

Abstract
Introduction
Factor VIII or FIX-deficient hemophilic patients display deficits in platelet and fibrin deposition under flow detectable in microfluidics. Compared to fibrin generation, decreased platelet deposition in hemophilic blood flow is more easily rescued with recombinant factor VIIa (rFVIIa), whereas rFVIIa requires FXIIa participation to generate fibrin when tissue factor (TF) is absent.
Aims
Perfusion of hemophilic whole blood over collagen/TF surfaces was used to determine if rFVIIa/TF was sufficient to bypass poor FIXa/FVIIIa function in blood from hemophilia A and B patients.
Methods
Whole blood (WB) treated with high dose corn trypsin inhibitor (40 μg/ml) from 7 healthy donors and 10 patients was perfused over fibrillar collagen presenting low or high TF (TFlow or TFhigh) at wall shear rate of 100 s−1.
Results
With WB from healthy controls, platelet deposition and fibrin accumulation increased as TF was increased. Factor-deficient WB (1–3% of normal) displayed striking deficits in platelet deposition and fibrin formation at either TFlow or TFhigh. In contrast, mildly factor-deficient WB (14–32%) supported fibrin formation under flow on TFhigh/collagen. With either TFlow or TFhigh, exogenously added rFVIIa (20 nM) increased platelet deposition and fibrin accumulation in WB from factor-deficient patients (1–3% of normal) to levels commensurate with untreated healthy WB.
Conclusion
The absence of FIXa/FVIIIa in severe hemophilic patients results in deficits in fibrin formation that cannot be rescued by wall-derived TF ex vivo. The effects of rFVIIa on platelet adhesion and rFVIIa/TF can act together to reinforce thrombin generation, platelet deposition, and fibrin formation under flow.
Keywords: platelets, fibrin, microfluidics, hemophilia, recombinant FVIIa
INTRODUCTION
Hemophilia A and B are X-linked genetic disorders resulting in deficiencies in coagulation factor VIII (FVIII) or factor IX (FIX), respectively[1]. These deficiencies cause a wide range of bleeding phenotypes depending on severity [2–5]. Current treatment strategies for hemophilic patients include clotting factor replacement or bypass therapies. Bypass therapies such as activated prothrombin complex concentrates (aPCC) or recombinant factor VIIa (rFVIIa) were developed to treat hemophilic patients with inhibitors[6]. Though rFVIIa is approved, its mechanism of action is complex[7,8]. Past work has indicated that rFVIIa can exploit tissue factor to function and may outcompete endogenous FVII for TF[9,10]. Additional studies indicate that rFVIIa can also bind cellular surface to generate FXa independent of TF [11–15]. The cellular mechanism of FVIIa is well supported in the literature and motivates the development of rFVIIa mutants that have less sensitivity to TF as a cofactor (and potentially less thrombotic risk).
Microfluidic studies can help elucidate the roles of intrinsic tenase, extrinsic tenase, and rFVIIa under flow. In vitro models with perfusion of FVIII-deficient blood resulted in decreased platelet aggregation and fibrin formation at low shear conditions[16–18]. The extrinsic pathway is unable to completely mitigate deficits in hemophilia A and B, possibly because TF expression varies greatly in human tissues[19,20]. Bleeding in hemophilia is common in the joints where TF expression is considered to be low[20]. The contribution of FXa and thrombin stemming from residual intrinsic tenase in various TF-laden backgrounds has not been studied extensively. Previous studies only assessed severe cases of human FVIII or FIX deficiency (<1%) often with FXIIa function uncontrolled or blood from mice deficient in FVIII or FIX[17,18,21]. Few have examined how milder deficiencies in FVIII or FIX levels affect platelet adherence and fibrin formation in the presence of bypass therapy and surface TF[22].
Previously we have developed a contact pathway-driven microfluidic model (no exogenous TF) that evaluated the role of FVIII or FIX in patients with congenital bleeding disorders[23,24]. Furthermore, we previously observed that rFVIIa enhances platelet adhesion but not fibrin production when contact pathway is strongly inhibited, suggestive of low level generation of thrombin by rFVIIa on platelets (in the absence of TF)[24]. In Li et al, we also describe how FXIIa activity can work with rFVIIa to generate fibrin. In this study, we aimed to unify our previous two studies and establish the role of wall-presented TF and rFVIIa in the regulation of thrombus formation. We measured healthy or hemophilic WB perfusion over collagen type I surfaces bearing TFlow or TFhigh at a venous wall shear rate of 100 s−1. This study confirms that FIXa/FVIIIa is required for fibrin generation under healthy WB flow, even when TF is abundant at the wall.
MATERIALS AND METHODS
Blood collection and patient recruitment
Blood was drawn from healthy donors (n = 7) and hemophilic patients (n = 10) under Internal Review Board approval of the University of Pennsylvania and informed consent. Patient data was collected (Table 1). Residual FVIII and FIX activity was also measured. Healthy donors were self-reported free of oral medication for 7 days and abstained from alcohol 48 h prior to blood donations. WB was drawn into 40 μg/ml CTI (Haematologic Technologies, Essex Junction, VT).
Table 1.
10 patients were examined with respect to exogenous rFVIIa efficacy by perfusion of high CTI inhibited WB on TFlow and TFhigh collagen surfaces. Time since last hemostatic therapy, % residual coagulation factor activity, aPTT, PT, and platelet count are as reported.
| Patient ID# | Diagnosis | Residual Coagulation Factor %* | aPTT (Sec) | PT | Platelets (103/μL) | Timing of Most Recent Therapy | Replacement Therapy Dosage |
|---|---|---|---|---|---|---|---|
| 62 | Severe Hemophilia A | 3 | 56.1 | 13.3 | 172 | 3 days | rFVIII (35 IU/kg) |
| 63 | Severe Hemophilia A | 32 | 32.7 | 12.9 | 291 | 5 hrs | rVIII (25 IU/kg) |
| 64 | Severe Hemophilia B | 14 | 34.4 | 13.7 | 252 | 96 hrs | rFIX (unknown) |
| 65 | Severe Hemophilia B | 6 | 51.2 | 14.9 | 200 | 3 days | rFIX (47 IU/kg) |
| 67 | Mild Hemophilia A | 18 | 37.4 | 12.9 | 244 | None in past 5 years | rFVIII (unknown) |
| 68 | Moderate Hemophilia A | 6 | 50.8 | 14.1 | 173 | None in past 10 months | N/A |
| 69 | Moderate Hemophilia A | 2 | 53.5 | 12.5 | 217 | None since 2012 | N/A |
| 70 | Severe Hemophilia A | 24 | 40.1 | 12.8 | 273 | 26 hrs | rFVIII (50 IU/kg) |
| 71 | Moderate Hemophilia B | 5 | 51.1 | 15.9 | 208 | None in past 5 years | N/A |
| 29 | Severe Hemophilia B | **<1 | 133.6 | 14.5 | 125 | 8 hrs | rFIX (50 IU/kg) |
Reflects in some cases recent administration of factor concentrate.
Patient had active inhibitors titer of 2.0 Bethesda Units (BU) at time of blood draw but no bypassing agents within 96 hrs.
Preparation of TF bearing collagen surfaces
Glass slides were first functionalized with Sigmacote (Sigma, St. Louis, MO) to create a hydrophobic surface. Acid-insoluble equine type I collagen (Chrono-Par, Chrono-log, Havertown, PA) were diluted to 800 μg/ml in isotonic glucose solution (Chronopar) and introduced into patterning device (250 μm by 1 cm) [25,26]. This patterning device was then filled with 5 μL of Dade Innovin PT reagent (Siemens Healthcare USA, Malvern, PA. 20 nM stock concentration) diluted 300 or 5 fold with 2-[4-(2-hydroxyethyl)piperazin-1-yl] ethanesulfonic acid (HEPES)-buffered saline (HBS, 20 mM HEPES, 160 mM NaCl, pH 7.4) to obtain TFlow or TFhigh surface concentrations. Nominal low and high TF surface concentrations ([TF]wall) were ~0.1 and ~2 molecules per μm2, respectively, as previously measured[27]. In all experiments, TF bearing collagen surfaces were incubated for 30 minutes with no flow and then washed with 5 μl of 0.5% bovine serum albumin (BSA) in HBS. This single channel patterning device was then removed to allow placement of microfluidic flow.
Extrinsic pathway triggered microfluidic flow assay on TF bearing collagen surfaces
An 8-channel microfluidic device was fabricated and its dimensions and operations were previously described[25,28]. Blood samples were treated with fluorescently conjugated non-function blocking anti-CD41a antibody (clone VI-PL2, Becton Dickson, Franklin Lakes, NJ, 0.125 μg/ml final concentration) to label platelets and fluorescently conjugated anti-fibrin antibody (clone T2G1, Dr. Mortimer Poncz laboratory, Children’s Hospital of Philadelphia, 0.5 μg/ml final concentration) to label fibrin. WB samples were also treated with vehicle HBS or rFVIIa. Recombinant FVIIa (NovonSeven, Novo Nordisk, Plainsboro NJ, 1 mg/ml final concentration) was reconstituted in histidine diluent. WB samples were treated with detection antibodies and rFVIIa 5 min prior to initiation of microfluidic assays. Whole blood perfusion in devices occurred within 15 min of venipuncture. Blood samples were perfused at an initial local wall shear rate of 100s−1 (1 μL/min per channel) for 15 min.
Platelet, fibrin accumulation, occlusion time, and statistical analysis
Platelet and fibrin fluorescent intensities were imaged at 60 sec intervals for 900 sec using ImageJ software (NIH) with background subtraction and region of interest analysis conducted as previously described [25,26]. Full channel occlusion of the 60-micron high channel (indicated as 100% OCC) was detected during the microfluidic experiments with healthy whole blood when the flow stopped. Hemophilia patient blood (no added rFVIIa) typically never reached full occlusion in the 900 sec assays. Statistical comparisons (2-tailed student’s t-test for n = number of individual blood samples) of platelet and fibrin deposition were made for hemophilia cohorts relative to a healthy cohort (7 donors) at the 900 sec endpoint. Statistical comparisons were also made for responses of each rFVIIa-treated blood sample, internally normalized to its response with no added rFVIIa, via clotting tests run side-by-side on the microfluidic devices.
RESULTS
Surface-triggered extrinsic pathway under flow does not rescue platelet adherence or fibrin generation at 1–3% factor activity
In our previous work, we reported no fibrin formation under flow in two FVIII-deficient patients (1–3% factor level) when high CTI-anticoagulated hemophilic blood was perfused over collagen surfaces (no exogenous surface TF)[24]. We now sought to evaluate the hemostatic potential of surface TF to rescue such deficits through perfusion of hemophilic WB over collagen surfaces bearing TFlow or TFhigh. Aggregate analysis of high CTI-inhibited WB from one severely VIII-deficient patient (#62:3% factor level), one severely factor IX-deficient patient (#29:<1% factor level), and one moderately FVIII-deficient patient (#69:2% factor level) produced no detectable levels of fibrin at 900 sec assay end point (Figure 1B&D: red lines, p < 0.01) as opposed to healthy controls where robust platelet deposition and fibrin generation was measured regardless of surface TF concentration (Figure 1B&D: black lines). Interestingly, at < 3% factor activity in hemophilic blood, platelet deposition at 900 sec was significantly reduced compared to healthy blood independent of surface TF concentration (Figure 1A&C: red lines, p < 0.01 [TFlow collagen], p < 0.05 [TFhigh collagen]).
Figure 1. Comparison of 1–3% clotting factor activity patients against healthy donor response to TFlow or TFhigh bearing collagen surfaces.
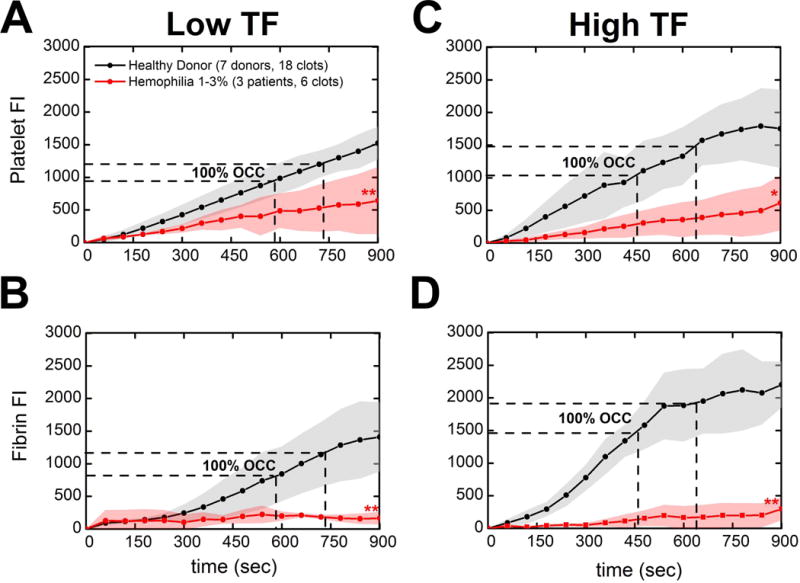
(A & B), Platelet adherence and fibrin deposition dynamics measured by platelet and fibrin fluorescence respectively. WB inhibited with 40 μg/ml CTI was perfused over TFlow bearing collagen surfaces at 100 s−1. (B & C), Platelet adherence and fibrin deposition dynamics over time. WB inhibited with 40 μg/ml CTI was perfused over TFhigh bearing collagen surfaces at 100 s−1. Lines with shaded traces and points are the mean and standard deviation of 6 clotting events measured in 60 sec intervals over 15 min for three distinct hemophilic patients (red) or 18 clotting events measured in 60 sec intervals over 15 min for seven distinct healthy subjects (black). Aggregate hemophilic data was from one severely VIII-deficient patient (#62:3% factor level), one severely factor IX-deficient patient (#29:<1% factor level), and one moderately FVIII-deficient patient (#69:2% factor level). (** p < 0.01, * p < 0.05, ns: non-significant)
Extrinsic pathway triggered coagulation under flow partially restores fibrin generation at 3–14% factor activity
Perfusion of 2 severely FIX-deficient patients WB (#64 & #65) recently treated with therapy to increase their FIX levels to 14% and 6% respectively, a moderately FVIII-deficient patient (#68:6% factor level), and a moderately FIX-deficient patient (#71:5% factor level) resulted in significantly reduced fibrin accumulation at 900 sec under flow as compared to healthy donors (Figure 2B,D: red lines, p < 0.05 [TFlow collagen], p < 0.01 [TFhigh collagen]). The wall TF had some detectable activity on fibrin under these moderate hemophilic conditions, however, since perfusion of CTI (40 μg/ml)-treated hemophilic whole blood on collagen surfaces alone (no TF) from four patients at 3–10% residual factor levels did not result in any fibrin accumulation by 900 sec[24]. Additionally, similar to the 1–3% factor activity group, small non-statistically significant deficits in platelet adhesion on TF collagen surfaces were again observed at 900 sec (Figure 2A&C: red lines, ns). These results indicate that engagement of the intrinsic tenase (FIXa/FVIIIa → FXa) in combination with FXa production from TF/FVIIa on TFhigh collagen surfaces was sufficient to produce low amounts of fibrin (Figure 2B&D: red lines).
Figure 2. Comparison of 3–14% clotting factor activity patients against healthy donor response to TFlow or TFhigh bearing collagen surfaces.

(A & B), Platelet adherence and fibrin deposition dynamics measured by platelet and fibrin fluorescence respectively. WB inhibited with 40 μg/ml CTI was perfused over TFlow bearing collagen surfaces at 100 s−1. (B & C), Platelet adherence and fibrin deposition dynamics over time. WB inhibited with 40 μg/ml CTI was perfused over TFhigh bearing collagen surfaces at 100 s−1. Lines with shaded traces and points are the mean and standard deviation of 8 clotting events measured in 60 sec intervals over 15 min for four distinct hemophilic patients (red) or 18 clotting events measured in 60 sec intervals over 15 min for seven distinct healthy subjects (black). Aggregate hemophilic data from 2 severely FIX-deficient patients WB (#64 & #65) recently treated with therapy to increase their FIX levels to 14% and 6% respectively, a moderately FVIII-deficient patient (#68:6% factor level), and a moderately FIX-deficient patient (#71:5% factor level). (** p < 0.01, * p < 0.05, ns: non-significant)
Platelet adhesion is rescued by the extrinsic pathway on TFlow collagen surfaces while fibrin formation is fully restored on TFlow and TFhigh collagen surfaces at >14% factor activity
Platelet deposition from WB of 2 severely FVIII-deficient patients (#63 & #70) recently treated with therapy to increase their FVIII levels to 32% and 24% respectively and one mildly FVIII-deficient patient (#67:18% factor level) was commensurate with healthy blood until occlusion and then decreased in comparison to the platelet deposition from healthy controls on TFhigh collagen surfaces at the assay end-point (Figure 3C: red line, p < 0.05), an effect not apparent with TFlow collagen surfaces (Figure 3A: red line, ns). We observed no statistically significant deficits in fibrin production at >14% factor activity at 900 sec in the presence of either surface concentration of TF on collagen when compared to healthy donors (Figure 3B&D: red lines, ns). This is in contrast to our previous work where two mildly FVIII-deficient patients (15% factor level) generated negligible to low amounts of fibrin with perfusion of CTI (40 μg/ml)-treated hemophilic blood on collagen surfaces (no surface TF) [24]. Time to fibrin initiation and total fibrin accumulation was fully restored with TFhigh collagen surfaces (Figure 3D: red line, ns).
Figure 3. Comparison of >14% clotting factor activity patients against healthy donor response to TFlow or TFhigh bearing collagen surfaces.
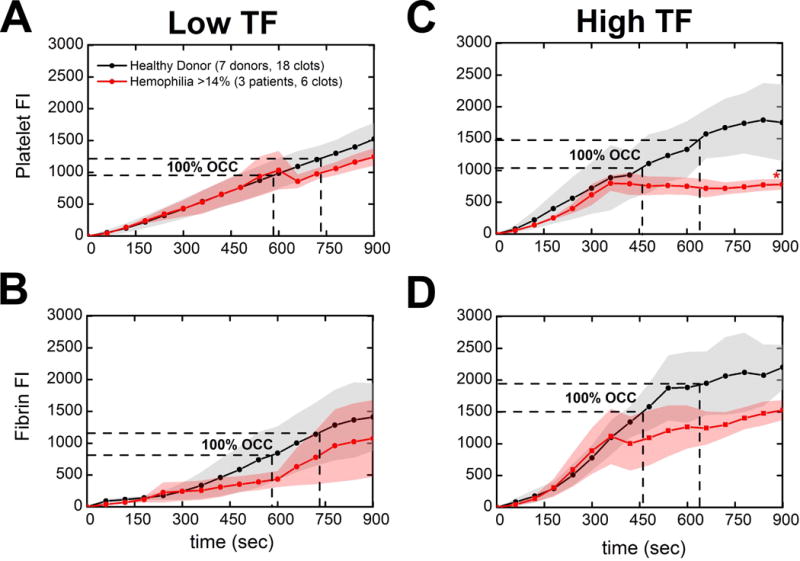
(A & B), Platelet adherence and fibrin deposition dynamics measured by platelet and fibrin fluorescence respectively. WB inhibited with 40 μg/ml CTI was perfused over TFlow bearing collagen surfaces at 100 s−1. (B & C), Platelet adherence and fibrin deposition dynamics over time. WB inhibited with 40 μg/ml CTI was perfused over TFhigh bearing collagen surfaces at 100 s−1. Lines with shaded traces and points are the mean and standard deviation of 6 clotting events measured in 60 sec intervals over 15 min for three distinct hemophilic patients (red) or 18 clotting events measured in 60 sec intervals over 15 min for seven distinct healthy subjects (black). Aggregate hemophilic data from 2 severely FVIII-deficient patients (#63 & #70) recently treated with therapy to increase their FVIII levels to 32% and 24% respectively and one mildly FVIII-deficient patient (#67:18% factor level). (** p < 0.01, * p < 0.05, ns: non-significant)
In examining total platelet and fibrin accumulation under flow at 15 min for all cohorts, platelet and fibrin accumulation was highly defective for blood from patients with 1–3% normal factor levels when compared to healthy blood (Figure 4A–D: p <0.01 [TFlow collagen], p < 0.05 [TFhigh collagen]). High levels of surface TF do not rescue fibrin formation at 1–3% factor levels in hemophilic patients (Figure 4D: 1–3%, p<0.01) whose blood generated essentially no fibrin.
Figure 4. Total platelet or fibrin accumulation for different factor deficiencies levels of hemophilia.
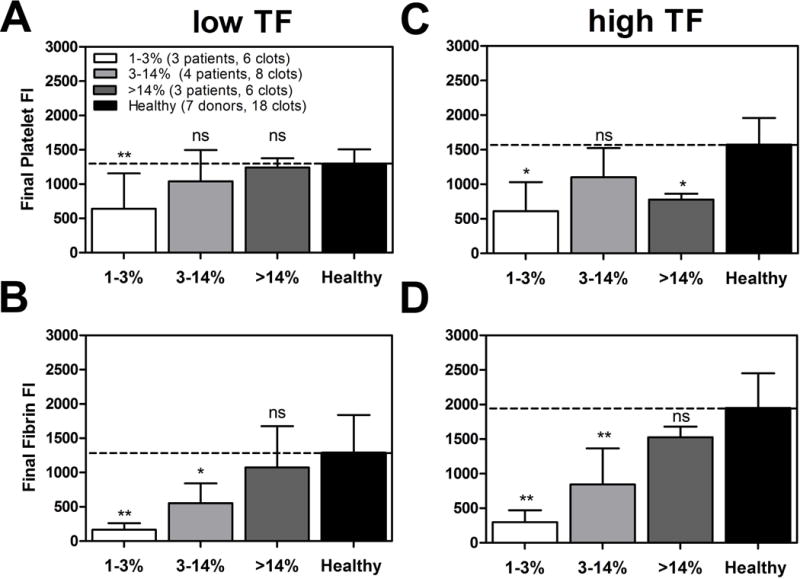
(A & B), Final platelet or fibrin accumulation on TFlow bearing collagen surfaces at 100 s−1 measured by platelet or fibrin fluorescence at t = 15 min or just prior to full channel occlusion. (C & D), Final platelet or fibrin accumulation on TFhigh bearing collagen surfaces at 100 s−1 measured by platelet or fibrin fluorescence at t = 15 min or just prior to full channel occlusion. All data is ordered into three factor level categories (1–3%, 3–14% & > 14%) and compared to mean healthy subject final platelet or fibrin accumulation. Error bars are the standard deviation of 6 clotting events for each factor level category and 18 clotting events for seven healthy subjects. (** p < 0.01, * p < 0.05, ns: non-significant)
Exogenous rFVIIa significantly enhances platelet deposition on TFhigh collagen surfaces, but non-significantly increases fibrin accumulation at 20 nM on TFlow or TFhigh collagen at 1–3% factor activity
Perfusion of WB from a severely FIX-deficient patient (#29, <1% FIX) showed negligible fibrin accumulation that increased to healthy donor levels with 20 nM exogenous rFVIIa on TFlow or TFhigh collagen (Figure 5B: black, gray & red lines). Addition of 20 nM rFVIIa also enhanced platelet deposition to healthy donor levels at either TF concentration (Fig. 5A,C: red lines). Fig. 5 demonstrated in a single severe hemophilic patient blood sample that rFVIIa enhanced platelet and fibrin production in a dose-dependent manner, especially at the high TF surface, to levels approaching the healthy donor cohort baseline.
Figure 5. Effect of in vitro rFVIIa (0–20 nM) in severely FVIII-deficient patient (#29, <1% factor level).
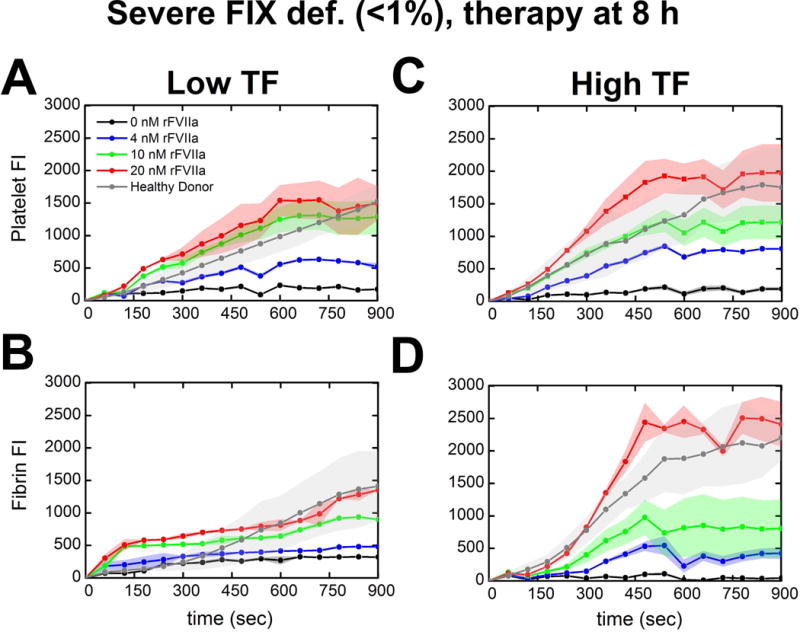
(A & B), Platelet adherence and fibrin deposition in the presence or absence of exogenous rFVIIa measured by platelet and fibrin fluorescence at t = 15 min or just prior to full channel occlusion. WB treated with exogenous rFVIIa (0–20 nM) and inhibited with 40 μg/ml CTI was perfused over TFlow bearing collagen surfaces at 100 s−1. (C & D), Platelet adherence and fibrin deposition in the presence or absence of exogenous rFVIIa measured by platelet and fibrin fluorescence. WB treated with exogenous rFVIIa (0–20 nM) and inhibited with 40 μg/ml CTI was perfused over TFhigh bearing collagen surfaces at 100 s−1. Lines with shaded traces and points are the mean and standard deviation of 2 clotting events measured in 60 sec intervals over 15 min for each concentration of rFVIIa (colored) or 18 clotting events measured in 60 sec intervals over 15 min for seven distinct healthy subjects (gray).
The effect of exogenously added rFVIIa was tested on all patients examined in Figures 1–4. At 1–3% factor level, exogenous 20 nM rFVIIa significantly increased final platelet accumulation versus 0 nM rFVIIa (Figure 6C: 1–3%, p < 0.05) to levels commensurate with that observed with untreated healthy blood on TFhigh collagen. Final fibrin accumulation with 20 nM rFVIIa was also increased versus 0 nM rFVIIa and trended towards statistical significance on TFhigh collagen surfaces at 1–3% factor activity (Figure 6D: 1–3% p = 0.0715). For the severe cohort (1–3% factor level) of 3 donors, the mean fold-enhancement of fibrin deposition at 20 nM rFVIIa (with each signal internally normalized by the 0 nM rFVIIa condition) was statistically significant (4.6 ± 2.1-fold over baseline at high TF, p<0.05). Also, for high TF surfaces (Fig. 6D), the average fibrin deposition with 20 nM rFVIIa treatment for the severe cohort (n = 3) was not statistically different (p=0.14) from that observed for the healthy cohort (n = 7), indicating pharmacological correction of the defect. With increasing rFVIIa supplementation, healthy whole blood also displayed statistically significant increases in platelet and fibrin deposition on TFlow or TFhigh collagen surfaces(Figure 6A–D: Healthy, p < 0.05).
Figure 6. Total platelet or fibrin accumulation in response to in vitro rFVIIa (0–20 nM) for different factor deficiency levels of hemophilia.
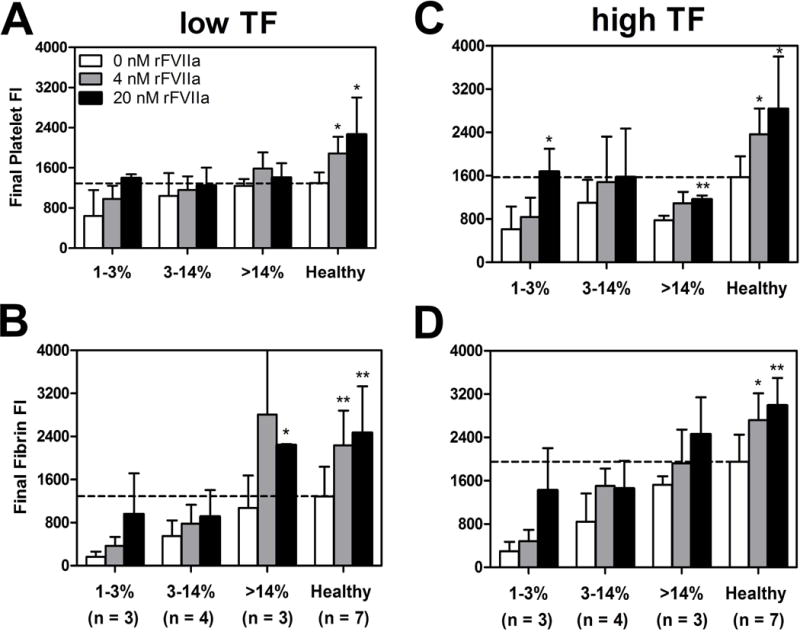
(A & B), Maximum platelet or fibrin accumulation in the presence or absence of exogenous rFVIIa measured by platelet or fibrin fluorescence. WB treated with exogenous rFVIIa (0–20 nM) and inhibited with 40 ug/ml CTI was perfused over TFlow bearing collagen surfaces at 100s−1. (C & D), Maximum platelet or fibrin accumulation in the presence or absence of exogenous rFVIIa measured by platelet or fibrin fluorescence. WB treated with exogenous rFVIIa (0–20 nM) and inhibited with 40 μg/ml CTI was perfused on TFhigh bearing collagen surfaces at 100s−1. All data is ordered into three factor level categories (1–3%, 3–14% & > 14%) compared to mean healthy subject final platelet or fibrin accumulation. Error bars are the standard deviation of 6 clotting events for each factor level category and 18 clotting events for seven healthy subjects. N indicates the number of patients or donors (** p < 0.01, * p < 0.05, ns: non-significant)
DISCUSSION AND CONCLUSION
Microfluidic assays consisting of high CTI (40 μg/ml)-inhibited WB perfusion on TFlow or TFhigh/collagen surfaces at 100 s−1 were used to evaluate platelet deposition and fibrin formation in hemophilic patients. A broad cohort of patients were tested representing a large spectrum of clinical phenotypes of hemophilia A or B (Table 1). Under these ex vivo hemodynamic conditions, we observed significantly decreased platelet deposition and fibrin formation at 900 sec on TFlow and TFhigh collagen surfaces in hemophilic samples with 1–3% residual factor levels (Figure 1A–D). Taken together, these results indicate that in healthy blood clotting under flow conditions over TF, most Factor Xa comes from the intrinsic tenase (FIXa/FVIIIa) even when some FXa can be initially generated by the extrinsic tenase TF/FVIIa (Figure 1–4, Figure 7).
Figure 7. Systems Approach to Hemophilic Blood Clotting.
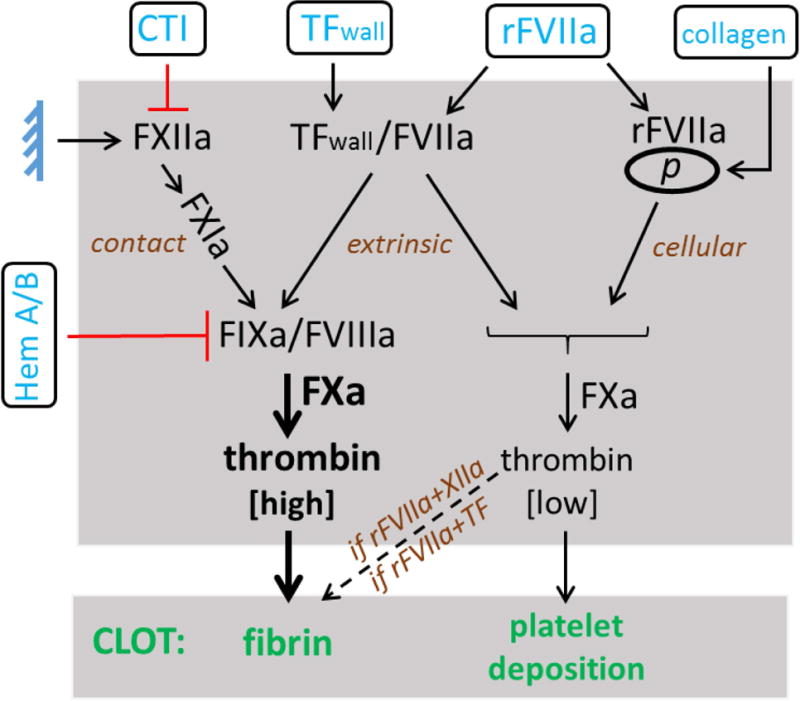
Modulation of the contact pathway [±CTI], TFWall, exogenous concentrations of rFVIIa, and collagen and its resultant effects on platelet deposition and fibrin formation. Results from perfusion of WB from Hemophilia A or B patients over TF/collagen indicate a crucial role of FIXa/FVIIIa in driving high FXa levels and subsequent thrombin production that produces fibrin under flow. Recombinant FVIIa localization to the platelet surface in combination with contact pathway engagement (FXIIa→FXIa) also drives FXa production, which then results in fibrin formation under flow. Thrombin feedback activation of platelets (not shown) would be expected to reinforce both platelet activation (and procoagulant activity) and platelet deposition to some extent through PAR1/4 signaling.
Healthy blood responded to higher TF surfaces with a faster occlusion time and with more fibrin made (Fig. 1), whereas severely deficient blood (1–3% clotting factor) was largely insensitive to TF level and never reached full occlusion. For patients with moderate hemophilia (3–14% clotting factor), the blood displayed some ability to utilize surface TF and generate some fibrin with increased platelet deposition, albeit less than healthy blood. For patients with mild deficiency (>14% clotting factor), the blood performs in a manner approaching that of healthy blood in this microfluidic assay, at either low or high TF levels.
With the intrinsic tenase severely attenuated as in the case of severe FVIII or FIX-deficiency, surface TF-triggered coagulation failed to produce adequate levels of thrombin for detectable fibrin formation (Figure 1B,D, p < 0.01). At high surface TF concentrations of ~ 2 TF molecules/μm2 as measured in a prior study [27], the extrinsic tenase could not support fibrin formation out to 900 sec when critical residual clotting factor activity was <3% (Figure 1D, p < 0.01).
We also assessed the role of rFVIIa in platelet deposition and fibrin formation under flow in hemophilic and healthy WB samples. Results from a severe hemophilic patient indicate that rFVIIa amplifies platelet deposition and fibrin formation in the presence of surface TF (Figure 5). Analysis of final platelet and fibrin deposit size in the presence of exogenous rFVIIa indicates enhancement of platelet and fibrin accumulation at 1–3% normal factor levels (Figure 6). The number (mass) of platelets on the surface is strongly correlated with the height of the clot. Importantly, rFVIIa increases the growth rate and height of the platelet mass. Fibrin accumulation commences after platelet accumulation has begun indicative of a protective low shear zone for fibrin polymerization within the platelet deposit.
Numerous and complex couplings exist between platelets, TF, thrombin and fibrin. In general for healthy blood, we have found platelet deposition to be sensitive to thrombin, particularly when comparing zero thrombin to low thrombin, where thrombin can enhance platelet deposition by about ~2 or 3-fold. However, the collagen used in the experiment is potent and evokes a reasonable amount of platelet deposition even for severe hemophilic blood. In contrast, fibrin deposition with healthy blood is extremely responsive to thrombin production and can increase >10 to 20-fold with TF. Based on a separate study [29], we conclude fibrin formation is more dependent on TF (and thrombin) and somewhat less dependent on platelets during the first 500 sec of clotting, once the first layer of platelets have deposited on the surface. For healthy blood, the first layer of platelets is largely sufficient for thrombin production by surface TF during the first 500 sec. After 500 sec, thrombin production is more platelet dependent since platelets facilitate thrombin feedback activation of FXIa (potentially via polyphosphate [27]. Such pathways remain to be studied in hemophilic blood in microfluidic assay.
Through flow experiments, we sought to better understand the relation between the nonlinear thrombin kinetics in a tube assay (i.e. initiation, amplification, propagation) and fibrin formation under flow. In our earlier work [23], a PTT clotting time of > 40 sec was fully predictive of poor fibrin formation under flow over collagen (no TF). Yet, recent measurements of thrombin generation during whole blood flow (using TAT assay) indicate a different kinetics may be in place under flow where thrombin flux increases linearly with time to about 0.5 × 10−12 nmole/μm2-sec by 500 sec while dense intraclot fibrin is a powerful inhibitor of thrombin [29]. Viewed from a “cell-based” perspective of rFVIIa function, the microfluidic data supports a mechanism for: (1) initiation of coagulation (TF or FXIIa), and (2) the ability to propagate FXa and thrombin generation on the platelet surface (via FIXa/FVIIIa or rFVIIa) to support platelets and especially fibrin deposition (Fig. 7).
In summary, we took a systems approach by modulating the following inputs: contact pathway engagement, the procoagulant surface trigger, and exogenous concentrations of rFVIIa. We then explored the impact of these inputs and found distinct changes in platelet deposition and fibrin generation as a function of these inputs (Figure 7, Table 2). Based on this study and our earlier study[24], endogenous FVII(a) or rFVIIa cannot fully rescue fibrin deposition via the cellular pathway alone in severe hemophilia, unless FXIIa or TF participates. However, the deconvolution of rFVIIa function on platelets in the presence of wall-bound TF requires further study as it is complicated in part by thrombin feedback mechanisms that may cross-enhance the two pathways, as was seen for net thrombin production by rFVIIa and FXIIa in an earlier study[24].
Table 2. Clotting potential of severe or moderate hemophilic A or B blood when triggered by the low or high levels of contact activation or extrinsic activation in the presence and absence of rFVIIa.
Rescue indicates return to response levels of healthy blood. Low CTI: 4 μg/mL corn trypsin inhibitor; High CTI: 40 μg/mL corn trypsin inhibitor. Mild hemophilic blood (>14–15%) performs without large functional deficit when compared to healthy blood in this assay under conditions of contact or extrinsic activation with reduced sensitivity to added rFVIIa.
| Condition | Clotting at 100 s−1 | |||||
|---|---|---|---|---|---|---|
| Phenotype | Surface | Contact pathway | rFVIIa | Platelets | Fibrin | [ref.] |
| Severe (< 3%) |
Collagen | No (high CTI) | 0 | low | none | [24] |
| No (high CTI) | + | modest rescue | none | |||
| Yes (low CTI) | 0 | Low | none | |||
| Yes (low CTI) | + | slight rescue | ~none | |||
| Moderate (3–14%) |
Collagen | No (high CTI) | 0 | modest | none | [24] |
| No (high CTI) | + | rescue | none | |||
| Yes (low CTI) | 0 | modest | very low | |||
| Yes (low CTI) | + | rescue | rescue | |||
| Severe (<3 %) |
Collagen + [TF]low |
No (high CTI) |
0 | very low | none | * |
| Collagen + [TF]low | + | rescue | rescue | |||
| Collagen + [TF]high | 0 | very low | none | |||
| Collagen + [TF]high | + | rescue | rescue | |||
| Moderate (3–14%) |
Collagen + [TF]low |
No (high CTI) |
0 | modest | modest | * |
| Collagen + [TF]low | + | modest | modest | |||
| Collagen + [TF]high | 0 | moderate | moderate | |||
| Collagen + [TF]high | + | moderate | moderate | |||
this study.
In conclusion, our microfluidic assay results from WB of FVIII or FIX-deficient patients indicate a critical role of the intrinsic tenase in driving platelet adhesion and fibrin formation on TF-laden collagen substrates at venous shear rates (Figure 7). FVIII/FIX dependent thrombin production on the platelet surface and the subsequent thrombin activation of platelets along with fibrin formation are potent, key pathways in thrombus formation under flow.
Acknowledgments
R. Li and S.L. Diamond designed the experiments. R. Li and K.A. Panckeri collected the data. K.A. Panckeri, P. Fogarty, and A. Cuker were responsible for patient enrollment. R. Li and S.L. Diamond analyzed the data. R. Li, P. Fogarty, A. Cuker, and S.L. Diamond wrote the manuscript.
P. Fogarty has received advisory board fees from Bayer Healthcare, Baxter/Baxalta, Biogen, Chugai, CSL Behring, Novo Nordisk, and Pfizer, and research support from Bayer Healthcare, Baxter/Baxalta, Biogen, CLS Behring, Pfzer and Spark Therapeutics, and is an employee of Pfizer. A. Cuker has served as a consultant for Amgen, Biogen, and Genzyme and has received research funding from Biogen and T2 Biosystems.
This work was supported by the National Institute of Health R01 HL103419 (S.L.D.), NIH UM HL120877 TACTIC Consortium, NIH U01-HL131053 (S.L.D.), and HHS Federal Region III Hemophilia Treatment Centers MCHB #H30MC24050 (A.C.).
References
- 1.Mannucci P, Tuddenham E. The Hemophilias-From Royal Genes to Gene Therapy. N Engl J Med. 2011;344:1773–9. doi: 10.1056/NEJM200106073442307. [DOI] [PubMed] [Google Scholar]
- 2.Sørensen B, Ingerslev J. Whole blood clot formation phenotypes in hemophilia A and rare coagulation disorders. Patterns of response to recombinant factor VIIa. J Thromb Haemost. 2004;2:102–10. doi: 10.1111/j.1538-7836.2004.00528.x. [DOI] [PubMed] [Google Scholar]
- 3.Carcao MD, van den Berg HM, Ljung R, Mancuso ME, PedNet Correlation between phenotype and genotype in a large unselected cohort of children with severe hemophilia A. Blood. 2013;121:3946–53. doi: 10.1182/blood-2012-11-469403. [DOI] [PubMed] [Google Scholar]
- 4.Santagostino E, Mancuso ME, Tripodi A, Chantarangkul V, Clerici M, Garagiola I, Mannucci PM. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8:737–43. doi: 10.1111/j.1538-7836.2010.03767.x. [DOI] [PubMed] [Google Scholar]
- 5.van den Berg HM, De Groot PHG, Fischer K. Phenotypic heterogeneity in severe hemophilia. J Thromb Haemost. 2007;5:151–6. doi: 10.1111/j.1538-7836.2007.02503.x. [DOI] [PubMed] [Google Scholar]
- 6.Astermark J, Donfield SM, DiMichele DM, Gringeri A, Gilbert SA, Waters J, Berntorp E. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: The FEIBA NovoSeven Comparative (FENOC) study. Blood. 2007;109:546–51. doi: 10.1182/blood-2006-04-017988. [DOI] [PubMed] [Google Scholar]
- 7.Lisman T, de Groot PG. The role of cell surfaces and cellular receptors in the mode of action of recombinant factor VIIa. Blood Rev. 2015;29:223–9. doi: 10.1016/j.blre.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Rao VM, Rapaport SI. Factor VIIa-Catalyzed Activation of Factor X Independent. Blood. 1990;75:1069–73. [PubMed] [Google Scholar]
- 9.van ’t Veer C, Golden N, Mann K. Inhibition of thrombin generation by the zymogen factor VII: implications for the treatment of hemophilia A by factor VIIa. Blood. 2000;95:1330–5. [PubMed] [Google Scholar]
- 10.Francisco S, Butenas S, Brummel KE, Branda RF, Paradis SG, Mann KG. Mechanism of factor VIIa – dependent coagulation in hemophilia blood: Presented in part at the 42nd Annual Meeting of the American Society of. Vascular. 2012;99:923–30. doi: 10.1182/blood.v99.3.923. [DOI] [PubMed] [Google Scholar]
- 11.Shibeko AM, Woodle SA, Lee TK, Ovanesov MV. Unifying the mechanism of recombinant FVIIa action: Dose dependence is regulated differently by tissue factor and phospholipids. Blood. 2012;120:891–9. doi: 10.1182/blood-2011-11-393371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Augustsson C, Persson E. In vitro evidence of a tissue factor-independent mode of action of recombinant factor VIIa in hemophilia. Blood. 2014;124:3172–4. doi: 10.1182/blood-2014-05-576892. [DOI] [PubMed] [Google Scholar]
- 13.Aljamali MN, Kjalke M, Hedner U, Ezban M, Tranholm M. Thrombin generation and platelet activation induced by rFVIIa (NovoSeven) and NN1731 in a reconstituted cell-based model mimicking haemophilia conditions. Haemophilia. 2009;15:1318–26. doi: 10.1111/j.1365-2516.2009.02073.x. [DOI] [PubMed] [Google Scholar]
- 14.Monroe DM, Hoffman M, Oliver JA, Roberts HR. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol. 1997;99:542–7. doi: 10.1046/j.1365-2141.1997.4463256.x. [DOI] [PubMed] [Google Scholar]
- 15.Feng D, Whinna H, Monroe D, Stafford DW. FVIIa as used pharmacologically is not TF dependent in hemophilia B mice. Blood. 2014;123:1754–6. doi: 10.1182/blood-2013-08-522987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fressinaud E, Sakariassen KS, Rothschild C, Baumgartner HR, Meyer D. Shear rate-dependent impairment of thrombus growth on collagen in nonanticoagulated blood from patients with von Willebrand disease and hemophilia A. Blood. 1992;80:988–94. [PubMed] [Google Scholar]
- 17.Weiss HJ, Turitto VT, Vicic WJ, Baumgartner HR. Fibrin formation, fibrinopeptide A release, and platelet thrombus dimensions on subendothelium exposed to flowing native blood: greater in factor XII and XI than in factor VIII and IX deficiency. Blood. 1984;63:1004–14. [PubMed] [Google Scholar]
- 18.Ogawa S, Szlam F, Dunn A, Bolliger D, Ohnishi T, Hosokawa K, Tanaka K. Evaluation of a novel flow chamber system to assess clot formation in factor VIII-deficient mouse and anti-factor IXa-treated human blood. Haemophilia. 2012;18:926–32. doi: 10.1111/j.1365-2516.2012.02867.x. [DOI] [PubMed] [Google Scholar]
- 19.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–97. [PMC free article] [PubMed] [Google Scholar]
- 20.Fleck A, Rao LVM, Rapaport SI. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;59:421–37. doi: 10.1016/0049-3848(90)90148-6. [DOI] [PubMed] [Google Scholar]
- 21.Swieringa F, Kuijpers MJE, Lamers MME, van der Meijden PEJ, Heemskerk JWM. Rate-limiting roles of the tenase complex of factors VIII and IX in platelet procoagulant activity and formation of platelet-fibrin thrombi under flow. Haematologica. 2015;100:748–56. doi: 10.3324/haematol.2014.116863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onasoga-Jarvis AA, Leiderman K, Fogelson AL, Wang M, Manco-Johnson MJ, Di Paola JA, Neeves KB. The Effect of Factor VIII Deficiencies and Replacement and Bypass Therapies on Thrombus Formation under Venous Flow Conditions in Microfluidic and Computational Models. PLoS One. 2013;8:e78732. doi: 10.1371/journal.pone.0078732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colace T, Fogarty PF, Panckeri KA, Li R, Diamond SL. Microfluidic assay of hemophilic blood clotting: Distinct deficits in platelet and fibrin deposition at low factor levels. J Thromb Haemost. 2014;12:147–58. doi: 10.1111/jth.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li R, Panckeri K, Fogarty P, Diamond S. Recombinant factor VIIa enhances platelet deposition from flowing haemophilic blood but requires the contact pathway to promote fibrin deposition. Haemophilia. 2015;21:266–74. doi: 10.1111/hae.12558. [DOI] [PubMed] [Google Scholar]
- 25.Neeves KB, Maloney SF, Fong KP, Schmaier A, Kahn ML, Brass LF, Diamond SL. Microfluidic focal thrombosis model for measuring murine platelet deposition and stability: PAR4 signaling enhances shear-resistance of platelet aggregates. J Thromb Haemost. 2008;6:2193–201. doi: 10.1111/j.1538-7836.2008.03188.x. [DOI] [PubMed] [Google Scholar]
- 26.Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol (Camb) 2010;2:183–92. doi: 10.1039/b919728a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu S, Travers RJ, Morrissey JH, Diamond SL. FXIa and platelet polyphosphate as therapeutic targets during human blood clotting on collagen/tissue factor surfaces under flow. Blood. 2015;126:1494–502. doi: 10.1182/blood-2015-04-641472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li R, Fries S, Li X, Grosser T, Diamond SL. Microfluidic assay of platelet deposition on collagen by perfusion of whole blood from healthy individuals taking aspirin. Clin Chem. 2013;59:1195–204. doi: 10.1373/clinchem.2012.198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu S, Lu Y, Sinno T, Diamond SL. Dynamics of thrombin generation and flux from clots during whole human blood flow over collagen/tissue factor surfaces. J Biol Chem. 2016;291:23027–35. doi: 10.1074/jbc.M116.754671. [DOI] [PMC free article] [PubMed] [Google Scholar]