

Abstract
Bone metastases (BoM) are a significant cause of morbidity in patients with estrogen receptor–positive (ER-positive) breast cancer; yet, characterizations of human specimens are limited. In this study, exome-capture RNA sequencing (ecRNA-seq) on aged (8–12 years), formalin-fixed, paraffin-embedded (FFPE), and decalcified cancer specimens was evaluated. Gene expression values and ecRNA-seq quality metrics from FFPE or decalcified tumor RNA showed minimal differences when compared with matched flash-frozen or nondecalcified tumors. ecRNA-seq was then applied on a longitudinal collection of 11 primary breast cancers and patient-matched synchronous or recurrent BoMs. Overtime, BoMs exhibited gene expression shifts to more Her2 and LumB PAM50 subtype profiles, temporally influenced expression evolution, recurrently dysregulated prognostic gene sets, and longitudinal expression alterations of clinically actionable genes, particularly in the CDK/Rb/E2F and FGFR signaling pathways. Taken together, this study demonstrates the use of ecRNA-seq on decade-old and decalcified specimens and defines recurrent longitudinal transcriptional remodeling events in estrogen-deprived breast cancers.
Keywords: Genetics, Oncology
Keywords: Breast cancer
Exome-capture RNA sequencing is performed on decade-old and decalcified tumors to define longitudinal transcriptional changes in ER-positive breast cancer bone metastases.
Introduction
Bone metastases (BoMs) occur in approximately 65%–75% of breast cancer patients with relapsed disease, resulting in significant comorbidities, such as fractures and chronic pain (1). Following colonization to the bone, breast cancer cells exploit the local microenvironment by activating osteoclasts, which in turn provides proliferative fuel for tumor cells (2). This process is targeted clinically using antiosteoclast agents, such as bisphosphonates and RANKL inhibitors, yet these therapies do not confer significant survival benefits (3).
Importantly, the majority of breast cancers that metastasize to bone are estrogen receptor (ER) positive and present clinically in the context of long-term endocrine therapies, such as selective ER modulators and aromatase inhibitors (4). In vivo models of BoM have unfortunately been somewhat restricted to ER-negative disease due to the more indolent characteristics of ER-positive cell lines (5). Molecular characteristics of ER-positive specimens that have recurred in an estrogen-blunted system, which represents the major burden of breast cancer BoM, are thus essential to reinforce the significant scientific contributions made using in vivo bone metastasis models (6–9). Nonetheless, data sets are currently limited, in part due to the practical difficulties of obtaining and processing human BoM specimens (10).
Large-scale molecular characterizations of patient-matched samples — primary tumors and synchronous or asynchronous matched metastases — show that metastatic lesions acquire features distinct from primary tumors that are clinically actionable or confer therapy resistance (11–13). Indeed, current treatment guidelines in breast cancer recommend a metastatic biopsy to guide therapy in advanced disease if possible (14). Unfortunately, BoMs often undergo harsh decalcification procedures with strong acids to eliminate calcium deposits prior to specimen sectioning. Decalcification degrades nucleic acids and can alter results of IHC (15–17). Furthermore, formalin-fixed, paraffin embedding (FFPE) — often performed in concert with decalcification — causes severe degradation and hydrolysis of RNA (18). In light of this, new capture-based methods of nucleic acid sequencing on aged FFPE specimens have shown efficacy in identifying DNA variants and even guiding care in academic centers (19–21). Exome-capture RNA sequencing (ecRNA-seq) is less well characterized in aged tumor samples, although recent studies on FFPE specimens have shown promising expression correlations with flash-frozen tissues (22–24).
Because of the untapped potential of archived, decalcified BoM specimens; the burden of BoMs in breast cancer patients; and the lack of long-term endocrine-treated tumor data sets, the performance of ecRNA-seq from decade-old, degraded, and decalcified tumor samples was assessed. Following this evaluation, ecRNA-seq was then applied to a collection of 11 ER-positive patient-matched primary breast cancers and BoMs to define transcriptional evolution in breast cancer cells following metastatic colonization in the bone and years of endocrine therapy.
Results
ecRNA-seq of aged and decalcified breast cancers.
To determine the feasibility of sequencing an aged, FFPE, and decalcified tumor cohort, ecRNA-seq on two separate sample sets was performed. The first sample set included four cases of primary breast tumors that, at the time of resection, were split in two. One section was flash frozen and stored at –80°C, and the other tumor section was FFPE and stored at room temperature. Storage times ranged from 8.2 to 12.3 years. RNA-sequencing quality control (QC) analyses after alignment showed differences in GC content and insert size, yet gene body coverage and transcript diversity assignments were largely similar (Figure 1A). After quantifying and normalizing gene abundances, expression correlations between frozen and FFPE-matched samples were assessed using log2-transformed trimmed mean of M values–normalized (TMM-normalized) CPM (log2normCPM) values. Pearson r correlations ranged from 0.929 to 0.963, with an average correlation of 0.953 (Figure 1B). The same analysis was performed using a second sample set of matched FFPE-decalcified and FFPE-nondecalcified samples. Again, no concerning deviations in ecRNA-seq quality metrics were observed between the two differently processed sample groups (Figure 1C), and Pearson r expression correlations ranged from 0.936 to 0.969 (Figure 1D). Furthermore, correlation matrices of the two sample sets showed that matched tumor sample expression values were more similar to each other than expression values from tumors with equivalent processing and storage (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.95703DS1). Full ecRNA-seq metrics from the QC analysis did reveal differences in some metrics between FFPE and flash-frozen tissue (i.e., splice junction loci number) that may be informative for other applications (Supplemental Tables 1 and 2). In summary, ecRNA-seq shows outstanding quality metrics for analysis of aged FFPE and decalcified BoMs samples.
Figure 1. Exome-capture RNA sequencing of aged, FFPE, and decalcified tumors.

(A) ecRNA-seq quality metrics (GC content, insert size, gene body coverage, and cumulative gene assignment diversity) of aged and tumor-matched, formalin-fixed, paraffin-embedded (FFPE) and flash-frozen (FF) samples. FF samples in blue, FFPE samples in red (n = 4 pairs). (B) Expression value correlations between four sets of matched tumor samples (FF vs. FFPE), along with Pearson r correlations and sample ages. (C) ecRNA-seq quality metrics of matched nondecalcified and decalcified samples. Nondecalcified samples in blue, decalcified samples in red (n = 3 pairs). (D) Expression correlations between three sets of matched tumor samples (nondecalified vs. decalcified), along with Pearson r correlations.
ecRNA-seq of breast cancer BoMs.
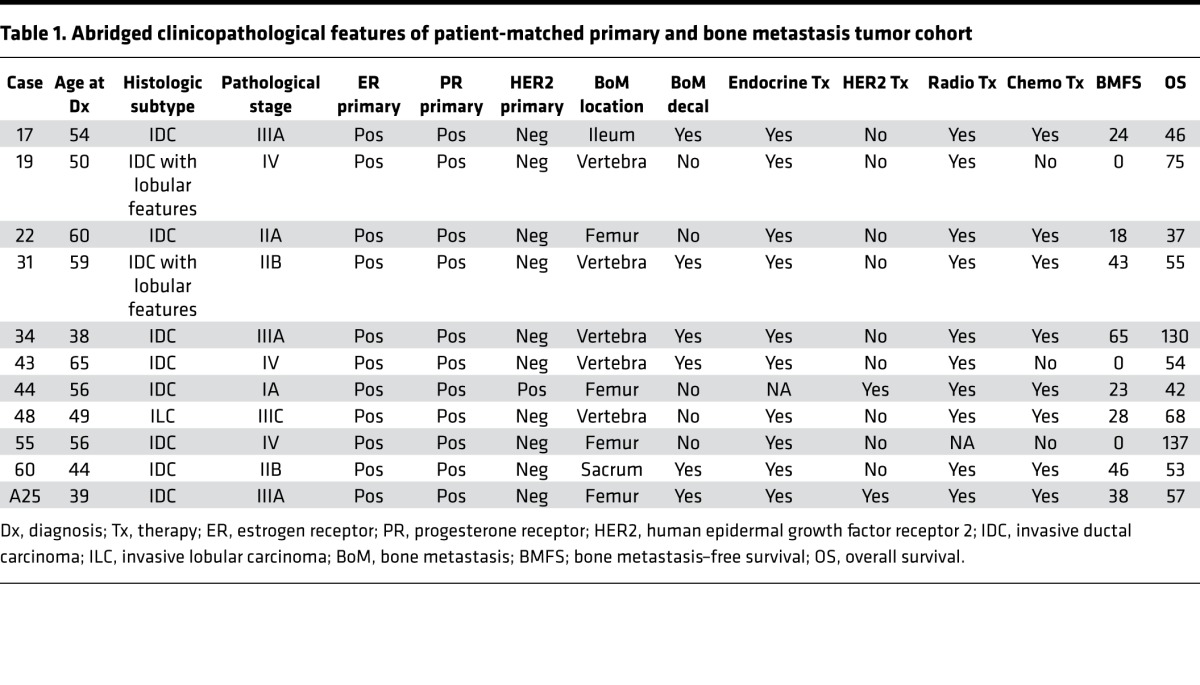
Following the validation of ecRNA-seq, a cohort of 11 ER-positive patient-matched primary tumors and BoMs was acquired through the University of Pittsburgh Health Science Tissue Bank (Table 1 and Supplemental Table 3). Abstracted clinical records showed that nearly all patients (10 of 11) were documented as having received adjuvant endocrine therapy, and bone metastasis–free survival ranged from 0 (synchronous) to greater than 5 years, with the most common site of bone metastasis being the vertebral column. ecRNA-seq was performed on the 22 samples, yielding an average read count of 59,570,288 and an average Salmon transcript-mapping rate of 92.9% (Supplemental Table 4). Consistent with the initial QC studies above, quality metrics on these samples showed consistent gene body coverage, GC content, insert sizes, and transcript diversity, regardless of decalcification status (Supplemental Figure 2 and Supplemental Table 5). Furthermore, since samples within the cohort had been surgically excised and banked many years apart, all paired specimens underwent an analysis of shared variants, which confirmed tumor pairs were patient matched (Supplemental Figure 3).
Table 1. Abridged clinicopathological features of patient-matched primary and bone metastasis tumor cohort.
Clustering and temporal expression shifts.
Unsupervised hierarchical clustering of patient-matched pairs revealed that decalcification of BoMs did not produce independent clades, with 5 of 11 BoMs clustering in the same doublet clade as their matched primary tumor (denoted with an asterisk in Figure 2A). Notably, 3 of 5 doublet-clustering cases were synchronous metastases. Discrete PAM50 intrinsic subtype assignments were identical in 7 of 11 pairs. Three pairs switched from LumA to LumB in the metastasis and another was classified as normal subtype in the primary tumor and LumB in the BoM (Figure 2B). To obtain more granularity than discrete PAM50 calls, probability scores for each PAM50 subtype were assigned (Figure 2B and Supplemental Table 6). Her2 and LumB profile gains (defined as a probability gain of >10% in a matched BoM) were the most common — both being observed in 5 of 11 cases (Figure 2B). Given shifts in expression profiles of BoMs and doublet clustering of synchronous BoMs, temporal influence on transcriptional evolution was analyzed. Pearson r correlations between each patient-matched pair using log2normCPM expression values were utilized as a metric for transcriptional similarity. Expression pair similarity was significantly correlated (Pearson r = –0.864, P < 0.001) with time from primary tumor diagnosis to bone metastasis (Figure 2C).
Figure 2. Unsupervised clustering, intrinsic subtype shifts, and temporal evolution of ER-positive bone metastases.

(A) Unsupervised hierarchical clustering heatmap (red, high relative expression; blue, low relative expression) of patient-matched pairs (n = 11) using the top 5% most variable genes (n = 1,096) across the cohort. Tumor (primary in blue, metastasis in red) and decalcification status (positive in green, negative in black) are indicated. Asterisks below heatmap designate patient-matched pairs that cluster in a single doublet clade. (B) Discrete PAM50 assignments (red, basal; green, HER2; blue, LumA; purple, LumB; yellow, normal) and PAM50 probabilities for patient-matched pairs. PAM50 probability shifts in metastases (if greater than 10%) are marked with black diamonds. (C) Correlation of patient-matched tumor expression similarity versus clinical time to metastasis, with Pearson r value and correlation P value.
Differentially expressed genes in BoMs.
To determine genes consistently upregulated or downregulated in BoMs, a paired DESeq2 differential gene expression analysis was performed. 207 genes were differentially expressed (FDR-adjusted P < 0.10) — 80 genes with increased and 127 genes with decreased expression in BoMs (Figure 3A and Supplemental Table 7). Gene ontology analysis was performed to determine biological processes represented in the upregulated and downregulated gene sets. Generally, genes within osteogenic programs showed the most significant increases in expression, while muscle-related, adhesion, and motility gene sets were found to be significantly lost in BoMs (Supplemental Table 8 and Supplemental Figure 4). Given that a subset of these genes may be mediating therapy resistance and/or distant metastases, single-sample gene set enrichment analysis (ssGSEA) scores (25) were calculated using tumor expression data from patients with long-term outcomes in METABRIC (26). Two separate gene lists were created to build the signatures, representing the most significantly upregulated (boneMetSigUp) and downregulated (boneMetSigDown) genes in BoMs (Supplemental Table 9). Tumors intrinsically expressing higher boneMetSigUp and lower boneMetSigDown ssGSEA scores conferred worse (log-rank P < 0.001) disease-specific survival (DSS) outcomes (Figure 3B). To increase the power of discerning gene expression effects due to long-term estrogen deprivation, a differential gene expression analysis was performed excluding the treatment-naive, synchronous BoMs (n = 8 pairs). This yielded a list of 612 differentially expressed genes (DEGs) (Supplemental Table 10), some of which were not detected as differentially expressed with treatment-naive synchronous bone metastasis cases included.
Figure 3. Differentially expressed genes in patient-matched bone metastases.

(A) Heatmap (red, high relative expression; blue, low relative expression) of log2normCPM values from 207 differentially expressed genes (FDR-adjusted P value [padj] < 0.10, DESeq2) between primary tumors and patient-matched bone metastases. Heatmap is segregated into two sections; genes with log2 fold change >0 on top and genes with log2 fold change <0 on bottom. Each section is gene sorted by adjusted P values. (B) Disease-specific survival (DSS) outcome differences in ER-positive METABRIC tumors using boneMetSigUp (top) and boneMetSigDown (bottom) expression scores as strata. 95% confidence intervals are highlighted along with log-rank P values and associated risk tables.
Dysregulated gene sets and RBBP8 expression loss.
To determine pathway level changes in breast cancer BoMs, a preranked GSEA was performed. All genes were ranked by DESeq2-calculated log2 fold changes (metastasis vs. primary, Supplemental Table 11) and then analyzed for enrichments using Molecular Signature Database (MsigDB) gene sets (http://software.broadinstitute.org/gsea/msigdb; H: hallmark gene sets; C6: oncogenic signatures) (27). This yielded several significantly metastasis-enriched and metastasis-diminished gene sets (FDR q < 0.10, Supplemental Table 12). The three most significantly enriched gene sets in metastases involved E2F transcription factor targets, genes mediating the G2M checkpoint, and an experimental perturbation gene set consisting of genes upregulated with knockdown of RBBP8 in a breast cell line (Figure 4A). Other upregulated gene sets included hedgehog signaling and gene sets associated with Rb loss and KRAS gains. The three most significantly negatively correlated gene sets consisted of an NF-κB/TNF gene set, genes involved in epithelial mesenchymal transition, and an embryonic development gene set. We further interrogated RBBP8 due to it being the most significant gene set enriched in bone metastasis. As predicted by the enrichment, BoMs carried significant RBBP8 expression loss (Wilcoxon signed-rank P = 0.02), with 5 of 11 metastases (45%) having at least a 2-fold decrease in expression versus patient-matched primaries (Figure 4B). Tumors intrinsically expressing lower levels of RBBP8 showed worse disease-specific and bone metastasis–free survival outcomes (Figure 4C).
Figure 4. Dysregulated gene sets and RBBP8 loss in breast cancer bone metastases.

(A) Top three enriched and depleted gene sets (by FDR q value) in bone metastases from ranked GSEA analysis (n = 11 pairs). Gene list ranking was performed using log2 fold change values from DESeq2 differential expression output, where a positive log2 fold change represents increased expression in metastasis (red) and a negative log2 fold change represents decreased expression in metastasis (blue). Green lines show running enrichment scores. Black vertical lines below curve show where genes within the query gene set are represented in the ranked list. Normalized enrichment score (NES) and FDR q values (derived from GSEA tool) are noted below gene set names. (B) RBBP8 expression values (log2normCPMs) in primary tumors (blue) and bone metastasis (red). Pairs are connected with a line and Wilcoxon signed-rank P value is shown. (C) Disease-specific survival (DSS) outcome differences in ER-positive tumors (METABRIC) and bone metastasis–free survival (BMFS) differences (GSE12276) using normalized RBBP8 expression values as strata. 95% confidence intervals are highlighted along with log-rank P values and risk tables.
Expression gains and losses in clinically actionable genes.
Because of the observed acquisition of clinically actionable targets reported in other studies of paired primary and recurrent tumors (12, 13), a paired expression analysis to define clinically actionable expression changes in ER-positive BoMs was performed (Supplemental Table 13). Using stringent, case-informed cutoffs for expression alterations (Supplemental Figure 5), the genes that most commonly exhibited a longitudinal loss of expression included PIK3C2G (8 of 11 cases, 73%), ESR1 (7 of 11 cases, 64%), and TUBB3 (6 of 11 cases, 55%) (Figure 5A and Supplemental Figure 6). Other notable losses were found in GREM1, PTPRT, CDKN2A, KIT, and GATA3. The most recurrent longitudinal expression gains were seen in FGFR3 (7 of 11 cases, 64%) and EPHA3 and PTPRD (6 of 11 cases, 55%). PDGFRA, PTCH1, ALK, HGF, FGFR1, and FGFR4 also showed highly recurrent gains (Figure 5B). Interestingly, some expression gains were absent in synchronous bone metastasis cases (cases 19, 43, and 55), yet highly recurrent in long-term endocrine-deprived cases (EPHA3, PTPRD, PDGFRA, PTCH1), suggesting clinically actionable, treatment-driven gains in endocrine-resistant breast cancer recurrences.
Figure 5. Recurrent, clinically actionable expression gains and losses in ER-positive bone metastasis.

(A) Recurrent expression alteration losses, ranked by frequency, for each patient-matched case (columns, n = 11 cases). Each blue tile represents a bone metastasis with a lower log2 fold change vs. its matched primary than the case-specific expression loss threshold. Expression values (log2normCPMs) for most recurrent losses (PIK3C2G, ESR1) are pair plotted, with corresponding Wilcoxon signed-rank test P values noted. (B) Recurrent expression alteration gains, ranked by frequency. Red tiles represent bone metastases with higher log2 fold change than the case-specific expression gain thresholds. The two most recurrent expression gains (FGFR3, EPHA3) are also plotted with Wilcoxon signed-rank test P values.
Discussion
Bone is the most common site of distant recurrence for patients with ER-positive breast cancer, yet comprehensive sequencing data sets of endocrine therapy–treated, metastatic samples are currently limited. This is in part due to the challenge of obtaining tissue and degradation of nucleic acids caused by decalcification. In this study, we found that aged FFPE and FFPE-decalcified tumors showed highly similar transcript quantification values as matched flash-frozen and FFPE-nondecalcified tumors. As a proof of concept, we then applied ecRNA-seq to a cohort of patient-matched primary and BoMs collected over a period of 5 years. We identified subtle shifts in intrinsic subtypes and found a strong temporal influence on transcriptional evolution in breast cancer recurrences. Furthermore, we created several DEG sets/signatures that are prognostic and point toward acquired RBBP8 loss and CDK/Rb/E2F and FGFR pathway gains as mediators of ER-positive breast cancer progression. Finally, we found BoMs commonly gain or lose expression in clinically actionable genes, which may be distinct from primary tumors.
ecRNA-seq is an effective method for quantifying expression on aged, FFPE, and decalcified tumor specimens. Previous work has assessed nucleic acid amplification success, DNA sequencing, and RNA integrity metrics using decalcified samples (17, 28, 29); however, a comprehensive analysis of RNA sequencing, to our knowledge, has not yet been performed. Consistent with only very minor differences between GC content, insert sizes, and other QC metrics, gene expression values between aged-matched FFPE/flash-frozen and FFPE-decalcified/FFPE-nondecalcified tumors are highly correlated (Pearson r range 0.929–0.969). This study reinforces and should encourage the use of capture-hybridization approaches to sequence RNA from retrospectively collected, low-yield, highly degraded, and decalcified archival specimens (Supplemental Table 14 and refs. 22–24). Expanding sample sets and modalities for genome-wide characterization, especially for rare specimen cohorts that may be impractical to obtain prospectively in large numbers, will accelerate translational discoveries.
Given promising results from our evaluation, we applied ecRNA-seq in a proof-of-concept effort to characterize the transcriptome of 11 archival patient-matched ER-positive primary and recurrent metastases — 3 cases having treatment-naive, synchronous BoMs and 8 recurrent cases harboring long-term endocrine-therapy treated metastases. In the recurrent cases, bone metastasis–free survival ranged from 18 to 65 months. Despite a large portion of the BoMs being decalcified, global transcriptome QC metrics showed similar features (i.e., GC content, insert sizes, gene body coverage, and transcript assignment diversity) and no outliers. Consistent with this, unsupervised hierarchical clustering showed no distinct clusters of decalcified samples, with 5 BoMs clustering in the same doublet clade as their patient-matched primary breast cancer. Interestingly, 3 of these doublet-clustering pairs were clinically synchronous, treatment-naive BoMs, implying limited transcriptional evolution from the primary tumor in synchronous metastases. This was further corroborated with a striking negative correlation between patient-matched expression similarity and time to bone metastasis, suggesting metachronous metastases that present later clinically in their treatment course are more dissimilar from their derived primary lesions. Intrinsic subtyping revealed 4 of the 11 cases changed PAM50 subtypes, with all 4 cases switching to LumB in the metastasis. Subtle Her2 and LumB profile shifts were also the most common when observing continuous PAM50 probability scores, even in samples that remained concordant in their discrete PAM50 assignments. A recent targeted expression study analyzed PAM50 assignments in 123 matched breast cancer metastases, and the authors found similar frequencies of LumB and Her2 acquisitions in ER-positive metastatic tumors (30). Given this transcriptional evolution to more LumB and Her2 profiles, a thoughtful reevaluation of therapy selection in the advanced and perhaps the adjuvant setting may be necessary — especially considering HER2-targeted therapies are generally reserved for patients with HER2-positive primary disease.
We found 207 genes to be differentially expressed between primary tumors and patient-matched BoMs. The top upregulated genes — BGLAP, RANKL, and PTH1R — belonged to osteogenic gene sets and all showed significant expression gains. This supports in vivo modeling observations of breast cancer osteomimicry and hijacking of the bone microenvironment (31). Downregulated gene sets included genes involved in broad categories, such as cellular adhesion, hemidesmosome assembly, and epithelium development, pointing toward specific biological programs lost following metastatic colonization. Moreover, when either the upregulated or downregulated genes are expressed coordinately in primary tumors, we found that they confer worse and better outcomes, respectively, in ER-positive tumors, suggesting some tumors may develop these transcriptional programs early in their evolution. Finally, a differential expression analysis between endocrine-naive primary tumors and long-term endocrine-treated BoMs identified a larger list of DEGs. Importantly, known mediators of endocrine resistance are represented in the list, including dysregulated expression of Wnt family members (32), expression gains in FGFR1 (33) and FOXC1 (34), and loss of ESR1 expression (35). Notably, many of these genes do not overlap with the differential expression analysis that included the synchronous metastases, suggesting expression alterations specific to late recurrent therapy-treated tumors. This nonoverlapping gene set included a greater than 2-fold average expression gain of ABCG2 — a multidrug resistance protein shown to be active in breast cancer (36, 37) — in therapy-exposed metastases and loss of CDKN2A. CDKN2A encodes p16, a negative regulator of CDK4/CDK6 and is located on a common somatically deleted region (9p21) in cancer (38). Given the recent success of CDK4/CDK6-inhibiting compounds (palbociclib and ribociclib) in treating ER-positive breast cancers, this recurrent, acquired, metastatic-specific loss of CDK2NA is a clinically important observation (39–41).
Following significant gene-level changes, a GSEA defined enriched and diminished pathways in breast cancer BoMs. Enriched genes included those involved in G2M checkpoint and E2F targets. Consistent with the observed LumB enrichments, our data suggest breast cancer cells may develop a more proliferative phenotype following bone colonization, and the strong enrichment of E2F signature in metastatic disease again highlights the CDK/Rb/E2F pathway as a prime target. Interestingly, another study that utilized a targeted gene expression platform found proliferative gene signatures in ER-positive metastases may be more accurate at predicting overall survival than signatures in the primary tumor (30). A survival analysis for this work was impractical given the small set of patient-matched pairs, but future meta-analyses are warranted to determine if gene expression signatures in metastases are better predictors of overall survival in the advanced setting, especially given the significant transcriptomic shifts observed in this study.
The most significant gene set enriched in bone metastasis was an experimental perturbation gene set involving the knockdown of the tumor suppressor RBBP8 (42). RBBP8 (also known as CtIP) binds directly to Rb, mediates cell cycle regulation, and helps maintain genomic stability, and loss of RBBP8 incurs tamoxifen resistance and sensitizes breast cancer cells to PARP inhibition in vitro (43–46). Concordant with the GSEA analysis, BoMs have significant expression loss of RBBP8, with 45% of cases showing a greater than 2-fold decrease in expression. We found that low RBBP8 expression in ER-positive tumors confers poorer DSS and bone metastasis–free survival outcomes. These observations point to RBBP8 loss in metastatic breast cancers as being a compelling, perhaps therapeutically relevant candidate for further preclinical investigations.
Finally, considering that we have previously shown that brain metastases acquire highly recurrent gains in clinically actionable genes following colonization (13), particularly in HER2, we analyzed an expanded set of these genes in BoMs. All tumors harbored significant gains and losses, some of which were highly recurrent. PIK3C2G, a relatively uncharacterized gene in the PI3K pathway, was the most recurrent gene expression loss. Other notable losses included ESR1, CDKN2A, and GATA3. Intriguingly, GATA3 is one of the most recurrently mutated genes in breast cancer, being particularly enriched in ER-positive disease (47). Moreover, GATA3 inhibits breast cancer metastasis in various model systems, and given that losses of GATA3 in ER-positive BoMs are common, further evaluation of GATA3 as a potentially targetable breast cancer metastasis suppressor gene should be encouraged (34, 48, 49). Metastatic expression gains were found in FGFR family members (FGFR3, FGFR4, FGFR1), ALK, and KDR — all protein products having small molecules currently in clinical trials. Some highly recurrent expression gains (i.e., EPHA3, PTPRD, PDGFRA, PTCH1) were exclusive to long-term endocrine-treated BoMs, suggesting them as clinically actionable candidate mediators of therapy resistance. Collectively, these observations provide further evidence of acquired transcriptional remodeling in metastatic lesions and suggest that precision care in advanced cancers may benefit from defining longitudinal changes in tumor transcriptomes. Further research into these longitudinal transcriptional remodeling events is demanded — especially given their high recurrence rates — including identifying events that may be specific or more likely to occur in metastases from certain anatomic sites, such as HER2 gains in brain metastases (13).
Although this study points toward ecRNA-seq as being a viable option to characterize the transcriptome of archived, decalcified specimens, there are limitations. First, multiple methods are used for decalcification with varying effects on nucleic acids, and we were unaware of this information for the profiled specimens, as it is rarely recorded in clinical notes (17). Second, in metastasis-versus–primary tumor expression studies, it is difficult to deconvolute expression contributions from tumor cells and cells within the altered microenvironment of the distant organ site. To limit these artifacts in this study, regions of high tumor cellularity in the bone metastasis were cored by a trained molecular pathologist for RNA extraction, which was corroborated by ecRNA-seq–derived tumor purity estimates — as no significant tumor purity differences between primary and metastatic tumors (Supplemental Table 15) were observed (50). Nonetheless, single-cell sequencing approaches will be crucial to bring cell-level resolution to identifying transcriptional differences between primary and metastatic cells. Novel computational methods that deconvolute heterogeneous sample sets, until single-cell sequencing becomes more widely adopted, will also be essential (51–53). All of this withstanding, features of this data set are encouraging, such as patient-matched tumors clustering together, intuitive PAM50 assignments, corroboration of other groups’ findings, and treatment-specific gains and losses. A third limitation is performing an analysis on already colonized metastatic lesions, as this likely masks some of the intermediate steps involved in metastasis, such as epithelial mesenchymal transition and tumor-initiating programs. Finally, another limitation of this study is the small sample size. Hopefully, these results will encourage the use of ecRNA-seq to transcriptionally profile other highly degraded samples and begin a collection of genomic data from metastatic or rare tissues for integration. Importantly, deidentified clinical data should be provided alongside the sequencing, as in this study, to allow more fluid merging of data sets and inspire clinical phenotype-driven analyses.
In summary, this study both validates the use of ecRNA-seq to transcriptionally profile highly degraded RNA from decade-old and decalcified tumor specimens and defines multiple acquired and lost transcriptional programs in ER-positive BoMs. We highlight acquired changes in the CDK/Rb/E2F and FGFR pathways, particularly relevant given the recent clinical use of CDK4/6 inhibitors, and point toward RBBP8 as a particularly compelling candidate in breast cancer progression. We also found significant gains in clinically actionable genes that may have not been appreciated in primary tumors, reinforcing the need for longitudinal characterizations of tumor transcriptomes to guide clinical care.
Methods
Sample acquisition.
Eleven sets of FFPE primary breast tumors and patient-matched BoMs (total of 22 samples) were obtained from the Health Sciences Tissue Bank, a certified honest broker facility at the University of Pittsburgh that maintains an IRB-approved protocol for collecting excess tissue and biological materials. A molecular pathologist reviewed hematoxylin and eosin slides from each sample and then subsequently cut one to two 0.6- to 1-mm cores from the paraffin block exclusively from regions of high tumor cell purity for RNA extraction.
Tissue processing and RNA extraction.
Tissues were digested overnight with shaking at 300 rpm at 56°C in PKD buffer with the addition of proteinase K (Qiagen). RNA extraction was then performed with the FFPE RNeasy kit (Qiagen, catalog 73504) according to the manufacturer’s instructions under sterile RNase/DNase-free conditions. RNA concentration was determined with the Qubit 3.0 Fluorometer (ThermoFisher Scientific). Quality RNA integrity number scores, and fragment sizes (DV200 metrics) were obtained utilizing either the Agilent 2100 Bioanalyzer or the Agilent 4200 TapeStation.
ecRNA-seq.
Sequencing library preparation was performed using a minimum of 25 ng RNA according to Illumina’s TruSeq RNA Access Library Preparation protocol. Indexed, pooled libraries were then sequenced on the Illumina NextSeq 500 platform with high-output flow cell–producing stranded, paired-end reads (2 × 75 bp). A target count of 50 million reads per sample was used to plan indexing and sequencing runs.
RNA sequencing expression quantification and normalization.
RNA transcripts from paired-end FASTQ files were mapped and quantified using k-mer–based quasi-mapping with seqBias and gcBias corrections (Salmon v0.7.2, 31-kmer index built from GRCh38 Ensembl v82 transcript annotations) (54). Transcript-level abundance estimates were collapsed to gene-level estimates using tximport (55). To filter out nonexpressed genes or genes with low expression, only genes harboring a TPM value of more than 0.5 in at least 10% of samples were considered. Gene-level counts or log2normCPM values were implemented for subsequent analyses (56, 57).
Expression correlations and ecRNA-seq quality assessment.
Exome-capture ecRNA-seq was performed on two cohorts. A set of four aged (ranging from 8–12 years) primary breast cancer specimens that, at the time of surgical resection, were split in half and either immediately embedded in optimal cutting temperature compound and flash frozen for storage at –80°C or FFPE and stored at room temperature. A second cohort consisted of three breast cancer BoMs that, at the time of resection, were split in half and either decalcified or nondecalcified and processed to FFPE. These data sets were quantified and normalized as described above. Pearson r correlations between all samples were determined using log2normCPM values. Reads and mapping rates were obtained from Salmon. More detailed ecRNA-seq metrics were calculated and plotted using QoRTs (v1.1.8) following two-pass read alignment with STAR (v2.4.2a) for the 11 patient-matched cases (58, 59).
tumorMatch patient-matched sample identifier.
To confirm samples were patient-matched, variants from ecRNA-seq were called using GATK’s Best Practices for variant calling on ecRNA-seq (60). Output.vcf files were then provided to tumorMatch, a custom R script that analyzes a pool of.vcf files and calculates the proportion of shared variants (POSV) between each sample. These proportion values were visualized using corrplot in R (61).
Unsupervised hierarchical clustering and intrinsic subtyping.
Hierarchical clustering was performed using the heatmap.3 function (https://raw.githubusercontent.com/obigriffith/biostar-tutorials/master/Heatmaps/heatmap.3.R) in R on log2normCPM values of the top 5% most variable genes (defined by IQR) with 1 minus Pearson correlations as distance measurements and the “average” agglomeration method. PAM50 calls were generated using the molecular.subtyping function in genefu (62). A separate cohort of exome-capture RNA-sequencing expression data from primary tumors (n = 12 ER negative, 9 ER positive) was merged with the bone metastasis cohort to help account for test set bias and increase the stability of the PAM50 assignments (63). To call PAM50 subtypes, for each sample in the bone metastasis cohort, a random subset of primary tumor expression data were added to enforce a balanced distribution of ER-positive and ER-negative tumors. This was repeated 20 times, and the discrete PAM50 subtype was designated as the mode of this 20-fold PAM50 assignment test, while the final probability score was an average of all 20 probability scores from genefu.
Differential gene expression.
Salmon gene-level counts with effective lengths of target transcripts were used to call DEGs between primary tumors and BoMs using DESeq (64). Given that samples were patient matched, a multifactor design was implemented (~patient + tumor [i.e., metastasis vs. primary]). Genes with an FDR-adjusted P value of less than 0.10 were assigned as differentially expressed. An unclustered heatmap using log2normCPM values from the 207 DEGs, first segregated by metastatic log2 fold change gains and losses and then sorted by DESeq2-adjusted P values, was created in R using heatmap.3. DEGs within the MsigDB database that were gained or lost in BoMs were separately interrogated for gene ontology (GO: Biological Process) enrichment by computing significant (top 10 gene sets) gene overlaps using the MsigDB online tool (27).
ssGSEA signatures and METABRIC survival analyses.
Microarray expression along with DSS data were obtained from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) through Synapse (https://www.synapse.org/, Synapse ID: syn1688369), following IRB approval for data access from the University of Pittsburgh (26). Normalized expression values from IHC-confirmed ER-positive tumors were used to develop a ssGSEA for strongly DEGs (adjusted P < 0.05) between primary tumors and BoMs (25). 48 genes that carried positive log2 fold change values and had a corresponding gene expression value in METABRIC were assigned to the “boneMetSigUp” signature; 74 genes with negative log2 fold change values were assigned to the “boneMetSigDown” signature. A ssGSEA score for each sample from both gene sets was calculated using the ssGSEA method implemented in the GSVA R package (65). Binary dichotomization of samples (low vs. high) based on ssGSEA signature score strata (10th, 25th, 50th, 75th, 90th percentiles) and log-rank testing were used to assess significant differences in DSS (66). The strata with the most significant log-rank P values were plotted using survminer from CRAN (67).
Ranked GSEA.
To determine pathways significantly enriched or lost in breast cancer BoMs versus patient-matched primary tumors, GSEA analyses were performed using gene sets with coordinately expressed genes representing specific biological and cancer-related pathways (MSigDB: H and C6 sets). Input into GSEA was a ranked list (DESeq2 log2 fold change values) of 21,702 genes. Enrichment scores, significance values, and plots were generated using default settings of the Broad Institute’s javaGSEA Desktop Application (v2.2.3).
RBBP8 survival analysis.
RBBP8 expression was further interrogated and plotted using log2normCPM values from patient-matched tumors. RBBP8 expression influence on DSS in METABRIC ER-positive patients was interrogated as described above. RBBP8 expression influence on bone metastasis–free survival was assessed by querying a GCRMA-normalized microarray expression data set (GSE12276) from 204 primary tumors and associated survival data as described above (68).
Gains and losses in clinically actionable genes.
The clinically actionable gene set was obtained using the Drug Gene Interaction Database (DGBIdB 2.0) (69). Considering that metastatic fold change distributions calculated from log2normCPM values for all genes were slightly different for each case, stringent case-specific fold change thresholds were used to transform continuous fold change values into discrete “expression alterations.” More specifically, if the fold change value for a clinically actionable Gene_X was greater than the 95th percentile of all gene fold change values, in that case, Gene_X would be designated as having a significant, case-specific expression gain. If the fold change value for Gene_Y was lower than the 5th percentile, Gene_Y was designated as having a significant, case-specific expression loss (Supplemental Figure 6 and Supplemental Table 13). After assigning discrete expression alteration calls to clinically actionable genes, data were visualized using the oncoprint function in ComplexHeatmap (70).
Data and code.
Raw expression values for all samples, as well as R code related to this study, are deposited in GitHub (https://github.com/npriedig/).
Statistics.
To determine DEGs between patient-matched primary tumors and BoMs, DESeq2 was used. DESeq2 is designed for ecRNA-seq gene-based count abundance estimates and assigns differential expression P values based on a negative binomial distribution. For Kaplan-Meier curves, the log-rank test was used to determine statistically significant differences in event probabilities (i.e., death or time to metastasis) based on binary expression or signature strata. For single-gene queries, paired Wilcoxon signed-rank tests on log2normCPM values were used. A P value of less than 0.05 was considered significant. If error bars are shown, they represent mean ± SD. All statistical tests are 2 tailed, unless otherwise specified.
Study approval.
A protocol for this study was reviewed and approved by the University of Pittsburgh IRB Office. Tissue and associated data were obtained from the Health Sciences Tissue Bank, a certified honest broker facility at the University of Pittsburgh that maintains an IRB-approved protocol for collecting excess tissue and biological materials. Requirement for informed consent was waived, considering all samples were deidentified, there was no more than minimal risk to human subjects, and all tissue was obtained as part of routine clinical care.
Author contributions
NP, RJW, SO, and AVL provided the study concept and design; NP, RJW, PCL, AB, RB, WH, JKK, ZF, MQR, AMB, KRW, SO, AVL acquired, analyzed, or interpreted data; NP, RJW, SO, and AVL drafted the manuscript; NP, RJW, PCL, AB, RB, WH, JKK, ZF, MQR, AMB, KRW, SO, AVL provided critical revision of the manuscript for important intellectual content; and PCL, AB, RB, KRW, WH, JKK, MQR, ZF, and AMB provided administrative, technical, or material support.
Supplementary Material
Acknowledgments
This project used the University of Pittsburgh Health Sciences Core Research Facilities Genomics Research Core and Health Sciences Tissue Bank and the UPMC Hillman Cancer Center Tissue and Research Pathology Services, supported in part by award NIH grant P30CA047904. The authors would like to thank the patients who contributed samples to this study as well as Lori Miller (University of Pittsburgh), Alma E. Heyl (UPMC), and Jorge A. Rios (UPMC) for their efforts in collecting tissues. This research was also supported in part by the University of Pittsburgh Center for Research Computing and the University of Pittsburgh Center for Simulation and Modeling through the resources provided. Research funding for this project was provided in part by Susan G. Komen Scholar awards (SAC110021 to AVL and SAC160073 to SO), the Breast Cancer Research Foundation (to AVL and SO), the Fashion Footwear Association of New York, the Magee-Womens Research Institute and Foundation, the Nicole Meloche Foundation, the Penguins Foundation, the Penguins Alumni Foundation, and the Mario Lemieux Foundation as well as through a postdoctoral fellowship awarded to RJW from the Department of Defense (BC123242). NP was supported by a training grant from the NIH/National Institute of General Medical Sciences (2T32GM008424-21) and an individual fellowship from the NIH/National Cancer Institute (5F30CA203095).
Version 1. 08/31/2017
Electronic publication
Funding Statement
Awarded to AVL
Awarded to SO
Awarded to SO and AVL
Awarded to RW
Awarded to NP
Awarded to SO and AVL
Awarded to SO and AVL
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(17): e95703. https://doi.org/10.1172/jci.insight.95703.
Contributor Information
Nolan Priedigkeit, Email: priedigkeitn@upmc.edu.
Peter C. Lucas, Email: lucaspc@upmc.edu.
Ahmed Basudan, Email: basudana@upmc.edu.
Rohit Bhargava, Email: rbhargava@mail.magee.edu.
William Horne, Email: william.horne@chp.edu.
Jay K. Kolls, Email: jay.kolls@chp.edu.
Zhou Fang, Email: fangz.ark@gmail.com.
Steffi Oesterreich, Email: oesterreichs@upmc.edu.
References
- 1.Ibrahim T, Mercatali L, Amadori D. A new emergency in oncology: Bone metastases in breast cancer patients (Review) Oncol Lett. 2013;6(2):306–310. doi: 10.3892/ol.2013.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11(6):411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Moos R, Haynes I. Where do bone-targeted agents RANK in breast cancer treatment? J Clin Med. 2013;2(3):89–102. doi: 10.3390/jcm2030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.James JJ, et al. Bone metastases from breast carcinoma: histopathological - radiological correlations and prognostic features. Br J Cancer. 2003;89(4):660–665. doi: 10.1038/sj.bjc.6601198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang XH, Giuliano M, Trivedi MV, Schiff R, Osborne CK. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin Cancer Res. 2013;19(23):6389–6397. doi: 10.1158/1078-0432.CCR-13-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/S1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 7.Minn AJ, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115(1):44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu X, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell. 2011;20(6):701–714. doi: 10.1016/j.ccr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27(2):193–210. doi: 10.1016/j.ccell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosol TJ. Pathogenesis of bone metastases: role of tumor-related proteins. J Bone Miner Res. 2000;15(5):844–850. doi: 10.1359/jbmr.2000.15.5.844. [DOI] [PubMed] [Google Scholar]
- 11.Hugo W, et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell. 2015;162(6):1271–1285. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brastianos PK, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5(11):1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Priedigkeit N, et al. Intrinsic subtype switching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA Oncol. 2017;3(5):666–671. doi: 10.1001/jamaoncol.2016.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Poznak C, et al. Use of biomarkers to guide decisions on systemic therapy for women with metastatic breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2015;33(24):2695–2704. doi: 10.1200/JCO.2015.61.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto-Fukud T, et al. Effects of various decalcification protocols on detection of DNA strand breaks by terminal dUTP nick end labelling. Histochem J. 2000;32(11):697–702. doi: 10.1023/A:1004171517639. [DOI] [PubMed] [Google Scholar]
- 16.Gertych A, et al. Effects of tissue decalcification on the quantification of breast cancer biomarkers by digital image analysis. Diagn Pathol. 2014;9:213. doi: 10.1186/s13000-014-0213-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrijver WA, et al. Influence of decalcification procedures on immunohistochemistry and molecular pathology in breast cancer. Mod Pathol. 2016;29(12):1460–1470. doi: 10.1038/modpathol.2016.116. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol. 2002;161(6):1961–1971. doi: 10.1016/S0002-9440(10)64472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Allen EM, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20(6):682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munchel S, et al. Targeted or whole genome sequencing of formalin fixed tissue samples: potential applications in cancer genomics. Oncotarget. 2015;6(28):25943–25961. doi: 10.18632/oncotarget.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greytak SR, Engel KB, Bass BP, Moore HM. Accuracy of molecular data generated with FFPE biospecimens: lessons from the literature. Cancer Res. 2015;75(8):1541–1547. doi: 10.1158/0008-5472.CAN-14-2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cieslik M, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. 2015;25(9):1372–1381. doi: 10.1101/gr.189621.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graw S, et al. Robust gene expression and mutation analyses of RNA-sequencing of formalin-fixed diagnostic tumor samples. Sci Rep. 2015;5:12335. doi: 10.1038/srep12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabanski CR, et al. cDNA hybrid capture improves transcriptome analysis on low-input and archived samples. J Mol Diagn. 2014;16(4):440–451. doi: 10.1016/j.jmoldx.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh VM, et al. Analysis of the effect of various decalcification agents on the quantity and quality of nucleic acid (DNA and RNA) recovered from bone biopsies. Ann Diagn Pathol. 2013;17(4):322–326. doi: 10.1016/j.anndiagpath.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Zheng G, et al. Clinical mutational profiling of bone metastases of lung and colon carcinoma and malignant melanoma using next-generation sequencing. Cancer. 2016;124(10):744–753. doi: 10.1002/cncy.21743. [DOI] [PubMed] [Google Scholar]
- 30.Cejalvo JM, et al. Intrinsic subtypes and gene expression profiles in primary and metastatic breast cancer. Cancer Res. 2017;77(9):2213–2221. doi: 10.1158/0008-5472.CAN-16-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu GC, Chung LW. RANK-mediated signaling network and cancer metastasis. Cancer Metastasis Rev. 2014;33(2-3):497–509. doi: 10.1007/s10555-013-9488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loh YN, Hedditch EL, Baker LA, Jary E, Ward RL, Ford CE. The Wnt signalling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer. 2013;13:174. doi: 10.1186/1471-2407-13-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner N, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu-Rice Y, et al. FOXC1 is involved in ERα silencing by counteracting GATA3 binding and is implicated in endocrine resistance. Oncogene. 2016;35(41):5400–5411. doi: 10.1038/onc.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle LA, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95(26):15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mo W, Zhang JT. Human ABCG2: structure, function, and its role in multidrug resistance. Int J Biochem Mol Biol. 2012;3(1):1–27. [PMC free article] [PubMed] [Google Scholar]
- 38.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368(6473):753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 39.Turner NC, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209–219. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 40.Finn RS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–1936. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 41.Hortobagyi GN, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–1748. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 42.Furuta S, et al. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell. 2006;10(1):13–24. doi: 10.1016/j.ccr.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 43.Liu F, Lee WH. CtIP activates its own and cyclin D1 promoters via the E2F/RB pathway during G1/S progression. Mol Cell Biol. 2006;26(8):3124–3134. doi: 10.1128/MCB.26.8.3124-3134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu M, et al. CtIP silencing as a novel mechanism of tamoxifen resistance in breast cancer. Mol Cancer Res. 2007;5(12):1285–1295. doi: 10.1158/1541-7786.MCR-07-0126. [DOI] [PubMed] [Google Scholar]
- 45.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459(7245):460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Ding Q, Fujimori H, Motegi A, Miki Y, Masutani M. Loss of CtIP disturbs homologous recombination repair and sensitizes breast cancer cells to PARP inhibitors. Oncotarget. 2016;7(7):7701–7714. doi: 10.18632/oncotarget.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J Biol Chem. 2010;285(18):14042–14051. doi: 10.1074/jbc.M110.105262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15(2):201–213. doi: 10.1038/ncb2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshihara K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Xie X. A mixture model for expression deconvolution from RNA-seq in heterogeneous tissues. BMC Bioinformatics. 2013;14(suppl 5):S11. doi: 10.1186/1471-2105-14-S5-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gong T, Szustakowski JD. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics. 2013;29(8):1083–1085. doi: 10.1093/bioinformatics/btt090. [DOI] [PubMed] [Google Scholar]
- 53.Onuchic V, et al. Epigenomic deconvolution of breast tumors reveals metabolic coupling between constituent cell types. Cell Rep. 2016;17(8):2075–2086. doi: 10.1016/j.celrep.2016.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartley SW, Mullikin JC. QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics. 2015;16:224. doi: 10.1186/s12859-015-0670-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.1033. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wei T, Simko V. corrplot: Visualization of a Correlation Matrix. R package version 0.77. https://cran.r-project.org/web/packages/corrplot/index.html Updated April 21, 2016. Accessed August 8, 2017.
- 62.Gendoo DM, et al. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics. 2016;32(7):1097–1099. doi: 10.1093/bioinformatics/btv693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patil P, Bachant-Winner PO, Haibe-Kains B, Leek JT. Test set bias affects reproducibility of gene signatures. Bioinformatics. 2015;31(14):2318–2323. doi: 10.1093/bioinformatics/btv157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bland JM, Altman DG. The logrank test. BMJ. 2004;328(7447):1073. doi: 10.1136/bmj.328.7447.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kassambara A, Kosinski M. survminer: drawing survival curves using ‘ggplot2’. R package version 0.2.4. https://CRAN.R-project.org/package=survminer Updated June 7, 2017. Accessed August 8, 2017.
- 68.Bos PD, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagner AH, et al. DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016;44(D1):D1036–D1044. doi: 10.1093/nar/gkv1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.