

Abstract
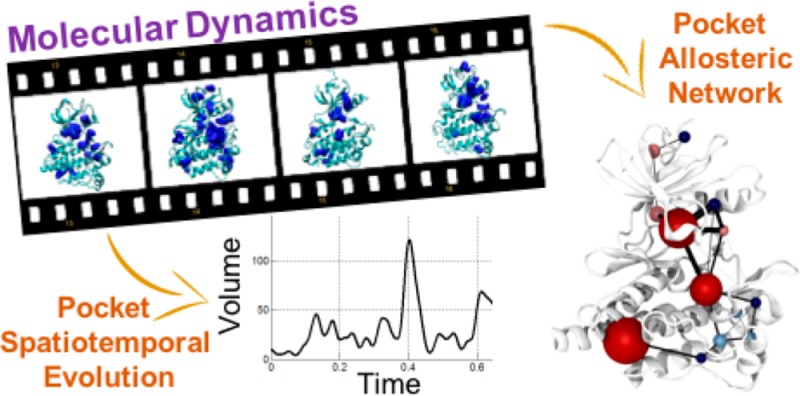
The detection and characterization of binding pockets and allosteric communication in proteins is crucial for studying biological regulation and performing drug design. Nowadays, ever-longer molecular dynamics (MD) simulations are routinely used to investigate the spatiotemporal evolution of proteins. Yet, there is no computational tool that can automatically detect all the pockets and potential allosteric communication networks along these extended MD simulations. Here, we use a novel and fully automated algorithm that examines pocket formation, dynamics, and allosteric communication embedded in microsecond-long MD simulations of three pharmaceutically relevant proteins, namely, PNP, A2A, and Abl kinase. This dynamic analysis uses pocket crosstalk, defined as the temporal exchange of atoms between adjacent pockets, along the MD trajectories as a fingerprint of hidden allosteric communication networks. Importantly, this study indicates that dynamic pocket crosstalk analysis provides new mechanistic understandings on allosteric communication networks, enriching the available experimental data. Thus, our results suggest the prospective use of this unprecedented dynamic analysis to characterize transient binding pockets for structure-based drug design.
Short abstract
Allosteric communication is revealed via protein pocket crosstalk networks, obtained by a novel and fully automated algorithm that examines pockets’ spatiotemporal evolution from extended MD simulations.
Introduction
Binding pockets are often crucial for modulating the function of biomolecules, such as those in protein enzymes and ion channels. For example, small molecule drugs exert their beneficial action by binding to a functional pocket of the protein target(s).1 Detecting and characterizing these functional binding pockets is therefore of paramount importance for biochemistry and drug discovery.2 In this regard, molecular dynamics (MD) is a useful tool for studying the appearance, evolution, and structural modifications of binding pockets in large biomolecules, along trajectories of hundreds of nanoseconds to even a few milliseconds.3,4 MD can also describe the plasticity of those superficial and shallow transient cavities,5 which are often involved in protein function because they interact with a small substrate or another partner protein.6,7
As MD trajectories of large structural ensembles increase in length, they create massive data files. These files can be hundreds of gigabytes in size and are expected to reach tens of terabytes in the near future.8 Therefore, there is a major need for algorithms that can automatically extract the embedded information from these massive data sets and produce intelligible reports on the spatiotemporal evolution of the targeted protein, including its potentially druggable binding pockets.9
There are already a number of algorithms that can detect protein binding pockets in static structures.10,11 Some of these rely on the Voronoi diagrams12 (e.g., MolAxis,13 MOLE14), grids15 (e.g., POCKET,16 PocketFinder,17 POVME18,19), and molecular surfaces and probes (e.g., HOLE,20 SURFNET21). Other algorithms analyze ensembles of structures, but usually require a preliminary structural alignment (e.g., MDpocket,22 PocketAnalizerPCA,23 Epock, Trj_cavity,24 and TRAPP25). In this case, the resulting information may depend on the specific reference structure used for the alignment. Atom-based algorithms (e.g., PROVAR26 and EPOSBP27) avoid the alignment step. Nevertheless, most of these methods are limited to analyzing a priori defined pocket(s) of interest only. Another key aspect is that pockets can sometime take part in protein allosteric signaling.2,28 Indeed, a number of theoretical approaches already exist to investigate allosteric signaling, such as bioinformatics methods that rely on the analysis of protein sequences under the assumption that evolutionarily conserved residues are likely to have a functional role.29 Vibrational motions of proteins examined through normal-mode analysis (NMA) can also offer insights into potential allosteric mechanisms. In this case, low frequency modes define functionally relevant movements often triggered by the binding of an allosteric effector.30,31 Alternatively, allosteric signaling is often investigated through protein conformational ensembles generated via molecular dynamics (MD). These conformational ensembles are mapped into a graph-based representation, which is composed of interconnected nodes. The degree of a node’s interdependence, which reflects correlation of motions of distant allosteric parts of the protein, can be calculated, for example, via a mutual information analysis32,33 or using the analysis of atomic positional fluctuations.34−39
Here, we present an original algorithm for efficiently analyzing extended MD trajectories. Differently from all previous methods, this algorithm detects the formation and spatiotemporal evolution of all the protein pockets. In addition, it monitors pocket crosstalk, defined as the temporal exchange of atoms between adjacent pockets, which we propose as a means to identify allosteric signaling (see Theory). In particular, our algorithm automatically executes (a) an alignment-independent identification of all the pockets on the protein surface; (b) a quantification and visualization of the volume and surface area of all the pockets found in the protein, for each structural frame of the MD trajectory; (c) a report of crosstalk between pockets, described as merging and splitting events; and (d) a detection of allosteric signal transmission networks across the protein surface, defined as interconnected pocket motions (see Theory).
We have applied this new algorithm to long MD simulations of a few selected pharmaceutically relevant targets: (i) the purine nucleoside phosphorylase (PNP) enzyme, (ii) the adenosinic receptor (A2A) in POPC membrane, and (iii) the Abelson (Abl) kinase. The algorithm produced a detailed analysis of the structure–dynamics–function relationships, which it automatically extracted from these MD simulations. Our results demonstrate the power of this dynamical analysis, particularly as a prospective tool for characterizing binding pockets in structure-based drug design.
Theory
Outline of the Method
The algorithm identifies pockets and tracks their evolution over time along an MD trajectory. NanoShaper 0.7 (freely available at www.electrostaticszone.eu),40 is used as a preliminary utility to detect the pockets on the protein surface on individual frames, with particular attention given to those that could be druggable sites. In a single frame of a protein of ∼2000 atoms, the execution time for detecting the binding pockets is in the range of 2–6 seconds on a standard workstation. The main part of the method is then dedicated to characterizing the pocket dynamics along the MD trajectory through the assignment of a unique and dynamics-consistent identifier for every pocket. The spatiotemporal evolution of each pocket is thus monitored over the entire MD trajectory. Both algorithms are described in the following sections. This method is called Pocketron and is a module implemented in the BiKi Life Sciences software suite (www.bikitechnologies.com).
Static Pocket Detection
The static pocket detection algorithm is based on the concept of the solvent excluded surface (SES),41 or Connolly–Richards surface,42 which is defined as the surface obtained by rolling a spherical probe over the van der Waals surface of the molecular system. The analytical computation of the SES is done via the Alpha Shapes theory, as described by Decherchi and Rocchia.40 Then, pockets are identified by calculating the volumetric difference between the regions enclosed by the SESs, obtained with two different probe radii (Supporting Figure 4). The smaller rolling spherical probe has a radius of 1.4 Å, which corresponds to the spherical approximation of a water molecule. Conversely, the larger rolling spherical probe has a default radius of 3 Å. The size values can be modified at will. However, we found that these specific values, together with a subsequent filter selecting only pockets with a volume of at least 34.5 Å3 (∼3 water molecules), provide a reasonable identification of potential binding sites. This information can be used to analyze the entire protein surface, list the identified pockets, and store their calculated volume and the list of contributing atoms.
Dynamical Pocket-Tracking Algorithm and Pocket Crosstalk Detection
The algorithm tracks the atoms forming each pocket along an MD trajectory. These data constitute the fundamental basis for how the algorithm monitors the pocket dynamics. The algorithm monitors all the atom-based events occurring along an MD trajectory, without requiring any structural alignment or prior knowledge of the region to be searched. It thus tracks the exchange of atoms between adjacent pockets, which is our indicator of “pocket crosstalk”. With “exchange of atoms”, we refer to the fact that the same atom may belong to different pockets at different times (i.e., frames) during the simulation. This analysis considers “merging” and “splitting” events to be significant. These events are calculated via simple operations on the atom sets that form each pocket. More specifically, a “merge” event occurs when atoms belonging to the same pocket in the current frame have belonged to separate pockets in a previous frame. Similarly, a “split” event occurs when atoms of a single pocket divide into two or more distinct pockets. To guarantee consistency in pocket tracking, the algorithm initially stores the list of pockets found in the first frame, as defined by their constituent atoms, and assigns a unique pocket identifier (pID) to each stored pocket. Then, for every additional frame, the new set of pockets is computed and compared with the stored ones. When there is a mismatch with respect to the stored pockets, the current pocket is added as a new entry in the stored pocket’s list and assigned a new pID. The matching algorithm relies on the Jaccard index to measure the degree of similarity between two pockets. For example, let A and B be the sets of atoms belonging to two pockets. Then, the Jaccard index between them is defined as
where the modulus symbol indicates the cardinality of the atom sets. If A and B are identical (i.e., have the same constituent atoms), then the index reaches its maximum value of 1. If only a fraction of the atoms are shared between A and B, then the index provides the number of matching atoms (numerator) divided by the cardinality of the union of the atom sets. If there are no matching atoms, the index is null.
Pocket identification is therefore
performed with the similarity matrix M. Each entry M(i,k) is the Jaccard index
between the atoms of the ith stored pocket and those
of the kth pocket (detected in the current frame).
For each frame, each pocket is compared to all of the stored ones.
If M(i,k) = 0 ∀i, then the kth pocket is considered to
be a new pocket and is stored. Otherwise, the kth
pocket is connected to the stored entry that maximizes the Jaccard
index. In mathematical terms, pi is the ith stored pocket and 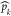 (t) is the kth detected pocket on the frame at time t; therefore,
the M matrix is the following:
(t) is the kth detected pocket on the frame at time t; therefore,
the M matrix is the following:
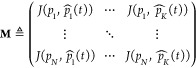 |
where K is the total number of detected pockets in the current frame and N is the total number of stored pockets so far. As already mentioned, the pocket ID assignment is executed by finding the maximum entry for every column of M:
where the index wk is corresponding to the stored pocket to which k is assigned. If M(wk,k) = 0, a new entry is created and stored in the pocket list with its own new pID.
Similarly, to detect merging and splitting events, the algorithm builds the matrix F, so that F(k,j) is the Jaccard index of the kth pocket at instant t and the jth pocket found in the previous frame at instant t – Δt. Then, the F matrix is
 |
where J is now the total number of pockets at the t – Δt instant (i.e., the previous frame presented to the algorithm). Traversing this matrix allows the easy detection of merge and split events. Indeed, if we let “NZ” be the operator that returns all the nonzero entries of a row/column vector, and let “(i,:) ” and “(:,i)” be the operators that return all the elements of the ith row/column, respectively, then we have
where Merge(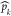 (t)) is the set
containing
all the indexes of the pockets detected at the time (t – Δt) that shared some atoms with
pocket k. If this set is empty, then no merge event
involved
(t)) is the set
containing
all the indexes of the pockets detected at the time (t – Δt) that shared some atoms with
pocket k. If this set is empty, then no merge event
involved 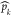 (t). Similarly, Split(
(t). Similarly, Split(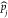 (t – Δt)) is the set containing all the indexes of the pockets
detected at the at time t that shared some atoms
with the pocket labeled as j at time (t – Δt). If this set is empty, then
no split event involved
(t – Δt)) is the set containing all the indexes of the pockets
detected at the at time t that shared some atoms
with the pocket labeled as j at time (t – Δt). If this set is empty, then
no split event involved  (t – Δt). On a technical note, the entries of the F matrix involve only those pockets detected at the t and t – Δt instants.
However, once the merging and splitting events have been detected,
the M matrix is once again used to connect these pockets
to the stored ones and to make the tracking consistent, as described
above.
(t – Δt). On a technical note, the entries of the F matrix involve only those pockets detected at the t and t – Δt instants.
However, once the merging and splitting events have been detected,
the M matrix is once again used to connect these pockets
to the stored ones and to make the tracking consistent, as described
above.
Pocket Crosstalk Analysis and Allosteric Signaling
With the above-mentioned ability to track pockets over time, the volume and surface area of all pockets are stored for each frame. Thus, the algorithm generates the full history of the volume, area, splitting, and merging events of each pocket, along the MD trajectory. It also generates the list of contributing atoms and residues for all the unique pockets detected during the MD run. This allows the estimation of the probability (estimated as a relative frequency) that each residue belongs to a given pocket. Moreover, for each pocket, all the merging and splitting events with other pockets are stored. Indeed, one of the final results of the tracking process is a square matrix called NM, whose (i,j)th entry represents the merging probability between two pockets, expressed as the number of times they merge over the total number of frames. Similarly, the square matrix NS expresses the corresponding statistics for the splitting events. Empirically, we found that the corresponding entries of NS and NM very often coincide in magnitude, indicating that the splitting and merging events often occur at the same time. For this reason, we define a matrix of aggregate statistics N, which contains the maximum value of the corresponding entries in NM and NS (Figure 1). The nature of the information stored in N calls for effective graphical methods to represent the pocket dynamics, particularly since N is usually very sparse. The algorithm translates the merging and splitting frequency matrix N into a 3D network graph, where the nodes represent pockets, and the edges indicate communication between two pockets or, in other words, how often two pockets exchange atoms. In detail, the position of each node is the geometric center of the atoms that form the corresponding pocket. The color of the node indicates its persistency over the simulation time. The red color indicates high persistency whereas blue indicates low persistency. The thickness of the edge is directly proportional to the frequency of the merging and splitting events between the two connected pockets, according to the corresponding entry in the N matrix. The dimension of the sphere represents the pocket volume (Figure 1 and Figure 6). Together with the merging and splitting of adjacent pockets, the N matrix returns indirectly also the long-range crosstalk network connecting distant pockets. That is, a crosstalk network can connect distant pockets through a sequence of neighboring pockets that exchange their atoms during the MD run. In this way, a 3D representation of the crosstalk network can be observed, which can be used to identify putative allosteric signaling network.
Figure 1.

(A) Representation of the merging and splitting matrix F, calculated using all the detected pockets. The matrix F allows retrieving information on merging and splitting events. For each frame along the MD trajectory, pockets at time t are compared with pockets at time t – Δt, using the Jaccard index. In this example, at time t, the pockets 1, 2, 3, and 4 (in rows) have been detected and stored. At this point, the Jaccard index is computed with all pockets detected in the previous frame at times t – Δt, i.e., with pockets 1, 2, and 3 (in columns). Moving from t – Δt to t, this example shows that pocket 1 split into two pockets, forming the new pocket 4. Concomitantly, pockets 2 and 3 merged, forming a larger pocket that is still identified as pocket 3, according to its Jaccard index. (B) Schematic example of the conversion of the aggregate merging/splitting statistics N into an undirected network graph. In the matrix N, the off-diagonal red numbers indicate the frequency of the merging and splitting events, which is then reflected by the size of the edge connecting two pockets.
Figure 6.

Networks of the most persistent pockets found in the KDin, T315I-KDin, Myr/KDin, and Myr/T315I-KDin trajectories. Each pocket (i.e., network node) is represented as a sphere, with the different colors indicating the pocket’s persistency. The pockets are connected via black lines (i.e., network edges). The width of each edge is proportional to the communication frequency. The networks connect the ATP and the myristate binding sites in all systems except T315I-KDin. We performed our analysis considering only pockets having a persistency of at least 20% and above, along the simulation time.
Results
Protein Binding Pocket Detection and Dynamics
First, we tested the ability of our new algorithm to identify and monitor protein pocket dynamics. We used ∼700 ns long MD simulations of the purine nucleoside phosphorylase (PNP), a homotrimeric enzyme that is involved in purine metabolism and T-cell function.43 PNP inhibition is a strategy for treating T-cell-mediated diseases, such as leukemia and lymphoma.44,45
The algorithm detected a total of 22 pockets that exist for more than 30% of the overall simulations describing the dynamic docking of DADMe-ImmH into PNP. Among the most persistent ones, our algorithm identified three large pockets, namely, pocket ID (pID) 4, pID 9, and pID 12. The persistency is ∼79% for pID 4 and ∼96% for both pID 9 and pID 12. These three pockets lie at the edge between the adjacent PNP monomers (Figure 2 and Supporting Figure 1). Each of them corresponds to the known orthosteric binding site that is targeted by the endogenous substrate as well as by known PNP inhibitors such as Immucillin-H46 and DADMe-ImmH.47 These pockets have an average volume of 367, 894, and 977 Å3, respectively. These volumes are larger than those observed in the holo PNP crystal, where they are 332, 327, and 409 Å3, respectively. This suggests an elevated plasticity of the orthosteric pockets. Notably, the reduced volume of pID 4 is explained by the existence of a nearby stable pocket, pID 13, which has a persistency of 94% and an average volume of 313 Å3 (Supporting Figure 1). These two pockets are merged for ∼26% of the simulation time, resulting in a single larger binding pocket with an average volume of 677 Å3, which is comparable with the other two orthosteric pockets pID 9 and pID 12. Furthermore, our algorithm detected two smaller pockets, pID 3 and pID 14, located in proximity of the orthosteric binding pockets pID 4 and pID 12 (Figure 2). These pockets are present for 71% and 73% of the simulation time and have an average volume of 133 Å3 and 144 Å3, respectively, in agreement with their value in the PNP X-ray structure (203 Å3 and 232 Å3, respectively). Interestingly, these pockets have been shown to constitute a prebinding site where the inhibitor DADMe-ImmH transiently binds before accessing one of the orthosteric sites.48 Notably, this prebinding site was not found in proximity of the orthosteric pocket pID 9. Likely, this is because pID 9 already embeds the prebinding site during our simulations, as suggested by its larger volume compared to pID 4 and pID 12.
Figure 2.

(A) Localization of the main pockets computed for the PNP X-ray structure 3K8O. On the right, the orthosteric ligand DADMe-ImmH located in the orthosteric pocket (in orange), as in the X-ray structure, and in the prebinding pocket (in yellow), as found in our MD simulations. (B) Volume over time of the three orthosteric sites pID 4, pID 9, and pID 12. The volumes have been smoothed employing a Gaussian filter.
Another pocket, namely, pID 22, is detected at the center of the trimerization interface, with an average volume of 398 Å3 (248 Å3 in the X-ray structure) and a time persistency of 88%. Intriguingly, this pocket shows a marked crosstalk pattern with the orthosteric binding site of each monomer during the MD simulations (Supporting Figure 2). This suggests a possible signaling transmission network between the different monomers, mediated by this common interface pocket. This may explain the negative cooperativity between PNP subunits observed by Schramm and co-workers49,50 (see Discussion).
Identification of Interaction Patterns between Pockets in Proteins
Our algorithm detects pocket interactions by observing pocket splitting and merging during an MD simulation (see Theory). Here, we demonstrate this feature on two proteins with multiple binding pockets, which are well-characterized with biochemical and structural data (see below). These proteins are (1) the adenosinic receptor A2A and (2) the Abelson (Abl) tyrosine kinase. The adenosinic receptor A2A is a G-protein coupled receptor (GPCR) with a recognized key role in several pathophysiological processes.51 It is a promising target for pain, depression,52 and neurological diseases such as Parkinson’s disease.53 The Abelson (Abl) tyrosine kinase is involved in cell growth and survival.54 It is a validated target for treating several types of cancer.55
Adenosinic Receptor A2A
The analysis performed on 100 ns long A2A trajectories revealed 13 pockets that were present for more than 30% of the simulations. Of these, pID 15 and pID 22 coincide with the structurally characterized orthosteric and allosteric binding sites, respectively.56,57 In particular, pID 15 is located on the extracellular side of the receptor and is targeted by both agonist and antagonist drugs, such as adenosine58 and ZM241385.59 In contrast, pID 22 is located in the core of the transmembrane bundle and is normally occupied by a sodium ion, which can be displaced by allosteric drugs, such as amiloride and HMA (Figure 3).60,61 Our analysis revealed that, although pID 15 and pID 22 were well-separated during most of the MD simulations, they sometimes communicated via merging and splitting events. This means that these pockets share a set of residues at their interface. These residues are Val 84, Ala 88, Phe 242, and Trp 246 (Figure 3A). They are involved in a crosstalk between these pockets. Interestingly, both MD simulations and experimental studies have demonstrated a ligand-dependent A2A signaling, which involves an allosteric effect via these two pockets.60,62,63
Figure 3.

(A) Time persistency of residues that define the orthosteric pocket (blue stem, pID 15) and the allosteric pocket (red stem, pID 22). (B) Volume plot of pID 15 and pID 22 in selected frames and representation of merging and splitting events.
For example, at frame 844 (out of 1000 frames analyzed), the larger orthosteric pocket pID 15 joins the allosteric pID 22 (Figure 3B). Interestingly, when these two pockets are merged, they allow a small channel to form (Figure 3 and Figure 4). As suggested by thermal stability studies60 and MD simulations,64 this channel permits the passage of a sodium ion from the extracellular side to the allosteric pocket. The residues at the edge between the two pockets may therefore act as a gate, mediating this channel’s opening and closing, and thus modulating the entrance of the sodium ion. This would also account for the conformation flexibility of Trp 246, known as the “toggle switch” residue, which is associated with the activation mechanism of GPCRs.56,59,65,66 Notably, the conformational flexibility of residues that lie between two pockets has been shown to be crucial also in other enzymes, such as those processing lipids (e.g., fatty acid amide hydrolase67,68 and monoacylglycerol lipase7).
Figure 4.
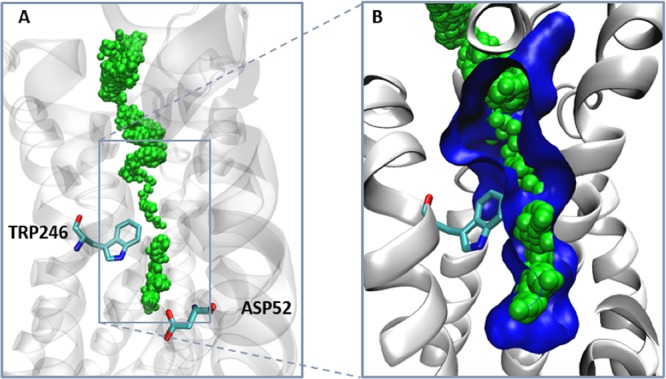
Pocket crosstalk analysis reveals that when pID 15 merges with the allosteric pID 22, a small channel is formed (Figure 3). We used adiabatic biased simulations to characterize the passage of a sodium ion through the transient channel detected by our algorithm (see Supporting Information). (A) The green spheres indicate the pathway followed by the sodium ion along the simulations, toward the extracellular site. The two conserved residues Trp246 and Asp52 are shown. In panel B we show the narrowest section of the channel, with the gating Trp246 partially flipped so as to allow ion passage.
We used adiabatic bias simulations to test the passage of the sodium ion through the transiently formed channel detected via pocket crosstalk analysis (Figure 4); although qualitative, our results indicate that this channel can allow the sodium ion to access the protein through a continuous and sterically accessible pathway (Figure 4). While the calculation of the associated thermodynamics would require more extended simulations, it is interesting to note that this pathway well resembles the one shown by Yuan et al., identified along 9.6 μs of plain MD simulation.69
Abelson (Abl) Tyrosine Kinase
Here, we analyzed the MD simulations of the kinase domain (KD) of Abl, both in the catalytically active DFG-in (KDin) and in the catalytically inactive DFG-out (KDout) conformations (see Methods for details).70 Among the most persistent pockets in KDin, our algorithm successfully identified the large ATP site (pID 5, see Supporting Figure 3).71 In addition to pID 5, we found another nearby pocket located between the DFG motif and the αC helix (pID 3, see Supporting Figure 3), which is known to be an allosteric site located in the KD.72 Interestingly, pID 5 and pID 3 present a marked crosstalk in our MD simulations, sharing a large set of residues, which are listed in Supporting Table 1. For ∼63% of the simulation, they remain separate, with volumes of 439 Å3 and 167 Å3, respectively. However, for ∼35% of the simulations, the two pockets merge into a single larger pocket with an average volume of 497 Å3 (Figure 5).
Figure 5.
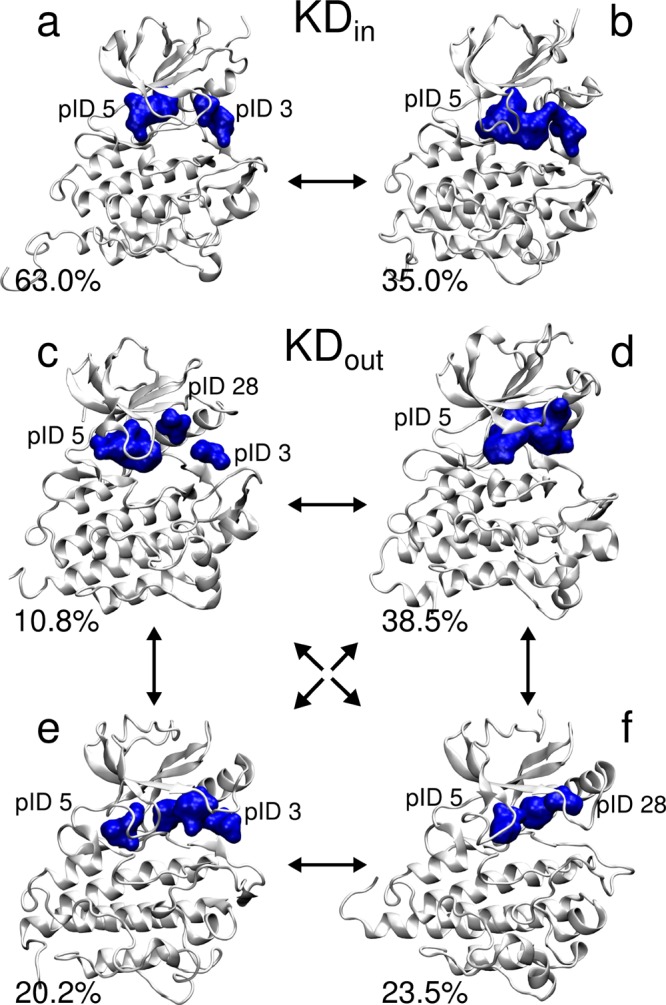
Representation of the dynamical behavior of the ATP pockets during MD simulations of the KDin (a, b) and KDout systems (c–f). In KDin, the ATP pocket pID 5 coexists with the nearby pID 3 for 63.0% of the simulations (a), while pID 5 is the only pocket for 35% of the simulations (b). In KDout, the ATP pocket coexists with pID 3 and pID 28 for 11% of the MD simulations (c). It is the only emerging pocket for 38% of the simulations (d), and it coexists with pID 3 for 20% of the simulations (e) and with pID 28 for 23% of the simulations (f).
In KDout, our algorithm again identified the ATP pocket pID 5 and the nearby allosteric pocket pID 3 (Supporting Figure 3), with an average volume of 339 Å3 and 222 Å3, respectively. However, the algorithm identified also an additional pocket, pID 28, which is located between the P-loop and the β3-αC-helix loop (Supporting Figure 3), and which has a smaller volume of 150 Å3. Notably, the region containing this new pocket is targeted by a new class of diphenylamine-derived allosteric inhibitors of MEK 1 and MEK 2 kinases.73 Here too, the three pockets (pID 3, pID 5, and pID 28) share a set of residues (listed in Supporting Table 1), suggesting a communication network. Interestingly, in this case, these three pockets exist as individual entities for only ∼11% of the overall simulated time. This is because merging events are more frequent than in the catalytically active KDin system (Figure 5).
According to our analysis, the DFG-in conformation limits the crosstalk between the ATP pocket and the nearby pocket pID 3 in the catalytically active KDin system. In contrast, in KDout, the DFG-out conformation favors communication with the surrounding pockets pID 3 and pID 28. Indeed, the DFG-out conformation has been demonstrated to increase the flexibility of the ATP binding site, which leads to a loss of KD activity.74,75 Thus, our analysis suggests that the specific DFG conformation (in or out) affects the formation of transient pockets and their communication network. These, in turn, modulate the ATP binding site’s overall shape and the resulting KD activity.
Allosteric Signal Transmission Networks Inferred from Pocket Crosstalk Analysis
Here, we tested the capability of our algorithm to reveal allosteric signal propagation pathways in proteins, starting from an MD-generated equilibrium ensemble of structures. As a paradigmatic example, we investigated the Abl kinase, which is one of the most characterized systems in terms of molecular determinants for protein allostery.74−77 We performed a comparative analysis of four different Abl systems: (1) the wild-type KDin form, (2) KD bound to myristate (Myr/KDin), (3) the KD apo form of the T315I mutation (T315I-KDin), and (4) system T315I-KDin bound to myristate (Myr/T315I-KDin).
In all systems, our analysis detected both the orthosteric ATP pocket and the allosteric myristate pocket, which is located in the C-lobe of the KD.78 The analysis also returned an intensive crosstalk between pockets distributed on the protein surface. This was detected by looking at the off-diagonal elements of the square matrix N (see Theory). Notably, this crosstalk network connects the distal orthosteric ATP and the allosteric myristate binding sites (Figure 6), which is known to be central for the allosteric signal propagation in Abl.79−82 In particular, in both KDin and Myr/KDin, the ATP and the myristate pockets are connected via a long-range crosstalk network composed of a number (∼3 to 5) of small transient pockets (average volume ∼90 Å3, and persistency between 20% and 70%). These pockets are mainly located in the C-lobe and mostly involve the αG and αI helices (Figure 6). Notably, the location of the pockets corresponds to that of the new allosteric pockets reported by Shan et al. in their study on binding pathways in Src kinase.83 Moreover, in the MD simulations of Abl with the drug-resistant T315I point mutation (T315I-KDin system), the ATP ↔ myristate long-range crosstalk network is actually disrupted (Figure 6). The perturbation of this communication pathway in the T315I-KDin system might explain the dysregulation of the T315I kinase form.84,85 Remarkably, however, the ATP ↔ myristate pockets’ long-range communication network is restored in our simulations when the myristate binds to the T315I Abl mutated form (i.e., the Myr/T315I-KDin system). This is also observed in the wild-type system and is in line with other experimental and computational studies showing that myristate mimesis elicits a structural rearrangement on the ATP pocket and influences the binding affinity of orthosteric inhibitors (see Supporting Information).77,79 This allosteric communication network could also explain the results of HX MS experiments, which further detailed the presence of an allosteric-signal-propagation pathway from myristate to the ATP pockets, passing through KD structural elements such as the αI-helix.79
As a following step, we aimed at connecting such a mechanism for allosteric signaling with useful information for ligand design. Toward this end, we performed enhanced sampling MD simulations to test whether this network for allosteric signaling affects ligand binding at the orthosteric site. Thus, we performed a set of scaled-MD simulations86 on the T315I mutant of Abl, either in complex with dasatinib bound at the orthosteric site (i.e., Das/T315I-KDin) or in complex with both dasatinib and the myristate, at the distal myristate binding pocket (i.e., Das-Myr/T315I-KDin). Scaled-MD simulations can be used to accelerate the unbinding process by reducing the atomic interactions in the potential energy of the system. While facilitating the unbinding from the pocket during the MD run, scaled-MD simulations allow also a qualitative and relative estimation of the ligand residence time.86,87 Here, we used scaled-MD simulations to estimate the residence time of dasatinib in Das-Myr/T315I-KDin and Das/T315I-KDin systems, which only differ in the presence of the myristate (Figure 6). In these simulations, dasatinib showed an average scaled residence time ∼2 times longer when unbinding from Das-Myr/T315I-KDin compared to when it unbinds from Das/T315I-KDin (see Supporting Table 2). Converted back to nonscaled residence times, (see ref (86) for further details) dasatinib in the two systems returns a ratio value of more than 4. Although only qualitative, this result further indicates that the presence of the myristate, which coincides with the formation of the crosstalk signaling network connecting the orthosteric ATP and the allosteric myristate pockets (Figure 6), stabilizes the complex with dasatinib. Notably, biochemical assays confirm that dasatinib is active against the T315I Abl form only in the presence of an allosteric binder.79 Moreover, the residence time of a drug often correlates with its binding affinity, due to the relationship between the rate of dissociation (koff) and the thermodynamic dissociation constant (Kd). Hence, a longer residence time suggests a greater affinity of the drug for its target. Taken together, these results verify binding cooperativity effects in the Abl system, in which the presence of an allosteric binder at the myristate pocket is shown to impact on ligand binding at the orthosteric site. This example suggests that the information generated by dynamic pocket crosstalk analysis can help identify suitable conformational states for ligand binding thermodynamics obtained through subsequent calculations.
Discussion
In this work, we provide new mechanistic insights on pocket dynamics and allosteric communication in three pharmaceutically relevant proteins, namely, the purine nucleoside phosphorylase (PNP) enzyme, the adenosinic receptor (A2A), and the Abelson (Abl) kinase. Importantly, these findings are obtained through the use of a novel algorithm that analyzes pocket crosstalk events along the dynamics of a given biomolecular system. In this study, we demonstrate that this method can effectively reveal allosteric communication networks embedded in the ever-longer available MD trajectories. In addition, this new method characterizes all binding pockets through site-centered descriptors, which can be used in machine-learning-based virtual screening protocols with enhanced predictivity.3
First, the algorithm identified all three experimentally known orthosteric binding sites in the PNP homotrimeric enzyme (pID 4, pID 9, and pID 12 in Figure 2).44 As expected, these are the largest and most persistent pockets. The dynamical analysis also identified smaller pockets in proximity of these three sites. Notably, these auxiliary pockets were involved in a prebinding state of DADMe-ImmH, a PNP inhibitor,47 which was found to transiently interact with this small pocket before entering the main orthosteric site.48 Analysis of the volume variation over time pointed to an elevated plasticity of the orthosteric binding sites. In fact, their average volume is higher than the crystallographic corresponding value. This suggests that the pockets undergo a structural rearrangement, which likely facilitates the binding of inhibitors observed during the simulations.48 Our analysis also revealed a marked pattern of communication between each orthosteric pocket and its nearby prebinding site, reflected by a high frequency of merge and split events. Interestingly, this evidence is in line with structural studies, which show that two of the binding site’s structural motifs (the His 257 helix and His 64 loop) undergo a major conformational alteration upon the binding of the inhibitor. This alteration affects the binding pocket’s size and shape.49 In addition, the high flexibility of this structural region in PNP was recently used to demonstrate that MD can be effectively deployed to improve virtual screening results.3
Our algorithm also identified a long-range communication pathway connecting the orthosteric pockets and another pocket at the center of the trimerization interface (pID 22). This pathway suggests a possible allosteric connection between the three orthosteric pockets that passes through pID 22, at the trimerization interface. Intriguingly, this allosteric signal may help explain the negative cooperation between PNP subunits, as observed by Schramm and collaborators.49,50 In H/D exchange experiments, dynamical coupling has also been observed between the orthosteric sites and the pocket located at the trimerization interface.88 This further corroborates our hypothesis, according to which pID 22 could be the hub of the allosteric signal transmission between PNP monomers.
We then analyzed the MD trajectory of A2A, simulated in POPC membrane. This analysis captured the experimentally known pockets, which are the orthosteric binding site, located close to the extracellular side, and the underlying sodium allosteric site (Figure 3).56,57,59 Our analysis also showed that, during the simulation, the two pockets communicated through a set of residues located at the interface of the two sites (i.e., Val 84, Ala 88, Phe 242, and Trp 246). This result is in line with earlier experimental and computational studies, which hypothesized the presence of an allosteric signal between the two cavities, which is crucial for modulating A2A function.60,64,89 In this regard, we found that the conformational flexibility of Trp 246, also known as the “toggle switch”, appears to be crucial for the formation of a small channel during a two-pocket merging event. Via this channel, the Na+ ion may access the inner part of the protein from the extracellular space, as experimentally reported (Figure 3 and Figure 4).56,60,90
Merging and splitting events were also shown in our MD simulations of the Abl kinase. Here, we focused on the orthosteric ATP site, comparing the results obtained from analyzing the catalytically active DFG-in kinase domain and the catalytically inactive DFG-out kinase domain (KDin and KDout, respectively). Together with the ATP site, our algorithm correctly detected two additional smaller adjacent pockets. One is an allosteric pocket often exploited by type II kinase inhibitors, while the other is present only in KDout, lying between the P-loop and the β3-αC-helix loop (Figure 5). Notably, this region is targeted by a new class of MEK1 and 2 kinase inhibitors.73 This pocket also accommodates the piperazine–phenyl–pyrimidine moiety of SCH772984 in both ERK1 and ERK2 kinases.91 This confirms the ability of our algorithm to identify small hidden cavities that can be exploited in the design of new selective inhibitors. Notably, our analysis captured even subtle protein conformational changes, such as split and merge events between the large ATP site and the two nearby subpockets. These events are more frequent in the inactive KDout than in the active KDin form. This intensive crosstalk reflects the high plasticity of the ATP binding site in KDout, as suggested by previous computational studies,74,75 which results in a diminished catalytic activity.
Lastly, the method was able to reveal a long-range connection between the orthosteric ATP binding site and the allosteric myristate pocket. This has frequently been reported in the literature as a crucial allosteric mechanism for kinase function.79−82 Our analysis reveals a pocket crosstalk network, which passes through a set of small pockets located close to the αG and αI helices of the C-lobe, in a region that can be targeted by allosteric kinase inhibitors, as suggested by a recent computational study (Figure 5).83 Computational and experimental studies76,77,79 suggest that the integrity of this communication network coincides with a proper functioning of Abl. Targeting this pocket communication network may thus offer a new way for modulating the activity of the protein, as exemplified here for the binding of dasatinib to Abl, and its allosteric modulation.79 Indeed, both HX MS experiments and MD simulations indicated that the αG and αI helices are involved in the allosteric signal propagation in Abl.79 Our analysis detected a perturbation of this pocket communication network in the dysregulated T315I Abl form only, which occurs at the level of the αG and αI helices of the C-lobe (Figure 6). That is, we found that the T315I point mutation in the absence of the myristate interrupts this pocket crosstalk network, which might explain the dysregulation of the T315I Abl form.84 Ultimately, within the sampling limitations of the input trajectories, this new algorithm’s dynamical analysis detects interaction networks, which involve rearrangements that may reveal new binding sites on the protein surface.92 It can also point to possible mechanisms for allosteric signal propagation, spotting protein configurations that may be used as target structures for ligand binding thermodynamics through computations for structure-based drug design.93
Conclusions
In this work, we provide new mechanistic understandings about pocket formation and allosteric communication in three relevant drug discovery targets, namely, PNP, A2A, and Abl kinase. Importantly, these findings are obtained through the use of an original and fully automated method for analyzing pocket crosstalk along microsecond-long MD simulations, which are performed to examine the spatiotemporal evolution of proteins. We demonstrate that this unprecedented dynamical analysis can reveal otherwise hidden connections between pockets, which may also underlie allosteric communication networks in proteins, as discussed for the biomolecular systems here investigated. Ultimately, we propose dynamic pocket crosstalk analysis for a more detailed understanding of the structural dynamics of proteins and biological regulation through allosteric communication, suggesting a prospective use of this method in structure-based drug design.
Methods
Structural Models
In the present work, we analyzed the MD simulations of three different systems, namely, purine nucleoside phosphorylase (PNP), the adenosinic receptor (A2A), and the Abelson (Abl) kinase. The homotrimeric construct of PNP was modeled using 3K8O X-ray structures and was simulated in the presence of nine DADME-ImmH ligands and phosphate ions, retrieved from 1RSZ and 1RR6(94) PDB structures, respectively. The A2A receptor was mostly built using the 3UZC X-ray structure,95 while the 4EIY X-ray structure56 was used as a template for the missing ECL2 in 3UZC. The apo protein was embedded in POPC membrane. For Abl, we built five different model systems. Two included the apo wild-type KD alone in both the DFG-in (KDin) and DFG-out conformations (KDout). Two comprised the KD mutated at the gatekeeper residue T315 (located at the ATP binding site) with an isoleucine residue. These mutated forms are either in the apo form (T315I-KDin) or in complex with myristate (Myr/T315I-KDin). The last system is the wild-type KD in complex with myristate (Myr/KDin). The KDout system was built starting from the 1OPL chain B X-ray structure96 after removal of the SH2 domain. The remaining systems were modeled starting from the 2F4J X-ray structure.97 For further details, see the Supporting Information.
MD Simulations
PNP was parametrized using the Amber ff99SB-ILDN force field,98 while the General Amber Force Field (GAFF)99 was used to parametrize the ligands and the ions. Ligand partial charges were fitted using the RESP procedure via Antechamber.100 The system was immersed in a TIP3P water box101 and comprised ∼100,000 atoms. After 350 ns of equilibration, which led the system to a temperature of 300 K and to a pressure of 1 bar, we ran the production in NVT ensemble. For the analysis, we collected ∼700 ns of simulation stored in ∼1800 frames.
A2A was parametrized using the Amber ff99SB-ILDN force field.98 The protein and the POPC membrane were immersed in a TIP3P water box101 reaching a total of 65,802 atoms. Here too, the system was first equilibrated to reach a temperature of 300 K and a pressure of 1 bar, while the production was run for 100 ns in NPT ensemble. We extracted ∼1000 frames from the trajectory for analysis. For adiabatic bias simulations, we used Plumed2.102 The reaction coordinate was the distance between the sodium ion and the geometric center of the alpha carbons of Leu267, Asp175, and Gln163 (pdb 3EML). The line connecting this geometric center and the sodium ion is quasi parallel to the principal axis of the GPCR and thus is a reasonable steering coordinate. The spring constant was set to 500 kJ/mol/ Å2.
The five Abl kinase systems were parametrized using the Amber ff99SB force field.103 For Myr/T315I-KDin and Myr/KDin models, the myristate was parametrized using the General Amber Force Field (GAFF)99 after computing the point charges at the HF/6-31G* level of theory. The systems were solvated in TIP3P water and comprised ∼45000 atoms. After 5 ns of equilibration, the systems reached a temperature of 300 K and a pressure of 1 bar, and we then ran the production in NPT ensemble for a simulation time that ranged from ∼0.8 μs to ∼2.5 μs. Here, our algorithm was used to analyze ∼16,000 to ∼50,000 frames. See the Supporting Information for more details.
Algorithm Parameters
For each analysis, we set the small and large probe radii to 1.4 and 3 Å, respectively. To avoid unnecessary noise due to the detection of very small pockets, we set the minimum volume of a detectable pocket to the equivalent of 5 water molecules for PNP and A2A, and 4 water molecules for Abl kinase. Finally, for tracking purposes, we removed all the ligands, ions, membrane, and water molecules from each trajectory, taking into account the sole protein.
Acknowledgments
M.D.V. is supported by the Italian Association for Cancer Research (AIRC) [“MFAG” Grant No. 18883]. We acknowledge the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support. We thank Grace Fox for proofreading and copyediting the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00211.
Experimental details (PDF)
Author Contributions
‡ G.L.S. and S.D. contributed equally.
The authors declare the following competing financial interest(s): M.D.V. is scientific advisor, while S.D. and W.R. are shares owners of BiKi Technologies s.r.l., a company that commercializes software solutions for medicinal chemistry including the presented method.
Supplementary Material
References
- Bruce A.; Johnson A.; Lewis J.; Raff M.; Roberts K.; Walter P.. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, 2002. [Google Scholar]
- Stank A.; Kokh D. B.; Fuller J. C.; Wade R. C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49 (5), 809–815. 10.1021/acs.accounts.5b00516. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Benedetti P.; Decherchi S.; Rocchia W.; Cavalli A.; Alcaro S.; Ortuso F.; Baroni M.; Cruciani G. A Pipeline To Enhance Ligand Virtual Screening: Integrating Molecular Dynamics and Fingerprints for Ligand and Proteins. J. Chem. Inf. Model. 2015, 55 (10), 2256–2274. 10.1021/acs.jcim.5b00169. [DOI] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59 (9), 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- Oleinikovas V.; Saladino G.; Cossins B. P.; Gervasio F. L. Understanding Cryptic Pocket Formation in Protein Targets by Enhanced Sampling Simulations. J. Am. Chem. Soc. 2016, 138 (43), 14257–14263. 10.1021/jacs.6b05425. [DOI] [PubMed] [Google Scholar]
- Eyrisch S.; Helms V. What Induces Pocket Openings on Protein Surface Patches Involved in Protein–protein Interactions?. J. Comput.-Aided Mol. Des. 2009, 23 (2), 73–86. 10.1007/s10822-008-9239-y. [DOI] [PubMed] [Google Scholar]
- Riccardi L.; Arencibia J. M.; Bono L.; Armirotti A.; Girotto S.; De Vivo M. Lid Domain Plasticity and Lipid Flexibility Modulate Enzyme Specificity in Human Monoacylglycerol Lipase. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862 (5), 441–451. 10.1016/j.bbalip.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Stone J. E.; Vandivort K. L.; Schulten K.. Immersive Out-of-Core Visualization of Large-Size and Long-Timescale Molecular Dynamics Trajectories. In Lecture Notes in Computer Science (LNCS); Bebis G., Boyle R., Parvin B., Koracin D., Wang S., Kyungnam K., Benes B., Moreland K., Borst C., DiVerdi S., Yi-Jen C., Ming J., Eds.; Springer: Berlin, Heidelberg, 2011; pp 1–12. [Google Scholar]
- Ribeiro A. A. S. T.; Ortiz V. A Chemical Perspective on Allostery. Chem. Rev. 2016, 116 (11), 6488–6502. 10.1021/acs.chemrev.5b00543. [DOI] [PubMed] [Google Scholar]
- Krone M.; Kozlíková B.; Lindow N.; Baaden M.; Baum D.; Parulek J.; Hege H.-C.; Viola I. Visual Analysis of Biomolecular Cavities: State of the Art. Comput. Graph. Forum 2016, 35 (3), 527–551. 10.1111/cgf.12928. [DOI] [Google Scholar]
- Pérot S.; Sperandio O.; Miteva M. A.; Camproux A.-C.; Villoutreix B. O. Druggable Pockets and Binding Site Centric Chemical Space: A Paradigm Shift in Drug Discovery. Drug Discovery Today 2010, 15 (15), 656–667. 10.1016/j.drudis.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Manak M.; Zemek M.; Szkandera J.; Kolingerova I.; Papaleo E.; Lambrughi M. Hybrid Voronoi Diagrams, Their Computation and Reduction for Applications in Computational Biochemistry. J. Mol. Graphics Modell. 2017, 74, 225–233. 10.1016/j.jmgm.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Yaffe E.; Fishelovitch D.; Wolfson H. J.; Halperin D.; Nussinov R. MolAxis: Efficient and Accurate Identification of Channels in Macromolecules. Proteins: Struct., Funct., Genet. 2008, 73 (1), 72–86. 10.1002/prot.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petřek M.; Košinová P.; Koča J.; Otyepka M. MOLE: A Voronoi Diagram-Based Explorer of Molecular Channels, Pores, and Tunnels. Structure 2007, 15 (11), 1357–1363. 10.1016/j.str.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Goodford P. J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28 (7), 849–857. 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Levitt D. G.; Banaszak L. J. POCKET: A Computer Graphies Method for Identifying and Displaying Protein Cavities and Their Surrounding Amino Acids. J. Mol. Graphics 1992, 10 (4), 229–234. 10.1016/0263-7855(92)80074-N. [DOI] [PubMed] [Google Scholar]
- An J.; Totrov M.; Abagyan R. Pocketome via Comprehensive Identification and Classification of Ligand Binding Envelopes. Mol. Cell. Proteomics 2005, 4 (6), 752–761. 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; de Oliveira C. A. F.; McCammon J. A. POVME: An Algorithm for Measuring Binding-Pocket Volumes. J. Mol. Graphics Modell. 2011, 29 (5), 773–776. 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; Votapka L.; Sørensen J.; Amaro R. E. POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics. J. Chem. Theory Comput. 2014, 10 (11), 5047–5056. 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S.; Neduvelil J. G.; Wang X.; Wallace B. A.; Sansom M. S. P. HOLE: A Program for the Analysis of the Pore Dimensions of Ion Channel Structural Models. J. Mol. Graphics 1996, 14 (6), 354–360. 10.1016/S0263-7855(97)00009-X. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A. SURFNET: A Program for Visualizing Molecular Surfaces, Cavities, and Intermolecular Interactions. J. Mol. Graphics 1995, 13 (5), 323–330. 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- Schmidtke P.; Bidon-Chanal A.; Luque F. J.; Barril X. MDpocket: Open-Source Cavity Detection and Characterization on Molecular Dynamics Trajectories. Bioinformatics 2011, 27 (23), 3276–3285. 10.1093/bioinformatics/btr550. [DOI] [PubMed] [Google Scholar]
- Craig I. R.; Pfleger C.; Gohlke H.; Essex J. W.; Spiegel K. Pocket-Space Maps to Identify Novel Binding-Site Conformations in Proteins. J. Chem. Inf. Model. 2011, 51 (10), 2666–2679. 10.1021/ci200168b. [DOI] [PubMed] [Google Scholar]
- Paramo T.; East A.; Garzón D.; Ulmschneider M. B.; Bond P. J. Efficient Characterization of Protein Cavities within Molecular Simulation Trajectories: Trj_cavity. J. Chem. Theory Comput. 2014, 10 (5), 2151–2164. 10.1021/ct401098b. [DOI] [PubMed] [Google Scholar]
- Kokh D. B.; Richter S.; Henrich S.; Czodrowski P.; Rippmann F.; Wade R. C. TRAPP: A Tool for Analysis of Transient Binding Pockets in Proteins. J. Chem. Inf. Model. 2013, 53 (5), 1235–1252. 10.1021/ci4000294. [DOI] [PubMed] [Google Scholar]
- Ashford P.; Moss D. S.; Alex A.; Yeap S. K.; Povia A.; Nobeli I.; Williams M. A. Visualisation of Variable Binding Pockets on Protein Surfaces by Probabilistic Analysis of Related Structure Sets. BMC Bioinf. 2012, 13 (1), 39. 10.1186/1471-2105-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyrisch S.; Helms V. Transient Pockets on Protein Surfaces Involved in Protein-Protein Interaction. J. Med. Chem. 2007, 50 (15), 3457–3464. 10.1021/jm070095g. [DOI] [PubMed] [Google Scholar]
- Nussinov R. Introduction to Protein Ensembles and Allostery. Chem. Rev. 2016, 116 (11), 6263–6266. 10.1021/acs.chemrev.6b00283. [DOI] [PubMed] [Google Scholar]
- Lockless S. W.; Ranganathan R. Evolutionarily Conserved Pathways of Energetic Connectivity in Protein Families. Science (Washington, DC, U. S.) 1999, 286 (5438), 295–299. 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Panjkovich A.; Daura X. Exploiting Protein Flexibility to Predict the Location of Allosteric Sites. BMC Bioinf. 2012, 13 (1), 273. 10.1186/1471-2105-13-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fenza A.; Rocchia W.; Tozzini V. Complexes of HIV-1 Integrase with HAT Proteins: Multiscale Models, Dynamics, and Hypotheses on Allosteric Sites of Inhibition. Proteins: Struct., Funct., Genet. 2009, 76 (4), 946–958. 10.1002/prot.22399. [DOI] [PubMed] [Google Scholar]
- McClendon C. L.; Friedland G.; Mobley D. L.; Amirkhani H.; Jacobson M. P. Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles. J. Chem. Theory Comput. 2009, 5 (9), 2486–2502. 10.1021/ct9001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivalta I.; Sultan M. M.; Lee N.-S.; Manley G. A.; Loria J. P.; Batista V. S. Allosteric Pathways in Imidazole Glycerol Phosphate Synthase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (22), E1428–E1436. 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A.; Eargle J.; Black A. A.; Luthey-Schulten Z. Dynamical Networks in tRNA:protein Complexes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (16), 6620–6625. 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanWart A. T.; Eargle J.; Luthey-Schulten Z.; Amaro R. E. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J. Chem. Theory Comput. 2012, 8 (8), 2949–2961. 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A. T.; Durrant J.; Votapka L.; Amaro R. E. Weighted Implementation of Suboptimal Paths (WISP): An Optimized Algorithm and Tool for Dynamical Network Analysis. J. Chem. Theory Comput. 2014, 10 (2), 511–517. 10.1021/ct4008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetz G.; Verkhivker G. M. Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLoS Comput. Biol. 2017, 13 (1), e1005299. 10.1371/journal.pcbi.1005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley M. J.; Chivers P. T.; Baker N. A. Molecular Dynamics Simulation of the Escherichia Coli NikR Protein: Equilibrium Conformational Fluctuations Reveal Interdomain Allosteric Communication Pathways. J. Mol. Biol. 2008, 378 (5), 1155–1173. 10.1016/j.jmb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G.; Verkhivker G.; Colombo G. Modeling Signal Propagation Mechanisms and Ligand-Based Conformational Dynamics of the Hsp90 Molecular Chaperone Full-Length Dimer. PLoS Comput. Biol. 2009, 5 (3), e1000323. 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decherchi S.; Rocchia W. A General and Robust Ray-Casting-Based Algorithm for Triangulating Surfaces at the Nanoscale. PLoS One 2013, 8 (4), e59744. 10.1371/journal.pone.0059744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly M. L. IUCr. Analytical Molecular Surface Calculation. J. Appl. Crystallogr. 1983, 16 (5), 548–558. 10.1107/S0021889883010985. [DOI] [Google Scholar]
- Richards F. M. Areas, Volumes, Packing, and Protein Structure. Annu. Rev. Biophys. Bioeng. 1977, 6 (1), 151–176. 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Ealick S. E.; Rule S. A.; Carter D. C.; Greenhough T. J.; Babu Y. S.; Cook W. J.; Habash J.; Helliwell J. R.; Stoeckler J. D.; Parks R. E. Three-Dimensional Structure of Human Erythrocytic Purine Nucleoside Phosphorylase at 3.2 A Resolution. J. Biol. Chem. 1990, 265 (3), 1812–1820. [DOI] [PubMed] [Google Scholar]
- Schramm V. L. Development of Transition State Analogues of Purine Nucleoside Phosphorylase as Anti-T-Cell Agents. Biochim. Biophys. Acta, Mol. Basis Dis. 2002, 1587 (2–3), 107–117. 10.1016/S0925-4439(02)00073-X. [DOI] [PubMed] [Google Scholar]
- Furman R. R.; Hoelzer D. Purine Nucleoside Phosphorylase Inhibition as a Novel Therapeutic Approach for B-Cell Lymphoid Malignancies. Semin. Oncol. 2007, 34, S29–S34. 10.1053/j.seminoncol.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Kicska G. A.; Long L.; Horig H.; Fairchild C.; Tyler P. C.; Furneaux R. H.; Schramm V. L.; Kaufman H. L. Immucillin H, a Powerful Transition-State Analog Inhibitor of Purine Nucleoside Phosphorylase, Selectively Inhibits Human T Lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (8), 4593–4598. 10.1073/pnas.071050798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans G. B.; Furneaux R. H.; Lewandowicz A.; Schramm V. L.; Tyler P. C. Synthesis of Second-Generation Transition State Analogues of Human Purine Nucleoside Phosphorylase. J. Med. Chem. 2003, 46 (24), 5271–5276. 10.1021/jm030305z. [DOI] [PubMed] [Google Scholar]
- Decherchi S.; Berteotti A.; Bottegoni G.; Rocchia W.; Cavalli A. The Ligand Binding Mechanism to Purine Nucleoside Phosphorylase Elucidated via Molecular Dynamics and Machine Learning. Nat. Commun. 2015, 6, 6155. 10.1038/ncomms7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez J.; Haapalainen A. M.; Cahill S. M.; Ho M. C.; Yan F.; Almo S. C.; Schramm V. L. Catalytic Site Conformations in Human PNP by 19F-NMR and Crystallography. Chem. Biol. 2013, 20 (2), 212–222. 10.1016/j.chembiol.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem M.; Zhadin N.; Callender R.; Schramm V. L. Loop-Tryptophan Human Purine Nucleoside Phosphorylase Reveals Submillisecond Protein Dynamics. Biochemistry 2009, 48 (16), 3658–3668. 10.1021/bi802339c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth S.; Brito R.; Mukherjea D.; Rybak L. P.; Ramkumar V. Adenosine Receptors: Expression. Int. J. Mol. Sci. 2014, 15, 2024–2052. 10.3390/ijms15022024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha R. A.; Ferre S.; Vaugeois J. M.; Chen J. F. Potential Therapeutic Interest of Adenosine A2A Receptors in Psychiatric Disorders. Curr. Pharm. Des. 2008, 14 (15), 1512–1524. 10.2174/138161208784480090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli M.; Carta A. R.; Jenner P. Adenosine A2A Receptors and Parkinson’s Disease. Handb. Exp. Pharmacol. 2009, 193, 589–615. 10.1007/978-3-540-89615-9_18. [DOI] [PubMed] [Google Scholar]
- Van Etten R. A. Cycling, Stressed-out and Nervous: Cellular Functions of c-Abl. Trends Cell Biol. 1999, 9 (5), 179–186. 10.1016/S0962-8924(99)01549-4. [DOI] [PubMed] [Google Scholar]
- Greuber E. K.; Smith-Pearson P.; Wang J.; Pendergast A. M. Role of ABL Family Kinases in Cancer: From Leukaemia to Solid Tumours. Nat. Rev. Cancer 2013, 13 (8), 559–571. 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherezov V.; Stevens R. C. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science (Washington, DC, U. S.) 2012, 337 (6091), 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massink A.; Louvel J.; Adlere I.; Van Veen C.; Huisman B. J. H.; Dijksteel G. S.; Guo D.; Lenselink E. B.; Buckley B. J.; Matthews H.; Ranson M.; Kelso M.; Ijzerman A. P. 5′-Substituted Amiloride Derivatives as Allosteric Modulators Binding in the Sodium Ion Pocket of the Adenosine A2A Receptor. J. Med. Chem. 2016, 59 (10), 4769–4777. 10.1021/acs.jmedchem.6b00142. [DOI] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G. W.; Tate C. G. Agonist-Bound Adenosine A2A Receptor Structures Reveal Common Features of GPCR Activation. Nature 2011, 474 (7352), 521–525. 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V.-P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; IJzerman A. P.; Stevens R. C. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science (Washington, DC, U. S.) 2008, 322 (5905), 1211–1217. 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-de-Terán H.; Massink A.; Rodríguez D.; Liu W.; Han G. W.; Joseph J. S.; Katritch I.; Heitman L. H.; Xia L.; Ijzerman A. P.; Cherezov V.; Katritch V.; Stevens R. C. The Role of a Sodium Ion Binding Site in the Allosteric Modulation of the A2A Adenosine G Protein-Coupled Receptor. Structure 2013, 21 (12), 2175–2185. 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L.; Van Eps N.; Zimmer M.; Ernst O. P.; Scott Prosser R. Activation of the A2A Adenosine G-Protein-Coupled Receptor by Conformational Selection. Nature 2016, 533 (7602), 265–268. 10.1038/nature17668. [DOI] [PubMed] [Google Scholar]
- Gao Z.-G.; Ijzerman A. P. Allosteric Modulation of A2A Adenosine Receptors by Amiloride Analogues and Sodium Ions. Biochem. Pharmacol. 2000, 60 (5), 669–676. 10.1016/S0006-2952(00)00360-9. [DOI] [PubMed] [Google Scholar]
- Katritch V.; Fenalti G.; Abola E. E.; Roth B. L.; Cherezov V.; Stevens R. C. Allosteric Sodium in Class A GPCR Signaling. Trends Biochem. Sci. 2014, 39 (5), 233–244. 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selent J.; Sanz F.; Pastor M.; De Fabritiis G. Induced Effects of Sodium Ions on Dopaminergic G-Protein Coupled Receptors. PLoS Comput. Biol. 2010, 6 (8), e1000884. 10.1371/journal.pcbi.1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Parnot C.; Deupi X.; Ratnala V. R. P.; Swaminath G.; Farrens D.; Kobilka B. Coupling Ligand Structure to Specific Conformational Switches in the β2-Adrenoceptor. Nat. Chem. Biol. 2006, 2 (8), 417–422. 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- Audet M.; Bouvier M. Insights into Signaling from the β2-Adrenergic Receptor Structure. Nat. Chem. Biol. 2008, 4 (7), 397–403. 10.1038/nchembio.97. [DOI] [PubMed] [Google Scholar]
- Palermo G.; Bauer I.; Campomanes P.; Cavalli A.; Armirotti A.; Girotto S.; Rothlisberger U.; De Vivo M. Keys to Lipid Selection in Fatty Acid Amide Hydrolase Catalysis: Structural Flexibility, Gating Residues and Multiple Binding Pockets. PLoS Comput. Biol. 2015, 11 (6), e1004231. 10.1371/journal.pcbi.1004231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo G.; Campomanes P.; Neri M.; Piomelli D.; Cavalli A.; Rothlisberger U.; De Vivo M. Wagging the Tail: Essential Role of Substrate Flexibility in FAAH Catalysis. J. Chem. Theory Comput. 2013, 9 (2), 1202–1213. 10.1021/ct300611q. [DOI] [PubMed] [Google Scholar]
- Yuan S.; Vogel H.; Filipek S. The Role of Water and Sodium Ions in the Activation of the μ-Opioid Receptor. Angew. Chem., Int. Ed. 2013, 52 (38), 10112–10115. 10.1002/anie.201302244. [DOI] [PubMed] [Google Scholar]
- Reddy E. P.; Aggarwal A. K. The Ins and Outs of Bcr-Abl Inhibition. Genes Cancer 2012, 3 (5–6), 447–454. 10.1177/1947601912462126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O.; Grebien F.; Superti-Furga G. The Growing Arsenal of ATP-Competitive and Allosteric Inhibitors of BCR–ABL. Cancer Res. 2012, 72 (19), 4890–4895. 10.1158/0008-5472.CAN-12-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccotto F.; Ardini E.; Casale E.; Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53 (7), 2681–2694. 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- Ohren J. F.; Chen H.; Pavlovsky A.; Whitehead C.; Zhang E.; Kuffa P.; Yan C.; McConnell P.; Spessard C.; Banotai C.; Mueller W. T.; Delaney A.; Omer C.; Sebolt-Leopold J.; Dudley D. T.; Leung I. K.; Flamme C.; Warmus J.; Kaufman M.; Barrett S.; Tecle H.; Hasemann C. A. Structures of Human MAP Kinase Kinase 1 (MEK1) and MEK2 Describe Novel Noncompetitive Kinase Inhibition. Nat. Struct. Mol. Biol. 2004, 11 (12), 1192–1197. 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- La Sala G.; Riccardi L.; Gaspari R.; Cavalli A.; Hantschel O.; De Vivo M. HRD Motif as the Central Hub of the Signaling Network for Activation Loop Autophosphorylation in Abl Kinase. J. Chem. Theory Comput. 2016, 12 (11), 5563–5574. 10.1021/acs.jctc.6b00600. [DOI] [PubMed] [Google Scholar]
- Dolker N.; Gorna M. W.; Sutto L.; Torralba A. S.; Superti-Furga G.; Gervasio F. L. The SH2 Domain Regulates c-Abl Kinase Activation by a Cyclin-like Mechanism and Remodulation of the Hinge Motion. PLoS Comput. Biol. 2014, 10 (10), e1003863. 10.1371/journal.pcbi.1003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse A.; Verkhivker G. M. Molecular Dynamics Simulations and Structural Network Analysis of c-Abl and c-Src Kinase Core Proteins: Capturing Allosteric Mechanisms and Communication Pathways from Residue Centrality. J. Chem. Inf. Model. 2015, 55 (8), 1645–1662. 10.1021/acs.jcim.5b00240. [DOI] [PubMed] [Google Scholar]
- Dixit A.; Verkhivker G. M. Computational Modeling of Allosteric Communication Reveals Organizing Principles of Mutation-Induced Signaling in ABL and EGFR Kinases. PLoS Comput. Biol. 2011, 7 (10), e1002179. 10.1371/journal.pcbi.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O.; Superti-Furga G. Regulation of the c-Abl and Bcr-Abl Tyrosine Kinases. Nat. Rev. Mol. Cell Biol. 2004, 5 (1), 33–44. 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- Iacob R. E.; Zhang J.; Gray N. S.; Engen J. R. Allosteric Interactions between the Myristate- and ATP-Site of the Abl Kinase. PLoS One 2011, 6 (1), e15929. 10.1371/journal.pone.0015929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Adrián F. J.; Jahnke W.; Cowan-Jacob S. W.; Li A. G.; Iacob R. E.; Sim T.; Powers J.; Dierks C.; Sun F.; Guo G.-R.; Ding Q.; Okram B.; Choi Y.; Wojciechowski A.; Deng X.; Liu G.; Fendrich G.; Strauss A.; Vajpai N.; Grzesiek S.; Tuntland T.; Liu Y.; Bursulaya B.; Azam M.; Manley P. W.; Engen J. R.; Daley G. Q.; Warmuth M.; Gray N. S. Targeting Bcr-Abl by Combining Allosteric with ATP-Binding-Site Inhibitors. Nature 2010, 463 (7280), 501–506. 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flight M. H. Kinase Inhibitors: A Winning Combination against BCR–ABL. Nat. Rev. Drug Discovery 2010, 9 (3), 194. 10.1038/nrd3119. [DOI] [Google Scholar]
- Fabbro D.; Manley P. W.; Jahnke W.; Liebetanz J.; Szyttenholm A.; Fendrich G.; Strauss A.; Zhang J.; Gray N. S.; Adrian F.; Warmuth M.; Pelle X.; Grotzfeld R.; Berst F.; Marzinzik A.; Cowan-Jacob S. W.; Furet P.; Mestan J. Inhibitors of the Abl Kinase Directed at Either the ATP- or Myristate-Binding Site. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804 (3), 454–462. 10.1016/j.bbapap.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Shan Y.; Kim E. T.; Eastwood M. P.; Dror R. O.; Seeliger M. A.; Shaw D. E. How Does a Drug Molecule Find Its Target Binding Site?. J. Am. Chem. Soc. 2011, 133 (24), 9181–9183. 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob R. E.; Pene-Dumitrescu T.; Zhang J.; Gray N. S.; Smithgall T. E.; Engen J. R. Conformational Disturbance in Abl Kinase upon Mutation and Deregulation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (5), 1386–1391. 10.1073/pnas.0811912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M.; Seeliger M. A.; Gray N. S.; Kuriyan J.; Daley G. Q. Activation of Tyrosine Kinases by Mutation of the Gatekeeper Threonine. Nat. Struct. Mol. Biol. 2008, 15 (10), 1109–1118. 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica L.; Decherchi S.; Zia S. R.; Gaspari R.; Cavalli A.; Rocchia W. Kinetics of Protein-Ligand Unbinding via Smoothed Potential Molecular Dynamics Simulations. Sci. Rep. 2015, 5 (1), 11539. 10.1038/srep11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica L.; Theret I.; Antoine M.; Perron-Sierra F.; Charton Y.; Fourquez J.-M.; Wierzbicki M.; Boutin J. A.; Ferry G.; Decherchi S.; Bottegoni G.; Ducrot P.; Cavalli A. Molecular Dynamics Simulations and Kinetic Measurements to Estimate and Predict Protein–Ligand Residence Times. J. Med. Chem. 2016, 59 (15), 7167–7176. 10.1021/acs.jmedchem.6b00632. [DOI] [PubMed] [Google Scholar]
- Wang F.; Miles R. W.; Kicska G.; Nieves E.; Schramm V. L.; Angeletti R. H. Immucillin-H Binding to Purine Nucleoside Phosphorylase Reduces Dynamic Solvent Exchange. Protein Sci. 2000, 9 (9), 1660–1668. 10.1110/ps.9.9.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T. W.; Frimurer T. M.; Holst B.; Rosenkilde M. M.; Elling C. E. Molecular Mechanism of 7TM Receptor activation—A Global Toogle Switch Model. Annu. Rev. Pharmacol. Toxicol. 2006, 46 (1), 481–519. 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Trzaskowski B.; Latek D.; Yuan S.; Ghoshdastider U.; Debinski A.; Filipek S. Action of Molecular Switches in GPCRs--Theoretical and Experimental Studies. Curr. Med. Chem. 2012, 19 (8), 1090–1109. 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Tacconi E. M. C.; Zimmer J.; Liang Y.; Gray N. S.; Tarsounas M.; Knapp S. A Unique Inhibitor Binding Site in ERK1/2 Is Associated with Slow Binding Kinetics. Nat. Chem. Biol. 2014, 10 (10), 853–860. 10.1038/nchembio.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds K. A.; McLaughlin R. N.; Ranganathan R. Hot Spots for Allosteric Regulation on Protein Surfaces. Cell 2011, 147 (7), 1564–1575. 10.1016/j.cell.2011.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wu Y.; Deng Y.; Kim B.; Pierce L.; Krilov G.; Lupyan D.; Robinson S.; Dahlgren M. K.; Greenwood J.; Romero D. L.; Masse C.; Knight J. L.; Steinbrecher T.; Beuming T.; Damm W.; Harder E.; Sherman W.; Brewer M.; Wester R.; Murcko M.; Frye L.; Farid R.; Lin T.; Mobley D. L.; Jorgensen W. L.; Berne B. J.; Friesner R. A.; Abel R. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137 (7), 2695–2703. 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- Shi W.; Ting L. M.; Kicska G. A.; Lewandowicz A.; Tyler P. C.; Evans G. B.; Furneaux R. H.; Kim K.; Almo S. C.; Schramm V. L. Plasmodium Falciparum Purine Nucleoside Phosphorylase: Crystal Structures, Immucillin Inhibitors, and Dual Catalytic Function. J. Biol. Chem. 2004, 279 (18), 18103–18106. 10.1074/jbc.C400068200. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Andrews S. P.; Doré A. S.; Hollenstein K.; Hurrell E.; Langmead C. J.; Mason J. S.; Ng I. W.; Tehan B.; Zhukov A.; Weir M.; Marshall F. H. Discovery of 1,2,4-Triazine Derivatives as Adenosine A 2A Antagonists Using Structure Based Drug Design. J. Med. Chem. 2012, 55 (5), 1898–1903. 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B.; Hantschel O.; Young M. A.; Scheffzek K.; Veach D.; Bornmann W.; Clarkson B.; Superti-Furga G.; Kuriyan J. Structural Basis for the Autoinhibition of c-Abl Tyrosine Kinase. Cell 2003, 112 (6), 859–871. 10.1016/S0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Young M. A.; Shah N. P.; Chao L. H.; Seeliger M.; Milanov Z. V.; Biggs W. H.; Treiber D. K.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J.; Sawyers C. L.; Kuriyan J. Structure of the Kinase Domain of an Imatinib-Resistant Abl Mutant in Complex with the Aurora Kinase Inhibitor VX-680. Cancer Res. 2006, 66 (2), 1007–1014. 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins: Struct., Funct., Genet. 2010, 78 (8), 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graphics Modell. 2006, 25 (2), 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185 (2), 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struct., Funct., Genet. 2006, 65 (3), 712–725. 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.