cell differentiation is driven by myocardin (MYOCD), a co-activator for the transcription factor serum-responsive factor (SRF) that is required for the re-expression of many contractile markers in VSM cells 23. However, we found that ectopic expression of MYOCD in sub-cultured rat aortic VSM cells was unable to rescue CYGB expression in sub-cultured rat VSM cells despite increased expression of CNN1 and MYH11 (data not shown). Our results showing re-expression of CYGB within 3-7 days in the rat carotid injury model suggested that CYGB protein levels could alternatively increase in an environment rich in proliferative and inflammatory signals, independent of a strict association with differentiated VSM. Thus, we tested the effect of selected growth factors and mediators of innate immunity on CYGB expression. Cultured rat aortic VSM cells were incubated with concentrations of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and platelet-derived growth factor (PDGF) optimal for known response of VSM cells to these agents. We found that IL-1β and IFN-γ but not PDGF or TNF-α increased Cygb mRNA and protein levels (Figure VIIA and B in the online-only Data Supplement). Significantly, the combination of IL-1β or IFN-γ with hypoxia (1% O2) - a known inducer of CYGB expression16 – increased Cygb mRNA levels by almost 10-fold over the normoxic control (21% O2; Figure 5A), an effect that was mirrored at the protein level by an approximately 7-fold increase (Figure 5B).
Figure 5. Increase in cell death upon loss of CYGB is O2 and NO-dependent in rat aortic vascular smooth muscle.
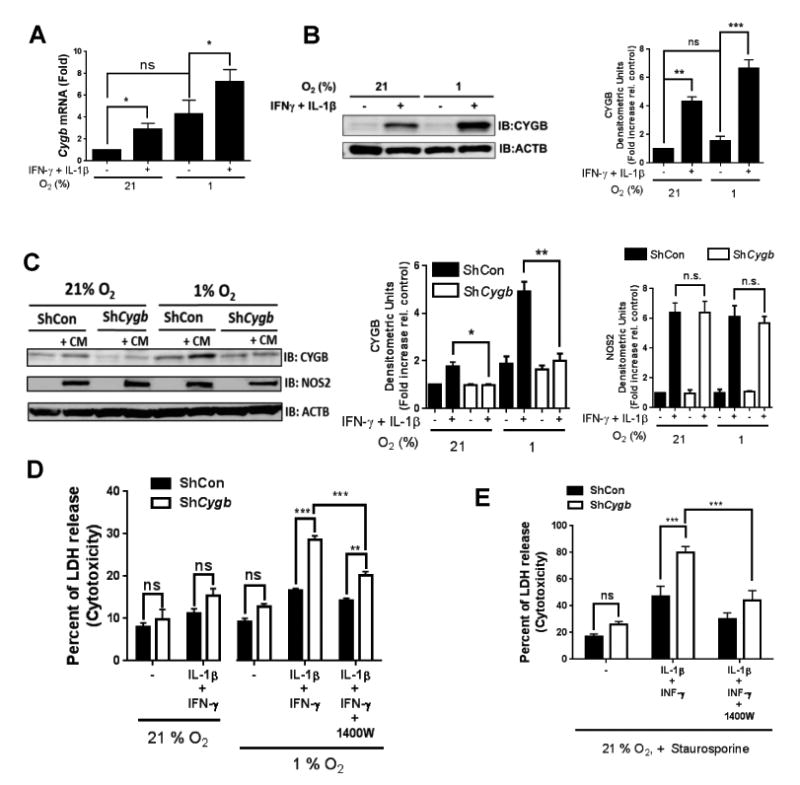
A) qRT-PCR results showing mRNA levels of CYGB in sub-cultured rat aortic vascular smooth muscle (RAVSM) cells. The cells were stimulated for 48 hours with interleukin-1β (IL-1β; 10 ng/ml) and interferon-γ (IFN-γ; 200 U/ml) at 21 or 1% O2. Mean ± SEM, n = 8. B) WB results showing protein levels of CYGB in sub-cultured RAVSM using the same conditions described in (A). Densitometric analysis is shown on the right panel. Mean ± SEM, n ≥ 6; for (A) and (B) *P <0.05, **P<0.01, and ***P<0.001, as determined by single or paired sample t-test with Bonferroni correction. C) Western blot results showing protein levels of CYGB and NOS2 in rat aortic VSM cells in the presence of a cytokine mix (CM, IL-1β + IFN-γ) with or without adenoviral silencing of CYGB.. Densitometric analysis is shown on the right panels. Mean ± SEM, n = 4;*P <0.05, **P<0.01, as determined by paired sample t-test with Bonferroni correction. D) Cells were stimulated with IL-1β and IFN-γ for 48 hours at 21 or 1% O2 with or without silencing of CYGB and cytotoxicity was determined by measuring LDH release. Mean ± SEM, n =3 E) cells were stimulated with IL-1β and IFN-γ for 48 hours at 21% O2 with or without silencing of CYGB and incubated with staurosporine for 24 hours before cytotoxicity was determined by measuring LDH release. Mean ± SEM, n =5. for (D) and (E) **P<0.01, and ***P<0.001, as determined by 2-way ANOVA.
We evaluated VSM cell death using the release of lactate dehydrogenase (LDH) in the cell culture media as an indicator of cell death, independent of specific pathways such as necrosis or apoptosis. We confirmed that treatment with adenoviral ShCygb effectively suppressed the expression of CYGB proteins after stimulation with IL-1β and IFN-γ, under normoxic and hypoxic conditions (Figure 5C). Decrease in CYGB content in VSM cells using this adenoviral strategy was not sufficient to increase LDH release above background levels at 21% O2, including after treatment with the cytokine mix (Figure 5D). When the same experiment was performed at 1% O2, decrease CYGB levels resulted in a statistically significant increase in cell cytotoxicity such that LDH release after cytokine treatment attained approximately 30% of maximum, from 15% in the controlled adenovirus treatment with cytokine mix (Figure 5D). Inflammatory cytokines including IL-1β and IFN-γ strongly increase NO production in rodents through the upregulation of NOS2 24 and we confirmed the upregulation of NOS2 by the cytokine mix (Figure 5C). We found that pre-treatment of the cells with the NOS2 specific inhibitor 1400W reversed the cytotoxic effect associated with decrease in CYGB content and cytokine treatment at 1% O2 (Figure 5D). Significantly, pre-treatment with IL-1β and IFN-γ under normoxic conditions strongly sensitized VSM cells to the ATP analog staurosporine (STS), a universal and potent activator of apoptosis. The effect of STS was potentiated through decrease CYGB expression and inhibited by pretreatment with 1400W (Figure 5E). We conclude that CYGB may be re-expressed in de-differentiated VSM cells upon cytokine and hypoxia exposure and in this case, the loss of CYGB is sufficient to sensitize cells to NO-dependent cytotoxicity.
Loss of CYGB induces autonomous and redox-sensitive apoptosis in human aortic VSM cells
Previous studies indicated that depletion of CYGB alone was sufficient to trigger cell death 12, an effect that was potentiated in response to oxidative stress. In contrast to rat aortic VSM cells - sub-cultured human aortic smooth muscle (HAoVSM) cells retained some levels of CYGB expression despite little to no expression of the smooth muscle cell markers MYH11 and LMOD1 (Figure 6A). HAoVSM cells were electroporated with CYGB specific small interfering RNAs resulting in an approximately 65% decrease in CYGB protein expression compared to controls. We confirmed the absence of NOS1 and 2 expression in these cells (Figure 6B) and found no change in proliferative capacity (Figure 6C) or cytototoxicity upon silencing of CYGB alone (Figure 6D).
Figure 6. CYGB inhibits staurosporine induced apoptosis in human aortic SMCs independent of cell proliferation.
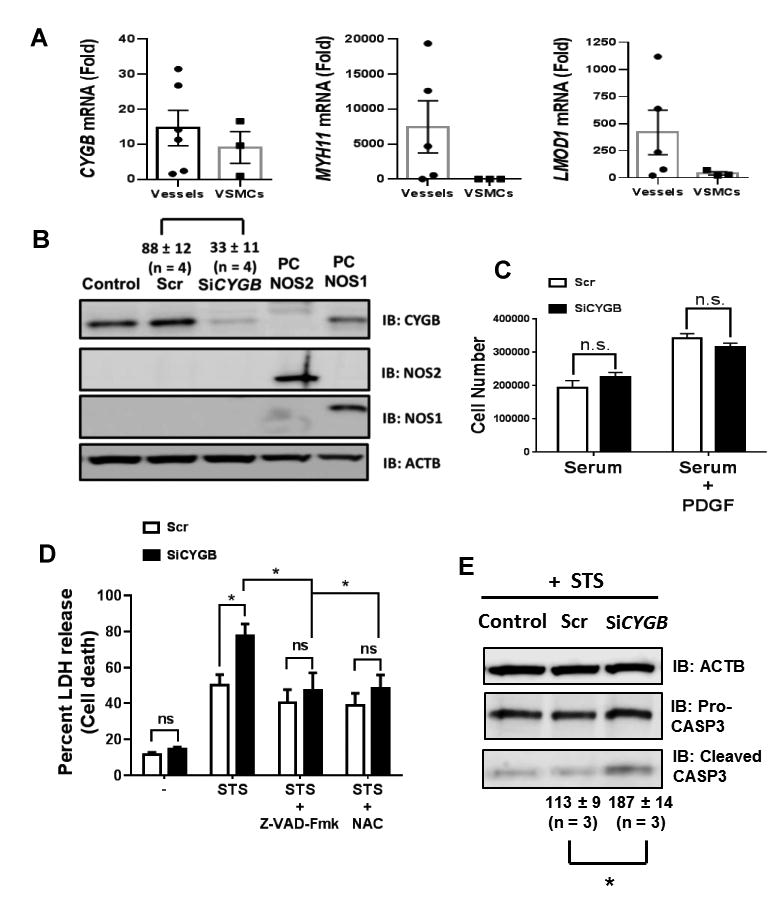
A) qRT-PCR results showing relative mRNA levels for CYGB, MYH11, and LMOD1 in six human aortas and sub-cultured human aortic smooth muscle cells (VSMCs) derived from 3 different dispersions. B) Sub-cultured human aortic SMCs were electroporated with siRNA mix targeting CYGB (SiCYGB) or scrambled siRNA (Scr). Western blots show protein levels of CYGB, NOS2, and NOS1. Values represent the mean ± SEM of the percentage change in CYGB immunoreactivity compared to non-transfected conditions (Control) using ACTB as an internal reference; n = 4, *P<0.05 C) Human Aortic VSMCs were incubated with serum or serum + PDGF for 3 days and cell count was determined. There was no difference between SiCYGB and Scr treated cells. Mean ± SEM with n = 4. D) Sub-cultured human aortic SMCs were treated with staurosporine (STS, 1 μM) for 24 hours in the presence or absence of the pan-caspase inhibitor (z-VAD-Fmk) or N-acetyl cysteine (NAC). Cytotoxicity was determined by measuring LDH release. Mean ± SEM, with n = 4-6. *P<0.05. E) Western blot analysis for Caspase-3 (Pro-CASP3) and cleaved CASP3 from human aortic SMCs incubated with staurosporine (1 μM for 6 hours) showing increased in cleaved caspase-3 upon silencing of CYGB. Values represent the mean ± SEM of the percentage change in cleaved CASP3 immunoreactivity in SiCYGB and Scr treated cells compared to non-transfected conditions(Control) using ACTB as an internal reference; n = 3, *P<0.05.
We examined the cytotoxic response of HAoVSM cells to hydrogen peroxide (H2O2) and staurosporine (STS) using final concentrations of 250 μM and 400 nM, respectively. These concentrations were close to their respective IC50, which were calculated from cytotoxicity dose-response curves (Figure VIII in the online-only Data Supplement). Surprisingly, there was no change in H2O2 mediated cytotoxicity, upon loss of CYGB (Figure IX in the online-only Data Supplement). However, CYGB silencing potentiated the cytotoxic effect of STS by approximately 30 percentage points (Figure 6D). The sensitization of VSM cells to the loss of CYGB was completely abrogated upon pre-treatment with the pan-caspase inhibitor z-VAD-fmk. There was also a fraction of STS-induced cytotoxicity, which was insensitive to z-VAD-fmk as previously shown in other systems25 (Figure 6D). Most importantly, over-sensitization to cell death following CYGB silencing was inhibited by the antioxidant N-acetyl cysteine (NAC) (Figure 6D) and occurrence of apoptosis was confirmed by showing that cleaved caspase-3 was increased upon silencing of CYGB and treatment with STS (Figure 6E). Finally, stable overexpression of human CYGB in HEK293 cells that lack endogenous CYGB suppressed STS-induced cytotoxicity (Figure X in the online-only Data Supplement). All together, these results indicated that directly modulating cellular levels of CYGB was sufficient to alter cytotoxicity in human VSM cells in a caspase and redox-dependent manner.
Discussion
The functions of hemoglobin (HB) and myoglobin (MB) in the cardiovascular system are relatively well understood and include O2 transport and storage, and NO and nitrite (NO2-) signaling1, 2, 21, 26, 27. Their expression is not restricted to erythrocytes (for HB) or cardiomyocytes and striated skeletal muscle cells (for MB) but also extends to endothelial and vascular smooth muscle (VSM) cells1, 2. The present study provides a hypothesis for how CYGB might contribute to vascular remodeling. Our results suggest that it regulates the hyperplastic response associated with vascular injury through modulation of apoptosis. We show that CYGB is abundantly expressed in mature VSM cells and lost after de-differentiation. Most significantly, CYGB protects human and rodent cultured VSM cells from programed cell death and loss of CYGB in vivo leads to medial cell loss and inhibition of neointima formation during vascular injury. Our results indicate that the absence of CYGB over-sensitizes medial VSM cells to caspase-3 activation and triggers inopportune apoptosis during injury.
Vascular smooth muscle (VSM) cells are the primary cell type in the tunica media. They are characterized by low turnover and primarily regulate tension through active contraction and relaxation. Under diseased conditions, mature medial VSM cells can undergo a phenotypic switch characterized by the downregulation of contractile genes, alteration in survival capacity, and increased proliferation, migration, extracellular matrix deposition, and cytokine production28, 29. Past studies have established that a large fraction of the proliferating cells populating the neointima in experimental models of restenosis or after atherosclerosis is derived from mature medial VSM cells30. More recently, it has been shown that VSM cells may give rise to a macrophage-like phenotype in atherosclerotic lesions31-33 and also may undergo reprogramming to produce a subpopulation of adventitial progenitor cells that contribute to vessel remodeling34. All of these studies indicate that VSM cells are critical contributors to the hyperplastic response associated with many types of vasculopathies. In the present study, we show that CYGB is expressed in mature (contractile) VSM cells in the media. In addition to rodent vessels, we found that CYGB was expressed in human arteries in amounts that always exceeded those of MB. Our results also indicate that CYGB expression may increase in human veins derived from revised arteriovenous fistula (Figure I). In these vessels, CYGB would seem primarily expressed in the media and to a lesser extent in the neointima (Figure III in the Online-only Data Supplement). Because these vessels have undergone maturation before secondary failure, these results would suggest that increase CYGB expression might be associated with arterialization of veins. This will need to be explored further.
In the rat carotid injury model, CYGB protein levels are decreased acutely but slowly recovered such that two weeks after surgery, it was primarily associated with CNN1+ cells in outer medial layers (Figure 2E). We also found that CYGB expression was lost in vitro upon de-differentiation of freshly isolated rat aortic VSM cells coincident with contractile VSM markers. An important driver of VSM contractile marker gene expression such as CNN1 is myocardin (MYOCD), a myogenic transcriptional co-activator that acts through stabilization of the binding of serum response factor (SRF) to CArG elements within the regulatory regions of most contractile VSM cell marker genes23. However, we found no evidence that CYGB expression may be directly or indirectly dependent on MYOCD based on forced expression of MYOCD in cultured rat aortic VSM cells in vitro (not shown). Our in vitro studies suggest that CYGB may be re-expressed in de-differentiated VSM cells exposed to inflammatory cytokines. To our knowledge, the stimulatory effect of IL-1β and IFN- γ on CYGB expression and synergism with hypoxia has not been documented previously. While we have not examined the specifics underlying this effect, our results indicate that some of the regulation occurs at the transcriptional level. It is noteworthy that the promoter region of CYGB contains binding elements recognizing the transcription factors AP-1, NFAT, and HIF1-α, all of which could be directly implicated with the IL-1β and IFN- γ mediated upregulation of CYGB in VSM cells16. Synergism of inflammatory cytokines in regulating gene expression is also well-documented in VSM cells24, 35. We found that the loss of CYGB under these conditions sensitized rat aortic VSM cells to NOS2-dependent cytotoxicity (Figure 5). Whether the hypoxic conditions used for some of these experiments can be directly extrapolated to the in vivo models we have tested is questionable. However, they might reflect conditions associated with other vascular pathologies such as atherosclerosis, and provide direct evidence for a role for CYGB in regulating VSM cell death.
The significance of CYGB expression in the vasculature has been recently associated with the regulation of NO bio-availability and its NO dioxygenase activity18. In the liver, CYGB is preferentially expressed in stellate cells and confers cytoprotection in multiple liver injury models including carbon tetrachloride administration36, hepatosteatosis 37, and acetaminophen-induced hepatotoxicity 38. Silencing of CYGB in myoblasts is sufficient to induce apoptosis and potentiated the pro-apoptotic effects of hypoxia and oxidants 12. Modulation of CYGB levels in neonatal rat brains through adenoviral delivery strategies also regulated caspase levels and activation in response to ischemia reperfusion 39. Our results are reminiscent of the aforementioned study, as indicated by a strong association between the loss of CYGB and caspase-3 activation (Figure 4D). One possibility is a direct effect of CYGB on caspase-3 through redox or nitrosation/nitrosylation-based mechansims40, 41. We found that decreasing levels of CYGB was not sufficient to over-sensitize HuAoVSM cells to H2O2; however, the increase in cytotoxicity to STS associated with loss of CYGB was inhibited with the antioxidant N-acetyl cysteine (NAC; Figure 6D). These results suggested that – while CYGB may not exert direct antioxidant effects – the signaling network associated with its function is redox sensitive. We also found that in cultured rat aortic VSM cells the sensitization to CYGB silencing was dependent upon NOS2 activity, further reinforcing the concept of one or several redox dependent steps that are perturbed upon manipulation of CYGB levels.
The occurrence of VSM apoptosis is well established in flow-induced and balloon angioplasty models as well as in aortic aneurysms42, 43 and atherosclerotic plaques 44. Here, we show that the loss of CYGB in vitro and in vivo is associated with an increase in apoptosis and that ectopic expression of CYGB in cultured cells is sufficient to inhibit staurosporine-mediated cell death. Vascular remodeling after injury is a dynamic process that involves programed cell death, as well as cell migration, and proliferation. We found no evidence that decreasing CYGB alters VSM proliferation and migration and propose that increase VSM apoptosis is the major determinant of the inhibitory effect of CYGB silencing on neointima formation. Apoptosis has been documented in the rabbit and rat as early as 30 min after arterial injury followed primarily by migration and proliferation of resident cells 22, 45. It is unlikely however that site targeted adenoviral delivery of shCYGB affects the first few hours of the injury where VSM apoptosis is most prominent because adenoviral-mediated silencing requires 2-3 days to be effective. Instead, our results suggest that silencing of CYGB might limit the survival capacity of a pre-existing cell population that still expresses CYGB at the 2-3 day time point. Alternatively, adenoviral-mediated silencing of CYGB could prevent the re-expression of cytoprotective CYGB in new populations of VSM cells that re-populate the media and contribute to the expansion of the neointima. Our findings are consistent with previous studies showing that decrease in the expression of a number of genes that normally inhibit apoptosis is conducive to medial VSM cell death and is also associated with the reduction of neointimal hyperplasia 46-48. In contrast, in flow-induced remodeling, strategies that increase VSM apoptosis after initiation of remodeling promote rather than inhibit neointimal formation 49. Although additional investigations are warranted including additional experimental models, our results showing that Cygb knockout mice developed little to no neointimal lesions after carotid ligation suggest important functions for CYGB during proliferative vasculopathies, beyond the rat balloon angioplasty model.
Why VSM would maintain high levels of CYGB in intact vessels? Cytoglobin could contribute to the tight regulation of programed cell death in healthy vessels to avoid the loss of contractile cells with a low proliferation rate and turnover. In contrast, the vascular response to injury requires high cell plasticity where proliferation, migration, and programed cell death need to maximize repair. In this case, regulatory pathways that couple cell proliferation and survival such as activation of survivin might be better suited early on during the repair process47. It is still possible that CYGB has functions unrelated to apoptosis that necessitate high levels of expression in mature VSM cells. For example, globins regulate nitrite signaling through their nitrite reductase activity2, 50. Liu et al. recently provided some evidence that in intact vessels, CYGB is a sink for NO and impact vascular tone18. Alternatively, we would like to propose that CYGB might represent an important O2, NO, and nitrite sensor to regulate VSM function. The present study reveals a role for CYGB in vascular remodeling and suggests possible regulatory roles in vascular proliferative syndromes associated with programed cell death such as vascular injury, atherosclerosis, and aneurysm.
Supplementary Material
Highlights.
Cytoglobin is expressed in human and rodent vessels, primarily in differentiated medial smooth muscle cells.
Cytoglobin expression is lost upon de-differentiation of smooth muscle cells and re-expressed in vitro upon hypoxia and cytokine stimulation.
Loss of cytoglobin inhibits neointima formation in two distinct models of vascular injury in rodents.
Cytoglobin inhibits smooth muscle cell apoptosis in vivo and in vitro, through redox and nitric oxide-dependent mechanisms.
Acknowledgments
None
Sources of funding: This study was supported by AHA Grant #16GRNT31280002 (to D.J.), Dialysis Clinic, Inc. Paul Teschan Research Fund (to A. A. and H.A.S., and to X. L.), and DCI Reserve Fund Project grant #C-3804 (A.A. and H.A.S.), RO1HL49426 (H.A.S.), and RO1HL122686 (X. L.).
Non standard abbreviations and acronyms
- AP-1
activator protein-1
- AVF
arteriovenous fistula
- HAoVSM
human aortic vascular smooth muscle
- HIF-1
hypoxia inducible factor-1
- IL-1β
Interleukin-1β
- IFN-γ
Interferon- γ
- LDH
lactate dehydrogenase
- NFAT
nuclear factor of activated T cell
- RAVSM
rat aortic vascular smooth muscle
- RT-PCR
reverse transcriptase polymerase chain reaction
- siRNA
small interfering RNA
- STS
staurosporine
- TNF-α
tumor necrosis α
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- TUNEL
terminal uridine nick-end labeling
- VSM
vascular smooth muscle
- WB
Western blot
Footnotes
Disclosures: None.
References
- 1.Straub AC, Lohman AW, Billaud M, Johnstone SR, Dwyer ST, Lee MY, Bortz PS, Best AK, Columbus L, Gaston B, Isakson BE. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature. 2012;491:473–7. doi: 10.1038/nature11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Totzeck M, Hendgen-Cotta UB, Luedike P, et al. Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation. 2012;126:325–34. doi: 10.1161/CIRCULATIONAHA.111.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 4.Burmester T, Ebner B, Weich B, Hankeln T. Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Molecular biology and evolution. 2002;19:416–21. doi: 10.1093/oxfordjournals.molbev.a004096. [DOI] [PubMed] [Google Scholar]
- 5.Kawada N, Kristensen DB, Asahina K, Nakatani K, Minamiyama Y, Seki S, Yoshizato K. Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. J Biol Chem. 2001;276:25318–23. doi: 10.1074/jbc.M102630200. [DOI] [PubMed] [Google Scholar]
- 6.Trent JT, 3rd, Hargrove MS. A ubiquitously expressed human hexacoordinate hemoglobin. J Biol Chem. 2002;277:19538–45. doi: 10.1074/jbc.M201934200. [DOI] [PubMed] [Google Scholar]
- 7.Hoogewijs D, Ebner B, Germani F, Hoffmann FG, Fabrizius A, Moens L, Burmester T, Dewilde S, Storz JF, Vinogradov SN, Hankeln T. Androglobin: a chimeric globin in metazoans that is preferentially expressed in Mammalian testes. Molecular biology and evolution. 2012;29:1105–14. doi: 10.1093/molbev/msr246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hankeln T, Ebner B, Fuchs C, et al. Neuroglobin and cytoglobin in search of their role in the vertebrate globin family. J Inorg Biochem. 2005;99:110–9. doi: 10.1016/j.jinorgbio.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Burmester T, Hankeln T. Function and evolution of vertebrate globins. Acta Physiol (Oxf) 2014;211:501–14. doi: 10.1111/apha.12312. [DOI] [PubMed] [Google Scholar]
- 10.Mimura I, Nangaku M, Nishi H, Inagi R, Tanaka T, Fujita T. Cytoglobin, a novel globin, plays an antifibrotic role in the kidney. Am J Physiol Renal Physiol. 2010;299:F1120–33. doi: 10.1152/ajprenal.00145.2010. [DOI] [PubMed] [Google Scholar]
- 11.Shaw RJ, Omar MM, Rokadiya S, Kogera FA, Lowe D, Hall GL, Woolgar JA, Homer J, Liloglou T, Field JK, Risk JM. Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. Br J Cancer. 2009;101:139–44. doi: 10.1038/sj.bjc.6605121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh S, Canseco DC, Manda SM, Shelton JM, Chirumamilla RR, Goetsch SC, Ye Q, Gerard RD, Schneider JW, Richardson JA, Rothermel BA, Mammen PP. Cytoglobin modulates myogenic progenitor cell viability and muscle regeneration. Proc Natl Acad Sci U S A. 2014;111:E129–38. doi: 10.1073/pnas.1314962111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fordel E, Thijs L, Martinet W, Schrijvers D, Moens L, Dewilde S. Anoxia or oxygen and glucose deprivation in SH-SY5Y cells: a step closer to the unraveling of neuroglobin and cytoglobin functions. Gene. 2007;398:114–22. doi: 10.1016/j.gene.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 14.Fordel E, Thijs L, Martinet W, Lenjou M, Laufs T, Van Bockstaele D, Moens L, Dewilde S. Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci Lett. 2006;410:146–51. doi: 10.1016/j.neulet.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 15.Latina A, Viticchie G, Lena AM, Piro MC, Annicchiarico-Petruzzelli M, Melino G, Candi E. DeltaNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene. 2016;35:1493–503. doi: 10.1038/onc.2015.222. [DOI] [PubMed] [Google Scholar]
- 16.Singh S, Manda SM, Sikder D, Birrer MJ, Rothermel BA, Garry DJ, Mammen PP. Calcineurin activates cytoglobin transcription in hypoxic myocytes. J Biol Chem. 2009;284:10409–21. doi: 10.1074/jbc.M809572200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halligan KE, Jourd'heuil FL, Jourd'heuil D. Cytoglobin is expressed in the vasculature and regulates cell respiration and proliferation via nitric oxide dioxygenation. J Biol Chem. 2009;284:8539–47. doi: 10.1074/jbc.M808231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, El-Mahdy MA, Boslett J, Varadharaj S, Hemann C, Abdelghany TM, Ismail RS, Little SC, Zhou D, Thuy LT, Kawada N, Zweier JL. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nat Commun. 2017;8:14807. doi: 10.1038/ncomms14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Thuy TT, Thuy LT, Yoshizato K, Kawada N. Possible Involvement of Nitric Oxide in Enhanced Liver Injury and Fibrogenesis during Cholestasis in Cytoglobin-deficient Mice. Sci Rep. 2017;7:41888. doi: 10.1038/srep41888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thuy le TT, Morita T, Yoshida K, Wakasa K, Iizuka M, Ogawa T, Mori M, Sekiya Y, Momen S, Motoyama H, Ikeda K, Yoshizato K, Kawada N. Promotion of liver and lung tumorigenesis in DEN-treated cytoglobin-deficient mice. The American journal of pathology. 2011;179:1050–60. doi: 10.1016/j.ajpath.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu Y, Sutton L, Riggs AF. Identification of myoglobin in human smooth muscle. J Biol Chem. 1998;273:23426–32. doi: 10.1074/jbc.273.36.23426. [DOI] [PubMed] [Google Scholar]
- 22.Perlman H, Maillard L, Krasinski K, Walsh K. Evidence for the rapid onset of apoptosis in medial smooth muscle cells after balloon injury. Circulation. 1997;95:981–7. doi: 10.1161/01.cir.95.4.981. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–56. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Chen X, Teng X, Snead C, Catravas JD. Molecular cloning and analysis of the rat inducible nitric oxide synthase gene promoter in aortic smooth muscle cells. Biochem Pharmacol. 1998;55:1873–80. doi: 10.1016/s0006-2952(98)00078-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhang XD, Gillespie SK, Hersey P. Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol Cancer Ther. 2004;3:187–97. [PubMed] [Google Scholar]
- 26.Hendgen-Cotta UB, Kelm M, Rassaf T. Myoglobin functions in the heart. Free radical biology & medicine. 2014;73:252–9. doi: 10.1016/j.freeradbiomed.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Gladwin MT. How Red Blood Cells Process Nitric Oxide: Evidence for the Nitrite Hypothesis. Circulation. 2017;135:177–179. doi: 10.1161/CIRCULATIONAHA.116.024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 29.Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S, Weiser-Evans MC. SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol. 2011;31:1300–8. doi: 10.1161/ATVBAHA.111.223701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, Feil R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014;115:662–7. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 33.Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM, Fisher EA. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. 2015;35:535–46. doi: 10.1161/ATVBAHA.114.304029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi AK, Dong XR, Poczobutt JM, Nemenoff RA, Weiser-Evans MC. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by KLF4. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.116.309322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CN, Li YS, Yeh YT, Lee PL, Usami S, Chien S, Chiu JJ. Synergistic roles of platelet-derived growth factor-BB and interleukin-1beta in phenotypic modulation of human aortic smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:2665–70. doi: 10.1073/pnas.0510973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu R, Harrison PM, Chen M, Li L, Tsui TY, Fung PC, Cheung PT, Wang G, Li H, Diao Y, Krissansen GW, Xu S, Farzaneh F. Cytoglobin overexpression protects against damage-induced fibrosis. Mol Ther. 2006;13:1093–100. doi: 10.1016/j.ymthe.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 37.Thuy le TT, Matsumoto Y, Thuy TT, Hai H, Suoh M, Urahara Y, Motoyama H, Fujii H, Tamori A, Kubo S, Takemura S, Morita T, Yoshizato K, Kawada N. Cytoglobin deficiency promotes liver cancer development from hepatosteatosis through activation of the oxidative stress pathway. The American journal of pathology. 2015;185:1045–60. doi: 10.1016/j.ajpath.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 38.Teranishi Y, Matsubara T, Krausz KW, Le TT, Gonzalez FJ, Yoshizato K, Ikeda K, Kawada N. Involvement of hepatic stellate cell cytoglobin in acute hepatocyte damage through the regulation of CYP2E1-mediated xenobiotic metabolism. Lab Invest. 2015;95:515–24. doi: 10.1038/labinvest.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian SF, Yang HH, Xiao DP, Huang YJ, He GY, Ma HR, Xia F, Shi XC. Mechanisms of neuroprotection from hypoxia-ischemia (HI) brain injury by up-regulation of cytoglobin (CYGB) in a neonatal rat model. J Biol Chem. 2013;288:15988–6003. doi: 10.1074/jbc.M112.428789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan S, Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–9. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 41.Lai YC, Pan KT, Chang GF, Hsu CH, Khoo KH, Hung CH, Jiang YJ, Ho FM, Meng TC. Nitrite-mediated S-nitrosylation of caspase-3 prevents hypoxia-induced endothelial barrier dysfunction. Circ Res. 2011;109:1375–86. doi: 10.1161/CIRCRESAHA.111.256479. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. The American journal of pathology. 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 43.Nataatmadja M, West M, West J, Summers K, Walker P, Nagata M, Watanabe T. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation. 2003;108(Suppl 1):II329–34. doi: 10.1161/01.cir.0000087660.82721.15. [DOI] [PubMed] [Google Scholar]
- 44.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–80. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 45.Malik N, Francis SE, Holt CM, Gunn J, Thomas GL, Shepherd L, Chamberlain J, Newman CM, Cumberland DC, Crossman DC. Apoptosis and cell proliferation after porcine coronary angioplasty. Circulation. 1998;98:1657–65. doi: 10.1161/01.cir.98.16.1657. [DOI] [PubMed] [Google Scholar]
- 46.Leeper NJ, Raiesdana A, Kojima Y, et al. Loss of CDKN2B promotes p53-dependent smooth muscle cell apoptosis and aneurysm formation. Arterioscler Thromb Vasc Biol. 2013;33:e1–e10. doi: 10.1161/ATVBAHA.112.300399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanc-Brude OP, Yu J, Simosa H, Conte MS, Sessa WC, Altieri DC. Inhibitor of apoptosis protein survivin regulates vascular injury. Nat Med. 2002;8:987–94. doi: 10.1038/nm750. [DOI] [PubMed] [Google Scholar]
- 48.Pollman MJ, Hall JL, Mann MJ, Zhang L, Gibbons GH. Inhibition of neointimal cell bcl-x expression induces apoptosis and regression of vascular disease. Nat Med. 1998;4:222–7. doi: 10.1038/nm0298-222. [DOI] [PubMed] [Google Scholar]
- 49.Yu H, Clarke MC, Figg N, Littlewood TD, Bennett MR. Smooth muscle cell apoptosis promotes vessel remodeling and repair via activation of cell migration, proliferation, and collagen synthesis. Arterioscler Thromb Vasc Biol. 2011;31:2402–9. doi: 10.1161/ATVBAHA.111.235622. [DOI] [PubMed] [Google Scholar]
- 50.Alef MJ, Vallabhaneni R, Carchman E, Morris SM, Jr, Shiva S, Wang Y, Kelley EE, Tarpey MM, Gladwin MT, Tzeng E, Zuckerbraun BS. Nitrite-generated NO circumvents dysregulated arginine/NOS signaling to protect against intimal hyperplasia in Sprague-Dawley rats. J Clin Invest. 2011;121:1646–56. doi: 10.1172/JCI44079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.