

Findings reveal a novel basis for protecting CNS neurons against Aβ oligomers (AβOs), neurotoxins believed to instigate neural damage leading to Alzheimer’s dementia. Results with spatially separated cocultures of astrocytes and hippocampal neurons show an exosome-like mechanism by which insulin/IGF1 from astrocytes clear bound AβOs from neuronal surfaces.
Abstract
Synaptopathy underlying memory deficits in Alzheimer’s disease (AD) is increasingly thought to be instigated by toxic oligomers of the amyloid beta peptide (AβOs). Given the long latency and incomplete penetrance of AD dementia with respect to Aβ pathology, we hypothesized that factors present in the CNS may physiologically protect neurons from the deleterious impact of AβOs. Here we employed physically separated neuron–astrocyte cocultures to investigate potential non–cell autonomous neuroprotective factors influencing AβO toxicity. Neurons cultivated in the absence of an astrocyte feeder layer showed abundant AβO binding to dendritic processes and associated synapse deterioration. In contrast, neurons in the presence of astrocytes showed markedly reduced AβO binding and synaptopathy. Results identified the protective factors released by astrocytes as insulin and insulin-like growth factor-1 (IGF1). The protective mechanism involved release of newly bound AβOs into the extracellular medium dependent upon trafficking that was sensitive to exosome pathway inhibitors. Delaying insulin treatment led to AβO binding that was no longer releasable. The neuroprotective potential of astrocytes was itself sensitive to chronic AβO exposure, which reduced insulin/IGF1 expression. Our findings support the idea that physiological protection against synaptotoxic AβOs can be mediated by astrocyte-derived insulin/IGF1, but that this protection itself is vulnerable to AβO buildup.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia in the elderly (Alzheimer’s Association, 2013). Dementia correlates with synapse loss (e.g., Terry et al., 1991), and recent efforts to understand the mechanisms of synapse deterioration in AD have focused on the toxic impact of Aβ oligomers (AβOs; Ferreira and Klein, 2011; Mucke and Selkoe, 2012). AβOs are soluble, synaptotoxic Aβ assemblies that begin to accumulate before amyloid plaques (Hsia et al., 1999; Oddo et al., 2006). AβO distribution in AD brain can be distinct from amyloid plaques but still shows a diffuse, plaque-like appearance, attributable to association with dendritic arbors (Kayed et al., 2003; Lacor et al., 2007; Koffie et al., 2009). AβOs attach to synapses, inhibit synaptic plasticity, disrupt synaptic cytoskeletal proteins and receptor trafficking, and ultimately lead to synapse loss (Lambert et al., 1998; Walsh et al., 2002; Lacor et al., 2004, 2007; Koffie et al., 2009; Tomiyama et al., 2010; Zempel et al., 2010; Figueiredo et al., 2013).
Because AβOs are increasingly thought to instigate dementia, there is interest in identifying factors that protect neurons against their toxicity. One possibility is that protection is provided in healthy brain by endogenous cellular mechanisms. This idea is consistent with the fact that dementia requires decades to develop, despite the rapid formation of AβOs at very low Aβ concentrations (Chang et al., 2003; Velasco et al., 2012), and also with the fact that Aβ buildup in the brain precedes cognitive deficits by many years (Jack et al., 2013). Regulation of Aβ levels by glia (Koenigsknecht and Landreth, 2004; Yin et al., 2006) and its clearance by the glymphatic system (Peng et al., 2016) represent likely protective mechanisms. Another mechanism has been suggested in which neurons are protected against AβO-induced damage through activation of insulin/insulin-like growth factor 1 (IGF1) signaling pathways (De Felice et al., 2009; Zhao et al., 2009; Bomfim et al., 2012; Pitt et al., 2013). The latter possibility has provided support for ongoing clinical trials of intranasal insulin to treat early-AD patients (Craft et al., 2012). Defective insulin signaling is a risk factor for AD (Ott et al., 1999; Launer, 2005), and evidence from animal models indicates that insulin deficiency in the CNS promotes AβO formation and AD-type tau phosphorylation (Grunblatt et al., 2007; Bitel et al., 2012). Despite the putative relevance of insulin in protecting against AD progression, how insulin/IGF1 signaling prevents AβO-induced neuronal dysfunction and the source of neurotrophic insulin/IGF1 remain unknown.
In the current work, we investigated whether astrocytes are a source of factors that protect neurons against AβO synaptotoxicity, and the mechanisms that underlie their protective action. We focused on astrocytes because of their well-established trophic functions (Chernausek, 1993; Grunblatt et al., 2007; Eroglu and Barres, 2010; Diniz et al., 2014). To determine whether astrocytes impact AβO toxicity, we used physically isolated neuron–astrocyte cocultures, which allow convenient separation of the two cell types while allowing free diffusion of soluble factors between them (Kaech and Banker, 2006). We found that astrocytes greatly reduce AβO toxicity to hippocampal neurons without affecting AβO levels. To accomplish this, astrocytes secrete insulin and IGF1, which act on neurons to prevent synapse deterioration by stimulating release of newly bound AβOs. Results show that neurons are resistant to AβOs under conditions of healthy insulin/IGF1 signaling and suggest that robust chemical cross-talk between astrocytes and neurons may contribute to delaying AD progression.
RESULTS
Astrocytes increase neuronal resistance to AβO synaptotoxicity
We first used physically isolated hippocampal neuron–astrocyte cocultures (Figure 1A) to test whether the synaptotoxic impact of AβOs on hippocampal neurons was altered in the presence of astrocytes. Toxicity was assessed by the decrease in immunoreactivity of spinophilin, an actin-binding protein enriched at dendritic spines (Feng et al., 2000) and used as a proxy of spine integrity, following exposure of cultures to AβOs (500 nM, 24 h). In the absence of AβOs, neurons exhibited 0.57 ± 0.04 spinophilin-immunoreactive puncta per micrometer of dendritic segment, and this was unaffected (0.59 ± 0.04) by separation from the astrocyte feeder layer for 24 h (Supplemental Figure 1). In the absence of astrocytes, neurons exposed to AβOs exhibited 0.33 ± 0.03 spinophilin puncta per micrometer, a 44% decrease compared with control neurons (Figure 1). In contrast, no impact on dendritic spines was observed when neurons were exposed to AβOs in the presence of a physically isolated astrocyte feeder layer (Figure 1B).
FIGURE 1:

Astrocytes prevent AβO-induced spine loss and reduce dendritic AβO accumulation. (A) Hippocampal neurons were grown on coverslips above astrocyte feeder layers using drops of paraffin wax as spacers. Coverslips were either maintained above feeder layers or moved to astrocyte-free dishes, and AβOs were added. Spine loss and AβO binding were measured by immunocytochemistry. (B) When neurons were isolated from their astrocyte feeder layer (−Ac), spinophilin levels (green) along neurites (TuJ, red) were reduced by 44 ± 4% after treatment with 500 nM AβOs for 24 h. When astrocytes were present (+Ac), spinophilin levels were unaffected by addition of AβOs. (C) Under similar conditions of AβO treatment, neurons separated from their astrocyte feeder layer had prominent AβO labeling (NU4, green) along their neurites (TuJ, red), but the presence of astrocytes reduced AβO accumulation by 57 ± 4%. *, p < 0.0001, Mann-Whitney. Scale bars: 10 μm.
Consistent with their protective action against spine loss, astrocytes prevented the dendritic buildup of AβOs. In the absence of astrocytes, neurons exposed to AβOs (500 nM, 24 h) showed prominent AβO immunoreactivity (detectable by the AβO-sensitive antibody NU4) along dendrites (Figure 1C). The presence of an astrocyte feeder layer during exposure to AβOs significantly reduced AβO immunoreactivity (a decrease of 57 ± 4%; Figure 1C). These results showed that astrocytes prevent the accumulation and toxic impact of AβOs at synapses.
Because astrocytes actively clear molecules from the synaptic cleft, we asked whether their protective action might involve AβO clearance. We investigated this by using purified astrocyte cultures with no detectable levels of neuronal or microglial markers (Supplemental Figure 2). Intracellular levels of AβOs in astrocyte lysates were measured by a dot immunoblot assay using the AβO-sensitive antibody NU4. Under control conditions, total astrocyte lysates showed a low level of background labeling by NU4 (Figure 2, A and B). Exposure to 5 or 50 nM AβOs did not produce measurable changes in NU4 immunoreactivity, but exposure to 500 nM AβOs significantly increased NU4 immunoreactivity in astrocyte lysates (Figure 2A). The increase in NU4 immunoreactivity was time dependent (Figure 2B), consistent with oligomer uptake by astrocytes. However, analysis of the total AβO concentration remaining in the culture medium showed no significant decrease upon incubation with astrocytes (Figure 2C). This indicates that uptake by astrocytes removed only a very small fraction of AβOs from the medium, suggesting that depletion of oligomers from the medium cannot explain the marked neuroprotection by astrocytes described earlier.
FIGURE 2:
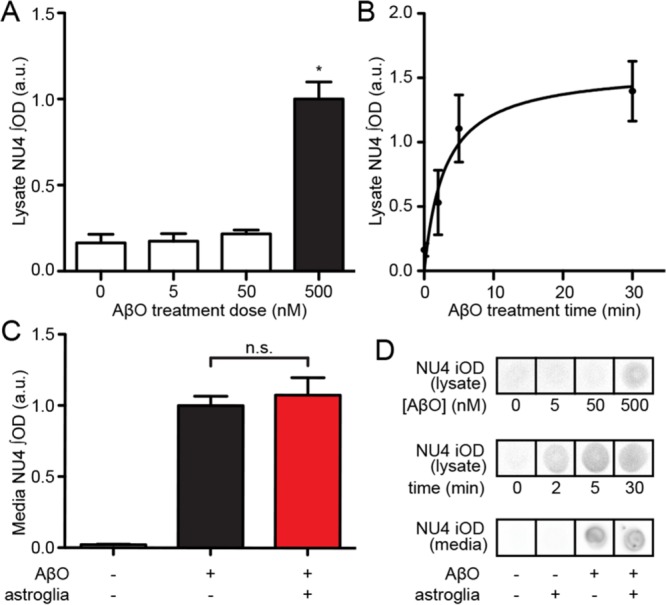
Protection by astrocytes is not due to removal of AβOs from media. (A) Dot blots of astrocyte lysates showed readily detectable oligomer immunoreactivity when obtained from cultures incubated with 500 nM AβOs. (B) Uptake is time dependent and plateaus at ∼5 min. (C) AβO levels in media (500 nM) were unchanged by the presence of astrocyte feeder layers over a 30-min period (red bar; astrocyte-free control conditions, black bar). MEM culture media contained 500 nM AβOs. *, p < 0.05, Mann-Whitney; n.s., p = 0.30, unpaired t test. (D) Examples of dot immunoblot signals quantified in A–C.
Astrocyte-secreted factors stimulate the release of AβOs previously bound to neurons
We hypothesized that soluble factors secreted by astrocytes might eliminate sites to which oligomers bind or, alternatively, stimulate detachment of oligomers from their binding sites on the neuronal surface. In support of the latter alternative, experiments with live neurons showed that astrocyte-derived factors stimulated the release of AβOs subsequent to their attachment to neurons. Released AβOs were detected in the culture medium by a dot immunoblot assay (Figure 3A), which offered three important advantages: 1) speed, important for detection of metastable assemblies such as AβOs; 2) high sensitivity, detecting AβOs at concentrations as low as 1 nM; and 3) detection of AβOs under nondenaturing conditions, unlike in Western blots following SDS–PAGE (Hepler et al., 2006). Coverslips with attached hippocampal neuronal cultures were transferred to fresh MEM containing 500 nM AβOs. After 15 min to allow AβO binding to neurons, coverslips were dip-rinsed in MEM and transferred to either astrocyte-conditioned MEM (ACM) or basal MEM (Figure 3A). Neurons placed in basal MEM released a small but detectable amount of AβOs to the medium. However, when neurons were incubated with ACM, the release of AβOs to the medium was increased at least 10-fold (Figure 3B). We thus concluded that astrocyte-secreted factors caused release of AβOs that had been previously attached to neurons.
FIGURE 3:

Neuron-bound AβOs are released into the media due to the action of astrocyte-derived insulin and IGF1. (A) Primary hippocampal neurons were grown on coverslips and exposed to AβOs. Unbound AβOs were quickly removed by submerging coverslips in excess MEM. Coverslips were transferred to new dishes containing MEM supplemented with growth factors. (B) Dot blot analysis of the media showed ACM contained AβOs released from neurons (red bar). This effect was also observed when MEM was supplemented with 300 nM insulin (blue bar) or IGF1 (green bar) or 10 μM demethylasterriquinone B1 (DB1). Significant release was not observed after supplementation with 300 nM EGF, NGF, or BDNF. Treatment with AG 1024, an antagonist of insulin and IGF1 receptors, prevented ACM from stimulating AβO release. (C) IDE treatment of ACM reduced its ability to stimulate AβO liberation by 56 ± 5%. (D) Insulin and IGF1 mRNAs were detectable in cultured astrocytes by RT-PCR at 1:100 and 1:1000 dilutions. RT-PCRs without cDNA did not yield a detectable product. (E, F) The detection of AβOs in the media was accompanied by a reduction in AβO immunofluorescence (NU1, green) along neurites (TuJ, red). ACM and insulin reduced neuritic AβO burden by 40 ± 15% and 37 ± 10%, respectively. For clarity, the AβO signal is shown in black and white next to each condition. (G) Half-maximal AβO liberation occurred at 1.6 ± 0.7 min and 3.4 ± 2.2 min for insulin and ACM treatments, respectively. (H) Insulin stimulated the removal of neuron-bound AβOs with an EC50 of 270 nM. *, p < 0.05, Mann-Whitney; **, p < 0.01, Mann-Whitney; ***, p < 0.0005, Mann-Whitney.
Release of bound AβOs is mediated by insulin/IGF1
Astrocytes constitute the main source of growth factors in the CNS and play major roles in brain morphogenesis, including neuronal survival and maturation, precursor proliferation, and neuronal circuitry formation (Araque et al., 1998; Gomes et al., 1999; Mauch et al., 2001; Beattie et al., 2002; Martinez and Gomes, 2002; Zhang et al., 2003; Christopherson et al., 2005; Elmariah et al., 2005; e Spohr et al., 2011; Allen et al., 2012; Diniz et al., 2012). To identify the neurotrophic factors responsible for the protective effects of astrocytes, we initially measured the release of neuron-bound AβOs in fresh MEM supplemented with EGF, NGF, BDNF, insulin, or IGF1 (300 nM of each). The effects of ACM in inducing oligomer release from hippocampal neurons were mimicked by both insulin and IGF1 treatments, while BDNF, EGF, and NGF failed to instigate release of AβOs (Figure 3B). Further, robust AβO release was induced by demethylasterriquinone B1, a small-molecule activator of the insulin and IGF1 receptor tyrosine kinases, indicating the involvement of insulin/IGF1 signaling in AβO release (Figure 3B). The concentration of Aβ released into the medium following insulin stimulation was estimated to be 20.5 ± 2.2 nM, corresponding to ∼1.5 fmol Aβ released per neuron.
We next asked whether insulin and IGF1 present in ACM were responsible for inducing AβO release from neurons. First, we treated hippocampal neurons with AG1024, an inhibitor of the tyrosine kinase activity of insulin/IGF1 receptors, and found that this blocked AβO release induced by ACM (Figure 3B). Next we treated ACM with insulin-degrading enzyme (IDE), which degrades both insulin and IGF1, before testing its ability to stimulate AβO release. Because Aβ, although not necessarily AβOs (Walsh et al., 2002), is a known target of IDE (Qiu et al., 1998), His-tagged IDE was removed from the ACM before its use in neuronal cultures to prevent potential degradation of AβOs. IDE significantly attenuated the release of AβOs induced by ACM (Figure 3C), further supporting the notion that insulin/IGF1 present in ACM triggered oligomer release from neurons. These findings are consistent with detection of insulin and IGF1 transcripts in cultured astrocytes using conventional, end-point reverse transcriptase PCR (RT-PCR; Figure 3D). Taken together, these results demonstrate that insulin and IGF1 secreted by astrocytes induce the release of oligomers to the extracellular medium after their initial attachment to dendritic binding sites.
We next used immunofluorescence microscopy to examine AβO accumulation on dendrites after stimulating oligomer release with exogenous insulin or ACM. Following treatments with ACM or insulin, dendritic AβO immunoreactivity was reduced by ∼40% (Figure 3, E and F), comparable to previous observations (Pitt et al., 2013). We then characterized the kinetics and insulin-concentration dependence of AβO release from neurons. Oligomer-bound neurons were treated with either ACM or insulin, and AβOs released to the medium were measured as a function of time. AβO release was fast and essentially complete ∼10 min after addition of either ACM or insulin (Figure 3G) and displayed an EC50 = 290 nM for insulin (Figure 3H). Quantification of AβO release into the medium showed that insulin induced an extracellular release of ∼20.5 pmols AβOs into 1 ml culture volume (Supplemental Figure 3). This is equivalent to release of 1.5 fmol AβOs/neuron. These results establish that release of attached oligomers contributes to the mechanism by which exogenous or astrocyte-derived insulin prevents toxic accumulation of AβOs at synapses.
Extracellular release of AβOs involves endocytosis
To determine the mechanism by which insulin caused the release of AβOs previously bound to neurons, we first asked whether insulin-induced oligomer release involved activation of surface proteases. Proteinaceous Aβ binding sites, including APP (Shaked et al., 2006; Fogel et al., 2014) and p75NTR (Knowles et al., 2009), are known to undergo proteolytic cleavage that could lead to the release of surface-bound AβOs (Sotthibundhu et al., 2008; Kenchappa et al., 2010). In initial experiments, we found that addition of a protease inhibitor cocktail reduced insulin-induced AβO release by 72%. Using more specific inhibitors, we ruled out the involvement of γ-secretase (100 nM BMS 299897 and 250 nM DAPT) or matrix metalloproteinases 1, 2, 3, 7, 9, 14, and 17 (0.1–1 μM batimastat and marimastat), as their inhibitors failed to alter AβO release (Figure 4A).
FIGURE 4:
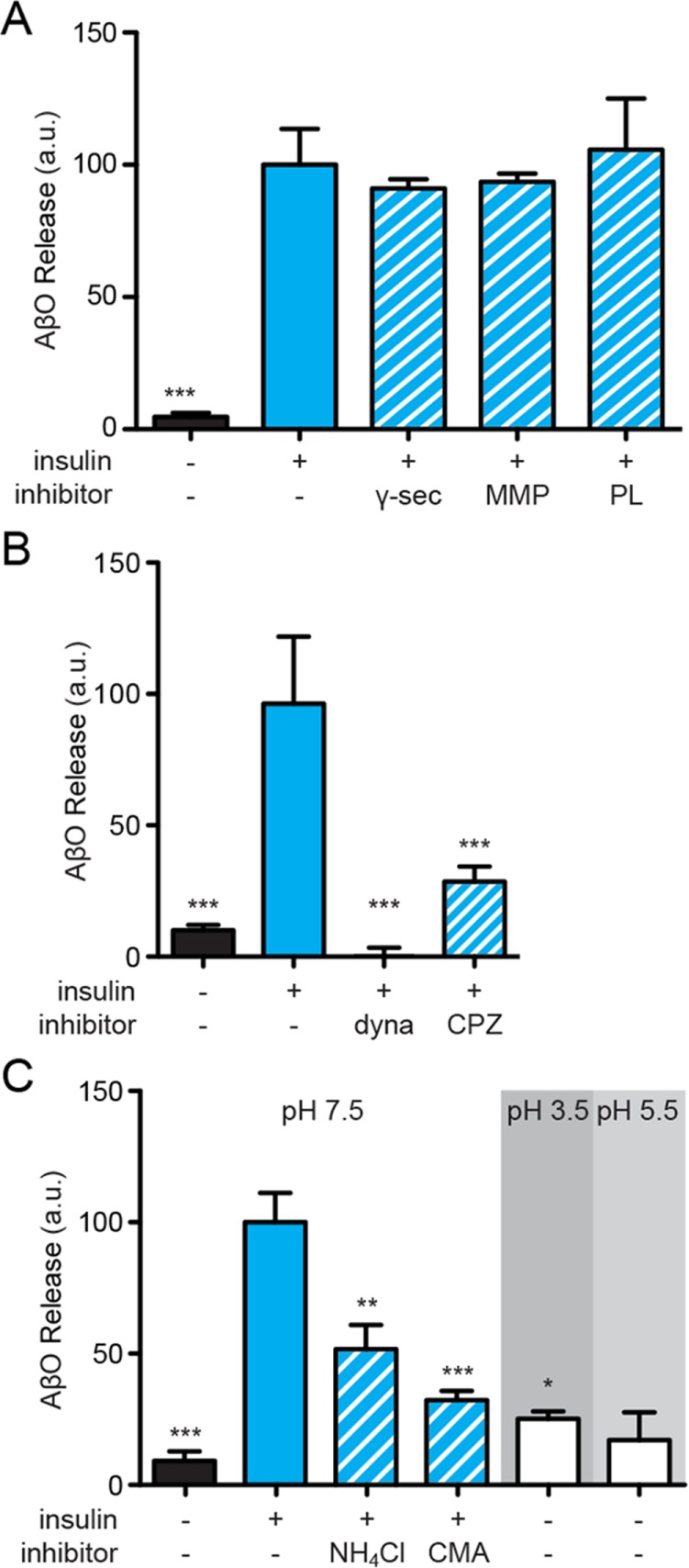
Surface-bound AβOs are endocytosed before release. (A) Inhibition of proteolytic enzymes did not prevent insulin-dependent AβO release. Statistical comparisons are relative to insulin treatments unless otherwise noted; *, p < 0.05, Mann-Whitney; **, p < 0.005, Mann-Whitney; ***, p < 0.0005, Mann-Whitney. (B) Pharmacological inhibition of clathrin-mediated endocytosis using 100 μM dynasore or 75 μM chlorpromazin reduced insulin-mediated AβO release by 100% or 68 ± 7%, respectively. (C) Treatment with 2 mM NH4Cl or 2 μM concanamycin A (CMA) reduced insulin-dependent AβO release by 48 ± 9% or 68 ± 3%, respectively. Minimal oligomer liberation occurs at pH 5.5 and 3.5 in the absence of insulin.
In our initial report demonstrating the neurotoxic action of AβOs, we found that their attachment to neurons involved a trypsin-sensitive binding site (Lambert et al., 1998). Because insulin stimulation activates a trypsin-like protease responsible for generating pyruvate dehydrogenase–activating secondary messengers (Seals and Czech, 1980), we tested whether trypsin inhibition impacted insulin-induced AβO release. In the presence of soybean trypsin inhibitor (SBTI; 0.1 mg/ml), AβO release from insulin-treated neurons was reduced by 71 ± 11%, consistent with the inhibition observed with the protease inhibitor cocktail (not shown). However, immunocytochemical analysis of neurons treated with AβOs in the presence of SBTI revealed an 87 ± 2% reduction of neuritic AβO binding (Supplemental Figure 4A). Therefore, while SBTI does reduce the number of AβOs in the media of insulin-treated neurons, it primarily acts by reducing the initial binding of oligomers to the neuronal surface rather than altering any subsequent step in their processing or release back into the media. These results are consistent with recent findings that SBTI binds to the surface of neurons and blocks AβO/receptor binding in both cellular and cell-free binding assays (Wilcox et al., 2015). In summary, results with SBTI and other protease inhibitors suggest that insulin-induced AβO release from neurons does not require activation of cell surface proteases.
Given the evidence that PrP represents a potential binding partner for AβOs (Lauren et al., 2009), we next asked whether cleavage of PrP and/or other glycosylphosphatidylinositol (GPI)-anchored proteins might constitute a potential mechanism of AβO release. To examine this possibility, we treated oligomer-bound neurons with phosphatidylinositol-specific phospholipase (PI-PLC) and measured AβO release compared with the release induced by insulin treatment. PI-PLC treatment released a small but measurable amount of AβOs (threefold greater than control), equivalent to 18% of the total amount of AβOs released following insulin treatment. Moreover, treatment with an array of phospholipase inhibitors (100 μM FIPI, OBAA, U 73122, or D609, 45 min) failed to attenuate insulin-induced AβO release (Figure 4A and Supplemental Figure 4B), suggesting that release of GPI-anchored proteins does not play a major role in insulin-induced AβO release from neurons.
It has been suggested that exosomes may be involved in the molecular mechanisms of AD (Rajendran et al., 2006; Yuyama et al., 2012; Dinkins et al., 2014). This prompted us to test whether release of surface-bound AβOs induced by insulin might involve uptake into endomembrane compartments, a feature of exosome trafficking. To this end, we performed experiments using chlorpromazine (75 μM, 45 min) and the dynamin-specific inhibitor dynasore (100 μM, 45 min), both of which block endocytosis (Wang et al., 1993; Kirchhausen et al., 2008). Interestingly, chlorpromazine reduced insulin-induced AβO release by 68 ± 6%, while dynasore completely blocked release (Figure 4B). Results therefore suggest that AβO release from neurons involves initial trafficking from the plasma membrane to intracellular compartments.
To better visualize the effect of insulin on AβO distribution, we imaged AβOs using structured illumination microscopy (SIM). Figure 5 shows that AβOs bound to dendritic spines appear more punctate when imaged by SIM than when imaged by confocal immunofluorescence (compare with Figure 1 and Supplemental Figure 1). SIM imaging also suggests an elongated nature of spines in AβO-exposed neurons. We next double-labeled neurons exposed to AβOs in the absence or presence of insulin to determine whether insulin might promote colocalization of AβOs with the endosome markers Rab11 and Rab4 (Sheff et al., 1999). Although there was no indication that AβOs colocalized with either marker at 15 min or at 4 h following exposure to AβOs, results showed that insulin stimulated the internalization of AβOs to compartments within dendrites (Figures 6 and 7). In addition, consistent with AβO release to the medium detected by immunoblot assays (Figures 2– 4; also see Figure 9 later in this article), insulin increased AβO levels in the culture substrate, as revealed by confocal immunofluorescence microscopy (Figure 8, A and B). Taken together, biochemical, pharmacological, and cellular data show that insulin stimulated the release of membrane-bound AβOs to the extracellular milieu in a manner that required endocytosis of oligomers.
FIGURE 5:
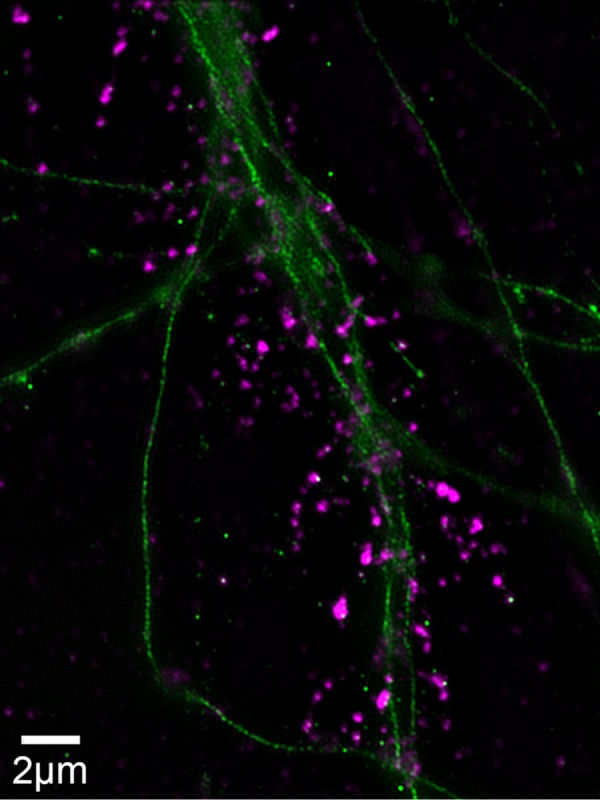
SIM enables precise imaging of AβOs bound to spines. SIM was used to determine the binding of AβOs to primary hippocampal neurons, using the N-SIM superresolution microscope with a lateral resolution at 100× of ∼100 nm, compared with the more typical 400 nm of most microscopes. Primary hippocampal cells, cultured for 19 d, were treated with 100 nM AβOs for 1 h and immunolabeled with anti-TuJ (green) and NU4 (magenta).
FIGURE 6:

Prolonged exposure shows a reduction of AβOs in spines. Hippocampal cells were pretreated with or without insulin for 1 h before addition of AβOs for 15 min or 4 h. Cells were then probed for the exosomal marker Rab11 (green) and AβOs (red). (A, B) Primary hippocampal neurons pretreated for 1 h without (A) or with (B) insulin before 15-min incubation with AβOs show a marked internalization of bound AβOs to the processes. (C, D) Pretreatment without (C) or with (D) insulin followed by a 4-h incubation with AβOs induces a reduction in AβO binding as well as an internalization AβOs. No significant colocalization of AβOs with the endosome marker Rab11 is seen.
FIGURE 7:

AβOs are internalized in response to insulin pretreatment. Hippocampal cells were pretreated with insulin for 1 h before addition of AβOs for 15 min. (A) or 4 h (B) Cells were then probed for the exosomal marker Rab4 (green) and AβOs (red). (A) Cells receiving insulin and AβOs for 15 min. (B) Cells receiving insulin and AβOs for 4 h. (C) Inset reveals that AβOs appear to be segregated into endosome-like compartments in dendritic spines.
FIGURE 9:

AβOs become resistant to insulin-dependent removal mechanisms. (A) Immediately after AβO exposure, neurons were placed into basal MEM for 0, 2, 5, 10, and 15 min before addition of 1 μM insulin to stimulate release. At 2 min, AβO removal is reduced ∼50%. At times longer than 5 min, insulin fails to liberate AβOs. (B) AβOs were releasable when there was no delay between AβO binding and insulin treatment. (C) A 30-min delay following AβO binding resulted in AβOs that were not releasable by insulin treatment. (D) Despite the continued presence of nonreleasable AβOs, a second application of AβOs immediately before insulin treatment proved to be fully releasable compared with B.
FIGURE 8:

AβOs induce a release of exosomes and pTau 231 to the substrate after pretreatment with insulin. (A, B) Confocal microscopy shows that pretreatment of primary hippocampal cells for 1 h without (A) or with (B) insulin caused levels of culture substrate–bound AβOs (red) and Rab4 (green) to be elevated. No colocalization was observed. (C, D) Wide-field fluorescence microscopy of hippocampal neurons pretreated for 4 h without (C) or with (D) insulin before 24-h incubation with AβOs (red) and a sphingomyelinase inhibitor shows that insulin increases the AβO-induced release of pTau 231 (green).
We next found that insulin in the presence of AβOs caused a buildup of substrate-associated Rab4 (Figure 8B, green) and Rab11 (unpublished data), both of which are reported to be present in exosomes (Vidal and Stahl, 1993; Savina et al., 2005). Without insulin, neither Rab11 nor Rab4 (Figure 8A) appear to be attached to the substratum. These exosome markers did not appear to colocalize with AβOs (red), raising the possibility that AβO externalization might occur by an exosome-independent mechanism. However, when cells were treated with an inhibitor of sphingomyelinase (GW4869), which has been reported to block exosome trafficking (Yuyama et al., 2012), AβOs (red) did not build up on the culture substrate, even after 24 h (Figure 8C). The data are consistent with the idea that insulin promotes the removal of cell surface–bound AβOs via a pathway dependent upon exosome trafficking. The cultures exposed for 24 h also were labeled for and phosphorylated tau-Ser231 (pTau231) (green). AβOs are known to stimulate this pathological tau phosphorylation (De Felice et al., 2008; Ma et al., 2009; Zempel et al., 2010) and may additionally stimulate pathological tau release from neurons (Pooler et al., 2015). We found that if cells were exposed to insulin as well as AβOs in these 24 h experiments, the culture substrate showed a marked buildup of punctate pTau231 immunoreactivity (Figure 8D). Interestingly, this pTau231 buildup was not promoted by AβO alone (Figure 8C) or by insulin alone (unpublished data).
Because specific endomembrane components (e.g., recycling endosomes or lysosomes) are more acidic than the extracellular milieu, we investigated the possibility that AβOs interact with membrane proteins in a pH-dependent manner in endomembrane compartments, becoming unbound at lower pH. To test this hypothesis, we asked whether AβO release was affected by inhibiting V-type ATPase, which is responsible for endosomal acidification. Interestingly, treatment with concanamycin A (2 μM, 4 h) or ammonium chloride (2 mM, 45 min, to elevate the pH of endomembrane compartments) reduced insulin-dependent AβO release (Figure 4C). The effects of concanamycin and ammonium chloride could be mediated by elevation of the pH of endomembrane compartments or by inhibition of endosomal vesicle formation (Aniento et al., 1996; Malikova et al., 2004). In the absence of added insulin, lowering the pH of the medium to 5.5 caused release of a small but measurable amount of AβOs from neurons (Figure 4C). However, this required nonphysiological manipulation of the extracellular pH, suggesting that, while highly acidic pH can indeed induce dissociation of a small fraction of AβOs from their receptors at the neuronal surface, proton gradients are more likely necessary for vesicle formation and AβO release within endomembrane compartments.
Inhibition of insulin signaling leads to irreversible AβO binding
Because AβOs progressively self-associate to form large extracellular complexes on the neuronal surface (Renner et al., 2010), we tested whether delaying insulin stimulation could make bound oligomers resistant to release induced by insulin. Indeed, a sharp decrease in the total amount of AβOs released from neurons was observed when insulin stimulation was delayed by as little as 2 min (Figure 9A). After 5 min, AβO release induced by insulin was minimal (Figure 9A), suggesting that oligomers had rapidly formed stable, release-resistant complexes on the surface of neurons.
Based on our previous findings that AβOs cause the removal of dendritic insulin receptors from the neuronal plasma membrane (Zhao et al., 2008; De Felice et al., 2009), the decrease in insulin-induced oligomer release could result from AβO-induced neuronal insulin resistance. To test this possibility, we repeated our time-delay experiments, adding an additional early predose of AβOs to distinguish between oligomer stabilization and neuronal insulin resistance (Figure 9, B–D). Neurons exposed to AβOs for 15 min immediately before insulin-induced oligomer release was measured showed the expected release behavior (Figure 9B). When neurons were exposed to AβOs for the same length of time (15 min) and then subjected to a 30 min delay period before insulin treatment, no release could be detected (Figure 9C). However, even after a 15 min delay period, sufficient to make previously added oligomers nonreleasable, AβOs reapplied for another 15 min could still be released upon stimulation by insulin (Figure 9D). These findings indicate that insulin insensitivity is not responsible for the inhibition of insulin-induced oligomer release caused by a time delay between AβO binding and insulin stimulation. Instead, results suggest that AβOs rapidly become trapped at the neuronal surface in the absence of insulin signaling.
Neural cells chronically exposed to AβOs show reduced insulin/IGF1 expression
Given the association of impaired insulin/IGF1 signaling with AD dementia (Rivera et al., 2005; Bomfim et al., 2012; Craft et al., 2012; Talbot et al., 2012), we investigated whether insulin/IGF1 expression in neural cells was disrupted by AβOs (500 nM, 8 d) using qRT-PCR. In cultured astrocytes, insulin expression was unaffected by AβOs (95% confidence interval [CI] = 52.62–128.3% compared with control). However, IGF1 expression was reduced by 72% (Figure 10A). In hippocampal neuronal cultures, treatment with AβOs (500 nM, 24 h) reduced insulin expression by 50% (Figure 10B). These results demonstrate that AβOs reduce the expression of insulin and IGF1 in neural cells.
FIGURE 10:
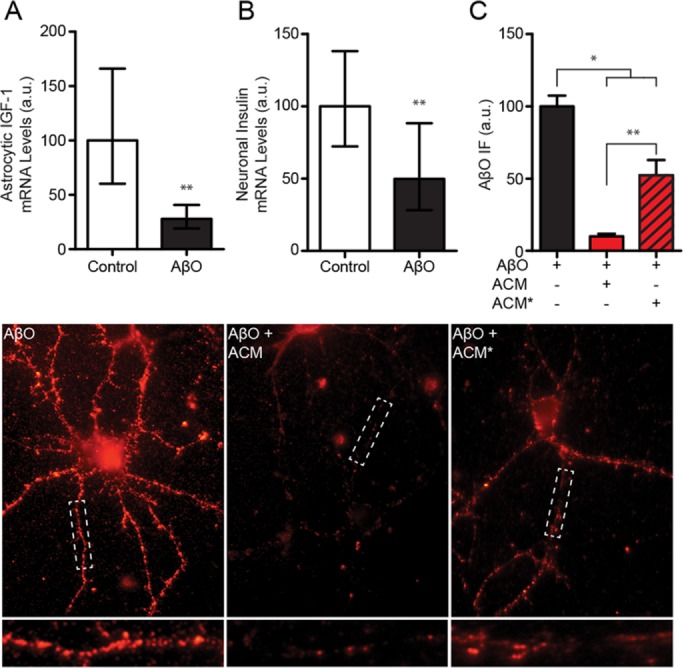
AβOs reduce insulin and IGF1 expression in astrocytes and neurons. (A) Treatment of cultured astrocytes with AβOs (500 nM) reduced IGF1 expression more than twofold (geometric mean = 28.0%; 95% CI = 19.1–40.8%) compared with control (geometric mean = 100%; 95% CI = 60.2–166%). (B) Treatment of cultured neurons with AβOs reduced insulin expression in neurons approximately twofold (geometric mean = 49.9%; 95% CI = 28.2–88.3%) compared with control (geometric mean = 100%; 95% CI = 72.3–138%). (C) Treatment of cultured astrocytes reduced the protective efficacy of conditioned media (based on images below). While conditioned media from untreated astrocytes (ACM) reduced neuronal AβO accumulation ∼90% (red bar), accumulation was down only ∼45% using media from astrocytes previously exposed to AβOs (AβO-ACM*; red and black striped bar). Geometric means and 95% CIs are plotted in A and B. Arithmetic means and standard errors are plotted in C. *, p < 0.05, Mann-Whitney; **, p < 0.01, Mann-Whitney.
Finally, having found that AβOs decrease IGF1 expression in astrocytes, we tested whether AβO treatment reduced the protective efficacy of astrocytes. Mouse astrocyte cultures were exposed to AβOs (500 nM, 24 h) or vehicle. After being rinsed thoroughly with DMEM/F12 to remove residual AβOs, ACM was collected for a period of 24 h and tested for its ability to prevent accumulation of AβOs (500 nM, 3 h) along the dendrites of cultured hippocampal neurons. Consistent with our observations described earlier, ACM from vehicle-treated astrocytes reduced AβO accumulation by 90% (Figure 10C). However, conditioned medium from AβO-exposed astrocytes reduced dendritic AβO accumulation by only 48% compared with control (ACM from nonexposed astrocytes) (Figure 10C). These results show that previous exposure to AβOs reduces the protective capacity of astrocytes.
DISCUSSION
AβOs are soluble toxins that accumulate in the AD brain and bind to dendritic spines when added to cultured hippocampal neurons. Accumulation of AβOs leads to spine deterioration, synapse failure, and, eventually, synapse loss (Lacor et al., 2007; Shankar et al., 2007; Koffie et al., 2009; Wilcox et al., 2011; Sivanesan et al., 2013). We report here that robust protection against the synaptoxicity of AβOs is conferred by soluble factors released from astrocytes. Compared with astrocyte-free cultures, neurons maintained in the presence of an astrocyte feeder layer or supplied with ACM showed greatly reduced dendritic binding of exogenously added AβOs. Astrocyte-derived protective factors were found to comprise insulin and IGF1. The mechanism of protection by insulin and IGF1 involves release of recently attached AβOs to the extracellular milieu, a process that exhibits features of exosome trafficking (Figure 11). Interestingly, in addition to extracellular release of AβOs, there also was release of tau phosphorylated at a prototypic AD epitope. This raises the possibility that insulin might help neurons eliminate both AβOs and pathological tau, but it also suggests that cell-to-cell propagation of toxic forms of pTau could potentially be stimulated by the presence of insulin and high levels of AβOs. In the absence of insulin/IGF1 signaling, even for relatively short times, neuron-bound AβOs transitioned to a state that was refractory to release upon subsequent insulin/IGF1 treatment (Figure 11). These findings are consistent with the hypothesis that sustained insulin/IGF1, perhaps derived from astrocytes, plays an important role in warding off dementia associated with the buildup of synaptotoxic AβOs in aging brain.
FIGURE 11:

Proposed model for insulin/IGF1-stimulated AβO release. After AβO attachment to the neuronal surface, stimulation of insulin/IGF1 signaling leads to AβO internalization. AβOs are detached from their binding targets and shuttled back to the neuronal surface, where they are released to the extracellular space. When insulin/IGF1 signaling is deficient, AβOs are rapidly trapped at the neuronal surface and become resistant to insulin/IGF1-induced release.
There has been considerable controversy regarding the involvement of Aβ-derived toxins in AD pathogenesis (Karran et al., 2011). Substantial evidence from human genetics and pathology, however, indicates they play a key role (Selkoe and Hardy, 2016). This is strongly supported by the discovery of the Icelandic A673T mutation in APP; this mutation decreases Aβ production and protects carriers against AD onset (Jonsson et al., 2012). The major Aβ species implicated in AD pathogenesis comprise soluble AβOs (Mucke and Selkoe, 2012; Selkoe and Hardy, 2016; DiChiara et al., 2017). Experimentally, AβOs instigate memory failure (Lesne et al., 2006) and AD neuropathology, including tau hyperphosphorylation and synapse dysfunction and deterioration (De Felice et al., 2007, 2008; Lacor et al., 2007; Ma et al., 2009; Balducci et al., 2010; Nimmrich et al., 2010; Tomiyama et al., 2010; Sebollela et al., 2012; Figueiredo et al., 2013). The putative primary role of AβOs in AD pathogenesis is substantiated by the dementia and neuropathology caused by the E693Δ APP “Osaka” mutation (Tomiyama et al., 2008, 2010), carriers of which manifest abundant AβOs but no amyloid plaques.
In AD patients and mouse models, AβOs accumulate early, before plaque buildup (Jacobsen et al., 2006; Oddo et al., 2006; Lacor et al., 2007) and possibly decades before clinical symptoms develop (Jack et al., 2010). How dementia can be successfully postponed until older ages is of considerable interest, as AβOs self-assemble at extremely low levels of Aβ (Chang et al., 2003; Velasco et al., 2012), which exists at substantial concentrations in the brain and is released in response to neural activity (Bero et al., 2011). Several mechanisms may play roles in this. First, buildup of AβOs is slowed by Aβ degradation (Jiang et al., 2008; Cramer et al., 2012), which can be mediated by astrocytes and microglia (Mandrekar-Colucci et al., 2012), and stimulated by peroxisome proliferator-activated receptor-γ, a known insulin-sensitizing factor. AβO levels also can be reduced by clearance from interstitial fluid (Mawuenyega et al., 2010; Takeda et al., 2013). Aβ pathology, however, appears to be present 10–20 yr before clinical symptoms manifest (Price and Morris, 1999; Jansen et al., 2015). Evidence presented here suggests that, if degradation and clearance are insufficient, and AβOs begin to accumulate, neurons could be made resistant to their toxicity by the neuroprotective activity of astrocyte-derived insulin/IGF1.
Insulin and IGF1 secreted by astrocytes were found here to promote the release of AβOs bound to neuronal surfaces. Insulin/IGF1 could thus help hold off AD by removing AβOs from neurons and by subsequently promoting their degradation by glia. These findings confirm and extend previous indications that insulin signaling protects neurons against AβOs and AD pathogenesis (Townsend et al., 2007; Jolivalt et al., 2008; De Felice et al., 2009; Zhao et al., 2009; Craft et al., 2012; Long-Smith et al., 2013). No reduction in AβO binding to neurons was afforded by NGF, EGF, or BDNF, which are also expressed in astrocytes (Zafra et al., 1992). Results add insulin/IGF1 to the growing list of neuroactive compounds released from astrocytes, which includes glutamate (Parpura et al., 1994; Cavelier and Attwell, 2005), adenosine/ATP (Panatier et al., 2011; Schmitt et al., 2012), transforming growth factor beta 1 (Diniz et al., 2012, 2014, 2017), thrombospondin (Christopherson et al., 2005), and d-serine (Henneberger et al., 2010).
The presence of insulin/IGF1 originating from CNS cells is consistent with findings that peripheral but not brain insulin is reduced by systemic treatment with streptozotocin (Havrankova et al., 1979), which is blood–brain barrier impermeable. It seems plausible that constitutive release of insulin/IGF1 by astrocytes (and/or neurons) complements the function of metabolically regulated insulin originating in the pancreas. This would be analogous to the glutamate tone provided by astrocytes (Cavelier and Attwell, 2005), which establishes a basal level of excitation in the striatum and hippocampus. A constitutive insulin tone may be essential to the mechanism of protection, as we found that AβOs become resistant to release induced by insulin/IGF1 if they remain attached to the neuronal surface in the absence of insulin for as little as 15 min. This phenomenon is consistent with single-molecule tracking data (Renner et al., 2010), which showed that surface-bound AβOs transition from a free-moving to an immobilized state embedded in detergent-resistant domains a few minutes after binding to the neuronal plasma membrane.
The simplest possible mechanisms we could envisage to explain the reduction induced by insulin in neuron-bound AβOs are not supported by the current data. For example, competitive binding between AβOs and insulin to a common neuronal receptor is ruled out by the fact that insulin is without effect if the kinase activity of its receptor is inhibited by AG1024 (De Felice et al., 2009; present study). Insulin-induced down-regulation of the receptor proteins to which AβOs might bind also appears as an incomplete explanation, as results showed insulin signaling acts to release AβOs after they had attached to neurons. Further, removal of bound AβOs is not mediated by their proteolytic cleavage, as AβOs released to the medium appear to be intact, as they are recognized by a conformation-specific antibody. Finally, insulin-induced proteolysis of neuronal surface proteins that act as oligomer receptors also appears unlikely, given the lack of effect of a number of specific protease inhibitors (including inhibitors of beta secretase 1 and various metalloproteinases) in blocking insulin-induced AβO release from neurons.
A salient finding relevant to the oligomer release mechanism is that it is blocked by dynasore and chlorpromazine, two inhibitors of endocytosis. Release of AβOs into the extracellular milieu thus depends upon intracellular trafficking. This is consistent with high-resolution imaging (Figure 5), which shows AβOs within dendrites and, possibly, within spines. The net impact of insulin on AβO trafficking and the relatively rapid transition of bound AβOs to an insulin-resistant state are illustrated in Figure 11. The ability of insulin to stimulate endocytosis in neurons is well known, including the endocytosis of potential AβO-binding proteins (Zhao et al., 2010). Further, vesicle acidification by V-ATPase appears essential in shuttling endocytosed AβOs back to the surface and into the extracellular space, as release was prevented by concanamycin A, a V-ATPase inhibitor (Malikova et al., 2004), and was attenuated by ammonium chloride. Acidification of the extracellular medium per se, however, did not substantially stimulate release. In addition to release to the medium, it also appeared that AβOs were deposited in particulate form onto the culture substrate. Although not yet proven, the data are consistent with a mechanism in which removal of bound AβOs is a consequence of insulin-stimulated exosome trafficking (Aoki et al., 2007; Muller et al., 2009). In harmony with this interpretation, an inhibitor of exosome trafficking was found to block AβO release from insulin-treated neurons.
Interestingly, insulin treatment of AβO-exposed neurons caused externalization and substrate attachment of tau phosphorylated at serine residue 231, an AD-associated epitope (Modrego, 2006). It remains to be determined whether removal of both AβOs and a pathological form of tau is a completely beneficial effect of insulin, or whether removal potentially might be harmful due to increased potential for cell-to-cell transmission of AD-linked pTau. Of note, recent microfluidics experiments strongly indicate that cellular transmission of AD-type tau can be propagated by exosomes (Usenovic et al., 2015).
The relationship between AβOs and CNS insulin signaling overall is surprisingly complex (Ferreira and Klein, 2011; De Felice, 2013). Impaired insulin/IGF1 function not only makes it possible for toxic AβOs to accumulate on neurons, but is itself a consequence of AβO accumulation, as bound AβOs down-regulate insulin receptors and inhibit IRS-1 (Zhao et al., 2008; Bomfim et al., 2012; Talbot et al., 2012), thereby rendering neurons insulin resistant. Moreover, expression of insulin and IGF1 in CNS cells exposed to AβOs is reduced, as found here. This decrease is consistent with findings that insulin and IGF1 expression in the CNS is reduced in AD patients (Rivera et al., 2005; Gil-Bea et al., 2010; Moloney et al., 2010). These phenomena have the potential to create a vicious cycle in which 1) brain cell expression of insulin/IGF1 is reduced by exposure to AβOs; 2) reduced levels of insulin/IGF1 make it easier for AβOs to bind and accumulate at synapses; 3) increasingly elevated AβO binding (to neurons and astrocytes) reduces insulin signaling further by reducing insulin/IGF-1 expression (as found here) or by instigating removal of insulin receptors and inhibition of IRS-1 (Zhao et al., 2008; Bomfim et al., 2012); and 4) the resulting major dysfunction in insulin/IGF1 signaling allows oligomer binding to reach toxic levels (De Felice et al., 2009; Zhao et al., 2009). Compounding the problem, diabetes likely is a factor that instigates AβO buildup in the brain, as observed experimentally in studies of diabetes in wild-type rabbits (Bitel et al., 2012). Intriguingly, the most important AD risk factor, age, itself manifests with compromised brain insulin signaling (Fernandes et al., 2001).
Maintaining healthy CNS insulin signaling should be considered an important factor in preventing AD progression. Loss of robust CNS insulin signaling may account, at least in part, for the fact that type II diabetes, which can present with reduced brain insulin (Hu et al., 2013), is an important AD risk factor (Ott et al., 1999; Launer, 2005). Reduced brain insulin signaling, whatever the origin, would be expected to accelerate the vicious cycle of pathogenesis described earlier. As proposed (De Felice et al., 2009), such an accelerating feedback loop would likely require several levels of therapeutic intervention, optimally combining anti-AβO therapy using antibodies capable of recognizing oligomers, such as Aducanumab (Sevigny et al., 2016), together with CNS-targeted insulin therapy (Craft et al., 2012) and/or drugs that activate CNS insulin-signaling pathways (Gault and Holscher, 2008; Bomfim et al., 2012; De Felice, 2013; Lourenco et al., 2013; Pitt et al., 2013). Results here suggest that neuronal resistance to AβO toxicity could also be raised by enhancing the natural release of insulin/IGF1 from aging astrocytes.
MATERIALS AND METHODS
Materials
Reagents and chemicals were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO) unless otherwise specified. MEM with Earle’s salts and l-glutamine (Invitrogen; 11095-080), N2 Supplement (Invitrogen; 17502-048), Neurobasal media (Invitrogen; 21103-049), B-27 supplement (Invitrogen; 17504-044), horse serum (Invitrogen; 16050), Aβ1-42 (American Peptide; 62-0-80), FAM-Aβ1-42 (AnaSpec; 23525-05), insulin (Sigma; I9278), IGF1 (Genway; GWB-4E7F14), epidermal growth factor (Sigma; E4127), nerve growth factor (Millipore; GF028), brain-derived neurotrophic factor (Millipore; GF029), demethylasterriquinone B1 (Tocris; 1819), PI-PLC (Sigma; P5542), insulin-degrading enzyme, His-Tag, rat recombinant (Calbiochem; 407241), Dynabeads His-Tag isolation and pull-down (Invitrogen; 101.03D), dynasore (Tocris; 2897), edelfosine (Tocris; 3022), AG 1024 (Calbiochem; 121767), chlorpromazine (Sigma; C8138), BMS 299897 (2-[(1R)-1-[[(4-chlorophenyl)sulfonyl](2, 5-difluorophenyl)amino]ethyl-5-fluorobenzenebutanoic acid; Tocris; 2870), DAPT (N-[(3,5-difluorophenyl)acetyl]-l-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester; Tocris; 2634), batimastat (Tocris; 2961), marimastat (Tocris; 2631), FIPI (N-[2-[4-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)-1-piperidinyl]ethy]-5-fluoro-1H-indole-2-carboxamide hydrochloride; Tocris; 3600), OBAA (4-(4-octadecylphenyl)-4-oxobutenoic acid; Tocris; 0606), U 73122 (1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; Tocris; 1268), D609 (O-(octahydro-4,7-methano-1H-inden-5-yl) carbonopotassium dithioate; Tocris; 1437), and SBTI (Sigma; T9128).
Hippocampal neuron cultures
Primary hippocampal cultures were prepared from E18 rat embryos as previously described (Kaech and Banker, 2006). Neurons were plated at 150,000 cells per 60-mm dish and maintained in N2 medium (N2 supplement and 0.6% glucose in MEM). Experiments were carried out at 18–21 days in vitro (DIV).
Cortical astrocyte cultures
Secondary cortical astrocyte cultures were prepared from E18 rat embryos as previously described (Kaech and Banker, 2006). Astrocytes were grown in 75 cm2 flasks containing astrocyte medium (0.6% glucose, 10% horse serum, and 1% penicillin–streptomycin in MEM). Astrocytes were isolated by mechanical dissociation of poorly adherent, presumably nonastrocytic cells. Briefly, each flask was hit briskly on the side before change of media to remove loosely attached, nonastrocytic cells (e.g., microglia). Dislodged cells were discarded. After reaching confluence, astrocytes were split into 60-mm dishes at 100,000 cells per dish and grown to ∼70% confluency before use as feeder layers. Astrocyte medium was exchanged for N2 medium 1 d before neuronal culture preparation.
AβO preparations
AβOs and FAM-AβOs were prepared as previously described (Pitt et al., 2009).
AβO accumulation and toxicity
Following any pretreatments, primary cell cultures were exposed to 500 nM AβOs (molarity based on Aβ monomers) for 24 h. After treatment, cultures were fixed for 10 min at room temperature with 4% paraformaldehyde/4% sucrose in phosphate-buffered saline (PBS). Cultures were washed 5 times with PBS and stored at 4°C until immunolabeled (maximum of 1 wk). In other experiments, astrocyte cultures were pre-exposed to AβOs and ACM was tested for its impact on AβO binding to neurons. Primary mouse astrocyte cultures were prepared as previously described (Gomes et al., 1999). Secondary astrocytes were plated at 3 million cells per 25 cm2 culture flask and maintained in DMEM/F12 with 10% bovine calf serum. After reaching confluence, astrocytes were exposed for 24 h to 500 nM AβOs in DMEM/F12 without serum. Cultures were washed to remove AβOs, and fresh DMEM/F12 was conditioned for 24 h before use in protection assays.
Mouse primary dissociated hippocampal neurons were plated at 100,000 cells per 13-mm dish and maintained in neurobasal media with B-27 supplement. At 19–21 DIV, neurons were placed in fresh or astrocyte-conditioned DMEM/F12 and exposed for 3 h to 500 nM AβOs. After treatment, cells were fixed by adding an equal volume of 3.7% formaldehyde (in PBS buffer) to the medium for 5 min; this was followed by removal of the entire fix/media solution and replacement with 3.7% formaldehyde for 10 min. Cultures were washed three times with PBS and stored at 4°C until immunolabeled (maximum of 1 wk).
AβO release assay
Hippocampal neurons cultured on 18-mm coverslips were moved into individual wells in 12-well plates containing 1 ml MEM. For AβO attachment, 500 nM AβOs were added for 15 min. Release of AβOs was stimulated by moving coverslips to new wells with fresh MEM supplemented with factors of interest. Coverslips were washed with MEM in between wells to remove any unbound AβOs. MEM was analyzed for AβO content by dot immunoblot (described in the following section) using NU1, an AβO-sensitive antibody (Lambert et al., 2007). Pharmacological inhibitors, when used, were added 30 min before AβO attachment.
AβO dot immunoblotting
Media samples from AβO release assays were applied to nitrocellulose film in triplicate. Each dot contained 1 μl of media. All membranes also included a positive control (50 nM AβO; concentration based on monomers) and a negative control (PBS), both in triplicate. In a subset of experiments, a standard curve of AβOs (1–500 nM) was spotted onto the membrane to estimate the amount of AβO released. After spots were dry, membranes were blocked in immunoblocking buffer (Tris-buffered saline [TBS] with 0.05% Tween-20, 5% nonfat dry milk) for 45 min at room temperature. Primary stains were carried out overnight at 4°C using the AβO-sensitive antibody NU1 (mouse; 1.5 μg/ml in immunoblocking buffer). Membranes were washed four times (5 min each wash) with TBS containing 0.05% Tween-20. Membranes were then incubated with an anti-mouse, horseradish peroxidase–conjugated secondary antibody (1:5000 in immunoblocking buffer) for 90 min at room temperature. Membranes were washed as described above and developed using the appropriate chemiluminescent reagents.
Glial conditioning of MEM and insulin-degrading enzyme treatment
For release assays, conditioned medium was swapped for MEM on secondary astrocyte cultures. ACM was collected after 12–24 h. For IDE treatment, 1.5 ml ACM was treated with 1 U IDE for 1 h at 37°C. His-tagged IDE was removed using Dynabeads His-tag isolation and pull-down following the manufacturer’s instructions.
Immunostaining
Antibodies against the following antigens were used: TuJ1 (1:1000; Promega; G7121), TuJ1 (1:2000; Covance; MRB-435P), GFAP (1:1000; Promega; G560A), and spinophilin (1:250; Abcam; ab18561). Anti-AβO antibodies NU1 (1.5 μg/ml) and NU4 (1.5 μg/ml) are monoclonal antibodies derived from mice immunized with AβOs (Lambert et al., 2007). Both NU1 and NU4 interact strongly with oligomeric forms of Aβ. Immunostaining was carried out as previously described (Pitt et al., 2009).
qRT-PCR
Desalted primers were custom synthesized (Integrated DNA Technologies) against the following genes in Rattus norvegicus: 18S rRNA (forward: gcttgcgttgattaagtccctg; reverse: agtcaagttcgaccgtcttctc), β-actin (forward: ccctgaagtaccccattgaaca; reverse: ctgggtcatcttttcacggttg), GAPDH (forward: cctggagaaacctgccaagtat; reverse: caccctgttgctgtagccata), insulin-1 (forward: ccctaagtgaccagctacaatc; reverse: ccacaaaggtgctgtttgac), and IGF1 (forward: acatgcccaagactcagaag; reverse: ggtgttccgatgttttgcag). Total RNA was extracted with RNeasy Mini Kit (Qiagen; 74104). cDNA was synthesized using qScript cDNA Synthesis Kit (Quanta Biosciences; 95047-100). Conventional PCR was carried out with the following thermal-cycling conditions: 94°C for 5 min, followed by 44 cycles at 94°C for 45 s, 56°C for 45 s, and 72°C for 70 s. RT-PCR products were visualized by electrophoresis with Gel Red (Biotium). Real-time PCR (qPCR) was carried out using SYBR Green Master Mix (Invitrogen; 4385612) in a StepOne Plus thermocycler (Applied Biosystems), following the manufacturers’ protocols. Weighted CTs for three reference genes (actin, GAPDH, and 18S rRNA) were calculated using the RefFinder tool provided by the EST Database of Cotton (www.leonxie.com/referencegene.php). Relative gene expression was calculated for 13 control samples and 14 AβO-treated samples across four separate experiments by the ΔCT method. Statistical analysis was carried out in Prism 5 (GraphPad).
Imaging and data analysis
Images were acquired using a 60× objective on a Nikon Eclipse TE2000-U epifluorescence microscope and exported into CellProfiler (Carpenter et al., 2006) to analyze the number of pixels positive for each antibody normalized by neurite length. To avoid potential biases in results related to distance from the soma or dendritic order, we quantified the signal along neurite segments at various distances from the cell body and averaged. Manual analysis of microscopy data was performed in MetaMorph. N-SIM images were captured on a Nikon N-SIM Structured Illumination superresolution microscope. Confocal images were captured on a Nikon A1R+ confocal laser microscope system. Wide-field fluorescent images were captured using a Molecular Devices ImageXpress confocal microscope at 40×. Western blots were quantified using ImageJ (National Institutes of Health). Numerical data from each experimental repetition were exported and pooled for descriptive and statistical analysis in Prism 5 (GraphPad). All experiments were carried out a minimum of three times. In each experiment, each experimental condition contained at least triplicate samples. qPCR data are reported as geometric means ± 95% confidence intervals. All other data are reported as means ± SEM.
Supplementary Material
Acknowledgments
This research was funded by the National Institutes of Health (NIH) (grant 1F31AG039216 to J.P.); the Alzheimer’s Association (grant ZEN09133875 to W.L.K.); and the National Institute for Translational Neuroscience/Brazil, CNPq/Brazil, and FAPERJ/Brazil (to S.T.F., F.G.D.F., and F.C.A.G.). Imaging work with the N-SIM and A1R microscopes was performed at the Northwestern University Center for Advanced Microscopy, generously supported by National Cancer Institute (NCI) grant CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Structured illumination microscopy was performed on a Nikon N-SIM system, purchased through the support of NIH 1S10OD016342-01. Wide-field fluorescent imaging was performed at the Northwestern University High-Throughput Analysis Laboratory generously supported by NCI grant CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
Abbreviations used:
- ACM
astrocyte-conditioned MEM
- AD
Alzheimer’s disease
- AβO
Aβ oligomer
- APP
amyloid precursor protein
- BDNF
brain-derived neurotrophic factor
- CI
confidence interval
- EGF
epidermal growth factor
- IDE
insulin-degrading enzyme
- IGF1
insulin-like growth factor-1
- NGF
nerve growth factor
- PBS
phosphate-buffered saline
- PI-PLC
phosphatidylinositol-specific phospholipase C
- RT-PCR
reverse transcriptase PCR
- SBTI
soybean trypsin inhibitor
- SIM
structured illumination microscopy.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-06-0416) on August 9, 2017.
REFERENCES
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486:410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013;9:208–245. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal beta COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki N, Jin-no S, Nakagawa Y, Asai N, Arakawa E, Tamura N, Tamura T, Matsuda T. Identification and characterization of microvesicles secreted by 3T3-L1 adipocytes: redox- and hormone-dependent induction of milk fat globule-epidermal growth factor 8-associated microvesicles. Endocrinology. 2007;148:3850–3862. doi: 10.1210/en.2006-1479. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci USA. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitel CL, Kasinathan C, Kaswala RH, Klein WL, Frederikse PH. Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimers Dis. 2012;32:291–305. doi: 10.3233/JAD-2012-120571. [DOI] [PubMed] [Google Scholar]
- Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J Clin Invest. 2012;122:1339–1353. doi: 10.1172/JCI57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Bakhos L, Wang Z, Venton DL, Klein WL. Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening. J Mol Neurosci. 2003;20:305–313. doi: 10.1385/JMN:20:3:305. [DOI] [PubMed] [Google Scholar]
- Chernausek SD. Insulin-like growth factor-I (IGF-I) production by astroglial cells: regulation and importance for epidermal growth factor-induced cell replication. J Neurosci Res. 1993;34:189–197. doi: 10.1002/jnr.490340206. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG. Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J Clin Invest. 2013;123:531–539. doi: 10.1172/JCI64595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Natl Acad Sci USA. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiChiara T, DiNunno N, Clark J, Bu RL, Cline EN, Rollins MG, Gong Y, Brody DL, Sligar SG, Velasco PT, et al. Alzheimer’s toxic amyloid beta oligomers: unwelcome visitors to the Na/K ATPase alpha3 docking station. Yale J Biol Med. 2017;90:45–61. [PMC free article] [PubMed] [Google Scholar]
- Diniz LP, Almeida JC, Tortelli V, Vargas Lopes C, Setti-Perdigao P, Stipursky J, Kahn SA, Romao LF, de Miranda J, Alves-Leon SV, et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor beta signaling through modulation of D-serine levels in cerebral cortex neurons. J Biol Chem. 2012;287:41432–41445. doi: 10.1074/jbc.M112.380824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz LP, Tortelli V, Garcia MN, Araujo AP, Melo HM, Silva GS, Felice FG, Alves-Leon SV, Souza JM, Romao LF, et al. Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia. 2014;62:1917–1931. doi: 10.1002/glia.22713. [DOI] [PubMed] [Google Scholar]
- Diniz LP, Tortelli V, Matias I, Morgado J, Bergamo Araujo AP, Melo HM, Seixas da Silva GS, Alves-Leon SV, de Souza JM, Ferreira ST, et al. Astrocyte transforming growth factor beta 1 protects synapses against Aβ oligomers in Alzheimer’s disease model. J Neurosci. 2017;37:6797–6809. doi: 10.1523/JNEUROSCI.3351-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2014;35:1792–1800. doi: 10.1016/j.neurobiolaging.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- e Spohr TC, Dezonne RS, Rehen SK, Gomes FC. Astrocytes treated by lysophosphatidic acid induce axonal outgrowth of cortical progenitors through extracellular matrix protein and epidermal growth factor signaling pathway. J Neurochem. 2011;119:113–123. doi: 10.1111/j.1471-4159.2011.07421.x. [DOI] [PubMed] [Google Scholar]
- Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci USA. 2000;97:9287–9292. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes ML, Saad MJ, Velloso LA. Effects of age on elements of insulin-signaling pathway in central nervous system of rats. Endocrine. 2001;16:227–234. doi: 10.1385/endo:16:3:227. [DOI] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight Aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci. 2013;33:9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O’Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, et al. APP homodimers transduce an amyloid-beta-mediated increase in release probability at excitatory synapses. Cell Rep. 2014;7:1560–1576. doi: 10.1016/j.celrep.2014.04.024. [DOI] [PubMed] [Google Scholar]
- Gault VA, Holscher C. Protease-resistant glucose-dependent insulinotropic polypeptide agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. J Neurophysiol. 2008;99:1590–1595. doi: 10.1152/jn.01161.2007. [DOI] [PubMed] [Google Scholar]
- Gil-Bea FJ, Solas M, Solomon A, Mugueta C, Winblad B, Kivipelto M, Ramirez MJ, Cedazo-Minguez A. Insulin levels are decreased in the cerebrospinal fluid of women with prodomal Alzheimer’s disease. J Alzheimers Dis. 2010;22:405–413. doi: 10.3233/JAD-2010-100795. [DOI] [PubMed] [Google Scholar]
- Gomes FC, Maia CG, de Menezes JR, Neto VM. Cerebellar astrocytes treated by thyroid hormone modulate neuronal proliferation. Glia. 1999;25:247–255. [PubMed] [Google Scholar]
- Grunblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem. 2007;101:757–770. doi: 10.1111/j.1471-4159.2006.04368.x. [DOI] [PubMed] [Google Scholar]
- Havrankova J, Roth J, Brownstein MJ. Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest. 1979;64:636–642. doi: 10.1172/JCI109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P, et al. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry. 2006;45:15157–15167. doi: 10.1021/bi061850f. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SH, Jiang T, Yang SS, Yang Y. Pioglitazone ameliorates intracerebral insulin resistance and tau-protein hyperphosphorylation in rats with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2013;121:220–224. doi: 10.1055/s-0032-1333277. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, Schuff N, Krueger G, Killiany RJ, Decarli CS, et al. Update on the magnetic resonance imaging core of the Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 2010;6:212–220. doi: 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study G, Aalten P, Aarsland D, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. J Am Med Assoc. 2015;313:1924–1938. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, et al. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86:3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kenchappa RS, Tep C, Korade Z, Urra S, Bronfman FC, Yoon SO, Carter BD. p75 neurotrophin receptor-mediated apoptosis in sympathetic neurons involves a biphasic activation of JNK and up-regulation of tumor necrosis factor-alpha-converting enzyme/ADAM17. J Biol Chem. 2010;285:20358–20368. doi: 10.1074/jbc.M109.082834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T, Macia E, Pelish HE. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol. 2008;438:77–93. doi: 10.1016/S0076-6879(07)38006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JK, Rajadas J, Nguyen TV, Yang T, LeMieux MC, Vander Griend L, Ishikawa C, Massa SM, Wyss-Coray T, Longo FM. The p75 neurotrophin receptor promotes amyloid-beta(1–42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci. 2009;29:10627–10637. doi: 10.1523/JNEUROSCI.0620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, et al. Monoclonal antibodies that target pathological assemblies of Aβ. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Launer LJ. Diabetes and brain aging: epidemiologic evidence. Curr Diab Rep. 2005;5:59–63. doi: 10.1007/s11892-005-0069-1. [DOI] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, O’Neill C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. NeuroMol Med. 2013;15:102–114. doi: 10.1007/s12017-012-8199-5. [DOI] [PubMed] [Google Scholar]
- Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metabolism. 2013;18:831–843. doi: 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, Chen PP, Hudspeth B, Chen C, Zhao Y, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikova M, Shi J, Kandror KV. V-type ATPase is involved in biogenesis of GLUT4 vesicles. Am J Physiol Endocrinol Metab. 2004;287:E547–E552. doi: 10.1152/ajpendo.00571.2003. [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez R, Gomes FC. Neuritogenesis induced by thyroid hormone-treated astrocytes is mediated by epidermal growth factor/mitogen-activated protein kinase-phosphatidylinositol 3-kinase pathways and involves modulation of extracellular matrix proteins. J Biol Chem. 2002;277:49311–49318. doi: 10.1074/jbc.M209284200. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrego PJ. Predictors of conversion to dementia of probable Alzheimer type in patients with mild cognitive impairment. Curr Alzheimer Res. 2006;3:161–170. doi: 10.2174/156720506776383103. [DOI] [PubMed] [Google Scholar]
- Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller G, Jung C, Straub J, Wied S, Kramer W. Induced release of membrane vesicles from rat adipocytes containing glycosylphosphatidylinositol-anchored microdomain and lipid droplet signalling proteins. Cell Signal. 2009;21:324–338. doi: 10.1016/j.cellsig.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Nimmrich V, Reymann KG, Strassburger M, Schoder UH, Gross G, Hahn A, Schoemaker H, Wicke K, Moller A. Inhibition of calpain prevents NMDA-induced cell death and beta-amyloid-induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol. 2010;159:1523–1531. doi: 10.1111/j.1476-5381.2010.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Aβ) oligomerization in an in vivo model of Alzheimer disease. A link between Aβ and tau pathology. J Biol Chem. 2006;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- Panatier A, Vallee J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, Regan S, Kasper T, Peng S, Ding F, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2016;93:215–225. doi: 10.1016/j.nbd.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt J, Roth W, Lacor P, Smith AB, III, Blankenship M, Velasco P, De Felice F, Breslin P, Klein WL. Alzheimer’s-associated Aβ oligomers show altered structure, immunoreactivity and synaptotoxicity with low doses of oleocanthal. Toxicol Appl Pharmacol. 2009;240:189–197. doi: 10.1016/j.taap.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt J, Thorner M, Brautigan D, Larner J, Klein WL. Protection against the synaptic targeting and toxicity of Alzheimer’s-associated Aβ oligomers by insulin mimetic chiro-inositols. FASEB J. 2013;27:199–207. doi: 10.1096/fj.12-211896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, William C, Saqran L, Cagsal-Getkin O, Pitstick R, et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun. 2015;3:14. doi: 10.1186/s40478-015-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- Savina A, Fader CM, Damiani MT, Colombo MI. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic. 2005;6:131–143. doi: 10.1111/j.1600-0854.2004.00257.x. [DOI] [PubMed] [Google Scholar]
- Schmitt LI, Sims RE, Dale N, Haydon PG. Wakefulness affects synaptic and network activity by increasing extracellular astrocyte-derived adenosine. J Neurosci. 2012;32:4417–4425. doi: 10.1523/JNEUROSCI.5689-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals JR, Czech MP. Evidence that insulin activates an intrinsic plasma membrane protease in generating a secondary chemical mediator. J Biol Chem. 1980;255:6529–6531. [PubMed] [Google Scholar]
- Sebollela A, Freitas-Correa L, Oliveira FF, Paula-Lima AC, Saraiva LM, Martins SM, Mota LD, Torres C, Alves-Leon S, de Souza JM, et al. Amyloid-beta oligomers induce differential gene expression in adult human brain slices. J Biol Chem. 2012;287:7436–7445. doi: 10.1074/jbc.M111.298471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Aβ induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597–624) FASEB J. 2006;20:1254–1256. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanesan S, Tan A, Rajadas J. Pathogenesis of Aβ oligomers in synaptic failure. Curr Alzheimer Res. 2013;10:316–323. doi: 10.2174/1567205011310030011. [DOI] [PubMed] [Google Scholar]
- Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008;28:3941–3946. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Hashimoto T, Roe AD, Hori Y, Spires-Jones TL, Hyman BT. Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease. FASEB J. 2013;27:3239–3248. doi: 10.1096/fj.13-229666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, et al. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- Townsend M, Mehta T, Selkoe DJ. Soluble Aβ inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC, Hatcher NG, Schachter J, Renger JJ, Parmentier-Batteur S. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. 2015;35:14234–14250. doi: 10.1523/JNEUROSCI.1523-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco PT, Heffern MC, Sebollela A, Popova IA, Lacor PN, Lee KB, Sun X, Tiano BN, Viola KL, Eckermann AL, et al. Synapse-binding subpopulations of Aβ oligomers sensitive to peptide assembly blockers and scFv antibodies. ACS Chem Neurosci. 2012;3:972–981. doi: 10.1021/cn300122k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal MJ, Stahl PD. The small GTP-binding proteins Rab4 and ARF are associated with released exosomes during reticulocyte maturation. Eur J Cell Biol. 1993;60:261–267. [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–1117. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox KC, Lacor PN, Pitt J, Klein WL. Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol Neurobiol. 2011;31:939–948. doi: 10.1007/s10571-011-9691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox KC, Marunde MR, Das A, Velasco PT, Kuhns BD, Marty MT, Jiang H, Luan CH, Sligar SG, Klein WL. Nanoscale synaptic membrane mimetic allows unbiased high throughput screen that targets binding sites for Alzheimer’s-associated Aβ oligomers. PLoS One. 2015;10:e0125263. doi: 10.1371/journal.pone.0125263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci. 2006;26:10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J Biol Chem. 2012;287:10977–10989. doi: 10.1074/jbc.M111.324616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci. 1992;12:4793–4799. doi: 10.1523/JNEUROSCI.12-12-04793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Lacor PN, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric aβ. J Biol Chem. 2009;284:18742–18753. doi: 10.1074/jbc.M109.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, et al. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.