

Abstract
It has been proposed that xanthone derivatives with anticancer potential act as topoisomerase II inhibitors because they interfere with the ability of the enzyme to bind its ATP cofactor. In order to further characterize xanthone mechanism and generate compounds with potential as anticancer drugs, we synthesized a series of derivatives in which position 3 was substituted with different polyamine chains. As determined by DNA relaxation and decatenation assays, the resulting compounds are potent topoisomerase IIα inhibitors. Although xanthone derivatives inhibit topoisomerase IIα-catalyzed ATP hydrolysis, mechanistic studies indicate that they do not act at the ATPase site. Rather, they appear to function by blocking the ability of DNA to stimulate ATP hydrolysis. On the basis of activity, competition, and modeling studies, we propose that xanthones interact with the DNA cleavage/ligation active site of topoisomerase IIα and inhibit the catalytic activity of the enzyme by interfering with the DNA strand passage step.
Keywords: Xanthone derivative, polyamines, DNA topoisomerase IIα, anticancer drug, catalytic inhibitor, DNA cleavage
Graphical Abstract
Xanthones are a class of heterotricyclic planar compounds that were originally identified as secondary metabolites in plants and microorganisms.1–3 These compounds display a wide range of biological activities and represent a prominent scaffold for agents with medicinal properties. Several xanthones currently are under development as anticancer, anti-inflammatory or anti-thrombotic agents.2, 4–11 A number of these compounds display activity against type II topoisomerases.11–16 Furthermore, the primary cellular target of gambogic acid, a xanthone derivative in human phase II clinical trials as an anticancer drug, appears to be topoisomerase IIα.12, 13
Human type II topoisomerases play critical roles in a number of nucleic acid processes, including DNA replication, transcription, recombination, and chromosome segregation.17–22 These enzymes alter the topological state of the genetic material by transiently cleaving both strands of the double helix and passing a second double helix through the enzyme-generated DNA gate. This process is dependent on the presence of an ATP cofactor. ATP binding promotes the DNA strand passage event, while hydrolysis of the cofactor is required for enzyme turnover. Type II topoisomerases regulate DNA under- and overwinding, and more importantly, remove tangles and knots from the human genome.17–22 Humans encode two enzyme isoforms, topoisomerase IIα and IIβ. Topoisomerase IIα is essential for the survival of proliferating cells and is the isoform that is involved in DNA replication and the segregation of daughter chromosomes. In contrast, topoisomerase IIβ is not essential for cells survival, but is important for neurological development. Although the precise function of topoisomerase IIβ is not well defined, it appears to play an important role in the expression of hormonally regulated genes.17–22
Type II topoisomerases are the target for several commonly prescribed anticancer drugs, including etoposide, doxorubicin and mitoxantrone.17, 18, 22–27 These drugs act by increasing levels of covalent topoisomerase II-cleaved DNA complexes (i.e., cleavage complexes), which are intermediates in the catalytic cycle of the enzyme. As result of drug action, the type II enzymes are converted to cellular toxins that fragment the genome. Drugs that act by this mechanism are called topoisomerase II poisons.17, 18, 22–27
A second group of cytotoxic drugs that includes novobiocin, merbarone, aclarubicin, and ICRF-193 also targets type II topoisomerases. In contrast to the topoisomerase II poisons, these compounds are catalytic inhibitors that kill cells by robbing them of the essential activities of the type II enzyme. Catalytic inhibitors can block topoisomerase II activity by a variety of mechanisms, which can have important consequences for cells. Whereas drugs that block ATP binding (novobiocin), DNA cleavage, or strand passage (merbarone) inhibit the catalytic reactions of topoisomerase II, those that disrupt topoisomerase II-DNA binding (aclarubicin) impair both the structural and catalytic roles of the enzyme. Furthermore, drugs that allow ATP binding but block the hydrolysis of the high-energy cofactor (ICRF-193), trap the enzyme on the DNA.28–37
Gambogic acid and several other xanthone derivatives are believed to inhibit human topoisomerase IIα by binding to the ATPase active site of the enzyme and blocking interactions between the enzyme and the ATP cofactor.12, 15, 16, 38 This hypothesis is based on i) modeling studies that docked compounds to the ATPase site of topoisomerase IIα, ii) in vitro experiments that demonstrated the inhibition of ATP hydrolysis by xanthone-based compounds, and iii) surface plasmon resonance studies that suggested that gambogic acid could bind the ATP domain of the human enzyme.
However, several lines of evidence suggest that the inhibition of topoisomerase IIα by xanthone derivatives may be more complex. First, all of the ATPase studies reported for xanthone-based compounds were carried out in the presence of DNA.12, 15, 16 Because the ATPase activity of type II topoisomerases is stimulated by DNA binding and strand passage,39–41 interfering with DNA interactions could manifest itself as an indirect inhibition of ATP hydrolysis. Second, many xanthone-based compounds bind to DNA.13, 16 Thus, they may be able to interact with the DNA cleavage/ligation active site of type II topoisomerases. Third, some previously described xanthone derivatives display an IC50 for inhibition of ATP hydrolysis that is >10-fold higher than observed for the inhibition of relaxation.15, 16 This makes it unlikely that the loss of overall catalytic activity could have resulted from interference with ATP interactions. Fourth, some xanthone-based compounds inhibit the DNA relaxation reaction of topoisomerase I. This is despite the fact that the type I enzyme has no binding site for ATP.42
Therefore, to further examine the mechanism by which xanthones inhibit topoisomerase IIα, we synthesized a series of new xanthone polyamine conjugates, 2-5, by inserting at the 3 position a side chain containing different polyamine moieties, including propandiamine (compound 2), butandiamine (compound 3), spermidine (compound 4), and spermine (compound 5) (Fig. 1). Substitution at the 3 position is favored over the 1 position because of the proximity of the carbonyl.16 These polyamines were chosen because a previous study found that the presence of a secondary amine group in the side chain plays an important role in mediating topoisomerase II-drug interactions.43–45 Furthermore, the addition of a spermine side chain to the core of etoposide (generating F14512) greatly enhanced the ability of the drug to act as a topoisomerase II poison and to be taken up by cancer cells with active polyamine transport systems.43–47
Fig. 1.

Structures and synthetic pathway of the compounds utilized in this study. Reagents and Conditions: (a) ZnCl2, POCl3, 70 °C, 3 hours, 69% yield; (b) epichlorohydrin, K2CO3, DMF, 80 °C, 5 hours, mw, 32% yield; (c) DMF, 50 °C, 26 hours, 32-80% yield; (d) CF3COOH, CH2Cl2, 0 °C, 2 hours or HCl in dioxane, 0 °C, 2-5 hours, 33-60%yield. Boc = (CH3)3COCO. * = hydrochloride salt; ** = trifluoroacetate salt.
Compounds 1-5 were synthesized using the generalized scheme shown in Fig. 1. The key intermediate, 1-hydroxy-3-(oxiran-2-ylmethoxy)-9H-xanthen-9-one (7), was synthesized by an O- alkylation reaction of compound 6. The synthesis of compound 7 was performed under microwave irradiation in order to shorten the reaction time. To join the nucleophilic chains to the xanthone core, intermediate 7 was coupled with butylamine and the N-Boc protected polyamines 12-15 to generate compounds 1 and 8-11, respectively. In order to synthesize the final compounds 2 and 5 or 3 and 4, tert-butyloxycarbonyl (Boc) groups were removed with 4 M HCl in dioxane or with trifluoroacetic acid (TFA) in CH2Cl2, respectively. All of the compounds were synthesized as racemic mixtures. The detailed syntheses and physical and chemical characterizations of the compounds are described in the accompanying Supplementary Data.
As a first step toward characterizing the activities of the xanthone derivatives shown in Fig.1 against human topoisomerase IIα, the effects of compounds 1-6 on enzyme-mediated DNA cleavage were determined (Fig. 2). Consistent with previous reports,12, 15, 16 none of the compounds displayed a significant ability to enhance DNA cleavage. Thus, these xanthone derivatives do not appear to act primarily as topoisomerase II poisons.
Fig. 2.

Effects of xanthone derivatives on DNA cleavage mediated by topoisomerase IIα. Results for compounds 1-6 (2.5 µM, blue; 10 µM, red; 50 µM, yellow; 100 µM, green) on the generation of enzyme-mediated double-stranded DNA breaks are shown. Due to solubility issues, compound 5 was only used up to 10 µM. DNA cleavage in the presence of 100 µM etoposide (purple) is shown for comparison. DNA cleavage levels were calculated relative to control reactions that contained no drug (TII, orange) and were set to 1. Error bars represent standard deviations for 2-3 independent experiments. The inset shows an ethidium bromide-stained gel of a typical DNA cleavage experiment carried out in the presence of 10 µM compounds 1-6 and 100 µM etoposide. The positions of negatively supercoiled (form I, FI), nicked (form II, FII) and linear (form III, FIII) DNA are indicated.
Next, we examined the abilities of compounds 1-6 to inhibit the overall catalytic activity of topoisomerase IIα using a DNA relaxation assay (Fig. 3). All of the compounds inhibited DNA relaxation and fell into three groups regarding potency: compounds 2 and 5 displayed an IC50 ≈ 1 µM, compounds 3 and 4 displayed an IC50 ≈ 2.5-5 µM, and compounds 1 and 6 displayed an IC50 ≈ 100 µM. From these results, it appears that the presence of the side chain containing a primary and secondary amine is critical. However, the number of secondary amines in the chain and their distance from the primary amine appear to be less important. These results notwithstanding, it should be noted that a previous study reported that compound 1 displayed an IC50 ≈ 9 µM for the inhibition of DNA relaxation catalyzed by topoisomerase IIα.16 We could not recapitulate this finding in the present study. Given the low activity of compound 1 in our hands and the fact that the activity of compound 3 was similar to other derivatives we tested, we focused on compounds 2 and 4-6 for more detailed studies.
Fig. 3.

Effects of xanthone derivatives on DNA relaxation catalyzed by topoisomerase IIα. Results for compounds 1-6 are shown. Assays containing intact negatively supercoiled DNA in the absence of topoisomerase IIα (DNA) or negatively supercoiled DNA treated with topoisomerase IIα in the absence of xanthone derivatives (TII) are shown as controls. The positions of negatively supercoiled (form I, FI) and nicked (form II, FII) DNA are indicated. Gels are representative of 2-4 independent experiments.
The ability of xanthone derivatives to inhibit the catalytic activity of topoisomerase IIα was confirmed using a decatenation assay (Fig. 4). Although IC50 values were ~2- to 3- fold higher than observed in the relaxation assay, the order of potency remained similar, with compounds 2, 4, 5 being much more potent than compound 6.
Fig. 4.

Effects of xanthone derivatives on DNA decatenation catalyzed by topoisomerase IIα. Results for compounds 2, 4-6 are shown. Assays containing intact kDNA in the absence of topoisomerase IIα (DNA) or kDNA treated with topoisomerase IIα in the absence of xanthone derivatives (TII) are shown as controls. The positions of intact kDNA at the origin (kDNA), decatenated nicked kDNA minicircles, and decatenated supercoiled (SC) kDNA minicircles are indicated. Gels are representative of 3 independent experiments. Quantification of results is shown in the bar graphs at the bottom. Levels of decatenation in the absence of xanthone derivatives was set to 100%. Error bars represent standard deviations for 3 independent experiments.
Polyamines have the potential to bind to the double helix and alter topoisomerase II-DNA interactions in the absence of a specific interaction with the enzyme.48–50 Consequently, is possible that the xanthone derivatives inhibit the activity of topoisomerase IIα in a bimodal fashion with the aromatic core of one molecule binding to the protein and the polyamine tail of another acting through a general effect on the DNA. Therefore, the importance of the linkage between the spermidine/spermine polyamine tails and the xanthone core (compound 6) in a single molecule was examined.51 Independently, the IC50 values for the inhibition of DNA relaxation by topoisomerase IIα for spermidine and spermine were >1 mM (data not shown) and that of compound 6 was >100 µM. As seen in Fig. 5, a 1:1 mixture of compound 6 and spermidine or spermine showed no ability to inhibit DNA relaxation at 10 µM. This is compared to compounds 4 and 5 (which are essentially compound 6 coupled to spermidine and spermine, respectively), that displayed IC50 values <5 µM. Thus, the linkage between the xanthone core and the polyamine tails is critical for the potent inhibitory activity of these compounds.
Fig. 5.

Covalent linkage of the C3 polyamine moiety to the 1,3-dyhydroxy-9h-xanthen-9-one core is critical for the inhibition of DNA relaxation catalyzed by topoisomerase IIα. The effects of a 1:1 mixture of 1,3- dyhydroxy-9h-xanthen-9-one (compound 6) + spermidine (left panel) or + spermine (right panel) on the DNA relaxation activity of human topoisomerase IIα is shown. Assays containing intact negatively supercoiled DNA in the absence of topoisomerase IIα (DNA) or negatively supercoiled DNA treated with topoisomerase IIα in the absence of xanthone derivatives (TII) are shown as controls. The positions of DNA markers are as shown in Figure 3. Gels are representative of 2 independent experiments. Quantification of results is shown in the graphs at the bottom. Data for compound 6 + spermidine (Spmd, left) and compound 6 + spermine (Spm, right) are shown as open circles. Control reactions in the presence of compound 4 (left) and 5 (right) are shown as closed circles. Error bars represent standard error of the mean for 2 independent experiments.
As discussed above, the mechanism by which a compound inhibits the activity of topoisomerase II can have profound cellular consequences. Thus, it is important to understand which step of the topoisomerase II catalytic cycle is affected by xanthones. This catalytic cycle can be divided into six discrete steps: 1) DNA binding, 2) DNA bending, 3) DNA cleavage, 4) strand passage, 5) DNA religation, 6) release of the DNA substrate and enzyme turnover.17, 18, 52 ATP binding drives the strand passage step and ATP hydrolysis is required for enzyme turnover and the completion of the catalytic cycle.17, 18, 20, 21, 32, 53
To determine whether the xanthone derivatives affect topoisomerase II activity at steps up to and including DNA scission, we reassessed the effects of the compounds 2, 4-6 on enzyme-mediated DNA cleavage (Fig. 6). Normally, topoisomerase IIα maintains very low levels of cleavage complexes in the presence of Mg2+, its physiological divalent cation.54, 55 This makes it difficult to determine the ability of the drug to inhibit this reaction step. Consequently, the experiments shown in Fig. 6 were carried out in the presence of Ca2+, which raises baseline levels of DNA cleavage ~10- to 15-fold.54, 56 No substantial inhibition was observed for xanthone concentrations up to 10 µM, the range in which compounds inhibited overall catalytic activity. This finding strongly suggests that these compounds do not impair the overall catalytic activity (DNA relaxation or decatenation) of topoisomerase IIα by inhibiting any of the reaction steps through DNA cleavage. However, this conclusion comes with the caveat that the DNA cleavage assay utilizes 150 nM enzyme as compared to the relaxation assay, which uses 3 nM. Thus, it could take higher concentrations of xanthone derivatives to inhibit the catalytic activity of topoisomerase IIα under conditions of the cleavage assay. Therefore, as a control, the effects of compounds 2, 4-6 on DNA relaxation catalyzed by 150 nM topoisomerase IIα were determined (Fig. 7). Similar to the results of Fig. 3, compounds 2, 4, and 5 displayed complete (or near complete) inhibition of enzyme activity by 10 µM. (Note that 10 µM compound 6 displayed no ability to inhibit DNA relaxation under either assay condition).
Fig. 6.

Inhibition of topoisomerase IIα-mediated DNA cleavage by xanthone derivatives. Results for compounds 2, 4-6 are shown. Reactions were carried out in the presence of CaCl2 to enhance baseline levels of DNA cleavage by the enzyme. Assays containing negatively supercoiled DNA in the absence of topoisomerase IIα (DNA) or negatively supercoiled DNA treated with topoisomerase IIα in the absence of xanthone derivatives (TII) are shown as controls. The positions of DNA markers are as shown in Figure 2 inset. Gels are representative of 3 independent experiments. Quantification of results is shown in the graphs at the bottom. Levels of DNA cleavage in the absence of xanthone derivatives was set to 100%. Error bars represent standard deviations for 2-3 independent experiments.
Fig. 7.

Effects of xanthone derivatives on DNA relaxation catalyzed by 150 nM topoisomerase IIα. Results for compounds 2, 4-6 are shown. Assays containing intact negatively supercoiled DNA in the absence of topoisomerase IIα (DNA) or negatively supercoiled DNA treated with topoisomerase IIα in the absence of xanthone derivatives (TII) are shown as controls. The positions of DNA markers are as in Figure 3. Gels are representative of 2-4 independent experiments.
Taken together, the results shown in Figs. 6 and 7 indicate that the xanthone derivatives act by inhibiting topoisomerase IIα in a reaction step that follows DNA cleavage. Consequently, we examined the effects of compounds 4, and 5 on DNA strand passage mediated by the enzyme (Fig. 8). A DNA catenation assay was employed that utilizes a high concentration (150 nM) of topoisomerase IIα and replaces the ATP cofactor with the non-hydrolyzable analogue, APP(NH)P. Because APP(NH)P cannot be hydrolyzed by topoisomerase IIα, this assay monitors enzyme activity through the strand passage step.52, 57 By using a DNA catenation assay, a single catalytic event moves the DNA substrate away from the bands of relaxed DNA. Relaxed, rather than negatively supercoiled, DNA was used for this assay because it is a preferred substrate for catenation. Similar to the results in Fig. 7, compounds 4 and 5 all displayed IC50 values <10 µM. Because these compounds showed little ability to inhibit DNA cleavage (Fig. 6), the step that immediately precedes the strand passage step, we conclude that the xanthone derivatives decrease the overall catalytic activity of topoisomerase IIα by inhibiting the ability of the enzyme to carry out DNA strand passage.
Fig. 8.

Effects of xanthone derivatives on DNA strand passage mediated by topoisomerase IIα. Results for compounds 4 and 5 are shown. Assays monitored the catenation of relaxed DNA in the presence of the non- hydrolyzable ATP analogue APP(NH)P so that the enzyme could only carry out one round of DNA strand passage. Assays containing relaxed DNA in the absence of topoisomerase IIα (DNA) or relaxed DNA treated with topoisomerase IIα in the absence of xanthone derivatives (TII) are shown as controls. The positions of relaxed DNA (Rel) and catenated DNA at the origin (Cat) are indicated. Gels are representative of at least 3 independent experiments. Quantification of results is shown in the graphs at the bottom. Levels of catenation in the absence of xanthone derivatives was set to 100%. Error bars represent standard deviations for at least 3 independent experiments.
Xanthones can inhibit the strand passage step in three different ways: they can bind at the ATP active site and interfere with topoisomerase IIα-ATP interactions, they can bind at the DNA cleavage/ligation active site and interfere directly with DNA movement, or they may bind outside of either active site and cause deleterious conformational changes in the enzyme. On the basis of modeling studies and the ability of compounds to inhibit enzyme-catalyzed ATP hydrolysis, previous studies suggested that xanthone derivatives acted by inhibiting topoisomerase IIα-ATP interactions.12, 15, 16, 38 Therefore, we assessed the effects of compound 4 on ATP hydrolysis catalyzed by topoisomerase IIα. As seen in Fig. 9, compound 4 inhibited ATP hydrolysis with an IC50 ≈10 µM. However, as with earlier studies, ATPase assay mixtures contained DNA. As discussed above (and shown in Fig. 9), rates of ATP hydrolysis are stimulated ~ 5-fold by the presence of DNA. Thus, decreased rates of ATP hydrolysis observed in the presence of compound 4 could be due to interactions of the xanthone derivative at either the DNA or ATP sites. In order to distinguish between these two possibilities, the effects of compound 4 on ATP hydrolysis were examined in the absence of DNA (Fig. 9). Under this condition, no inhibition was observed up to 50 µM xanthone. Thus, we conclude that the xanthone-induced decrease in the rate of ATP hydrolysis in the presence of DNA is not due to a direct inhibition of ATP binding. Rather, it is observed because xanthone derivatives inhibit the ability of DNA to stimulate the rate of ATP hydrolysis. Consistent with this conclusion, ATPase rates generated in the presence of supercoiled plasmid asymptotically approached those seen in the absence of DNA as the concentration of compound 4 increased (Fig. 9).
Fig. 9.

Effects of compound 4 on ATP hydrolysis catalyzed by topoisomerase IIα. Assays carried out in the presence or absence of negatively supercoiled DNA are shown as closed or open circles, respectively. ATPase activity rate in the presence of DNA and in the absence of compound 4 was set to 100%. Error bars represent standard error of the mean for 2 independent experiments.
To further define the site of interaction of xanthone derivatives on topoisomerase IIα, we used a competition assay to determine whether these compounds could be inhibiting DNA strand passage (and ATP hydrolysis) by acting at the DNA cleavage/ligation active site. The competition assay determined the ability of compounds 2, 4 and 5 (which do not inhibit the DNA cleavage step) to block DNA cleavage enhancement by etoposide. This drug has been shown to bind at the DNA cleavage/ligation active site of human type II topoisomerases.58 As seen in Fig. 10, all three compounds competed with etoposide, diminishing the ability of the anticancer drug to induce topoisomerase IIα-mediated DNA scission. These data strongly suggest that the xanthone derivatives interact in the vicinity of the DNA cleavage/ligation active site of the human type II enzyme, despite the fact that they have little effect on the cleavage reaction. A similar conclusion has been drawn for the binding of some quinolone-derivatives that do not enhance DNA cleavage to eukaryotic or prokaryotic type II topoisomerases.59, 60
Fig. 10.

Ability of xanthone derivatives to inhibit the enhancement of topoisomerase IIα-mediated DNA cleavage by 100 µM etoposide. Results for compounds 2, 4, and 5 (5 µM, blue; 10 µM, red; 25 µM, green; 50 µM, yellow; 100 µM, orange) on the generation of enzyme-mediated double-stranded DNA breaks are shown. Due to solubility issues, compound 5 was only used up to 10 µM. DNA cleavage in the presence of 100 µM etoposide (purple) in the absence of xanthone derivatives set to 100%. Error bars represent standard deviations for 2-3 independent experiments.
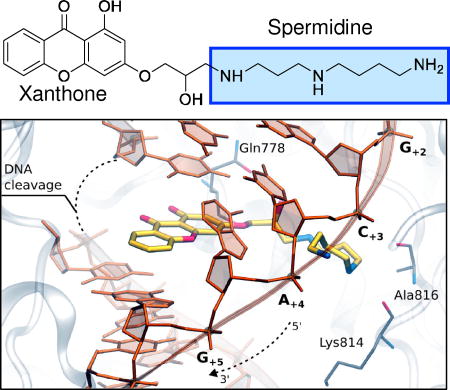
Finally, to determine the feasibility of xanthone binding at the cleavage/ligation active site of topoisomerase IIα, compound 4 was docked into the topoisomerase II cleavage complex by molecular modeling. Both the R and S enantiomers were used for modeling studies. Fig. 11 shows the results with the R enantiomer. Our findings suggest that the heterocyclic moiety of the xanthone derivative preferentially locates in a region similar to that of the 4’-demethylepipodophyllotoxin core of etoposide.58 The polyamine chain extends toward the DNA major groove, as was observed in previous computations of F14512 and other polyamine-conjugates.45 Compound 4 has the potential to form several favorable interactions with both the enzyme and the DNA. The hydroxyl group along the polyamine chain points toward Gln778, while the central nitrogen atom is in close enough proximity to form hydrogen bonds with the backbone of either Lys814 or Ala816. The terminal amine is stabilized by the DNA, as it lays in between the phosphates of DNA bases C+3 and A+4.
Fig. 11.

Molecular modeling of compound 4. Docking of compound 4 into the DNA cleavage/ligation active site of topoisomerase IIα (Top). Superposition between etoposide (pdb: 3QX3) and the docking pose of compound 4 into the topoisomerase II DNA cleavage complex (Bottom).
In conclusion, xanthone derivatives represent a potent class of topoisomerase II inhibitors with anticancer potential. Previously, these compounds were believed to inhibit enzyme activity by interfering with the binding of the high-energy ATP cofactor. However, results of the present study strongly suggest that xanthone-polyamine conjugates act at the DNA cleavage/ligation active site of human topoisomerase IIα and impair catalytic activity by blocking the DNA strand passage step of the topoisomerase II catalytic cycle. Having a greater understanding of xanthone mechanism may allow for future development of more active derivatives that utilize this important medicinal scaffold.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grant GM033944 to N.O., Italian Association for Cancer Research (AIRC) grant IG n. 18883 to M.D.V., and funds from the University of Bologna (RFO) to A.M. We are grateful to Elizabeth G. Gibson for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interests.
Experimental details for the synthesis and characterization of the xanthone derivatives, as well as sources of materials and methods for biochemical assays and modeling studies, are available in the accompanying Supplementary Data.
References
- 1.Peres V, Nagem TJ, de Oliveira FF. Phytochemistry. 2000;55:683. doi: 10.1016/s0031-9422(00)00303-4. [DOI] [PubMed] [Google Scholar]
- 2.Pinto MM, Sousa ME, Nascimento MS. Curr Med Chem. 2005;12:2517. doi: 10.2174/092986705774370691. [DOI] [PubMed] [Google Scholar]
- 3.Sousa ME, Pinto MM. Curr Med Chem. 2005;12:2447. doi: 10.2174/092986705774370736. [DOI] [PubMed] [Google Scholar]
- 4.Liou SS, Shieh WL, Cheng TH, Won SJ, Lin CN. J Pharm Pharmacol. 1993;45:791. doi: 10.1111/j.2042-7158.1993.tb05686.x. [DOI] [PubMed] [Google Scholar]
- 5.Lin CN, Liou SJ, Lee TH, Chuang YC, Won SJ. J Pharm Pharmacol. 1996;48:539. doi: 10.1111/j.2042-7158.1996.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 6.Lin CN, Hsieh HK, Liou SJ, Ko HH, Lin HC, Chung MI, Ko FN, Liu HW, Teng CM. J Pharm Pharmacol. 1996;48:887. doi: 10.1111/j.2042-7158.1996.tb05994.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang LW, Kang JJ, Chen IJ, Teng CM, Lin CN. Bioorg Med Chem. 2002;10:567. doi: 10.1016/s0968-0896(01)00315-7. [DOI] [PubMed] [Google Scholar]
- 8.Riscoe M, Kelly JX, Winter R. Curr Med Chem. 2005;12:2539. doi: 10.2174/092986705774370709. [DOI] [PubMed] [Google Scholar]
- 9.Marona H, Pekala E, Antkiewicz-Michaluk L, Walczak M, Szneler E. Bioorg Med Chem. 2008;16:7234. doi: 10.1016/j.bmc.2008.06.039. [DOI] [PubMed] [Google Scholar]
- 10.Na Y. J Pharm Pharmacol. 2009;61:707. doi: 10.1211/jpp/61.06.0002. [DOI] [PubMed] [Google Scholar]
- 11.Shagufta, Ahmad I. Eur J Med Chem. 2016;116:267. doi: 10.1016/j.ejmech.2016.03.058. [DOI] [PubMed] [Google Scholar]
- 12.Qin Y, Meng L, Hu C, Duan W, Zuo Z, Lin L, Zhang X, Ding J. Mol Cancer Ther. 2007;6:2429. doi: 10.1158/1535-7163.MCT-07-0147. [DOI] [PubMed] [Google Scholar]
- 13.Woo S, Jung J, Lee C, Kwon Y, Na Y. Bioorg Med Chem Lett. 2007;17:1163. doi: 10.1016/j.bmcl.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 14.Su QG, Liu Y, Cai YC, Sun YL, Wang B, Xian LJ. Invest New Drugs. 2011;29:1230. doi: 10.1007/s10637-010-9468-5. [DOI] [PubMed] [Google Scholar]
- 15.Jun KY, Lee EY, Jung MJ, Lee OH, Lee ES, Park Choo HY, Na Y, Kwon Y. Eur J Med Chem. 2011;46:1964. doi: 10.1016/j.ejmech.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 16.Park SE, Chang IH, Jun KY, Lee E, Lee ES, Na Y, Kwon Y. Eur J Med Chem. 2013;69:139. doi: 10.1016/j.ejmech.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 17.Deweese JE, Osheroff MA, Osheroff N. Biochem Mol Biol Educ. 2008;37:2. doi: 10.1002/bmb.20244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deweese JE, Osheroff N. Nucleic Acids Res. 2009;37:738. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nitiss JL. Nat Rev Cancer. 2009;9:338. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vos SM, Tretter EM, Schmidt BH, Berger JM. Nat Rev Mol Cell Biol. 2011;12:827. doi: 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen SH, Chan N-L, Hsieh T-S. Ann Rev Biochem. 2013;82:139. doi: 10.1146/annurev-biochem-061809-100002. [DOI] [PubMed] [Google Scholar]
- 22.Pommier Y, Sun Y, Huang S-yN, Nitiss JL. Nat Rev Mol Cell Biol. 2016;17:703. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nitiss JL. Nat Rev Cancer. 2009;9:327. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pommier Y, Leo E, Zhang H, Marchand C. Chem Biol. 2010;17:421. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pommier Y. ACS Chem Biol. 2013;8:82. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pogorelcnik B, Perdih A, Solmajer T. Curr Pharm Des. 2013;19:2474. doi: 10.2174/1381612811319130016. [DOI] [PubMed] [Google Scholar]
- 27.Pendleton M, Lindsey RH, Jr, Felix CA, Grimwade D, Osheroff N. Ann NY Acad Sci. 2014;1310:98. doi: 10.1111/nyas.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishida R, Hamatake M, Wasserman RA, Nitiss JL, Wang JC, Andoh T. Cancer Res. 1995;55:2299. [PubMed] [Google Scholar]
- 29.Andoh T, Ishida R. Biochim Biophys Acta. 1998;1400:155. doi: 10.1016/s0167-4781(98)00133-x. [DOI] [PubMed] [Google Scholar]
- 30.Jensen LH, Nitiss KC, Rose A, Dong J, Zhou J, Hu T, Osheroff N, Jensen PB, Sehested M, Nitiss JL. J Biol Chem. 2000;275:2137. doi: 10.1074/jbc.275.3.2137. [DOI] [PubMed] [Google Scholar]
- 31.Fortune JM, Osheroff N. J Biol Chem. 1998;273:17643. doi: 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- 32.Fortune JM, Osheroff N. Prog Nucleic Acid Res Mol Biol. 2000;64:221. doi: 10.1016/s0079-6603(00)64006-0. [DOI] [PubMed] [Google Scholar]
- 33.Larsen AK, Escargueil AE, Skladanowski A. Pharmacol Ther. 2003;99:167. doi: 10.1016/s0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa T, Hayashita Y, Maeno K, Masuda A, Sugito N, Osada H, Yanagisawa K, Ebi H, Shimokata K, Takahashi T. Cancer Res. 2004;64:4826. doi: 10.1158/0008-5472.CAN-04-0871. [DOI] [PubMed] [Google Scholar]
- 35.Hajji N, Mateos S, Pastor N, Dominguez I, Cortes F. Mutat Res. 2005;583:26. doi: 10.1016/j.mrgentox.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 36.Chene P, Rudloff J, Schoepfer J, Furet P, Meier P, Qian Z, Schlaeppi JM, Schmitz R, Radimerski T. BMC Chem Biol. 2009;9:1. doi: 10.1186/1472-6769-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ketron AC, Osheroff N. Phytochem. Rev. 2014;13:19. doi: 10.1007/s11101-013-9291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alam S, Khan F. Drug Des Devel Ther. 2014;8:183. doi: 10.2147/DDDT.S51577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osheroff N, Shelton ER, Brutlag DL. J Biol Chem. 1983;258:9536. [PubMed] [Google Scholar]
- 40.Lindsley JE, Wang JC. J Biol Chem. 1993;268:8096. [PubMed] [Google Scholar]
- 41.Bjergbaek L, Kingma P, Nielsen IS, Wang Y, Westergaard O, Osheroff N, Andersen AH. J Biol Chem. 2000;275:13041. doi: 10.1074/jbc.275.17.13041. [DOI] [PubMed] [Google Scholar]
- 42.Wu G, Yu G, Kurtan T, Mandi A, Peng J, Mo X, Liu M, Li H, Sun X, Li J, Zhu T, Gu Q, Li D. J Nat Prod. 2015;78:2691. doi: 10.1021/acs.jnatprod.5b00636. [DOI] [PubMed] [Google Scholar]
- 43.Barret JM, Kruczynski A, Vispe S, Annereau JP, Brel V, Guminski Y, Delcros JG, Lansiaux A, Guilbaud N, Imbert T, Bailly C. Cancer Res. 2008;68:9845. doi: 10.1158/0008-5472.CAN-08-2748. [DOI] [PubMed] [Google Scholar]
- 44.Gentry AC, Pitts SL, Jablonsky MJ, Bailly C, Graves DE, Osheroff N. Biochemistry. 2011;50:3240. doi: 10.1021/bi200094z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palermo G, Minniti E, Greco ML, Riccardi L, Simoni E, Convertino M, Marchetti C, Rosini M, Sissi C, Minarini A, De Vivo M. Chem Commun. 2015;51:14310. doi: 10.1039/c5cc05065k. [DOI] [PubMed] [Google Scholar]
- 46.Annereau JP, Brel V, Dumontet C, Guminski Y, Imbert T, Broussas M, Vispe S, Breand S, Guilbaud N, Barret JM, Bailly C. Leuk Res. 2010;34:1383. doi: 10.1016/j.leukres.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 47.Kruczynski A, Vandenberghe I, Pillon A, Pesnel S, Goetsch L, Barret JM, Guminski Y, Le Pape A, Imbert T, Bailly C, Guilbaud N. Invest New Drugs. 2011;29:9. doi: 10.1007/s10637-009-9328-3. [DOI] [PubMed] [Google Scholar]
- 48.Shao Q, Goyal S, Finzi L, Dunlap D. Macromolecules. 2012;45:3188. doi: 10.1021/ma300211t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas TJ, Tajmir-Riahi HA, Thomas T. Amino Acids. 2016;48:2423. doi: 10.1007/s00726-016-2246-8. [DOI] [PubMed] [Google Scholar]
- 50.Ashley RE, Dittmore A, McPherson SA, Turnbough CL, Jr, Neuman KC, Osheroff N. Nucleic Acids Res. 2017 doi: 10.1093/nar/gkx649. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gentry AC, Osheroff N. DNA topoisomerases: Type II, Encyc Biol Chem. Academic Press; Waltham, MA: 2013. p. 163. [Google Scholar]
- 52.Osheroff N. J Biol Chem. 1986;261:9944. [PubMed] [Google Scholar]
- 53.McClendon AK, Osheroff N. Mutat Res. 2007;623:83. doi: 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deweese JE, Osheroff N. Metallomics. 2010;2:450. doi: 10.1039/c003759a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palermo G, Cavalli A, Klein ML, Alfonso-Prieto M, Dal Peraro M, De Vivo M. Acc Chem Res. 2015;48:220. doi: 10.1021/ar500314j. [DOI] [PubMed] [Google Scholar]
- 56.Osheroff N, Zechiedrich EL. Biochemistry. 1987;26:4303. doi: 10.1021/bi00388a018. [DOI] [PubMed] [Google Scholar]
- 57.Baird CL, Harkins TT, Morris SK, Lindsley JE. Proc Natl Acad Sci U S A. 1999;96:13685. doi: 10.1073/pnas.96.24.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, Chan NL. Science. 2011;333:459. doi: 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- 59.Elsea SH, Westergaard M, Burden DA, Lomenick JP, Osheroff N. Biochemistry. 1997;36:2919. doi: 10.1021/bi962488f. [DOI] [PubMed] [Google Scholar]
- 60.Aldred KJ, Breland EJ, Vlckova V, Strub MP, Neuman KC, Kerns RJ, Osheroff N. Biochemistry. 2014;53:5558. doi: 10.1021/bi500682e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.