

Abstract
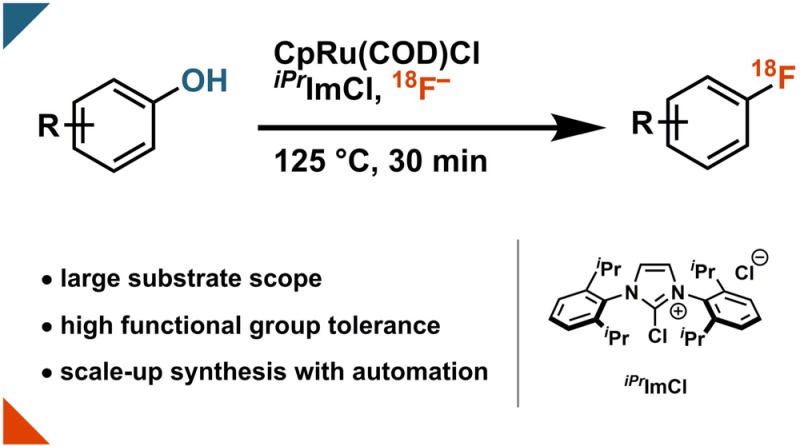
The deficiency of robust and practical methods for 18F-radiofluorination is a bottleneck for positron emission tomography (PET) tracer development. Here, we report the first transition-metal-assisted 18F-deoxyfluorination of phenols. The transformation benefits from readily available phenols as starting materials, tolerance of moisture and ambient atmosphere, large substrate scope, and translatability to generate doses appropriate for PET imaging.
Short abstract
A practical transition-metal-assisted 18F-deoxyfluorination of phenols with large substrate scope empowers the production of radiotracers appropriate for positron emission tomography imaging.
Introduction
Fluorination reactions with 18F of drug-like molecules enable the study of drug disposition and biochemical interactions in humans using positron emission tomography (PET).1,2 As a PET-active nucleus, 18F is attractive for radiotracer design due to the prevalence of aryl fluorides in pharmaceuticals, the metabolic stability of the C–F bond, and the appropriate half-life (109.8 min) for small molecules when compared to other PET isotopes. Although several modern late-stage 18F-fluorination methods have successfully expanded PET radiotracer synthesis, general methods, especially with large substrate scope and functional group tolerance, are still scarce.3 Here we report the first 18F-deoxyfluorination reaction of phenols activated through η6 π-coordination to a ruthenium complex. The method combines deoxyfluorination through a PhenoFluor4,5-like mechanism,6 which ensures large functional group tolerance, with π-activation by ruthenium, which expands the substrate scope to even the most electron-rich phenols. As a consequence, the new 18F-fluorination reaction has the potential to afford previously inaccessible electron-rich aryl fluorides via traditional SNAr reactivity, without some of the intrinsic limitations to other transition metal-mediated fluorination reactions. We further show that the transformation can be automated and scaled to provide labeled materials suitable for human PET imaging (Scheme 1).
Scheme 1. Synthesis of β-CFT via Ruthenium-Mediated Deoxyfluorination.
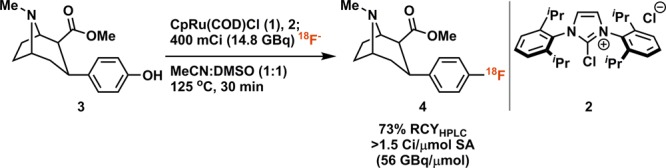
RCYHPLC = radiochemical yield determined as fraction of product radioactivity of total counts by radio-HPLC.
Traditional nucleophilic aromatic substitution (SNAr) reactions, including those with 18F-fluoride as nucleophile, require arene substrates with strong electron-withdrawing substituents.7 More modern SNAr reactions based on diaryl iodonium salts pioneered by the Pike group,8−11 or related reagents,12,13 enabled access to a wider range of fluoroarenes, including electron-rich arenes, but the synthesis of the diaryliodonium substrates can be challenging, especially for complex molecules. We reported high-valent Pd-14 and Ni-based1518F-fluorination reactions, in which electrophilic 18F-reagents were generated in situ from 18F-fluoride. While both methods could be translated to nonhuman primate PET imaging,16,17 the nontrivial synthesis of a new transition metal complex for each new desired 18F-labeled molecule rendered the methods cumbersome to be implemented broadly. Sanford and Scott developed copper-mediated 18F-fluorination reactions from aryliodonium mesylates18 and demonstrated the viability of copper-mediated fluorination of aryl stannanes19 for radiochemistry, including the preparation of clinical doses.20 Gouverneur reported an 18F-fluorination reaction of arylboronic acid derivatives that proceeds through high-valent copper intermediates.21 The method is practical, easy to execute, suitable for automation and has a large substrate scope.22 The functional group tolerance is reminiscent of what can be expected for high-valent Cu complexes: high, but unprotected alcohols and amines can give rise to undesired C–N and C–O bond formation, respectively. Over the past five years, the field of late-stage fluorination has made several fundamental advances,3 yet the development of general, reliable, scalable, and translatable 18F-fluorination methods to achieve a measurable impact on clinical PET imaging is still outstanding.23 The data we report here suggest that Ru-mediated deoxyfluorination may develop into such a method.
Cognizant of the challenges we have observed with high-valent metal redox chemistry in 18F-fluorination, we developed a metal-free 18F-deoxyfluorination of phenols.6 The method is exceptionally functional group tolerant, to the best of our knowledge, more so than any other late-stage fluorination reaction. The functional group tolerance could be rationalized through a mechanism analysis:6 The key tetrahedral intermediate features a nonbasic, neutral organofluoride (B), which rearranges to product (Scheme 2a). The tetrahedral intermediate (B) is in equilibrium with its corresponding uronium fluoride ion pair (A). The equilibrium is inconsequential for reactions with excess 19F-fluoride.24 But because 18F-fluoride is used as the limiting reagent, detrimental side reactions from the ion pair (A) sequester 18F-fluoride irreversibly, especially if the equilibrium constant K1 is small, as is expected for electron-rich phenols. As a consequence, although highly functional group tolerant, the substrate scope for current 18F-deoxyfluorination is limited, and electron-rich phenols cannot be 18F-deoxyfluorinated, despite the productive reaction with 19F. Our strategy to devise a more general reaction entailed to increase the equilibrium constant K, specifically K2 ≫ K1. It is well established that η6 π-coordination of arenes to certain transition metal complexes reduces the electron density of the arene and activates the arene for nucleophilic aromatic substitution,25−31 although such a strategy has not been successfully implemented for general 18F-fluorination reactions (Scheme 2b). Here we disclose the successful implementation of a practical, robust ruthenium-mediated 18F-deoxyfluorination of both electron-rich and electron-poor phenols. The reaction retains the desirable features of deoxyfluorination without ruthenium and can tolerate nucleophilic functional groups such as amines. In contrast to the redox active 18F-fluorination reactions with Pd, Ni, and Cu, the Ru center does not undergo redox chemistry, nor does the reaction proceed through high-valent complexes that could engage in undesired side reactions with amines or other nucleophilic groups.
Scheme 2. (a) Concerted Nucleophilic Aromatic Substitution (cSNAr) via B To Furnish the Fluorinated Arene (K1: Equilibrium Constant, Ar = 2,6-Diisopropylphenyl) and (b) The Ruthenium Fragment Decreases the Electron Density of the Phenol, Which Renders the Tetrahedral Intermediate Energetically More Favorable — K2 ≫ K1.

Isotopologue [19F]6 was synthesized from 5 in 75% yield to confirm the identity of [18F]6.
Results and Discussion
Heating the air and moisture stable Ru complex CpRu(COD)Cl (1, Scheme 3) with a phenol in ethanol for 30 min at 85 °C affords a solution of ruthenium phenol complex, e.g., 5, Scheme 2b; subsequent addition of chloroimidazolium chloride 2 provides a solution, which is used to elute 18F-fluoride off an anion exchange cartridge. After addition of a 1:1 DMSO/acetonitrile (v/v) solution, the reaction mixture is heated for 30 min to afford aryl fluorides. No precautions to exclude moisture or air are necessary at any point in the process.
Scheme 3. Substrate Table for Ruthenium-Mediated 18F-Deoxyfluorination of Phenols.

RCYTLC = Radiochemical yield determined as fraction of product radioactivity of total counts by radio-TLC; * 26% activity yield.
In line with our goal, electron-rich substrates like anisole (7a) and dialkylaniline derivatives (7c) show high radiochemical yields by TLC (RCYTLC). A variety of functional groups is tolerated, most importantly basic amines (7b, c, i, l) which can present a major limitation to several available radiofluorination methods.14,15,19 Protic functional groups (7e, j, n) are unproblematic and phenolic hydroxyl groups can be selectively deoxyfluorinated in the presence of unprotected aliphatic alcohols without affecting carbinol stereochemistry (7h, l). Additionally, several heterocyclic scaffolds, including pyrimidines (7m), indoles (7o), and quinolines (7k) are good substrates for the reaction. Ortho-substitution is tolerated (7k, o).
The overall yield of the reaction is affected by the RCY and elution efficiency (EE) of 18F-fluoride off the anion exchange cartridge, and both were addressed when optimizing reaction conditions. Chloride as X-type ligand for CpRu(COD)X gave higher yields than other evaluated ligands. EE and RCY were improved by higher concentration and molar excess of both 1 and 2. The minimal increases in yield obtained by more than three equivalents of 1 and 2 did not justify the expenditure of reagents and additional purification difficulties. Both EE and RCY were higher when ethanol was present in the reaction mixture. While more than 30% ethanol was detrimental to the fluorination, small solvent volumes were impractical to handle, and we continued our work with 50 μL of ethanol in 400 μL total reaction volume. Although several salt additives increased elution efficiency, the gain was offset by a reduction in RCY. None of the additives investigated improved the overall yield. Reaction temperatures below 125 °C and reaction times less than 30 min afforded lower yields of product.
For any 18F-radiolabeling methodology to be practically useful, it needs to be amenable to automation on commercial radiosynthesis modules. On an Elixys (Sofie Biosciences) radiosynthesizer, we were able to perform the reaction in a fully automated fashion: From 461 mCi (17.1 GBq) of 18F-fluoride obtained in aqueous solution from a cyclotron, we were able to isolate 111 mCi (4.11 GBq) of purified, protected 18F-fluorophenylalanine derivative 9 within 80 min (Scheme 4). Additional reformulation and filtration, as is required for human PET tracer synthesis, formally resulted in 82.3 mCi (3.05 GBq) of reformulated, purified 9 (see Supporting Information for details). The stereochemical purity of the starting material was maintained throughout the reaction. Initially, yields of the automated syntheses were more than 10-fold lower than in manual experiments. The main factors responsible for the lower yields were identified as vial size and the associated headspace, and the use of 4 mL instead of 10 mL reactors resolved the issue. It is crucial to ensure that an internal temperature of 125 °C is reached when using a vial adapter. In commercial radiosynthesis systems for which smaller vials are not commonly available, high pressure is an alternative strategy to counteract the larger headspace. For example, in a Siemens Explora FDG4 system, an increase in pressure from 30 to 205 kPa during the reaction improved the RCY toward protected fluorophenylalanine 5-fold (see Supporting Information for details).
Scheme 4. Fully Automated Ruthenium-Mediated 18F-Deoxyfluorination of Tyrosine Derivative.

111 mCi (4.11 GBq) activity yield AY (24.1%) is non-decay-corrected.
The PhenoFluor reagent is known to facilitate a concerted nucleophilic aromatic substitution mechanism, which enables deoxyfluorination of electron-rich phenol substrates.6 Our computational results (see Figure 1 and Supporting Information) suggest that coordination of the RuCp fragment withdraws sufficient electron density from the phenol substrate that a classic SNAr mechanism via a Meisenheimer complex (MHC) is preferred. Density functional theory (DFT)-calculated reaction barriers are 5.3 kcal·mol–1 for fluoride attack (TS1) and 7.0 kcal·mol–1 for leaving group loss (TS2). We consider the barriers remarkably low for C–F bond formation, and, hence, the potential of η6-coordination for radiofluorination of other arene electrophiles may be a promising research endeavor. Attempts to isolate reaction intermediates in the deoxyfluorination of phenols with 19F-fluoride only yielded the aryl fluoride product complexed to Ru (e.g., 6), which corroborates the low deoxyfluorination barriers obtained computationally. Decomplexation of the aryl fluoride product from the Ru fragment requires elevated reaction temperatures, and, currently, is the rate-limiting step in the sequence. Because 18F-fluoroarene dissociation is rate-limiting, we note that 18F-fluoroarene complexed to the RuCp fragment can be observed as a byproduct if the reaction times are too short.
Figure 1.
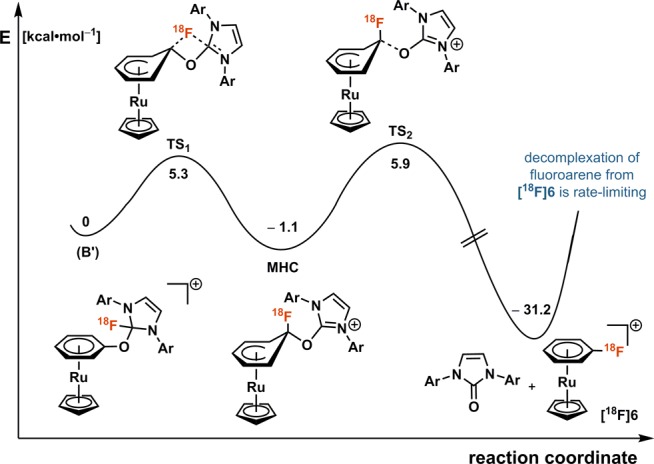
Proposed mechanism for ruthenium-mediated 18F-deoxyfluorination (Ar = 2,6-diisopropylphenyl, MHC = Meisenheimer complex).
The ruthenium-mediated dexoyfluorination presents a valuable addition to the radiochemical toolbox. Very electron-rich substrates are challenging to fluorinate with conventional radiofluorination reactions but are unproblematic for this approach. Basic amines and ortho-substitution are fully compatible. The substrates are readily accessible and stable phenols. The reaction is operationally simple and can be executed in air in the presence of moisture, and automation is established. The method is broadly applicable and easy to adapt in a radiopharmaceutical production environment. A current technical limitation of the reaction is the requirement for 30–40 mg of total mass of reagents (combination of phenol, 1 and 2), which can complicate purification and is not economic. Although several heterocyclic substrates are efficiently radiofluorinated (7k, 7m, and 7o), we have found that sometimes the presence of heterocycles will interfere with efficient ruthenium complex formation. We attribute this fact to their ability to coordinate to the ruthenium center in modes detrimental to product formation. Mechanistically, the slowest step in the sequence is product decomplexation. The reaction requires 30 min of heating, which reduces the activity yield by 20% merely through radioactive decay. Decomplexation could potentially be improved by more sophisticated design of the ruthenium ligand environment.32−35 The elution efficiency can be as low as 49% in some cases, which is an additional factor that limits the yield. Higher ionic strength of the eluent solution is a possible remedy, and efforts are ongoing in our laboratory to develop improved ruthenium complexes and imidazolium reagents to expand the utility and generality of the promising 18F-deoxyfluorination reaction.
Acknowledgments
We thank the NIH NIGMS (GM088237), the Phelps foundation, the Harvard Blavatnik Biochemical Accelerator, and the Max-Planck-Institut fuer Kohlenforschung for funding. We thank Dr. Shao-Liang Zheng (Harvard) for help with the X-ray and Rocco Policarpo (Harvard) for help with HPLC purification.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00195.
Author Contributions
# M.H.B., D.M., and M.G.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Fowler J.; Wolf A. Working against Time: Rapid Radiotracer Synthesis and Imaging the Human Brain. Acc. Chem. Res. 1997, 30, 181–188. 10.1021/ar960068c. [DOI] [Google Scholar]
- Phelps M. PET: The Merging of Biology and Imaging into Molecular Imaging. J. Nucl. Med. 2000, 41, 661–681. [PubMed] [Google Scholar]
- Preshlock S.; Tredwell M.; Gouverneur V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- Tang P.; Wang W.; Ritter T. Deoxyfluorination of Phenols. J. Am. Chem. Soc. 2011, 133, 11482–11484. 10.1021/ja2048072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T.; Becker F.; Ritter T. PhenoFluor: Practical Synthesis, New Formulation, and Deoxyfluorination of Heteroaromatics. Org. Process Res. Dev. 2014, 18, 1041–1044. 10.1021/op500121w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann C. N.; Hooker J. M.; Ritter T. Concerted Nucleophilic aromatic Substitution with 19F– and 18F–. Nature 2016, 534, 369–373. 10.1038/nature17667. [DOI] [PubMed] [Google Scholar]
- Bunnett J.; Zahler E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. 10.1021/cr60153a002. [DOI] [Google Scholar]
- Pike V. W.; Aigbirhio F. I. Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts−a novel single-step route to no-carrier-added [18F]fluoroarenses. J. Chem. Soc., Chem. Commun. 1995, 2215–2216. 10.1039/C39950002215. [DOI] [Google Scholar]
- Shah A.; Pike V. W.; Widdowson D. A. The synthesis of [18F]fluoroarenes from the reaction of cyclotron-produced [18F]fluoride ion with diaryliodonium salts. J. Chem. Soc., Perkin Trans. 1 1998, 2043–2046. 10.1039/a802349b. [DOI] [Google Scholar]
- Chun J.; Telu S.; Lu S.; Pike V. W. Radiofluorination of diaryliodonium tosylates under aqueous-organic and cryptand-free conditions. Org. Biomol. Chem. 2013, 11, 5094–5099. 10.1039/c3ob40742j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J.; Pike V. W. Single-step syntheses of no-carrier-added functionalized [18F]fluoroarenes as labeling synthons from diaryliodonium salts. Org. Biomol. Chem. 2013, 11, 6300–6306. 10.1039/c3ob41353e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale J.; Ermert J.; Humpert S.; Coenen H. H. Iodonium ylides for one-step, no-carrier-added radiofluorination of electron rich arenes, exemplified with 4-(([18F]fluorophenoxy)-phenylmethyl)piperidine NET and SERT ligands. RSC Adv. 2014, 4, 17293–17299. 10.1039/C4RA00674G. [DOI] [Google Scholar]
- Rotstein B. H.; Stephenson N. A.; Vasdev N.; Liang S. H. Spirocyclic hypervalent iodine (III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun. 2014, 5, 4365. 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]
- Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. A fluoride-derived electrophilic late-stage fluorination reagents for PET imaging. Science 2011, 334, 639–642. 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.; Hooker J. M.; Ritter T. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F]Fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamlet A. S.; Neumann C. N.; Lee E.; Carlin S. M.; Moseley C. K.; Stephenson N.; Hooker J. M.; Ritter T. Application of Palladium-Mediated 18F-Fluorination to PET Radiotracer Development: Overcoming Hurdles to translation. PLoS One 2013, 8, e59187. 10.1371/journal.pone.0059187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H.; Wey H.-Y.; Strebl M.; Neelamegam R.; Ritter T.; Hooker J. M. A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluoroucil from [18F]Fluoride for Human PET Imaging. ACS Chem. Neurosci. 2014, 5, 611–615. 10.1021/cn500078e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiishi N.; Brooks A. F.; Topczewski J. J.; Rodnick M. E.; Sanford M. S.; Scott P. J. H. Cu-catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts with K[18F]F. Org. Lett. 2014, 16, 3224–3227. 10.1021/ol501243g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamache R. F.; Waldmann C.; Murphy J. M. Copper-mediated oxidative fluorination of aryl stannanes with fluoride. Org. Lett. 2016, 18, 4522–4525. 10.1021/acs.orglett.6b02125. [DOI] [PubMed] [Google Scholar]
- Makaravage K. J.; Brooks A. F.; Mossine A. V.; Sanford M. S.; Scott P. J. H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. 10.1021/acs.orglett.6b02911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tredwell M.; Preshlock S. M.; Taylor N. J.; Gruber S.; Huiban M.; Passchier J.; Mercier J.; Génicot C.; Gouverneur V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem., Int. Ed. 2014, 53, 7751–7755. 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]
- Preshlock S.; Calderwood S.; Verhoog S.; Tredwell M.; Huiban M.; Hienzsch A.; Gruber S.; Wilson T. C.; Taylor N. J.; Cailly T.; Schedler M.; Collier T. L.; Passchier J.; Smits R.; Mollitor J.; Hoepping A.; Mueller M.; Genicot C.; Mercier J.; Gouverneur V. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun. 2016, 52, 8361–8364. 10.1039/C6CC03295H. [DOI] [PubMed] [Google Scholar]
- Campbell M. G.; Mercier J.; Genicot C.; Gouverneur V.; Hooker J. M.; Ritter T. Bridging the gaps in 18F PET tracer development. Nat. Chem. 2017, 9, 1–3. 10.1038/nchem.2693. [DOI] [PubMed] [Google Scholar]
- Fujimoto T.; Ritter T. PhenoFluorMix: Practical Chemoselective Deoxyfluorination of Phenols. Org. Lett. 2015, 17, 544–547. 10.1021/ol5035518. [DOI] [PubMed] [Google Scholar]
- Kane-Maguire L. A. P.; Honig E. D.; Sweigart D. A. Nucleophilic addition to coordinated cyclic π-hydrocarbons: mechanistic and synthetic studies. Chem. Rev. 1984, 84, 525–543. 10.1021/cr00064a001. [DOI] [Google Scholar]
- Rose-Munch F.; Gagliardini V.; Renard C.; Rose E. (η6-Arene)tricarbonylchromium and (η5-cyclohexadienyl) tricarbonylmanganese complexes: indirect nucleophilic substitutions. Coord. Chem. Rev. 1998, 178–180, 249–268. 10.1016/S0010-8545(98)00078-2. [DOI] [Google Scholar]
- Pike R. D.; Sweigart D. A. Electrophilic reactivity of coordinated cyclic π-hydrocarbons. Coord. Chem. Rev. 1999, 187, 183–222. 10.1016/S0010-8545(98)00231-8. [DOI] [Google Scholar]
- Pape A. R.; Kaliappan K. P.; Kündig E. P. Transition-Metal-Mediated Dearomatization Reactions. Chem. Rev. 2000, 100, 2917–2940. 10.1021/cr9902852. [DOI] [PubMed] [Google Scholar]
- Semmelhack M. F.; Chlenov A. (Arene) Cr(CO)3 Complexes: Aromatic Nucleophilic Substitution. Top. Organomet. Chem. 2004, 7, 43–69. 10.1007/b94491. [DOI] [Google Scholar]
- Walton J. W.; Williams J. M. J. Catalytic SNAr of unactivated aryl chlorides. Chem. Commun. 2015, 51, 2786–2789. 10.1039/C4CC07116F. [DOI] [PubMed] [Google Scholar]
- Konovalov A. I.; Gorbacheva E. O.; Miloserdov F. M.; Grushin V. V. Ruthenium-catalyzed nucleophilic fluorination of halobenezenes. Chem. Commun. 2015, 51, 13527–13530. 10.1039/C5CC05436B. [DOI] [PubMed] [Google Scholar]
- Muetterties E. L.; Bleeke J. R.; Sievert A. C. Arene transition metal chemistry: III. Arene exchange phenomena. J. Organomet. Chem. 1979, 178, 197–216. 10.1016/S0022-328X(00)87871-7. [DOI] [Google Scholar]
- Semmelhack M. F.; Chlenov A.; Ho D. M. Accelerated Arene Ligand Exchange in the (Arene)Cr(CO)2L Series. J. Am. Chem. Soc. 2005, 127, 7759–7773. 10.1021/ja042705x. [DOI] [PubMed] [Google Scholar]
- Okazaki M.; Yamahira N.; Minglana J. J. G.; Komuro T.; Ogino H.; Tobita H. [Ru(xantsil)(CO)(η6-toluene)]: Synthon for a Highly Unsaturated Ruthenium (I) Complex through Facile Dissociation of the Toluene Ligand [xantsil = (9,9-dimethylxanthene-4,5-diy)bis(dimethylsilyl)]. Organometallics 2008, 27, 918–926. 10.1021/om701194e. [DOI] [Google Scholar]
- Otsuka M.; Yokoyama H.; Endo K.; Shibata T. Facile Catalytic SNAr reaction of nonactivated fluoroarenes with amines using η6-benzene ruthenium (II) complex. Synlett 2010, 2601–2606. 10.1055/s-0030-1258774. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.